
Abstract
The pyruvate kinase M2 (PKM2)-mediated aerobic glycolysis has been shown to play a critical role in promoting cell survival and proliferation. However, little is known about the function of intestinal epithelial PKM2 in intestine homeostasis. Here we investigate whether and how intestinal epithelial PKM2 modulates the morphology and function of the adult intestine in experimental colitis. Analyzing colonoscopic biopsies from Crohn’s disease and ulcerative colitis patients, we found significantly decreased level of intestinal epithelial PKM2 in patients compared to that in non-inflamed tissues. Similar reduction of intestinal epithelial PKM2 was observed in mice with dextran sulfate sodium-induced colitis. Moreover, intestinal epithelial-specific PKM2-knockout (Pkm2−/−) mice displayed more severe intestinal inflammation, as evidenced by a shortened colon, disruption of epithelial tight junctions, an increase in inflammatory cytokine levels, and immune cell infiltration, when compared to wild-type mice. Gene profiling, western blot, and function analyses indicated that cell survival signals, particularly the Wnt/β-catenin pathways, were associated with PKM2 activity. Increasing mouse intestinal epithelial PKM2 expression via delivery of a PKM2-expressing plasmid attenuated experimental colitis. In conclusion, our studies demonstrate that intestinal epithelial PKM2 increases cell survival and wound healing under the colitic condition via activating the Wnt/β-catenin signaling.
Similar content being viewed by others


Introduction
The gastrointestinal epithelium plays a critical role in preventing the access of luminal pathogens to the mucosal and systemic immune system.1,2 Disruption of this dynamic and selective barrier often results in compromised barrier function and mucosal inflammation. Gastrointestinal epithelial homeostasis is generally decided by two factors: gastrointestinal epithelium damage induced by inflammatory responses of various infiltrating immune cells and the robustness of gastrointestinal epithelium repair. Therefore, controlling intestinal inflammation and promoting wound healing of the gastrointestinal epithelium is beneficial for gastrointestinal epithelium integrity. Given the importance of gastrointestinal epithelium, humans and other animals have developed different mechanisms to maintain the gastrointestinal epithelial integrity. Many studies have highlighted the critical role of secreted lipids and proteins, including lipoxins, resolvins, protectins, maresins, and prostaglandins, in facilitating epithelial wound repair,3,4,5 suggesting that these pro-resolving mediators of inflammation are part of a protective mechanism for the gastrointestinal epithelium. Injuries of the gastrointestinal epithelium are also associated with metabolic reprograming induced by proinflammatory activation. Certain intermediate products of carbon metabolites, such as lactate, have been shown to play a critical role in preventing intestinal inflammation in 2, 4, 6-trinitrobenzene sulfonic acid (TNBS)-induced mouse colitis6 and for maintaining intestinal barrier function.7
Pyruvate kinase M2 (PKM2) has been recently demonstrated to act as a bridge between energy metabolism and inflammatory cellular processes.8 PKM2 activity is a determinant of cell proliferation through the shunting of metabolites into the pentose phosphate pathway to support nucleotide biosynthesis required by rapidly dividing cells.9 As the rate-limiting enzyme controlling the final step of glycolysis, PKM2 plays a key role in the Warburg effect in cancer cells, as well as immune cells.10,11,12,13 Interestingly, the non-metabolic functions of PKM2 have been reported in various cell types recently.13,14,15,16 Yang et al.14 have revealed a novel non-metabolic role for PKM2, wherein PKM2 translocates to the nucleus and binds with nuclear β-catenin to induce the expression of cyclin D1. In the nucleus, activated β-catenin can bind with T cell factor-4 to form a complex that induces transcription of a group of target genes involved in cell proliferation, migration, sorting, and Paneth cell differentiation.17 In fact, the nuclear translocation and function of PKM2 is associated with other transcription factors including HIF-1α18 and OCT4.19 Given that PKM2 can activate various transcription factors, the role of PKM2 may be quite different in various cell types. For instance, although the abundance of PKM2 in innate immune cells such as neutrophils and macrophages implicates a critical role of PKM2 in immune cell recruitment and inflammatory responses,15 PKM2 upregulation in the intestinal epithelium may repress apoptosis and facilitate cell survival.16 Recently, enhanced levels of PKM2 were found in the feces20 and sera21 from patients with active inflammatory bowel disease (IBD) and the detection of this cell-free PKM2 was correlated with the severity of intestinal inflammation. However, the functions of epithelial-specific PKM2 in modulating gastrointestinal inflammation and epithelial repair remain unknown.
In the present study, we report that PKM2 expression in intestinal epithelial cells (IECs) is significantly reduced in IBD patients, as well as in the dextran sulfate sodium ()-induced colitis mouse model. By generating intestinal epithelial-specific Pkm2−/− mice, we show that Pkm2−/− mice display more severe inflammation and intestinal epithelium damage in DSS-induced colitis compared to wild-type (WT) littermates. RNA-sequencing (RNA-Seq) and gene set enrichment analysis (GSEA) identify that cell survival Wnt/β-catenin signaling in intestinal epithelial cells (IECs) is significantly suppressed by Pkm2 gene knockout, which is confirmed by western blot (WB) analysis. In line with this, in vitro cell wound healing assay shows that, via a loss of Wnt/β-catenin signaling, Pkm2 knockdown decreases the wound healing of colonic epithelial cells. Finally, increasing intestinal epithelial Pkm2 level in DSS-treated mice via delivery of a PKM2-expressing plasmid effectively mitigates DSS-induced colitis. Taken together, our studies demonstrate for the first time that intestinal epithelial-specific PKM2 serves as a safeguard against experimental colitis via activation of the β-catenin signaling pathway.
