
Abstract
Myh1 is a mouse deafness gene with an unknown function in the auditory system. Hearing loss in Myh1-knockout mice is characterized by an elevated threshold for the auditory brainstem response and the absence of a threshold for distortion product otoacoustic emission. Here, we investigated the role of MYH1 in outer hair cells (OHCs), crucial structures in the organ of Corti responsible for regulating cochlear amplification. Direct whole-cell voltage-clamp recordings of OHCs revealed that prestin activity was lower in Myh1-knockout mice than in wild-type mice, indicating abnormal OHC electromotility. We analyzed whole-exome sequencing data from 437 patients with hearing loss of unknown genetic causes and identified biallelic missense variants of MYH1 in five unrelated families. Hearing loss in individuals harboring biallelic MYH1 variants was non-progressive, with an onset ranging from congenital to childhood. Three of five individuals with MYH1 variants displayed osteopenia. Structural prediction by AlphaFold2 followed by molecular dynamic simulations revealed that the identified variants presented structural abnormalities compared with wild-type MYH1. In a heterogeneous overexpression system, MYH1 variants, particularly those in the head domain, abolished MYH1 functions, such as by increasing prestin activity and modulating the membrane traction force. Overall, our findings suggest an essential function of MYH1 in OHCs, as observed in Myh1-deficient mice, and provide genetic evidence linking biallelic MYH1 variants to autosomal recessive hearing loss in humans.
Similar content being viewed by others

Introduction
Hearing loss (HL) is a common sensory disorder affecting an estimated 1–2 of every 1000 newborns in the populations1,2. Hearing loss either occurs as an isolated condition (nonsyndromic, 70%) or presents with additional phenotypes (syndromic, 30%)3. Almost half of all congenital hearing loss cases have a genetic basis. Nonsyndromic hearing loss (NSHL), which accounts for 70% of genetic hearing loss, is associated with 150 loci (DFNA, DFNB, and DFNX) and more than 124 genes (Hereditary Hearing Loss Homepage; refer to Web Sources)4,5. Although several genes are associated with hearing loss, the genetic causes of ~60% of hearing loss cases remain unidentified, particularly those occurring in childhood and young adulthood6,7,8.
Large-scale mouse phenotype screening studies have revealed that at least 450 genes are required for murine hearing function9,10, suggesting that a multitude of additional genes may be linked to hearing loss in humans. These studies identified 261 deafness genes in mice that have not been detected in human patients and whose roles in the auditory system have not been elucidated, making them excellent candidates for deafness genes in humans.
Myh1, which encodes myosin heavy chain 1, is a mouse deafness gene and constitutes a crucial component of myosin II, a hexameric actin-based motor protein that transforms chemical energy into mechanical force through ATP hydrolysis. Within eukaryotic cells, MYH1 is one of the two myosin heavy chains in myosin II and plays a pivotal role in various motility-related processes11. Our research focused on elucidating the involvement of MYH1 in the auditory system and providing evidence on how MYH1 deficiency leads to hearing loss.
Prestin, a transmembrane motor protein located in the outer hair cells (OHCs) of the cochlea, promotes the electromotility of OHCs in the auditory system12,13,14. Prestin is critical for the remarkable sensitivity and frequency selectivity of the auditory pathway15. Prestin belongs to the SLC26 family of anion transporters but uniquely functions as a motor protein, driving the rapid electromotile response of OHCs16,17. This electromotility is essential for the amplification of sound vibrations, thereby greatly enhancing auditory capabilities. Mutations in the gene encoding Prestin are linked to hearing impairments, underscoring its crucial role18. However, little is known about the factors that regulate or impact the activity of prestin, which may be important in hearing.
In this study, we explored the role of Myh1 in OHCs, focusing on cellular motility, using Myh1-KO mice and assessing its association with prestin. Additionally, we presented five unrelated human cases segregating with biallelic missense variants of MYH1, providing the first genetic evidence in humans.
Materials and methods
Audiometric characterization of Myh1-knockout mice
C57BL/6N-Myh1tm1b(KOMP)Wtsi/JMmucd mice were purchased from the Mutant Mouse Resource and Research Center (MMRRC; Davis, CA, USA; Cat. # 048699-UCD) and designated Myh1−/− mice. Auditory brainstem response (ABR) thresholds were measured in a soundproof chamber using Tucker-Davis Technologies (TDT) RZ6 digital signal processing hardware and BioSigRZ software (Alachua, FL, USA). Subdermal needles (electrodes) were positioned at the vertex and ventrolateral to the right and left ears of anesthetized mice. A calibrated click stimulus (10 µs duration) or tone burst stimuli (5 ms duration) were produced at 4, 6, 8, 10, 12, 18, 24, 30, and 42 kHz using SigGenRZ software and an RZ6 digital signal processor and were delivered to the ear canal by a multifield 1 (MF1) magnetic speaker (TDT). The stimulus intensity was increased from 10 to 95 dB SPL in 5 dB steps. The ABR signals were fed into a low-impedance Medusa Biological Amplifier System (RA4LI, TDT), which delivered the signal to the RZ6 digital signal processing hardware. The recorded signals were filtered using a 0.5–1 kHz bandpass filter, and the ABR waveforms in response to 512 tone bursts were averaged. ABR thresholds for each frequency were determined using BioSigRZ software. Peak amplitudes (mV) and peak latencies (ms) were calculated from the waveform signal of the click-evoked ABRs as input/output (I/O) functions with an increasing stimulus level (20–90 dB SPL). Distortion product otoacoustic emission (DPOAE) was measured using a combination TDT microphone–speaker system. Primary stimulus tones were produced using an RZ6 digital signal processor with SigGenRZ software and delivered by a custom probe with an ER 10B+ microphone (Etymotic, Elk Grove Village, IL, USA) and MF1 speakers positioned in the ear canal. The primary tones were set at a frequency ratio (f2/f1) of 1.2 with target frequencies of 6, 8, 10, 12, 16, 18, 24, and 30 kHz. The f2 intensity levels were the same as the f1 intensity levels (L1 = L2). The sounds resulting from the primary tones were received by the ER 10B+ microphone and recorded using an RZ6 digital signal processor. The DPOAE I/O functions were determined at specific frequencies (6 and 30 kHz) with a frequency ratio (f2/f1) of 1.2 and equal intensity levels (L1 = L2). The intensity level of the primary tones was increased from 20 to 80 dB SPL in 5 dB SPL increments. Fast Fourier transform (FFT) was performed at each primary tone for the DP grams and each intensity for the I/O functions using BioSigRZ software to determine the average spectra of the two primaries, the 2f1–f2 distortion products, and the noise floors.
Scanning electron microscopy (SEM)
Inner ear samples were prepared for SEM analysis as described previously19. Inner ears were dissected from 4-week-old Myh1−/− mice and fixed with 2% paraformaldehyde containing 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h at room temperature. After fixation, the cochlear epithelium and tectorial membrane were separated and fixed overnight at 4 °C with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2 mM calcium chloride and 3.5% sucrose. The fixed samples were washed three times with 0.1 M sodium cacodylate buffer containing 2 mM calcium chloride for 20 min at 4 °C and then postfixed using an osmium tetroxide (OsO4)/thiocarbohydrazide (OTO) protocol. The samples were dehydrated using a graded ethanol series, dried using a critical point dryer (Leica EM CPD300, Wetzlar, Germany), fixed on a stub, and coated with platinum to a thickness of 20–30 nm using a sputter coater (ACE600; Leica Microsystems). The platinum-coated samples were mounted on a stub holder and imaged using a Schottky emission scanning electron microscope (JSM-IT500, JEOL, Tokyo, Japan).
