
Abstract
The capacity of CD4 T cells to protect against Mycobacterium tuberculosis (Mtb) is governed by their ability to localize to the lung site of infection. Subunit vaccine H56/CAF01, a liposome-adjuvanted fusion protein of Mtb antigens Ag85B, ESAT-6, and Rv2660, conferred durable protection and elicited polyfunctional CD4 T cells that preferentially localized to the lung parenchyma. These lung-resident T cells had reduced KLRG1 and increased CXCR3 expression, an intermediate state of Th1 differentiation that has been associated with Mtb protection. Importantly, KLGR1− CXCR3+ cells were also enriched in the lung vasculature and peripheral circulation of vaccinated animals, but not controls. Moreover, S1P1R blockade rapidly cleared this population from the blood and adoptive transfer of T cells recovered from the vasculature of vaccinated, but not control, mice efficiently trafficked into the Mtb-infected lung parenchyma. Thus, durable immunity elicited by H56/CAF01 vaccination is associated with the maintenance of circulating CD4 T cells that selectively home to the lung parenchyma.
Similar content being viewed by others


Introduction
The precise nature of a protective T-cell response to Mycobacterium tuberculosis (Mtb) infection remains unclear. Although a Th1 response is required to control infection in both animals and humans, many individuals that mount strong Th1 responses against Mtb still progress to disease.1, 2, 3 In contrast, the requirement for Th17 cells in Mtb control is less absolute and depends on the infecting Mtb strain.4, 5, 6 However, vaccine-promoted Th17 cells can improve Mtb control in animal models. BCG vaccine-primed Th17 cells improve control of Mtb infection in the absence of Th1 cells.7 Moreover, peptide vaccine-induced Th17 cells promote early Th1 cell recruitment to the lung following aerosol Mtb infection and reduce bacterial burden.4 Similarly, early Th1 and Th17 cell recruitment and improved protection were associated with a recombinant BCG vaccine that promoted an enhanced Th17 response.8 Thus, the interaction and cooperation of Th1 and Th17 cells can enhance control of Mtb infection.
Recent studies in the mouse model have also shown that protection is linked to the ability of Mtb-specific CD4 T cells to localize to the lung parenchyma9, 10 and to interact directly with infected antigen-presenting cells.11 Thus, early and sustained localization of Mtb-specific T cells within infected lung tissue is likely integral for controlling infection. However, identifying T cells that truly reside in the lung parenchyma has been confounded because the common practice of lung perfusion fails to remove many T cells that actually reside in the lung vasculature.9, 12 These vasculature-localized T cells can be identified by in vivo labeling with intravenously injected fluorescent antibody shortly before euthanasia. Using this technique, two recent studies have shown that Mtb-specific T cells in the lung parenchyma and those in the lung vasculature represent distinct populations.9, 10 Whereas, KLRG1− CD4 T cells with intermediate levels of the Th1 transcription factor T-bet preferentially localize to the lung parenchyma, terminally differentiated KLRG1+ CD4 T cells with high T-bet expression accumulate in the lung vasculature.9, 10 When transferred into T cell-deficient mice recently infected with Mtb, KLRG1− lung parenchyma-localized T cells readily re-trafficked into the lung parenchyma of recipients and controlled Mtb growth more efficiently than T cells transferred from the lung vasculature. Thus, optimal Mtb control is mediated by Th1 cells that are not terminally differentiated but retain the capacity to traffic into the parenchyma of the Mtb-infected lung.
Several studies have shown that vaccine-primed Mtb-specific CD4 T cells maintain distinct phenotypes from their infection-primed counterparts.13, 14 Indeed, sustained control of chronic Mtb infection by a subunit vaccine delivered with the cationic liposome adjuvant CAF01 is associated with a population of vaccine-specific interleukin (IL)-2-producing, KLRG1− CD4 central memory-like T cells (Tcm) that persists throughout infection.13 Given that protective, lung parenchyma-localized CD4 T cells in Mtb-infected mice share this KLRG1− phenotype, we hypothesized that vaccination-primed Tcm mediate their protective effect by establishing a stable population of lung parenchyma-homing T cells.
Here we investigated vaccine-induced CD4 T-cell responses to the H56 candidate fusion protein (Ag85B-ESAT-6-Rv2660) adjuvanted with CAF01 liposomes. H56/CAF01 provides prolonged control of infection in mice, enhances BCG-mediated protection in non-human primates, and is currently in clinical development.15, 16 We found that, compared with infection-driven responses, vaccine-specific CD4 T cells in H56/CAF01-immunized mice trafficked more efficiently to the Mtb-infected lung and preferentially localized to the parenchyma. Polyfunctional vaccine-specific CD4 T cells producing IL-2 and/or IL-17 were enhanced in both the lung parenchyma and vasculature compartments of vaccinated mice and displayed a non-terminally differentiated phenotype (KLRG1− CXCR3+). Blockade of the S1P1R pathway rapidly cleared these cells from the vasculature, suggesting that these T cells represent recent emigrants from lymphoid tissues whose presence in circulation is relatively transient. Upon adoptive transfer into infection-matched recipients, these cells preferentially trafficked into the lung parenchyma. These studies show that durable vaccine-induced immunity against Mtb infection is associated with the presence of a circulating population of lung-homing CD4 T cells.