Materials and methods
Human colon tissues, cell lines, and reagents
Colonoscopic biopsies from five subjects without IBD, as well as five Crohn’s disease (CD) patients and five ulcerative colitis (UC) patients, were obtained from Nanjing General Hospital and Nanjing Drum Tower Hospital, Nanjing University School of Medicine (Nanjing, China). Ethical approval was obtained from the ethics committee of Nanjing University School of Medicine. Non-transformed intestinal epithelial IEC-6 cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium with 5% fetal bovine serum (FBS) (Life Technologies), 2 mM glutamine (Life Technologies), and 0.1 U/mL insulin. For isolation of mouse colon epithelial cells, murine normal colon was washed with cold Hank’s balanced salt solution (HBSS) buffer (Life Technologies) until the supernatant was clear. Next, the tissue fragments were incubated in 2 mM EDTA cold chelation buffer (100 mM NaCl, 5 mM Na2HPO4, 10 mM KH2PO4, 2 mM KCl, 40 mM sucrose, 55 mM d-sorbitol, 0.5 mM dithiothreitol) for 1 h on ice. After removal of the EDTA buffer, the tissue fragments were vigorously re-suspended in cold HBSS buffer using a 10 mL pipette to isolate intestinal crypts. The tissue fragments were allowed to precipitate under normal gravity and the supernatant was removed for inspection by inverted microscopy. Isolated crypts were pelleted, washed with cold HBSS buffer, and centrifuged (150–200 × g, 3 min) to separate the crypts from single cells. The phosphotyrosine peptide (GGAVDDDpYAQFANGG) was purchased from GenScript Corporation (Nanjing, China). DASA-58 and TEPP-46 were purchased from MCE (Monmouth Junction, USA). Dimethyl sulfoxide was used as vehicle control for TEPP/DASA in all assays.
Mice
B6.129S-Pkmtm1.1Mgvh/J transgenic mouse were crossed with B6.Cg-Tg (Vil1-cre) 1000 Gum/J transgenic (both from Jackson Laboratory). Mice were backcrossed to C57BL/6J for at least 10 generations and fed under specific pathogen-free conditions at Nanjing University. For experiments, the 6–8-week-old male epithelial cell-specific Pkm2-knockout mice were used, and their WT littermates not carrying the Vil1-cre transgene (fl/fl) served as controls. All mouse protocols followed the National Institutes of Health guide for the care and use of mice and were approved by IACUC, Nanjing University (Nanjing, China). Mouse genotypes were determined by PCR with primer for Vil-Cre (common: 5′-GCCTTCTCCTCTAGGCTCGT-3′; internal positive control reverse: 5′-TATAGGGCAGAGCTGGAGGA-3′; transgene reverse: 5′-AGGCAAATTTTGGTGTACGG-3′) and Pkm2 flox (forward: 5′-AGGTAGGAGGCGGCGTG-3′; reverse: 5′-CCACTCACTCTTGGCATCC-3′). For the other experiments, the 6–8-week-old male C57BL/6J mice (20–22 g) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China) and fed under specific pathogen-free conditions at Nanjing University.
DSS-induced experimental colitis
DSS (2.5%) (36,000–50,000 Da, MP Biomedicals, Solon, OH, USA) was administered in drinking water to mice for 7 days, and bodyweight, stool softness, and blood in the stool of mice were assessed daily. The disease activity index was used to evaluate the grade of intestinal inflammation in DSS-induced colitis. To increase colonic epithelial PKM2 expression in mice treated with DSS, 0.2 mg Pkm2-expressing plasmid was mixed with polyetherimine (PEI)22 and dissolved in 200 μl saline, and slowly administered into the lumen of the colon via a catheter inserted 4 cm into the colon through the anus.23
In vivo barrier function experiments
Mice (Pkm2−/− or fl/fl) at 8–10 weeks of age were gavage fed with 0.6 g/kg bodyweight of a 100 mg/mL24 solution of 4 kDa dextran-FITC (fluorescein isothiocyanate) (Sigma-Aldrich), and serum was collected after 4 h. A standard curve was prepared using serial dilutions of dextran in phosphate-buffered saline (PBS). Fluorescence emission was measured on a Tecan Safire2 multimode microplate reader (Tecan, Switzerland) at an excitation of 485 nm and an emission of 521 nm.
Histopathological analysis
For histological evaluation of intestinal inflammation, mice were sacrificed at indicated time points whereupon tissue sections were fixed in buffered 4% paraformaldehyde and paraffin embedded. We conducted an histological analysis on the hematoxylin and eosin (H&E) staining sections of intestinal inflammation as described.25 Briefly, histological score of H&E staining sections was graded from 0 to 4 as previously described.26 Scoring was performed in a blinded fashion. For immunofluorescence of colon, human and mouse intestines were frozen in OCT for sectioning. Intestinal sections with 10 μm were fixed in 4% paraformaldehyde and blocked in 10% donkey serum for 30 min. We used anti-PKM2 (Cell Signaling, D78A4, 4053), anti-E-cadherin (Abcam, ab76055), anti-CD11b (R&D, MAB1124), anti-F4/80 (R&D, MAB5580), anti-β-catenin (Cell Signaling, D10A8, 8480), and anti-β-catenin (Ser675) (Cell Signaling, D2F1, 4176) as primary antibodies. After incubation with antibodies for overnight at 4 °C, tissue sections were incubated with fluorescently tagged secondary antibodies (488 and 594 nm) (Invitrogen) at room temperature for 1 h. After washing three times, tissue sections were stained briefly by 4′,6-diamidino-2-phenylindole dye (Santa Cruz) and then mounted by AntiFade mountant (Invitrogen). Confocal microscopy study was performed on the Nikon C2 Plus confocal microscope (Nikon Corp, Japan).
