
Abstract
Autoimmune disorders are characterized by aberrant T cell and B cell reactivity to the body’s own components, resulting in tissue destruction and organ dysfunction. Autoimmune diseases affect a wide range of people in many parts of the world and have become one of the major concerns in public health. In recent years, there have been substantial progress in our understanding of the epidemiology, risk factors, pathogenesis and mechanisms of autoimmune diseases. Current approved therapeutic interventions for autoimmune diseases are mainly non-specific immunomodulators and may cause broad immunosuppression that leads to serious adverse effects. To overcome the limitations of immunosuppressive drugs in treating autoimmune diseases, precise and target-specific strategies are urgently needed. To date, significant advances have been made in our understanding of the mechanisms of immune tolerance, offering a new avenue for developing antigen-specific immunotherapies for autoimmune diseases. These antigen-specific approaches have shown great potential in various preclinical animal models and recently been evaluated in clinical trials. This review describes the common epidemiology, clinical manifestation and mechanisms of autoimmune diseases, with a focus on typical autoimmune diseases including multiple sclerosis, type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus, and sjögren’s syndrome. We discuss the current therapeutics developed in this field, highlight the recent advances in the use of nanomaterials and mRNA vaccine techniques to induce antigen-specific immune tolerance.
Similar content being viewed by others

Introduction
Autoimmune disorders such as multiple sclerosis (MS), type 1 diabetes (T1D) and rheumatoid arthritis (RA) occur when autoreactive immune cells, especially T cells and B cells are overactivated and recruited to cause self-tissue damage.1,2 By far, researchers have discovered about 150 types of autoimmune diseases and adopted a series of treatment measures.3 The diversity and rapid rise of autoimmune diseases challenge the health care system and the entire pharmaceutical industry. Current drugs available for the treatment of autoimmune diseases are non-specific and have side-effects such as infection, allergy and malignant disease.4 Instead, antigen-specific immunotherapies for autoimmune diseases aim to induce tolerization toward autoantigens without suppressing the systemic immunity.
New therapies are developed based on a detailed understanding of the mechanisms of autoimmune diseases.4 In this review, we describe the epidemiology, clinical diagnosis, pathogenesis, mechanisms and therapies of autoimmune diseases. We provide a timeline to summarize the significant advances in the field of antigen-specific immunotherapy for the treatment of autoimmune diseases. We describe the different strategies developed for non-specific biotherapeutics as well as antigen-specific immunotherapy, and the delivery methods to induce immune tolerance. We also summarize the Food and Drug Administration (FDA) approved drugs for autoimmune diseases and antigen-specific therapies that have entered clinical trials. The most recent biomaterial-based and mRNA vaccine strategies for inducing antigen-specific tolerance are highlighted.
Basic information of autoimmune diseases
Common epidemiology
Autoimmune diseases have been shown to affect 3–5% of the population and become one of the most important public health problems.5,6 Recently, Conrad et al. reported a population-based cohort study of 19 autoimmune diseases in the UK about 22,009,375 individuals from 2000 to 2019.7 During this period, 978,872 individuals were newly diagnosed with autoimmune diseases and the average age of these individuals was 54, however, autoimmune diseases can occur in almost all age groups (0–95 years). Besides, 63.9% of these newly diagnosed patients are female, and the age and sex standardized incidence rates increased. The incidence of celiac disease and Sjogren’s syndrome increased. Autoimmune diseases affect about 10% of the population in this study and consume considerable social resources.7 In addition, some autoimmune diseases show seasonal and regional variations which may provide a guidance direction for autoimmune disease prevention and therapy.8,9
Immune dysregulation
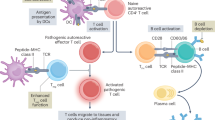
Autoimmune diseases are characterized by immune disturbances that cause the aberrant activation of autoreactive immune cells, resulting in tissue damage. Immune tolerance is established both centrally and peripherally.10,11 As we all know, T cells undergo positive and negative selection in the thymus before entering the periphery to perform immune functions. The negative selection of autoreactive T cells in the thymus is the major mechanism of central immune tolerance12 (Fig. 1). Besides, peripheral tolerance-related mechanisms can further limit the expansion of autoreactive cells through clonal deletion, immune anergy or the induction of regulatory T cells.13,14 Peripheral clonal deletion is mainly through activation-induced cell death or restimulation-induced cell death (RICD).15,16 Immune anergy mainly exerts its mechanism through various costimulatory molecules (like CTLA-4) and immune regulatory-related cells.17 Besides, follicular DCs and helper T cells can also affect the immune tolerance condition.18

Pattern diagram of the mechanisms of autoimmune diseases. After differentiation of hematopoietic stem cells, progenitor T cell (pro-T cell) will leave the bone marrow and enter the thymus, and differentiate from double-negative (DN) T cells into double-positive (DP) T cells. Under death by neglect, negative selection, and positive selection via thymic epithelial cells, single positive T cells with low avidity to autoantigens-MHC complexes survive and differentiate into CD4 or CD8 and enter the periphery. However, some autoreactive T cells can avoid these select clearance effects and enter the peripheral. These autoreactive T cells include three types: (1) molecular mimicry, TCR can recognize the autoantigens and foreign antigens similar to autoantigens such as viruses and some bacteria. (2) dual TCRs, one TCR can recognize the non-autoantigens and another can recognize the autoantigens. (3) chimeric TCR, different Vα and Vβ combinations can recognize the autoantigens and non-autoantigens. Viruses, bacteria, and other autoantigens lead to the necrosis of autologous cells and result in the release of autoantigens. Some bacteria similar to autoantigens can induce the activation of these T cells susceptible to autoantigens and promote the autoimmune disease. Besides, the stimulation of external antigens can promote the continuous inflammatory environment and lead to the highly activated immune state of T and B cells. These T cells can secrete various inflammatory cytokines, activate B cells and recruit many immune cells, and induce inflammatory reaction. Eventually this will lead to the occurrence and development of autoimmune diseases. (Part of the figure was modified from Servier Medical Art(http://smart.servier.com/), licensed under a Creative Common Attribution 4.0 Generic License. (https://creativecommons.org/licenses/by/4.0/)
T cells and B cells have been well investigated for their role in initiating and sustaining of autoimmune diseases. During autoimmunity, autoreactive T cells infiltrated into the target tissue. CD8+ cytotoxic T cells can directly contact and kill the targeted cells. CD4+ T cells can release large amounts of proinflammatory factors or provide activation signals to B cells. These proinflammatory factors recruit many myeloid inflammatory cells to specific tissue and executive-related immune response. Mature B cells can differentiate into plasma cells and secrete a large number of autoantigen-targeting antibodies (Fig. 1). Autoantibodies activate the complement system or kill the targeting cells by antibody-dependent cell-mediated cytotoxicity. Besides, the formation of antigen-antibody complexes is critical for some autoimmune diseases such as SLE. In SLE, these complexes deposit in the kidney and stimulate the inflammatory response in local tissue to cause tissue damage.19
Genetic factors
The breakdown of immune tolerance is based on genetic susceptibility. Human leukocyte antigens (HLA) gene fragment is the most relevant gene to immune system, and variation of some loci in this region may promote the occurrence of autoimmune diseases.20 PTPN22 gene outside the major histocompatibility complex (MHC) region plays an important role in many autoimmune diseases including RA, Systemic lupus erythematosus (SLE), etc.21,22 Besides, the variation of gene coding antigens can also promote the occurrence of autoimmune diseases.23 Although most autoimmune diseases are polygenetic, some monogenic variations also have a strong impact on autoimmune diseases such as complement-related genes, nuclease hydrolysis-related genes and immune regulation-related genes.24,25,26 Researchers also emphasize the epigenetic factors for autoimmune diseases.27,28,29 Females are more frequently affected by autoimmune diseases than males. This gender bias is associated with hormones and X chromosome.30,31,32
Environmental triggers
Many environmental factors have been associated with the development of autoimmune diseases. Meanwhile, these factors also reflect the pathogenesis of autoimmune diseases. Molecular mimicry hypothesis suggests that molecular mimicry is one of the environmental factors that leads to the break of tolerance and elicits autoimmune responses. It occurs when exogenous antigens similar to autoantigens induce the activation of autoreactive T cells or B cells in a susceptible individual.33 In addition, the models of dual TCRs and chimeric TCR also raise other possibilities34,35 (Fig. 1). Researchers also considered the exposure of pathogen-associated molecular patterns such as endotoxin or lipopolysaccharide repeatedly can stimulate the innate immune responses and enhance the adaptive immune responses. T and B cells will be in a state of highly activated immune state in this case.36 Multiple infectious agents have been suggested to play a role in autoimmune diseases. For example, Epstein-Barr virus (EBV) can stimulate the innate and adaptive immune responses simultaneously because the protein structure of this virus is similar to RNA binding proteins.37,38,39 EBV has been associated with many autoimmune diseases such as MS,40 SLE41, and RA.42 The disturbance of the composition of microbiota (Fungi, bacteria, viruses, etc.) located in gut, mouth and skin of the host can affect the host immune system43 (Fig. 1). Besides, these coexisting microorganisms can translocate in blood circulation and locate in the tissue to trigger immune responses locally.44,45 Some microorganisms may regulate biological metabolic process to promote immunity.46,47 The nutrition change in some Western countries coincides with the rise in autoimmune diseases. This may be explained by interactions among dietary, gut microbiota, metabolites and immune cells.48 Smoking has also been reported to affect the progress of autoimmune diseases but the mechanism is still not clear.49
Molecular signaling pathways related with autoimmune diseases
The activation of immune cells requires the involvement of several molecular pathways and membrane surface molecules, which are closely related with autoimmune disease pathogenesis50 (Fig. 2). Here we also make a general description of some signal pathways and related molecules about T and B cell activation. CD28 system-related molecular pathways including CD28, CTLA4, and the shared ligands (CD80 and CD86) mainly are associated with the activation, proliferation and survival of T cells and this pathway is PI3K dependent. The YMNM sequence at the tail of CD28 is activated, and then the p85 subunit is combined with it subsequently. Activated PI3K will recruit proteins such as PDK1 and PKB/AKT, and then induce the activation of downstream targets, including mTOR, IκB, GSK3β and Bad, which can regulate the activity of transcription factors.51,52 CD28-deficient mice show the impaired germinal center and fail to generate normal levels of immunoglobulin.53,54,55 CD28 deficiency can delay disease progression and reduce disease severity in various autoimmune disease models including EAE,56 MRL/lpr model of SLE57 and collagen-induced arthritis model of RA.58 CTLA4 pathway can inhibit the CD28 pathway by binding the same ligands (CD80 and CD86).59,60,61 Targeting CTLA4 drugs have been applied in clinical trials in psoriasis and juvenile idiopathic arthritis.62,63 ICOS pathway will be upregulated after activation of CD4+ T cells and it can also mediate PI3K-AKT signal pathway for cell activation.64,65 ICOS is closely related to T follicular helper (Tfh) cells via IL-21 and IL-4 secretion.66 Hence, autoantibodies-related autoimmune diseases mentioned above are greatly influenced by ICOS pathway.67,68,69 Other CD28 superfamily members also include PD1 and BTLA which can inhibit immune activation.70,71 PD1 agonists can effectively reduce the severity of collagen-induced arthritis72,73 and colitis models induced by dextran sodium sulfate or T cell transfer.74

Related molecular pathways and membrane surface markers. OX40-OX40L, TRAF2/TRAF5/TRAF6 will induce the form of IKKα/β/γ which further leads to NF-κB entering the nucleus. Besides, OX40-OX40L can promote PI3K/Akt pathway and cause STAT5 to enter the nucleus. CD40-CD40L will recruit various downstream molecules. TRAF1, TRAF2, TRAF3, and TRAF5 bind competitively the one CD40 tail site and TRAF6 can bind to another individually. They can promote the Ras/ERK pathway and the non-classical NF-κB pathway, NIK pathway. Besides, it can promote the TAK1 and MKKs/p38 pathways. CD40-CD40L can start the JAK3/STATs pathway. CD28-B7-1/B7-2 also provides the activation signal. After the tyrosine phosphorylation of the YMNM fragment, the subunit p85 of PI3K binds to YMNM. PI3K will recruit PDK1 and PKB/Akt, and PKB can phosphorylate downstream targets such as mTOR, IκB, GSK3β and Bad after PKB is phosphorylated by PDK1 which leads to an increase of the transcriptional activity of NF-κB and NFAT. Besides, CD28 signal will recruit GRB2/GADs and increase NF-κB, NFAT, and AP1 by Vav catalysis. CTLA-4 also binds B7-1/B7-2, but it transmits the suppression signal to downstream. The specific process is through the inhibition of ZAP70 and PI3K/Akt pathway by recruitment of SHP2 and inhibition of PI3K/Akt pathway by PP2A. The combination with PD-1 and PD-L1 leads to the activation of the tyrosine phosphorylation of the ITIM and ITSM at the tail of PD1. SHP-1 or SHP-2 can bind the ITSM and promote the expression of PTEN which can further inhibit the activation of PI3K/Akt pathways and ZAP70. The SHP2 can also promote the BATF to enter the nucleus. It leads to the inhibition of T cell proliferation and inflammatory progression. This inhibitory process may be somewhat similar to the CTLA-4 pathway
The binding of CD40 on T cells and CD40L on B cells can promote B cells interior recruits the TNFR-associated factors (TRAFs), and reaction molecules include NIK, inhibitor of NF-κB kinase and TPL2 which lead to the activation of transcription factors such as NF-κB and AP1 at last.75 CD40-CD40L is a universal signal for various immune cells to induce widespread downstream immune function, especially in humoral immunity76 including T cell-dependent antibody production, formation of germinal centers and differentiation of memory B cells.77,78 In addition to antibody induction, CD40 pathway can also result in inflammatory factors including TNF and matrix metalloproteinases (MMPs) for joint destruction in RA.79 CD40 is also continuously expressed on salivary gland ductal epithelial cells and endothelial cells80 to up-regulate adhesion molecules for inflammatory progression in Sjögren’s syndrome (SS).81,82 Blocking CD40 pathway can decrease disease activity and clinical remission in a RA clinical trial.83 The similar therapeutic effect also appears in SS model treated with anti-CD40L.84 Besides, the binding of OX40 on T cells and OX40L on antigen presenting cells (APCs) is another important pathway for immune activation. The downstream of OX40 will induce many signal pathways such as PI3K-AKT, NF-κB, and MAPK by recruiting TRAF2, TRAF5, and other molecules.85,86 TNF receptor family also includes TNF, BAFF, APRIL, RANK, etc.50 OX40-OX40L mainly promotes the differentiation of helper T cell subsets, cell proliferation and activation, and the secretion of immune-related cytokines.85 Polymorphisms of the OX40L corresponding gene (TNFSF4) have been especially correlated with SLE,87,88,89 and have a general correlation with SS,90 system sclerosis91,92 and sleep disorder narcolepsy.93 The inhibition of OX40 showed the potential for atopic dermatitis treatment in a phase 2a clinical trial.94 Each signaling pathway interacts with each other to form a complex and multidimensional signaling network to maintain immune homeostasis. Researchers tried to treat autoimmune diseases by targeting these pathways to inhibit the expansion of inflammatory effects.
T cells and autoimmune disorders
T cells are the main cell type responsible for maintaining tolerance and play a key role in many autoimmune diseases. In this section, we describe autoimmune diseases that are characterized by inappropriate activation of autoreactive T cells and break of T cell tolerance. We review the clinical-related information and pathogenesis of T cell-mediated diseases including MS and T1D.
Multiple sclerosis
Epidemiology, genetic factors, and environmental triggers
MS is an inflammatory demyelinating disease that affects the central nervous system (CNS) and is the most common cause of non-traumatic disability in young people.95,96,97,98 There are about 2.8 million people living with MS and a new patient appears every 5 min all over the world.99 The incidence rate for females is twice that of males, but the ratio can even reach 4:1 in some countries.97,99 MS is a complex autoimmune disease with substantial heterogeneity among patients. Researchers have discovered that the HLA-DR15 haplotype may be the major consideration for MS risk genetically.100 Besides, another large genetic research established a reference map about susceptibility genes of MS based on big data processing, which includes 200 autosomal susceptibility variants outside the MHC region, 32 variants in the MHC region and 1 variant in chromosome X.101 The MS risk-related gene map can help us to continue to deeply investigate the mechanisms of MS. For environmental factors, researchers have shown that the commensal microbiota in the human intestine may affect the occurrence of MS, based on their role in maintaining immune cell homeostasis. Disturbance in the composition of microbiota may trigger MS.102,103 Some researchers also point that EBV infection is essential for MS, and there is evidence to support that the MS prevalence of people with EBV infection is 32 times more likely than that of other virus infections.104 The mechanism of EBV infection to cause MS is still not clear, current studies suggest the role of molecular mimicry mentioned above in causing the break of immune tolerance and the development of autoimmune disorders.105 People in higher latitudes are more likely to have MS and researchers inferred that stronger ultraviolet light in high latitudes will affect the level of Vitamin D which can further affect the onset and prevalence of MS.106,107 Obesity and smoking have also been reported to have a certain correlation with MS.108,109
Clinical manifestation and diagnosis
The majority of MS patients will experience a relapse remission phase called Relapsing-Remitting MS (Rel-Rem MS, RRMS) characterized by acute relapses followed by partial recovery. Over time, about 80% of patients with RRMS will develop to the secondary process called secondary-progressive MS (SPMS) at which time the patient’s condition will deteriorate suddenly.97 Primary-progressive MS (PPMS) accounts for around 15% of MS patients which is characterized by a progressive disease course without a relapsing-remitting phase onset. The clinical manifestations of MS include cognitive impairment, motor impairment, fatigue, visual disorders and sensory disorders.110,111
For MS diagnosis, the combination of clinical, imaging and laboratory evidence is used. The diagnosis of MS via the detection of CNS lesions by T2-weighted scans or the contrast agent gadolinium from magnetic resonance imaging (MRI) and some other diagnostic methods are in continuous development such as positron emission tomography (PET) imaging technology.112,113,114 In addition, the detection analysis of cells and IgG antibodies, protein concentration, pleocytosis, and some immune cells in cerebrospinal fluid (CSF) and CSF oligoclonal bands are equally important to provide evidence for clinical diagnosis of MS.115,116 However, there are still no clear blood biochemical indicators available that can reflect the development of MS accurately. In addition, temporal and spatial development of clinical manifestations can provide the diagnostic basis for MS.116
Immune dysregulation in MS
The cause of MS remains elusive. The development of MS may start from the dysregulation of peripheral immune tolerance or CNS intrinsic events. The autoreactive T cells activated at peripheral traffic to the CNS through the blood-brain barrier (BBB) via some adhesion molecules (VCAM-1 and ICAM-1) to attack the myelin sheath formed by oligodendrocytes in CNS, meanwhile trigger more immune-activated cells infiltration to CNS, up-regulate the inflammatory signaling pathways and induce more inflammatory cytokines. Myelin-reactive T cells can migrate into the bone marrow in a CXCR4-dependent manner to skew hematopoietic stem cells (HSCs) toward myeloid lineage and augment CNS inflammatory injury and demyelination.117 Researchers suggested that the activation of memory B cells can drive the autoproliferation of Th1 brain-homing cells via HLA-DR.118 This work provides an explanation for the efficacy of anti-CD20 therapy for MS. Epitope spreading causes the change of autoantigens during the disease progression and gives rise to pathogenic T cell clones that evade regulation by Treg cells and trigger more damage.95,97 This is also the key and difficult point of treatment (Fig. 3a).

Pattern diagram of some typical autoimmune diseases. a Mechanism diagram of MS. Autoreactive T cells enter the CNS through the adhesion molecules on the BBB and trigger local inflammation of the CNS which causes the demyelination reaction and neuronal cell death. b Mechanism diagram of T1D. DCs induce the generation of autoreactive T cells which promote the local inflammation of the pancreas and cause the death of pancreatic β cells which lead to impaired glucose metabolism. c Mechanism diagram of RA. After the activation of induced autoreactive T cells by DCs, various immune cells in the joint cavity begin to execute abnormal programs and fibroblasts will proliferate. The autoreactive antibodies released by B cells can form immune complexes which further expand local inflammation. It ultimately causes the death of osteocytes and osteoarticular injuries. d Mechanism diagram of SLE. It most often involves the kidney, and the pathological change is similar to RA. Immune complexes and complement will deposit in the glomerulus and promote the inflammatory reaction which causes kidney damage finally. e Mechanism diagram of SS. The mechanism of abnormal activation of immune cells is similar to the aforementioned diseases. But it mainly occurs in salivary and lacrimal glands which leads to the epithelial cell death and loss of the function. (Part of the figure was modified from Servier Medical Art(http://smart.servier.com/), licensed under a Creative Common Attribution 4.0 Generic License. (https://creativecommons.org/licenses/by/4.0/)
The cells involved in MS include T cells, B cells, APCs, myeloid cells and some glial cells. Th1 and Th17 play main roles in attacking the myelin sheath specifically by secreting inflammatory-related cytokines and CD8+ T cells contribute to disease pathogenesis via a FasL-dependent mechanism that promotes lesion formation.119,120,121 B cells secrete antibodies or inflammatory cytokines to attack the myelin sheath. Besides, other inflammatory cells also secrete proinflammatory factors such as IFN-γ, TNF-α, IL-17, IL-23, etc. Foxp3+CD4+ T cells, IL-10+ T cells (TR1),122,123 and some regulatory B cells (Bregs) can secrete IL-10, TGF-β, IL-35, and other anti-inflammatory factors.124,125 Astrocytes are the initiators to create the inflammatory environment by generating MMPs, ROS, TNF-α, and RNS. In this pathological environment, CNS will be severely damaged and eventually lead to disease-related features97,126,127,128 (Fig. 3a).
Type I diabetes
Epidemiology, genetic factors, and environmental triggers
Type I diabetes (T1D) is a common autoimmune disease closely related with pathological T cell activation which is characterized by T cell infiltration into pancreatic islets and triggers immune responses against β-cell antigen.129,130 Approximately 8.4 million patients suffer from this disease worldwide, and the total incidence rate is increasing by 2–3% annually.131,132 According to the Markov model approach, researchers predicted that the affected populations will reach about 13.5–17.4 million in 2040.131 Availability and affordability of medicines for diabetes are poor in lower-middle-income countries.133 Although T1D can be diagnosed in any age group, the common population are children and adolescents. The peak manifestation period of T1D is between the ages of 5 and 7, as well as the pre-puberty period.134,135 Unlike typical autoimmune diseases, T1D is not biased towards females in terms of gender and the incidence rate of males will be slightly higher.136
T1D is a typical polygenic hereditary disease and susceptibility of T1D is strongly associated with genes that encode classical HLA. HLA DRB1*0301-DQA1*0501-DQ*B10201 (DR3) and HLA DRB1*0401-DQA1*0301-DQB1*0301 (DR4-DQ8) have been shown to increase disease susceptibility by 50%. In addition, DRB1*1501-DQA1*0102-DQB1-0602 (DR15-DQ6) appears to be protective.137,138,139 MHC-I-related genes also have an impact on the development of diseases and the mechanism of the effect is independent of MHC-II. More than 60 genes outside the HLA loci region such as CTLA4, PTPN22, KIR, VNTR, IL2RA, INS, etc. also contribute to T1D.137,139 Environmental triggers, daily dietary habits, and related enterovirus infection are associated with the development of T1D.140 Susceptibility factors such as obesity, vitamin D levels, virus infection, and human microbiota are similar to other autoimmune diseases.
Clinical manifestation and diagnosis
Fatigue, weakness, and lethargy will run through the entire disease process for T1D patients. If not treated in a timely manner, it will trigger a series of microvascular complications such as blindness, kidney failure, amputation, terminal sensory impairment, myocardial infarction and cerebral infarction.141
For patients with classical symptoms, diagnosis is based on the fasting blood glucose above 7 mmol/L, and 2-h plasma glucose value (2-h PG) above 11.1 mmol/L during the oral glucose tolerance test. Besides, it may be diagnosed by A1C concentration above 48 mmol/mol.142 Acute onset of T1D should be diagnosed by plasma glucose rather than A1C assay. C-peptide concentration as the marker of endogenous insulin level can serve as a diagnostic reference. However, it’s not enough to distinguish type I and type II diabetes only via the above detection methods.129 Biomarkers such as insulin autoantibodies and glutamic acid decarboxylase autoantibodies should be detected.143,144 T1D is defined by the presence of one or more such biomarkers.
Immune dysregulation in T1D
The analysis of biomarkers indicating the process from disease susceptibility to active immunity, and finally to the loss of autoimmune regulation, leads to the comprehensive understanding of T1D disease pathogenesis.143 The onset of T1D is considered to be the presentation of β-cells-related peptides via APCs to naïve T cells in pancreatic lymph nodes. These naïve T cells contacted with APCs escaped to the periphery because of the abnormal genetic variation mentioned above when they undergo both positive and negative selection in thymus.145 The activated T cells will further differentiate into functional effector and memory T cells. Part of CD4+ T cells will assist B cell differentiation to plasma cells to produce multiple anti-β-cells antibodies and others will continue to secrete inflammatory cytokines.145 Various myeloid inflammatory immune cells can enter pancreatic islets via gradient changes of chemotactic factors and attraction of numerous inflammatory cytokines for inflammatory environment expansion.146 More CD8+ T cells can release killing factors such as perforin and granular enzymes when they contact with β-cells directly.147 These immune cells will lose the immune regulation function gradually with the T1D development.129,130,148 The entire pancreatic islet shows immune infiltration and overall pancreatic manifestations are reduced volume, morphological atrophy and loss of secretion function149 (Fig. 3b).
Autoantibodies and autoimmune disorders
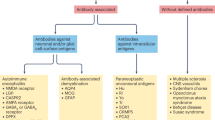
Autoimmune diseases mainly mediated by antibodies tend to be more like systemic syndrome caused by immune system disorders. From the initial immune imbalance of a single organ or tissue, almost all kinds of organs in the body will be affected because of the occurrence of epitope diffusion. Here we review the clinical-related information and pathogenesis of autoantibodies-related diseases including RA, SLE and SS.
