
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder characterized by early molecular events that influence disease progression. Still, the molecular mechanisms caused by different mutations of AD are not understood. We have performed a multidisciplinary study to investigate and compare the early stages of the pathology in two transgenic AD mouse models: P301S and 5xFAD. Using SNOTRAP-based mass spectrometry, we assessed changes in S-nitrosylation, a nitric oxide-mediated post-translational modification, of proteins in both models during their juvenile age. The increased levels of 3-nitrotyrosine confirmed nitrosative stress in the mutant mice. Systems biology analysis revealed shared processes between the models, particularly in the γ-aminobutyric acid (GABA)ergic and glutamatergic neurotransmission processes. In the P301S model, we identified 273 S-nitrosylated (SNOed) proteins in the cortex, with 244 proteins uniquely SNOed in the diseased mice. In the 5xFAD model, 309 SNOed proteins were identified. We have found altered proteins expression of different glutamate/GABA-related markers in the cortex and hippocampus of both AD mouse models. Additionally, the phosphorylation levels of the mTOR signaling components revealed hyperactivation of this pathway in P301S mice. Conversely, 5xFAD mice showed no significant changes in mTOR signaling except for elevated phosphorylation of the ribosomal protein S6 in the cortex. Our findings revealed key molecular mechanisms in the two AD mouse models during their early stages. These mechanisms could serve as potential biomarkers and therapeutic targets for early-stage AD.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder characterized by the gradual decline of cognitive functions, including memory loss [1,2,3]. It is the most common form of Dementia and the 7th leading cause of death worldwide. At least 55 million people live with AD or other types of Dementia [3,4,5]. AD is a progressive disease characterized by memory loss due to synaptic dysfunction and disrupted communication of neural circuits [6, 7]. Gradually, symptoms become more severe, causing cognitive decline, including difficulties in performing normal daily activities due to cortical neuron loss [8]. The underlying pathophysiology of this disease is still elusive. No cure or early diagnostic tools for the risks of developing this disease have been found. Clinical studies have revealed the accumulation of extracellular misfolded amyloid-beta peptides as the first specific hallmark of AD, followed by the accumulation of intracellular hyper-phosphorylated tau protein starting from the hippocampus and cortex regions as another neuropathological hallmark of AD brains [9,10,11,12].
Studies have shown several mutations in the deterministic and risk genes that contribute to AD and other neurodegenerative diseases [13]. Animal models that express human-based mutations are utilized to study the development and progression of diseases [14]. In the current study, we used two well-established and widely used mouse models of AD: 5xFAD and P301S. The 5xFAD model expresses three human-based mutations in the APP gene: the Swedish (K670N/M671L), Florida (I716V), and London (V717I) mutations, and two mutations in the PSEN1 gene: M146L and L286V mutations, leading to altered production of Aβ [15]. 5xFAD mice exhibit severe amyloid pathology and show neuropathology consistent with AD. The extracellular amyloid deposition appears around two months, and cognitive impairment and synaptic loss occur by four months of [16]. Recent studies revealed that in 5xFAD mice, tau levels and phosphorylation rates significantly increase [17,18,19,20]. Furthermore, Aβ is believed to induce the hyper-phosphorylation of tau protein - the later toxic event contributing to the pathology - through activating several neuronal signaling responses [21].
The P301S is the other model used in our study. It is a tauopathy transgenic mouse model that overexpresses a coding missense human-based mutation in the microtubule-associated protein tau (MAPT) gene that changes the CCG codon to TCG and results in impaired ability to promote microtubule assembly. P301S mice exhibit intraneuronal neurofibrillary tangles (NFTs) in the cortex, hippocampus, and amygdala. Synaptic loss and microgliosis are detected at three months, and neuronal loss and brain atrophy occur later, at eight months of age [22, 23].