Western blot
Tissues and cultured cells were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), pH 7.4) containing a cocktail of protease inhibitors (Thermo Scientific), sonicated, and centrifuged at 12,000 × g for 10 min at 4 °C. The supernatant fraction was collected, and the protein concentration was determined by a BCA assay (Pierce, Rockford, CT), and then denaturated with 5× loading buffer (Beyotime) for 10 min at 95 °C. Aliquots of proteins were separated on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Roche). The membranes were blocked for 1 h with 5% non-fat milk in TBS plus 0.1% Tween-20 (TBST) followed by an overnight swing incubation at 4 °C with antibodies diluted in blocking buffer (1:1000). After three 10 min washes with TBST, blots were incubated at room temperature for 1 h with the appropriate secondary antibody conjugated to horseradish peroxidase, and protein expression was detected with an enhanced chemiluminescent reagent (SuperSignal, Rockford, CT). The above-mentioned primary antibodies were obtained from the following companies: anti-PKM2 (Cell Signaling, D78A4, 4053), anti-PKM1 (Proteintech, 15821-1-AP), anti-GAPDH (Santa Cruz, sc-32233), anti-E-cadherin (Abcam, ab76055), anti-occludin (Proteintech, 13409-1-AP), anti-junctional adhesion molecule-1 (JAM-1) (Abcam, ab180821), anti-β-catenin (Cell Signaling, D10A8, 8480), anti-phosphorylated Akt (p-AKT)(S473) (Cell Signaling, D9E, 4060), anti-AKT (Cell Signaling, 11E7, 4685), anti-WIF1 (Absin, abs116623), anti-p-GSK-3β (S9) (Cell Signaling, D85E12, 5558), anti-GSK-3β (Cell Signaling, D5C5Z, 12456), anti-β-actin (Santa Cruz, sc-47778), and anti-cyclin D1 (Abcam, ab134175). The autoradiographic intensity of each band was scanned and calculated with the Image J software.
RNA-Seq data processing
The total RNA was extracted from control and Pkm2-deficient colon epithelial cells from mice. The RNA-Seq experiments were performed for assay from Vazyme Biotech (Nanjing, China). Next, the transcriptome library for sequencing was created using VAHTS mRNA-Seq V2 Library Prep Kit for Illumina (Nanjing, China) according to the manufacturer’s recommendations. Fore clustering of messenger RNA (mRNA) samples VAHTSHERNA Adapters set1/set2 for Illumina (Vazyme Biotech Co.) was used. After the clustering, the libraries were ranked by Illumina Hiseq X Ten platform with (2 × 150 bp) paired-end module. Genes with different expressions were identified with a q value <0.05 and a fold change of ≥2 between two samples. The unbiased gene set enrichment was analyzed by GSEA (software.roadinstitute.org/gsea/index.jsp).
ELISA and Immunoprecipitation
Serum from mice with indicated treatments were isolated from blood, settled for 30 min at room temperature, and centrifuged at 3000 r.p.m. for 10 min. Then, supernatants were analyzed by ELISA kit (4A Biotech Co., Beijing, China) to detect different cytokines, including interleukin-1β (IL-1β), IL-17A, IL-6, and IL-12/23. For immunoprecipitation, cells were lysed with lysis buffer for 30 min on ice and the lysates were obtained after centrifugation (16,000 × g, 10 min, 4 °C). Cell lysate was then incubated with anti-β-catenin, anti-PKM2 antibodies, or normal rabbit immunoglobulin G (IgG) (Beyotime), followed by incubation with Protein G-Agarose beads (Santa Cruz). After extensive washing, proteins were eluted and further analyzed by WB using anti-β-catenin and anti-PKM2 antibodies.
Cell treatment
IEC-6 cells were transfected with PKM1/PKM2-expressing plasmids or specific small interfering RNAs (siRNAs) of PKM1, PKM2, and β-catenin to test the effects on cell wound healing and proliferation. In separate experiment, cells were incubated in the presence of 10% serum obtained from colitic mice. The concentration of chemical molecules used in cell processing was as follows: DASA-58, 50 μM; TEPP-46, 50 μM; pTyr, 50 μM. Pkm2-expressing plasmid with mCherry was purchased from GeneCopoeia (Guangzhou, China). The siRNA for each target gene was designed and synthesized (Ribobio, Guangzhou, China). The siRNA sequences were as follows: PKM1 siRNA, 5′-CCTCCAGTCAATCCACAGATT-3′; PKM2 siRNA, 5′-CCATTATCGTGCTCACCAATT-3′; β-catenin siRNA, 5′-CCATGGAGCCAGACAGAAATT-3′.
Wound healing assay
IEC-6 cell migration was assessed using a scratch wound assay as previously published with minor modification.27 Confluent cell monolayers grown in 6-well tissue culture plates were transfected with PKM1/2-expressing plasmid or control plasmid 6 h prior to the mechanical wound injury. Wounded monolayers were washed with PBS and incubated for 24 h in the presence of Wnt3a for wound healing. In some experiments, wounded cell monolayers were incubated in the medium containing 50 μM DASA-58, TEPP-46, or pTyr. The rate of cell migration was determined by measuring the entire wound area immediately after wounding as well as at the indicated time points, and by normalizing to the control condition.
Cell proliferation assay
To measure the proliferation rate of IEC-6 cells, we conducted a 5-ethynyl-2′-deoxyuridine (EdU) assay according to protocols described elsewhere. Briefly, IEC-6 cells were seeded in 6-well plates and transfected with small RNA oligonucleotides or plasmids, after 24 h stimulation by Wnt3a following transfection, cells were harvested and reseeded in 48-well plates for EdU assay. In some experiments, IEC-6 monolayers were incubated for 24 h in the medium containing 50 μM DASA-58, 50 μM TEPP-46, or 50 μM pTyr. For the EdU assay, an EdU assay kit (RiBoBio, China) was used to determine the proliferation rate of cells according to the manufacturer’s instructions.
Statistical analysis
Data derived from at least three independent experiments were presented as the mean ± SEM. All the images including WB, immunofluorescence, and cell migration experiments are representative of at least three independent experiments. Statistical comparisons between two groups were performed by Student’s t test. Multiple-group comparisons were determined using two-way analysis of variance (ANOVA). P value < 0.05 was considered statistically significant.