Electrophysiology
Voltage-clamp experiments in mouse OHCs or human embryonic kidney (HEK) 293T cells (American Type Culture Collection, USA; #CRL-3216) were performed in standard whole-cell configurations. All recordings were performed at room temperature (22–25 °C). Microglass pipettes (World Precision Instruments, USA) were fabricated using a PP-830 single-stage glass microelectrode puller (Narishige, Japan) with a resistance of 2–5 MΩ. The liquid junction potential was rectified using an offset circuit prior to each recording. Currents were recorded using an Axopatch 700B amplifier and Digidata 1550 A interface, digitized at 100 kHz, and lowpass filtered at 10 kHz using pClamp software 10.7 (Molecular Devices, USA). The whole-cell voltage-clamp configuration was verified by measuring the series resistance to less than 10 MΩ, which was not compensated before each recording.
In the whole-cell voltage-clamp configuration, a sine wave stimulus with a 10 mV amplitude and 1 kHz frequency was overlapped with a ramp pulse protocol from −150 mV to 100 mV for a 250 ms duration. The holding potential was −70 mV. For each cell, at least two recordings were made to ensure stable measurement conditions.
The calculation of nonlinear capacitance (NLC) was adopted from previous studies17,20. The admittance (Y(ω)) of the system was determined through a spectral analysis, whereas the DC conductance (b) was extracted from the steady-state current preceding the sine wave stimulus. The circuit parameters, including capacitance (C), membrane resistance (Rm), and series resistance (Rs), were calculated as follows:
FFT is the fast Fourier transform, and Re(Y) and Im(Y) are the real and imaginary components, respectively. The capacitance was fit with a derivative of the Boltzmann equation as follows:
where Vh is the maximal activation voltage, Qmax is the maximal charge transfer between the plasma membrane, α is the slope factor for the voltage-dependent charge transfer, z is the charge valence, e is the electron charge, k is Boltzmann’s constant, and T is the absolute temperature. An in-house Python script was used for the NLC calculation and analysis.
Preparation of the organ of Corti for electrophysiology
Organs of Corti were acutely isolated from mice at 21–28 days after birth. The animals were anesthetized with isoflurane (Sigma-Aldrich) and euthanized via decapitation. The bone surrounding the apical turn of the cochlea was removed, and the apical turn of the organ of Corti was carefully detached using forceps and subsequently separated from the lateral cochlear wall, stria vascularis, modiolus, and tectorial membrane. The whole dissection procedure was performed in a standard extracellular solution containing 144 mM NaCl, 5.8 mM KCl, 10 mM HEPES, 5.6 mM glucose, 0.7 mM NaH2PO4, 1.3 mM CaCl2, 0.9 mM MgCl2, and 10 mM sorbitol, adjusted to pH 7.4 using NaOH. The cochlea was immobilized onto a 12 mm diameter coverslip with a stainless steel pin and Sylgard and mounted on an upright microscope ECLIPSE FN1 (Nikon, Japan).
Solutions
The whole-cell voltage-clamp experiment was conducted using a standard extracellular bath solution containing 144 mM NaCl, 5.8 mM KCl, 10 mM HEPES, 5.6 mM glucose, 0.7 mM NaH2PO4, 1.3 mM CaCl2, 0.9 mM MgCl2, and 10 mM sorbitol, adjusted to pH 7.4 with NaOH. The standard pipette solution used to record electromotility contained 140 mM CsCl, 10 mM HEPES, 1 mM EGTA, 3 mM Mg-ATP, and 1 mM MgCl2 and was adjusted to pH 7.2 with KOH.
Immunofluorescence staining and whole-mount assay
The cochleae were isolated from Myh1+/+, Myh1+/−, or Myh1−/− C57BL/6 mice at postnatal Day 30, fixed with 4% paraformaldehyde overnight, washed with phosphate-buffered saline (PBS), decalcified with 10% ethylenediaminetetraacetic acid (pH 7.2) for 7 days, transferred to a series of gradient ethanol solutions, immersed in dimethylbenzene, and finally embedded in paraffin for sectioning. For immunostaining, the sections were deparaffinized in xylene and rehydrated in gradient ethanol solutions. The cells were incubated in blocking buffer containing 10% donkey serum and 1% bovine serum albumin for 1 h at room temperature. The samples were incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. After being washed with PBS, the samples were incubated with secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) for 30 min at room temperature, washed, and covered with mounting medium and cover slips. Images were obtained with an LSM 780 microscope (Carl Zeiss). Anti-MYH1 (LSbio, LS-C346145), anti-MYO7A (Santa Cruz Biotechnology, sc-74516), and anti-Prestin (Santa Cruz Biotechnology, sc-293212) antibodies were obtained from commercial sources. Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies and DAPI were obtained from Invitrogen.
OHC circuit modeling
We assumed that three channels are responsible for major ionic conductance in OHCs: a mechanoelectrical transducer (MET) channel, a small conductance K+ channel, and a large conductance K+ channel, IK,n, whose molecular identity is KCNQ4.
The closed OHC circuit can be expressed using the following equation:
We implemented the NLC of the OHC for the membrane capacitance as follows:
The gating of the MET channel was as follows21,22:
The gating of the Ks and Kn channels was as follows23:
The power of electromotility was calculated using the following equation:
The differential equation was solved by an in-house Python script using the NumPy and SciPy modules24,25.
Research subjects
This study was approved by the Institutional Review Board of Severance Hospital, Yonsei University Health System (IRB #4-2015-0659). We obtained written informed consent from individuals with hearing loss for their participation in this study and for the publication of their clinical data. The participants were enrolled in the Yonsei University Hearing Loss (YUHL) cohort based on the following inclusion criterion: hearing loss without congenital CMV infection or other medical diseases primarily affecting hearing function. We prescreened for pathogenic biallelic variants in SLC26A4 and GJB2 or copy number variations (CNVs) in STRC, which are the most common genetic causes in East Asians26,27. In total, probands from 437 families with no pathogenic variants in these three known genes were enrolled in the YUHL cohort for exome sequencing, and the majority had NSHL. Among those 437 families, 151 (151/437, 34.5%) presented either pathogenic variants or CNVs within deafness-causing genes. Otherwise, the exome sequencing analysis of 286 families revealed no other pathogenic variants in known hearing loss genes.
Subject evaluation
Pure tone and speech audiograms were obtained for all patients and their affected and unaffected family members. Pure tone air (250–8000 Hz) and bone conduction (250–4000 Hz) thresholds were measured using a clinical audiometer in a double-walled audio booth. The degree of hearing loss was determined by averaging the air conduction thresholds at 500, 1000, 2000, and 4000 Hz. Steady-state auditory responses were also determined for young babies. Temporal bone computed tomography and magnetic resonance imaging were performed to evaluate inner ear abnormalities.
Whole-exome sequencing (WES)
WES was performed as described previously. Briefly, genomic DNA was extracted from the peripheral blood or saliva samples of the affected individuals and their parents (where available) and subjected to exome capture using an Agilent SureSelect V5 enrichment capture kit (Agilent Technologies). The enriched library was then sequenced on an Illumina NovaSeq 6000 sequencing platform (151 bases, paired-end), and sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19) using CLC Genomic Workbench (version 9.5.3) software (Qiagen). After alignment, variants with a minimum coverage of two were subjected to a variant calling process using the “Basic Variant Caller” function in CLC and then annotated by Variant Effect Predictor. Single nucleotide polymorphisms reported in the dbSNP (version 147) database were excluded. Nonsynonymous variants or variants located within splice sites were then subjected to further genetic evaluation.
Three-dimensional structural modeling
ColabFold and AlphaFold2 were used to construct the structures of the WT and mutant MYH1 proteins28,29. Due to the flexibility of the C-terminal region in MYH1 and the limited total length of the input sequences, we focused only on the head domain of MYH1, which corresponds to residues 1–843. The ColabFold program was downloaded from the website https://github.com/sokrypton/ColabFold and run locally. For protein modeling, the number of cycles was set to six, five models were predicted, and the top-ranked structure was selected for further analysis. The model type variable was AlphaFold2-multimer-v2. ChimeraX software was used for the three-dimensional visualization of the proteins30.