Results
H56-specific CD4 T cells are rapidly recruited to the lung parenchyma after Mtb infection
Consistent with previous reports, H56/CAF01 vaccination significantly limited bacterial growth in mice as early as 16 days post aerosol Mtb infection and maintained enhanced control through later phases of infection, confirming its efficacy as a prophylactic vaccine (Figure 1a).15 We investigated the CD4 T-cell responses in these mice by determining how vaccine-induced T cells localized within the lungs following aerosol infection. By combining intravenous (IV) anti-CD45 staining with ex vivo intracellular cytokine staining (ICS) of H56-stimulated lung cells, we found stark differences in the localization of cytokine-producing H56-specific T cells in vaccinated and adjuvant-only controls (Figure 1b and c). In adjuvant control mice, Mtb (H56)-specific T cells were first detectable in the lung parenchyma 13 days post aerosol infection and did not peak in numbers until 28 days after infection (Figure 1c). In contrast, in H56/CAF01-vaccinated mice, H56-specific CD4 T cells were found in increased numbers in the lung parenchyma prior to infection. This population rapidly expanded during infection, reaching numbers >100-fold above unvaccinated animals and peaking 16 days post Mtb challenge (Figure 1c).

H56/CAF01-mediated protection is accompanied by accelerated recruitment of Mtb-specific T cells to the lung parenchyma. sCB6F1 mice were vaccinated and rested 6 weeks prior to aerosol Mtb challenge. (a) Total Lung CFU from adjuvant-only control (Ctrl,○) and H56/CAF01-vaccinated (H56,●) CB6F1 mice. Symbol, mean±s.d. of 5–6 mice. (b) Mice were stained i.v. with anti-CD45-FITC to distinguish parenchymal (IV–) from vasculature (IV+) cells, followed by ex vivo stimulation of lung cells with H56 protein and ICS staining for IFNγ/TNFα/IL- 2/IL-17 production to enumerate H56-specific CD4 T cells. H56-specific IFNγ production by IV+ and IV- CD4 lung cells from a control (left) and a H56/CAF01-vaccaninated (right) mouse 3 weeks after aerosol Mtb infection. (c) Total number of IV- H56-specific CD4 lung cells from adjuvant only control (○) and H56/CAF01-vaccinated (●) CB6F1 mice before and after aerosol Mtb infection, based on ICS staining for cells producing any cytokine (IFNγ,TNFα,IL-2, or IL-17). Data are pooled from three independent experiments each with 3–6 mice per time point. Symbol, mean±s.e.m. Dotted line represents assay background, i.e., mean preinfection H56-specific response in adjuvant-only control mice. *P<0.05,**P<0.01, ***P<0.001.
Vaccination enhances sustained IL-2- and IL-17-producing lung CD4 T cells
CAF01 is a Th1/Th17-promoting adjuvant,17, 18 and prophylactic H56/CAF01 immunization has been shown to promote IL-2-producing CD4 T-cell populations, including multifunctional IFNγ+TNFα+IL-2+ and TNFα+IL-2+ cells.15, 18 We therefore sought to determine the tissue localization of vaccine-specific Th1/Th17 cytokine-producing cells in the lungs of immunized mice after Mtb infection. We focused on responses to the H56 component ESAT-6 (EsxA) that is continually expressed throughout Mtb infection and primes a strong T-cell response in infected mice.10, 19 We compared these ESAT-6 responses to the T-cell response with a related Mtb protein, TB10.4 (EsxH), which has been reported to have similar immunogenicity in the mouse model but is not present in the H56 vaccine.20, 21 This strategy allowed us to monitor vaccine-induced T-cell responses (ESAT-6) while controlling for inherent differences in infection-driven responses (TB10.4) caused by reductions in bacterial burden (Figure 2a). The number of TB10.4-specific lung CD4 T cells was similar in H56/CAF01-vaccinated and control mice. TB10.4-specific CD4 T numbers in vaccinated mice were slightly above controls only at a single time point (day 16) and were then below controls at days 28 and 42, perhaps owing to the reduced bacterial load in immunized mice (Figure 1a). In contrast, the number of ESAT-6-specific CD4 T cells in the lungs of H56/CAF01-vaccinated mice was >100-fold higher than control mice at early time points after Mtb infection but became similar to control mice as infection progressed (Figure 2a).

H56/CAF01 sustains population of IL-2 and IL-17-producing vaccine-specific CD4 T cells in lung parenchyma during Mtb infection. CB6F1 mice were assayed before and after aerosol Mtb infection using i.v. staining to identify parenchymal (IV–) T cells. Lung cells were stimulated ex vivo with ESAT-61-15 (a) or TB10.471-88 (b) and stained by ICS to determine the total number of antigen-specific CD4 cells (IFNγ, TNFα, IL-2, or IL-17-producing). (c) The total number of lung parenchyma (IV-) ESAT-6-specific CD4 T cells expressing the indicated cytokine from adjuvant control (Ctrl,○) and H56/CAF01-vaccinated (●) mice is shown . Symbol, mean±s.e.m. of 3–6 mice. *P< 0.05, **P<0.01 ***P<0.001 vs. Ctrl. (d) Frequency of IV–ESAT-6-specific Lung CD4 T cells expressing each cytokine combination, based on combinatorial Boolean gating analysis, in adjuvant control (Ctrl, □) and H56/CAF01-vaccinated (▪) mice 42 days post Mtb infection. Bar, mean±s.d. of five(□) or six(▪) mice.