Results
Loss of IEC PKM2 during development of colitis
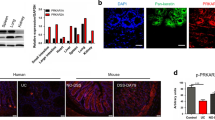
Previous studies have detected PKM2 in feces20 and serum21 of IBD patients, suggesting that PKM2 may serve as an indicator for intestinal epithelial damage. However, PKM2 is expressed in various cell types and the source of fecal or serum PKM2 remains unknown. To monitor whether the intestinal epithelial PKM2 level is altered during development of colitis, we established a DSS-induced experimental murine colitis model as previously described.22 As shown in Supplementary Fig. S1, mice administered with 2.5% DSS in drinking water displayed significant weight loss (Fig. S1A) and aggravation of colitis measured by disease index on day 7 (Fig. S1B) compared to control mice without DSS treatment. Histochemical analysis of mouse colon tissue sections clearly showed that intestinal epithelial structure was disrupted by DSS treatment (Fig. S1C). Mouse colon tissue sections were examined for the expression and localization of Pkm2 by immunofluorescence labeling. As shown in Fig. 1, mouse colon tissue sections were co-labeled with anti-Pkm2 and anti-E-cadherin antibodies. The chicken-wire labeling pattern of E-cadherin marked the structure of IEC monolayers. Compared to control mice without DSS treatment, mice treated with DSS displayed significantly less Pkm2 in IECs, which were labeled by E-cadherin, by day 7 post treatment (Fig. 1b).

Reduction of intestinal epithelial Pkm2 in DSS-induced colitic mice and IBD patients. a Immunofluorescence staining of E-cadherin (green) and PKM2 (red) in colon tissue sections from healthy subject and patients with CD or UC (n = 5 each). b Immunofluorescence staining of E-cadherin (green) and PKM2 (red) in colon tissue sections from DSS--induced colitic mice and healthy mice (n = 6 mice per experiment). Bar, 20 μm. Data are presented as the mean ± SEM. All p values are determined by the Student’s t test. ***p < 0.001; **p < 0.01
The reduction of intestinal epithelial PKM2 was further confirmed in patients with a chronic colitic condition. For this experiment, we collected colonoscopic biopsies (five each) from CD and UC patients and subjects without chronic intestinal inflammatory condition. Immunofluorescence labeling showed that, compared to control subjects without intestinal inflammatory condition, patients with active CD and UC both had a significantly decreased PKM2 expression in intestinal epithelium (Fig. 1a).
Intestinal epithelial-specific Pkm2 deficiency aggravates intestinal injury during DSS-induced colitis
To analyze the role of intestinal epithelial Pkm2 in experimental murine colitis, we crossed B6.129S-Pkmtm1.1Mgvh/J transgenic mice with B6.Cg-Tg (Vil1-cre) 1000 Gum/J transgenic mice (both from Jackson Laboratory) to generate transgenic mice lacking Pkm2 specific in IECs (Pkm2−/−) (Fig. 2a). Mice were then backcrossed to C57BL/6J for at least 10 generations. As shown in Fig. 2b, genotype (left), immunofluorescence (middle), and WB (right) clearly indicated that Pkm2 was specifically deleted in the IECs in Pkm2−/− mice. Interestingly, we found that Pkm1, another isoform of pyruvate kinase, was significantly upregulated in mouse IECs in which Pkm2 was deleted. This observation is in agreement with previous finding that shows PKM2 deficiency causes increased PKM1 expression.28

Pkm2−/− mice are more susceptible to DSS-induced colitis compared to fl/fl mice. a Schema illustrating generation of intestinal-specific Pkm2−/− mice. b Representative PCR, immunofluorescence, and blot images of Pkm2 and Pkm1 protein in IECs isolated from fl/fl and Pkm2−/− mice. c Percentage reduction in bodyweight in fl/fl and Pkm2−/− mice treated with 2.5% DSS for 7 days (fl/fl, n = 6; Pkm2−/−, n = 9. d Clinical disease activity index of mice subjected to DSS-induced colitis for 7 days (fl/fl, n = 6; Pkm2−/−, n = 9). e Kaplan–Meier survival analysis of fl/fl and Pkm2−/− mice (each 12 total) following DSS treatment. f Representative pictures of colons on day 0, 3, 7 and colon length of fl/fl and Pkm2−/− mice on days 0, 3, and 7 post DSS treatment (day 0, n = 5; days 3 and 7, n = 6). g H&E staining of colon sections of fl/fl and Pkm2−/− mice on days 3 and 7 post DSS treatment. Bars, 100 μm. Histological examination and disease scores of respective groups were shown on days 3 and day 7. All p values are determined by the Student’s t test and two-way ANOVA analysis. ***p < 0.001; **p < 0.01; *p < 0.05
Next, we compared the colitis disease condition induced by 2.5% DSS treatment in Pkm2−/− mice with that in WT littermates (fl/fl). In this experiment, mice were given drinking water with or without 2.5% DSS. Mouse bodyweight and a disease index (based on factors such as weight, occult blood, and fecal continuity) were daily monitored. After sacrificing on different days, mouse colon tissues were collected for histological assessment of intestinal inflammation. As shown, Pkm2−/− mice displayed a faster weight loss (Fig. 2c), higher disease index (Fig. 2d), and poorer survival rate (Fig. 2e) than fl/fl mice after DSS treatment. Although DSS treatment shortened colon length in Pkm2−/− mice and fl/fl littermates compared to that in non-treated mice, the length of colon in Pkm2−/− mice was significantly shorter than that in fl/fl mice after DSS treatment (Fig. 2f). Histological examination and scoring of distal colon tissue section on day 7 revealed a significantly higher pathology score characterized by increased epithelial disruption, follicle aggregation, crypt loss, and enhanced erosion in Pkm2−/− mice than in fl/fl mice (Fig. 2g). In line with this, in vivo cell proliferation assay clearly showed that DSS treatment markedly decreased IEC proliferation in Pkm2−/− mice relative to their fl/fl counterparts (Supplementary Fig. 2).
Given that colitis is often associated with production of inflammatory cytokines and infiltration of innate immune cells,29 we examined the circulating cytokine level and infiltration of CD11b- or F4/80-positive (CD11b+ or F4/80+) immune cells. Immunofluorescence labeling showed that DSS treatment caused more infiltration of CD11b+ or F4/80+ cells into colon tissues in Pkm2−/− mice compared to those in fl/fl mice (Fig. 3a). As shown in Fig. 3b, the serum levels of inflammatory cytokines, including IL-17, characteristically elevated cytokine in DSS-induced colitis,30 as well as IL-1β, IL-6, and IL-12/23 were significantly higher in the Pkm2−/− mice than in the fl/fl mice after DSS treatment.