Molecular dynamics simulations
The predicted structures of WT and mutant MYH1 were used as templates for molecular dynamics (MD) simulations. The system was initially neutralized and solvated in a 150 mM KCl environment using a CHARMM-GUI web server31,32. The final system was contained in an ~120 × 120 × 240 Å3 box, with ~105,000 water molecules and a total of ~330,000 atoms. All the simulations were performed with the CHARMM36m force field and GROMACS software33,34.
The particle mesh Ewald algorithm was used to evaluate the long-distance electrostatic interactions. The van der Waals interactions for a smoothing function were between 10 and 12 Å. The system was minimized for 5000 steps using the steepest descent algorithm. Then, 125 ps equilibration steps were applied to the protein, and the restraints were gradually reduced to zero. A complete electronic evaluation was performed every 2 fs using the SHAKE algorithm35. The pressure and temperature were, respectively, maintained at 1 atm and 298 K throughout the simulation using the Nosé–Hoover Langevin piston method and Langevin dynamics, respectively36,37. No anisotropic cell fluctuations were observed. For each system, a 100 ns simulation was performed, and snapshots were saved at 100 ps intervals for further analysis.
The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were calculated using Visual Molecular Dynamics (VMD) software38. For principal component analysis (PCA), protein trajectories were extracted every 100 ps and calculated using Bio3D software in R39,40.
Surface biotinylation and immunoprecipitation
The experiments were performed as described previously41,42. Anti-Myc (sc-40) and anti-aldolase A1 (sc-12059; Santa Cruz Biotechnology, Dallas, TX, USA) antibodies were purchased from commercial sources. Surface biotinylation was performed with 0.3 mg/mL EZ-Link Sulfo-NHS-SS-Biotin and streptavidin (Thermo Fisher Scientific). Immunoprecipitation and coimmunoprecipitation (co-IP) were performed using EZview Red Anti-FLAG M2 Affinity Gel (Sigma) and Pierce Anti-c-Myc Magnetic Beads (Thermo Fisher Scientific).
Immunoblotting
Immunoblotting was performed using primary antibodies, including anti-MYH1 (LS-C346145, LSBio), anti-actin (sc-18262, Santa Cruz Biotechnology), anti-Myc (2276T, Cell Signaling Technology, Danvers, MA, USA) and anti-Flag (14793T, Cell Signaling Technology) antibodies at a 1:1000 dilution, followed by the corresponding anti-isotype secondary antibodies (Santa Cruz Biotechnology) at a 1:2000 dilution. The signals were visualized using the SuperSignal West Pico Kit (Thermo Fisher Scientific).
Fourier transform traction microscopy (FTTM)
Traction force microscopy was performed as described previously43,44,45. Briefly, transfected/untransfected COS-7 cells were plated sparsely on a polyacrylamide (PAA) gel coated with fibronectin (Thermo Fisher Scientific) and allowed to adhere and stabilize for 24 h. Phase contrast and fluorescence images of microbeads (Molecular Probes, Eugene, OR, USA) embedded near the gel apical surface of each cell were captured, while a fluorescence image of the same region and focal plane in the gel was captured after cell detachment with trypsin (traction-free reference). The microbead displacement between the two images was measured by identifying the coordinates of the peak of the cross-correlation function43,44. The traction field was computed using the elastic property of the gel (Young’s modulus of 20 kPa and Poisson’s ratio of 0.48), and the displacement field was computed using a MATLAB code.
Statistical analysis
All the quantitative data were obtained from at least three independent experiments. Significant differences between two groups were examined using Student’s t-tests or two-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons using GraphPad Prism 7.0 software (GraphPad Software, San Diego, CA, USA). Two-sided p values of <0.05 were considered significant.
Results
Myh1 −/− mice exhibit an impaired hearing phenotype due to outer hair cell dysfunction
We generated Myh1−/− mice (Fig. 1a) to understand the role of MYH1 in the inner ear; the mice were viable and fertile but smaller in size with a significantly lower body weight than Myh1+/− and Myh1+/+ mice at postnatal Day 30 (Fig. 1b). We determined the hearing sensitivity of Myh1−/− mice by measuring the ABR threshold in response to click stimuli of mixed broadband or pure tone sounds at individual frequencies on postnatal Day 30 (Fig. 1c, d). The thresholds for both click and pure tone stimuli at all frequencies were significantly increased in the Myh1−/−, but not in the Myh1+/− mice, compared with those in the Myh1+/+ mice (Fig. 1d). ABR wave I amplitudes, which reflect the summed responses of auditory nerve fibers, were significantly lower in response to click stimuli up to 70 dB in the Myh1−/− mice than in the Myh1+/+ or Myh1+/− mice (Fig. 1e). However, the ABR wave I amplitudes evoked by higher-intensity stimuli (>80 dB) did not differ significantly among any of the genotypes. These results suggest that the machinery by which inner hair cells (IHCs) stimulate spiral ganglion neurons is intact and that the reduced hearing sensitivity indicated by elevated ABR thresholds is likely due to OHCs failing to act as cochlear amplifiers. We tested this hypothesis by analyzing DPOAE thresholds, which reflect the cochlear amplification function of OHCs. Compared with Myh1+/+ or Myh1+/− mice, Myh1−/− mice presented significantly higher DPOAE thresholds at all frequencies (Fig. 1f). In addition, DPOAE amplitudes were significantly reduced at both low (6 kHz) and high (30 kHz) frequencies in Myh1−/− mice (Fig. 1g, h), suggesting that OHC dysfunction is the main cause of hearing loss in Myh1−/− mice. SEM revealed that the stereociliary bundles of OHCs in 4-week-old Myh1−/− mice were comparable to those in Myh1+/− mice (Supplementary Fig. 1). Consistently, the imprints on the undersurface of the tectorial membrane, which represent connections between the tallest stereocilia and tectorial membrane, also appeared normal in the Myh1−/− mice (Supplementary Fig. 1). These results suggest that Myh1 is required for normal hearing, most likely by contributing to OHC function rather than stereociliary morphology. Furthermore, these findings confirm that MYH1 is linked to hearing loss with autosomal recessive inheritance in both humans and mice.

a Overall appearance of Myh1+/+, Myh1+/−, and Myh1−/− mice at postnatal Day 30. b Body weights of the Myh1+/+, Myh1+/−, and Myh1−/− mice. c Tone-pip ABR thresholds, d click ABR thresholds, and e ABR I/O functions of the Myh1+/+, Myh1+/−, and Myh1−/− mice at postnatal Day 30. f DPOAE thresholds and g DPOAE I/O functions at 6 kHz and h 30 kHz. b–h Myh1+/+ (black, n = 5), Myh1+/− (blue, n = 7), and Myh1−/− (red, n = 7) mice. The data are presented as the means ± SEMs. Statistical comparisons were performed using two-way ANOVA with Bonferroni correction for multiple comparisons in (b–h). **P < 0.01 and ***P < 0.001.
Myh1 is expressed in multiple cell types in the cochlea
In mammals, at least ten different myosin heavy chain isoforms, which are encoded by different genes of the MYH family, have been described in striated, smooth, and nonmuscle cells11. Given that the expression of Myh1 has not been described in the inner ear, we performed immunofluorescence staining and a whole-mount assay using cochleae from mice at postnatal Day 30. Myh1 was detected in multiple cochlear structures and strongly expressed in spiral ganglion neurons with axonal sprouts and supporting cells around hair cells (Fig. 2a). Whole-mount imaging of the organ of Corti revealed that Myh1 was expressed not only in supporting cells but also in inner and OHCs (Fig. 2b).

a Cross section of the inner ear from C57BL6/N mice at postnatal Day 30. MYH1, red; MYO7A, green. b Whole-mount images of the inner ears of C57BL6/N mice at postnatal Day 30. MYH1, red; phalloidin, green.