Rheumatoid arthritis
Epidemiology, genetic factors, and environmental triggers
RA is a common systemic autoimmune disease and chronic inflammatory arthritis characterized by symmetric and polyarticular pain. RA mainly accumulates synovium and surrounding soft tissue at the synovium.150,151,152 The incidence of RA varies widely across the world which is reflected in the higher incidence in Europe and North America, and lower incidence in Southeast Asia region.153 The age-standardized prevalence and incidence increased by 7.4% and 8.2%, respectively.154 All ages are at RA risk, but the risk increases significantly after the age of 40.155 The male-to-female sex ratio also increases with age from 1:2 in the young to 1:4 in the old, which may be caused by the decline of estrogen levels after menopause in women.153,156
HLA-DR locus is the most important genetic risk factor for RA. Researchers found the key 5 amino acid sequences (70–74) of the HLA-DRβ chain called a shared epitope.157,158 Other genetics regions such as PTPN22, PADI4 and TNFRSF11A in non-MHC regions also contribute to the RA occurrence even if the contribution is not particularly significant.159,160,161 Besides, epigenetic modifications such as DNA methylation will increase RA susceptibility.162 For environmental factors, smoke seems to be the most important for RA.163 The reason may be that exposure to cigarette smoke promotes the pulmonary mucosal and draining lymph nodes prior to inflammation and then induces immune disorder inside the organism.164,165 Besides, some infection factors such as EBVs, retroviruses and bacteria especially in the oral cavity and the interaction of many microorganisms influence RA occurrence, but the specific mechanism is still unclear.166,167,168 Obesity and sodas are also important for RA.169 However, it is worth noting that alcohol intake seems to provide protection against RA, and some groups considered this may be related to the change of the microbial structure composition by alcohol.170 Some trials also demonstrated that long-term supplements of Vitamin D and omega-3 fatty acids can decline the RA incidence.171,172
Clinical manifestation and diagnosis
For most patients, the clinical symptoms show the gradual pain and swelling of joints early and the chronic inflammation of almost whole-body joints at later RA stages. The wrists and finger facet joint usually have an obvious manifestation at an early stage. With the development of RA, the large joints such as shoulders and knees will show corresponding symptoms. Affected joints will become bloated, and even develop into deformity and cause limited movement in severe cases.150 The affected joints will progress from active inflammation to irreversible lifelong damage without treatment. Morning stiffness is the characteristic performance of RA, and it usually lasts 30 min or longer time with fatigue and weakness simultaneously. Serious patients may have a high level of C-reactive proteins (CRP) and erythrocyte sedimentation rate (ESR), and some patients may have a fever and weight loss. Furthermore, RA can increase the incidence of cardiovascular disease, and it is mainly manifested in a functional lesion of the coronary artery. Some patients also developed pulmonary fibrosis and inflammation of the respiratory system with RA expansion.173,174,175
The American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) revised the diagnostic criteria in 2010. The new diagnosis was made by the overall score of the 4 dimensions which include the number counting of joint involvement, rheumatoid factor (RF) antibody and anti-cyclic citrullinated peptide antibodies (ACPAs) titers in serum, CRP and ESR of acute phase reactants and whether the duration of symptoms lasts for 6 weeks. When the score is more than 6, RA can be confirmed clinically.176,177
Immune dysregulation in RA
A variety of autoantibodies, mainly ACPAs and RF antibodies, are the initiators of this disease.178,179 The cell-cell interaction of specific immune cells within the synovium is the basis for RA occurrence. APCs represented by DCs present the RA-related antigens such as citrullinated peptides to T cells with the major phenotypes as CD4+ PD1+CXCR5-, and they are also called peripheral helper T (Tph) cells that generate IL-21 primarily within the synovium.180 Besides, some CD8+GZMK+ T cells also appear to generate IFN-γ.181 Tph cells can assist B cells to differentiate into plasm cells and generate a large number of antibodies along with IL-6 and GM-CSF to attack the tissue in the synovium. In this process, macrophages, neutrophils and other myeloid cells can provide the inflammatory environment. Numerous fibroblasts also emerge under the action of TNF-α, IL-12, IL-13, IL-17 and TGF-β, and amplify inflammatory effects.182,183,184 Monocytes will further differentiate into osteoclasts to release related proteases for bone erosion and cartilage loss. Researchers found a distinct population of CX3CR1+ tissue-resident macrophages that exert immune regulatory function by maintaining a tight-junction-mediated barrier and restricting inflammation.185 These multiple pathways and mechanisms expand into a systemic autoimmune response without effective treatment151,186,187 (Fig. 3c).
Systemic lupus erythematosus
Epidemiology, genetic factors, and environmental triggers
SLE is an autoimmune disease characterized by producing anti-nuclear autoantibodies and causing the immune complex deposition in various organs, and it leads to chronic and systemic diffuse connective tissue disease that mainly affects young women.188,189,190 The overall global prevalence and the incidence of SLE are about 0.3–0.5% and 0.0022–0.0231%, respectively.188 The annual age-standardized mortality rate of patients is higher than many other autoimmune diseases, and about 2.7 deaths per million inhabitants in 2014.191 The mortality rate of women is much higher than that of men. Black, Asian, and Spanish populations have a higher risk than the white population for SLE and the clinical manifestation of diseases is more serious.191,192,193,194 It is worth noting that about 90% of patients are women, and most of them are of childbearing age and presenting diversity in SLE performance can significantly affect fertility function.195
HLA-II gene region is the susceptible locus of SLE, and HLA-DRB1 has the strongest correlation with SLE. Studies have shown that HLA-DRB1*03:01 is related with the generation of anti-Ro and anti-La autoantibodies and HLA-DR3 has a strong connection with anti-dsDNA antibodies. A high-density case-control single nucleotide polymorphism research in the MHC region identified the independent and interacting sites of HLA-DPB1, HLA-G and MSH5.196 Besides, mutations in complement pathway-related genes are a high risk for SLE because of the obstacle to cleaning the cellular debris. Monogenic influence on SLE to cause high-IFN levels is also undeniable, and these monogenic groups include DNASE1/DNASE1L3, PRKCD, TREX1, STING, SAMHD1, etc.196 Epigenetic modification is also an important genetic reason.197 Like many other autoimmune diseases, smoking and EBV infection can induce the pathogenesis and moderate drinking provides a protective mechanism.198,199 The difference is that mercury and silica exposure are important environmental factors for SLE because of their function as an adjuvant to induce the transcription of proinflammatory cytokines and T-cell responses.200,201
Clinical manifestation and diagnosis
The clinical features of SLE are heterogeneous and various organs are affected. The patients usually have constitutional symptoms and fevers, and many patients also exhibit skin and mucosal symptoms such as butterfly erythema, mucosal ulcer (usually appearance in oral and nasal cavities) and alopecia. Butterfly erythema is a typical symptom appearing as red patches located on the bridge of the nose or both sides of the cheekbones.190,192 Many patients have joint and bone pain complications similar to RA, which are also symmetrical and have morning stiffness. Some patients also have chest, pericardium, and peritoneal fluid once the serosal inflammation progresses to a certain extent. Lupus nephritis is a major visceral manifestation of RA, the patients will experience hematuria, proteinuria, and possible systemic edema at last.202 Besides, SLE can affect the cardiovascular system to cause pericarditis, endocarditis and coronary artery lesions, the respiratory system to cause pulmonary arterial hypertension and pulmonary fibrosis, and the digestive system to cause pancreatitis and a series of intestinal diseases.190,203,204 Therefore, early identification and intervention are necessary to prevent serious and irreversible pathological damage.
EULAR and ACR developed new classification criteria in 2019 that include positive antinuclear antibodies (ANA) followed by 7 clinical (constitutional, hematological, neuropsychiatric, mucocutaneous, serosal, musculoskeletal, renal) and 3 immunological (anti-phospholipid antibodies, complement proteins and SLE-specific antibodies inspection) items.205 Anti-nuclear antibodies at a titer of ≥1:80* on HEp-2 cells or an equivalent positive ANA test should be used as the entry criterion.205
Immune dysregulation in SLE
The pathogenesis of SLE is complex, with non-immune cells, innate immune responses and adaptive immune responses participating in the disease process. Endogenous nucleic acid combined with autoantibodies in the form of immune complexes (ICs) has the potential to drive the production of IFN-α in plasmacytoid dendritic cells which is pivotal in the pathogenesis of SLE.206 Besides, Janus kinase (JAK)-signal transducer activator of transcription (STAT) pathway and Bruton’s tyrosine kinase (BTK) pathway have been shown to play important roles in the pathogenesis of SLE.207,208 The inflammatory environment promotes adaptive immune response, and APCs dominated by DC can present autoantigens to T cells. These activated T cells further expand inflammatory response by releasing more inflammatory cytokines (TNF, B lymphocyte stimulator, etc.) and simultaneously assist in the activation of B cells.209 B cells undergo differentiation to plasma cells to produce massive autoantibodies and form complexes with numerous nucleic acids and related proteins. ICs can deposit and promote an intense inflammatory response to damage the corresponding organs and tissues, and ultimately lead to the development of SLE206 (Fig. 3d).
Sjögren’s syndrome
Epidemiology, genetic factors, and environmental triggers
Sjögren’s syndrome (SS) is a systemic and chronic autoimmune disease characterized by inflammatory reaction of exocrine organs including but not limited to lacrimal and salivary glands that lead to the drying of the mouth, eyes, respiratory tract, and vagina eventually.210,211,212 The prevalence and incidence of SS is about 0.01–0.72% and 0.003–0.011% in the population, respectively.210 The gender difference and clinical features are obvious for SS, the ratio of female to male patients is about 10:1 and female patients have more serious clinical manifestations.213 A study for the epidemiology of SS in a French multiracial population discovered that the non-European race has a higher SS prevalence and disease profile than the European race, and another study discovered a higher prevalence for white females.214,215 Although SS can occur at all ages, children are rarely diagnosed and the population in 30–50 age are mainly diagnosed.210
HLA gene region is also the key to SS occurrence, and a recent review has summarized detailed research about the genetics and epigenetics of SS. Genes significantly associated with SS and exhibiting pathogenicity include HLA-DQA1, HLA-DQB1, HLA-DRA (rs115575857), HLA-DRB1 (rs116232857),216 HLA-B (rs2523607)217 and MICA (MICA*008)218 in MHC region, TNF (rs1800629),219 STAT4(rs10168266)220 and IL12A (rs485497)216 in non-MHC region. In addition, IKZF1 (rs4917129),221 OAS1 (rs10774671)16 and MAPT (rs7210219)222 may possess SS protective mechanisms. The epigenetic modification also affects the occurrence and development of SS.223 In addition to common environmental factors which can induce autoimmune diseases, silicone breast implants also lead to a high risk for SS.224 Virus infection seems to be particularly important for SS and many studies have demonstrated that EBV protein EBNA2 can bind with related high-risk sites of SS.222 Unlike other autoimmune diseases, smoking is not associated with the development of primary SS.225,226
Clinical manifestation and diagnosis
There is typical heterogeneity in the clinical manifestations of primary SS, similar to SLE, and the patients have various performances because of different organ involvement. Almost 85% of patients will have glandular symptoms manifested as ocular dryness (major symptom), ocular inflammation, oral drying (major symptom), dysphagia, pruritus in the ear canal, vaginal pruritus or dyspareunia. Approximately 50% of patients will have cutaneous features such as cutaneous vasculitis, including purpura and urticarial papules that depend on the condition of the blood vessel lesion.215 Some patients show the nonspecific phenomenon such as Raynaud’s phenomenon of skeletal muscle pain and fatigue.227 Almost half of primary SS patients can develop into systemic performance and invasion of the kidney, lung, liver, and other organs.228
SS can be diagnosed via a series of exocrine gland tests and laboratory examinations. Patients will have assessment tests such as unstimulated salivary flow rates, stimulated salivary flow rates, and salivary scintigraphy for evaluating the main salivary glands. Schirmer’s test I, Schirmer’s test II, and Corneal staining can be used to evaluate the lacrimal gland function.210 Autoantibodies detection is very sensitive and can be detected even 20 years before SS occurrence.229 Antinuclear antibodies (ANAs) are the most common for the majority of patients. Anti-RNA-related protein antibodies (anti-Ro/SSA antibodies) are representative of different clinical stages, histological changes and immunopathological changes. In addition, anti-La/SSB antibodies are also specific antibodies for SS patients.230 For laboratory abnormalities, the samples from SS patients show normocytic anemia, leukopenia, and thrombocytopaenia, and some advanced patients will show the elevation of visceral damage-related enzymes. Salivary gland biopsy is the most specific detection method, and clinical pathologists can make the final diagnosis of SS via checking the distribution and number of antibodies and lymphocyte infiltration.231
Immune dysregulation in SS
The pathogenesis of autoimmune epithelitis is an explanation for the immunopathology of SS. The TLRs molecular pathway activation of glandular epithelial cells such as salivary gland epithelial cells leads to the production of autoantigen that can be presented to immune cells. Furthermore, activation of TLR signaling leads to the upregulation of immune-competent molecules such as HLA molecules, FAS receptors and ligands, chemokines, and cytokines. Immune cells and inflammatory microenvironment create a circle of interaction between epithelial cells and immune cells that promotes the development of SS210,223 (Fig. 3e).
New therapeutic strategies for autoimmune disorders
Here we mainly summarize antibody therapy, RNA interference (RNAi) therapy, and Hematopoietic stem cell transplantation (HSCT) therapy for autoimmune diseases. We review the outcome of these approaches and discuss their translational potential.
Antibody therapy
Combination of targeted antibody therapies
It is undeniable that single antibody treatment may have some effect on autoimmune diseases, however, combined treatment may target two or more signaling pathways and achieve synergistic treatment effects.
In a 68-week phase II double-blind study for primary SS treatment (GSK study 201842, NCT02631538), researchers used the combination of belimumab and rituximab to achieve more effective results than single rituximab treatment. Almost all CD20+ B cells in salivary glands are exhausted and the phenomenon also occurs in peripheral CD19+ B cells simultaneously. The regulative effect is more intense and lasting for the combination of belimumab and rituximab than single rituximab treatment. In addition, there are no new side effects added.232 Another randomized controlled trial (ISRCTN: 47873003) tried belimumab after rituximab treatment mode for SLE patients via the score of the IgG anti-dsDNA antibody level in serum.233 Other groups also proved the low-dose rituximab and alemtuzumab combination treatment for autoimmune cytopenias can achieve a 100% overall remission rate and 58% complete response but there are still 6 patients developing infection (NCT00749112).234 Anti-CD22 monoclonal antibody conjugated with calicheamicin (anti-CD22/cal) and CTLA4-Ig combination therapy can suppress autoimmunity in NOD mice and prolong the allograft survival time.235 Recently, researchers in the Hospital for Special Surgery (New York) also conducted a clinical trial to detect the treatment effect of belimumab and rituximab combination in diffuse cutaneous systemic sclerosis (NCT03844061). However, not all combination therapies can have significant therapeutic effects, Atisha-Fregoso et al. demonstrated that the combination of belimumab and rituximab did not alleviate symptoms of general Lupus Nephritis patients (NCT02260934).236 The rituximab and alemtuzumab combination therapy trial (NCT03312907) for SLE by GlaxoSmithKline started in 2019.237 Regrettably, the result of combination therapy illustrates that it cannot improve disease conditions and even cause more serious infections.238
Compared with therapeutic measures of multiple antibody combinations, therapeutic monoclonal antibodies combined with some chemotherapeutics or other immunosuppressive biologics seem to be more widely applied. A study about Certolizumab pegol and methotrexate (MTX) combination treatment (NCT01519791) for RA showed a significant therapeutic effect without extra side effects compared with placebo + MTX.239 In some earlier studies, researchers also tried to treat relapsing MS with natalizumab plus IFNβ-1a (NCT00030966) and followed up on the patient’s recurrence and MRI images. Although the therapy results are encouraging, there are still unavoidable adverse reactions such as anxiety, congestion and edema.240 Glatiramer acetate and natalizumab combination also have significant therapeutic effects and are well tolerated.241 Ocrelizumab (200 mg) with MTX can reduce the development of RA, but ocrelizumab (500 mg) with MTX will lead to ascending levels of serious infections (NCT00485589).242 The type I interferon receptor antibody, anifrolumab, combined with oral glucocorticoids and mycophenolate mofetil (MMF) achieved some success in complete renal response (CRR), however, the incidence of herpes zoster in the combination group was twice that in the placebo group (NCT02547922).243 Belimumab with MMF or cyclophosphamide-azathioprine combination trial (NCT01639339) for Lupus Nephritis also confirmed the effectiveness of combination therapy.244 Rituximab and prednisone combination for warm autoimmune hemolytic anemia in adults (NCT01181154) showed more effective and safer than placebo with prednisone.245 Burmester et al. also specially studied the influence of the combined MTX dose on side effects and explained the correlation between dose effect and clinical efficacy (NCT01185301).246 Studies also propose less certain treatment effects in monoclonal antibodies combined with other immunosuppressive drugs. Rituximab + MMF + corticosteroids combination (NCT00282347) did not show more excellent therapy results compared with rituximab treatment alone.247
In sum, there is still uncertainty in antibody combination therapy, and no simple superposition of therapeutic effects through several targeted drugs and antibodies combination. Meanwhile, the drug side effects may be strengthened by medicine combination. In addition, a large number of clinical trials are needed to explore the dose of drugs used in combination therapy.
Bispecific antibodies therapies
Bispecific antibodies (BsAbs) are a new class of antibodies that can identify two different antigens or two different epitopes of the same antigen (Fig. 4a). The successful generating of more than 100 BsAbs formats benefit from the significant advances in antibody engineering and antibody biology.248 Thanks to their strong multitargeting, high binding potency, bridging cell action, and cascade amplification effect,249,250 they have been applied to the treatment of complex tumors and autoimmune diseases.251,252,253,254,255,256

Other new therapeutic strategies to autoimmune diseases. a Some examples of bispecific antibodies in clinical trials. b The schematic diagram of intracellular mechanisms of siRNA. siRNA consists of a guide (antisense) strand and passenger (sense) strand. The former is a functional segment for siRNA and the latter is responsible for transportation and loading. siRNA can combine with RNA-induced silencing complex (RISC) consisting of Argonaute 2 (AGO2), trans-activation response RNA binding protein 2 and DICER1. After the degradation of the passenger strand, the target RNA sequence can be recognized by the guide strand. Eventually, it can induce the silence of the target RNA. c The schematic diagram of hematopoietic stem cell transplantation (HSCT). Before determining transplantation, transplanted patients should be identified. Besides, patients are monitored to prevent flares. Generally, G-CSF and cyclophosphamide (2–4 g/m2) plus uromitexan are applied to the mobilization of HSCs in patients. About 4 or 5 days after mobilization, we collect the peripheral blood stem cells by leukapheresis and these cells are CD34+ in general. The patients can be discharged and wait for the immune conditioning after 1 or 2 weeks. The conditioning process may last for about 10 days. Then HSCs can be infused back into the patients. Patients accepting HSCs are left to observe in the hospital until the neutrophil level returns to normal. After HSCs infusion, the patients’ lymphocytes may decrease extremely but their immune systems can rebuild
Bimekizumab which can selectively inhibit IL-17A and IL-17F simultaneously is the first BsAbs approved by the FDA in 2021. In two studies for the treatment of plaque psoriasis, adalimumab (NCT03412747) and secukinumab (NCT03536884) were compared with bimekizumab, respectively to evaluate the treatment effect of bimekizumab.257,258 bimekizumab showed non-inferior therapeutic ability to adalimumab in reducing symptoms and signs of plaque psoriasis but had adverse events including higher frequency of oral candidiasis and diarrhea.257 Besides, bimekizumab is also applied in moderate-to-severe plaque psoriasis (NCT03025542, NCT03410992),259,260 hidradenitis suppurativa (NCT03248531),261 RA (NCT02430909)262 and ankylosing spondylitis (NCT02963506, NCT03928704, NCT03928743),263,264 and has achieved good curative effects, but infections and infestations still persist.
Tibulizumab (LY3090106) is another novel tetravalent BsAb which can target and inhibit the B cell activating factor (BAFF) and IL-17, and it is synthesized by the link of anti-IL-17 single-chain variable fragment from ixekizumab and the anti-BAFF fragment from tabalumab265 (Fig. 4a). And in vivo mouse models and cynomolgus monkey, tibulizumab can effectively inhibit the development and survival of B cells for a long time in mouse models and cynomolgus monkey.265 Relevant clinical trials (NCT03736772, NCT01925157, NCT02614716) have been initiated, but no results have been disclosed yet.
Rozibafusp alfa (AMG 570) BsAb composed of the AMG 557 antigen providing anti-ICOSL sequence and the BAFF-binding peptides from AMG 623 linking with the C-terminus of AMG 557 heavy chain,266 can target and inhibit the BAFF and ICOSL (Fig. 4a). The treatment effect is more significant than single inhibitor in mouse NZB/NZW lupus model and arthritis (CIA) model. It can also inhibit the development of B cells in cynomolgus monkeys.266 Clinical studies (NCT02618967, NCT03156023) have been initiated to investigate the pharmacokinetics (PK) and pharmacodynamics (PD) of rozibafusp alfa.267
Anti-CD22/CD20 bispecific hexavalent antibody (bsHexAb), 22*-(20)-(20) is developed by Rossi et al., which is composed of Ck-AD2-IgG-epratuzumab (anti-CD22) and two dimeric CH1- DDD2-Fab-veltuzumab units (anti-CD20)268,269 (Fig. 4a). This BsAb may be inspired by the previous BsAb therapy for lymphoma.270,271 The researchers have tried to treat SLE with 22*-(20)-(20), and demonstrated the enhanced trogocytosis resulting in reductions of many B cell surface marker levels. In addition, the 22*-(20)-(20) used alone showed a better treatment effect than the combination therapy of the two parental antibodies.269
Notably, many BsAbs such as obexelimab (XmAb5871) targeting CD19 and FcyRIIb to inhibit B cells line,272,273,274 MT-6194 targeting both IL-17A and IL-6R to inhibit the development of inflammatory environment275 (Fig. 4a), JNJ-61178104 targeting TNF and IL-17A276 and romilkimab (SAR156597) targeting both IL-4 and IL-13277 have been generated and tested in clinical trials. Clinical trials about these BsAbs reflect the broad clinical application potential (NCT02758392, NCT02725515, NCT02725476, NCT02921971).278,279,280,281
Compared with application in tumor therapy, the research of BsAbs in autoimmune diseases is still in its infancy, and there are many challenges. In a clinical trial of lutikizumab (ABT-981)282 (Fig. 4a) for the treatment of arthritis with synovitis (NCT02087904), the pain and arthritis symptom improvement are not obvious.283,284 Another phase II clinical trial about SAR156597 for idiopathic pulmonary fibrosis (NCT02345070) has also failed.285 Although the results of clinical trials might vary, BsAbs still have many advantages and offer new therapeutic options for autoimmune diseases.286
RNA interference therapy
RNAi was first discovered in Caenorhabditis Elegans by Fire and Mello in 1998.287 After that, researchers further studied these small mRNA (sRNA) and found small-interfering RNAs (siRNAs).288 Although there are many sRNA types including siRNAs, microRNA (miRNA) mimics, short hairpin RNAs (shRNAs) and Dicer substrate RNAs (DsiRNAs), the research on siRNA is more in-depth and shows more direct effects in translation.289,290,291 Hence, in this review, we emphasize the siRNA application for autoimmune diseases. siRNA usually is 15–30 bp in overall length. siRNAs can trigger efficient target gene silence by inhibiting mRNA translation and promoting mRNA degradation (Fig. 4b). Pharmaceutical companies have been devoted to developing the siRNA therapeutics and major breakthroughs were being made that paved the way to successful clinical translation.292,293 In 2018, the FDA approved the first liposome complex for siRNA binding (Patisiran) for the treatment of a rare disease called hereditary transthyretin-mediated amyloidosis (hATTR).294,295,296 Indeed, the rapid development of siRNA is benefit from lipid nanoparticles (LNPs) technology progress and related nucleic acid modification methods.297,298,299 Researchers also use siRNAs for the treatment of autoimmune diseases and achieved some progress.300,301
Herman et al. delivered siRNA based on the LNP system to two types of mouse models of RA for hnRNP A2/B1 silence and downregulate the expression of proinflammatory cytokines in macrophages.302 The noncovalent binding of siRNA targeting the p65 subunit of NF-κB (p5RHH-p65) and melittin-derived cationic amphipathic peptide can also control inflammation and protect the integrity of cartilages in RA.303 Other groups also tried PEG-PLL-PLLeu nanoparticle,304 polycaprolactone-polyethylenimine (PCL-PEI)/polycaprolactone-polyethyleneglycol (PCL-PEG),305 folate conjugated liposome-based hybrid carrier,306 etc., to deliver siRNA targeting NF-κB for the treatment of autoimmune disorders.
Lee et al. designed a nanocomposite composed of poly-siRNA targeting TNF-α and thiolated glycol chitosan (tGC) for RA treatment. The related inflammatory genes were effectively silenced in the macrophage stimulation culture test and mouse RA model.307 Besides, Different nanomaterial carriers are used to deliver the siRNA targeting TNF-α including Lipid-polymer hybrid nanoparticles (LPNs),308 degradable cationic polymer (PDAPEI),309 sheddable PEGylated solid-lipid nanoparticle,310 folate-PEG-chitosan DEAE nanoparticle,311 etc.
Poly-siRNA targeting Notch1 combined with tGC also has good performance in RA.312 In addition, siRNA is designed to target complement fragment 5 (C5),313 MMP-9,314 BTK,315 IFN regulatory factor 5 (IRF5)/ B cell-activating factor (BLYSS)316 and other inflammation-related genes.
Currently, research about siRNA for the treatment of autoimmune diseases has been mainly focused on RA, it is essential to investigate its potential treatment effects on other autoimmune diseases. With the rapid development of targeted drug delivery technology, siRNA-based therapy will undoubtedly be used to treat many other diseases.