DSS treatment induced more severe colonic inflammatory condition in Pkm2−/− mice compared to fl/fl mice. a Immunofluorescence labeling of CD11b-positive or F4/80-positive (green) immune cells and E-cadherin-positive epithelial cell in the colon of fl/fl and Pkm2−/− mice on days 3 and 7 post DSS treatment (n = 5). Bars, 100 μm. b The IL-1β, IL-17A, IL-6, and IL-12/23 concentrations in the serum from mice on day 0 (n = 5), day 3 (n = 5), and day 7 (n = 5) post DSS treatment. All p value was determined by the Student’s t test. **p < 0.01; *p < 0.05; n.s. not significant
Next, we compared the barrier function of gastrointestinal epithelium in Pkm2−/− mice with that in fl/fl mice during DSS treatment. FITC-dextran assay27 showed that Pkm2−/− mice had higher permeability in their gastrointestinal epithelium than fl/fl littermates on day 3 post DSS treatment (Fig. 4a). In line with this, on day 3 post DSS treatment, the expression of multiple cell junction-associated proteins, including E-cadherin, JAM-1, and occludin, was significantly reduced in the IECs from Pkm2−/− mice compared to that from fl/fl mice (Fig. 4b, c). Taken together, these results show that mice with IEC-specific Pkm2 deletion are more susceptible to DSS-induced experimental colitis than fl/fl mice.

Pkm2−/− mice displayed higher permeability in gastrointestinal epithelium at early stages of DSS-induced colitis than fl/fl mice. a Mouse permeability of gastrointestinal epithelium detected by FITC-dextran (n = 6). b, c Western blot analysis of junctional protein levels in mouse IECs. Data were presented as the mean ± SEM from three independent experiments and p values are determined by the Student’s t test. ****p < 0.0001; ***p < 0.001
PKM2 facilitates intestinal epithelial survival and promotes wound healing through activating the Akt/β-catenin signal pathway
To identify signal pathways differing between mice with IEC-specific Pkm2 deletion versus fl/fl mice after DSS treatment, we next performed RNA-Seq and principal components analysis using dissected mouse intestinal epithelium. In this experiment, mouse intestinal epithelial monolayers were isolated on day 3 post DSS treatment and total RNA was extracted for RNA-Seq analysis. As shown in Fig. 5a, the expression of a large number of genes was altered in Pkm2−/− mouse intestinal epithelium compared to those in fl/fl mouse intestinal epithelium. The upregulated genes included Akt1, Akt3, Sfrp1, Axin1-2, and Senp2, and so on, while the downregulated genes included Mvp, Dvl1, Fstl1, and Wif1, and so on. The differential expression of many genes in mouse intestinal epithelium suggests that Pkm2 deletion reprogramed mouse IEC. Close examination of differentially regulated genes (Fig. 5b) and GSEA31 (Fig. 5c) revealed a dramatic enrichment in genes and pathways integral to Wnt/β-catenin signaling. This finding is consistent with the previous findings that showed, following epidermal growth factor receptor activation, PKM2 relocates to the nucleus where it binds to β-catenin in human cancer cells and induces expression of cyclin D1, thereby promoting tumor proliferation.14 Supporting the notion that Pkm2 deletion affects Wnt/β-catenin signaling in mouse IECs, WB analysis showed that, on day 3 post DSS treatment, the levels of β-catenin and WIF1 were reduced, while the levels of p-Akt and Gsk (S9) (p-Gsk) were increased in Pkm2−/− mouse intestinal epithelium compared to those in fl/fl mouse intestinal epithelium (Fig. 5d). The significant reduction of both total β-catenin and phosphorylated β-catenin (Y675) in Pkm2−/− mouse intestinal epithelium, compared to fl/fl mouse intestinal epithelium, was confirmed by immunofluorescence labeling of mouse colon tissue sections (Fig. 5e). Moreover, cross-immunoprecipitation and immunoblot analysis, in which normal IgG served as a control, showed the specific binding of Pkm2 with β-catenin under the stimulation of Wnt3a (Fig. 5f). To test the effect of PKM2 on β-catenin-mediated signaling, we silenced Pkm2 in rat intestinal epithelial IEC-6 cells via Pkm2 siRNA. As shown in Fig. 5g, in the presence of 10% serum obtained from colitic mice, silencing PKM2 via Pkm2 siRNA reduced cellular β-catenin levels. As protein kinase activity of PKM2 is dependent on its switch between tetramer and dimer formation,32 we also determined the level of β-catenin and its associated signaling molecules in IEC-6 cells after modulating PKM2 activity using pTyr, a peptide that selectively enhances PKM2 phosphorylation and thus dimerize PKM2,33 or DASA-58 and TEPP-46,34 two reagents that strengthen the tetramer formation of PKM2. The results showed that increasing PKM2 tetramer formation by DASA-58 and TEPP-46 decreased cellular β-catenin levels, whereas promoting PKM2 dimer form by pTyr enhanced the β-catenin levels, confirming that β-catenin level in IECs is dependent on PKM2 activity.

