
Abstract
Thioredoxin reductase (TrxR) is overpressed in many human tumors and has a key role in regulating intracellular redox balance. Recently, thioredoxin system has emerged as a valuable target for anticancer drug development. Herein we demonstrate that selenocystine (SeC) could enhance auranofin (AF)-induced A549 human lung adenocarcinoma cell apoptosis in vitro and in vivo through synergetic inhibition of TrxR1. SeC pretreatment significantly enhanced AF-induced loss of mitochondrial membrane potential (Δψm) by regulating Bcl-2 family proteins. The combined treatment with SeC and AF also resulted in enhanced intracellular reactive oxygen species (ROS) accumulation, DNA damage, and inactivation of ERK and AKT. Inhibitors of ERK and AKT effectively enhanced combined treatment-induced apoptotic cell death. However, inhibition of ROS reversed the apoptosis induced by SeC and AF, and recovered the inactivation of ERK and AKT, which revealed the importance of ROS in cell apoptosis and regulation of ERK and AKT pathways. Moreover, xenograft lung tumor growth in nude mice was more effectively inhibited by combined treatment with SeC and AF by induction of apoptosis through targeting TrxR1 in vivo. Taken together, our results suggest the strategy to use SeC and AF in combination could be a highly efficient way to achieve anticancer synergism by targeting TrxR1.
Similar content being viewed by others


Main
Non-small-cell lung cancer, accounting for about 80% of human lung cancers, is one of the leading causes of cancer deaths worldwide. Among histological types of non-small-cell lung cancer, the adenocarcinoma is the main one found in nonsmoker.1 Single-agent chemotherapy is no longer appropriate for treatment of human adenocarcinoma, because of its poor prognosis, metastasis, and drug resistance.2 Recently, combined (rather than single-agent) chemotherapy has been found to be a superior treatment strategy. Hence, searching of effective chemosensitizers that could augment the efficiency of anticancer drugs and simultaneously overcome multi-drug resistance and side effects is urgently needed.3
The mammalian thioredoxin reductases (TrxRs) are a family of selenium-containing pyridine nucleotide-disulphide oxidoreductases.4 There are currently two confirmed forms of mammalian TrxRs,TrxR1 and TrxR2, which are found in cytoplasm and mitochondria, respectively.4 TrxR is overpressed in many human tumors and has a key role in regulating intracellular redox balance, because that it can catalyze the NADPH-dependent reduction of the redox protein thioredoxin (Trx), as well as of other endogenous and exogenous compounds.5 Hence, the Trx system has emerged as a valuable target for anticancer drug development.6, 7 Auranofin (AF) has been found to react with selenol-containing residues and to inhibit TrxR activity, induce oxidative stress and lead to apoptotic cell death.8 In particular, AF exerts marked sensitive to various types of resistant cancer cells, including cisplatin-resistant phenotypes.9
Selenium, in the form of selenocysteine, has an essential part in several important redox enzymes, such as glutathione peroxidase and TrxR.5 Selenocystine (SeC), a naturally available selenoamino acid, displays broad-spectrum anticancer activities in our previous studies. SeC-induced antiproliferative effect against human cancer cells involves oxidative stress-induced apoptosis, than that of selenomethionine, Se-methyl-seleno-cysteine, selenite, and selenate.10 Despite this potency, SeC showed less toxic to human normal cells, indicating that SeC possesses great selectivity between human cancer and normal cells and displays potential application in cancer chemoprevention.10 Furthermore, SeC shows potential therapeutic effect in vivo in inhibition of A375 melanoma cells through induction of apoptosis.11
In the present study, we evaluated the ability of SeC to enhance AF-induced A549 human lung adenocarcinoma cell killing in vitro and in vivo. Mechanistic investigation revealed that, as a potential chemosensitizer, SeC could dramatically enhance the AF-induced apoptosis in A549 human lung adenocarcinoma cells by synergistically targeting TrxR1 in vitro and in vivo. This study indicates that our strategy to use SeC and AF in combination could be a highly efficient way to achieve anticancer synergism by synergetic inhibition of TrxR1.
Results
SeC enhances AF-induced cell apoptosis by initiating mitochondria dysfunction
Preliminary screening by 3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay was performed to ascertain the time and dose of the combined treatment of SeC with AF on several human cancer cells and normal cell. The results showed that treatment of human cancer cells with SeC for 48 h or AF for 6 h significantly decreased the cell viability in a dose-dependent manner (Supplementary Figure 1). In addition, the cells were pretreated with SeC (0, 3, 6, 12 or 24 h) and then co-treated with AF (6 h), the combined treatment displayed higher anti-proliferative activities on A549 cells than that of single agent (Figures 1a and b). However, single treatment or combined treatment showed less cytotoxicity toward HK-2 human normal proximal tubular cells (Supplementary Figure 1), indicating the selectivity of SeC in combination AF.

SeC enhances AF-induced growth inhibition and apoptosis in A549 cells. Experimental scheme (a) and enhanced growth inhibition against A549 cells (b) induced by SeC and AF. Preliminary screen were conducted to ascertain the optimal dose and time of combined treatment as described in section of Materilas and Methods. Briefly, A549 cells were pre-treated with 8 μM SeC (0, 3, 6, 12, and 24 h) and co-incubated with 6 μM AF for 6 h. Cell viability was determined by MTT assay. (c) SeC enhances AF-induced apoptosis in A549 cells. Cell cycle distribution was analyzed by flow cytometric analysis. Cells after treatment were collected and stained with PI solution after fixation by 70% ethanol. Apoptotic cells with hypodiploid DNA content were measured by quantifying the sub-G1 peak. (d) Caspases activities. Cells after treatment were lysed and cell proteins were used to determine caspase-3, -8, and -9 activities with a fluorescence assay kit using specific substrates. (e) Western blot analysis of expression levels of PARP and caspases in A549 cells. Cells after treatment were collected and lysed, cell proteins (40 μg) were separated by SDS-PAGE and immunoblotted with specific primary antibodies. Equal loading was confirmed by stripping immunoblots and reprobing for β-actin. All data here are expressed as means±S.D. of triplicates. All images shown here are representative of three independent experiments with similar results. Bars with different characters are statistically different at P<0.05 level
Specifically, as shown in Supplementary Figure 1, treatment of A549 cells with 8 μM SeC for 48 h reduced cell viability by 27.8%, and treatment of the cells with 6 μM AF for 6 h showed no cytotoxicity. Nevertheless, combined treatment of cells with 8 μM SeC pretreatment for 24 h and 6 μM AF co-treatment for 6 h significantly decreased the cell viability by 66.3%, implying that SeC pretreatment dramatically enhanced AF-induced growth inhibition against A549 cells. The result showed that the interaction index: γ=0.56<1, meaning that the combined effects between SeC and AF were strongly synergistic. In the phase-contrast observation, pretreatment of cells with SeC obviously enhanced AF-induced reduction in cell numbers, cell shrinkage and loss of cell-to-cell contact, which further confirm this synergistic effect (figures not shown).