Hematopoietic stem cell transplantation
As previously discussed, the fundamental mechanism of autoimmune diseases is the break of autoimmune tolerance because of the environment and genetic factors. HSCT provides a treatment option to restore immune tolerance by replacing or resetting immune responses.317 During the immune reconstitution process, NK cells and B cells recovering faster than T cells, with CD4+ T cells recovered slowly compared to CD8+ T cells based on a study in MS patients after HSCT transplantation.318 The pre-existing T cells with pathological and autoimmune reactions will be replaced by newly formed T cells.319 After autologous HSCT transplantation in MS patients, B cells shifted from a predominantly transitional to naïve phenotype, and memory B cells recovered slowly with reduced repertoire diversity.320 Altogether, these processes can quench the pre-existing autoimmune responses and reestablish immune tolerance. However, complete deletion of all autoimmune pathogenic cells is impossible, and immune cells with regulatory capacity control the homeostasis of the repopulated immune system.318 Tregs play an important role in balancing the body’s immune axis.321,322,323 Besides, other cells represented by tolerogenic DCs (tolDCs) with tolerance characteristics have beneficial roles.324,325 TolDCs have enormous potential for the treatment of autoimmune diseases due to their ability to induce immune tolerance.326,327,328 These tolDCs express low co-stimulatory molecules and high levels of immunosuppressive membrane surface molecules including programmed cell death ligand (PD-L1)329 and inhibitory Ig-like transcripts (ILTs),330 which leads to the T cell clonal anergy and expansion of regulatory T cells eventually.327 In the antigen-specific treatment of autoimmune diseases, researchers regard induced tolDCs as a standard of treatment success and we will discuss them in more detail later.
The study of bone marrow transplantation for improving RA in rat models seems to be groundbreaking to HSCT therapy.331 Afterwards, related technologies developed rapidly and researchers have applied HSCT to various autoimmune diseases. From the European Society for Blood and Marrow Transplantation (EBMT) autoimmune diseases working party database, we can acquire the earliest therapy information started in 1994.332 In 1996, Tamm et al. reported the first treatment with HSCT for autoimmune disease.333 In 1997, Fassa et al. reported the first results of the treatment with HSCT for MS and preliminarily verified its feasibility.334
Autologous HSC may be derived from peripheral blood or bone marrow and the process is as follows335,336 (1) Mobilization of stem cells by treatment with cyclophosphamide and granulocyte colony-stimulating factor (G-CSF). Stem cells can be collected 4–5 days after the treatment. (2) Conditioning. 1 or 2 weeks after cell collection, the patients will accept the immunoablative conditioning including anti-thymocyte globulin and cyclophosphamide. The different ways will be implemented in different individual patients. (3) Infusion of autologous CD34+ stem cells and hospitalized for observation. Patients will continue to be hospitalized to prevent the sudden occurrence of adverse events after infusion for 1–3 weeks until the recovery of neutrophil numbers (Fig. 4c).
We mainly describe the therapeutic effect and development of HSCT in MS. In 2006, a retrospective survey of 183 MS patients from EBMT reported 5.3% transplant-related mortality (TRM) and 63% of MS patients improved the disease development or stabilized the mental state during the median follow-up of 41.7 months.337 Afterwards, a group reported that the HSCT can be effective for aggressive MS failing to respond to conventional treatment according to the Italian multi-center experience.338 In 2015, Burt et al. reported a significant improvement in the quality of MS patient life scores and the significant reduction of MRI T2 lesion area.339 Compared with standard immunotherapy, HSCT therapy promotes the continuous improvement of active secondary progression.340 Compared with alemtuzumab for RRMS, HSCT also seems to have more treatment feedback but it also leads to more adverse events in the first 100 days after transplantation in an observational study (NCT03477500).341 Similar results also appear in the comparison of HSCT with Fingolimod and natalizumab.342 A long-term clinical outcome and an observational cohort study in Sweden also affirmed the role of HSCT for most MS patients with certain efficacy and safety.343,344 However, a recent matched observational study did not support the use of autologous HSCT to control disability in progressive MS with advanced disability and low relapse activity.345
HSCT might also become a treatment option for other autoimmune diseases. It has been reported that children with refractory juvenile idiopathic arthritis (JIA) gradually recovered after reduced toxicity conditioning HSCT therapy. In this report, all the patients alleviated disease progression and improved their quality of life, 11 children of them even achieved complete drug-free remission.346 A clinical trial (NCT00742300) reported the disappearance of pathogenic dsDNA and resetting of the adaptive immune system, the regeneration of Foxp3+ Tregs from thymus in refractory SLE patients accepting HSCT after depletion of pre-immune system.347 Recently, researchers also found that HSCT favorably changed the antibody reservoir in systemic sclerosis patients.348 The C-peptide levels also increased significantly and most of the patients achieved insulin independence under good control of blood sugar level after HSCT,349 but a report demonstrated 52% of patients experienced adverse effects despite a complete immune system recovery.350 This study suggested an urgent need for safer HSCT options.
It is undeniable that there are many adverse effects of HSCT in autoimmune disease treatment. Researchers must consider the possible infertility, early menopause and heart damage in MS and systemic sclerosis patients.335,351,352 The use of immunosuppressants will also increase the risk of various infections and malignancies.335,353 Incomplete rearrangement of immune cells after HSCT will lay a hidden danger for the recurrence of autoimmune disease. A report showed nearly 10% of secondary autoimmune disease after HSCT.354 Besides, improved risk estimates and supportive care are particularly important for patients who received allogeneic HSCT.355,356,357 Nevertheless, the HSCT is still a reasonable option for the treatment of autoimmune diseases based on its capability to reset or rebalance the immune system to restore immune tolerance.
Emerging therapeutic strategies based on antigen-specific immunotherapy
Due to the fact that antigen-specific immunotherapy can target the disease-causing immune cells without suppressing the whole immune system, there has been an urgent need to develop new immunotherapies that induce long-term antigen-specific immune tolerance for the treatment of autoimmune diseases.358 Although significant advances have been made in this field, successful clinical application is still limited. Here we will discuss the current strategies developed in this field, and highlight the recent advances in the use of nanomaterials and mRNA vaccine techniques to induce antigen-specific immune tolerance. Besides, we also provide a timeline to summarize the significant advances in the field of antigen-specific immunotherapy for the treatment of autoimmune diseases based on MS and T1D (Fig. 5).

Timeline of the significant advances in the field of antigen-specific therapy for autoimmune diseases. In 1960, researchers discovered that encephalitogenic protein can suppress EAE progression. Then researchers tried to use modified autoantigens or MHC conjugated autoantigens to treat animal models of autoimmune diseases. In 1998, researchers have tried to use the DNA coding autoantigens to treat EAE. Afterward, the application of nanomaterials gradually emerged in autoantigens transportation, and antigen-specific therapy has experienced a rapid development over the past 20 years. Some researchers also tried to apply the combination of immunosuppressive factors, autoantigens, and nanoparticles for treatment. In 2021, mRNA-LNP technology has been applied for the first time in autoimmune disease models
Autoantigen-based therapies
Whole antigen or modified peptides
It has long been known that the damage of CNS can be prevented in animals by the administration of a mixture of encephalitogenic substances before the experimental autoimmune encephalomyelitis (EAE) model establishment. In 1960, SHAW et al. found that the combination of Freund’s adjuvants and encephalitogenic proteins extracted from the homologous brain can suppress the EAE progression, meanwhile, the suppressing effect is closely related to protein injection dose.359 They attributed this phenomenon to the specific desensitization, deflection, antibody neutralization reaction, or disability of antibody-forming mechanisms359 (Fig. 6). Early studies have shown that the combination of Freund incomplete adjuvant and myelin basic protein (MBP) inhibit EAE.360 However, the administration of MBP whole antigen has been shown to be ineffective treatments or major exacerbations have emerged both in clinical trials and in animal models.361,362 For T1D, insulin administration has been shown to prevent NOD mice from developing the disease.363 Multiple clinical trials using insulin immunotherapy have been conducted to prevent or treat T1D, but the results are still uncertain.364,365,366,367

Approaches to deliver autoantigen for the treatment of autoimmune diseases. (1) Whole antigens, peptides, and APL are administered through subcutaneous injection, intravenous injection, intramuscular injection, oral and inhalation. (2) Autoantigens are transported by microbes such as Lactococcus lactis. (3) Microneedles loading antigens target DC cells in the skin. (4) Autoantigens are delivered by hyperbranched polymers. (5) Nanoparticles for delivering autoantigen or pMHC; (6) Combination of autoantigen, Nanoparticles, and immunosuppressive drugs. (7) Gel vaccine with immunosuppressive drugs. (8) Autoantigen transported by extracellular vesicles. (9) Engineered cells modified by autoantigen specificity. (10) Autoantigen-specific tolerogenic cells adoptive transfer. (11) Gene therapies based on DNA-plasmid coding autoantigens. (12) Gene therapies based on mRNA coding autoantigens. Abbreviations: i.m.= intramuscular injection; i.v. intravenous injection, s.c. subcutaneous injection. (Part of the figure was modified from Servier Medical Art(http://smart.servier.com/), licensed under a Creative Common Attribution 4.0 Generic License. (https://creativecommons.org/licenses/by/4.0/)
The mechanism of antigen-specific immunotherapy is through induction of immune tolerance by injection of autoantigens with high and repeat dose that leads to T cell anergy or results in RICD and generation of Tregs.16,368 The RICD process is closely related to TCR recognizing antigens and FAS-FASL inducing apoptosis and it is an antigen-specific immune regulatory induction process.16 Investigators found that stronger immune suppression can be induced by high-dose, oligomerized, linear, and soluble epitope peptides.369,370,371 The change of protein structure and modification of certain amino acids in the peptide can induce immune suppression for the treatment of autoimmune diseases more efficiently.372 Early researchers have demonstrated that MBP coupled with diazotized arsanilic and sulfanilic acid (Ars-Sulf-MBP) as well as modification of arginine, lysine, and tryptophan residues of MBP selectively can suppress EAE development.373,374 MBP modified by bromide was shown to be effective for EAE treatment.375 Furthermore, researchers mixed MBP and hapten for EAE suppression.376 Recently, our group designed novel fusion proteins to treat EAE and revealed related mechanisms about how cognate antigens suppress CNS inflammation and EAE progression.377
The route of administration is particularly important to achieve better immune tolerance effects. Oral administration and inhalation of MBP were reported in 1988 and 1993 respectively.378,379 It is worth mentioning that drugs can enter the CNS directly through the olfactory nervous and trigeminal nerves, and indirectly through the nasal mucosa by intranasal or inhalational administered for better CNS drug delivery.380,381
Altered peptide ligands (APL)
Altered peptide ligands (APL) are natural peptide analogs with at least one amino acid substitution at TCR positions (Fig. 6). Different substitutions in particular residues may induce different T cells responses,382,383 even though APL possess similar binding between MHC and TCR to natural peptide. Some APL cannot induce a complete signal for T cell proliferation, hence the immune anergy can be induced in this way384,385 (Fig. 7). Early researchers have attempted to utilize single amino acid substitution in peptide segments of MBP for the prevention and treatment of EAE.386,387,388 Corresponding APL can also be synthesized by molecular mimicry techniques of microbes to prevent EAE.389 Besides, some groups designed the MHC anchor-substituted variant of PLP139-151 (145D, HSLGKWDGHPDKF) that the seventh amino acid was replaced by aspartic acid and demonstrated the 145D will not induce the acute hypersensitivity reaction.390

Altered Peptide Ligands (APL) for tolerance induction in TCR-peptides-MHCII. Several amino acid substitutions in key TCR identification positions can cause the signal transmission process obstacles which can affect the immune activation and induce immune tolerance. The yellow circles represent natural amino acids; the red circles represent altered amino acids
In recent years, APL is still being used for the treatment of autoimmune diseases. The novel 3aza-MBP APL which contains aza substitutions increased protease resistance property and effectively suppressed EAE disease progression.391 Besides, some investigators also demonstrated that MBP87-99 (Ala91, Ala96) APL cyclo can promote the bond to HLA-DR4 and induce antigen-specific immune regulation392 and others validated cyclic MOG35-55 can reduce the pathological process of EAE.393
APL of p55-70 of Imogen38 (Imogen38p55-70 APL) can inhibit the proliferation of β-cell reactive T-cell clone but fail to induce classical β-cell reactive T cells anergy. In addition, the APL cannot down-regulate TCR/CD3 complexes.394 Treatment of NOD mice with IGRP206-214 APLs is inefficient for T1D. Thus, it is necessary to test the dose of APL as well as the affinity between APL, MHC and TCR.395 In some clinical trials, APL possesses a certain potential for induction of immunosuppression.396,397,398 However, a small portion of the patients show hypersensitivity reactions which lead to disease progression in certain early clinical trials using APL for MS treatment.399,400 It was considered that the APL therapy is more appropriate for Th1-mediated autoimmune diseases because APL can promote the shift away from Th1 cytokines to Th2 cytokines and this can be an explanation for hypersensitivity reactions.400 Besides, these APLs all are used in RRMS, APLs for other types of MS have not been reported yet.
MHC-autoantigen peptides
Naïve T cell activation relies on 3 signals: (1) interaction between TCR and peptide/MHC (signal 1); (2) co-stimulatory molecules (signal 2); (3) cytokines and chemokines (signal 3).401,402,403,404,405 Rather than autoantigen being uptake and presented by APCs, soluble peptide/MHC (pMHC) can directly interact with T cells without co-stimulatory signals. Anergy T cells will thus be induced if only the existence of the first signal while the co-stimulator is missing, and it can facilitate further immune suppression or inhibit the avidity maturation of pathogenic T cells.406,407
Accordingly, pMHC complexes are applied in autoimmune disease treatment and Sharma et al. reported the first strategy of I-As protein-MBP91-103/ PLP139-151 for EAE therapy in 1991.408 Studies using MHC II linking acetylcholine receptor α chain (AChRα100-116 or AChRα144-163) effectively inhibited experimental autoimmune myasthenia gravis (EAMG)409,410 and DR2-MOG35-55 can suppress EAE development.411 The stable complexes composed of two-domain MHC II and MBP69-89 can inhibit and detect encephalitogenic T cells.412 Subsequently, investigators validated that the I-As/PLP139-151 peptide (RTL401) can induce cytokine switch, promote the Th2-related cytokines expression in CNS, and inhibit the encephalitogenic potential of specific pathogenic T cells.413 Peptides-MHC II dimer was also designed for T1D and achieved the expected effect.414,415 Recently, Urbonaviciute et al. reported that MHC II- galactosylated collagen type II (COL2) can target the antigen-specific TCR via positively charged tags to expand VISTA-positive nonconventional Tregs for RA.416
Biomaterials-based new strategies for autoantigen delivery
The induction of immune tolerance is affected by several factors including antigen dosage, antigen administration route, and delivery system.417 Biomaterials facilitate new strategies to induce immune tolerance by providing accurate delivery of autoantigens to the target organs and controlled release of therapeutics.418,419,420
Microparticles delivery systems
Nanoparticles have been used for drug delivery and disease treatment, and some nanoparticles have expanded into extensive clinical applications.421,422 The size, surface charge, shape, hydrophobicity, and constituent materials co-determine the drug loading efficacy and organs/cell targeting ability.423,424 Some nanoparticles themselves have inflammatory inhibitory effects.425,426,427 Nanoparticles have been extensively investigated in autoimmunity disease treatment.428,429,430
Investigators developed a dual peptide nanoparticle platform which delivers antigen peptides for primary signal and other peptides (LABL, binding with ICAM-1) for inhibitory of co-stimulatory signal. The NPsLABL+MOG is designed for EAE treatment by this platform, which is more effective than NPsMOG for the reduction of myelin sheath inflammatory infiltration and induction of immunosuppression.431 Polystyrene or biodegradable poly(lactide-co-glycolide) (PLG) microparticles bearing PLP139-151 can be taken up by macrophages expressing the MARCO receptor and this process is mediated by Tregs, T cell anergy and the activation of abortive T cell. These microparticles carrying PLP139-151 can suppress the autoimmune progress and prevent epitope spreading via apoptotic clearance pathways to inactive pathogenic T cells.432 Based on this principle, low-cost, safe and good biodegradable PLG coupled with PLP139-151 has also verified that it can reduce a series of inflammatory cells and inhibit the epitope spreading in the relapsing-remitting EAE model.433 In another further study, PLG NPs-PLP139-151 significantly downregulates the positive co-stimulatory molecules and remains high in negative co-stimulatory molecules.434,435 Selective targeting of liver sinusoidal endothelial cells (LSEC) using NPs delivering autoantigen peptides can induce antigen-specific Tregs and protect mice from autoimmune diseases.436 Phospholipid phosphatidyl serine-liposomes (PS-lipo) loading Insulin A and B peptides can also induce tolerance APCs and prevent T1D.437 Wilson et al. modified the autoantigens by synthetic glycosylation (N-acetylgalactosamine or N-acetylglucosamine) which can target the liver and induce tolerance more easily. Besides, these modified autoantigens can expand the specific Tregs in T1D, MS, and other autoimmune diseases mice models.438,439,440
Some investigators packaged autoantigen into gold nanoparticles (AuNPs) owing to their features of ease of synthesis, ease of shaping, ease of functionalization and facilitating internalization.441,442,443 In addition, AuNPs are validated to have an anti-inflammatory effect by inhibition of leukocyte migration and cytokines secretion which attracts us to use it to treat autoimmune diseases.444 Wegmann et al. designed multiple Ag peptides (MAPs) containing eight PLP139-151 peptides around dendrimeric branched lysine core for the treatment of relapsing EAE.445 Functional amphiphilic hyperbranched (HB) polymers can precisely control molecular weight and chemical composition to achieve good biocompatibility, expected drug metabolism and accurate targeting for drug delivery.446,447,448 Our group has shown that functional amphiphilic HB polymers can efficiently deliver autoantigen and induce immune tolerance by inducing autoreactive T cell deletion449 (Fig. 6).
Researchers also attempted to improve treatment strategies based on pMHC for antigen-specific therapy by nanoparticles in recent years.450,451 It has been reported that systemic delivery of nanoparticles coated with pMHC II (pMHC II-NPs) can up-regulate IL-10 and TR1-related markers in TR1 poised, antigen-experienced CD4+ T cells (Fig. 6). The group showed that pMHC II-NPs triggered the expansion of TR1 like cells to promote the formation of immune regulatory networks and can restore motor function in EAE mice.452 Regulatory B cells are also a potential immune regulatory cells population124,125 and play a pivotal role in the antigen-specific regulatory network induced by pMHC II-NPs452 (Fig. 8).

The framework of the establishment of antigen-specific immune regulatory networks by pMHC II-NPs. pMHC II-NPs can be recognized by pathogenic T cells when enter the lymph node through high endothelial venule (HEV) in the T cell zone. Owing to the absence of costimulatory molecules and the action of IL-10, the pathogenic IFN+CD4+ Th1 will differentiate into memory TR1. The TR1 cells can be amplified and migrate to the specified location before interacting with DCs and cognate B cells. B cells can differentiate into regulatory B cells (Bregs). DCs may dampen the ability of activating pathogenic T cells assisted by relevant anti-inflammatory factors. Meanwhile, the Bregs and TR1 can further regulate the antigen-specific regulatory networks and blunt the autoantigenic and pathogenic cells. The suppression induced by pMHC II-NPs is disease-specific and self-limiting
By analyzing transcriptional markers, Solé et al. pointed out that the production of FOXP3-IL-10+ Treg1 cells originates from the Tfh cells via BLIMP1-dependent manner and furthermore confirmed the important role of the pMHC therapy method for autoimmune diseases.453 Vacchio et al. reported transcription factor Thpok was necessary for driving Bcl6 and Maf expression to promote differentiation from CD4+T cells to Tfh cells.454
Targeting IL-2 to induce Tregs for the treatment of autoimmune diseases attracted more attention in recent years.455,456 Investigators tried to apply this method to pMHC and then designed tolerogenic microparticles (tol-MPs) loaded with rapamycin (RAPA), biased fusion IL-2 protein and peptide-MHC II tetramers for EAE treatment.457 The designed tol-MPs supported Treg expansion and promoted sustained disease reversal of EAE mice.457 Umeshappa et al. showed that broad liver autoimmune disease suppression can be induced by TR1 cell formation via pMHC II-NPs displaying autoantigen epitopes in an organ rather than disease-specific manner.458 For the single-chain pMHC complex (scKd.IGRP) designed by another group, the peptides are covalently attached with β2-microglobulin (β2m) which is linked with MHC I H-2Kd. The group suggested that pMHC I can induce the apoptosis of CTLs.459
Tsai et al. utilized NPs to deliver T1D-related peptides-MHC complexes for monospecific resistance of the T1D development, and demonstrated that pMHC can expand the memory-like and autoregulatory CD8+ T cells.460 NPs coated with pMHC can blunt T1D progression and restore normoglycemia in diabetic animals.460
Transdermal microneedle patches
The skin has abundant APCs and other immune cells (Dermal DCs, Langerhans cells, macrophages, dermal γδ T cells, etc.) which makes it an attractive target for antigen-specific immunotherapy461,462,463 (Fig. 9). Hence, microneedle (MN) administration can effectively promote APCs in the skin to engulf these autoantigens and induce immune regulatory response.464,465,466 Researchers reported a dry-coated MN binding with the topical steroid which can promote longer-retention in the skin. This delivery way can transport autoantigen to the skin for T1D treatment and it promotes the antigen presentation for tolerogenic APCs more strongly than ID injection.467

The sketch of antigen peptides delivery by microneedle patch. The microneedle delivery system can deliver antigen peptides to the dermis where there are multiple types of APCs including Langerhans cells, macrophages, and DCs. The abundance of APCs located in the dermis layers makes it an attractive location to deliver antigen peptides for induction of immune tolerance. (Part of the figure was modified from Servier Medical Art(http://smart.servier.com/), licensed under a Creative Common Attribution 4.0 Generic License. (https://creativecommons.org/licenses/by/4.0/)
Dul M at el. have employed the MN delivery system, MicronJet600, to target the Langerhans cells in the skin for delivering peptides coupled with gold nanoparticles468 (Fig. 6). The addition of gold nanoparticles is validated to expand the distribution of poorly-soluble peptides in lymphoid organs.469 MN-gold nanoparticles conjugated with proinsulin peptide (C19-A3 GNP) were designed for T1D treatment.470 Another group designed a MN delivery system which includes peptides, diluents, and surfactants, and reported that 86% of therapeutic payload can be delivered to local skin tissue just in 150 s.471 A similar study was also reported for RA treatment.472
Overall, MN can cause fewer lesions as well as no skin layer distension compared with traditional needles, and furthermore, it can target the APCs in the skin to present the autoantigen peptide efficiently for a longer time with safety and painlessness.473,474,475,476 Some transdermal patch is currently applied in clinical trials for MS and has shown safety and well toleration.477
Soluble antigen arrays
Soluble antigen arrays (SAgAs) are new antigen-specific immunotherapies strategies that contain small hyaluronic acid (HA) chains backbone. The peptides can be conjugated onto HA by hydrolysable linkers (hSAgAs) or stable click chemistry linkers (cSAgAs) and delivered to the body via the multivalent, soluble and linear form.478 Investigators combined a hybrid insulin peptide and a mimotope as SAgAs and showed efficacy for T1D prevention.478 The group also reported that SAgAs can direct the response of epitope-specific T cells.479
SAgAs are also validated to induce the desensitization of pathogenic B cell populations and the restoration of the healthy phenotype of autopathogenic APCs in the EAE model.480,481 Furthermore, the cSAgAs had a better performance in the antigen presentation process.478,479,480,481
Biomaterials co-delivering autoantigen, immunoregulatory molecules and drugs
Biomaterials loaded with the combination of autoantigen peptides, a series of immune suppression cytokines, and immunosuppressive drugs can inhibit the progression of autoimmune diseases (Fig. 6).
Poly(lactic-co-glycolic acid) (PLGA) and poly(lactic acid) (PLA) have good biocompatibility, immunosuppressive drug loading capacity and appropriate size for application in tolerogenic vaccination. Some researchers demonstrated that PLGA itself can down-regulate the expression of MHCII, CD80 and CD86, and resist DC maturity after lipopolysaccharide (LPS) stimulation which is related with that PLGA can derive lactic acid to inhibit the phosphorylation of TAK1 and then suppress NF-κB activation. Significantly, the immune suppression effect depends on the molecular weight of PLGA, and the higher the molecular weight, the longer the time to immune tolerance induction.427 Meanwhile, PLGA can promote the continuous release of antigens and immune regulatory cytokines,482 which is beneficial for Treg induction.483 Biomaterials based on the PLA system also are potential tools for immune modulation.484 Biomaterials combined with immune suppression cytokines and immunosuppressive drugs can further reduce immunogenicity and induce the tolDCs in vivo.327,482 Besides, PLGA/PLA-NPs have held approval for many applications in clinical diagnosis and treatment by the FDA.482,485,486,487 Cappellano et al. designed an inverse vaccine containing PLGA NP loaded with MOG35-55 and IL-10 for EAE treatment.488 Nanoparticles containing PLGA, CD22L, autoantigen glucose-6-phosphate-isomerase (GPI) and RAPA were shown to induce B cell tolerance (measured by the low anti-GPI antibodies and decreased antibody-secreting plasma cells) as well as T cell tolerance (measured by the expansion of Tregs).489 In another report, PLGA-NPs-PLP139-151 coupled with RAPA inhibited the activation of antigen-specific T cells and B cells, meanwhile induced Tregs and Bregs in SJL mice and protected from EAE development by s.c. or i.v. administration.490 Further study demonstrated the robustness of induced tolerance even under antigen rechallenge with TLR7/8 agonist or complete Freund’s adjuvant (CFA) and the transferrable tolerance of antigen-specific Tregs to EAE.491
Antigen-specific PLGA dual microparticle (dMP) system which contained two sizes of MPs, one is phagocytosable MPs about 1 μm for antigen delivery and the other is non-phagocytosable about 50 μm for encapsulating factors delivery, was designed for the treatment of mouse model for MS and showed complete protection against disease.492,493 A similar study is also reported about acPLG-PLP-TGF-β,494 PLGA NPs-MOG/MHC-TGF-β1 coupled with PD-L1 Fc and CD47 fragments495,496 and PLG- BDC peptide binding GM-CSF.497 In NOD mice, the dMP system induces immature phenotype and LPS-activated resistance phenotype of DC and also prevents the T1D development to a certain extent.498,499
Moreover, studies have shown that colloidal gel vaccine containing alginate, chitosan and autoantigen peptide can induce long-term suppression of EAE500 (Fig. 6). Park et al. developed a tolerogenic nanovaccine to deliver MOG peptide and dexamethasone loaded on an abatacept-modified polydopamine core nanoparticle (AbaLDPN-MOG). AbaLDPN-MOG can reduce IFN-γ secretion by blocking the interaction between CD80/CD86 and CD28.501 NPs-MOG35-55 coupled with 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) also induce the FoxP3+ Treg differentiation in vitro and inhibit the EAE model.502 Yeste A at el. have designed nanoparticles to deliver both a tolerogenic molecule and β-cell antigen proinsulin to induce tolerogenic phenotype in DCs through induction of SOCS2 and suppressed autoimmune diabetes in the nonobese diabetic mice model.503 Similarly, the NP-allergen epitope fragment coupled with adjuvant R848 (TLR7 ligand for reducing the allergy symptoms) protected mice from food allergic responses.504 At an early stage, Capini et al. also reported that RA-related Ag-NF-κB inhibitor-egg phosphatidylcholine liposomes can induce Ag-specific FoxP3+ Tregs and inhibit the clinical symptoms of RA.505 Upregulation of PD-L1 is observed in mice treated with calcitriol-antigenic peptide-liposomes, and disease development has been alleviated in the corresponding RA model and vasculitis models.506 Besides, Multiepitope citrullinated peptide (Cit-ME)-Rapa-Lipid coating calcium phosphate nanoparticles (LCPs) can inhibit the consistent inflammation in RA models.507 A similar formula drug was also validated in another experiment for RA treatment.508
Luo et al. did not choose tolerogenic drugs but used CRISPR-Cas9 plasmid (pCas9) combined with antigens and nanoparticles, which can present the antigens and block the CD80, CD86, and CD40 simultaneously. It also promoted the generation and expansion of antigen-specific Tregs.509
Thus, co-delivery of autoantigen peptides with other tolerogenic agents is essential to combine multiple signals to induce long-term immune tolerance for antigen-specific immunotherapy.