Pkm2 deficiency in IECs significantly reduced the pro-survival Wnt/β-catenin signaling. a Alteration of gene expression in Pkm2−/− mouse IECs compared to that in fl/fl mouse IECs on day 3 post DSS treatment. b Selected genes that were altered in Pkm2−/− mouse IECs compared to that in fl/fl mouse IECs on day 3 post DSS treatment. c Gene ontology gene analysis in fl/fl and Pkm2−/− mouse IECs. In red were pathways enriched in Pkm2−/− IECs. d WB analysis of levels of various proteins associated with Wnt/β-catenin signaling pathway. e Distribution and level of β-catenin and phosphorylated β-catenin (Y675) in Pkm2−/− or fl/fl mouse IECs on day 3 post DSS treatment. f Under the stimulation of Wnt3a, cross-IP and cross-blot of PKM2 and β-catenin in IEC-6 cells using anti-β-catenin and anti-PKM2 antibodies, respectively. Immunoprecipitation using normal IgG served as controls. g β-Catenin levels in IEC-6 cells treated with Pkm2 siRNA or Pkm2 inhibitor/activator in the presence of 10% serum from colitic mice. Data are presented as the mean ± SEM from three independent experiments and p values are determined by the Student’s t test. ***p < 0.001; **p < 0.01; *p < 0.05; n.s. not significant
Previous reports suggest that Wnt/β-catenin signaling is not only involved in promoting cell survival but also the wound healing process.29 We next tested whether PKM2 plays a critical role in modulating IEC wound healing and cell proliferation. In the scratch wound healing assay, confluent IEC-6 cell monolayers grown in 6-well tissue culture plates were treated with Pkm2 siRNA, Pkm1 siRNA, or control siRNA, and then scraped in the presence of Wnt3a stimulation.35 As shown in Fig. 6a, compared to control siRNA, treatment with Pkm2 siRNA markedly inhibited the healing process of wounded IEC-6 monolayers (upper panel) and cell proliferation (lower panel). In contrast, overexpression of Pkm2 in IEC-6 cells via transfecting cells with PKM2-expressing plasmid effectively promoted the wound closure of IEC-6 monolayers (Fig. 6b, upper panel) and cell proliferation (Fig. 6b, lower panel) compared to empty vector transfection. Interestingly, treatment with Pkm1 siRNA also inhibited the healing process of wounded IEC-6 monolayers (upper panel) and cell proliferation (lower panel) albeit to a lesser degree compared to Pkm2 siRNA treatment, but Pkm1 overexpression in IEC-6 cells via transfection with a Pkm1-expressing plasmid did not promote the wound closure of IEC-6 monolayers (Fig. 6b, upper panel) and cell proliferation (Fig. 6b, lower panel) compared to empty vector transfection. These results suggest that, unlike Pkm2, Pkm1 is not directly involved in IEC wound healing and proliferation process, and the moderate inhibition of PKM1 knockdown on IEC wound healing and proliferation may be simply due to the reduction of cellular energy production. In line with the notion that PKM2 modulates IEC wound healing and proliferation, an increase in PKM2 tetramer formation by DASA-58 and TEPP-46 delayed the wound healing of IEC-6 monolayers (Fig. 6c, upper panel) and cell proliferation (Fig. 6c, lower panel), whereas treatment with pTyr strongly enhanced the healing process of wounded IEC-6 monolayers (Fig. 6c, upper panel) and cell proliferation (Fig. 6c, lower panel). To further test whether PKM2 executes its non-metabolic function through the β-catenin signal pathway, we determined the effect of PKM2 on IEC-6 cells wound healing and proliferation after knocking down β-catenin. As shown in Fig. 6d, knockdown of β-catenin in IEC-6 cells via transfection with β-catenin siRNA largely abolished the effect of PKM2 overexpression on promoting cell wound healing (upper panel) and proliferation (lower panel).

Effect of Pkm2 on wound healing of IEC-6 monolayers under the stimulation of Wnt3a. a Treatment with Pkm2 siRNA and Pkm1 siRNA analyzed the healing process of wounded IEC-6 monolayers (upper panel) and cell proliferation (lower panel). b Overexpression of Pkm2 and Pkm1 in IEC-6 cells via transfecting cells with plasmid analyzed the wound closure of IEC-6 monolayers (upper panel) and cell proliferation (lower panel) compared to empty vector transfection. c Effect of Pkm2 inhibitor and activator on IEC-6 cell wound healing (upper panel) and cell proliferation (lower panel). d Overexpression of Pkm2 plasmid in IEC-6 cells via transfecting cells under the β-catenin siRNA analyzed the wound closure of IEC-6 monolayers (upper panel) and cell proliferation (lower panel) compared to empty vector transfection. Data are presented as the mean ± SEM from three independent experiments and p values are determined by the Student’s t test. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; n.s. not significant
Enhancing Pkm2 level in IECs via delivery of Pkm2-expressing plasmid attenuates experimental murine colitis
To further examine the protective role of intestinal epithelial Pkm2 in DSS-induced colitis mouse model, we employed an effective strategy23 to increase the expression level of Pkm2 in mouse colonic epithelium. In this experiment, 3 days prior to DSS treatment, 0.2 mg of Pkm2-expressing plasmid was mixed with PEI22 in 200 μl saline, and slowly administered into the lumen of the colon via a catheter inserted 4 cm into the colon through the anus (Fig. 7a). As shown in Fig. 7b, a significant fluorescence of mCherry (red) associated with the plasmid was observed in mouse colonic IECs, suggesting a successful delivery of protein into mouse colonic epithelial cells. On day 7 post DSS treatment, mice administered with Pkm2 plasmid/PEI displayed significantly decreased weight loss (Fig. 7c) and a colitic disease index (Fig. 7d) compared to mice administered with control plasmid/PEI. Increase of mouse colonic epithelial Pkm2 by delivering Pkm2-expressing plasmid/PEI also mitigated the progression of DSS-induced colitis, as assessed by overall colon length (Fig. 7E) and histological scoring of distal colon section (Fig. 7f).

Increase of colonic epithelial Pkm2 level via delivery of Pkm2-expressing plasmid attenuated mouse DSS-induced colitis. a Experimental design. b Successful delivery of mCherry Pkm2-expressing plasmid/PEI to mouse IECs detected by mCherry fluorescence. c WB analysis of Pkm2 level in mouse IECs with or without administration of Pkm2-expressing plasmid/PEI. d–g Delivery of Pkm2-expressing plasmid/PEI mitigated mouse colitic disease condition caused by DSS treatment: d percentage reduction in bodyweight; e disease index; f colon length; g colon section H&E staining and histological analysis. For all experiments, control (CTL) plasmid group and Pkm2 plasmid group consisted of nine and eight mice, respectively. Data are presented as the mean ± SEM. P values are determined by the Student’s t test and two-way ANOVA. **p < 0.01; *p < 0.05
Discussion
Previous studies by us36 and others37,38,39 suggest a non-metabolic role of PKM2 in various biological processes, including increasing cell proliferation, promoting exosomal secretion, and enhancing cell survival. These findings prompted us to investigate the role of intestinal epithelial PKM2 during intestinal inflammation. Through establishing IEC-specific Pkm2-deficient mice and comparing the development of DSS experimental colitis in Pkm2-deficient mice and fl/fl mice, we found that IEC-specific Pkm2 deficiency led to more severe inflammation and intestinal epithelium disruption in DSS-induced colitis. Cell wound healing assays showed that Pkm2 knockdown significantly delayed the healing process of wounded intestinal IEC-6 monolayers. Mechanistic studies further suggested that PKM2 may execute its non-metabolic function in protecting colonic epithelial against DSS-induced colitis by activating the β-catenin-mediated signaling pathway.