Prestin activity is lower in outer hair cells from Myh1 −/− mice
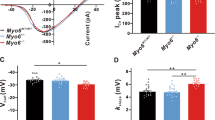
Prestin, encoded by the SLC26A5 gene, is a motor protein responsible for OHC electromotility and serves as the molecular basis of cochlear amplification12,13. We hypothesized that MYH1 may regulate prestin activity based on several lines of evidence: (i) the elevated DPOAE threshold in Myh1−/− mice with intact tip links (Fig. 1 and Supplementary Fig. 1), indicating aberrant OHC electromotility; (ii) the direct interaction of MYH1 with the actin cytoskeleton; and (iii) the importance of the actin organization and distribution for electromotility in OHCs14,46. We thus examined prestin activity in the OHCs of postnatal Day 21 Myh1+/+ and Myh1−/− mice using whole-cell voltage-clamp recordings. Surprisingly, when stimulated with the sine wave ramp pulse protocol (Fig. 3a), the current amplitude shift was greatly compromised in the OHCs of the Myh1−/− mice (Fig. 3b, c). We then calculated the NLC from the current traces using the FFT method, as the NLC component is considered a direct measurement of prestin activity. Indeed, normalized NLC activity was impaired in Myh1−/− mice, which was ~1/3 that of wild-type mice (Fig. 3d). Fitting the normalized NLC to the derivative of the Boltzmann equation allowed us to compare the maximal activation voltage (Vh) and maximal charge transfer; Vh was significantly elevated, whereas the charge transfer was significantly decreased (Fig. 3e, f).

a Input voltage-clamp pulse protocol used for stimulating prestin in the outer hair cells. A 1 kHz sine wave with a 10 mV amplitude was simultaneously applied at −150 to 100 mV for a 250 ms duration. b, c Representative traces of Myh1+/+ and Myh1−/− mouse outer hair cell recordings at postnatal Day 21. d Voltage-normalized nonlinear capacitance (NLC) relationship of Myh1+/+ (n = 6) and Myh1−/− (n = 8) mouse outer hair cells. The bell-shaped curve was fit to the derivative of the Boltzmann equation. e Vh values calculated from the outer hair cells of Myh1+/+ and Myh1−/− mice. f Normalized charge transfer density calculated from the outer hair cells of Myh1+/+ and Myh1−/− mice. Data are presented as the means ± SEMs. Statistical comparisons were performed using two-sample independent t-tests in (e) and (f). *P < 0.05 and **P < 0.01.
Myh1 accounts for the majority of the electromotility power
We next aimed to determine the extent of the decrease in electromotility power caused by impaired prestin activity in OHCs from Myh1−/− mice. For this purpose, we constructed a model of the OHC circuit based on previous studies21,22,23 (Supplementary Table 4). We hypothesized that three ion channels are responsible for the majority of ion conduction in OHCs, namely, the MET channel, small conductance voltage-activated K+ channel, and large conductance voltage-activated K+ channel, which corresponds to IK,n and is mediated by KCNQ447,48 (Fig. 4a). We also implemented NLC caused by prestin activity based on our electrophysiology experiments (Fig. 3). When the MET channel was stimulated with a 1000 Hz wave of 32 nm in amplitude (Fig. 4b), we observed fluctuations in the membrane potential with the corresponding frequency (Fig. 4c). Interestingly, although the resting membrane potential was near −73 mV, which was similar to the reported value49, the fluctuation in the membrane potential was greater in Myh1−/− OHCs, implying that the Myh1−/− OHC plasma membrane was less capable of storing electric charges. The gating of MET and Kn channels was similar between Myh1+/+ and Myh1−/− OHCs (Fig. 4d, e), whereas Ks channel gating was greater in Myh1−/− OHCs. Surprisingly, the membrane capacitance of Myh1−/− OHCs was significantly lower in both the resting and fluctuating states (Fig. 4f). We next calculated the power generated by the capacitance of Myh1+/+ and Myh1−/− OHCs, which was significantly lower in Myh1−/− OHCs than in Myh1+/+ OHCs during the stimulation timeline (Fig. 4g). We next expanded our calculations across different stimulation frequencies from 100 to 10,000 Hz and observed that Myh1−/− OHCs presented decreased electromotility power across all frequencies (Fig. 4h). When normalized to Myh1+/+ OHCs, Myh1−/− OHCs had 16.2% and 29.8% electromotility power at 100 and 1000 Hz, respectively (Fig. 4i).

a Illustration of the outer hair cell (OHC) circuit. gMET conductance of the mechanoelectrical transducer (MET) channel, gK,s conductance of the small conductance K+ channel, gK,n conductance of IK,n, corresponding to the KCNQ4 channel, Cm cell membrane capacitance, EP endocochlear potential, EK K+ reversal potential, Vm basolateral membrane potential. b Stimulation of stereocilia at 1000 Hz for 200 ms. c Membrane potential oscillation by 1000 Hz stereocilia stimulation. d MET channel gating induced by 1000 Hz stereocilia stimulation. e K+ channel gating induced by 1000 Hz stereocilia stimulation. f Membrane capacitance oscillation induced by 1000 Hz stereocilia stimulation. g OHC electromotility power at 1000 Hz stereocilia stimulation. h OHC electromotility power across different frequencies of stereocilia stimulation. i Normalized electromotility power of Myh1−/− OHCs compared to Myh1+/+ OHCs across different frequencies. In (b–h), Myh1+/+ OHCs are indicated by black lines, and Myh1−/− OHCs are indicated by red lines.
MYH1 variants identified from patients with hearing loss
We performed WES of a cohort of 437 families with hearing loss. A total of 151 of 437 families (34.6%) were molecularly diagnosed with either single nucleotide variants and insertions and deletions or CNVs in genes linked to nonsyndromic HL (https://hereditaryhearingloss.org/) or syndromic HL (https://www.omim.org/) (Supplementary Fig. 2a)50,51,52,53,54,55,56. Pathogenic variants in any of the previously reported genes were not detected in 286 families. After applying the variant filtering criteria (Supplementary Information; Materials and Methods, Supplementary Fig. 3, and Supplementary Table 1), we identified eight different biallelic variants in MYH1 (myosin heavy chain 1; MIM: 16073) in five probands who were diagnosed with a recessive form of sensorineural hearing loss. The coverages of the targeted regions in the exome data for the 15-fold read depth were 94.4% (YUHL100-21), 96.9% (YUHL105-21), 97.4% (YUHL110-21), 92.5% (YUHL162-21) and 98.0% (YUHL624-21), and the average read depths were 54×, 68×, 73×, 94×, and 57×, respectively. These individuals also had biallelic variants in other genes (Supplementary Table 2). The eight identified variants of MYH1 were missense variants, which are rare in both gnomAD v2.1.1 (https://gnomad.broadinstitute.org/) and Korean Variant Archive 2 (KOVA2; approximately n = 5305 individuals), with minor allele frequencies <0.00557. Sanger sequencing confirmed that the variants cosegregated with hearing loss as an autosomal recessive trait in five families (Supplementary Fig. 4). MYH1 variants, including c.582G > C (p.Gln194His) and c.2977A > G (p.Ile993Val), c.2231A > C (p.Lys744Thr) and c.5416C > A (p.Gln1806Lys), c.1379T > C (p.Ile460Thr) and c.2438 A > T (p.Glu813Val), were present in the compound heterozygous state in probands from the YUHL110, YUHL105, and YUHL100 families, respectively (Supplementary Figs. 2b and 4). In the YUHL100 family, the affected status was segregated into another family member (YUHL100-22) with a shared audiological phenotype (Supplementary Fig. 5). Two homozygous variants of MYH1, c.2495C > G (p.Pro832Arg) and c.4617A > T (p.Gln1539His), were identified and segregated in the YUHL162 and YUHL624 families (Supplementary Figs. 2b and 4). However, according to the ACMG guidelines58, the clinical significance of MYH1 variants identified in the cohort remains uncertain and requires further evaluation of the patients’ phenotypes (Supplementary Table 1). According to the International Mouse Phenotyping Consortium (IMPC; https://www.mousephenotype.org/), Myh1-knockout mice also present decreased bone mineral density, abnormal vocalization, decreased grip strength, and decreased urine creatinine levels9.