To elucidate the mechanisms of combined treatment-induced growth inhibition, we examine the effect of SeC or/and AF on cell apoptosis and cell cycle distribution by flow cytometry in A549 cells. As shown in Figure 1c, treatment of cells with SeC in combination with AF resulted in a marked increase in the proportion of apoptotic cells compared with that of SeC or AF alone, as reflected by the increase in sub-G1 peaks from 14.1% (AF) and 21.9% (SeC) to 64.5% (combination). The results of flow cytometric analysis clearly indicated that SeC pretreatment noticeably enhanced AF-induced apoptosis in A549 cells. These results suggest that SeC can act as an enhancer to sensitize AF-induced cell growth inhibition against A549 cells by induction of apoptosis.
Apoptotic signaling can be conducted by two central mechanisms, the extrinsic (death receptor-mediated) and the intrinsic (mitochondrial-mediated) pathways.11 To investigate the molecular events initiated by SeC or/and AF, we first examined the contribution of caspases for the apoptotic program. As shown in Figure 1e, pretreatment of A549 cells with 8 μM SeC distinctly enhanced AF-induced activation of caspase-3, -8, and -9, indicating the activation of both the intrinsic and extrinsic apoptosis pathways. Moreover, activation of caspase-9, as the predominant initiator in mitochondria-mediated apoptotic pathway, is more prominently than that of caspase-8 induced by combined treatment, indicating that combined treatment-induced apoptosis is mainly initiated by the mitochondria-mediated apoptosis pathway. Activated caspases subsequently induced proteolytic cleavage of PARP and finally resulted in cell apoptosis. The hypothesis was further confirmed by western blotting at protein level. As shown in Figure 1f, treatment with SeC and AF in combination resulted in noticeable elevation in activation of caspases-3, -7, -8, -9, and -10, and PARP cleavage. Taken together, these results suggest that SeC as a potent chemosensitizer enhances the AF-induced cell growth inhibition and apoptosis in A549 cells mainly by initiating the mitochondria-mediated apoptosis pathway.
Enhancement of AF-induced mitochondrial dysfunction by SeC through regulation of Bcl-2 family proteins
Loss of mitochondrial membrane potential (Δψm) is catastrophic for cells and leads to the release of cytochrome c into the cytosol.12 Mitochondrial dysfunction has been shown to participate in the induction of apoptosis and has been suggested to activate the apoptotic pathway.13 Therefore, flow cytometric analysis was used to confirm whether combined treatment-induced apoptosis occurred through destroying mitochondrial homeostasis using JC-1 as a molecular probe. As shown in Figure 2a, treatment of cell with 8 μM SeC or 6 μM AF alone obviously decreased the Δψm, as evidenced by the elevation of green fluorescence from the red to green. However, 8 μM SeC and 6 μM AF in combination resulted in an enhanced depletion of Δψm from 27.4% (AF) and 31.2% (SeC) to 72.9% (combination).

SeC enhances AF-induced loss of mitochondrial membrane potential by regulating the expression of Bcl-2 family proteins. (a) SeC enhances AF-induced disruption of mitochondrial membrane potential (Δψm). Cells after treatment were stained with JC-1 and then analyzed by flow cytometry. The number presented in each histogram represents the percentage of cells that lost Δψm. (b) Western blot analysis effects of SeC in combination with AF on expression of Bcl-2 family proteins in A549 cells. (c) Time-dependent effects of SeC on expression of Bcl-2 family proteins in A549 cells. Western blotting method was used to detect the proteins expression in treated A549 cells. All experiments were done at least in triplicates
Bcl-2 family proteins could either promote cell survival (Bcl-2 and Bcl-XL) or induce apoptosis (Bax and Bad). The imbalance of pro-apoptotic and anti-apoptotic Bcl-2 family proteins will lead to the loss of mitochondrial membrane potential and finally result in the induction of apoptosis.14, 15 Hence, it was of interest to identify the Bcl-2 family members involved in the combined treatment-induced apoptosis. As shown in Figure 2b, combined treatment of SeC with AF remarkably decreased the expression rates of Bcl-2/Bax and Bcl-xL/Bad in treated A549 cells. The result demonstrated that the activation of mitochondrial apoptosis pathway was fulfilled by regulating Bcl-2 family proteins. To further determine the effect of SeC, the time-course effects of SeC on expression of Bcl-2 family proteins was evaluated. The western blotting results showed that SeC treatment for indicated time notably increased expression of Bax, but decreased expression of Bcl-2 in a time-dependent manner (Figure 2c). Taken together, these results all suggest that SeC as a potent regulator of Bcl-2 family protein enhances AF-induced mitochondrial apoptosis pathway by depletion of Δψm through regulation of Bcl-2 family protein expression.