Further analysis of the individual cytokines produced by ESAT-6-specific T cells in the lung parenchyma revealed two distinct patterns. T cells producing the classic Th1 cytokines interferon (IFN)γ or tumor necrosis factor (TNF)α reached peak numbers by day 16 post infection in vaccinated animals, whereas control animals had increasing levels until day 28. This resulted in equivalent numbers between vaccinated and control animals at days 28 and 42 as the animals progressed into chronic infection (Figure 2b, upper panels). In contrast, IL-2- and IL-17-producing ESAT-6-specific CD4 T cells were maintained in higher numbers in the lung parenchyma of H56/CAF01-vaccinated mice relative to control mice at all time points post infection (Figure 2b, lower panels). Combinatorial Boolean gating analysis of ESAT-6-specific lung CD4 T cells during the early chronic stage further showed that the majority of IL-2- and IL-17-producing cells in vaccinated mice were polyfunctional as they co-expressed IFNγ and/or TNFα, a cytokine profile associated with H1/CAF01-mediated protection (Figure 2c).22 A similar pattern of IFNγ/TNFα- and IL-2/IL-17-producing CD4 T cells was also seen within the lung vasculature (IV+) of these mice (Supplementary Figure 1 online).
Lung CD4 T cells in H56/CAF01-vaccinated mice are less differentiated and express increased parenchymal homing markers
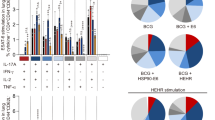
KLRG1 expression on Mtb-specific CD4 T cells has been linked to terminal differentiation, localization to the lung-associated vasculature, and decreased protective capacity during infection.9, 10, 23 H1/CAF01 vaccination promotes vaccine-specific KLRG1− CD4 T cells, but whether these vaccine-induced cells localize to the lung parenchyma or vasculature has not been examined.13 Using I-Ad:TB10.474-88 and I-Ab:ESAT-64-17 tetramers, we monitored the phenotypes of Mtb-primed versus vaccine-induced T cells, respectively, in the IV- and IV+ lung compartments during chronic Mtb infection. Whereas the relative distribution of I-Ad:TB10.474-88-tetramer+ cells with the parenchyma (IV-) versus the vasculature (IV+) was similar between H56/CAF01-vaccinated and control mice, the proportion of I-Ab:ESAT-64-17 cells that were in the parenchyma (IV-) was markedly increased in H56/CAF01-vaccinated mice (Figure 3a). In control animals, at least 80% of the ESAT-6- and TB10.4-specific CD4 T cells in the lung vasculature (IV+) expressed KLRG1, whereas the majority of lung parenchymal (IV-) CD4 T cells were KLRG1− (Figure 3a and b). The similar phenotypes of ESAT-6- and TB10.4-specific CD4 T cells in controls support the idea that that T cells recognizing these related antigens respond similarly to infection. In H56/CAF01-vaccinated mice, TB10.4-specific CD4 T cells had similar KLRG1 expression as control mice in both the parenchyma and vasculature, whereas ESAT-6-specific CD4 T cells exhibited markedly reduced KLRG1 expression in both compartments (Figure 3a and b). Thus, vaccine-induced CD4 T cells maintain a less-differentiated phenotype in both the lung parenchyma and vasculature during chronic Mtb infection.

H56/CAF01 promotes a vaccine-specific population of KLRG1-CXCR3+ CD4 T cells in the lung parenchyma and vasculature. CB6F1 mice chronically infected with Mtb were injected i.v. with anti-CD45-FITC prior to tissue harvest to characterize parenchymal (IV–) and intravascular (IV+) lung CD4 T cells in adjuvant control (Ctrl) and H56/CAF01-vaccinated (H56) mice. (a) Representive plots of KLRG1 expression and IV labelling of I-Ad:TB10.474-88 tetramer+ (left) and I-Ab:ESAT-64-17 tetramer+ (right) CD4 lung cells from a control (top) and H56/CAF01-vaccinated (bottom) mouse 6 wks post infection. (b) Compiled %KLRG1+ of I-Ad:TB10.474-88 and I-Ab:ESAT-64-17 tetramer+ CD4 T cells from the lung parenchyma (IV–, left) and vasculature (IV+, right) from (a). Bar, mean±s.d. of 4–6 mice with >15 tetramer+ events. *p<0.05, ***p<0.001 by paired t-test. (c–d) Surface staining of I-Ab:ESAT-64-17 tetramer+lung CD4 T cells from 14 wk Mtb-infected adjuvant control (top) and H56/CAF01-vaccinated (bottom) mice, located in the tissue parenchyma (IV–, left) and vasculature (IV+, right). Plots are electronically concatenated from four mice, and % expression of individual mice plotted below. Bar, mean±s.d. **P<P< 0.01, ****P<0.0001 by t-test. All data representative of ⩾2.
We also investigated the expression of Th1-associated chemokine receptors on ESAT-6-specific T cells in control and H56/CAF01-vaccinated mice during chronic Mtb infection. In adjuvant controls, CX3CR1 expression on ESAT-6-specific CD4 T cells was restricted to the KLRG1+ population, primarily located in the lung vasculature (Figure 3c, upper panels), whereas expression of the inflammatory chemokine receptor, CXCR3, was largely restricted to KLRG1− parenchyma-localized cells (Figure 3d, upper panels). In H56/CAF01-vaccinated mice, as in controls, most ESAT-6-specific T cells expressing CXCR1 were located in the lung vasculature and co-expressed KLRG1 (Figure 3c). Furthermore, the majority of ESAT-6-specific CD4 T cells in the lung parenchyma did not express KLRG1, and as in controls, a significant proportion of these cells expressed CXCR3 (Figure 3c and d lower left). Compared with controls, H56/CAF01-vaccinated mice had a significantly increased population of KLRG1− cells that co-expressed CXCR3 in the lung vasculature (Figure 3d, right), accompanied by a reciprocal significant decrease in CX3CR1+ KLRG1+ cells (Figure 3c). A similar population of KLRG1− CXCR3+ ESAT-6-specific CD4 T cells was also observed in the peripheral blood of H56/CAF01-vaccinated mice (Supplementary Figure 2). Taken together, these results show that H56/CAF01 promotes a population of vaccine-induced KLRG1−CXCR3+ CD4 T cells in the circulation that resemble the phenotype of lung parenchyma-localized CD4 T cells.