Autoantigen coupled probiotics and extracellular vesicles
Lactococcus lactis as a versatile and mucosa-targeted vehicle has been applied to carry a series of drugs including peptides in recent years.510,511,512,513 It has been reported that genetically engineered Lactococcus lactis can induce antigen-specific tolerance through oral administration and the utilization of genetically modified Lactococcus lactis for celiac disease514,515 (Fig. 6). The main advantage of the therapeutic approach is to induce intestinal Tregs (iTregs) differentiation by CD103+ DC after the antigen uptake516 and facilitate bystander immunosuppression effects by secreting anti-inflammatory cytokines.517 The co-delivery of IL-10 and proinsulin via oral administration of Lactococcus lactis combined with low-dose anti-CD3 therapy can induce infiltration of autoimmune CD8+ T cells and promote the accumulation of Tregs in the pancreas.518
Furthermore, extracellular vesicles (EVs) are the cell-natural nanoparticles released from all eucaryotic and procaryotic cells and play a vital role in intercellular communication and material transport, and show great potential in drug or peptide delivery.519,520,521 Meanwhile, EVs contain multiple intracellular proteins and cell surface proteins that are similar to the source cells.522,523 These characteristics of EVs are extremely useful for antigen-specific therapy. Oligodendrocyte-derived EVs (OI-EVs) containing multiple myelin peptides naturally have been shown to induce up-regulation of PD-L1 in monocytes as well as IL-10 in immune microenvironment to suppress EAE development524 (Fig. 6). Another group designed engineered EVs coupled with HLA-PPI15-24 (pre-proinsulin peptide) and PD-L1 to negatively regulate the activation of T cells in T1D.525
Cell-based antigen-specific immunotherapy
Chimeric antigen receptor T cells
Chimeric antigen receptor T cells (CAR-T) is a technology biased towards cell engineering by importing a manually designed CAR molecule to the surface of T cells to enable these cells’ efficient stress recognition with targeting cells in the MHC-independent manner.526,527,528 The original intention of this therapy was to achieve precise tumor treatment and it achieved breakthrough results.529,530 Due to the precise targeting mechanism, researchers have tried to promote it to other fields such as autoimmune disease treatment531,532,533,534 (Fig. 6).
The CAR structure is composed of extracellular structures, transmembrane domains and intracellular domains. The extracellular portion is usually a single-chain variable fragment (scFv) and spacer, and the former is connected by heavy and light chain ligands of monoclonal antibodies and can combine the specific antigen.535 The transmembrane domains usually come from CD8 or CD28, and these domains are used to connect the extracellular antigen binding domains and intracellular signal transduction domain. The intracellular domains are usually CD28, CD3ζ, and other co-stimulatory molecule domains for T cell activation.536
Zhang et al. chose the 4 citrullinated peptide epitopes as the ligands targeting autoreactive B cells to generate engineering T cells. These engineering T cells can kill the hybridoma cells induced by antigenic peptides and the autoreactive B cells from RA patients specifically.537 A recent report about a young woman with severe SLE and serious complications accepted the CAR-T targeting CD19 and other B cell epitopes after the failure of treatment with other monoclonal antibodies and glucocorticoids. During the next 7 weeks after treatment, CAR-T cell numbers rapidly increased and the patient did not have any adverse events related with CAR-T.538 Zhang et al. also reported the mAb287 CAR (287-CAR) which can target the critical I-Ag7-B:9- 23(R3) complex for attacking the pathogenic CD4+ T cells in NOD mice and demonstrated that these 287-CAR-T can gather in the pancreatic lymph nodes. However, they also reported that a single dose of injection can only delay but cannot prevent the progression of the disease because of the exhaustion of transferred 287-CAR-T.539 HLA-DR1 CAR CD8+ T cells are designed to target the pathogenic autoreactive CD4+ T cells and restrict RA development.540 In addition, it has been reported that the engineered CAR-T cells produced by importing mRNA encoding InsB15-23/β2m/CD3-ζ into the CD8+ T cell can target the pathogenic CD8+ T cells and it offers a new approach to treat T1D diseases.541
Ellebrecht et al. designed the chimeric autoantibody receptor (CAAR) expressed human T cells, and CAAR can aim to target the pemphigus vulgaris (PV) autoantigen, desmoglein (Dsg) 3. It has been confirmed that Dsg3 CAAR-T can specifically kill the B cells expressing Dsg3 on BCR, and they can proliferate to prolong the killing effect.542 Other groups also designed the NMDA receptor (NMDAR)-CAAR-T which can identify and eliminate the autoantibodies originating from B cell lines in NMDAR encephalitis.543
Compared with the engineering of effective T cells, the engineered Tregs have a wider range of applications in autoimmune diseases.544 In previous studies, polyclonal and broad-spectrum T cells were also used for autoimmune disease treatment, however, the effectiveness of these polyclonal T cells is not satisfactory.545,546,547,548 The emerging engineered CAR-Treg can effectively solve this problem. Actually, researchers developed Tregs redirected by antigen-specific chimeric receptor targeting specific antigens and the therapeutic effect has been validated.549,550,551 Fransson et al. tried to engineer the CD4+ T cells with CAR targeting MOG in trans with the murine FoxP3 gene which can drive Tregs differentiation and suppress EAE when administered by intranasal cell delivery.552 Tenspolde et al. redirected the specificity of T cells to insulin by CAR technology and induced effective T cells to differentiate into Tregs. These CAR Tregs have stable expression, effective inhibition, and long-term existence in NOD mice, but they cannot prevent the disease development in female NOD/Ltj mice significantly.553 Other groups also reported that engineering Tregs with anti-InsB10-23(InsB-g7 CAR Treg) can down-regulate BDC2.5T effector cells in the pancreas and peripheral lymphoid organs and induce bystander immunosuppression for T1D.554
However, CAR-T also has some potential safety hazards including cytokine release syndrome (CRS)555 and neurological toxicity.556 CRS is the most prevalent adverse effect after CAR-T therapy which can manifest as a strong immune activation and powerful inflammatory storm. Neurological toxicity usually manifests as confusion, myoclonus, and expressive aphasia.557 Some researchers found that CAR Treg will change the cellular phenotype with regulatory function and convert to pathogenic autoreactive T cells which is undoubtedly devastating for autoimmune disease patients accepting CAR-T therapy. It is uncertain whether side effects like CRS and neurological toxicity will occur in CAR Treg, but it still needs more attention.531
Cell engineering beyond CAR-T technology
In addition to the engineering of T cells, other immune cells can be engineered for autoimmune disease treatment. As mentioned above, tolDCs can efficiently induce T cell tolerance and they are also a key target of many therapeutic drugs so that they can be engineered to treat autoimmune diseases326,327,328 (Fig. 6). A group developed the engineering tolDCs by importing lentiviral vectors carrying some specific antigens and IL-10 sequence. These engineering tolDCs can secrete IL-10 and inhibit the autoreactive CD4+ and CD8+ T cells from celiac disease patients. Besides, these engineering tolDCs can induce antigen-specific Tr1 and prevent the development of T1D in NOD mice.558 Gudi et al. engineered DC to express B7.1wa, PD-L1, HVEM-CRD1 or multi-ligand combination which can prevent the CD4+ T cells proliferation and related inflammatory cytokines secretion. Researchers use DCs loading mouse thyroglobulin to prevent the development in experimental autoimmune thyroiditis.559 In a phase 1b trial, the engineering tolDCs loading with myelin proteins and aquaporin-4 (AQP4) to treat MS patients and induced increase of Tr1 and IL-10 levels successfully without serious adverse events and therapy-related reactions.560 VitD3-antigen-specific tolDCs pulsed with MOG40-55 ameliorated EAE.561,562 OVA-pulsed DCs activated by LPS also alleviated inflammation in OVA-sensitized mice.563
Investigators generated MOG mRNA-electroporated tolDCs presenting autoantigen via electroporation with mRNA encoding MOG and demonstrated its capability to stabilize the clinical score in EAE mice.564 Besides, engineered bi-specific Tregs expressing TCR cross-reactive to MOG and neurofilament-medium (NF-M) had superior protective properties than engineered Tregs expressing MOG mono-specific TCR.565 Other researchers synthesized engineered MBP-specific human Tregs to suppress the development of EAE and demonstrated the induction of bystander suppression.566 Qian et al. engineered naïve T cells by importing a retroviral expression system connected with related antigens and verified that these engineered Tregs can exhibit different abilities compared with traditional Tregs.567
By cell engineering, the specific antigen can also couple with some other cells for tolerance induction568,569,570,571,572 (Fig. 6). Erythrocytes covalently linked to antigenic peptides via the interaction between RBCs endogenous proteins and LPET-sortase covalent intermediate, are designed for protecting against EAE and T1D in an antigen-specific manner.573 Peripheral blood mononuclear cells (PBMCs) coupled with 7 myelin-related peptides have been investigated in a phase 1 trial in patients with MS and the results indicated good safety and tolerance of this strategy.574 The engineered cells can also originate from the location of inflammation, and Au et al. reported the bioengineering PD-L1 and CD86 functionalized Schwann cells for EAE tolerance treatment.575
TolDCs cell for antigen-specific therapy
Harry et al. harvested monocytes from RA patients and healthy donors and induced the cells to differentiate into tolDCs using immunosuppressive drugs, immunomodulatory, and vitamin D3 (VitD3). TolDCs established from patients with RA exhibited typical tolerogenic phenotypes and are comparable to those induced from healthy controls.576 TolDCs can also be generated from MS patients and T1D patients with the aim of developing therapeutics for these diseases.577,578 Recently, VitD3-tolDCs generated from healthy donors and MS patients combined with IFN-β decreased the percentage of activated T cells and induced a shift towards the Th2 profile to inhibit EAE.579 These tolDCs derived from the patients themselves may also have a certain antigen-specific inhibitory effect and are safer for transplantation.576 Some groups also chose to culture tolDCs derived from patients in vitro with autoantigens which may further increase antigen specificity.580 Other groups obtained tolDCs derived from healthy mice and cultured with 2-deoxy glucose (2-DG), and inhibited the experimental autoimmune uveoretinitis (EAU) in vivo581 (Fig. 6).
Subsequent studies implicated that the mature induction is required for tolDCs to maintain immune tolerance.582 Another study reported that efficient treatment will be achieved only when tolDCs are coupled with disease-related autoantigen peptides.583 Boks et al. compared clinical-grade tolDCs generated by coculture with different cytokines (VitD3, IL-10, dexamethasone, TGF-β or RAPA) and demonstrated that clinical-grade IL-10 generated DCs were optimal in tolerance induction.584
Treg cell therapy
In a 2015 report, the polyclonal Tregs are effective for T1D patients by adoptive transplantation without infusion reactions and related serious adverse events.585 The combination of IL-2 and autologous polyclonal Tregs also showed therapeutic effects.586 Investigators reported a SLE patient who accepted autologous Tregs enhanced and expanded in vitro can keep the disease stable. These transplanted cells can accumulate in the skin and induce IFN reduction, and enhance Th17-related pathways.587 In addition, some researchers directly extracted the MBP-reactive Tregs from Tg4 mice expressing transgenic MBP-reactive TCR and expanded them in vitro. Adoptive transplantation of these cells can improve the EAE condition588 (Fig. 6).
Gene therapy
Gene therapy can overcome the limitation of duplicate injection and some side effects caused by antibodies and cytokines therapy and has enormous potential in autoimmune diseases.589,590,591 We mainly introduce the treatment methods related to nucleic acid vaccines here.
DNA vaccine
DNA vaccines have been developed for a long time in numerous medical fields and today’s technologies of DNA vaccine have reached a high level for disease therapy.592,593 For autoimmune diseases, DNA vaccine encoding autoantigen/peptides have been used in antigen-specific immunotherapy594,595,596 (Fig. 6). We summarized the preparation process and drawn a flowchart (Fig. 10).

The flowchart of LNP-mRNA and plasmid-DNA vaccines for autoimmune diseases. The encoding of autoantigen is designed by protein and gene databases. DNA sequence fragments encoding the target antigen peptides are inserted into the plasmid vector to synthesize the recombinant plasmids. These plasmids can be used as DNA vaccines after quality control (QC) and purification. Plasmid DNA is transcribed into mRNA by incorporation of the modified bases. Therapeutic mRNA contains 5’cap, 5’UTR, ORF encoding the target protein/peptides, 3’UTR, and Poly(A) tail. Purified mRNA is mixed with LNP (its components are PEG-Lipids, ionizable lipids, helper lipids, and cholesterol) in a Microfluidic mixer to produce the mRNA-LNP vaccines. (Part of the figure was modified from Servier Medical Art(http://smart.servier.com/), licensed under a Creative Common Attribution 4.0 Generic License. (https://creativecommons.org/licenses/by/4.0/)
Recently, several strategies have been tested to improve the efficacy of this approach. To target hepatocytes for immune suppression induction, Akbarpour et al. designed ICLV.ET.InsB9-23.142T, which consisted of DNA sequence coding antigen peptides, integrase-competent lentiviral vectors (ICLVs), the enhanced transthyretin (ET) hepatocyte-specific promoter, and 142T regulatory elements.597 This DNA vaccine showed long-term existence and continuous expression in hepatocytes, and induced specific immune anergy to antigen peptides and bystander suppression to other antigens via Tregs generation.597 The adeno-associated virus-related antigen-specific vaccine targeting the liver can also restore the tolerance to EAE.598 Some groups also use plant virus nanoparticles such as cowpea mosaic virus and tomato bushy stunt virus to express the p524 in T1D or pLip1 and pFADK2 in RA to treat corresponding autoimmune diseases. They emphasized the peptide scaffold and adjuvant effect of the plant virus nanoparticles and it can provide experience in preclinical testing.599
The use of a single β-cell antigen to induce antigen-specific tolerance for the treatment of T1D patients has so far not been successful. Investigators designed DNA vaccines to deliver multi-epitopes from several β-cell antigens. This approach resulted in a broad engagement of antigen-specific CD4+ and CD8+ diabetogenic T cells and delayed the development of T1D diseases.600 In another report, this group showed that DNA vaccines can expand the regulatory CD4+ cell phenotype and achieve the best therapy effect via long-term intradermal injection.601 In a clinical trial, DNA vaccine encoding proinsulin (BHT-3021) has been shown to reduce the frequency of Proinsulin-reactive CD8+ T cells.602 However, DNA vaccines encoding GAD glutamic acid decarboxylase (GAD) showed mixed results in NOD mice for the treatment of T1D.603,604 More DNA vaccines for the treatment of autoimmune diseases have been put into clinical trials and the efficacy is still uncertain.605,606
Besides, DNA vaccines also have the risk of causing insertional mutations because DNA works by entering the nucleus which leads to the exposure to exogenous genes for vaccines.593 The DNA vaccines also may promote the generation of anti-DNA IgG autoantibodies, which will lead to the aggravation of autoimmune diseases.607
mRNA vaccine
In 1961, researchers discovered mRNA and tried to use protamine for mRNA delivery.608,609 Unlike DNA vaccines, mRNA-based vaccine has no potential risk to enter the host genome because it does not need to enter the nucleus. However, the characteristics of ease of decay, instability and immunogenicity restrict the application of mRNA to a large extent.610,611,612 Karikó et al. found that naturally occurring modified nucleosides can suppress the immunostimulatory activity of RNA.613,614 Furthermore, the modified nucleosides can promote the translational capacity and enhance the biological stability more effectively.615 This discovery allowed the fast development of various mRNA-based vaccines and therapeutics. Investigators tried to develop mRNA-based DCs vaccines.564,616,617 mRNA can code and produce any protein/peptides and this advantage makes it an ideal strategy to treat diseases that need protein/peptide expression. Moreover, a single mRNA strand can encode several antigens or tandem constructs that contain several epitopes from different antigens (Fig. 6). In 2016, Dastagir et al. have reported the delivery of mRNA with tandem multiple diabetes-associated antigen epitopes by DCs for T1D treatment.618
As mentioned earlier, the FDA approved the first liposome complex for small interfering RNA (siRNA) binding for the treatment of a rare disease called hereditary transthyretin-mediated amyloidosis (hATTR) in 2018294,295 and it is suggested that liposomes are feasible for transformation in the clinical application of RNA delivery.
Thanks to the great efforts made by several research groups and companies to develop efficient delivery systems and methods to decrease mRNA immunogenicity and improve the transportation efficiency over the past decades, mRNA technology has made a major breakthrough during the COVID-19 pandemic. Pfizer-BioNTech utilized LNPs to prepare the BNT162b2 mRNA vaccine against COVID-19 and achieved great success.619,620,621 LNPs are the most advanced mRNA delivery systems and have shown unique advantages.622,623,624,625 It is precisely because LNPs promote endosomal escape and thus enhance mRNA translation efficiency.626,627 mRNA structure consists of a 5’cap, a 5’ untranslated region (5’UTR), an open reading frame (ORF), a 3’untranslated region (3’UTR), and a poly (A) tail. Each part of mRNA has specific structures and composition to maintain mRNA stability.628,629 We summarized the preparation process of the LNP-mRNA vaccines and described the structure of mRNA sequence and the components of LNP (Fig. 10).
Krienke et al. designed nanoparticle formulated 1 methylpseudouridine-modified noninflammatory mRNA (m1Ψ mRNA) vaccine coding autoantigens and tested its efficacy to treat EAE630 (Fig. 6). They showed that autoantigen encoding m1Ψ mRNA treatment suppressed disease progress in several mouse models of MS via the expansion of Treg cells and the reduction of effector T cells.630 Furthermore, epitope spreading is suppressed via Treg cell-mediated bystander tolerance induced by LPX-m1Ψ mRNA encoding MOG35-55.630
There is growing interest in designing new LNPs to target different organs and cells for mRNA-based vaccines and therapeutics. Researchers designed a liver-targeting LNP platform to deliver mRNA-encoding allergen epitopes to treat peanut-induced anaphylaxis.631 When comparing mRNA delivered by LNPs and mRNA electroporated DCs, LNPs can stimulate T cell responses within a wider antigen-specific T cell subpopulation. Furthermore, nanoparticle-delivered mRNA localized in the spleen preferentially while mRNA electroporated DCs primarily localized in the lung after intravenous injection.632 Some researchers designed LNPs containing an anionic phospholipid, phosphatidylserine (PS) to deliver mRNA in the spleen for EAE treatment and achieved a promising efficacy.633 Microbubble-assisted focused ultrasound (FUS) technology can increase the BBB permeability for LNP-mRNA and may be more beneficial for mRNA-LNP therapy for MS.634
Recent studies reported that EVs extend the function of mRNA-LNPs, protect them from degradation and promote the transport of mRNA between cells.635,636 It can improve the efficiency of mRNA transmission as well as the cure rate of autoimmune diseases.
Although the application of mRNA technology is in full swing, we should still pay attention to the future challenges for mRNA development and application in the clinic, which include the delivery of mRNA macromolecules, improvement of the stability of mRNA-delivery carrier and the regulation of mRNA-delivery carrier for immune system.637
Clinical progress of therapeutic drugs
In recent years, we have witnessed the clinical translation of novel therapies for the treatment of autoimmune disorders. Here we overview the FDA-approved drugs and clinical pipelines of antigen-specific immunotherapy for autoimmune diseases. Major breakthroughs have been made in this field, which may pave the way for successful clinical translation of antigen-specific immunotherapies.
The FDA-approved drugs
Currently, available drugs for autoimmune diseases focus on the targeted blockade of immune inflammation-related membrane surface molecules or cytokines by monoclonal antibody (mAb). Main targets for autoimmune disease treatment include IL-23, IL-17, integrin, TNF, CD20, IL-1, IL-5, IL-6, BAFF/APRIL, etc., and their related receptors or ligands638 (Table 1).
Here we describe several mAb-targeting drugs that have achieved significant clinical treatment effects and some possible side effects.638 DUPIXENT (dupilumab) and ADBRY (tralokinumab) which target IL-4/13 can effectively treat atopic dermatitis and asthma. Rituximab targeting CD20, TYRUKO (natalizumab-sztn) and TYSABRI (natalizumab) targeting α4β1 and α4β7 integrins have been found to be efficient in treating MS. Anakinra, canakinumab, rilonacept targeting IL-1 and related loci can be effective for systemic autoinflammatory disease. STELARA (ustekinumab) and WEZLANA (ustekinumab-auub) targeting IL-12/23 (p40) can treat Crohn’s disease effectively. TREMFYA (guselkumab), SKYRIZI (risankizumab-rzaa) and ILUMYA (tildrakizumab-asmn) targeting IL-23 (p19) and COSENTYX (secukinumab), TALTZ (ixekizumab), BIMZELX (bimekizumab-bkzx) and SILIQ (brodalumab) targeting IL-17 or related loci are surprisingly effective for the treatment of plaque psoriasis.638
mAbs have some side effects related to their specific targets.639,640 For example, serious infections usually occur because of the removal of the target ligand for the mAbs. Patients will experience symptoms including cough, weight loss and low-grade fever. Allergic reaction is another common side effect and this symptom can be very dangerous once it occurs.
In addition, we also emphasize an antigen-mimetic drug, glatiramer acetate, which is a synthetic copolymer, and the component is based on the structure of MBP641,642 (Table 1). It has shown astonishing therapeutic effects in animal models and has been applied to clinical MS treatment.643,644,645
Antigen-specific immunotherapy clinical research progress
Antigen-specific immunotherapy has the high specificity and possesses the potential to induce bystander immune regulation, it holds great potential for the treatment of autoimmune diseases compared with systemic immunosuppressive therapy. There are many different approaches for antigen-specific immunotherapy including whole antigen or peptides, material-based delivery, modified peptides, MHC-peptides, cell-based therapy, and DNA vaccines. Antigen-specific therapies for autoimmune diseases are still in the early stages of clinical application50,646,647,648 and these new approaches hold great promise for successful clinical translation. Here we mainly summarize some related drug designs, progress, outcomes, etc., in clinical trials conducted on antigen-specific immunotherapy (Table 2). Some MS-related clinical trials show hypersensitivity reactions and disease deterioration in individual patients.399,400,649 Other clinical trials also show inadequate therapeutic effects.650,651,652 Researchers demonstrated the strong immunogenicity of MBP83-99 APL which can induce the cross-reactive with native autoantigen and lead to inflammatory differentiation of naïve T cells. The weak effect of specific treatment occurs not only in MS, but also in T1D.653,654,655 Admittedly, dose and route of administration are the key factors for treatment effects and side effects.399,653 However, these conditions are variable and adjustable. The fundamental reason is our limited understanding of the breadth of human autoantigen repertoire and the strategy to deal with the epitope spreading.358 For MS, although the role of some myelin has been verified in animal EAE models, the relation with MS is still debatable.399 Differences in the epitopes of autoreaction in different patients may provide an explanation for the different treatment effects and side effects in patients while they received the same medicine and routes of administration. Hence, the identification of relevant antigens and personalized autoantigen design need to be addressed urgently for the heterogeneity in individual patients.358,399 The combination of autoantigens with immunomodulatory drugs and nanomaterials has made a great progress in animal models,430,486,656 however, the toxicity and limitation of nanomaterials and immunomodulatory drugs should also deserve adequate attention in the process of clinical transformation.657 Besides, effective biomarkers are urgently essential for the establishment of preclinical diagnosis and long-term monitoring of the disease progress after administration.358
Conclusion
This review summarized the epidemiology, mechanisms, and new therapeutic strategies for autoimmune diseases. Continued surveillance of epidemiologic data around the world is needed to improve our understanding of disease risk and disease burden. The development of autoimmune diseases is driven by genetic and environmental factors. With regard to drug development, the treatments of autoimmune diseases have achieved great progress in both antigen-specific immunotherapy and biotherapeutics. The former is still in its infancy in clinical translation, but it has great potential in precise treatment without affecting the whole immune system. Biotherapeutics especially mAbs have successfully applied in clinics for the treatment of autoimmune diseases. The identification of new targets and related biomarkers will enable the development of new biotherapeutics.
Cumulative findings have shown that nanomaterials are promising approaches to deliver autoantigen protein/peptides, DNA, and mRNA for induction of immune tolerance for the treatment of autoimmune diseases.658,659,660,661 One of the mechanisms behind it is that specific antigens can induce the generation and differentiation of tolerogenic APCs, which will drive the anergy, deletion, and apoptosis of pathogenic CD4+/CD8+ T cells and induction of Foxp3+ Tregs. Furthermore, tolDCs can secrete a series of immunosuppressive cytokines including TGF-β and IL-10 to promote immune anergy.662 The additional immunomodulatory molecules/drugs (dexamethasone, ITE, RAPA, etc.) co-delivered by nanomaterials are also a highly efficient approach for antigen-specific therapy by providing multiple suppression-related signals.663,664,665,666 In addition, these compounds also promote the differentiation of tolDCs and exhibit immunomodulatory effects. Nowadays, many nanomaterials applied in antigen-specific therapy are in the preclinical stage. We believe that the practice of these nanomaterials in clinical trials can further promote the antigen-specific therapy.