Among the probands from the five unrelated families, YUHL110-21 was a 17-year-old female with moderate-to-severe sensorineural hearing loss (Supplementary Fig. 2c) that occurred in the first decade of her life. She did not have any cochlear anomalies (Supplementary Fig. 2d). In dual-energy X-ray absorptiometry, the z scores for the femur neck and total hip (−1.4 and −1.6, respectively) indicated osteopenia, according to the 2013 Pediatric ISCD Positions (Supplementary Table 2)59. Her urine creatinine level (45.6 mg/dL) was normal, indicating no muscle myopathy. YUHL105-21 was a 3-year-old male with congenital hearing loss, which was more severe in the left ear (Supplementary Fig. 2c). He had no perinatal medical problems and no syndromic features. He did not have any inner ear anomalies, but had severe-to-profound hearing loss in the more severe ear (Supplementary Fig. 2d). He underwent cochlear implantation in the left ear, and the performance was good, with an aided threshold of 20–30 dB HL. Although his muscular development was normal and his urine creatinine level was within the normal range (68.7 mg/dL), the z scores determined from dual-energy X-ray absorptiometry were −1.6 and −1.7 for the femoral neck and total hip, respectively, indicating osteopenia (Supplementary Table 3). YUHL162-21 was a 2-year-old male with bilateral severe-to-profound congenital hearing loss, which manifested more severely in the left ear (Supplementary Fig. 2c). With no other clinical manifestations, the patient presented with bilateral cochlear anomalies classified as incomplete partition type I (Supplementary Fig. 2d; red arrows). Nonetheless, the laminar layers in the cochlea were well preserved, and residual hearing was detected bilaterally (Supplementary Fig. 2d). The individual underwent cochlear implantation in the left ear, with a good outcome (as high as a score of 6 in the categorized auditory performance) 1 year after implantation. As YUHL162-21 was only 2 years old, the muscular phenotype and bone mineral density values needed to be determined at follow-up. YUHL100-21 was a 63-year-old male with bilateral moderate hearing loss of uncertain onset (Supplementary Fig. 2c). Given that he had poor pronunciation and speaking proficiency, hearing loss was presumed to have initiated during childhood. His word recognition scores were 68 and 56% for the right and left ears, respectively. He did not exhibit any other syndromic features. YUHL100-22 was a 75-year-old female with moderate-to-severe hearing loss (Supplementary Fig. 5) and poor speech intelligibility (word recognition scores of 20 and 60% for the right and left ears, respectively). In the simplex YUHL624 family, YUHL624-21 had profound bilateral congenital sensorineural hearing loss (Supplementary Fig. 2c). YUHL624-21 also had cochleovestibular anomalies such as incomplete partition type I. She underwent cochlear implantation surgery at the age of 8 months, in which a cerebrospinal fluid gusher was noted. The outcome after 6 months was successful, with a score of 5 for categorized auditory performance. Based on these clinical manifestations, myopathy may be present at the subclinical level, as decreased muscle tone may lead to a decreased bone mineral density; however, the severity of osteopathy in this patient was clinically uncertain.
MYH1 variants are structurally different from the wild-type protein
MYH1 is a myosin heavy chain that is predominantly expressed in adult skeletal muscle and contains an N-terminal globular head, SH3, and IQ domains, followed by a C-terminal coiled-coil rod-like tail60. Among the identified missense variants, three affected the motor domain, two affected the IQ domain, and the other three affected the tail domain of MYH1 (Fig. 5a). We questioned whether the MYH1 variants identified in individuals with hearing loss were structurally different from the wild-type protein. No crystal structure is available for MYH1; therefore, ColabFold28 and AlphaFold229 were used to predict the structure of the head domain of MYH1 corresponding to residues 1–843 (Fig. 5b). The overall prediction score was reasonably high for the majority of the residues, except for the N- and C-terminal loops, which were expected to be flexible (Supplementary Fig. 6). We then questioned whether MYH1 variants were dynamically different from the wild-type protein, and thus modeled p.Gln194His, p.Ile460Thr, p.Lys744Thr, p.Glu813Val, and p.Pro832Arg variants with AlphaFold229. Each protein was subsequently solvated, energetically minimized, and equilibrated for MD simulations. We included the benign variants p.Met360Ile, p.Tyr435His, and p.Met688Val as negative controls for the structural analysis, which were selected based on their allele frequency in the gnomAD database. During a 100 ns simulation, the root mean square deviation (RMSD) was lower in the last 50 ns for the p.Gln194His, p.Ile460Thr, p.Lys744Thr, and p.Glu813Val variants than for the wild-type protein or benign variants, indicating that these variants were structurally rigid (Fig. 5c). Notably, the p.Pro832Arg variant presented higher RMSD values in the last 50 ns of the simulation. A subsequent RMSF analysis of each amino acid revealed no major differences, except for the C-terminus of the p.Pro832Arg variant, which was consistent with its high RMSD and variant location (Fig. 5d). PCA of wild-type MYH1 revealed two major clusters, which were analyzed via the K-means clustering algorithm (Supplementary Fig. 7a). A comparison of these clusters highlighted the head domain loop and linker domain as the primary sources of structural differences (Supplementary Fig. 7b). Plotting the principal components of each variant against the wild-type protein revealed that the p.Gln194His, p.Ile460Thr, p.Lys744Thr, and p.Glu813Val variants clustered around a single cluster, indicating reduced flexibility of the linker domain (Fig. 5e).

a Location of the MYH1 variants in terms of the domain structure and amino acid conservation across other vertebrates. b The overall architecture of MYH1 (residues 1–843) predicted using AlphaFold2. The variant residues are indicated. c Root mean square deviation (RMSD) analysis of a 100 ns MD simulation of the wild-type and mutant MYH1 proteins. d Root mean square fluctuation (RMSF) analysis of the last 50 ns. e Principal component analysis (PCA) of MD simulation trajectories.
MYH1 variants do not increase prestin activity compared to the wild-type protein
Next, we investigated whether MYH1 variants are also associated with impaired prestin activity. We expressed prestin with or without the MYH1 head domain in HEK293T cells. Unlike mock-transfected cells, cells expressing prestin presented robust signs of electromotility (Fig. 6a). Interestingly, prestin and MYH1 double-transfected cells exhibited greater electromotility than the prestin-only transfected cells (Fig. 6a). Vh was lower in the cotransfected cells, whereas the charge transfer was significantly higher (Fig. 6b, c). These results were consistent with the OHCs from the Myh1+/+ and Myh1−/− mice (Fig. 3e, f). Next, we performed co-IP experiments in HEK293T cells to investigate the physical interaction between prestin and MYH1 (Fig. 6d). Interestingly, co-IP using both MYC and FLAG beads revealed a strong physical interaction between prestin and MYH1. While their direct interaction remains unclear, both prestin and MYH1 are likely regulated by actin cytoskeletal networks. We next transfected the mutant MYH1 with prestin and measured its electromotility. The p.Ile460Thr and p.Lys744Thr variants showed similar electromotility to the prestin-only transfected cells, whereas the p.Glu813Val variant exhibited moderately decreased electromotility compared with the wild-type MYH1 protein, and the electromotility of the p.Gln194His variant was similar to that of the wild-type protein (Fig. 6e). Vh and charge transfer also showed similar tendencies (Fig. 6f, g).