KLRG1− CD4 T cells in the lung vasculature are recent lymphoid emigrants
Given the distinct chemokine receptor expression pattern exhibited by these vaccine-induced, circulating KLRG1− CD4 T cells, we next investigated whether their requirements for entry into the blood differed from those of KLRG1+ CD4 T cells typically found in the vasculature of unimmunized Mtb-infected mice. To enable future adoptive transfer experiments, we performed these studies in C57BL/6 mice because of the availability of congenic strains on this genetic background. Mtb-infected C57BL/6 mice, either unimmunized or previously immunized with H56/CAF01, were treated with FTY720, an S1PR1 agonist that blocks egress of lymphocytes from lymphoid tissue into the blood. IV-labeling was performed 48 h later, and the phenotype of ESAT-6-specific cells in the lung vasculature was assessed. Consistent with our earlier findings in CB6F1 mice (Figure 3c and d), there was a significantly higher proportion of KLRG1− cells among ESAT-6-specific CD4 T cells in the lung vasculature (IV+) of H56/CAF01-vaccinated compared with adjuvant control mice (Figure 4). In both groups of FTY720-treated mice, however, we observed a marked and selective reduction in the number of vasculature-localized KLRG1− ESAT-6-specific CD4 T cells. Interestingly, KLRG1+ ESAT-6-specific CD4 T cells in the lung vasculature of H56-vaccinated Mtb-infected CB6F1 mice were primarily co-producers of IFNγ and TNFα (Supplementary Figure 3). In contrast, polyfunctional IL-2+IL-17+ cells were highly represented within the KLRG1− ESAT-6-specific CD4 T cell population (Supplementary Figure 3). These results support the hypothesis that circulating KLRG1− CD4 T cells induced by H56/CAF01 vaccination, but not KLRG1+ T cells that predominate in the vasculature of non-vaccinated mice, represent a population that has recently entered the blood in an S1P1R-dependent manner.

S1PR1 blockade reduces KLRG1-CD4 T cells in the lung vasculature. (a) Representative FACS plots of lung I-Ab:ESAT-64-17 tetramer+ CD4 T cells from H56/CAF01-vaccinated (lower plots) and adjuvant control (upper plots) C57BL/6 mice infected for 7 weeks with Mtb followed by 48 hr treatment with (right) or without (left) FTY720. (b) Bar graph shows the proportion of KLRG1– of I-Ab:ESAT-64-17 tetramer+ CD4 T cells in the lung vasculature (IV+) for each treatment group. Bar, mean±s.e.m. of n=3 mice per group. *P<0.05.
Circulating KLRG1− CD4 T cells readily traffic into the Mtb-infected lung parenchyma
The fact that KLRG1− CD4 T cells were quickly cleared from the lung vasculature by interfering with S1P1R signaling suggested that these cells are only transiently present in the circulation, exiting the circulation during the 48 h FTY720 treatment period. Given the phenotypic similarities between these circulating T cells and those that reside in the Mtb-infected lung parenchyma (KLRG1− CXCR3+ CX3CR1−), we hypothesized that they rapidly trafficked into the lung. To test this idea, IV+ lung CD4 T cells from Mtb-infected C57BL/6 mice (either adjuvant controls or vaccinated with H56/CAF01) were isolated and transferred into infection-matched, unimmunized congenic recipients (Figure 5a). Fourteen hours later, recipient mice were IV labeled, and lung cells were recovered. ESAT-6-specific donor cells were identified by their congenic marker and tetramer binding, and their homing into the lung parenchyma was determined by the absence of IV-labeling within the recipient mouse (Figure 5a). Consistent with our earlier findings (Figure 3a–d), ESAT-6-specific lung IV+ CD4 T cells from H56/CAF01-immunized donor mice expressed significantly less KLRG1 than those from control mice (Figure 5b). One day after transfer, only ∼30% of ESAT-6-specific donor cells from unvaccinated mice had trafficked to the lung parenchyma of recipient mice. In contrast, the vast majority (∼70%) of ESAT-6 specific donor cells from H56/CAF01-immunized mice trafficked to the Mtb-infected lung parenchyma (Figure 5c). In both groups, the donor cells that homed to the parenchyma were almost entirely KLRG1−, whereas KLRG1+ cells were largely excluded (Figure 5c). Similarly, ESAT-6-specific T cells isolated from the blood of the same H56/CAF01-vaccinated donor mice also had enhanced parenchymal homing upon transfer into an Mtb-infected recipient (Supplementary Figure 2). In a subsequent experiment to control for any variation among individual recipients, congenic CD4 T cells from infected adjuvant controls and H56/CAF01-vaccinated mice were co-transferred into the same Mtb-infected mice. Again, donor cells from H56/CAF01-vaccinated mice had superior lung parenchyma homing within each individual recipient (Supplementary Figure 4). Taken together, our findings indicate that H56/CAF01 vaccination promotes a population of circulating KLRG1− ESAT-6-specific T cells with enhanced ability to traffic into the Mtb-infected lung parenchyma. This population continually recirculates during chronic stages of Mtb infection, which may help explain the capacity of this vaccine to promote T cells that are refractory to terminal differentiation and that mediate long-term control in the murine TB model.