The fast development of mRNA-based therapies has attracted people’s attention to many diseases, especially cancer and autoimmune diseases.637 Investigators validated that mRNA vaccines have the capacity of inducing Treg cells which execute bystander immunosuppression in animal models for MS.630 mRNA technique has several advantages over protein or DNA drugs, including faster manufacturing, lower insertion risk, lower cost and controllable immunogenicity by nucleotide modification. In brief, the mRNA-LNP vaccine has infinite potential in the treatment of many difficult-to-treat diseases, including autoimmune diseases in the future, and now this is just the beginning.667 Novel ionizable lipids for mRNA delivery are continuously developed for efficient delivery, better therapeutic effect and safety.668,669 Besides, the mRNA-LNP vaccine has been approved for marketing for COVID-19, so it also can provide certain clinical transformation guidance for autoimmune diseases.
The administration routes are also key factors in tolerance induction. For MS, some groups have used the inhalation administration and intranasal delivery strategy for the EAE model and achieved the expected results.670 The next research direction can focus on synthesizing drug delivery vehicles for intranasal drug delivery routes. We also emphasize that the effectiveness of drugs varies at different stages of the disease. For MS, almost all drugs only target RRMS.671 For example, Tysabri and Fingolimod are approved drugs for RRMS, however, these therapies are ineffective for PMS. Therefore, it is essential to develop new effective therapies for all stages and types of the disease. Besides, the definition of optimal dose conversion, selection of route of administration, and the establishment of effective biomarkers are also huge challenges for individual optimization therapy and disease surveillance in clinical application.
In sum, we emphasized the development and future prospects of highly potential antigen-specific therapies for autoimmune diseases. We summarized antigen-specific therapy including whole antigen protein therapy, antigen modification methods, APL strategies, pMHCs, biomaterials-based delivery methods, tolerogenic cell-based therapy, and gene techniques treatment. Although significant advances have been made in this field, the treatment efficacy of antigen-specific therapy in humans is still uncertain. The development of a tolerable biomaterial delivery system, accurate prediction of specific antigen epitopes, and combination therapy with other immunomodulatory drugs is necessary in both animal research and human clinical trials. Meanwhile, this is also a fundamental challenge we will face in the future. In particular, the decoding of the autoantigen repertoire and epitope prediction can help us better understand the mechanism and origin of autoimmune diseases, and select the corresponding antigen for specific therapy. For the induction of bystander immunity to deal with epitope spreading, researchers must integrate multiple disciplines such as immunology, materials science, biotechnology, bioinformatics, etc. We expect that antigen-specific immunotherapy will soon have clinical application and benefit the patients with various autoimmune diseases.
Change history
24 December 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41392-025-02525-z
References
Davidson, A. & Diamond, B. Autoimmune diseases. N. Engl. J. Med. 345, 340–350 (2001).
Pisetsky, D. S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 19, 509–524 (2023).
Autoimmune Disease List, <https://autoimmune.org/disease-information/> (2024).
Fugger, L., Jensen, L. T. & Rossjohn, J. Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell 181, 63–80 (2020).
Wang, L., Wang, F. S. & Gershwin, M. E. Human autoimmune diseases: a comprehensive update. J. Intern. Med. 278, 369–395 (2015).
Scherlinger, M. et al. Worldwide trends in all-cause mortality of auto-immune systemic diseases between 2001 and 2014. Autoimmun. Rev. 19, 102531 (2020).
Conrad, N. et al. Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: a population-based cohort study of 22 million individuals in the UK. Lancet 401, 1878–1890 (2023).
Hensvold, A. & Klareskog, L. Towards prevention of autoimmune diseases: the example of rheumatoid arthritis. Eur. J. Immunol. 51, 1921–1933 (2021).
Wen, X. & Li, B. A population-based study on autoimmune disease. Lancet 401, 1829–1831 (2023).
Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 17, 281–294 (2017).
Koch, U. & Radtke, F. Mechanisms of T cell development and transformation. Annu Rev. Cell Dev. Biol. 27, 539–562 (2011).
Takaba, H. & Takayanagi, H. The mechanisms of T cell selection in the thymus. Trends Immunol. 38, 805–816 (2017).
Mueller, D. L. Mechanisms maintaining peripheral tolerance. Nat. Immunol. 11, 21–27 (2010).
ElTanbouly, M. A. & Noelle, R. J. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat. Rev. Immunol. 21, 257–267 (2021).
Green, D. R., Droin, N. & Pinkoski, M. Activation-induced cell death in T cells. Immunol. Rev. 193, 70–81 (2003).
Zheng, L., Li, J. & Lenardo, M. Restimulation-induced cell death: new medical and research perspectives. Immunol. Rev. 277, 44–60 (2017).
Schwartz, R. H. T cell anergy. Annu Rev. Immunol. 21, 305–334 (2003).
Kranich, J. & Krautler, N. J. How follicular dendritic cells shape the B-cell antigenome. Front. Immunol. 7, 225 (2016).
Kant, S., Kronbichler, A., Sharma, P. & Geetha, D. Advances in understanding of pathogenesis and treatment of immune-mediated kidney disease: a review. Am. J. Kidney Dis. 79, 582–600 (2022).
Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018).
Stanford, S. M. & Bottini, N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat. Rev. Rheumatol. 10, 602–611 (2014).
Tizaoui, K. et al. The role of PTPN22 in the pathogenesis of autoimmune diseases: a comprehensive review. Semin Arthritis Rheum. 51, 513–522 (2021).
Xie, J. et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat. Commun. 11, 1600 (2020).
Omarjee, O. et al. Monogenic lupus: dissecting heterogeneity. Autoimmun. Rev. 18, 102361 (2019).
Zipfel, P. F., Wiech, T., Stea, E. D. & Skerka, C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J. Am. Soc. Nephrol. 31, 241–256 (2020).
Crow, Y. J. & Stetson, D. B. The type I interferonopathies: 10 years on. Nat. Rev. Immunol. 22, 471–483 (2022).
Coit, P. et al. Epigenetic reprogramming in naive CD4+T cells favoring T cell activation and non-Th1 effector T cell immune response as an early event in lupus flares. Arthritis Rheumatol. 68, 2200–2209 (2016).
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Wang, Z. et al. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: a comprehensive overview. Cell Mol. Life Sci. 75, 3353–3369 (2018).
Furman, D. et al. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc. Natl Acad. Sci. USA 111, 869–874 (2014).
Fischinger, S. et al. Sex differences in vaccine-induced humoral immunity. Semin. Immunopathol. 41, 239–249 (2019).
Scofield, R. H. et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 58, 2511–2517 (2008).
Rojas, M. et al. Molecular mimicry and autoimmunity. J. Autoimmun. 95, 100–123 (2018).
Libbey, J. E., Cusick, M. F., Tsunoda, I. & Fujinami, R. S. Antiviral CD8( + ) T cells cause an experimental autoimmune encephalomyelitis-like disease in naive mice. J. Neurovirol. 18, 45–54 (2012).
Cusick, M. F., Libbey, J. E. & Fujinami, R. S. Multiple sclerosis: autoimmunity and viruses. Curr. Opin. Rheumatol. 25, 496–501 (2013).
Ogunrinde, E. et al. A link between plasma microbial translocation, microbiome, and autoantibody development in first-degree relatives of systemic lupus erythematosus patients. Arthritis Rheumatol. 71, 1858–1868 (2019).
Jog, N. R. & James, J. A. Epstein Barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol. 11, 623944 (2020).
Soldan, S. S. & Lieberman, P. M. Epstein-Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 21, 51–64 (2023).
Houen, G. & Trier, N. H. Epstein-Barr virus and systemic autoimmune diseases. Front Immunol. 11, 587380 (2020).
Holmoy, T., Kvale, E. O. & Vartdal, F. Cerebrospinal fluid CD4+T cells from a multiple sclerosis patient cross-recognize Epstein-Barr virus and myelin basic protein. J. Neurovirol. 10, 278–283 (2004).
Barzilai, O., Ram, M. & Shoenfeld, Y. Viral infection can induce the production of autoantibodies. Curr. Opin. Rheumatol. 19, 636–643 (2007).
Cao, Y. et al. Targeting the signaling in Epstein-Barr virus-associated diseases: mechanism, regulation, and clinical study. Signal Transduct. Target Ther. 6, 15 (2021).
Ruff, W. E., Greiling, T. M. & Kriegel, M. A. Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 18, 521–538 (2020).
Manfredo Vieira, S. et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161 (2018).
Yang, Y. et al. Within-host evolution of a gut pathobiont facilitates liver translocation. Nature 607, 563–570 (2022).
Marino, E. et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 18, 552–562 (2017).
Brown, J., Robusto, B. & Morel, L. Intestinal dysbiosis and tryptophan metabolism in autoimmunity. Front. Immunol. 11, 1741 (2020).
Thorburn, A. N., Macia, L. & Mackay, C. R. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40, 833–842 (2014).
Arnson, Y., Shoenfeld, Y. & Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 34, J258–J265 (2010).
Edner, N. M., Carlesso, G., Rush, J. S. & Walker, L. S. K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 19, 860–883 (2020).
Boomer, J. S. & Green, J. M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2, a002436 (2010).
Esensten, J. H. et al. CD28 costimulation: from mechanism to therapy. Immunity 44, 973–988 (2016).
Shahinian, A. et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science 261, 609–612 (1993).
Ferguson, S. E., Han, S., Kelsoe, G. & Thompson, C. B. CD28 is required for germinal center formation. J. Immunol. 156, 4576–4581 (1996).
Walker, L. S. et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J. Exp. Med. 190, 1115–1122 (1999).
Chang, T. T. et al. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J. Exp. Med. 190, 733–740 (1999).
Tada, Y. et al. Role of the costimulatory molecule CD28 in the development of lupus in MRL/lpr mice. J. Immunol. 163, 3153–3159 (1999).
Webb, L. M., Walmsley, M. J. & Feldmann, M. Prevention and amelioration of collagen-induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7-1 and B7-2. Eur. J. Immunol. 26, 2320–2328 (1996).
Hossen, M. M. et al. Current understanding of CTLA-4: from mechanism to autoimmune diseases. Front. Immunol. 14, 1198365 (2023).
Yu, C. et al. Rigid-body ligand recognition drives cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor triggering. J. Biol. Chem. 286, 6685–6696 (2011).
Hosseini, A. et al. CTLA-4: from mechanism to autoimmune therapy. Int Immunopharmacol. 80, 106221 (2020).
Abrams, J. R. et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J. Clin. Investig. 103, 1243–1252 (1999).
Brunner, H. I. et al. Subcutaneous abatacept in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase III open-label study. Arthritis Rheumatol. 70, 1144–1154 (2018).
Weber, J. P. et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J. Exp. Med. 212, 217–233 (2015).
Xu, H. et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 496, 523–527 (2013).
Gigoux, M. et al. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc. Natl Acad. Sci. USA 106, 20371–20376 (2009).
Simpson, N. et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 62, 234–244 (2010).
Szabo, K. et al. Follicular helper T cells may play an important role in the severity of primary Sjogren’s syndrome. Clin. Immunol. 147, 95–104 (2013).
Wang, J. et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 174, 212–220 (2013).
Francisco, L. M., Sage, P. T. & Sharpe, A. H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 (2010).
Ning, Z., Liu, K. & Xiong, H. Roles of BTLA in immunity and immune disorders. Front Immunol. 12, 654960 (2021).
Raptopoulou, A. P. et al. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. 62, 1870–1880 (2010).
Wang, G. et al. The effects of PDL-Ig on collagen-induced arthritis. Rheumatol. Int 31, 513–519 (2011).
Song, M. Y. et al. Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut. 64, 260–271 (2015).
Tang, T. et al. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharm. Ther. 219, 107709 (2021).
Karnell, J. L., Rieder, S. A., Ettinger, R. & Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv. Drug Deliv. Rev. 141, 92–103 (2019).
Foy, T. M. et al. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J. Exp. Med. 180, 157–163 (1994).
Kawabe, T. et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1, 167–178 (1994).
Harigai, M. et al. Ligation of CD40 induced tumor necrosis factor-alpha in rheumatoid arthritis: a novel mechanism of activation of synoviocytes. J. Rheumatol. 26, 1035–1043 (1999).
Tsunawaki, S. et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J. Rheumatol. 29, 1884–1896 (2002).
Goules, A. et al. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J. Autoimmun. 26, 165–171 (2006).
Dimitriou, I. D., Kapsogeorgou, E. K., Moutsopoulos, H. M. & Manoussakis, M. N. CD40 on salivary gland epithelial cells: high constitutive expression by cultured cells from Sjogren’s syndrome patients indicating their intrinsic activation. Clin. Exp. Immunol. 127, 386–392 (2002).
Karnell, J. L. et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 11, eaar6584 (2019).
Mahmoud, T. I. et al. Autoimmune manifestations in aged mice arise from early-life immune dysregulation. Sci. Transl. Med. 8, 361ra137 (2016).
Webb, G. J., Hirschfield, G. M. & Lane, P. J. OX40, OX40L and autoimmunity: a comprehensive review. Clin. Rev. Allergy Immunol. 50, 312–332 (2016).
Fu, Y., Lin, Q., Zhang, Z. & Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 10, 414–433 (2020).
Cunninghame Graham, D. S. et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 40, 83–89 (2008).
Lee, Y. H. & Song, G. G. Associations between TNFSF4 and TRAF1-C5 gene polymorphisms and systemic lupus erythematosus: a meta-analysis. Hum. Immunol. 73, 1050–1054 (2012).
Manku, H. et al. Trans-ancestral studies fine map the SLE-susceptibility locus TNFSF4. PLoS Genet 9, e1003554 (2013).
Nordmark, G. et al. Association of EBF1, FAM167A(C8orf13)-BLK and TNFSF4 gene variants with primary Sjogren’s syndrome. Genes Immun. 12, 100–109 (2011).
Bossini-Castillo, L. et al. A replication study confirms the association of TNFSF4 (OX40L) polymorphisms with systemic sclerosis in a large European cohort. Ann. Rheum. Dis. 70, 638–641 (2011).
Gourh, P. et al. Association of TNFSF4 (OX40L) polymorphisms with susceptibility to systemic sclerosis. Ann. Rheum. Dis. 69, 550–555 (2010).
Faraco, J. et al. ImmunoChip study implicates antigen presentation to T cells in narcolepsy. PLoS Genet. 9, e1003270 (2013).
Guttman-Yassky, E. et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 482–493.e487 (2019).
Charabati, M., Wheeler, M. A., Weiner, H. L. & Quintana, F. J. Multiple sclerosis: neuroimmune crosstalk and therapeutic targeting. Cell 186, 1309–1327 (2023).
Reich, D. S., Lucchinetti, C. F. & Calabresi, P. A. Multiple sclerosis. N. Engl. J. Med. 378, 169–180 (2018).
Rodriguez Murua, S., Farez, M. F. & Quintana, F. J. The immune response in multiple sclerosis. Annu. Rev. Pathol. 17, 121–139 (2022).
Compston, A. & Coles, A. Multiple sclerosis. Lancet 372, 1502–1517 (2008).
Walton, C. et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 26, 1816–1821 (2020).
Wang, J. et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell 183, 1264–1281 e1220 (2020).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 365, eaav7188 (2019).
Berer, K. et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479, 538–541 (2011).
Schnell, A. et al. Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 184, 6281–6298.e6223 (2021).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Lang, H. L. et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 3, 940–943 (2002).
Simpson, S. Jr. et al. Latitude continues to be significantly associated with the prevalence of multiple sclerosis: an updated meta-analysis. J. Neurol. Neurosurg. Psychiatry 90, 1193–1200 (2019).
Fitzgerald, K. C. et al. Association of vitamin D levels with multiple sclerosis activity and progression in patients receiving interferon beta-1b. JAMA Neurol. 72, 1458–1465 (2015).
Mokry, L. E. et al. Obesity and multiple sclerosis: a mendelian randomization study. PLoS Med. 13, e1002053 (2016).
Rosso, M. & Chitnis, T. Association between cigarette smoking and multiple sclerosis: a review. JAMA Neurol. 77, 245–253 (2020).
Dendrou, C. A., Fugger, L. & Friese, M. A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558 (2015).
Solomon, A. J. et al. Differential diagnosis of suspected multiple sclerosis: an updated consensus approach. Lancet Neurol. 22, 750–768 (2023).
Kolb, H. et al. 7T MRI differentiates remyelinated from demyelinated multiple sclerosis lesions. Ann. Neurol. 90, 612–626 (2021).
Eshaghi, A. et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat. Commun. 12, 2078 (2021).
Bodini, B., Tonietto, M., Airas, L. & Stankoff, B. Positron emission tomography in multiple sclerosis—straight to the target. Nat. Rev. Neurol. 17, 663–675 (2021).
Huang, J. et al. Inflammation-related plasma and CSF biomarkers for multiple sclerosis. Proc. Natl Acad. Sci. USA 117, 12952–12960 (2020).
Thompson, A. J. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 17, 162–173 (2018).
Shi, K. et al. Bone marrow hematopoiesis drives multiple sclerosis progression. Cell 185, 2234–2247.e2217 (2022).
Jelcic, I. et al. Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 175, 85–100.e123 (2018).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Murphy, A. C., Lalor, S. J., Lynch, M. A. & Mills, K. H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 24, 641–651 (2010).
Wagner, C. A. et al. Myelin-specific CD8+T cells exacerbate brain inflammation in CNS autoimmunity. J. Clin. Investig. 130, 203–213 (2020).
Machado-Santos, J. et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8 + T lymphocytes and B cells. Brain 141, 2066–2082 (2018).
Li, R., Patterson, K. R. & Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 19, 696–707 (2018).
Yoshizaki, A. et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 491, 264–268 (2012).
Matsushita, T. et al. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Invest 118, 3420–3430 (2008).
Yong, V. W., Power, C., Forsyth, P. & Edwards, D. R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2, 502–511 (2001).
Waxman, S. G. Nitric oxide and the axonal death cascade. Ann. Neurol. 53, 150–153 (2003).
Selmaj, K. W. & Raine, C. S. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol. 23, 339–346 (1988).
DiMeglio, L. A., Evans-Molina, C. & Oram, R. A. Type 1 diabetes. Lancet 391, 2449–2462 (2018).
Atkinson, M. A., Eisenbarth, G. S. & Michels, A. W. Type 1 diabetes. Lancet 383, 69–82 (2014).
Gregory, G. A. et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. 10, 741–760 (2022).
Mayer-Davis, E. J. et al. Incidence trends of type 1 and type 2 diabetes among youths, 2002-2012. N. Engl. J. Med. 376, 1419–1429 (2017).
Chow, C. K. et al. Availability and affordability of essential medicines for diabetes across high-income, middle-income, and low-income countries: a prospective epidemiological study. Lancet Diabetes Endocrinol. 6, 798–808 (2018).
Rogers, M. A. M., Kim, C., Banerjee, T. & Lee, J. M. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: a longitudinal study. BMC Med. 15, 199 (2017).
Harjutsalo, V., Sjoberg, L. & Tuomilehto, J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 371, 1777–1782 (2008).
Ostman, J. et al. Gender differences and temporal variation in the incidence of type 1 diabetes: results of 8012 cases in the nationwide Diabetes Incidence Study in Sweden 1983-2002. J. Intern Med. 263, 386–394 (2008).
Noble, J. A. Immunogenetics of type 1 diabetes: A comprehensive review. J. Autoimmun. 64, 101–112 (2015).
Erlich, H. et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57, 1084–1092 (2008).
Concannon, P., Rich, S. S. & Nepom, G. T. Genetics of type 1A diabetes. N. Engl. J. Med 360, 1646–1654 (2009).
Makela, M. et al. Enteral virus infections in early childhood and an enhanced type 1 diabetes-associated antibody response to dietary insulin. J. Autoimmun. 27, 54–61 (2006).
Hippisley-Cox, J. & Coupland, C. Development and validation of risk prediction equations to estimate future risk of blindness and lower limb amputation in patients with diabetes: cohort study. BMJ 351, h5441 (2015).
American Diabetes, A. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2018. Diabetes Care 41, S13–S27 (2018).
Ziegler, A. G. & Nepom, G. T. Prediction and pathogenesis in type 1 diabetes. Immunity 32, 468–478 (2010).
Bingley, P. J. Clinical applications of diabetes antibody testing. J. Clin. Endocrinol. Metab. 95, 25–33 (2010).
Delong, T. et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 351, 711–714 (2016).
Eizirik, D. L. et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552 (2012).
Roep, B. O., Arden, S. D., de Vries, R. R. & Hutton, J. C. T-cell clones from a type-1 diabetes patient respond to insulin secretory granule proteins. Nature 345, 632–634 (1990).
Ilonen, J., Lempainen, J. & Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 15, 635–650 (2019).
Campbell-Thompson, M. L. et al. Relative pancreas volume is reduced in first-degree relatives of patients with type 1 diabetes. Diabetes Care 42, 281–287 (2019).
Di Matteo, A., Bathon, J. M. & Emery, P. Rheumatoid arthritis. Lancet 402, 2019–2033 (2023).
Gravallese, E. M. & Firestein, G. S. Rheumatoid arthritis—common origins, divergent mechanisms. N. Engl. J. Med. 388, 529–542 (2023).
Smith, M. H. & Berman, J. R. What is rheumatoid arthritis? JAMA 327, 1194 (2022).
Finckh, A. et al. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 18, 591–602 (2022).
Safiri, S. et al. Global, regional and national burden of rheumatoid arthritis 1990-2017: a systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 78, 1463–1471 (2019).
Scott, I. C. et al. Rheumatoid arthritis, psoriatic arthritis, and axial spondyloarthritis epidemiology in England from 2004 to 2020: an observational study using primary care electronic health record data. Lancet Reg. Health Eur. 23, 100519 (2022).
Cutolo, M. & Straub, R. H. Sex steroids and autoimmune rheumatic diseases: state of the art. Nat. Rev. Rheumatol. 16, 628–644 (2020).
Gregersen, P. K., Silver, J. & Winchester, R. J. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 30, 1205–1213 (1987).
Klareskog, L. et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 54, 38–46 (2006).
Karlson, E. W. et al. Associations between human leukocyte antigen, PTPN22, CTLA4 genotypes and rheumatoid arthritis phenotypes of autoantibody status, age at diagnosis and erosions in a large cohort study. Ann. Rheum. Dis. 67, 358–363 (2008).
Plenge, R. M. et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 77, 1044–1060 (2005).
Ishigaki, K. et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 54, 1640–1651 (2022).
Torres, A. et al. Epigenetic regulation of nutrient transporters in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 74, 1159–1171 (2022).
Klareskog, L. et al. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol. 23, 92–98 (2011).
Gianfrancesco, M. A. & Crowson, C. S. Where there’s smoke, there’s a joint: passive smoking and rheumatoid arthritis. Arthritis Rheumatol. 73, 2161–2162 (2021).
Reynisdottir, G. et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 66, 31–39 (2014).
Moller, B., Kollert, F., Sculean, A. & Villiger, P. M. Infectious triggers in periodontitis and the gut in rheumatoid arthritis (RA): a complex story about association and causality. Front. Immunol. 11, 1108 (2020).
Scher, J. U. et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202 (2013).
Kishikawa, T. et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann. Rheum. Dis. 79, 103–111 (2020).
Lu, B. et al. Being overweight or obese and risk of developing rheumatoid arthritis among women: a prospective cohort study. Ann. Rheum. Dis. 73, 1914–1922 (2014).
Lu, B., Solomon, D. H., Costenbader, K. H. & Karlson, E. W. Alcohol consumption and risk of incident rheumatoid arthritis in women: a prospective study. Arthritis Rheumatol. 66, 1998–2005 (2014).
Hahn, J. et al. Vitamin D and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial. BMJ 376, e066452 (2022).
Gan, R. W. et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope-positive subjects at risk for rheumatoid arthritis. Ann. Rheum. Dis. 76, 147–152 (2017).
Ford, J. A. et al. Asthma, chronic obstructive pulmonary disease, and subsequent risk for incident rheumatoid arthritis among women: a prospective cohort study. Arthritis Rheumatol. 72, 704–713 (2020).
Ytterberg, S. R. et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N. Engl. J. Med. 386, 316–326 (2022).
Smolen, J. S. et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 82, 3–18 (2023).
Aletaha, D. et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62, 2569–2581 (2010).
Aletaha, D. et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 69, 1580–1588 (2010).
van Venrooij, W. J., van Beers, J. J. & Pruijn, G. J. Anti-CCP antibodies: the past, the present and the future. Nat. Rev. Rheumatol. 7, 391–398 (2011).
van den Broek, M. et al. The association of treatment response and joint damage with ACPA-status in recent-onset RA: a subanalysis of the 8-year follow-up of the BeSt study. Ann. Rheum. Dis. 71, 245–248 (2012).
Rao, D. A. et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017).
Jonsson, A. H. et al. Granzyme K(+) CD8 T cells form a core population in inflamed human tissue. Sci. Transl. Med. 14, eabo0686 (2022).
Wei, K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264 (2020).
Yan, M. et al. ETS1 governs pathological tissue-remodeling programs in disease-associated fibroblasts. Nat. Immunol. 23, 1330–1341 (2022).
Orange, D. E. et al. RNA identification of PRIME cells predicting rheumatoid arthritis flares. N. Engl. J. Med. 383, 218–228 (2020).
Culemann, S. et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 572, 670–675 (2019).
Guo, Q. et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 6, 15 (2018).
Ding, Q. et al. Signaling pathways in rheumatoid arthritis: implications for targeted therapy. Signal Transduct. Target Ther. 8, 68 (2023).
Dorner, T. & Furie, R. Novel paradigms in systemic lupus erythematosus. Lancet 393, 2344–2358 (2019).
Durcan, L., O’Dwyer, T. & Petri, M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet. 393, 2332–2343 (2019).
Kaul, A. et al. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2, 16039 (2016).
Barber, M. R. W. et al. Global epidemiology of systemic lupus erythematosus. Nat. Rev. Rheumatol. 17, 515–532 (2021).
Kiriakidou, M. & Ching, C. L. Systemic lupus erythematosus. Ann. Intern. Med. 172, Itc81–itc96 (2020).
Pons-Estel, B. A. et al. First Latin American clinical practice guidelines for the treatment of systemic lupus erythematosus: Latin American Group for the Study of Lupus (GLADEL, Grupo Latino Americano de Estudio del Lupus)-Pan-American League of Associations of Rheumatology (PANLAR). Ann. Rheum. Dis. 77, 1549–1557 (2018).