PI3K/AKT and MEK/ERK pathways contribute to combined treatment-induced cell apoptosis
MAPKs (including JNK, p38, and ERK) and PI3K/Akt are both upstream kinases that have key roles in regulation of cell proliferation, cell growth, and survival.16, 17 Therefore, it was interested to investigate whether the MAPKs and PI3K/Akt pathways were involved in the induction of apoptosis by combined treatment using specific antibodies against the phosphorylated (activated) forms of the kinases. The results showed that SeC alone induced the obvious downregulation of phosphorylated AKT and phosphorylated ERK (Figure 3a). Moreover, SeC and AF in combination dramatically caused ERK and AKT inactivation. No alteration in phosphorylation status of p38 and JNK was detected in treated A549 cells (Supplementary Figure S2), indicating that JNK and p38 MAPK did not have important roles in regulating cell death of A549 cells in response to SeC and AF. To further confirm the role of AKT and ERK, cell viability was detected by MTT assay using specific ERK and AKT inhibitors. As shown in Figure 3b, pretreatment of A549 cells with LY2294002 (an AKT-upstream inhibitor) and U0126 (an ERK inhibitor) for 1 h markedly enhanced combined treatment-induced cell growth inhibition against A549 cells, indicating that combined treatment induced apoptotic cell death of A549 cells with ERK- and AKT-dependent manner. Enhanced activation of caspase-3 further confirmed the effects of ERK and AKT inhibitors on cell apoptosis (Supplementary Figure S3). Results in protein level further affirmed this conclusion. For instance, treatment of A549 cells with LY2294002 or U0126 caused significant inactivation of ERK and AKT, respectively. In addition, enhanced PARP cleavages were observed after treatment with these two inhibitors. Taken together, the results above suggest that SeC can act as potent chemosensitizer to enhance AF-induced growth inhibition and apoptosis of A549 cells through inhibition of PI3K/AKT and MEK/ERK pathways.

Contribution of ERK and AKT pathways to combined treatment-induced apoptosis. (a) SeC enhances AF-induced inactivation of ERK and AKT. The protein expression were detected by western blotting method. (b) ERK and AKT inhibitors enhance combined treatment-induced growth inhibition against A549 cells. Cells were pre-treated with 10 μM LY294002 (AKT-upstream inhibitor) or U0126 (ERK inhibitor) for 1 h before combined treatment. Cell viability was detected by MTT assay. (c) AKT inhibitor enhances combined treatment-induced PARP cleavage and AKT inactivation. (d) ERK inhibitor enhances combined treatment-induced PARP cleavage and ERK inactivation. All data here are expressed as means±S.D. of triplicates. All images shown here are representative of three independent experiments with similar results. Bars with different characters are statistically different at P<0.05 level
DNA damage was triggered by SeC and AF in combination
AF can induce ROS-mediated DNA damage by inhibiting TrxR activity.18 Our previous studies have showed that SeC can trigger DNA damage through ROS overproduction.11, 19 To examine whether combined treatment induce enhanced DNA damage, two DNA damage markers, phosphorylated p53 (Ser15) and phosphorylation histone (Ser139), were employed to investigate this combined effect. As expected, A549 cells treated with SeC and AF alone activated phosphorylated p53 and phosphorylated histone (Figure 4a). Moreover, SeC pretreatment significantly enhanced AF-induced DNA damage, as convinced by enhanced phosphorylated p53 and phosphorylated histone. DNA damage induced by combined treatment was further confirmed by comet assay and TUNEL-4′,6-diamidino-2-phenyindole (DAPI) co-staining. As shown in Figure 4b, treatment with SeC and AF alone notably triggered DNA damage, and SeC treatment obviously enhanced AF-induced DNA damage, as convinced by tail-DNA in single-cell level (Figure 4b). Furthermore, after TUNEL-DAPI co-staining, A549 cells exhibited enhanced apoptotic features, such as DNA fragmentation and nuclear condensation (Figure 1d). Based on the importance of SeC, a time course of SeC was investigated. As shown in Figure 4d, exposure of A549 cells to 8 μM SeC alone resulted in notable elevation of phosphorylated p53 and phosphorylated histone in a time-dependent manner. Taken together, these results indicate that SeC could enhance AF-induced apoptosis through enhancement of AF-induced DNA damage. Furthermore, time-course analysis was carried out to examine the temporal reference of different signaling in response to drug treatments. As shown in Figure 4e, phosphorylated AKT and ERK were detected as a sustained activation after treatment with SeC alone for 4 and 12 h, whereas AF treatment for 3 and 6 h only caused modest decline in phosphorylation of AKT and ERK. In addition, both SeC and AF slightly activated Ser15-p53. Interestingly, addition of AF to SeC-pretreated cells significantly enhanced the phosphorylation of p53 and inactivation of ERK and AKT. Taken together, these results indicate that SeC enhances AF-induced apoptosis through regulation of p53, ERK, and AKT pathways.

SeC enhances AF-induced DNA damage. (a) SeC enhances AF-induced phosphorylation of p53 and histone. Cells after treatment were examined via western blotting method. (b) SeC enhances AF-induced DNA damage as evidenced by comet assay at the single-cell level. Cells after treatment were immediately analyzed by Comet assay as described in section of Materials and Methods. The length of tail reflects the DNA damages in cells. (c) Representative images of DNA fragmentation and nuclear condensation in A549 cells exposed to 8 μM SeC and/or 6 μM AF. Cells cultured in chamber slides were treated with 8 μM SeC in combination with 6 μM AF and fixed with formaldehyde. Nuclear staining in A549 cells were detected by TUNEL and DAPI staining assay (magnification, × 200). (d) Time-dependent effects of SeC on expression of phosphorylated p53 and phosphorylated histone in A549 cells. Cells were treated with 8 μM SeC for indicated time and western blotting method was used to detect the protein expression. All data here are expressed as means±S.D. of triplicates. (e) Temporal changes in the expression levels of phosphorylated ERK, AKT, and p53 in cells exposed to SeC and AF. All images shown here are representative of three independent experiments with similar results
Inhibition of ROS prevents the combined treatment-induced cell apoptosis
ROS can cause DNA damage and have an important role in anticancer agents-induced apoptosis.20 Therefore, combined treatment-induced change of intracellular redox status was evaluated by measure of ROS generation using a fluorescein-labeled probe, DCFH-DA. As shown in Figure 5a, treatment of cell with SeC and AF alone induced obvious ROS accumulation in time-dependent manner, and combined treatment with SeC and AF resulted in enhanced ROS overproduction. For further evaluation of ROS importance, glutathione (GSH) and N-acetyl-L-cysteine (NAC), thiol-reducing antioxidant, were employed to examine the role of intracellular ROS in combined treatment-induced apoptotic cell death. As anticipated, addition of GSH and NAC effectively attenuated combined treatment-induced cell growth inhibition against A549 cells (Figure 5b). For instance, combined treatment with SeC (8 μM) and AF (6 μM) decreased the cell viability to 44.3%. However, pretreatment of cells with 10 mM GSH for 2 h effectively elevated the cell viability to 66.4%. GSH-ethyl-ester pre-treatment showed the similar protective effect with GSH (Supplementary Figure S4), indicating that elimination of intracellular ROS effectively block combined treatment-induced cell killing.