Vaccine-specific lung vasculature CD4 T cells from H56/CAF01-vaccinated mice efficiently traffic into the Mtb-infected lung parenchyma. (a) Schematic of adoptive transfer system to study the parenchymal homing ability of lung-circulating H56/CAF01-induced CD4 T cells. Total IV+ lung CD4 T cells were FACS-sorted from 7week Mtb-infected adjuvant control or H56/CAF01-immunized BL/6 mice (CD45.2) and transferred into infection-matched congenic recipients (CD45.1). After 14 h, the migration of donor cells into the lung parenchyma was determined by a second i.v. stain of recipients prior to lung cell isolation and surface staining. (b) KLRG1 expression of ESAT-64-17 tet+ cells from sorted IV+ donor CD4 T cells from individual adjuvant control (left) or H56/CAF01-vaccinated (right) mice prior to transfer. Bar, mean±SD of 4 mice. (c) KLRG1 expression and IV-labeling of CD45.2+ I-Ab:ESAT-64-17 tetramer+ donor CD4 T cells isolated from the lungs of recipient mice to assess trafficking and phenotype of transferred cells. Bar, mean±s.d. of 3–4 mice. Data representative of two experiments with similar results. *P<0.05, **P<0.01 by unpaired t-test.
Discussion
CD4 T cells require direct recognition of Mtb-infected macrophages to control bacterial burden at the cellular level,11 and proper localization within the infected lung is therefore a critical parameter of optimal disease control. Two recent studies have shown that KLRG1+ Mtb-specific CD4 T cells accumulate in the lung vasculature of Mtb-infected mice and do not traffic into the lung parenchyma when adoptively transferred into infection-matched recipients.9, 10 In contrast, KLRG1− parenchyma-localized lung CD4 T cells readily re-traffic into the lung parenchyma upon transfer and are more protective on a per cell basis.9, 10 In the present study, we demonstrate that H56/CAF01 vaccination promotes a sustained population of KLRG1− CXCR3+ CX3CR1− cytokine-producing CD4 T cells in the peripheral and lung vasculature that efficiently migrates into the Mtb-infected lung parenchyma.
The chemokine receptor profile of T cells is critical for their translocation into various organs, and CXCR3 is characteristically upregulated on vaccine-promoted T cells in the lung parenchyma. CXCR3 is a Th1-associated receptor that recruits cells into inflamed tissues,24 and the CXCR3 ligands (MIG, I-TAC, and IP-10) are expressed in the granulomas of Mtb-infected lungs and contribute to granuloma formation.25 Therefore, one model based on the chemokine receptor expression data presented here and elsewhere9 is that vaccine-primed T cells are recruited to the lung parenchyma upon Mtb infection in a CXCR3-dependent manner and are positioned within granulomas along a chemokine gradient. Interestingly, a significant population of KLGR1−CXCR3+ T cells was also found in the peripheral and lung vasculature of vaccinated, but not unvaccinated mice during infection, suggesting that these cells represent a circulating vaccine-promoted population. Our observation that short-term treatment with FTY270 rapidly depletes this population from the peripheral circulation has two important implications: (1) KLGR1− CXCR3+ CD4 T cells have only recently entered the blood in an S1P1R-dependent manner and (2) their presence in the circulation is transient. Our finding that KLRG1− CD4 T cells recovered from the peripheral and lung vasculature rapidly traffic into the lung parenchyma after transfer into Mtb-infected recipients provides a potential explanation for why these cells do not persist long-term in the vasculature. Thus, our results support the idea that H56/CAF01 immunization induces a population of non-terminally differentiated CD4 T cells that continually migrate into the lung during chronic Mtb infection. Expression of central lymphoid homing markers CCR7 and CD62L on KLRG1− cells in the lung vasculature was not enhanced in H56-vaccinated mice (data not shown), suggesting that these cells are not Tcm. However, future studies will be needed to determine whether these circulating T cells originate from progenitors in the peripheral lymph nodes and spleen or represent T cells re-circulating from the infected lung itself, as S1P1R blockade can also inhibit T-cell exit from non-lymphoid tissues via the lymphatics.26, 27
The level of terminal differentiation of CD4 T cells greatly influences their protective, or even pathogenic, capacity during Mtb infection. Observations by Reiley et al.23 support a developmental model whereby proliferative KLRG1− PD1+ cells terminally differentiate into IFNγ-producing KLRG1+PD-1− cells during chronic Mtb infection. More recent studies have shown that KLRG1+ CXCR3− CX3CR1+ T cells accumulate in the lung vasculature, unable to readily traffic into the infected lung tissue.9 The increased protective capacity of less terminally differentiated KLRG1− CD4 T cells may thus be dependent on their improved trafficking into the parenchyma.9, 10 Conversely, the absence of highly differentiated KLRG1+ T cells in the granuloma has been suggested to have a regulatory role in limiting disease pathology.28 KLRG1 itself may have a direct role in promoting cell differentiation and impeding protection, as KLRG1-deficient mice are more resistant to Mtb infection, and their improved bacterial control and survival are associated with increased numbers of activated CD4 T cells in the lung.29 In addition, IL-27Ra deficiency and T-bet haplo-insufficiency during Mtb infection lead to CD4 T cells with decreased KLRG1 expression, as well as increased lung parenchyma homing and improved control of infection.30 Interestingly, sustained production of IL-2 was also associated with the improved fitness of IL-27Ra-deficient CD4 T cells within Mtb-infected lungs,30 an observation consistent with the finding that only KLRG1− CD4 T cells are capable of producing IL-2 during chronic phases of Mtb infection.10 Here, we demonstrate that H56/CAF01 vaccination primes a population of CD4 T cells that is capable of sustaining this protective phenotype throughout chronic Mtb infection. Vaccine-induced Mtb-specific KLRG1− CD4 T cells were enhanced in the peripheral circulation and lung parenchyma, where they maintained the capacity to produce IL-17 and IL-2. The absence of this population in control mice is in agreement with our earlier observation that the H56/CAF01 vaccine maintains a stable population of IL-2- producing KLGR1- central/effector memory-like T cells that are completely absent during the late stages of TB infection in control mice.13 IL-17 production by vaccine-elicited CD4 T cells has previously been associated with accelerated CD4 T-cell responses into the Mtb-infected lung,4 whereas inferior long-term control of Mtb infection in IL-17RA-deficient mice suggests that a maintained IL-17-response in chronic stages of infection can contribute to disease control.5 More recently, IL-17 production has been associated with the positioning of T follicular helper -like cells within the infected lung, promoting Mtb protection.31, 32 Ongoing work is focused on evaluating typical T follicular helper markers, such as ICOS, on these parenchyma-localized T cells.