a Voltage-normalized nonlinear capacitance (NLC) relationship of overexpressed prestin with or without MYH1 in HEK293T cells. b Vh values measured in cells overexpressing prestin with or without MYH1. c Normalized charge transfer density measured in cells overexpressing prestin with or without MYH1. d Immunoprecipitation (IP) and coimmunoprecipitation (co-IP) of prestin- and MYH1-transfected HEK293T cells. The input lanes represent 1/10 of the immunoprecipitated cell lysate. e NLC relationship between overexpressed prestin and mutant MYH1. f Vh values and normalized charge transfer density measured from cells overexpressing prestin and mutant MYH1. g Normalized charge transfer density measured from cells overexpressing prestin and mutant MYH1. The data are presented as the means ± SEMs. Statistical comparisons were performed using two-sample independent t-tests in (b) and (c). *P < 0.05 and ****P < 0.0001.
MYH1 regulates the traction force of the cell membrane
We then investigated whether the increased prestin activity observed in the presence of MYH1 results from the direct modulation of prestin itself. However, the surface biotinylation of prestin was not affected by MYH1 in overexpressing HEK293T cells, and prestin localization in OHCs was similar between Myh1+/+ and Myh1−/− mice (Supplementary Fig. 8). Thus, we hypothesized that MYH1 may affect the plasma membrane traction force, potentially exerting physical control over prestin activity. This hypothesis was derived from the observation that other factors that regulate membrane tension, such as cholesterol, have been shown to influence prestin activity61,62. We transfected COS-7 cells with wild-type MYH1 or its variants (p.Gln194His, p.Ile460Thr, p.Lys744Thr, and p.Glu813Val) to explore their mechanical effects. Wild-type MYH1 and its variants were observed throughout the COS-7 cell body, except for the nucleus (Supplementary Figs. 9 and 10), whereas other members of the myosin superfamily associated with hearing loss, such as MYO3A and MYO15A, have been shown to be localized to the tips of filopodia in COS-7 cells and stereocilia in hair cells63,64. We measured large-scale traction stress at the interface between adherent cells and the elastic matrix using Fourier transform force microscopy to assess how wild-type and variant MYH1 proteins function in cell mechanics (Fig. 7a). The wild-type and variant MYH1 proteins did not differ in terms of cellular expression, and the overexpression of MYH1 proteins in COS-7 cells induced cellular traction regardless of the variant type. However, the average traction stress generated differed between the wild-type and variant MYH1 proteins (Fig. 7b). Traction stress did not differ significantly between the cells expressing the wild-type and p.Glu813Val MYH1 proteins, whereas the cells expressing the other three MYH1 proteins (p.Gln194His, p.Ile460Thr, and p.Lys744Thr) presented a remarkable decrease in traction force (Fig. 7b). The cells transfected with the MYH1 variants showed a similar level of spreading to the cells transfected with wild-type MYH1 when loaded onto a fibronectin-coated polyacrylamide gel with a Young’s modulus of ~20 kPa (Fig. 7c).

a Representative phase contrast (top panel) and traction field images (bottom panel) of COS-7 cells adhered to an elastic polyacrylamide gel coated with fibronectin (Young’s modulus of 20 kPa with a Poisson’s ratio of 0.48). Scale bar = 50 µm. b Projected cell area measured in cells overexpressing wild-type or mutant MYH1. c Root mean square (RMS) traction measured in cells overexpressing wild-type or mutant MYH1. The data are presented as the means ± SEMs. Statistical comparisons were performed using two-way ANOVA with Bonferroni correction for multiple comparisons in (b) and (c). ns not significant; *P < 0.05; **P < 0.01.
Discussion
In this study, we successfully provided evidence for the function of Myh1 in murine hearing by performing functional studies in vivo. Consistent with previous publications9, Myh1−/− mice presented impaired auditory function and noticeably elevated DPOAE thresholds across all frequencies. Our research revealed broad expression of MYH1 across multiple cochlear structures, including supporting cells and hair cells. We speculated that the hearing loss in Myh1−/− mice might be due to OHC dysfunction and performed electrophysiology studies on electromotility to elucidate the role of MYH1 in OHCs. Through our analysis, we confirmed that reduced prestin activity was a major pathological characteristic in Myh1−/− mice. Additionally, based on our simulations, MYH1 was responsible for at least 70% of the electromotility power generated in OHCs. These results establish an essential role for MYH1 in OHCs, which are responsible for cochlear amplification throughout the auditory system65.
Prestin is a motor protein responsible for generating electromotility in OHCs12,13,14. While in vitro expression of prestin successfully reproduces the electrophysiological properties of OHC electromotility, many extrinsic factors are involved in amplifying prestin activity. Examples include cholesterol, the actin–myosin complex, and temperature46,61,66. In addition to these factors, we propose that MYH1 also physically affects prestin activity by increasing the traction force of the cell membrane. Our experiments revealed that MYH1 interacts with prestin, resulting in several-fold higher prestin activity in OHCs than in the heterogenous expression system (Figs. 3 and 5). Although coexpression of MYH1 with prestin increased charge transfer and lowered Vh, similar to the OHCs, the absolute NLC amplitude was still higher in the OHCs. We assume that unidentified molecular mechanisms specific to OHCs other than MYH1 do exist. These pathways could be due to the additional beta subunit, stiffening of the OHC membrane14, or prestin alignment for electromotility amplification62.
Here, we report five patients with autosomal recessive hearing loss in whom biallelic missense variants of MYH1 were identified. According to the ACMG/AMP guidelines, the identified missense variants are of uncertain significance, and further genetic confirmation is required. Notably, three of the five patients also exhibited osteopenia, mirroring the reduced bone mineral density observed in Myh1-KO mice. Furthermore, variants of MYH1 in the head domain caused decreases in prestin activity (p.Ile460Thr, p.Lys744Thr, and p.Glu813Val) and traction force (p.Gln194His, p.Ile460Thr, and p.Lys744Thr) in vitro compared with those of wild-type MYH1. These results suggest the hypomorphic nature and potential deleteriousness of the MYH1 variants in the head domain.
The gnomAD database contains 197 MYH1 alleles with loss-of-function variants, each having minor allele frequencies <0.005, suggesting the presence of potential disease-causing alleles67. In addition, compound heterozygous events of MYH1 missense variants are expected to be rare. According to variant co-occurrence data in gnomAD, out of 125,748 individuals, 434 carried more than two MYH1 variants with an MAF of <0.005. Among these 434 individuals, 43 were unphased, 390 carried MYH1 variants in a cis configuration, and only 1 carried variants in a trans configuration. Compound heterozygous missense MYH1 variants were reported as a likely pathogenic cause of childhood absence epilepsy in a child who also exhibited global developmental delays68. However, whether the child also has hearing loss is unclear. Data provided in gnomAD revealed two South Asian individuals homozygous for one of the MYH1 variants (c.868C > T; p.Arg290Cys), suggesting that this variant is unlikely to cause a severe phenotype, such as global developmental delay.
Other members of the myosin heavy chain family are associated with hearing loss in humans. MYH9 (MIM: 1607750) and MYH14 (MIM: 608568), both of which are class II myosin heavy chains in nonmuscle cells, are implicated in autosomal dominant hearing loss, specifically DFNA17 (MIM: 603622) and DFNA4A (MIM: 600652), respectively69,70. In addition, unconventional myosins are implicated in hearing loss. Mutations in MYO3A (MIM: 606808) are responsible for DFNB30 (MIM: 607101)64, and MYO6 (MIM: 600970) is mutated in DFNA22 (MIM: 606346) and DFNB37 (MIM: 607821)71,72. Mutations in MYO7A (MIM: 276903) are implicated in two forms of NSHL, DFNA11 (MIM: 601317) and DFNB2 (MIM: 600060)73,74. MYO15A (MIM: 602666) is also implicated in DFNB3 (MIM: 600316)75. Mutations in MYO15A are currently the third most common cause of hearing loss, with ~192 mutations identified to date. These mutations mostly cause prelingual severe-to-profound hearing loss, although some result in postlingual progressive hearing loss, likely depending on the nature of the mutation76. By combining our phenotype evaluation in mice with the IMPC database, we found that Myh1-knockout mice presented not only hearing impairment but also a decreased bone mineral density, abnormal vocalization, reduced grip strength, and lower urinary creatinine levels. While individuals with recessive MYH1 variants did not consistently display all these phenotypes, three probands (YUHL624-21, YUHL110-21, and YUHL105-21) presented a decreased bone mineral density, which could be explained by a common myosin-related pathogenesis. Additional population-based studies are necessary to establish a strong genotype–phenotype correlation for MYH1.