ROS scavengers prevent A549 cells from combined treatment-induced cell growth inhibition and apoptosis. (a) SeC enhances AF-induced intracellular ROS accumulation. Cells were washed and suspended in PBS containing 10 μM of DCFH-DA. Then cells pretreated with SeC for 2 h were co-treated with AF for another 2 h. ROS generation was determined by measuring the fluorescence intensity on microplate reader. (b) Addition of NAC and GSH weaken SeC and AF-induced cell growth inhibition against A549 cells. Cells were pre-treated with 5 mM NAC or GSH for 1 h before combined treatment. Trypan blue staining was used to quantify cell death. (c) GSH addition protects A549 cells from combined treatment-induced apoptosis. Cell apoptosis were detected by flow cytometric analysis. (d) GSH supplement suppresses combined treatment-induced activation of caspase-3. (e) GSH addition reverses combined treatment-induced apoptosis, ERK and AKT inactivation, and DNA damage. All data here are expressed as means±S.D. of triplicates. All images shown here are representative of three independent experiments with similar results. Bars with different characters are statistically different at P<0.05 level
The protective mechanism of GSH against the cytotoxic effects of SeC and AF were also investigated in this study. The flow cytometric analysis revealed that elimination of intracellular ROS reversed combined treatment-induced apoptosis (Figure 5c). For instance, combined treatment of SeC and AF triggered cell apoptosis at 57.9%. However, pretreatment with 10 mM GSH for 2 h effectively decreased the sub-G1 peak to 4.6%, suggesting that elimination of intracellular ROS effectively block combined treatment-induced cell apoptosis. Furthermore, both declined caspase-3 activity (Figure 5d) and reduced caspase-3 cleavage (Figure 5e) detected by western blotting confirmed this protective effect of GSH. For further investigation of underlying mechanism, the Bcl-2 family, DNA damage, and upstream kinases were all examined by western blotting method. Interestingly, addition of ROS scavenger distinctly attenuated combined treatment-induced DNA damage and completely reversed combined treatment-induced the inactivation of ERK and AKT (Figure 5e), indicating that ROS serves as upstream mediator in regulating induction of DNA damage and inactivation of ERK and AKT. Mitochondrial respiratory chain represents the main source of intracellular ROS. Intracellular ROS released from mitochondria was closely regulated by Bcl-2 family. Enhanced Bcl-XL expression after GSH addition was observed in Figure 5e, which indicated the possibility that GSH blocked the ROS-mediated mitochondrial dysfunction by regulating Bcl-2 family proteins. Taken together, these results demonstrate that SeC could sensitize AF-induced apoptosis in A549 cell in ROS-dependent manner.
SeC enhances the inhibitory effects of AF on TrxR1 in vitro
To clarify the mechanism of intracellular ROS accumulation, both TrxR1 activity and TrxR1 expression in vitro were evaluated. The TrxR1 activity was measured by 5,5′-dithiobis (2-nitrobenzoic) acid assay with rat liver TrxR as positive control. The results showed that incubation of the cell lysate with SeC or AF alone inhibited the TrxR1 activity in vitro in time-dependent manner. However, the TrxR1 activity in vitro was more effectively inhibited by the combined treatment of SeC and AF (Figure 6a). The results of western blot analysis revealed that both SeC alone and the combined treatment decreased TrxR1 expression in cell level, but AF alone caused no significant change in TrxR1 expression (Figure 6b). The result indicate that SeC in combination with AF synergistically inhibit TrxR1 in vitro.

Synergetic inhibition of TrxR1 activity in vitro. (a) SeC synergistically enhances AF-induced inhibition of TrxR1 activity in vitro. Cells were lysed and 100 μg protein were used to determine the TrxR1 activity in the presence of SeC or/and AF. (b) SeC decreased TrxR1 expression in vitro in A549 cells. TrxR1 expression was detected by western blotting method. All data here are expressed as means±S.D. of triplicates. All images shown here are representative of three independent experiments with similar results
SeC enhances in vivo anticancer activity of AF by targeting TrxR1
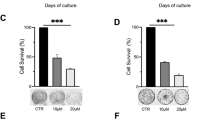
To investigate whether SeC or/and AF target TrxR1 in vivo, the therapeutic effect of combined treatment on immuno-deficient nude mice bearing A549 tumor xenografts was evaluated. After 16 days’ administration, treatment of SeC and AF alone slightly inhibited xenograft lung tumor growth. However, xenograft lung tumor growth in nude mice was more effectively inhibited by combined treatment with SeC and AF in vivo. For instance, combined treatment with SeC and AF significantly inhibited the tumor weight and tumor volume, but not affected body weight of mice (Figures 7a–d). The mechanism studies in vivo revealed that combined treatment inhibited tumor xenografts by induction of mitochondria-mediated apoptosis, as convinced by caspases activities (Figure 7e), cleaved PARP (Figure 7f), and cleaved caspase-3 staining. TrxR1 expression in tumor xenografts detected by western blotting was also evaluated, and the result indicates that SeC alone and combined treatment both reduced TrxR1 expression, but AF treatment alone caused no changes in TrxR1 expression. Furthermore, several cell markers using immunohistochemical (IHC) methods further confirmed that combined treatment with SeC and AF inhibited angiopoiesis (CD31 staining) in tumor xenografts, activated Ser15-p53 expression, and inhibited tumor xenograft cell proliferation (Ki67 staining). Taken together, these data support the conclusion that SeC can synergistically enhance AF-induced tumor growth inhibition in vivo by targeting TrxR1.