Overall, we find that H56/CAF01 vaccination promotes an early, protective lung CD4 T-cell response characterized by an expansion of KLRG1− Mtb-specific CD4 T cells with lung parenchyma-homing properties. As the infection progresses into a chronic state, an enhanced population of IL-2+ and IL-17+ ESAT-6-specific T cells is maintained in the lungs. It is tempting to speculate that this population may be sustained by the continual influx of KLRG1− CXCR3+ Mtb-specific CD4 T cells into the lungs, a population greatly enhanced in the peripheral circulation of vaccinated mice. Whether these cells are entering the lung for the first time from the peripheral lymphoid tissues or they are re-circulating and re-entering the lung, perhaps this trafficking provides a fresh population of CD4 T cells that either has not yet been exposed, or has had a hiatus from, the intense inflammatory environment of the Mtb-infected lung. This process of renewal may help keep the T cells from progressing to terminal differentiation and prevent them from becoming functionally exhausted. Determining the specific requirements for this T-cell population is crucial as we continue to unravel mechanisms of protective T-cell immunity during chronic TB infection. Moreover, as the current TB vaccine pipeline contains diverse candidates delivered in various adjuvant and live vector systems, it will be highly informative to investigate if this vaccine-induced population is common and has a role in protection promoted by these other vaccine systems.
Methods
Mice Six- to 8-wee ALB/c × C57BL/6, Harlan, Horst, Netherlands) rested 1 week were housed at Statens Serum Institut. All handling and procedures were performed in compliance with the European Community Directive 86/609 for the care and use of laboratory animals. C57BL/6, B6.PL/(84NS)CyJ (Thy1.1), and B6.SJL-PtprcaPep3bBoyJ (CD45.1) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred and maintained under specific pathogen-free conditions at the CIDR (Center for Infectious Disease Research Institute). All experimental procedures performed at CIDR involving animals were approved by the Institutional Animal Care and Use Committee.
Immunizations Mice were immunized subcutaneously three times at 2-week intervals with either CAF01 (250 mg DDA/50 mg TDB) alone or CAF01 mixed with 5ug H56 protein produced as previously described.18
Mtb infection and lung CFU enumeration Mice were rested at least 6 weeks following the final immunization before aerosol challenge with Mtb. Virulent Mtb Erdman (TMC 107, ATCC) grown to log-phase in Sauton medium (BD Pharmingen, San Diego, CA) stored at −80 °C. was suspended in PBS Tween 20 (0.05%) and aerosolized for inhalation using a Biaera exposure system controlled via AeroMP software. An average delivered dose of 50–150 colony-forming units (CFU)/animal was confirmed by CFU plating of total lung homogenates 1 day after infection. Infections for adoptive transfer experiments were performed using a Glas-Col aerosolization chamber (Terre Haute, IN, USA). Mice were infected with ∼100 CFU of Mtb Erdman, and two mice were sacrificed immediately after infection to determine the infectious dose for each experiment.
To enumerate bacteria after infection, left lung lobes from individual mice were homogenized using Milteny autoMACS dissociator and M-tubes (Miltenyi, Bergisch Gladbach, Germany), serially diluted in PBS, grown for 2 weeks at 37 °C, and CFU enumerated as described previously described.15
IV staining Anti-CD45.2 FITC (BD Biosciences, San Jose, CA), 2.5 μg per mouse was given intravenously 3 min prior to lung harvest to distinguished parenchymal-localized (IV-) and vasculature-associated (IV+) cells in CB6F1 mice. In C57BL/6 mice, anti-CD90.2 APC (BioLegend, San Diego, CA, USA), anti-CD45.2 APC (BioLegend) or CD4 FITC (RM4-4; BioLegend) was injected intravenously at 1 μg per mouse, 1 μg per mouse, and 2.5 μg per mouse, respectively, 10 min prior to lung harvest.
Tissue preparation Lungs aseptically removed from euthanized mice were perfused with cold RPMI or left un-perfused and kept at 4 °C until digested for 1 h at 37 °C with collagenase IV. Cells were forced through a 70 μm filter and washed twice before being stained directly for MHC II tetramers and surface markers or stimulated for ICS analysis.
Blood samples were taken by cardiac puncture and peripheral blood mononuclear cells isolated by density centrifugation over Lympholyte Mammal (Cedarlane Laboratories, Burlington, ON, Canada) as per manufacturer’s protocol.
Flow cytometry ICS for cytokines: A total of 1–2 × 106 lung MNCs were stimulated in vitro in V-bottom 96-well plates at 37 °C in media containing anti-CD49d (1 mg ml-1) and anti-CD28 (1 mg ml-1) without antigen or in the presence of protein or peptide antigen (5 μg ml-1) for 1 h, plus 6 h in the presence of 10 mg ml-1 brefeldin A (Sigma-Aldrich, Brønby, Denmark), after which cells were maintained at 4 °C for 2–8 h before staining.