Nitric oxide (NO•) is a crucial signaling molecule in the central nervous system that is highly implicated in neurological disorders, including AD [24]. NO• is a free radical gas molecule that targets proteins and affects their function. It is produced by the three isoforms of NO• synthases: neuronal (nNOS), endothelial (eNOS), and inducible nitric oxide synthases (iNOS) [25]. NO• can exert its effect through different pathways, including the posttranslational modification S-nitrosylation (SNO) and tyrosine nitration [26]. We have investigated the role of NO and SNO in different neurological disorders [2, 27,28,29,30,31,32,33,34,35,36]. In the brain, NO• is a multifunctional neurotransmitter participating in synaptic activity and plasticity regulation. Abnormal NO• activity affects learning processes and memory functions [26]. Previous work by Amal et al. [22] found significant changes in the S-nitrosoproteome of the P301S and wild-type (WT) mice. Recently, we have provided a link between NO• and the mechanistic target of the rapamycin (mTOR) signaling pathway in the P301S mouse model of AD [1].
S-nitrosylation is a reversible modification where NO• reacts with cysteine thiol, altering protein activity, localization, or interactions with other proteins. Aberrant SNO is implicated in neurodegenerative diseases [22, 37, 38]. A previous study demonstrated that pathways mediating a series of aberrant SNO reactions might play a crucial role in the etiology of neurodegenerative disorders [38]. Another study by Lipton et al. demonstrated a direct link between S-nitrosylation and Alzheimer’s disease (AD) pathology in human AD brains, tested in both male and female subjects [33].
We have also investigated the link between NO• and mTOR signaling, which is a kinase involved in the regulation of protein homeostasis within the cells [39]. Several studies have provided evidence of the mTOR implication in AD [40,41,42,43]. Studies have shown that the levels of phospho(p)-mTOR and its downstream targets are increased in the postmortem human AD brains compared to control, indicating overactivation of this pathway [44, 45].
In the current study, we aimed to investigate the role of NO• and S-nitrosylation in two AD models at the early stages of the disease progression using SNOTRAP-based mass spectrometry (MS) technology [46]. To look at the shared processes enriched in both models, we used systems biology analysis followed by biochemical validation. We focused on the γ-aminobutyric acid (GABA)ergic and glutamatergic systems and the mTOR pathway at 2 months of age [1]. The findings of this work contribute to our understanding of the SNO’s impact on AD pathology and may provide mechanistic insight into the altered pathways at the early stages of AD progression.
Materials and methods
The workflow of this study is graphically presented in Fig. 1.

*Tg transgenic mice.
Animals
Whole Cortex and hippocampus were isolated from two-month-old male P301S mice (Jackson Laboratory), [16] mice and respective wild-type control mice (n = 4/5 in each group). The tissues were collected on ice, immediately flash-frozen in liquid nitrogen, and stored at −80 °C. Approval was granted by the Authority for Biological and Biomedical Models of The Hebrew University and Tel-Aviv University. The approved ethical number for this study, by Committee at the Hebrew University of Jerusalem is MD-22-16988-3, and at Tel-Aviv University is 04-19-058.
All animal experiments were carried out in compliance with the guidelines set by the Institutional Animal Care and Use Committee at the Hebrew University of Jerusalem, Tel-Aviv University and the Association for Assessment and Accreditation of Laboratory Animal Care International. The study was conducted also in accordance with the ARRIVE guidelines.
Materials and reagents
High-capacity streptavidin was purchased from Thermo Scientific (Waltham, Massachusetts, US). SNOTRAP was acquired from Sigma-Aldrich (St. Louis, MO). Amicon ultra-10 centrifugal filter units were purchased from Merck (US). For MS, acetonitrile (ACN), protease inhibitors cocktail, and distilled water were purchased from Sigma-Aldrich. Biotin-PEG3-propionic acid was obtained from Chem Pep Inc. (Florida, USA). Sequencing-grade modified trypsin was purchased from Promega (Wisconsin, USA). The liquid chromatography-mass spectrometry (LC-MS) and high-performance liquid chromatography (HPLC) solvents were HPLC grade. SNOTRAP-biotin synthesis and nuclear magnetic resonance (NMR) analysis were performed as described previously [47]. A protease-phosphatase inhibitors cocktail (#5872) for Western blot sample preparation was purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies were purchased from CST, Abcam (Cambridge, UK), and Santa Cruz Biotechnology (Dallas, TX, USA).