SeC enhances AF-induced growth inhibition of tumor xenografts through targeting TrxR1 in vivo. Combined treatment inhibits tumor volume (a, b) and tumor weight (c) of A549 human lung adenocarcinoma xenografts in nude mice, but do not affect body weight (d) of mice. (e) Combined treatment activates caspase in vivo. (f) Combined treatment triggers PARP clevage and inactivation of TrxR1 in vivo. (g) Staining of tumor xenografts by hematoxylin and eosin and IHC method
Discussion
TrxR, overexpressed in many tumors cells, is the central redox protein controlling the cellular thiol redox state. Inhibition of Trx system may cause dramatic imbalance between the formation and the removal of ROS.5 Consequently, the Trx system has emerged as a novel target for anticancer drug development.6 In this study, we demonstrated that SeC can target TrxR1 in vitro and in vivo to enhance AF-mediated lung cancer cell killing through activating mitochondria-mediated apoptosis pathway. And this chemosensitization effect of SeC was achieved by triggering ROS-mediated DNA damage and inactivation of AKT and ERK. To our knowledge, this is the first study to demonstrate that SeC can target TrxR1 in vitro and in vivo. Our data suggest that the strategy to use SeC and AF in combination could be a highly efficient way to achieve anticancer synergism through synergistically targeting TrxR1 in vitro and in vivo.
Induction of cancer cell apoptosis is an effective method in inhibiting tumor growth in vitro and in vivo.11 Apoptotic cells exhibit typical apoptotic feature including series of morphology and biochemical changes.21, 22 Such as cell membrane blebbing, lost of connection in cell to cell, mitochondrial depolarization, caspase activation, apoptotic body formation, chromatin condensation, and DNA fragmentation. In the present study, SeC in combination of AF trigger apoptosis of A549 cells in vitro and in vivo, and the cells exhibit typical apoptotic feature. For instance, observation by optical microscope shows that combined treatment with SeC and AF dramatically enhanced cell shrinkage, loss of cell-to-cell contact, and cell reduction in cell numbers. Cells stained by TUNEL and DAPI appear enhanced DNA fragmentation and nuclear condensation. Furthermore, A549 cells in tumor xenografts also display apoptotic feature in vivo, such as the reduced-angiopoiesis and weaken-cell proliferation were observed after combined treatment. Caspase-3 activation and PARP cleavage both confirmed combined treatment-induced apoptosis in vitro and in vivo.
Mitochondria- and death receptor-mediated apoptosis both contribute to drug-induced cancer cell apoptosis.11 Caspase, a family of cysteine acid proteases, act important role in induction of apoptosis through the enzymolysis of series of substrates.23 In this study, SeC and AF in combination synergistically induced mitochondria-mediated apoptosis of A549 cell in vitro and in vivo, because of that the activation of caspase-9 is more obvious than that of caspase-8. Mitochondrial membrane potential (Δψm) has a key role in triggering mitochondria-mediated apoptosis and the permeabilization of mitochondria is closely regulated by Bcl-2 family expression,12, 13, 14 which could be classified into two groups, pro-apoptotic proteins such as Bax and Bad, and anti-apoptotic proteins such as Bcl-2 and Bcl-XL.24 Bcl-2 family proteins could bind to the membrane of mitochondria to regulate the Δψm in response to apoptotic stimulation.15 In this study, combined treatment with SeC and AF significantly decreased Bcl-2 and Bcl-X expression, but increased Bax and Bad expression. The imbalance of Bcl-2 family expression finally resulted in the mitochondrial dysfunction and induced mitochondria-mediated apoptosis in A549 cells. These results indicate that combined treatment-induced mitochondria-mediated apoptosis of A549 cells was fulfilled by regulation of Bcl-2 family expression.
PI3K/AKT and MEK/ERK pathways have pivotal role in fundamental cellular functions such as cell proliferation, cell growth, and survival by phosphorylating a variety of substrates.25 In recent years, it has been reported that components of PI3K/AKT and MEK/ERK signaling pathways are frequently altered in human tumors, which decisively contribute to the clinic drug resistance.25, 26 Hence, much effort has been made to develop treatment strategies that target these specific signaling molecules or their downstream effectors. Recently, many evidences support that seleno compounds can induce apoptosis and enhance chemosensitivity of anticancer drugs in cancer cells with involvement of AKT or/and ERK inactivation.27, 28 However, no information about the importance of MAPK and PI3K/Akt pathways as potential targets for the chemosensitization effects of SeC is available. The present study demonstrated that SeC synergistically enhanced AF-mediated human lung cancer cell killing and apoptosis involving AKT and ERK inactivation. Inhibitors of ERK and AKT significantly enhanced the combined treatment-induced apoptosis in A549 cells, indicating that combined treatment induce apoptosis of A549 cells with ERK- and AKT-dependent manner.
Accumulative evidence support that many anticancer drugs exhibit their therapeutic effects on cancers by induction of DNA damage and activation of downstream signaling pathways.29 Ser139-Histone, a DNA damage marker, can be activated in response to DNA damage. Activated histone can transfer the signal to upstream molecules, such as ataxia telangiectasia-mutated and ataxia telangiectasia and Rad3-related proteins, followed by CHK and p53.30 p53, a tumor suppressor, is a cell cycle checkpoint protein that contributes to the preservation of genetic stability in response to DNA damage.31, 32 Activation of p53 in response to DNA damage could trigger downstream signaling molecules, such as Bcl-2 family proteins and p21, to induce cell apoptosis or cell cycle arrest.33 The present study demonstrated that combined treatment triggered enhanced DNA damage, as convinced by upregulated phosphorylated p53 and phosphorylated histone. In single-cell level, we also observed obvious DNA damage, such as nuclear condensation and DNA fragmentation. These results indicate that induction of DNA damage contributed to combined treatment-induced apoptosis in A549 cells.