The notion that IEC-specific PKM2 served as a safeguard against colitis is supported by several pieces of evidence: first, reduction of IEC-specific PKM2 was observed in tissue samples from both IBD patients and mice with ongoing experimental colitis, which is in agreement with an inhibitory role of IEC PKM2 in intestinal inflammatory diseases. Second, Pkm2 deficiency in mouse IECs suppressed cell proliferation and knockdown of PKM2 in IEC-6 cells inhibited the healing process of wounded IEC-6 monolayer. Wound repairing of IEC-6 monolayer was also promoted by switching Pkm2 to its dimeric form with pTyr, but inhibited when Pkm2 was forced to remain as a tetramer by DASA-58 and TEPP-46. Finally, murine DSS colitis was mitigated when Pkm2 levels were increased in mouse IECs via direct delivery of Pkm2-expressing plasmid/PEI into mouse colon.
RNA-Seq and GSEA identified that Wnt/β-catenin signaling, an important cell survival signal pathway,40 in mouse IECs was significantly suppressed by Pkm2 deficiency. Moreover, the reduction of IEC β-catenin or phosphorylated β-catenin was confirmed by immunofluorescence analysis. This finding is consistent with several previous studies showing the role of the PKM2-β-catenin axis in cell survival and proliferation.14,41,42 In the present study, our functional assay also showed that β-catenin-mediated signaling is involved in PKM2-mediated protection of intestinal epithelium against DSS-induced colitis. As cell proliferation and migratory capacity are two essential processes for IEC wound healing, decrease of IEC proliferation (Fig. S2) and migration (Fig. 6) in PKM2-deficient mice and cultured IEC-6 monolayers with silencing PKM2, respectively, suggests that PKM2 may protect mouse intestinal epithelium against DSS-induced colitis through promoting cell proliferation and increasing migratory capacity, two synergistic processes of wound healing. In fact, a previous study has shown that curtailing the pyruvate kinase activity of PKM2 via dimerization of PKM2 can divert the metabolites of glycolysis from the production of ATP toward the supply of new building blocks for the anabolic process of wound repair of the epithelium.37 In addition to playing a critical role in cell proliferation, PKM2 has been shown to translocate into the mitochondria instead of nuclei under oxidative stress and inhibit apoptosis by stabilizing Bcl2.43 Therefore, PKM2 may execute its protection on IECs by inhibiting cell apoptosis under intestinal inflammatory condition.
Interestingly, in agreement with previous studies,28 we also observed the upregulation of PKM1 in mouse intestinal epithelium when PKM2 was knocked out (Fig. 2b). Although the exact mechanism remains undefined, it may be due to developmental compensation. PKM1 and PKM2 are two isoforms of glycolytic enzyme pyruvate kinase and are derived from the same primary RNA through alternative splicing. When PKM2 in IECs is knocked out, cells may increase PKM1 expression to meet the metabolic requirement. In general, PKM1 is constantly in tetramer form with a high glycolytic activity in catalyzing the irreversible transphosphorylation between phosphoenolpyruvate (PEP) and adenosine diphosphate, leading to the production of pyruvate and ATP. In contrast, under various conditions such as tumorigenesis and inflammation, PKM2 can switch from tetramer to dimer form and become a protein kinase to phosphorylate various molecules such as β-catenin14 and SNAP-23.36 By initiating the signal downstream of these molecules, PKM2 executes its non-metabolic role in modulating various aspects of cellular functions.
In conclusion, our studies demonstrate for the first time that, by activating Wnt/β-catenin signaling, intestinal epithelial PKM2 promotes cell proliferative and migratory response, leading to protection of IECs against DSS experimental colitis.
References
Okumura, R. & Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 38, 5 (2018).
Allaire, J. M. et al. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. 39, 677–696 (2018).
Gobbetti, T. et al. Protectin D1n-3 DPA and resolvin D5n-3 DPA are effectors of intestinal protection. Proc. Natl. Acad. Sci. USA 114, 3963–3968 (2017).
Marion-Letellier, R., Savoye, G. & Ghosh, S. Polyunsaturated fatty acids and inflammation. IUBMB Life 67, 659–667 (2015).
Stein, K. et al. A role for 12/15-lipoxygenase-derived proresolving mediators in postoperative ileus: protectin DX-regulated neutrophil extravasation. J. Leukoc. Biol. 99, 231–239 (2016).
Iraporda, C. et al. Local treatment with lactate prevents intestinal inflammation in the TNBS-induced colitis model. Front. Immunol. 7, 651 (2016).
Okada, T. et al. Microbiota-derived lactate accelerates colon epithelial cell turnover in starvation-refed mice. Nat. Commun. 4, 1654 (2013).
Alves-Filho, J. C. & Palsson-McDermott, E. M. Pyruvate kinase M2: a potential target for regulating inflammation. Front. Immunol. 7, 145 (2016).
Eigenbrodt, E., Reinacher, M., Scheefers-Borchel, U., Scheefers, H. & Friis, R. Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit. Rev. Oncog. 3, 91–115 (1992).
David, C. J., Chen, M., Assanah, M., Canoll, P. & Manley, J. L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368 (2010).
Vander Heiden, M. G. et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499 (2010).
Palsson-McDermott, E. M. & O’Neill, L. A. The Warburg effect then and now: from cancer to inflammatory diseases. BioEssays 35, 965–973 (2013).