SNOTRAP and mass spectrometry
Brain tissues were isolated from 2-month-old WT, P301S, and 5xFAD Tg mice. The brain samples were immediately transferred into liquid nitrogen and stored at − 80 °C for further analysis. For each biological replicate, 3 cortex and hippocampal tissue samples were prepared from 3 mice as described previously [46]. The tissues were homogenized on ice in freshly prepared lysis buffer: 250 mM HEPES–NaOH, 0.1 mM neocuproine, 1 mM EDTA, 1% TritonX, 20 mM Iodoacetamide (IAM), and 1% protease inhibitors cocktail, pH 7.7. The homogenates were centrifuged at 17,000 g for 45 min at 4 °C, the supernatant was collected, and protein concentration was estimated by BCA (Bicinchoninic Acid) protein assay (Sigma Aldrich). Samples were washed 3 times with 50 mM HEPES (pH 7.7) by centrifugation at 5000 g for 30 min at 4 °C with 10 K MWCO spin filters pre-rinsed once with 50Mm HEPES. After the centrifugation, the SNOTRAP labeling stock solution (in 50% ACN) was added to all samples to reach a final concentration of 1.5 mM. This was conducted to convert SNO to stable disulfide-iminophosphorane. All samples were incubated in the SNOTRAP solution at 25 °C for 2 h. After the SNOTRAP labeling, reagents were removed using 10 K filters by three consecutive washing with 50 mM HEPES (pH 7.7) buffer. After the ultrafiltration, each sample was incubated with 200 μl pre-rinsed streptavidin agarose beads (Thermo Fisher Scientific) for 2 h at room temperature with gentle shaking. The beads were washed with washing buffer (50 mM HEPES, 150 mM NaCl, 0.1% SDS, pH 7.7) three times and then with washing buffer (50 mM HEPES, pH 7.7) three times. After washing, proteins were eluted (with 10 mM TCEP in 50 mM HEPES, pH 7.7) and then alkylated with 10 mM IAM. Protein samples were then trypsinized (Promega) at 37 °C for 4 h and then desalted with C18 StageTips as described previously [48]. nanoLC-MS/MS Analysis was done to test the SNOtrap samples. MS analysis was performed using a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Waltham, MA USA) coupled online to a nanoflow UHPLC instrument, Ultimate 3000 Dionex (Thermo Fisher Scientific, Waltham, MA USA), as described previously [31]. Mass spectra data were processed using the MaxQuant computational platform, version 2.0.3.0, and analyzed as described previously [31].
Systems biology analysis
For the gene ontology (GO) analysis, we used two databases. First, the list of SNOed proteins was submitted to the search tool for the interacting protein (STRING) database (https://string-db.org) to check whether these proteins are involved in the biological processes implicated in AD pathology. Next, we used IPA software to analyze the pathways that might be regulated and governed by the SNOed proteins of each study group. Eventually, we looked at the processes shared between the two AD models. All terms with False Discovery Rates (FDR) values below 0.05 were accepted.
Western blots (WB)
Sample preparation and protein estimation
The cortex and hippocampus tissues were separately homogenized in RIPA buffer as described previously [31] 30 mm HEPES, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 5 mm EDTA, 1 mm NaV04, 50mmNaF, 1mmPMSF, and 1% protease/phosphatase inhibitors cocktail centrifuged for 45 min at the speed of 17,000 g at 4 °C, the supernatant was collected, and protein content was measured using the Bicinchoninic Acid (BCA) Protein Assay (Sigma-Aldrich).