Cervera, R. et al. Patterns of systemic lupus erythematosus expression in Europe. Autoimmun. Rev. 13, 621–629 (2014).
Buckley, L. et al. 2017 American College of Rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheumatol. 69, 1521–1537 (2017).
Ghodke-Puranik, Y. & Niewold, T. B. Immunogenetics of systemic lupus erythematosus: a comprehensive review. J. Autoimmun. 64, 125–136 (2015).
Coit, P. et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+T cells from lupus patients. J. Autoimmun. 43, 78–84 (2013).
James, J. A. et al. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthritis Rheum. 44, 1122–1126 (2001).
Barbhaiya, M. et al. Influence of alcohol consumption on the risk of systemic lupus erythematosus among women in the nurses’ health study cohorts. Arthritis Care Res. 69, 384–392 (2017).
Parks, C. G. et al. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: a population-based, case-control study in the southeastern United States. Arthritis Rheum. 46, 1840–1850 (2002).
Gardner, R. M. et al. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: a cross-sectional study. Environ. Res. 110, 345–354 (2010).
Mohan, C., Zhang, T. & Putterman, C. Pathogenic cellular and molecular mediators in lupus nephritis. Nat. Rev. Nephrol. 19, 491–508 (2023).
Lai, C. H. et al. Outcomes of percutaneous coronary intervention in patients with rheumatoid arthritis and systemic lupus erythematosus: an 11-year nationwide cohort study. Ann. Rheum. Dis. 75, 1350–1356 (2016).
Swigris, J. J. et al. Pulmonary and thrombotic manifestations of systemic lupus erythematosus. Chest 133, 271–280 (2008).
Aringer, M. et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1400–1412 (2019).
Lovgren, T. et al. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 50, 1861–1872 (2004).
Morand, E. F. et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-I). Lancet 401, 1001–1010 (2023).
Isenberg, D. et al. Efficacy, safety, and pharmacodynamic effects of the bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 73, 1835–1846 (2021).
Aringer, M. Inflammatory markers in systemic lupus erythematosus. J. Autoimmun. 110, 102374 (2020).
Brito-Zeron, P. et al. Sjogren syndrome. Nat. Rev. Dis. Prim. 2, 16047 (2016).
Mavragani, C. P. & Moutsopoulos, H. M. Sjogren’s syndrome. Annu Rev. Pathol. 9, 273–285 (2014).
Fox, R. I. Sjogren’s syndrome. Lancet 366, 321–331 (2005).
Ramos-Casals, M. et al. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun. Rev. 14, 670–679 (2015).
Maldini, C. et al. Epidemiology of primary Sjogren’s syndrome in a French multiracial/multiethnic area. Arthritis Care Res. 66, 454–463 (2014).
Jhorar, P., Torre, K. & Lu, J. Cutaneous features and diagnosis of primary Sjogren syndrome: an update and review. J. Am. Acad. Dermatol. 79, 736–745 (2018).
Lessard, C. J. et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat. Genet 45, 1284–1292 (2013).
Thorlacius, G. E. et al. Genetic and clinical basis for two distinct subtypes of primary Sjogren’s syndrome. Rheumatology 60, 837–848 (2021).
Carapito, R. et al. A new MHC-linked susceptibility locus for primary Sjogren’s syndrome: MICA. Hum. Mol. Genet 26, 2565–2576 (2017).
Bolstad, A. I. et al. Association between genetic variants in the tumour necrosis factor/lymphotoxin alpha/lymphotoxin beta locus and primary Sjogren’s syndrome in Scandinavian samples. Ann. Rheum. Dis. 71, 981–988 (2012).
Li, Y. et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjogren’s syndrome at 7q11.23. Nat. Genet 45, 1361–1365 (2013).
Qu, S. et al. Common variants near IKZF1 are associated with primary Sjogren’s syndrome in Han Chinese. PLoS ONE 12, e0177320 (2017).
Khatri, B. et al. Genome-wide association study identifies Sjogren’s risk loci with functional implications in immune and glandular cells. Nat. Commun. 13, 4287 (2022).
Thorlacius, G. E., Bjork, A. & Wahren-Herlenius, M. Genetics and epigenetics of primary Sjogren syndrome: implications for future therapies. Nat. Rev. Rheumatol. 19, 288–306 (2023).
Bjork, A., Mofors, J. & Wahren-Herlenius, M. Environmental factors in the pathogenesis of primary Sjogren’s syndrome. J. Intern Med. 287, 475–492 (2020).
Mofors, J. et al. Cigarette smoking patterns preceding primary Sjogren’s syndrome. RMD Open. 6, e001402 (2020).
Olsson, P. et al. Cigarette smoking and the risk of primary Sjogren’s syndrome: a nested case control study. Arthritis Res. Ther. 19, 50 (2017).
Skopouli, F. N., Dafni, U., Ioannidis, J. P. & Moutsopoulos, H. M. Clinical evolution, and morbidity and mortality of primary Sjogren’s syndrome. Semin. Arthritis Rheum. 29, 296–304 (2000).
Mavragani, C. P. & Moutsopoulos, H. M. The geoepidemiology of Sjogren’s syndrome. Autoimmun. Rev. 9, A305–A310 (2010).
Jonsson, R. et al. Autoantibodies present before symptom onset in primary Sjogren syndrome. JAMA 310, 1854–1855 (2013).
Tengner, P. et al. Detection of anti-Ro/SSA and anti-La/SSB autoantibody-producing cells in salivary glands from patients with Sjogren’s syndrome. Arthritis Rheum. 41, 2238–2248 (1998).
Daniels, T. E. et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjogren’s syndrome among 1,726 registry participants. Arthritis Rheum. 63, 2021–2030 (2011).
Mariette, X. et al. A randomized, phase II study of sequential belimumab and rituximab in primary Sjogren’s syndrome. JCI Insight. 7, e163030 (2022).
Shipa, M. et al. Effectiveness of belimumab after rituximab in systemic lupus erythematosus : a randomized controlled trial. Ann. Intern. Med. 174, 1647–1657 (2021).
Gomez-Almaguer, D. et al. Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood 116, 4783–4785 (2010).
Carvello, M. et al. Inotuzumab ozogamicin murine analog-mediated B-cell depletion reduces anti-islet allo- and autoimmune responses. Diabetes 61, 155–165 (2012).
Atisha-Fregoso, Y. et al. Phase II randomized trial of rituximab plus cyclophosphamide followed by belimumab for the treatment of lupus nephritis. Arthritis Rheumatol. 73, 121–131 (2021).
Teng, Y. K. O. et al. Phase III, multicentre, randomised, double-blind, placebo-controlled, 104-week study of subcutaneous belimumab administered in combination with rituximab in adults with systemic lupus erythematosus (SLE): BLISS-BELIEVE study protocol. BMJ Open 9, e025687 (2019).
Aranow, C. et al. Efficacy and Safety of Subcutaneous Belimumab (BEL) and Rituximab (RTX) Sequential Therapy in Patients with Systemic Lupus Erythematosus: The Phase 3, Randomized, Placebo-Controlled BLISS-BELIEVE Study, <https://acrabstracts.org/abstract/efficacy-and-safety-of-subcutaneous-belimumab-bel-and-rituximab-rtx-sequential-therapy-in-patients-with-systemic-lupus-erythematosus-the-phase-3-randomized-placebo-controlled-bliss-believe-stud/> (2021).
Emery, P. et al. Certolizumab pegol in combination with dose-optimised methotrexate in DMARD-naive patients with early, active rheumatoid arthritis with poor prognostic factors: 1-year results from C-EARLY, a randomised, double-blind, placebo-controlled phase III study. Ann. Rheum. Dis. 76, 96–104 (2017).
Rudick, R. A. et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 354, 911–923 (2006).
Goodman, A. D. et al. GLANCE: results of a phase 2, randomized, double-blind, placebo-controlled study. Neurology 72, 806–812 (2009).
Stohl, W. et al. Safety and efficacy of ocrelizumab in combination with methotrexate in MTX-naive subjects with rheumatoid arthritis: the phase III FILM trial. Ann. Rheum. Dis. 71, 1289–1296 (2012).
Jayne, D. et al. Phase II randomised trial of type I interferon inhibitor anifrolumab in patients with active lupus nephritis. Ann. Rheum. Dis. 81, 496–506 (2022).
Furie, R. et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N. Engl. J. Med. 383, 1117–1128 (2020).
Michel, M. et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am. J. Hematol. 92, 23–27 (2017).
Burmester, G. R. et al. Efficacy and safety of ascending methotrexate dose in combination with adalimumab: the randomised CONCERTO trial. Ann. Rheum. Dis. 74, 1037–1044 (2015).
Rovin, B. H. et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 64, 1215–1226 (2012).
Labrijn, A. F., Janmaat, M. L., Reichert, J. M. & Parren, P. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 18, 585–608 (2019).
Fan, G., Wang, Z., Hao, M. & Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 8, 130 (2015).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today 20, 838–847 (2015).
Kontermann, R. E. Dual targeting strategies with bispecific antibodies. MAbs 4, 182–197 (2012).
Klein, C., Brinkmann, U., Reichert, J. M. & Kontermann, R. E. The present and future of bispecific antibodies for cancer therapy. Nat. Rev. Drug Discov. 23, 301–319 (2024).
Peyrin-Biroulet, L., Demarest, S. & Nirula, A. Bispecific antibodies: The next generation of targeted inflammatory bowel disease therapies. Autoimmun. Rev. 18, 123–128 (2019).
Yasunaga, M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin. Cancer Biol. 64, 1–12 (2020).
Yang, B., Zhao, M., Wu, H. & Lu, Q. A comprehensive review of biological agents for lupus: beyond single target. Front. Immunol. 11, 539797 (2020).
Torres, T., Romanelli, M. & Chiricozzi, A. A revolutionary therapeutic approach for psoriasis: bispecific biological agents. Expert Opin. Investig. Drugs 25, 751–754 (2016).
Warren, R. B. et al. Bimekizumab versus adalimumab in plaque psoriasis. N. Engl. J. Med. 385, 130–141 (2021).
Reich, K. et al. Bimekizumab versus secukinumab in plaque psoriasis. N. Engl. J. Med. 385, 142–152 (2021).
Oliver, R. et al. Bimekizumab for the treatment of moderate-to-severe plaque psoriasis: efficacy, safety, pharmacokinetics, pharmacodynamics and transcriptomics from a phase IIa, randomized, double-blind multicentre study. Br. J. Dermatol. 186, 652–663 (2022).
Gordon, K. B. et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet 397, 475–486 (2021).
Glatt, S. et al. Efficacy and safety of bimekizumab in moderate to severe hidradenitis suppurativa: a phase 2, double-blind, placebo-controlled randomized clinical trial. JAMA Dermatol. 157, 1279–1288 (2021).
Glatt, S. et al. Efficacy and safety of bimekizumab as add-on therapy for rheumatoid arthritis in patients with inadequate response to certolizumab pegol: a proof-of-concept study. Ann. Rheum. Dis. 78, 1033–1040 (2019).
van der Heijde, D. et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann. Rheum. Dis. 79, 595–604 (2020).
van der Heijde, D. et al. Efficacy and safety of bimekizumab in axial spondyloarthritis: results of two parallel phase 3 randomised controlled trials. Ann. Rheum. Dis. 82, 515–526 (2023).
Benschop, R. J. et al. Development of tibulizumab, a tetravalent bispecific antibody targeting BAFF and IL-17A for the treatment of autoimmune disease. MAbs 11, 1175–1190 (2019).
Zhang, M. et al. Development of an ICOSL and BAFF bispecific inhibitor AMG 570 for systemic lupus erythematosus treatment. Clin. Exp. Rheumatol. 37, 906–914 (2019).
Abuqayyas, L. et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic properties of Rozibafusp Alfa, a bispecific inhibitor of BAFF and ICOSL: analyses of phase I clinical trials. Clin. Pharm. Ther. 114, 371–380 (2023).
Rossi, E. A., Chang, C. H., Cardillo, T. M. & Goldenberg, D. M. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjug. Chem. 24, 63–71 (2013).
Rossi, E. A., Chang, C. H. & Goldenberg, D. M. Anti-CD22/CD20 bispecific antibody with enhanced trogocytosis for treatment of Lupus. PLoS One 9, e98315 (2014).
Rossi, E. A. et al. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 113, 6161–6171 (2009).
Gupta, P., Goldenberg, D. M., Rossi, E. A. & Chang, C. H. Multiple signaling pathways induced by hexavalent, monospecific, anti-CD20 and hexavalent, bispecific, anti-CD20/CD22 humanized antibodies correlate with enhanced toxicity to B-cell lymphomas and leukemias. Blood 116, 3258–3267 (2010).
Horton, H. M. et al. Antibody-mediated coengagement of FcgammaRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J. Immunol. 186, 4223–4233 (2011).
Szili, D. et al. Suppression of innate and adaptive B cell activation pathways by antibody coengagement of FcgammaRIIb and CD19. MAbs 6, 991–999 (2014).
Chu, S. Y. et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcgamma receptor IIb inhibitory receptor. Arthritis Rheumatol. 66, 1153–1164 (2014).
Lyman, M. et al. A bispecific antibody that targets IL-6 receptor and IL-17A for the potential therapy of patients with autoimmune and inflammatory diseases. J. Biol. Chem. 293, 9326–9334 (2018).
Zheng, S. et al. Characterization of concurrent target suppression by JNJ-61178104, a bispecific antibody against human tumor necrosis factor and interleukin-17A. MAbs 12, 1770018 (2020).
Dhimolea, E. & Reichert, J. M. World bispecific antibody summit, September 27-28, 2011, Boston, MA. MAbs 4, 4–13 (2012).
Akpalu, D. E. et al. Pharmacokinetics, pharmacodynamics, immunogenicity, safety, and tolerability of JNJ-61178104, a novel tumor necrosis factor-alpha and interleukin-17A bispecific antibody, in healthy subjects. J. Clin. Pharm. 59, 968–978 (2019).
Merrill, J. T. et al. Obexelimab in systemic lupus erythematosus with exploration of response based on gene pathway co-expression patterns: a double-blind, randomized, placebo-controlled, phase 2 trial. Arthritis Rheumatol. 75, 2185–2194 (2023).
Perugino, C. A. et al. Evaluation of the safety, efficacy, and mechanism of action of obexelimab for the treatment of patients with IgG4-related disease: an open-label, single-arm, single centre, phase 2 pilot trial. Lancet Rheumatol. 5, e442–e450 (2023).
Allanore, Y. et al. A randomised, double-blind, placebo-controlled, 24-week, phase II, proof-of-concept study of romilkimab (SAR156597) in early diffuse cutaneous systemic sclerosis. Ann. Rheum. Dis. 79, 1600–1607 (2020).
Lacy, S. E. et al. Generation and characterization of ABT-981, a dual variable domain immunoglobulin (DVD-Ig(TM)) molecule that specifically and potently neutralizes both IL-1alpha and IL-1beta. MAbs 7, 605–619 (2015).
Fleischmann, R. M. et al. A Phase II Trial of Lutikizumab, an Anti-Interleukin-1alpha/beta Dual Variable Domain Immunoglobulin, in Knee Osteoarthritis Patients With Synovitis. Arthritis Rheumatol. 71, 1056–1069 (2019).
Kloppenburg, M. et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1alpha and anti-interleukin-1beta dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 78, 413–420 (2019).
Raghu, G. et al. SAR156597 in idiopathic pulmonary fibrosis: a phase 2 placebo-controlled study (DRI11772). Eur Respir J. 52, 1801130 (2018).
Zhao, Q. Bispecific antibodies for autoimmune and inflammatory diseases: clinical progress to date. BioDrugs 34, 111–119 (2020).
Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Bobbin, M. L. & Rossi, J. J. RNA interference (RNAi)-based therapeutics: delivering on the promise? Annu. Rev. Pharm. Toxicol. 56, 103–122 (2016).
Rosa, C., Kuo, Y. W., Wuriyanghan, H. & Falk, B. W. RNA interference mechanisms and applications in plant pathology. Annu Rev. Phytopathol. 56, 581–610 (2018).
Zhu, K. Y. & Palli, S. R. Mechanisms, applications, and challenges of insect RNA interference. Annu Rev. Entomol. 65, 293–311 (2020).
Setten, R. L., Rossi, J. J. & Han, S. P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 18, 421–446 (2019).
Jadhav, V., Vaishnaw, A., Fitzgerald, K. & Maier, M. A. RNA interference in the era of nucleic acid therapeutics. Nat. Biotechnol. 42, 394–405 (2024).
Wei, X. & Wei, Y. Opportunities and challenges in the nanoparticles for nucleic acid therapeutics: the first approval of an RNAi nanoparticle for treatment of a rare disease. Natl Sci. Rev. 6, 1105–1106 (2019).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med 379, 11–21 (2018).
Whitehead, K. A., Langer, R. & Anderson, D. G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009).
Tang, Q. & Khvorova, A. RNAi-based drug design: considerations and future directions. Nat. Rev. Drug Discov. 23, 341–364 (2024).
Bowie, A. G. & Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 (2008).
Gorabi, A. M. et al. Prospects for the potential of RNA interference in the treatment of autoimmune diseases: small interfering RNAs in the spotlight. J. Autoimmun. 114, 102529 (2020).
Feng, N. & Guo, F. Nanoparticle-siRNA: a potential strategy for rheumatoid arthritis therapy? J. Control. Release 325, 380–393 (2020).
Herman, S. et al. Inhibition of inflammation and bone erosion by RNA interference-mediated silencing of heterogeneous nuclear RNP A2/B1 in two experimental models of rheumatoid arthritis. Arthritis Rheumatol. 67, 2536–2546 (2015).
Zhou, H. F. et al. Peptide-siRNA nanocomplexes targeting NF-kappaB subunit p65 suppress nascent experimental arthritis. J. Clin. Investig. 124, 4363–4374 (2014).
Fan, T. et al. siRNA-mediated c-Rel knockdown ameliorates collagen-induced arthritis in mice. Int Immunopharmacol. 56, 9–17 (2018).
Wang, Q. et al. Targeting NF-kB signaling with polymeric hybrid micelles that co-deliver siRNA and dexamethasone for arthritis therapy. Biomaterials 122, 10–22 (2017).
Duan, W. & Li, H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J. Nanobiotechnology. 16, 58 (2018).
Lee, S. J. et al. TNF-alpha gene silencing using polymerized siRNA/thiolated glycol chitosan nanoparticles for rheumatoid arthritis. Mol. Ther. 22, 397–408 (2014).
Jansen, M. A. A. et al. Lipidoid-polymer hybrid nanoparticles loaded with TNF siRNA suppress inflammation after intra-articular administration in a murine experimental arthritis model. Eur. J. Pharm. Biopharm. 142, 38–48 (2019).
Song, J. et al. Efficient and non-toxic biological response carrier delivering TNF-alpha shRNA for gene silencing in a murine model of rheumatoid arthritis. Front. Immunol. 7, 305 (2016).
Aldayel, A. M. et al. Lipid nanoparticles with minimum burst release of TNF-alpha siRNA show strong activity against rheumatoid arthritis unresponsive to methotrexate. J. Control. Release 283, 280–289 (2018).
Shi, Q. et al. In vivo therapeutic efficacy of TNFalpha silencing by folate-PEG-chitosan-DEAE/siRNA nanoparticles in arthritic mice. Int. J. Nanomed. 13, 387–402 (2018).
Kim, M. J. et al. Notch1 targeting siRNA delivery nanoparticles for rheumatoid arthritis therapy. J. Control. Release 216, 140–148 (2015).
Mehta, G., Scheinman, R. I., Holers, V. M. & Banda, N. K. A new approach for the treatment of arthritis in mice with a novel conjugate of an anti-C5aR1 antibody and C5 small interfering RNA. J. Immunol. 194, 5446–5454 (2015).
Yin, N. et al. A novel indomethacin/methotrexate/MMP-9 siRNA in situ hydrogel with dual effects of anti-inflammatory activity and reversal of cartilage disruption for the synergistic treatment of rheumatoid arthritis. Nanoscale 12, 8546–8562 (2020).
Zhao, G. et al. Nanoparticle-delivered siRNA targeting Bruton’s tyrosine kinase for rheumatoid arthritis therapy. Biomater. Sci. 7, 4698–4707 (2019).
Guiteras, J. et al. The gene silencing of IRF5 and BLYSS effectively modulates the outcome of experimental lupus nephritis. Mol. Ther. Nucleic Acids 24, 807–821 (2021).
Alexander, T., Greco, R. & Snowden, J. A. Hematopoietic stem cell transplantation for autoimmune disease. Annu Rev. Med 72, 215–228 (2021).
Zeher, M., Papp, G., Nakken, B. & Szodoray, P. Hematopoietic stem cell transplantation in autoimmune disorders: from immune-regulatory processes to clinical implications. Autoimmun. Rev. 16, 817–825 (2017).
Ruder, J. et al. Dynamics of T cell repertoire renewal following autologous hematopoietic stem cell transplantation in multiple sclerosis. Sci. Transl. Med. 14, eabq1693 (2022).
von Niederhausern, V. et al. B-cell reconstitution after autologous hematopoietic stem cell transplantation in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 9, e200027 (2022).
Salomon, B. L., Sudres, M. & Cohen, J. L. Regulatory T cells in graft-versus-host disease. Springe. Semin. Immunopathol. 28, 25–29 (2006).
Muraro, P. A. et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J. Exp. Med. 201, 805–816 (2005).
Delemarre, E. M. et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood 127, 91–101 (2016).
Li, H. & Shi, B. Tolerogenic dendritic cells and their applications in transplantation. Cell Mol. Immunol. 12, 24–30 (2015).
Carretero-Iglesia, L. et al. Comparative study of the immunoregulatory capacity of in vitro generated tolerogenic dendritic cells, suppressor macrophages, and myeloid-derived suppressor cells. Transplantation 100, 2079–2089 (2016).
Thomson, A. W. & Robbins, P. D. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann. Rheum. Dis. 67(Suppl 3), iii90–iii96 (2008).
Passeri, L., Marta, F., Bassi, V. & Gregori, S. Tolerogenic dendritic cell-based approaches in autoimmunity. Int. J. Mol. Sci. 22, 8415 (2021).
Nam, J. H. et al. Functional ambivalence of dendritic cells: tolerogenicity and immunogenicity. Int. J. Mol. Sci. 22, 4430 (2021).
Keir, M. E., Francisco, L. M. & Sharpe, A. H. PD-1 and its ligands in T-cell immunity. Curr. Opin. Immunol. 19, 309–314 (2007).
Wu, J. & Horuzsko, A. Expression and function of immunoglobulin-like transcripts on tolerogenic dendritic cells. Hum. Immunol. 70, 353–356 (2009).
van Bekkum, D. W., Bohre, E. P., Houben, P. F. & Knaan-Shanzer, S. Regression of adjuvant-induced arthritis in rats following bone marrow transplantation. Proc. Natl Acad. Sci. USA 86, 10090–10094 (1989).
Snowden, J. A. et al. Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv. 1, 2742–2755 (2017).
Tamm, M. et al. Autologous haemopoietic stem cell transplantation in a patient with severe pulmonary hypertension complicating connective tissue disease. Ann. Rheum. Dis. 55, 779–780 (1996).
Fassas, A. et al. Peripheral blood stem cell transplantation in the treatment of progressive multiple sclerosis: first results of a pilot study. Bone Marrow Transpl. 20, 631–638 (1997).
Snowden, J. A. et al. Haematopoietic SCT in severe autoimmune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transpl. 47, 770–790 (2012).
Swart, J. F. et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat. Rev. Rheumatol. 13, 244–256 (2017).
Saccardi, R. et al. Autologous stem cell transplantation for progressive multiple sclerosis: update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult. Scler. 12, 814–823 (2006).
Mancardi, G. L. et al. Autologous haematopoietic stem cell transplantation with an intermediate intensity conditioning regimen in multiple sclerosis: the Italian multi-centre experience. Mult. Scler. 18, 835–842 (2012).
Burt, R. K. et al. Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing-remitting multiple sclerosis. JAMA 313, 275–284 (2015).
Boffa, G. et al. Hematopoietic stem cell transplantation in people with active secondary progressive multiple sclerosis. Neurology 100, e1109–e1122 (2023).
Zhukovsky, C. et al. Autologous haematopoietic stem cell transplantation compared with alemtuzumab for relapsing-remitting multiple sclerosis: an observational study. J. Neurol. Neurosurg. Psychiatry 92, 189–194 (2021).
Kalincik, T. et al. Comparative Effectiveness Of Autologous Hematopoietic Stem Cell Transplant Vs Fingolimod, Natalizumab, And Ocrelizumab In Highly Active Relapsing-remitting Multiple Sclerosis. JAMA Neurol. 80, 702–713 (2023).
Boffa, G. et al. Long-term clinical outcomes of hematopoietic stem cell transplantation in multiple sclerosis. Neurology 96, e1215–e1226 (2021).
Silfverberg, T. et al. Haematopoietic stem cell transplantation for treatment of relapsing-remitting multiple sclerosis in Sweden: an observational cohort study. J. Neurol. Neurosurg. Psychiatry 95, 125–133 (2024).
Kalincik, T. et al. Effectiveness of autologous haematopoietic stem cell transplantation versus natalizumab in progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 95, 775–783 (2024).
J, M. F. S. et al. Allogeneic hematopoietic stem cell transplantation for severe, refractory juvenile idiopathic arthritis. Blood Adv. 2, 777–786 (2018).
Alexander, T. et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood 113, 214–223 (2009).
Ayoglu, B. et al. Characterising the autoantibody repertoire in systemic sclerosis following myeloablative haematopoietic stem cell transplantation. Ann. Rheum. Dis. 82, 670–680 (2023).
Couri, C. E. et al. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 301, 1573–1579 (2009).
D’Addio, F. et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: a multicenter analysis. Diabetes 63, 3041–3046 (2014).
Daikeler, T., Tichelli, A. & Passweg, J. Complications of autologous hematopoietic stem cell transplantation for patients with autoimmune diseases. Pediatr. Res 71, 439–444 (2012).
Saccardi, R. et al. Consensus statement concerning cardiotoxicity occurring during haematopoietic stem cell transplantation in the treatment of autoimmune diseases, with special reference to systemic sclerosis and multiple sclerosis. Bone Marrow Transpl. 34, 877–881 (2004).