Intracellular ROS overproduction may injure lipids, proteins, and DNA, and finally caused cell apoptosis.10 Increasing evidence supports that seleno compounds induce cell apoptosis involving ROS overproduction.10, 34 In combating cancer cells, selenium acts as pro-oxidant rather than antioxidant by inducing apoptosis through the generation of oxidative stress.35 In our previous studies, SeC displays broad-spectrum anticancer activity against series of human cancer cells through ROS-mediated apoptosis. And the mechanism studies reveal that inhibition of ROS accumulation can effectively prevent cancer cells from SeC-induced apoptosis.10, 11, 19 The possibility is that GSH addition plenished intracellular stores of endogenous antioxidants and reversed apoptosis. Our previous studies showed that combined treatment with SeC and 5-fluorouracil can more effectively induce A375 human melanoma cell apoptosis by ROS overproduction.36 To character the importance of ROS in combined treatment, several antioxidants, such as GSH-ethylene-ester, NAC, diphenyleneiodonium chloride, DMSO, superoxide dismutase, and catalase, were employed to investigate the ROS origin. More importantly, activities of glutathione peroxidase, glutathione reductase, and TrxR were also measured, and the results indicated that SeC can enhance 5-fluorouracil-induced apoptosis in A375 cells by ROS overproduction through regulation of intracellular redox system.36 Therefore, based on these results, we proposed that SeC enhanced AF-induced A549 cell apoptosis by ROS accumulation through dysregulation of intracellular redox system.
The present study demonstrates that SeC can act as a natural inhibitor of TrxR1 in vitro and in vivo to enhance AF-induced human lung cancer cell killing and apoptosis through ROS-mediated DNA damage and inactivation of ERK and AKT pathways. It is reported that AF could bind to the SeC-containing C-terminal and the N-terminal redox center to inhibit TrxR activity and SeC probably acted as substrates to compete with Trx.37 We speculate the possibility that SeC inhibits TrxR1 activity by competing with Trx and caused ROS accumulation, which in turn oxidized intracellular thiol-containing antioxidant agents like GSH and Trx, thus sensitized the cancer cells to AF-induced apoptosis. In addition, decreased TrxR1 expression induced by SeC may contributed to combined treatment-induced A549 cell apoptosis. This finding predicts that SeC shows promising implications in improving the therapeutic efficacy when in combination with other anticancer drugs in clinic.
In summary, we showed the ability of SeC to enhance AF-induced human lung cancer cell killing in vitro and in vivo by mitochondria-mediated apoptosis through synergistically targeting TrxR1, and this sensitization can be achieved by triggering ROS-mediated DNA damage and inactivation of ERK and AKT (Figure 8). Taken together, our results suggest that the strategy to use SeC and AF in combination could be a highly efficient way to achieve anticancer synergism through synergistically targeting TrxR1.

Proposed signal pathway. SeC enhances AF-induced intracellular ROS accumulation through synergistic inhibition of TrxR1. ROS overproduction causes DNA damage, inactivation of AKT and ERK, and triggers p53 phosphorylation, which in turn triggers mitochondrial dysfunction to amplify the apoptotic signals
Materials and Methods
Materials
SeC, AF, propidium iodide (PI), solid JC-1, DAPI, 2′,7′-dichlorofluorescein diacetate, MTT, bicinchoninic acid kit for protein determination were purchased from Sigma (St. Louis, MO, USA). Reagent kit for single-cell gel electrophoresis assay (Comet Assay) was purchased from Trevigen (Gaithersburg, MD, USA). TrxR1 Assay Kit was bought from Cayman (Ann Arbor, MI, USA). Dulbecco’s modified Eagle’s medium, fetal bovine serum, and the antibiotic mixture (penicillin-streptomycin) were purchased from Invitrogen (Carlsbad, CA, USA). Caspase-3 substrate (Ac-DEVD-AMC), caspase-9 substrate (Ac-LEHD-AFC), and caspase-8 substrate (IETD-AFC) were purchased from Calbiochem (San Diego, CA, USA). U0126 and LY294002 were obtained from Calbiochem. All of the antibodies used in this study were purchased from Cell Signaling Technology (Beverly, MA, USA). All of the solvents used were of high-performance liquid chromatography grade. The water used for all experiments was supplied by a Milli-Q water purification system from Millipore (Billerica, MA, USA).
Cell culture
A549 human lung adenocarcinoma cell line was obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium supplemented with fetal bovine serum (10%), penicillin (100 U/ml) and streptomycin (50 U/ml) at 37 °C in a humidified incubator with 5% CO2 atmosphere.
Drug treatment
Preliminary screening was conducted to ascertain the time and dose of the combined treatment of SeC with AF. Briefly, A549 cells seeded in a 96-well microplate at 8 × 103 per well were pretreated with 2.5–40 μM SeC (0, 3, 6, 12, or 24 h) and then co-treated with 0.5–20 μM AF (0, 1, 2, 4, and 6 h). After treatment, MTT assay was used to ascertain the optimal dose and time of combined treatment.
Measurement of cell viability
A549 viability was measured by MTT assay and trypan blue staining. A549 cells (8 × 103 per well ) seeded in a 96-well microplate were pre-treated with 8 μM SeC for 24 h and co-incubated with 6 μM AF for 6 h. After incubation, the cell viability were detected by MTT assay or trypan blue staining.21 Cell viability was expressed as % of control. For the trypan blue staining assay, the cells after treatment were collected by trypsinization and resuspended in PBS buffer after centrifugation. An aliquot of the cell suspension was mixed with an equal volume of a 0.4% trypan blue solution. Each cell sample was immediately transferred to a hemacytometer for counting in triplicates. Stained (dead) and unstained (viable) cells were counted by phase-contrast microscopy.
Flow cytometric analysis
The cell cycle distribution was analyzed by flow cytometric analysis as previously described.19 Briefly, cells after treatment with SeC and AF were harvested by centrifugation and washed with PBS. Cells were stained with PI after fixation with 70% ethanol at −20 °C overnight. Labeled cells were washed with PBS and then analyzed by flow cytometer. The cell cycle distribution was analyzed using a MultiCycle software (Phoenix Flow Systems, San Diego, CA, USA). The proportions of cells in G0/G1, S, and G2/M phases were represented as DNA histograms. Apoptotic cells with hypodiploid DNA contents were measured by quantifying the sub-G1 peak. For each experiment, 10 000 events per sample were recorded.