Electrophoresis and immunoblotting
The samples were diluted with Laemmli sample buffer and electrophoresed according to the molecular weights of the proteins on 7.5% or 10% polyacrylamide gels (Bio-Rad Laboratories Ltd, Haifa, Israel), and transferred to PVDF membrane (Bio-Rad Laboratories Ltd). The membranes were blocked with 5% BSA or 5% skimmed milk in Tris-buffered saline with Twin 20 buffer (TBS-T) containing: 135 mM NaCl, 2.5 mM KCl, 50 mM Tris, and 0.1% Tween 20, pH 7.4 for 2 h at room temperature (RT). The membranes containing transferred proteins were incubated with primary antibodies overnight at 4 °C in shaking conditions. After three washes with TBS-T, the membranes were incubated with secondary antibodies for 2 h at RT in shaking conditions. A clarity max Western ECL Substrate (Bio-Rad Laboratories Ltd.) was used to visualize the protein bands. The bands were captured using the ChemiDoc imaging system (Bio-Rad Laboratories Ltd). The following antibodies were used: Primary antibodies: 3-nitrotyrosine (Abcam, ab110282, dilution 1:2000), iNOS (Santa Cruz, SC-7271, dilution 1/1000), nNOS (CST, 4231S, dilution 1:1000), TSC2 (CST, 4308S, dilution 1:2000), AKT (CST, 9272S, dilution 1:1000), p-AKT (Ser473) (CST, 9271S, dilution 1:1000), mTOR (CST, 2983S, dilution 1:1000), p-mTOR (Ser2448) (CST, 5536S, dilution 1:1000), RPS6 (CST, 2317S, dilution 1:1000), p-RPS6 (Ser235/236) (CST, 4858S), GAD1 (CST, 41318S, dilution 1:1000), NR1(Abcam, ab109182, dilution 1:1000), and VGAT (Abcam, ab235952, dilution 1:1000); Secondary antibodies: Anti-mouse IgG (CST, 7076S, 1/10000 dilution) and anti-rabbit IgG (CST, 7074S, 1/10000 dilution). Each group contained five biological replicates. Each biological replicate was comprised of individual cortex or hippocampus tissue.
Statistical analysis
The mean and standard error of the mean (SEM) were calculated for the WB. An unpaired/independent two-tailed test was used for the western blots. The data are presented as the mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ns = non‐significant. For the GO analysis we included only processes/terms with FDR < 0.05, as calculated using the Benjamini-Hochberg correction on the P-value according to the string database [49]. Bar graphs and statistical analysis were performed using Prism 9.3 (GraphPad Software, San Diego, CA).
Results
SNOTRAP analysis-based mass spectrometry to identify the S-nitrosylated proteins
The SNO data revealed shifts in the SNO levels, pattern, and enrichment of several processes implicated in AD. Some processes were exclusive to a specific AD model, and others were shared, pointing to the possible early stages of the disease progression.
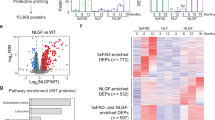
In the cortex of P301S mice, we identified 273 SNO proteins. Among them, 244 proteins were SNOed only in the P301S mice and 29 were shared between P301S and their WT counterparts. 82 proteins appear to be SNOed only in the WT mice, suggesting they were de-nitrosylated in the mutant mice. In the hippocampus, only 83 proteins were SNOed exclusively in P301S mice, and 6 SNO proteins were shared between the mutant and WT mice. 109 proteins were SNOed in the hippocampus of WT mice only (Fig. 2A).

A Shifts in the SNO pattern in P301S mice compared to their WT counterparts. The spheres contain the number of proteins S-nitrosylated in the mutant and WT mice or shared between the mutant and WT mice. B Biological processes related to the pathology which were enriched in the P301S brains. C Pathways analysis that might be regulated by the SNO-proteins in P301S brains. D Shifts in the SNO rates in 5xFAD mice compared to their WT counterparts. E Biological processes related to the pathology which were enriched in the 5xFAD brains. F Pathways analysis that might be regulated by the SNO-proteins in 5xFAD brains. G The shared processes related to AD that were enriched in both models.