Cells were stained for surface markers using anti–CD4-allophycocyanin (clone RM4-5; BD Pharmingen) before fixation and permeabilization using Cytoperm/Cytofix kit (BD Pharmingen) as per manufacturer’s instructions, and subsequently stained for intracellular cytokines with anti–IFN-γ PE-Cy7 (XMG1.2; BD Pharmingen), anti–TNF-α–PE (MP6-XT22; BD Pharmingen), IL-2–allophycocyanin–Cy7(JES6-5H4; BD Pharmingen), and IL-17A–PerCP–Cy5.5 (I7B7; eBiosciences, San Diego, CA). Antigen-specific cell numbers determined from Nucleocounter (ChemTec, Deerfield Beach. FL, USA) enumeration in combination with ICS data of % cytokine-producing cells after stimulation. Non-specific background cytokine values were determined for each combinatorial Boolean gate and subtracted.
Tetramer and surface staining: I-Ab:ESAT-64-17-APC, I-Ad:TB10.470-84-BV421, and I-Ab:hCLIP-APC, I-Ad:hCLIP-BV421-negative controls were provided by the NIH tetramer facility (Atlanta, USA). Cells were stained with tetramers for 30 min at 37 °C before surface staining at 4C with anti-CD4-BV786 (GK1.5, BD Pharmingen) or -allophycocyanin (RM4-5, eBiosciences), KLRG1-BV711 (2F1, BD Pharmingen) or -allophycocyanin (2F1, eBiosciences), CXCR3-PerCP/Cy5.5 (CXCR3-173 (eBiosciences), and CX3CR1-PE (Goat IgG, R&D Systems, Minneapolis MN,USA). Dead cells were excluded by the fixable viability e506 (eBioscience) or ZombieAqua (Biolegend). All Gates for surface markers are based on fluorescence-minus-one controls. All flow cytometry analyses including file concatenation and Boolean analysis was performed with FlowJo software v.X (Tree Star, Ashland, OR, USA).
FTY720 treatment Unimmunized C57BL/6 mice and H56/CAF01-immunized Thy1.1 mice were infected with 30 CFU Mtb by aerosol. After 7 weeks, half of the mice were injected i.p. with 1 mg kg−1 FTY720 (Sigma) 24 h and 48 h prior to harvest, whereas the other half served as untreated controls. Ten min prior to harvest, all mice were intravenously labeled with anti-CD4 (RM4-4) antibody to identify vasculature-resident cells. Mice were euthanized, and lungs were subsequently collected for flow cytometric analysis of Mtb-specific T cells.
Adoptive transfer of lung T cells Seven weeks post Mtb infection, H56/CAF01 or CAF01 (control)-immunized WT mice (CD45.2) were intravenously injected with 1 μg CD90.2 APC antibody. After 10 min, cardiac punctures were performed while mice were under lethal isoflurane anesthesia for the collection of peripheral blood. Peripheral blood mononuclear cells were isolated from the blood by layering over Histopaque reagent (Sigma-Aldrich). Lungs were then individually collected into HEPES buffer containing liberase blendzyme 3 (Roche) and DNAse (Sigma-Aldrich), homogenized using a gentleMacs dissociator (Miltenyi Biotec), and incubated at 37 °C for 30 min and passed through a 100 μM cell strainer to generate a single cell suspension. Red blood cells were lysed using RBC lysis buffer (eBioscience), and then CD4 T cells enriched by removal of CD19+ B cells using Magnisort negative selection magnetic beads (eBioscience). Lung vasculature-resident CD4 T cells were then sorted based on co-expression of the IV label (CD90.2 APC) and CD4 using a FACS Aria cell sorter (BD) maintained in a BSL3 facility. An aliquot was reserved to measure KLRG1 expression among IV+ESAT6+ CD4 T cells, and the remaining IV+ cells as well as the bulk eripheral blood mononuclear cells were adoptively transferred into individual congenic (CD45.1), infection-matched recipients. The following day, recipient mice were i.v. injected with 2.5 μg CD4 FITC (RM4-4), and lungs were harvested 10 min later for generation of single cell suspensions for analysis of translocation of adoptively transferred among congenically marked (CD45.2) ESAT6+ cells using flow cytometry (LSRII, BD).
Statistical analysis Prism software (v.5.6. GraphPad, San Diego, CA, USA) was used to perform all statistical analyses. CFU data and total cytokine-positive cells numbers were log-transformed to normalize variance before analysis. t-test was used to compare control and vaccinated groups, and results are described in the figure legends.
References
Andersen, P. & Urdahl, K.B. TB vaccines; promoting rapid and durable protection in the lung. Curr. Opin. Immunol. 35, 55–62 (2015).
Andersen, P. & Woodworth, J.S. Tuberculosis vaccines—rethinking the current paradigm. Trends Immunol. 35, 387–395 (2014).
Sakai, S., Mayer-Barber, K.D. & Barber, D.L. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 29, 137–142 (2014).
Khader, S.A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Freches, D., Korf, H., Denis, O., Havaux, X., Huygen, K. & Romano, M. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection. Immunology 140, 220–231 (2013).
Gopal, R. et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 10, e1004099 (2014).
Wozniak, T.M., Saunders, B.M., Ryan, A.A. & Britton, W.J. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect. Iimmun. 78, 4187–4194 (2010).
Desel, C., Dorhoi, A., Bandermann, S., Grode, L., Eisele, B. & Kaufmann, S.H. Recombinant BCG DeltaureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J. Iinfect. Dis. 204, 1573–1584 (2011).