TUNEL and DAPI co-staining
The induction of DNA fragmentation and nucleus condensation in A549 cells by SeC and AF were examined by TUNEL-DAPI co-staining assay as previously described.38
Evaluation of mitochondrial membrane potential (Δψm)
The effects of SeC and AF on the cell mitochondrial membrane potential (Δψm) were examined by flow cytometry using JC-1 as specific probe.36 The percentage of the green fluorescence from JC-1 monomers was used to represent the cells that lost Δψm.
Measurement of ROS generation
The effects of SeC and AF on intracellular ROS generation were evaluated by DCF fluorescence assay as previously described.10 The ROS levels were expressed as percentage of treated cells to that of control.
Single-cell gel electrophoresis (Comet Assay)
Single-cell gel electrophoresis for detection of DNA damage induced by SeC and AF was performed using Comet assay as previously described.39 Cellular DNA was stained with SYBR GreenI (Trevigen) and visualized under a fluorescence microscope (Nikon, Eclipse E600, Tokyo, Japan).
Caspase activity assay
Activation of caspases in A549 cells exposed to SeC and AF was examined by measuring the activities of caspase-3, -8, and -9 using specific substrates (Ac-DEVD-AMC for caspase-3, Ac-IETD-AMC for caspase-8, and Ac-LEHD-AMC for caspase-9).36
Western blot analysis
The effects of SeC and AF on the expression levels of proteins associated with different signaling pathways were examined by western blot analysis.10
Determination of TrxR1 activity
Cytosolic protein was extracted for TrxR1 activity using a Thioredoxin Reductase Assay Kit (Cayman). In this assay, TrxR1 catalyzes the reduction of 5,5′-dithiobis (2-nitrobenzoic) acid with NADPH to 5-thio-2-nitrobenzoic acid (TNB2−), which generates a strong yellow color (λmax=412 nm). Briefly, 100 μg of cytosolic protein was diluted by assay buffer and incubated with 8 μM SeC or/and 6 μM AF for 10 min at room temperature. The reaction was started by addition of NADPH and DNTB, and then the TrxR1 activity was detected in 1 h by measurement of TNB2− quantity at 412 nm. The TrxR1 activity was expressed as % of control.
In vivo study
In vivo study was administrated in male nude mice. Briefly, about 1 × 106 A549 human lung adenocarcinoma cells in 100 μl serum-free medium were subcutaneously injected into the right oxter of mice. When average tumor volume reached about 50–75 mm3 after 8 days, mice were randomly divided into four groups (eight mice/group): group 1 for PBS as control; group 2 for 5 mg/kg SeC; group 3 for 2 mg/kg AF; group 4 for 5 mg/kg SeC+2 mg/kg AF. Drugs were injected every other day, caudal vein, from the first day until the sixteenth day (eight times). At the termination of the experiments, tumors were collected, photographed, and weighed. Tumor dimensions were measured with calipers and the volume was calculated using the formula: volume=l × w2/2, with l being the maximal length and w being the width. A portion of the tumors from control and treated animals was used for preparation of tumor lysate used in further analysis. Another portion of tumors were removed, fixed in 10% buffered formalin, embedded with paraffin, and sectioned. The 4-μM sections were stained with hematoxylin and eosin staining for histological observation. Protein expression in sections was examined by IHC methods. All animal experiments were approved by the Animal Experimentation Ethics Committee.
Statistical analysis
Experiments were carried out at least in triplicate and repeated three times. All data were expressed as mean±S.D. Statistical analysis was performed using SPSS statistical package (SPSS 13.0 for Windows; SPSS, Inc. Chicago, IL, USA). The difference between two groups was analyzed by two-tailed Student’s t-test. The difference between three or more groups was analyzed by one-way analysis of variance multiple comparisons. Differences with P<0.05 (*) or P<0.01 (**) were considered statistically significant. Bars with different characters are statistically different at P<0.05 level.
The growth inhibitory effects of combination treatment (synergistic, addictive, or antagonistic) was evaluated by the interaction index.40 The index, denoted by γ, is defined by the isobolar relation: a/A+b/B=γ, where A and B are the doses of SeC (alone) and AF (alone), respectively, that give the 50% of cell killing and (a, b) is the combination dose that produces the same level. The quantities in equation are obtained from the dose–response curves of drugs A, B, and the combination. If γ=1, the interaction is additive; if γ<1, it is super-additive (synergistic), and if γ>1, it is sub-additive (antagonistic).
Abbreviations
- AF:
-
auranofin
- DAPI:
-
4′,6-diamidino-2-phenyindole
- GSH:
-
glutathione
- MTT:
-
3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NAC:
-
N-acetyl-L-cysteine
- PI:
-
propidium iodide
- ROS:
-
reactive oxygen species
- SeC:
-
selenocystine
- Trx:
-
thioredoxin
- TrxR:
-
thioredoxin reductase
References
Dragnev K, You M, Wang Y, Lubet R . Lung cancer chemoprevention: difficulties, promise and potential agents? Expert Opin Investig Drugs 2013; 22: 35–47.
Mulshine JL, Hirsch FR . Lung cancer chemoprevention: moving from concept to a reality. Lung cancer (Amsterdam, Netherlands) 2003; 41 (Suppl 1): S163–S174.
Kruh GD, Goldstein LJ . Doxorubicin and multidrug resistance. Curr Opin Oncol 1993; 5: 1029–1034.
Mustacich D, Powis G . Thioredoxin reductase. Biochem J 2000; 346 (Pt 1): 1–8.
Rigobello MP, Gandin V, Folda A, Rundlof AK, Fernandes AP, Bindoli A et al. Treatment of human cancer cells with selenite or tellurite in combination with auranofin enhances cell death due to redox shift. Free Radic Biol Med 2009; 47: 710–721.
Nguyen P, Awwad RT, Smart DD, Spitz DR, Gius D . Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett 2006; 236: 164–174.
Urig S, Becker K . On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin Cancer Biol 2006; 16: 452–465.
Liu J, Akahoshi T, Namai R, Matsui T, Kondo H . Effect of auranofin, an antirheumatic drug, on neutrophil apoptosis. Inflamm Res 2000; 49: 445–451.
Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP . Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med 2007; 42: 872–881.
Chen TF, Wong YS . Selenocystine induces reactive oxygen species-mediated apoptosis in human cancer cells. Biomed Pharmacother 2009; 63: 105–113.