In the 5xFAD mice, we identified the shared SNO-proteins between the diseased and the WT mice and quantified the levels of S-nitrosylation. In the hippocampus, we found different SNO pattern between the two groups. 226 SNO proteins were upregulated and 83 were downregulated in 5xFAD mice (Fig. 2D).
Biological processes and pathways analysis revealed enrichment of processes related to AD in P301S and 5xFAD mice
We performed GO analysis to provide biological insights to the SNO-proteins and to understand whether the SNOed proteins affect biological processes implicated in the pathology. Using the STRING software, we generated a list of biological processes affected by SNO in the two AD mouse models. The analysis revealed enrichment of the processes known to be altered in AD.
In P301S mice, SNO-proteins appeared to be involved in several processes related to the pathology, such as axonogenesis, neurogenesis, glutamic acid regulation, regulation of NMDA receptor, metabolic and cell communication processes, apoptosis, and cell survival (Fig. 2B). In 5xFAD mice, the analysis showed enrichment of the processes implicated in AD, such as neurogenesis, aging processes, apoptosis, glutamate metabolic processes, and response to misfolded proteins (Fig. 2E), revealing similarities between the two models.
The pathways analysis exposed the pathways that may be governed and regulated by the SNO proteins in the brains of AD mice. We found SNO-enrichment of several pathways implicated in the pathology in both models. In P301S, enriched processes including GABA receptor signaling, calcium signaling, axonal guidance, and synaptogenesis signaling pathway (Fig. 2C). While in 5xFAD, the calcium signaling pathway, degradation of GABA, autophagy of aggregates (aggrephagy), regulation of mTOR complex 1, and glutamate metabolism pathways were enriched (Fig. 2F).
Lastly, we looked at the shared biological features and found similar processes and pathways including regulation of the glutamatergic and GABAergic systems, apoptosis, and calcium signaling pathways. Based on our SNO data (Fig. 2G), and due to the critical role that GABAergic and glutamatergic systems play in AD [50], we tested the levels of key biomarkers of these systems using WB analysis to check whether, at the biochemical level, we can uncover specific differences and changes between the two models.
Increased nitrosative stress in AD mouse models
To examine other NO•-mediated effects in the brains of the AD mouse models, we tested the levels of 3-nitrotyrosine (3-Ntyr), a marker of NO• production and nitrosative/nitrative stress [51].
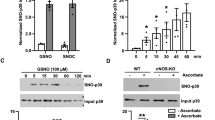
In P301S, we found a significant increase in 3-Ntyr levels in the cortex (Fig. 3A & D), and hippocampus regions (Fig. 4A & D) compared to the WT mice. A significant increase in the levels of iNOS in both regions (Fig. 3A & B, Fig. 4A & B) was also found in these mice. nNOS levels did not change in both regions, consistent with the previous findings (Fig. 3A & C, Fig. 4A & C) [22]. In 5xFAD mice, 3-Ntyr levels were significantly increased in the hippocampus (Fig. 4E & H) but not in the cortex region (Fig. 3E & H). iNOS levels were not changed in the cortex or the hippocampus (Fig. 3E & F, Fig. 4E & F).

A Representative WB for iNOS, nNOS and 3-Ntyr of the cortices of P301S mice and their WT counterparts. B The relative abundance of iNOS normalized to β-actin (a marker of protein loading). WT mice (n = 4), P301S mice (n = 4). C The relative abundance of nNOS normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). D The relative abundance of 3-Ntyr normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). E Representative WB for iNOS, nNOS, and 3-Ntyr prepared from the cortices of 5xFAD mice and their WT counterparts. F The relative abundance of iNOS normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). G The relative abundance of nNOS normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). H The relative abundance of 3-Ntyr normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). An unpaired/independent two-tailed test was used. *p ≤ 0.05, ns non significant.