In conclusion, our study elucidates the novel function of MYH1 in hearing. Its correlation with prestin activity implies that MYH1 plays an essential role in cochlear amplification and auditory functions in mice.
Data availability
All relevant data are included in the main manuscript and Supplementary Information. The Python script for simulating the OHC circuit is publicly available at https://github.com/doctroh/outer-hair-cell/tree/main.
References
Venkatesh, M., Moorchung, N. & Puri, B. Genetics of non syndromic hearing loss. Med. J. Armed Forces India 71, 363–368 (2015).
Vos, B., Noll, D., Pigeon, M., Bagatto, M. & Fitzpatrick, E. M. Risk factors for hearing loss in children: a systematic literature review and meta-analysis protocol. Syst. Rev. 8, 1–7 (2019).
Schrijver, I. Hereditary non-syndromic sensorineural hearing loss: transforming silence to sound. J. Mol. Diagn. 6, 275–284 (2004).
Angeli, S., Lin, X. & Liu, X. Z. Genetics of hearing and deafness. Anat. Rec. 295, 1812–1829, https://doi.org/10.1002/ar.22579 (2012).
Shearer, A. E., Hildebrand, M. S., Schaefer, A. M., & Smith, R. J. H. Genetic Hearing Loss Overview. In M.P. Adam (Eds.) et. al., GeneReviews®. University of Washington, Seattle (1999).
Investigators, G. P. P. et al. 100,000 genomes pilot on rare-disease diagnosis in health care - preliminary report. N. Engl. J. Med. 385, 1868–1880, https://doi.org/10.1056/NEJMoa2035790 (2021).
Shearer, A. E. et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 107, 21104–21109, https://doi.org/10.1073/pnas.1012989107 (2010).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450, https://doi.org/10.1007/s00439-016-1648-8 (2016).
Bowl, M. R. et al. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nat. Commun. 8, 886, https://doi.org/10.1038/s41467-017-00595-4 (2017).
Ingham, N. J. et al. Mouse screen reveals multiple new genes underlying mouse and human hearing loss. PLoS Biol. 17, e3000194, https://doi.org/10.1371/journal.pbio.3000194 (2019).
Masters, T. A, Kendrick-Jones, J & Buss, F. Myosins: domain organisation, motor properties, physiological roles and cellular functions. in Handbook of Experimental Pharmacology Vol. 235, 77–122 (2017). https://doi.org/10.1007/164_2016_29.
Zheng, J. et al. Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155, https://doi.org/10.1038/35012009 (2000).
Liberman, M. C. et al. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–304, https://doi.org/10.1038/nature01059 (2002).
He, D. Z., Jia, S. & Dallos, P. Prestin and the dynamic stiffness of cochlear outer hair cells. J. Neurosci. 23, 9089–9096, https://doi.org/10.1523/JNEUROSCI.23-27-09089.2003 (2003).
Takahashi, S., Zhou, Y., Kojima, T., Cheatham, M. A. & Homma, K. Prestin’s fast motor kinetics is essential for mammalian cochlear amplification. Proc. Natl. Acad. Sci. USA 120, e2217891120, https://doi.org/10.1073/pnas.2217891120 (2023).
Ge, J. et al. Molecular mechanism of prestin electromotive signal amplification. Cell 184, 4669–4679.e4613, https://doi.org/10.1016/j.cell.2021.07.034 (2021).
Bavi, N. et al. The conformational cycle of prestin underlies outer-hair cell electromotility. Nature 600, 553–558, https://doi.org/10.1038/s41586-021-04152-4 (2021).
Liu, X. Z. et al. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum. Mol. Genet. 12, 1155–1162, https://doi.org/10.1093/hmg/ddg127 (2003).
Koo, H. Y. et al. Follistatin regulates the specification of the apical cochlea responsible for low-frequency hearing in mammals. Proc. Natl. Acad. Sci. USA 120, e2213099120, https://doi.org/10.1073/pnas.2213099120 (2023).
Lindau, M. & Neher, E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflug. Arch. 411, 137–146, https://doi.org/10.1007/BF00582306 (1988).
Altoe, A., Pulkki, V. & Verhulst, S. The effects of the activation of the inner-hair-cell basolateral K+ channels on auditory nerve responses. Hear. Res. 364, 68–80, https://doi.org/10.1016/j.heares.2018.03.029 (2018).
Dierich, M. et al. Optimized tuning of auditory inner hair cells to encode complex sound through synergistic activity of six independent K+ current entities. Cell Rep. 32, 107869, https://doi.org/10.1016/j.celrep.2020.107869 (2020).
Mammano, F. & Ashmore, J. F. Differential expression of outer hair cell potassium currents in the isolated cochlea of the guinea-pig. J. Physiol. 496, 639–646, https://doi.org/10.1113/jphysiol.1996.sp021715 (1996).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272, https://doi.org/10.1038/s41592-019-0686-2 (2020).
Harris et al. Array programming with NumPy. Nature 585, 357–362, https://doi.org/10.1038/s41586-020-2649-2 (2020).
Jung, J. et al. Genetic predisposition to sporadic congenital hearing loss in a pediatric population. Sci. Rep. 7, 45973, https://doi.org/10.1038/srep45973 (2017).
Chang, M. Y. et al. Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing. Mol. Cells 38, 781–788, https://doi.org/10.14348/molcells.2015.0078 (2015).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682, https://doi.org/10.1038/s41592-022-01488-1 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, https://doi.org/10.1038/s41586-021-03819-2 (2021).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82, https://doi.org/10.1002/pro.3943 (2021).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865, https://doi.org/10.1002/jcc.20945 (2008).
Brooks, B. R. et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614, https://doi.org/10.1002/jcc.21287 (2009).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413, https://doi.org/10.1021/acs.jctc.5b00935 (2016).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695 (1985).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, https://doi.org/10.1016/0263-7855(96)00018-5 27–38 (1996).
Grant, B. J., Rodrigues, A. P., ElSawy, K. M., McCammon, J. A. & Caves, L. S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696, https://doi.org/10.1093/bioinformatics/btl461 (2006).
Grant, B. J., Skjaerven, L. & Yao, X. Q. The Bio3D packages for structural bioinformatics. Protein Sci. 30, 20–30, https://doi.org/10.1002/pro.3923 (2021).
Gee, H. Y., Tang, B. L., Kim, K. H. & Lee, M. G. Syntaxin 16 binds to cystic fibrosis transmembrane conductance regulator and regulates its membrane trafficking in epithelial cells. J. Biol. Chem. 285, 35519–35527, https://doi.org/10.1074/jbc.M110.162438 (2010).
Oh, K. S. et al. Overlooked KCNQ4 variants augment the risk of hearing loss. Exp. Mol. Med. 55, 844–859, https://doi.org/10.1038/s12276-023-00976-4 (2023).
Butler, J. P., Tolic-Norrelykke, I. M., Fabry, B. & Fredberg, J. J. Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 282, C595–C605, https://doi.org/10.1152/ajpcell.00270.2001 (2002).
Wang, N. et al. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am. J. Physiol. Cell Physiol. 282, C606–C616, https://doi.org/10.1152/ajpcell.00269.2001 (2002).