Chen TF, Wong YS . Selenocystine induces apoptosis of A375 human melanoma cells by activating ROS-mediated mitochondrial pathway and p53 phosphorylation. Cell Mol Life Sci 2008; 65: 2763–2775.
van Gurp M, Festjens N, van Loo G, Saelens X, Vandenabeele P . Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 2003; 304: 487–497.
Henry-Mowatt J, Dive C, Martinou JC, James D . Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 2004; 23: 2850–2860.
Harris MH, Thompson CB . The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ 2000; 7: 1182–1191.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–656.
Boldt S, Weidle UH, Kolch W . The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis 2002; 23: 1831–1838.
Lawlor MA, Alessi DR . PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 2001; 114: 2903–2910.
Reitz M, Lorei W, Lettko M . DNA single strand-breaks in human lymphocytes after therapy with antirheumatic drugs. Arzneimittelforschung 1991; 41: 1141–1144.
Chen TF, Zheng WJ, Wong YS, Yang F . Mitochondria-mediated apoptosis in human breast carcinoma MCF-7 cells induced by a novel selenadiazole derivative. Biomed Pharmacother 2008; 62: 77–84.
Mizutani H . [Mechanism of DNA damage and apoptosis induced by anticancer drugs through generation of reactive oxygen species]. Yakugaku Zasshi 2007; 127: 1837–1842.
Zhang YB, Li XL, Huang Z, Zheng WJ, Fan CD, Chen TF . Enhancement of cell permeabilization apoptosis-inducing activity of selenium nanoparticles by ATP surface decoration. Nanomedicine 2013; 9: 74–84.
Okada H, Mak TW . Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4: 592–603.
Riedl SJ, Shi Y . Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol l 2004; 5: 897–907.
Fecker LF, Geilen CC, Tchernev G, Trefzer U, Assaf C, Kurbanov BM et al. Loss of proapoptotic Bcl-2-related multidomain proteins in primary melanomas is associated with poor prognosis. J Invest Dermatol 2006; 126: 1366–1371.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–279.
Hu H, Jiang C, Li G, Lu J . PKB/AKT and ERK regulation of caspase-mediated apoptosis by methylseleninic acid in LNCaP prostate cancer cells. Carcinogenesis 2005; 26: 1374–1381.
Li S, Zhou Y, Wang R, Zhang H, Dong Y, Ip C . Selenium sensitizes MCF-7 breast cancer cells to doxorubicin-induced apoptosis through modulation of phospho-Akt and its downstream substrates. Mol Cancer Ther 2007; 6: 1031–1038.
Unni E, Koul D, Yung WK, Sinha R . Se-methylselenocysteine inhibits phosphatidylinositol 3-kinase activity of mouse mammary epithelial tumor cells in vitro. Breast Cancer Res 2005; 7: R699–R707.
Palchaudhuri R, Hergenrother PJ . DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Curr Opin Biotechnol 2007; 18: 497–503.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S . Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85.
Pellegata NS, Antoniono RJ, Redpath JL, Stanbridge EJ . DNA damage and p53-mediated cell cycle arrest: a reevaluation. Proc Natl Acad Sci USA 1996; 93: 15209–1514.
Ryan KM, Phillips AC, Vousden KH . Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 2001; 13: 332–337.
Blagosklonny MV . P53: an ubiquitous target of anticancer drugs. Int J Cancer 2002; 98: 161–166.
Zhong W, Oberley TD . Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res 2001; 61: 7071–7078.
Brozmanova J, Manikova D, Vlckova V, Chovanec M . Selenium: a double-edged sword for defense and offence in cancer. Arch Toxicol 2010; 84: 919–938.
Fan CD, Chen JJ, Wang Y, Wong YS, Zhang YB, Zheng WJ et al. Selenocystine potentiates cancer cell apoptosis induced by 5-fluorouracil by triggering reactive oxygen species-mediated DNA damage and inactivation of the ERK pathway. Free Radic Biol Med 2013; 65C: 305–316.
Rackham O, Shearwood AM, Thyer R, McNamara E, Davies SM, Callus BA et al. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radic Biol Med 2011; 50: 689–699.
Liu W, Li XL, Wong YS, Zheng WJ, Zhang YB, Cao WQ et al. Selenium nanoparticles as a carrier of 5-fluorouracil to achieve anticancer synergism. ACS Nano 2012; 6: 6578–6591.
Su J, Lai HQ, Chen JJ, Li L, Wong YS, Chen TF et al. Natural borneol, a monoterpenoid compound, potentiates selenocystine-induced apoptosis in human hepatocellular carcinoma cells by enhancement of cellular uptake and activation of ROS-mediated DNA damage. PLoS One 2013; 8: e63502.
Tallarida RJ . The interaction index: a measure of drug synergism. Pain 2002; 98: 163–168.
Acknowledgements
This work was supported by National Science and Technology Support Program (2012BAC07B05), Natural Science Foundation of China and Guangdong Province, Program for New Century Excellent Talents in University, Research Fund for the Doctoral Program of Higher Education of China and China Postdoctoral Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Fan, C., Zheng, W., Fu, X. et al. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis 5, e1191 (2014). https://doi.org/10.1038/cddis.2014.132
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/cddis.2014.132
Keywords
This article is cited by
-
Synthesis and screening for anticancer activity of two novel telluro-amino acids: 1,3-Tellurazolidine-4-carboxylic acid and tellurohomocystine
Amino Acids (2023)
-
Induction of synergistic apoptosis by tetramethoxystilbene and nutlin-3a in human cervical cancer cells
Toxicological Research (2022)
-
RETRACTED ARTICLE: Ethaselen synergizes with oxaliplatin in tumor growth inhibition by inducing ROS production and inhibiting TrxR1 activity in gastric cancer
Journal of Experimental & Clinical Cancer Research (2021)
-
Exploiting the reactive oxygen species imbalance in high-risk paediatric acute lymphoblastic leukaemia through auranofin
British Journal of Cancer (2021)
-
Anticancer effects against colorectal cancer models of chloro(triethylphosphine)gold(I) encapsulated in PLGA–PEG nanoparticles
BioMetals (2021)