A Representative WB for iNOS, nNOS and 3-Ntyr of the hippocampi of P301S mice and their WT counterparts. B The relative abundance of iNOS normalized to β-actin. WT mice (n = 4), P301S mice (n = 4). C The relative abundance of nNOS normalized to β-actin. WT mice (n = 5), P301S mice (n = 5) D The relative abundance of 3-Ntyr normalized to β-actin. WT mice (n = 4), P301S mice (n = 4). E Representative WB for iNOS, nNOS, and 3-Ntyr of the hippocampi of 5xFAD mice and their WT counterparts. F The relative abundance of iNOS normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). G The relative abundance of nNOS normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). H The relative abundance of 3-Ntyr normalized to β-actin. WT mice (n = 4), 5xFAD mice (n = 4). An unpaired/independent two-tailed test was used. *p ≤ 0.05, ns non significant.
GABAergic and glutamatergic alterations at early stages of the AD mouse models
Since we found that the GABAergic and glutamatergic systems were enriched by SNO using systems biology analysis, we have tested key biomarkers in both systems biochemically.
The glutamatergic marker, NR1, and the GABAergic markers, glutamic acid decarboxylase 1 (GAD1), and vesicular GABA transporter (VGAT), were tested. The levels of NR1 were altered in both models. In the P301S, the tauopathy model, NR1 levels in the cortex region were significantly increased (Fig. 5A & B), while in the hippocampus significantly decreased compared to the WT mice (Fig. 6A & B). In contrast, in the 5xFAD mice, NR1 levels were significantly reduced in both the cortex and the hippocampus regions compared to the WT mice (Fig. 5E & F, Fig. 6E & F). The downregulation of NR1 was also confirmed previously in the hippocampus of patients with sporadic AD [52].

A Representative WB for NR1, GAD1, and VGAT of the cortices of P301S and WT mice. B The relative abundance of NR1 normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). C The relative abundance of GAD1 normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). D The relative abundance of VGAT normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). E Representative WB for NR1, GAD1, and VGAT of the cortices of 5xFAD and WT mice. F The relative abundance of NR1 normalized to β-actin. WT mice (n = 4), 5xFAD mice (n = 4). G The relative abundance of GAD1 normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). H The relative abundance of VGAT normalized to β actin. WT mice (n = 4), 5xFAD mice (n = 4). An unpaired/independent two-tailed test was used, *p ≤ 0.05, ns non significant.

A Representative WB for NR1, GAD1, and VGAT of the hippocampi of P301S and WT mice. B The relative abundance of NR1 normalized to β-actin. WT mice (n = 4), P301S mice (n = 4). C The relative abundance of GAD1 normalized to β actin. WT mice (n = 5), P301S mice (n = 5). D The relative abundance of VGAT normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). E Representative WB for NR1, GAD1, and VGAT of the hippocampi of 5xFAD and WT mice. F The relative abundance of NR1 normalized to β-actin. WT mice (n = 4), 5xFAD mice (n = 4). G The relative abundance of GAD1 normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). H The relative abundance of VGAT normalized to β-actin. WT mice (n = 4), 5xFAD mice (n = 4). An unpaired/independent two -tailed test were used, *p ≤ 0.05, **p ≤ 0.01, ns non significant.*p < 0.05; **p < 0.01 vs. WT; ns non significant.
The levels of VGAT in P301S mice were significantly decreased only in the hippocampus (Fig. 6A & D), while in 5xFAD mice, downregulation of this transporter was observed in both regions compared to the WT mice (Fig. 5E & H, Fig. 6E & H). GAD1 levels didn’t change in the P301S or the 5xFAD mice compared to their WT counterparts (Fig. 5A & C, E & G Fig. 6A & C, E & G).