Greco, R. et al. Outcome of SARS-CoV2 infection in hematopoietic stem cell transplant recipients for autoimmune diseases. J. Autoimmun. 136, 103024 (2023).
Daikeler, T. et al. Secondary autoimmune diseases occurring after HSCT for an autoimmune disease: a retrospective study of the EBMT Autoimmune Disease Working Party. Blood 118, 1693–1698 (2011).
Pasquini, M. C. et al. Transplantation for autoimmune diseases in north and South America: a report of the Center for International Blood and Marrow Transplant Research. Biol. Blood Marrow Transpl. 18, 1471–1478 (2012).
Daikeler, T. et al. Allogeneic hematopoietic SCT for patients with autoimmune diseases. Bone Marrow Transpl. 44, 27–33 (2009).
Rizzo, J. D. et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation: joint recommendations of the European Group for Blood and Marrow Transplantation, the Center for International Blood and Marrow Transplant Research, and the American Society of Blood and Marrow Transplantation. Biol. Blood Marrow Transpl. 12, 138–151 (2006).
Kenison, J. E., Stevens, N. A. & Quintana, F. J. Therapeutic induction of antigen-specific immune tolerance. Nat. Rev. Immunol. 24, 338–357 (2024).
Shaw, C. M., Fahlberg, W. J., Kies, M. W. & Alvord, E. C. Jr. Suppression of experimental “allergic” encephalomyelitis in guinea pigs by encephalitogenic proteins extracted from homologous brain. J. Exp. Med 111, 171–180 (1960).
Raine, C. S., Traugott, U. & Stone, S. H. Suppression of chronic allergic encephalomyelitis: relevance to multiple sclerosis. Science 201, 445–448 (1978).
Weiner, H. L. et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 259, 1321–1324 (1993).
Hashim, G. A. Failure of myelin basic protein to prevent or suppress experimental allergic encephalomyelitis in guinea pigs. Neurochem Res. 5, 101–113 (1980).
Zhang, L., Nakayama, M. & Eisenbarth, G. S. Insulin as an autoantigen in NOD/human diabetes. Curr. Opin. Immunol. 20, 111–118 (2008).
Skyler, J. S. et al. Efficacy of inhaled human insulin in type 1 diabetes mellitus: a randomised proof-of-concept study. Lancet 357, 331–335 (2001).
Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study, G. et al. Effect of oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA 318, 1891–1902 (2017).
Karges, B. et al. Association of insulin pump therapy vs insulin injection therapy with severe hypoglycemia, ketoacidosis, and glycemic control among children, adolescents, and young adults with type 1 diabetes. JAMA 318, 1358–1366 (2017).
Heinemann, L. et al. Real-time continuous glucose monitoring in adults with type 1 diabetes and impaired hypoglycaemia awareness or severe hypoglycaemia treated with multiple daily insulin injections (HypoDE): a multicentre, randomised controlled trial. Lancet 391, 1367–1377 (2018).
Gaur, A. et al. Amelioration of autoimmune encephalomyelitis by myelin basic protein synthetic peptide-induced anergy. Science 258, 1491–1494 (1992).
Falk, K. et al. Induction and suppression of an autoimmune disease by oligomerized T cell epitopes: enhanced in vivo potency of encephalitogenic peptides. J. Exp. Med. 191, 717–730 (2000).
Critchfield, J. M. et al. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science 263, 1139–1143 (1994).
Stienekemeier, M. et al. Vaccination, prevention, and treatment of experimental autoimmune neuritis (EAN) by an oligomerized T cell epitope. Proc. Natl Acad. Sci. USA 98, 13872–13877 (2001).
Constant, S. L. & Bottomly, K. Induction of Th1 and Th2 CD4+T cell responses: the alternative approaches. Annu. Rev. Immunol. 15, 297–322 (1997).
Swanborg, R. H. Immunologic response to altered encephalitogenic protein in guinea pigs. J. Immunol. 102, 381–388 (1969).
Swanborg, R. H. The effect of selective modification of tryptophan, lysine and arginine residues of basic brain protein on encephalitogenic activity. J. Immunol. 105, 865–871 (1970).
Swanborg, R. H. Antigen-induced inhibition of experimental allergic encephalomyelitis. I. Inhibition in guinea pigs injected with non-encephalitogenic modified myelin basic protein. J. Immunol. 109, 540–546 (1972).
Traugott, U., Stone, S. H. & Raine, C. S. Chronic relapsing experimental autoimmune encephalomyelitis. treatment with combinations of myelin components promotes clinical and structural recovery. J. Neurol. Sci. 56, 65–73 (1982).
Li, J. et al. Mechanisms of antigen-induced reversal of CNS inflammation in experimental demyelinating disease. Sci. Adv. 9, eabo2810 (2023).
Higgins, P. J. & Weiner, H. L. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J. Immunol. 140, 440–445 (1988).
Metzler, B. & Wraith, D. C. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int. Immunol. 5, 1159–1165 (1993).
Duong, V. A., Nguyen, T. T. & Maeng, H. J. Recent advances in intranasal liposomes for drug, gene, and vaccine delivery. Pharmaceutics. 15, 207 (2023).
Meredith, M. E., Salameh, T. S. & Banks, W. A. Intranasal delivery of proteins and peptides in the treatment of neurodegenerative diseases. AAPS J. 17, 780–787 (2015).
Yachi, P. P., Ampudia, J., Zal, T. & Gascoigne, N. R. Altered peptide ligands induce delayed CD8-T cell receptor interaction–a role for CD8 in distinguishing antigen quality. Immunity 25, 203–211 (2006).
Kersh, G. J. & Allen, P. M. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J. Exp. Med 184, 1259–1268 (1996).
Sloan-Lancaster, J. & Allen, P. M. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev. Immunol. 14, 1–27 (1996).
Evavold, B. D., Sloan-Lancaster, J. & Allen, P. M. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol. Today 14, 602–609 (1993).
Kardys, E. & Hashim, G. A. Experimental allergic encephalomyelitis in Lewis rats: immunoregulation of disease by a single amino acid substitution in the disease-inducing determinant. J. Immunol. 127, 862–866 (1981).
Smilek, D. E. et al. A single amino acid change in a myelin basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 88, 9633–9637 (1991).
Karin, N. et al. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon gamma and tumor necrosis factor alpha production. J. Exp. Med. 180, 2227–2237 (1994).
Ruiz, P. J. et al. Microbial epitopes act as altered peptide ligands to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 189, 1275–1284 (1999).
Margot, C. D., Ford, M. L. & Evavold, B. D. Amelioration of established experimental autoimmune encephalomyelitis by an MHC anchor-substituted variant of proteolipid protein 139-151. J. Immunol. 174, 3352–3358 (2005).
Trager, N. N. M. et al. A novel aza-MBP altered peptide ligand for the treatment of experimental autoimmune encephalomyelitis. Mol. Neurobiol. 55, 267–275 (2018).
Deraos, G. et al. Properties of myelin altered peptide ligand cyclo(87-99)(Ala91,Ala96)MBP87-99 render it a promising drug lead for immunotherapy of multiple sclerosis. Eur. J. Med. Chem. 101, 13–23 (2015).
Lourbopoulos, A. et al. Cyclic MOG(35)(-)(55) ameliorates clinical and neuropathological features of experimental autoimmune encephalomyelitis. Bioorg. Med. Chem. 25, 4163–4174 (2017).
Geluk, A., van Meijgaarden, K. E., Roep, B. O. & Ottenhoff, T. H. Altered peptide ligands of islet autoantigen Imogen 38 inhibit antigen specific T cell reactivity in human type-1 diabetes. J. Autoimmun. 11, 353–361 (1998).
Han, B. et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat. Med. 11, 645–652 (2005).
Crowe, P. D., Qin, Y., Conlon, P. J. & Antel, J. P. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann. Neurol. 48, 758–765 (2000).
Kinnunen, T. et al. Potential of an altered peptide ligand of lipocalin allergen Bos d 2 for peptide immunotherapy. J. Allergy Clin. Immunol. 119, 965–972 (2007).
Walter, M. et al. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care 32, 2036–2040 (2009).
Bielekova, B. et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 6, 1167–1175 (2000).
Kappos, L. et al. Induction of a non-encephalitogenic type 2T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat. Med. 6, 1176–1182 (2000).
Zinkernagel, R. M. et al. Restriction specificities, alloreactivity, and allotolerance expressed by T cells from nude mice reconstituted with H-2-compatible or -incompatible thymus grafts. J. Exp. Med. 151, 376–399 (1980).
Zinkernagel, R. M. et al. On the thymus in the differentiation of “H-2 self-recognition” by T cells: evidence for dual recognition? J. Exp. Med. 147, 882–896 (1978).
Zinkernagel, R. M. et al. The lymphoreticular system in triggering virus plus self-specific cytotoxic T cells: evidence for T help. J. Exp. Med. 147, 897–911 (1978).
Gaud, G., Lesourne, R. & Love, P. E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 (2018).
Garcia, K. C., Adams, J. J., Feng, D. & Ely, L. K. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat. Immunol. 10, 143–147 (2009).
Amrani, A. et al. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 406, 739–742 (2000).
Casares, S., Bona, C. A. & Brumeanu, T. D. Modulation of CD4 T cell function by soluble MHC II-peptide chimeras. Int Rev. Immunol. 20, 547–573 (2001).
Sharma, S. D. et al. Antigen-specific therapy of experimental allergic encephalomyelitis by soluble class II major histocompatibility complex-peptide complexes. Proc. Natl Acad. Sci. USA 88, 11465–11469 (1991).
Spack, E. G. et al. Induction of tolerance in experimental autoimmune myasthenia gravis with solubilized MHC class II:acetylcholine receptor peptide complexes. J. Autoimmun. 8, 787–807 (1995).
Nicolle, M. W. et al. Specific tolerance to an acetylcholine receptor epitope induced in vitro in myasthenia gravis CD4+ lymphocytes by soluble major histocompatibility complex class II-peptide complexes. J. Clin. Investig. 93, 1361–1369 (1994).
Vandenbark, A. A. et al. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35-55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J. Immunol. 171, 127–133 (2003).
Burrows, G. G. et al. Two-domain MHC class II molecules form stable complexes with myelin basic protein 69-89 peptide that detect and inhibit rat encephalitogenic T cells and treat experimental autoimmune encephalomyelitis. J. Immunol. 161, 5987–5996 (1998).
Huan, J. et al. Monomeric recombinant TCR ligand reduces relapse rate and severity of experimental autoimmune encephalomyelitis in SJL/J mice through cytokine switch. J. Immunol. 172, 4556–4566 (2004).
Masteller, E. L. et al. Peptide-MHC class II dimers as therapeutics to modulate antigen-specific T cell responses in autoimmune diabetes. J. Immunol. 171, 5587–5595 (2003).
Li, L., Yi, Z., Wang, B. & Tisch, R. Suppression of ongoing T cell-mediated autoimmunity by peptide-MHC class II dimer vaccination. J. Immunol. 183, 4809–4816 (2009).
Urbonaviciute, V. et al. Therapy targeting antigen-specific T cells by a peptide-based tolerizing vaccine against autoimmune arthritis. Proc. Natl Acad. Sci. USA 120, e2218668120 (2023).
Kammona, O. & Kiparissides, C. Recent advances in antigen-specific immunotherapies for the treatment of multiple sclerosis. Brain Sci. 10, 333 (2020).
Gammon, J. M. & Jewell, C. M. Engineering immune tolerance with biomaterials. Adv. Health. Mater. 8, e1801419 (2019).
Rui, Y., Eppler, H. B., Yanes, A. A. & Jewell, C. M. Tissue-targeted drug delivery strategies to promote antigen-specific immune tolerance. Adv. Health. Mater. 12, e2202238 (2023).
Carey, S. T., Bridgeman, C. & Jewell, C. M. Biomaterial strategies for selective immune tolerance: advances and gaps. Adv. Sci. (Weinh.) 10, e2205105 (2023).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
Blau, R., Krivitsky, A., Epshtein, Y. & Satchi-Fainaro, R. Are nanotheranostics and nanodiagnostics-guided drug delivery stepping stones towards precision medicine? Drug Resist Updat 27, 39–58 (2016).
Waheed, S. et al. Engineering nano-drug biointerface to overcome biological barriers toward precision drug delivery. J. Nanobiotechnology. 20, 395 (2022).
Fu, L. et al. ‘Passive’ nanoparticles for organ-selective systemic delivery: design, mechanism and perspective. Chem. Soc. Rev. 52, 7579–7601 (2023).
Kenison, J. E. et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl Acad. Sci. USA 117, 32017–32028 (2020).
Elahi, E. et al. Immune modifying effect of drug free biodegradable nanoparticles on disease course of experimental autoimmune neuritis. Pharmaceutics. 14, 2410 (2022).
Allen, R. P. et al. Latent, immunosuppressive nature of poly(lactic-co-glycolic acid) microparticles. ACS Biomater. Sci. Eng. 4, 900–918 (2018).
Felten, R. et al. Novel therapeutic strategies for autoimmune and inflammatory rheumatic diseases. Drug Discov. Today 28, 103612 (2023).
Scotland, B. L. et al. Cell and biomaterial delivery strategies to induce immune tolerance. Adv. Drug Deliv. Rev. 203, 115141 (2023).
Liu, J., Liu, Z., Pang, Y. & Zhou, H. The interaction between nanoparticles and immune system: application in the treatment of inflammatory diseases. J. Nanobiotechnology 20, 127 (2022).
Wang, H. et al. Dual peptide nanoparticle platform for enhanced antigen-specific immune tolerance for the treatment of experimental autoimmune encephalomyelitis. Biomater. Sci. 10, 3878–3891 (2022).
Getts, D. R. et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 30, 1217–1224 (2012).
Hunter, Z. et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano 8, 2148–2160 (2014).
Kuo, R., Saito, E., Miller, S. D. & Shea, L. D. Peptide-conjugated nanoparticles reduce positive co-stimulatory expression and T cell activity to induce tolerance. Mol. Ther. 25, 1676–1685 (2017).
McCarthy, D. P. et al. An antigen-encapsulating nanoparticle platform for T(H)1/17 immune tolerance therapy. Nanomedicine 13, 191–200 (2017).
Carambia, A. et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J. Hepatol. 62, 1349–1356 (2015).
Pujol-Autonell, I. et al. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS ONE 10, e0127057 (2015).
Wilson, D. S. et al. Synthetically glycosylated antigens induce antigen-specific tolerance and prevent the onset of diabetes. Nat. Biomed. Eng. 3, 817–829 (2019).
Tremain, A. C. et al. Synthetically glycosylated antigens for the antigen-specific suppression of established immune responses. Nat. Biomed. Eng. 7, 1142–1155 (2023).
Thomson, A. W. & Knolle, P. A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 10, 753–766 (2010).
Arvizo, R. R. et al. Intrinsic therapeutic applications of noble metal nanoparticles: past, present and future. Chem. Soc. Rev. 41, 2943–2970 (2012).
Thakor, A. S. et al. Gold nanoparticles: a revival in precious metal administration to patients. Nano Lett. 11, 4029–4036 (2011).
Koushki, K. et al. Gold nanoparticles: multifaceted roles in the management of autoimmune disorders. Biomolecules. 11, 1289 (2021).
de Araujo, R. F. J. et al. Anti-inflammatory, analgesic and anti-tumor properties of gold nanoparticles. Pharm. Rep. 69, 119–129 (2017).
Wegmann, K. W., Wagner, C. R., Whitham, R. H. & Hinrichs, D. J. Synthetic Peptide dendrimers block the development and expression of experimental allergic encephalomyelitis. J. Immunol. 181, 3301–3309 (2008).
Lee, C. C., MacKay, J. A., Frechet, J. M. & Szoka, F. C. Designing dendrimers for biological applications. Nat. Biotechnol. 23, 1517–1526 (2005).
D’Emanuele, A. & Attwood, D. Dendrimer-drug interactions. Adv. Drug Deliv. Rev. 57, 2147–2162 (2005).
Zheng, Y., Li, S., Weng, Z. & Gao, C. Hyperbranched polymers: advances from synthesis to applications. Chem. Soc. Rev. 44, 4091–4130 (2015).
Li, J. et al. Cytomembrane infused polymer accelerating delivery of myelin antigen peptide to treat experimental autoimmune encephalomyelitis. ACS Nano 12, 11579–11590 (2018).
Serra, P. & Santamaria, P. Peptide-MHC-based nanomedicines for the treatment of autoimmunity: engineering, mechanisms, and diseases. Front Immunol. 11, 621774 (2020).
Singha, S. et al. Peptide-MHC-based nanomedicines for autoimmunity function as T-cell receptor microclustering devices. Nat. Nanotechnol. 12, 701–710 (2017).
Clemente-Casares, X. et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530, 434–440 (2016).
Sole, P. et al. A T follicular helper cell origin for T regulatory type 1 cells. Cell Mol. Immunol. 20, 489–511 (2023).
Vacchio, M. S. et al. A Thpok-directed transcriptional circuitry promotes Bcl6 and Maf Expression To Orchestrate T follicular helper differentiation. Immunity 51, 465–478.e466 (2019).
Zhang, B. et al. Proximity-enabled covalent binding of IL-2 to IL-2Ralpha selectively activates regulatory T cells and suppresses autoimmunity. Signal Transduct. Target Ther. 8, 28 (2023).
Pol, J. G. et al. Effects of interleukin-2 in immunostimulation and immunosuppression. J. Exp. Med. 217, e20191247 (2020).
Rhodes, K. R. et al. Bioengineered particles expand myelin-specific regulatory T cells and reverse autoreactivity in a mouse model of multiple sclerosis. Sci. Adv. 9, eadd8693 (2023).
Umeshappa, C. S. et al. Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat. Commun. 10, 2150 (2019).
Samanta, D. et al. Structural and functional characterization of a single-chain peptide-MHC molecule that modulates both naive and activated CD8+T cells. Proc. Natl Acad. Sci. USA 108, 13682–13687 (2011).
Tsai, S. et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 32, 568–580 (2010).
Kabashima, K., Honda, T., Ginhoux, F. & Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 19, 19–30 (2019).
Ho, A. W. & Kupper, T. S. T cells and the skin: from protective immunity to inflammatory skin disorders. Nat. Rev. Immunol. 19, 490–502 (2019).
Kobayashi, T., Naik, S. & Nagao, K. Choreographing immunity in the skin epithelial barrier. Immunity 50, 552–565 (2019).
Zhao, Z., Ukidve, A., Dasgupta, A. & Mitragotri, S. Transdermal immunomodulation: principles, advances and perspectives. Adv. Drug Deliv. Rev. 127, 3–19 (2018).
Dahri, M. et al. Biomaterial-based delivery platforms for transdermal immunotherapy. Biomed. Pharmacother. 165, 115048 (2023).
Amani, H. et al. Microneedles for painless transdermal immunotherapeutic applications. J. Control. Release 330, 185–217 (2021).
Zhao, X. et al. Microneedle delivery of autoantigen for immunotherapy in type 1 diabetes. J. Control. Release 223, 178–187 (2016).
Dul, M. et al. Conjugation of a peptide autoantigen to gold nanoparticles for intradermally administered antigen specific immunotherapy. Int. J. Pharm. 562, 303–312 (2019).
Singh, R. K. et al. Using gold nanoparticles for enhanced intradermal delivery of poorly soluble auto-antigenic peptides. Nanomedicine 32, 102321 (2021).
Tatovic, D. et al. Safety of the use of gold nanoparticles conjugated with proinsulin peptide and administered by hollow microneedles as an immunotherapy in type 1 diabetes. Immunother. Adv. 2, ltac002 (2022).
Arikat, F. et al. Targeting proinsulin to local immune cells using an intradermal microneedle delivery system; a potential antigen-specific immunotherapy for type 1 diabetes. J. Control. Release 322, 593–601 (2020).
Zhao, Y. et al. Transdermal microneedles alleviated rheumatoid arthritis by inducing immune tolerance via skin-resident antigen presenting cells. Small 20, e2307366 (2024).
Babiuk, S. et al. Cutaneous vaccination: the skin as an immunologically active tissue and the challenge of antigen delivery. J. Control. Release 66, 199–214 (2000).
Bal, S. M., Caussin, J., Pavel, S. & Bouwstra, J. A. In vivo assessment of safety of microneedle arrays in human skin. Eur. J. Pharm. Sci. 35, 193–202 (2008).
Gupta, J., Gill, H. S., Andrews, S. N. & Prausnitz, M. R. Kinetics of skin resealing after insertion of microneedles in human subjects. J. Control. Release 154, 148–155 (2011).
Zaric, M. et al. Skin dendritic cell targeting via microneedle arrays laden with antigen-encapsulated poly-D,L-lactide-co-glycolide nanoparticles induces efficient antitumor and antiviral immune responses. ACS Nano 7, 2042–2055 (2013).
Walczak, A. et al. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. 70, 1105–1109 (2013).
Firdessa-Fite, R. et al. Soluble antigen arrays efficiently deliver peptides and arrest spontaneous autoimmune diabetes. Diabetes 70, 1334–1346 (2021).
Leon, M. A. et al. Soluble antigen arrays displaying mimotopes direct the response of diabetogenic T cells. ACS Chem. Biol. 14, 1436–1448 (2019).
Hartwell, B. L. et al. Soluble antigen arrays disarm antigen-specific B cells to promote lasting immune tolerance in experimental autoimmune encephalomyelitis. J. Autoimmun. 93, 76–88 (2018).
Hartwell, B. L., Pickens, C. J., Leon, M. & Berkland, C. Multivalent soluble antigen arrays exhibit high avidity binding and modulation of B cell receptor-mediated signaling to drive efficacy against experimental autoimmune encephalomyelitis. Biomacromolecules 18, 1893–1907 (2017).
Cappellano, G., Comi, C., Chiocchetti, A. & Dianzani, U. Exploiting PLGA-based biocompatible nanoparticles for next-generation tolerogenic vaccines against autoimmune disease. Int. J. Mol. Sci. 20, 204 (2019).
Su, L. F. et al. Antigen exposure shapes the ratio between antigen-specific Tregs and conventional T cells in human peripheral blood. Proc. Natl Acad. Sci. USA 113, E6192–E6198 (2016).
Peres, C. et al. Poly(lactic acid)-based particulate systems are promising tools for immune modulation. Acta Biomater. 48, 41–57 (2017).
Danhier, F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release 161, 505–522 (2012).
Xiao, Q. et al. Biological drug and drug delivery-mediated immunotherapy. Acta Pharm. Sin. B 11, 941–960 (2021).
Elmowafy, E. M., Tiboni, M. & Soliman, M. E. Biocompatibility, biodegradation and biomedical applications of poly(lactic acid)/poly(lactic-co-glycolic acid) micro and nanoparticles. J. Pharm. Investig. 49, 347–380 (2019).
Cappellano, G. et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 32, 5681–5689 (2014).
Brzezicka, K. A. et al. Suppression of autoimmune rheumatoid arthritis with hybrid nanoparticles that induce B and T cell tolerance to self-antigen. ACS Nano 16, 20206–20221 (2022).
Maldonado, R. A. et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl Acad. Sci. USA 112, E156–E165 (2015).
LaMothe, R. A. et al. Tolerogenic nanoparticles induce antigen-specific regulatory T cells and provide therapeutic efficacy and transferrable tolerance against experimental autoimmune encephalomyelitis. Front. Immunol. 9, 281 (2018).
Cho, J. J. et al. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials 143, 79–92 (2017).
Kwiatkowski, A. J. et al. Treatment with an antigen-specific dual microparticle system reverses advanced multiple sclerosis in mice. Proc. Natl Acad. Sci. USA 119, e2205417119 (2022).
Casey, L. M. et al. Conjugation of transforming growth factor beta to antigen-loaded poly(lactide- co-glycolide) nanoparticles enhances efficiency of antigen-specific tolerance. Bioconjug. Chem. 29, 813–823 (2018).
Pei, W. et al. Direct modulation of myelin-autoreactive CD4(+) and CD8(+) T cells in EAE mice by a tolerogenic nanoparticle co-carrying myelin peptide-loaded major histocompatibility complexes, CD47 and multiple regulatory molecules. Int. J. Nanomed. 13, 3731–3750 (2018).
Wan, X. et al. A tolerogenic artificial APC durably ameliorates experimental autoimmune encephalomyelitis by directly and selectively modulating myelin peptide-autoreactive CD4(+) and CD8(+) T Cells. J. Immunol. 201, 1194–1210 (2018).
Verbeke, C. S. et al. Multicomponent injectable hydrogels for antigen-specific tolerogenic immune modulation. Adv. Healthc. Mater. 6, 1600773 (2017).
Lewis, J. S. et al. Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model. ACS Biomater. Sci. Eng. 5, 2631–2646 (2019).
Lewis, J. S. et al. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin. Immunol. 160, 90–102 (2015).
Buyuktimkin, B. et al. Vaccine-like controlled-release delivery of an immunomodulating peptide to treat experimental autoimmune encephalomyelitis. Mol. Pharm. 9, 979–985 (2012).
Park, J. et al. Tolerogenic nanovaccine for prevention and treatment of autoimmune encephalomyelitis. Adv. Mater. 35, e2202670 (2023).
Yeste, A. et al. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 109, 11270–11275 (2012).
Yeste, A. et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci. Signal. 9, ra61 (2016).
Hong, J. et al. Co-delivery of allergen epitope fragments and R848 inhibits food allergy by inducing tolerogenic dendritic cells and regulatory T cells. Int. J. Nanomed. 14, 7053–7064 (2019).
Capini, C. et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J. Immunol. 182, 3556–3565 (2009).
Galea, R. et al. PD-L1- and calcitriol-dependent liposomal antigen-specific regulation of systemic inflammatory autoimmune disease. JCI Insight. 4, e126025 (2019).
Chen, X. et al. Restoring immunological tolerance in established experimental arthritis by combinatorial citrullinated peptides and immunomodulatory signals. Nano Today 41, 101307 (2021).
Li, C. et al. Nanoemulsions target to ectopic lymphoids in inflamed joints to restore immune tolerance in rheumatoid arthritis. Nano Lett. 21, 2551–2561 (2021).
Luo, Y. L. et al. An all-in-one nanomedicine consisting of CRISPR-Cas9 and an autoantigen peptide for restoring specific immune tolerance. ACS Appl. Mater. Interfaces 12, 48259–48271 (2020).