Xie, M. et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280 (2016).
Yang, W. et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480, 118–122 (2011).
Rorvig, S., Ostergaard, O., Heegaard, N. H. & Borregaard, N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J. Leukoc. Biol. 94, 711–721 (2013).
Tang, Q. et al. Pyruvate kinase M2 regulates apoptosis of intestinal epithelial cells in Crohn’s disease. Dig. Dis. Sci. 60, 393–404 (2015).
Barker, N., van de Wetering, M. & Clevers, H. The intestinal stem cell. Genes Dev. 22, 1856–1864 (2008).
Yang, L. et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 5, 4436 (2014).
Morfouace, M. et al. Control of glioma cell death and differentiation by PKM2–Oct4 interaction. Cell Death Dis. 5, e1036 (2014).
Chung-Faye, G. et al. Fecal M2-pyruvate kinase (M2-PK): a novel marker of intestinal inflammation. Inflamm. Bowel Dis. 13, 1374–1378 (2007).
Almousa, A. A., Morris, M., Fowler, S., Jones, J. & Alcorn, J. Elevation of serum pyruvate kinase M2 (PKM2) in IBD and its relationship to IBD indices. Clin. Biochem. 53, 19–24 (2018).
Huang, Z. et al. miR-141 regulates colonic leukocytic trafficking by targeting CXCL12beta during murine colitis and human Crohn’s disease. Gut 63, 1247–1257 (2014).
Gu, H. et al. Salmonella produce microRNA-like RNA fragment Sal-1 in the infected cells to facilitate intracellular survival. Sci. Rep. 7, 2392 (2017).
Klepsch, V. et al. Nuclear orphan receptor NR2F6 as a safeguard against experimental murine colitis. Gut 67, 1434–1444 (2018).
Song-Zhao, G. X. & Maloy, K. J. Experimental mouse models of T cell-dependent inflammatory bowel disease. Methods Mol. Biol. 1193, 199–211 (2014).
Zuo, L. et al. Targeting delivery of anti-TNFalpha oligonucleotide into activated colonic macrophages protects against experimental colitis. Gut 59, 470–479 (2010).
Hung, Y. P. et al. Proton-pump inhibitor exposure aggravates Clostridium difficile-associated colitis: evidence from a mouse model. J. Infect. Dis. 212, 654–663 (2015).
Israelsen, W. J. et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 (2013).
Koch, S. et al. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology 141, 259–268 (2011).
Bian, Z. et al. Role of miR-150-targeting c-Myb in colonic epithelial disruption during dextran sulphate sodium-induced murine experimental colitis and human ulcerative colitis. J. Pathol. 225, 544–553 (2011).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Ashizawa, K., Willingham, M. C., Liang, C. M. & Cheng, S. Y. In vivo regulation of monomer–tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 266, 16842–16846 (1991).
Christofk, H. R., Vander Heiden, M. G., Wu, N., Asara, J. M. & Cantley, L. C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186 (2008).
Anastasiou, D. et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847 (2012).
Babbin, B. A. et al. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 179, 8112–8121 (2007).
Wei, Y. et al. Pyruvate kinase type M2 promotes tumour cell exosome release via phosphorylating synaptosome-associated protein 23. Nat. Commun. 8, 14041 (2017).
Zhang, J. et al. Nuclear translocation of PKM2 modulates astrocyte proliferation via p27 and -catenin pathway after spinal cord injury. Cell Cycle 14, 2609–2618 (2015).
Liu, F. et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 19, 1358–1370 (2017).
Prakasam, G. et al. Pyruvate kinase M knockdown-induced signaling via AMP-activated protein kinase promotes mitochondrial biogenesis, autophagy, and cancer cell survival. J. Biol. Chem. 292, 15561–15576 (2017).
Thaker, A. I. et al. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 145, 416–425 e411-414 (2013).
Zheng, Q. et al. Long noncoding RNA MEG3 suppresses liver cancer cells growth through inhibiting beta-catenin by activating PKM2 and inactivating PTEN. Cell Death Dis. 9, 253 (2018).
Yang, W. et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150, 685–696 (2012).
Liang, J. et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 27, 329–351 (2017).
Acknowledgements
We thank Drs. Jill Leslie Littrell and Koby Shane Kidder (Georgia State University, Atlanta, GA) for critical reading and constructive discussion of the manuscript. This work was supported by grants from the Ministry of Science and Technology of China (2018YFA0507100), National Natural Science Foundation of China (91640103, 31801088, 31670917, 31770981, 31571458, 31870821), Natural Science Foundation of Jiangsu Province (BK20170076), Six talent peaks project of Jiangsu Province, and Fundamental Research Funds for the Central Universities (020814380095, 020814380082).
Author information
Authors and Affiliations
Contributions
K.Z., F.W. and C.-Y.Z. designed the experiments. X.S., L.Y., D.W., L.Y., J.C., H.L., G.X., Y.W., L.L. and K.W. performed the experiments and analyzed results. X.S. and K.Z. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Sun, X., Yao, L., Liang, H. et al. Intestinal epithelial PKM2 serves as a safeguard against experimental colitis via activating β-catenin signaling. Mucosal Immunol 12, 1280–1290 (2019). https://doi.org/10.1038/s41385-019-0197-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41385-019-0197-6
This article is cited by
-
lncRNA PVT1 regulates chondrocyte proliferation and apoptosis through the glycolysis process mediated by miR-552-3p/PKM2 to promote osteoarthritis
In Vitro Cellular & Developmental Biology - Animal (2025)
-
The Heterochromatin protein 1 is a regulator in RNA splicing precision deficient in ulcerative colitis
Nature Communications (2022)
-
Diethyldithiocarbamate-copper complex (CuET) inhibits colorectal cancer progression via miR-16-5p and 15b-5p/ALDH1A3/PKM2 axis-mediated aerobic glycolysis pathway
Oncogenesis (2021)
-
TKT maintains intestinal ATP production and inhibits apoptosis-induced colitis
Cell Death & Disease (2021)