Altered mTOR signaling pathways in the AD mouse models
Our previous work has revealed a potential link between NO• and the mTOR signaling pathway in autism spectrum disorder and AD mouse models [1]. Here we aimed to investigate the activation pattern of the mTOR signaling pathway in the diseased mice by testing the levels of key critical components of this pathway: the positive regulator of mTOR p-AKT (Ser473), the negative regulator of mTOR, the catalytic subunit of the tuberous sclerosis complex 2 (TSC2), the activated form of mTOR p-mTOR (Ser2448), and the downstream target of p-mTOR phospho-ribosomal protein S6 (p-RPS6; Ser235) in the cortex (Fig. 7) and the hippocampus (Fig. 8).

A Representative WB for p-AKT, AKT, TSC2, p-mTOR, mTOR, p-RPS6, and RPS6 of the cortices of P301S and WT mice. B The relative abundance of p-AKT normalized for AKT and β-actin. WT mice (n = 5), P301S mice (n = 5). C The relative abundance of TSC2 normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). D The relative abundance of p-mTOR normalized to mTOR and β-actin. WT mice (n = 4), P301S mice (n = 4). E The relative abundance of p-RPS6 normalized to RPS6 and β-actin. WT mice (n = 4), P301S mice (n = 4). F Representative WB for p-AKT, AKT, TSC2, p-mTOR, mTOR, p-RPS6, and RPS6 of the cortices of 5xFAD and WT mice. G The relative abundance of p-AKT normalized to AKT and β-actin. WT mice (n = 5), 5xFAD mice (n = 5). H The relative abundance of TSC2 normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). I The relative abundance of p-mTOR normalized to mTOR and β-actin. WT mice (n = 5), 5xFAD mice (n = 5). J The relative abundance of p-RPS6 normalized to RPS6 and β-actin. WT mice (n = 4), 5xFAD mice (n = 4). An unpaired/independent two-tailed test were used, *p ≤ 0.05, **p ≤ 0.01, ns non significant.

A Representative WB for p-AKT, AKT, TSC2, p-mTOR, mTOR, p-RPS6, and RPS6 of the hippocampi of P301S and WT mice. B The relative abundance of p-AKT normalized to AKT and β-actin. WT mice (n = 4), P301S mice (n = 4). C The relative abundance of TSC2 normalized to β-actin. WT mice (n = 5), P301S mice (n = 5). D The relative abundance of p-mTOR normalized to mTOR and β-actin. WT mice (n = 5), P301S mice (n = 5). E The relative abundance of p-RPS6 normalized to RPS6 and β-actin. WT mice (n = 5), P301S mice (n = 5). F Representative WB for p-AKT, AKT, TSC2, p-mTOR, mTOR, p-RPS6, and RPS6 of the hippocampi of 5xFAD and WT mice. G The relative abundance of p-AKT normalized to AKT and β-actin. WT mice (n = 5), 5xFAD mice (n = 5). H The relative abundance of TSC2 normalized to β-actin. WT mice (n = 5), 5xFAD mice (n = 5). I The relative abundance of p-mTOR normalized to mTOR and β-actin. WT mice (n = 5), 5xFAD mice (n = 5). J The relative abundance of p-RPS6 normalized to RPS6 and β-actin. WT mice (n = 5), 5xFAD mice (n = 5). An unpaired/independent two-tailed test were used, *p ≤ 0.05, ns non significant.
We found a significant increase in the levels of p-AKT and p-mTOR and a significant decrease in TSC2 levels in both regions of the P301S mice compared to their WT counterparts (Figs. 7A–D & 8A–D). P-RPS6 levels were significantly increased in the cortex but not the hippocampus of these mice (Figs. 7E & 8E). However, in both regions of 5xFAD mice, we did not find significant changes in the levels of the mTOR pathway components (Figs. 7F–I & 8F–J), except for p-RPS6 whose levels were significantly increased in the cortex compared to the WT mice (Fig. 7J).