An, S. S., Fabry, B., Trepat, X., Wang, N. & Fredberg, J. J. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am. J. Respir. Cell Mol. Biol. 35, 55–64, https://doi.org/10.1165/rcmb.2005-0453OC (2006).
Zhang, X. D. et al. Prestin amplifies cardiac motor functions. Cell Rep. 35, 109097, https://doi.org/10.1016/j.celrep.2021.109097 (2021).
Kubisch, C. et al. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446, https://doi.org/10.1016/s0092-8674(00)80556-5 (1999).
Kharkovets, T. et al. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 25, 642–652, https://doi.org/10.1038/sj.emboj.7600951 (2006).
Housley, G. D. & Ashmore, J. F. Direct measurement of the action of acetylcholine on isolated outer hair cells of the guinea pig cochlea. Proc. Biol. Sci. 244, 161–167, https://doi.org/10.1098/rspb.1991.0065 (1991).
Shin, D. H. et al. A recurrent mutation in KCNQ4 in Korean families with nonsyndromic hearing loss and rescue of the channel activity by KCNQ activators. Hum. Mutat. 40, 335–346, https://doi.org/10.1002/humu.23698 (2019).
Yu, S. et al. A novel early truncation mutation in OTOG causes prelingual mild hearing loss without vestibular dysfunction. Eur. J. Med. Genet. 62, 81–84, https://doi.org/10.1016/j.ejmg.2018.05.018 (2019).
Hwang, D. Data speak how to treat disease big data-based precision medicine. Mol. Cells 45, 620–621, https://doi.org/10.14348/molcells.2022.0119 (2022).
Joo, S. Y. et al. Clinical heterogeneity associated with variants relies on affected domains. Biomedicines 10, 798 https://doi.org/10.3390/biomedicines10040798 (2022).
Koh, Y. I. et al. mutations impair autophagy and lead to hearing loss, potentially remedied by rapamycin. Autophagy 18, 2593–2614, https://doi.org/10.1080/15548627.2022.2040891 (2022).
Oh, K. S. et al. COCH-related autosomal dominant nonsyndromic hearing loss: a phenotype-genotype study. Hum. Genet. 141, 889–901, https://doi.org/10.1007/s00439-021-02368-y (2022).
Rim, J. H. et al. Differential genetic diagnoses of adult post-lingual hearing loss according to the audiogram pattern and novel candidate gene evaluation. Hum. Genet. 141, 915–927, https://doi.org/10.1007/s00439-021-02367-z (2022).
Lee, J. et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 54, 1862–1871, https://doi.org/10.1038/s12276-022-00871-4 (2022).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613, https://doi.org/10.1002/humu.23630 (2018).
Gordon, C. M., Leonard, M. B., Zemel, B. S. & International Society for Clinical Densitometry. 2013 Pediatric Position Development Conference: executive summary and reflections. J. Clin. Densitom. 17, 219–224, https://doi.org/10.1016/j.jocd.2014.01.007 (2014).
Oldfors, A., Tajsharghi, H., Darin, N. & Lindberg, C. Myopathies associated with myosin heavy chain mutations. Acta Myol. 23, 90–96 (2004).
Sfondouris, J., Rajagopalan, L., Pereira, F. A. & Brownell, W. E. Membrane composition modulates prestin-associated charge movement. J. Biol. Chem. 283, 22473–22481, https://doi.org/10.1074/jbc.M803722200 (2008).
Dehghani-Ghahnaviyeh, S., Zhao, Z. & Tajkhorshid, E. Lipid-mediated prestin organization in outer hair cell membranes and its implications in sound amplification. Nat. Commun. 13, 6877, https://doi.org/10.1038/s41467-022-34596-9 (2022).
Belyantseva, I. A., Boger, E. T. & Friedman, T. B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA 100, 13958–13963, https://doi.org/10.1073/pnas.2334417100 (2003).
Ebrahim, S. et al. Stereocilia-staircase spacing is influenced by myosin III motors and their cargos espin-1 and espin-like. Nat. Commun. 7, 10833, https://doi.org/10.1038/ncomms10833 (2016).
Dallos, P. Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol. 18, 370–376, https://doi.org/10.1016/j.conb.2008.08.016 (2008).
Meltzer, J. & Santos-Sacchi, J. Temperature dependence of non-linear capacitance in human embryonic kidney cells transfected with prestin, the outer hair cell motor protein. Neurosci. Lett. 313, 141–144, https://doi.org/10.1016/s0304-3940(01)02266-2 (2001).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, https://doi.org/10.1038/nature19057 (2016).
Wang, Y. et al. Genetic variants identified from epilepsy of unknown etiology in Chinese children by targeted exome sequencing. Sci. Rep. 7, 40319, https://doi.org/10.1038/srep40319 (2017).
Donaudy, F. et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 74, 770–776, https://doi.org/10.1086/383285 (2004).
Lalwani, A. K. et al. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am. J. Hum. Genet. 67, 1121–1128, https://doi.org/10.1016/S0002-9297(07)62942-5 (2000).
Melchionda, S. et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69, 635–640, https://doi.org/10.1086/323156 (2001).
Ahmed, Z. M. et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72, 1315–1322, https://doi.org/10.1086/375122 (2003).
Liu, X. Z. et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 16, 188–190, https://doi.org/10.1038/ng0697-188 (1997).
Liu, X. Z. et al. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 17, 268–269, https://doi.org/10.1038/ng1197-268 (1997).
Wang, A. et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280, 1447–1451, https://doi.org/10.1126/science.280.5368.1447 (1998).
Rehman, A. U. et al. Mutational Spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum. Mutat. 37, 991–1003, https://doi.org/10.1002/humu.23042 (2016).
Acknowledgements
We thank the families who participated in this study. We also thank the Yonsei Advanced Imaging Center along with Carl Zeiss Microscopy. The bioresources for this study were provided by the National Biobank of Korea, Centers for Disease Control and Prevention, Republic of Korea (4845-301, 4851-302, and -307). This work was supported by National Research Foundation (NRF) grants funded by the Korean government (RS-2024-00346485 to J.J., 2020R1A2C3005787 to J.Y.C., 2018R1A5A2025079 and RS-2023-00261905 to H.Y.G., 2022R1A2C3007281 to J.B., and Korea Mouse Phenotyping Project RS-2024-00400118 to J.B. and H.Y.G.), and the Team Science Award of Yonsei University College of Medicine (6-2021-0002 to J.J., 6-2021-0003 to H.Y.G., and 6-2021-0004 to J.B.).
Author information
Authors and Affiliations
Contributions
Conceptualization: J.J., H.Y.G., and J.Y.C. Formal analysis: S.Y.J., H.Y.M., J.W.R., J.H.R., J.A.K., S.J.K., and S.H.J. Investigation: J.J., S.Y.J., H.Y.M., J.W.R, K.A.K., J.H.M., Y.I.K, and B.C.K. Resources: J.J. and J.Y.C. Data curation: J.J., H.Y.K., and H.L. Writing—original draft preparation: J.J., S.Y.J., H.Y.M., and J.W.R. Writing—review and editing: S.Y.J., J.W.R., B.C.K., H.Y.G., and J.B. Visualization: J.J., S.Y.J., H.Y.M., and J.W.R. Supervision: H.Y.G., J.Y.C., J.B., and J.K.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jung, J., Joo, S.Y., Min, H. et al. MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss. Exp Mol Med 56, 2423–2435 (2024). https://doi.org/10.1038/s12276-024-01338-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-024-01338-4
This article is cited by
-
TMEM145 is a key stereociliary component in the link structures of outer hair cells and mediates the secretion of stereocilin and tubby
Nature Communications (2025)
-
Auditory genotype-phenotype correlation of patients with variants in STRC
Scientific Reports (2025)
-
Leveraging underrepresented population data improves interpretation of genetic variants associated with hearing loss
Scientific Reports (2025)