Sakai, S. et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 192, 2965–2969 (2014).
Moguche, A.O. et al. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J. Exp. Med. 212, 715–728 (2015).
Srivastava, S. & Ernst, J.D. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191, 1016–1020 (2013).
Anderson, K.G. et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol. 189, 2702–2706 (2012).
Lindenstrom, T., Knudsen, N.P., Agger, E.M. & Andersen, P. Control of chronic mycobacterium tuberculosis infection by CD4 KLRG1- IL-2-secreting central memory cells. J. Immunol. 190, 6311–6319 (2013).
Woodworth, J.S., Aagaard, C.S., Hansen, P.R., Cassidy, J.P., Agger, E.M. & Andersen, P. Protective CD4 T cells targeting cryptic epitopes of Mycobacterium tuberculosis resist infection-driven terminal differentiation. J. Immunol. 192, 3247–3258 (2014).
Aagaard, C. et al. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17, 189–194 (2011).
Lin, P.L. et al. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J. Clin. Invest. 122, 303–314 (2012).
Lindenstrom, T., Woodworth, J., Dietrich, J., Aagaard, C., Andersen, P. & Agger, E.M. Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect. Iimmun. 80, 3533–3544 (2012).
Knudsen, N.P. et al. Different human vaccine adjuvants promote distinct antigen-independent immunological signatures tailored to different pathogens. Sci. Rep. 6, 19570 (2016).
Shi, L., North, R. & Gennaro, M.L. Effect of growth state on transcription levels of genes encoding major secreted antigens of Mycobacterium tuberculosis in the mouse lung. Infect. Iimmun. 72, 2420–2424 (2004).
Skjot, R.L. et al. Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect. Iimmun. 68, 214–220 (2000).
Dietrich, J. et al. Exchanging ESAT6 with TB10.4 in an Ag85B fusion molecule-based tuberculosis subunit vaccine: efficient protection and ESAT6-based sensitive monitoring of vaccine efficacy. J. Immunol. 174, 6332–6339 (2005).
Lindenstrom, T. et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J. Immunol. 182, 8047–8055 (2009).
Reiley, W.W. et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 107, 19408–19413 (2010).
Xie, J.H. et al. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J. Leuk. Biol. 73, 771–780 (2003).
Seiler, P. et al. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur. J. Immunol. 33, 2676–2686 (2003).
Skon, C.N., Lee, J.Y., Anderson, K.G., Masopust, D., Hogquist, K.A. & Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14, 1285–1293 (2013).
Masopust, D. & Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 13, 309–320 (2013).
Orme, I.M., Robinson, R.T. & Cooper, A.M. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat. Immunol. 16, 57–63 (2015).
Cyktor, J.C., Carruthers, B., Stromberg, P., Flano, E., Pircher, H. & Turner, J. Killer cell lectin-like receptor G1 deficiency significantly enhances survival after Mycobacterium tuberculosis infection. Infect. Iimmun. 81, 1090–1099 (2013).
Torrado, E. et al. Interleukin 27R regulates CD4+ T cell phenotype and impacts protective immunity during Mycobacterium tuberculosis infection. J. Exp. Med. 212, 1449–1463 (2015).
Monin, L., Griffiths, K.L., Slight, S., Lin, Y., Rangel-Moreno, J. & Khader, S.A. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal. Immunol. 8, 1099–1109 (2015).
Slight, S.R. et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J. Clin. Invest. 123, 712–726 (2013).
Acknowledgements
This work was supported by the European Commission through the TBVAC2020 consortium contract H2020-PHC-2014-2015-643381 and the ADITEC consortium contract FP7-HEALTH-2011.1.4-4-2808, the Lundbeck Foundation (R171-2014- 830), the Danish Research Council (DFF-1331-00063), and by grants to KBU from the National Institutes of Health (U19 AI106761 and R01 AI076327). We thank Sandra Isling and Suk-Lin Zhou for excellent technical help, as well as the staff at the experimental animal facilities at Statens Serum Institut and the vivarium staff at the Center for Infectious Disease Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PA is co-inventor of patents relating to TB fusion protein Ag85B-ESAT-6-Rv2660 and cationic liposomes as vaccines adjuvants. All rights have been assigned to the Statens Serum Institut. The remaining authors declare no conflict of interest.
Additional information
Author contributions
JSW, SBC, AOM, EAG, KBU, and PA developed the concept and designed the experiments. JSW performed, analyzed and interpreted the protection and T-cell phenotype studies in CB6F1 mice. SBC, AOM, and CRP performed, analyzed, and interpreted the trafficking and adoptive transfer studies in B6 mice. JSW created the initial draft with critical input and revision for intellectual content from SBC, KBU, and PA All authors approved of the final version.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Woodworth, J., Cohen, S., Moguche, A. et al. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol 10, 555–564 (2017). https://doi.org/10.1038/mi.2016.70
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/mi.2016.70
This article is cited by
-
Adjunctive beneficial effect of c-di-GMP, a STING agonist, in enhancing protective efficacy of TLR4-adjuvanted tuberculosis subunit vaccine formulations
Journal of Biomedical Science (2025)
-
Novel fusion protein REA induces robust prime protection against tuberculosis in mice
npj Vaccines (2025)
-
Strong immune responses and robust protection following a novel protein in adjuvant tuberculosis vaccine candidate
Scientific Reports (2025)
-
Immune correlates of protection as a game changer in tuberculosis vaccine development
npj Vaccines (2024)
-
Vaccination with Mincle agonist UM-1098 and mycobacterial antigens induces protective Th1 and Th17 responses
npj Vaccines (2024)