Levit, R. et al. Use of genetically modified lactic acid bacteria and bifidobacteria as live delivery vectors for human and animal health. Gut Microbes 14, 2110821 (2022).
Wells, J. M. & Mercenier, A. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat. Rev. Microbiol. 6, 349–362 (2008).
Song, A. A., In, L. L. A., Lim, S. H. E. & Rahim, R. A. A review on Lactococcus lactis: from food to factory. Micro. Cell Fact. 16, 55 (2017).
Bron, P. A. & Kleerebezem, M. Lactic acid bacteria for delivery of endogenous or engineered therapeutic molecules. Front Microbiol 9, 1821 (2018).
Huibregtse, I. L. et al. Induction of antigen-specific tolerance by oral administration of Lactococcus lactis delivered immunodominant DQ8-restricted gliadin peptide in sensitized nonobese diabetic Abo Dq8 transgenic mice. J. Immunol. 183, 2390–2396 (2009).
Huibregtse, I. L. et al. Induction of ovalbumin-specific tolerance by oral administration of Lactococcus lactis secreting ovalbumin. Gastroenterology 133, 517–528 (2007).
Scott, C. L., Aumeunier, A. M. & Mowat, A. M. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol. 32, 412–419 (2011).
Cook, D. P., Gysemans, C. & Mathieu, C. Lactococcus lactis as a versatile vehicle for tolerogenic immunotherapy. Front Immunol. 8, 1961 (2017).
Cook, D. P. et al. Intestinal delivery of proinsulin and IL-10 via Lactococcus lactis combined with low-dose anti-CD3 restores tolerance outside the window of acute type 1 diabetes diagnosis. Front. Immunol. 11, 1103 (2020).
Simon, L., Lapinte, V. & Morille, M. Exploring the role of polymers to overcome ongoing challenges in the field of extracellular vesicles. J. Extracell. Vesicles 12, e12386 (2023).
Herrmann, I. K., Wood, M. J. A. & Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 16, 748–759 (2021).
Elsharkasy, O. M. et al. Extracellular vesicles as drug delivery systems: why and how? Adv. Drug Deliv. Rev. 159, 332–343 (2020).
Robbins, P. D. & Morelli, A. E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208 (2014).
Cocozza, F. et al. SnapShot: extracellular vesicles. Cell 182, 262–262.e261 (2020).
Casella, G. et al. Oligodendrocyte-derived extracellular vesicles as antigen-specific therapy for autoimmune neuroinflammation in mice. Sci. Transl. Med. 12, eaba0599 (2020).
Becker, M. W. et al. Immune engineered extracellular vesicles to modulate T cell activation in the context of type 1 diabetes. Sci. Adv. 9, eadg1082 (2023).
Hong, M., Clubb, J. D. & Chen, Y. Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 38, 473–488 (2020).
Lim, W. A. & June, C. H. The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017).
June, C. H. et al. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Bao, L. et al. Engineered T cells and their therapeutic applications in autoimmune diseases. Zool. Res. 43, 150–165 (2022).
Riet, T. & Chmielewski, M. Regulatory CAR-T cells in autoimmune diseases: progress and current challenges. Front. Immunol. 13, 934343 (2022).
Baker, D. J. & June, C. H. CAR T therapy extends its reach to autoimmune diseases. Cell 185, 4471–4473 (2022).
Orvain, C. et al. Is there a place for chimeric antigen receptor-T cells in the treatment of chronic autoimmune rheumatic diseases? Arthritis Rheumatol. 73, 1954–1965 (2021).
Lamers, C. H. et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117, 72–82 (2011).
Zhang, H., Zhao, P. & Huang, H. Engineering better chimeric antigen receptor T cells. Exp. Hematol. Oncol. 9, 34 (2020).
Zhang, B. et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann. Rheum. Dis. 80, 176–184 (2021).
Mougiakakos, D. et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 385, 567–569 (2021).
Zhang, L. et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 96, 50–58 (2019).
Whittington, K. B. et al. CD8(+) T cells expressing an HLA-DR1 chimeric antigen receptor target autoimmune CD4(+) T cells in an antigen-specific manner and inhibit the development of autoimmune arthritis. J. Immunol. 208, 16–26 (2022).
Fishman, S. et al. Adoptive transfer of mRNA-transfected T cells redirected against diabetogenic CD8 T cells can prevent diabetes. Mol. Ther. 25, 456–464 (2017).
Ellebrecht, C. T. et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016).
Reincke, S. M. et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell 186, 5084–5097.e5018 (2023).
Raffin, C., Vo, L. T. & Bluestone, J. A. T(reg) cell-based therapies: challenges and perspectives. Nat. Rev. Immunol. 20, 158–172 (2020).
Tang, Q. et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 199, 1455–1465 (2004).
Tarbell, K. V. et al. CD25+CD4+T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199, 1467–1477 (2004).
Veerapathran, A. et al. Ex vivo expansion of human Tregs specific for alloantigens presented directly or indirectly. Blood 118, 5671–5680 (2011).
Tuomela, K., Salim, K. & Levings, M. K. Eras of designer Tregs: harnessing synthetic biology for immune suppression. Immunol. Rev. 320, 250–267 (2023).
Elinav, E., Adam, N., Waks, T. & Eshhar, Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 136, 1721–1731 (2009).
Mekala, D. J. & Geiger, T. L. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+T lymphocytes. Blood 105, 2090–2092 (2005).
Moisini, I., Nguyen, P., Fugger, L. & Geiger, T. L. Redirecting therapeutic T cells against myelin-specific T lymphocytes using a humanized myelin basic protein-HLA-DR2-zeta chimeric receptor. J. Immunol. 180, 3601–3611 (2008).
Fransson, M. et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 9, 112 (2012).
Tenspolde, M. et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 103, 102289 (2019).
Spanier, J. A. et al. Tregs with an MHC class II peptide-specific chimeric antigen receptor prevent autoimmune diabetes in mice. J. Clin. Investig. 133, e168601 (2023).
Lee, D. W. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra225 (2014).
Bonifant, C. L., Jackson, H. J., Brentjens, R. J. & Curran, K. J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 3, 16011 (2016).
Passeri, L. et al. Tolerogenic IL-10-engineered dendritic cell-based therapy to restore antigen-specific tolerance in T cell mediated diseases. J. Autoimmun. 138, 103051 (2023).
Gudi, R. R. et al. Engineered dendritic cell-directed concurrent activation of multiple T cell inhibitory pathways induces robust immune tolerance. Sci. Rep. 9, 12065 (2019).
Zubizarreta, I. et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc. Natl Acad. Sci. USA 116, 8463–8470 (2019).
Mansilla, M. J. et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J. Neuroinflammation 13, 113 (2016).
Mansilla, M. J. et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci. Ther. 21, 222–230 (2015).
min, Z. et al. Lipopolysaccharide-activated bone marrow-derived dendritic cells suppress allergic airway inflammation by ameliorating the immune microenvironment. Front. Immunol. 12, 595369 (2021).
Derdelinckx, J. et al. Clinical and immunological control of experimental autoimmune encephalomyelitis by tolerogenic dendritic cells loaded with MOG-encoding mRNA. J. Neuroinflammation 16, 167 (2019).
Malviya, M. et al. Treatment of experimental autoimmune encephalomyelitis with engineered bi-specific Foxp3+ regulatory CD4+T cells. J. Autoimmun. 108, 102401 (2020).
Kim, Y. C. et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J. Autoimmun. 92, 77–86 (2018).
Qian, Z. et al. Engineered regulatory T cells coexpressing MHC class II:peptide complexes are efficient inhibitors of autoimmune T cell function and prevent the development of autoimmune arthritis. J. Immunol. 190, 5382–5391 (2013).
Braley-Mullen, H., Tompson, J. G., Sharp, G. C. & Kyriakos, M. Suppression of experimental autoimmune thyroiditis in guinea pigs by pretreatment with thyroglobulin-coupled spleen cells. Cell Immunol. 51, 408–413 (1980).
Kontos, S., Kourtis, I. C., Dane, K. Y. & Hubbell, J. A. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc. Natl Acad. Sci. USA 110, E60–E68 (2013).
Wang, F., Zong, R. & Chen, G. Erythrocyte-enabled immunomodulation for vaccine delivery. J. Control. Release 341, 314–328 (2022).
Raposo, C. J. et al. Engineered RBCs encapsulating antigen induce multi-modal antigen-specific tolerance and protect against type 1 diabetes. Front Immunol. 13, 869669 (2022).
Watkins, E. A. et al. Persistent antigen exposure via the eryptotic pathway drives terminal T cell dysfunction. Sci. Immunol. 6, eabe1801 (2021).
Pishesha, N. et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc. Natl Acad. Sci. USA 114, 3157–3162 (2017).
Lutterotti, A. et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci. Transl. Med. 5, 188ra175 (2013).
Au, K. M., Tisch, R. & Wang, A. Z. Immune checkpoint ligand bioengineered schwann cells as antigen-specific therapy for experimental autoimmune encephalomyelitis. Adv. Mater. 34, e2107392 (2022).
Harry, R. A., Anderson, A. E., Isaacs, J. D. & Hilkens, C. M. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann. Rheum. Dis. 69, 2042–2050 (2010).
Raiotach-Regue, D. et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 42, 771–782 (2012).
Giannoukakis, N. et al. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 34, 2026–2032 (2011).
Quirant-Sanchez, B. et al. Combined therapy of vitamin D3-tolerogenic dendritic cells and interferon-beta in a preclinical model of multiple sclerosis. Biomedicines. 9, 1758 (2021).
Zhou, X. et al. Adoptive transfer of GRP78-treated dendritic cells alleviates insulitis in NOD mice. J. Leukoc. Biol. 110, 1023–1031 (2021).
Christofi, M. et al. Low-dose 2-deoxy glucose stabilises tolerogenic dendritic cells and generates potent in vivo immunosuppressive effects. Cell Mol. Life Sci. 78, 2857–2876 (2021).
Jansen, M. A. A. et al. Matured tolerogenic dendritic cells effectively inhibit autoantigen specific CD4(+) T cells in a murine arthritis model. Front. Immunol. 10, 2068 (2019).
Mansilla, M. J. et al. Paving the way towards an effective treatment for multiple sclerosis: advances in cell therapy. Cell Mol. Immunol. 18, 1353–1374 (2021).
Boks, M. A. et al. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction–a comparative study of human clinical-applicable DC. Clin. Immunol. 142, 332–342 (2012).
Bluestone, J. A. et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 7, 315ra189 (2015).
Dong, S. et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight. 6, e147474 (2021).
Dall’Era, M. et al. Adoptive Treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol. 71, 431–440 (2019).
Stephens, L. A., Malpass, K. H. & Anderton, S. M. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur. J. Immunol. 39, 1108–1117 (2009).
Chernajovsky, Y., Gould, D. J. & Podhajcer, O. L. Gene therapy for autoimmune diseases: quo vadis? Nat. Rev. Immunol. 4, 800–811 (2004).
Shu, S. A., Wang, J., Tao, M. H. & Leung, P. S. Gene therapy for autoimmune disease. Clin. Rev. Allergy Immunol. 49, 163–176 (2015).
Chellappan, D. K. et al. Gene therapy and type 1 diabetes mellitus. Biomed. Pharmacother. 108, 1188–1200 (2018).
Gary, E. N. & Weiner, D. B. DNA vaccines: prime time is now. Curr. Opin. Immunol. 65, 21–27 (2020).
Pagliari, S. et al. DNA vaccines: history, molecular mechanisms and future perspectives. J. Mol. Biol. 435, 168297 (2023).
Coon, B., An, L. L., Whitton, J. L. & von Herrath, M. G. DNA immunization to prevent autoimmune diabetes. J. Clin. Investig. 104, 189–194 (1999).
Lobell, A. et al. Vaccination with DNA encoding an immunodominant myelin basic protein peptide targeted to Fc of immunoglobulin G suppresses experimental autoimmune encephalomyelitis. J. Exp. Med. 187, 1543–1548 (1998).
Garren, H. et al. Combination of gene delivery and DNA vaccination to protect from and reverse Th1 autoimmune disease via deviation to the Th2 pathway. Immunity 15, 15–22 (2001).
Akbarpour, M. et al. Insulin B chain 9-23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag-specific FoxP3+ Tregs. Sci. Transl. Med. 7, 289ra281 (2015).
Keeler, G. D. et al. Induction of antigen-specific tolerance by hepatic AAV immunotherapy regardless of T cell epitope usage or mouse strain background. Mol. Ther. Methods Clin. Dev. 28, 177–189 (2023).
Zampieri, R. et al. Prevention and treatment of autoimmune diseases with plant virus nanoparticles. Sci. Adv. 6, eaaz0295 (2020).
Postigo-Fernandez, J. & Creusot, R. J. A multi-epitope DNA vaccine enables a broad engagement of diabetogenic T cells for tolerance in type 1 diabetes. J. Autoimmun. 98, 13–23 (2019).
Postigo-Fernandez, J., Firdessa-Fite, R. & Creusot, R. J. Preclinical evaluation of a precision medicine approach to DNA vaccination in type 1 diabetes. Proc. Natl Acad. Sci. USA 119, e2110987119 (2022).
Roep, B. O. et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci. Transl. Med. 5, 191ra182 (2013).
Tsunoda, I. et al. Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J. Neuropathol. Exp. Neurol. 57, 758–767 (1998).
Filippova, M., Liu, J. & Escher, A. Effects of plasmid DNA injection on cyclophosphamide-accelerated diabetes in NOD mice. DNA Cell Biol. 20, 175–181 (2001).
Garren, H. et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann. Neurol. 63, 611–620 (2008).
Bar-Or, A. et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch. Neurol. 64, 1407–1415 (2007).
Mor, G. et al. Do DNA vaccines induce autoimmune disease? Hum. Gene Ther. 8, 293–300 (1997).
Brenner, S., Jacob, F. & Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 190, 576–581 (1961).
Smull, C. E., Mallette, M. F. & Ludwig, E. H. The use of basic proteins to increase the infectivity of enterovirus ribonucleic acid. Biochem Biophys. Res. Commun. 5, 247–249 (1961).
Pardi, N., Hogan, M. J. & Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 65, 14–20 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Wang, Y. S. et al. mRNA-based vaccines and therapeutics: an in-depth survey of current and upcoming clinical applications. J. Biomed. Sci. 30, 84 (2023).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Kariko, K. & Weissman, D. Naturally occurring nucleoside modifications suppress the immunostimulatory activity of RNA: implication for therapeutic RNA development. Curr. Opin. Drug Discov. Dev. 10, 523–532 (2007).
Kariko, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Kavanagh, D. G. et al. Expansion of HIV-specific CD4+ and CD8+T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood 107, 1963–1969 (2006).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Dastagir, S. R. et al. Efficient presentation of multiple endogenous epitopes to both CD4(+) and CD8(+) diabetogenic T cells for tolerance. Mol. Ther. Methods Clin. Dev. 4, 27–38 (2017).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Thomas, S. J. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine through 6 months. N. Engl. J. Med. 385, 1761–1773 (2021).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J. Pharm. 601, 120586 (2021).
Cheng, X. & Lee, R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 99, 129–137 (2016).
Eygeris, Y., Gupta, M., Kim, J. & Sahay, G. Chemistry of lipid nanoparticles for RNA delivery. Acc. Chem. Res 55, 2–12 (2022).
Sabnis, S. et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in Non-human Primates. Mol. Ther. 26, 1509–1519 (2018).
Zhang, Y. et al. Lipids and lipid derivatives for RNA delivery. Chem. Rev. 121, 12181–12277 (2021).
Paramasivam, P. et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 221, e202110137 (2022).
Herrera, M. et al. Illuminating endosomal escape of polymorphic lipid nanoparticles that boost mRNA delivery. Biomater. Sci. 9, 4289–4300 (2021).
Metkar, M., Pepin, C. S. & Moore, M. J. Tailor made: the art of therapeutic mRNA design. Nat. Rev. Drug Discov. 23, 67–83 (2024).
Ye, Z. et al. The mRNA vaccine revolution: COVID-19 has launched the future of vaccinology. ACS Nano 17, 15231–15253 (2023).
Krienke, C. et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 371, 145–153 (2021).
Xu, X. et al. Use of a liver-targeting immune-tolerogenic mRNA lipid nanoparticle platform to treat peanut-induced anaphylaxis by single- and multiple-epitope nucleotide sequence delivery. ACS Nano 17, 4942–4957 (2023).
Firdessa-Fite, R. & Creusot, R. J. Nanoparticles versus dendritic cells as vehicles to deliver mRNA Encoding Multiple Epitopes For Immunotherapy. Mol. Ther. Methods Clin. Dev. 16, 50–62 (2020).
Gomi, M. et al. Tolerogenic Lipid Nanoparticles For Delivering Self-antigen mRNA for the treatment of experimental autoimmune encephalomyelitis. Pharmaceuticals 16, 1270 (2023).
Ogawa, K. et al. Focused ultrasound/microbubbles-assisted BBB opening enhances LNP-mediated mRNA delivery to brain. J. Control Release 348, 34–41 (2022).
Nawaz, M. et al. Lipid nanoparticles deliver the therapeutic VEGFA mRNA in vitro and in vivo and transform extracellular vesicles for their functional extensions. Adv. Sci. 10, e2206187 (2023).
Maugeri, M. et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 10, 4333 (2019).
Qin, S. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target Ther. 7, 166 (2022).
Ghilardi, N., Pappu, R., Arron, J. R. & Chan, A. C. 30 years of biotherapeutics development-what have we learned? Annu. Rev. Immunol. 38, 249–287 (2020).
Hansel, T. T. et al. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 9, 325–338 (2010).
Johnson, D. E. Biotherapeutics: challenges and opportunities for predictive toxicology of monoclonal antibodies. Int. J. Mol. Sci. 19, 3685 (2018).
Dhib-Jalbut, S. Glatiramer acetate (Copaxone) therapy for multiple sclerosis. Pharm. Ther. 98, 245–255 (2003).
Schrempf, W. & Ziemssen, T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun. Rev. 6, 469–475 (2007).
Wolinsky, J. S. et al. Copaxone’s effect on MRI-monitored disease in relapsing MS is reproducible and sustained. Neurology 59, 1284–1286 (2002).
Comi, G., Filippi, M. & Wolinsky, J. S. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging–measured disease activity and burden in patients with relapsing multiple sclerosis. European/Canadian Glatiramer Acetate Study Group. Ann. Neurol. 49, 290–297 (2001).
Comi, G. et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet 374, 1503–1511 (2009).
Breedveld, F. C. Therapeutic monoclonal antibodies. Lancet 355, 735–740 (2000).
Wang, X. et al. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 9, 74–85 (2018).
Waldmann, H. & Cobbold, S. The use of monoclonal antibodies to achieve immunological tolerance. Trends Pharm. Sci. 14, 143–148 (1993).
Chataway, J. et al. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology 90, e955–e962 (2018).
Freedman, M. S. et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology 77, 1551–1560 (2011).
Warren, K. G., Catz, I., Ferenczi, L. Z. & Krantz, M. J. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur. J. Neurol. 13, 887–895 (2006).
Goodkin, D. E. et al. A phase I trial of solubilized DR2:MBP84-102 (AG284) in multiple sclerosis. Neurology 54, 1414–1420 (2000).
Nanto-Salonen, K. et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet 372, 1746–1755 (2008).
Skyler, J. S. et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial–type 1. Diabetes Care 28, 1068–1076 (2005).
Diabetes Prevention Trial–Type 1 Diabetes Study, G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 346, 1685–1691 (2002).
Feng, X. et al. Immunomodulatory nanosystems. Adv. Sci. 6, 1900101 (2019).
Mitarotonda, R. et al. Immunotherapeutic nanoparticles: from autoimmune disease control to the development of vaccines. Biomater. Adv. 135, 212726 (2022).
Serra, P. & Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 37, 238–251 (2019).
Yang, Y. & Santamaria, P. Antigen-specific nanomedicines for the treatment of autoimmune disease: target cell types, mechanisms and outcomes. Curr. Opin. Biotechnol. 74, 285–292 (2022).
Benne, N., Ter Braake, D., Stoppelenburg, A. J. & Broere, F. Nanoparticles for inducing antigen-specific T cell tolerance in autoimmune diseases. Front. Immunol. 13, 864403 (2022).
Willyard, C. Can autoimmune diseases be cured? Scientists see hope at last. Nature 625, 646–648 (2024).
Cifuentes-Rius, A. et al. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat. Nanotechnol. 16, 37–46 (2021).
Haddadi, A. et al. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J. Biomed. Mater. Res A 84, 885–898 (2008).
Das, S., Haddadi, A., Veniamin, S. & Samuel, J. Delivery of rapamycin-loaded nanoparticle down regulates ICAM-1 expression and maintains an immunosuppressive profile in human CD34+ progenitor-derived dendritic cells. J. Biomed. Mater. Res. A 85, 983–992 (2008).
Kim, S. H. et al. Induction of antigen-specific immune tolerance using biodegradable nanoparticles containing antigen and dexamethasone. Int. J. Nanomed. 14, 5229–5242 (2019).
Peine, K. J. et al. Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles. Mol. Pharm. 11, 828–835 (2014).
Wang, Y. et al. mRNA vaccine: a potential therapeutic strategy. Mol. Cancer 20, 33 (2021).
Chen, K. et al. mRNA vaccines against SARS-CoV-2 variants delivered by lipid nanoparticles based on novel ionizable lipids. Adv. Funct. Mater. 32, 2204692 (2022).
Kubara, K. et al. Lymph node macrophages drive innate immune responses to enhance the anti-tumor efficacy of mRNA vaccines. Mol. Ther. 32, 704–721 (2024).
Sharifian, A., Varshosaz, J., Aliomrani, M. & Kazemi, M. Nose to brain delivery of ibudilast micelles for treatment of multiple sclerosis in an experimental autoimmune encephalomyelitis animal model. Int J. Pharm. 638, 122936 (2023).
Dargahi, N. et al. Multiple sclerosis: immunopathology and treatment update. Brain Sci. 7, 78 (2017).
Alhadj Ali, M. et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 9, eaaf7779 (2017).
Fukaura, H. et al. Induction of circulating myelin basic protein and proteolipid protein-specific transforming growth factor-beta1-secreting Th3 T cells by oral administration of myelin in multiple sclerosis patients. J. Clin. Investig. 98, 70–77 (1996).
Streeter, H. B. et al. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol. Neuroimmunol. Neuroinflamm. 2, e93 (2015).
Van Rampelbergh, J. et al. First-in-human, double-blind, randomized phase 1b study of peptide immunotherapy IMCY-0098 in new-onset type 1 diabetes. BMC Med. 21, 190 (2023).
Norman, J. J. et al. Faster pharmacokinetics and increased patient acceptance of intradermal insulin delivery using a single hollow microneedle in children and adolescents with type 1 diabetes. Pediatr. Diabetes 14, 459–465 (2013).
Jurynczyk, M. et al. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann. Neurol. 68, 593–601 (2010).
Murray, J. A. et al. Safety and tolerability of KAN-101, a liver-targeted immune tolerance therapy, in patients with coeliac disease (ACeD): a phase 1 trial. Lancet Gastroenterol. Hepatol. 8, 735–747 (2023).
Belogurov, A. Jr. et al. CD206-targeted liposomal myelin basic protein peptides in patients with multiple sclerosis resistant to first-line disease-modifying therapies: a first-in-human, proof-of-concept dose-escalation study. Neurotherapeutics 13, 895–904 (2016).
Lomakin, Y. et al. Administration of Myelin Basic Protein Peptides Encapsulated in Mannosylated Liposomes Normalizes Level of Serum TNF-alpha and IL-2 and Chemoattractants CCL2 and CCL4 in Multiple Sclerosis Patients. Mediators Inflamm. 2016, 2847232 (2016).
Kazda, C. M. et al. Novel once-weekly basal insulin fc achieved similar glycemic control with a safety profile comparable to insulin degludec in patients with type 1 diabetes. Diabetes Care 46, 1052–1059 (2023).
Bue-Valleskey, J. M. et al. Once-weekly basal insulin Fc demonstrated similar glycemic control to once-daily insulin degludec in insulin-naive patients with type 2 diabetes: a phase 2 randomized control trial. Diabetes Care 46, 1060–1067 (2023).
Yadav, V. et al. Recombinant T-cell receptor ligand (RTL) for treatment of multiple sclerosis: a double-blind, placebo-controlled, phase 1, dose-escalation study. Autoimmune Dis. 2012, 954739 (2012).
Bell, G. M. et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 76, 227–234 (2017).
Marek-Trzonkowska, N. et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J. Transl. Med. 14, 332 (2016).
Marek-Trzonkowska, N. et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care 35, 1817–1820 (2012).
Nikolic, T. et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol. 8, 470–472 (2020).
ECTRIMS 2019. Late breaking news abstracts. Mult. Scler. J. 25, 890–938 (2019).
Acknowledgements
This work is supported by grant from Chongqing International Institute for Immunology (2022YZH02), the National Natural Science Foundation of China (32070912), Science and Technology Innovation Key R&D Program of Chongqing (CSTB2024TIAD-STX0001).
Author information
Authors and Affiliations
Contributions
Y.W. and J.L. conceived, supervised, and revised the manuscript. Y.S. organized figures and formatted the manuscript. All authors contributed to the writing and editing of the manuscript. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, Y., Li, J. & Wu, Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Sig Transduct Target Ther 9, 263 (2024). https://doi.org/10.1038/s41392-024-01952-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-024-01952-8
This article is cited by
-
Reassessing the role of watchful waiting strategy for IgG4-related disease: a real-world study
Arthritis Research & Therapy (2026)
-
Monoterpene indole alkaloids from the aerial parts of Ophiorrhiza brevidentata and their immunological activities
Natural Products and Bioprospecting (2026)
-
miRNA let-7f-5p-encapsulated labial gland MSC-derived EVs ameliorate experimental Sjögren’s syndrome by suppressing Th17 cells via targeting RORC/IL-17A signaling axis
Journal of Nanobiotechnology (2025)
-
Molecular mechanisms underlying the co-pathogenesis of abdominal aortic aneurysm and rheumatoid arthritis: evidence based on bioinformatics analysis and clinical data
BMC Medical Genomics (2025)
-
Challenges and strategies in clinical applications of CAR-T therapy for autoimmune diseases
Journal of Hematology & Oncology (2025)