Collectively, these results indicate an overactivation of the mTOR signaling pathway in both AD mouse models.
Discussion
In this work, we performed a comparative study of the molecular events corresponding to the early stages of AD in the P301S and 5xFAD AD models. We identified the S-nitrosylated (SNOed) proteins and found major SNO pattern changes in AD mice in the very early stages of the disease (Fig. 2A & D). Following the acquisition of the SNO-proteome, we performed biochemical validations using WB analysis to test the levels of 3-Ntyr [51]. Next, we identified the processes shared between the two transgenic mouse models using systems biology analysis. We focused on specific processes that were SNO-enriched in the AD models which included the GABAergic and glutamatergic neurotransmission processes, and the mTOR signaling pathway. The levels of critical components of the mTOR pathway and the two GABAergic and glutamatergic systems appeared to be significantly changed in the brains of the mutant mice.
Our work suggests new insight into the effects of NO• on brain functions in pathological conditions in addition to previous studies [53,54,55]. SNO, which targets cysteine thiols of the proteins [56], is a crucial posttranslational modification that manipulates the protein functions [56]. Dysregulation of NO• production and SNO is implicated in brain disorders such as AD, Parkinson’s disease [57, 58], autism spectrum disorder, and neurodevelopmental disorders [46, 59].
Our SNO data revealed shifts in the SNO pattern. In the cortex of P301S mice, we identified 244 proteins were SNOed exclusively in the diseased (mutant) mice. For example, clathrin coat assembly protein (AP180) was S-nitrosylated in the P301S mice but not in the WT. The decreased levels of AP180 have been linked to AD [60, 61]. This protein binds to synaptic vesicle proteins, including the vesicular glutamate transporters [62], that load glutamate into synaptic vesicles and maintain the homeostasis of the glutamatergic system [63]. AP180 inactivation modulates the excitatory neurotransmission by inhibiting synaptic vesicle recycling [62]. S-nitrosylation is a fundamental mechanism in the regulation of cellular signaling by the conformational changes, activation, or inhibition of the protein, and alteration of its localization [64]. Likewise, S-nitrosylation of AP180 may alter its function, implicating it in the regulatory mechanisms of the glutamatergic system.
We found that 82 proteins were SNOed in the cortex and 109 in the hippocampus of the WT counterparts of P301S mice, suggesting that these proteins underwent SNO in physiological conditions but were being denitrosylated in the AD brains. For example, neurogranin was SNOed only in the WT brains. It is a postsynaptic protein that binds to Ca2+-free form of calmodulin [65]. This protein is considered an AD marker once its levels are significantly reduced in the brain [66]. Neurogranin modulates Ca2+/calmodulin-dependent pathways, affecting activity-dependent synaptic plasticity and long-term potentiation, which are critical for learning and memory formation [67]. Previously, we showed that neurogranin is SNOed in the juvenile WT male mouse cortex, striatum, and hippocampus [68]. This protein is found in the cell bodies and dendrites of neurons of the cerebral cortex, hippocampus, and striatum of the rats’ brains [69]. Neurogranin has a highly specialized function in the CNS due to its specific localization [69]. We suggest that denitrosylation of neurogranin prevents its SNO-dependent conformation and thus, affects its localization and function [70], contributing to synaptic dysfunction characteristic of AD.
In 5xFAD mice, we identified 226 SNO proteins that were upregulated, and 83 others were downregulated in 5xFAD compared to the WT mice. The up- or downregulation of the SNO-proteins depend on either altered S-nitrosylation or changes in the abundance of this protein. Glutamine synthase was SNOed in the 5xFAD mutant mice significantly higher than in the WT animals. It has been shown that the SNO of glutamine synthase increases its activity [71], and more glutamate is converted to glutamine, affecting the glutamine/glutamate cycle. The levels of SNOed glutamate dehydrogenase (Glud1, also known as GDH) were also elevated in the diseased mice. The increased SNO of this protein is known to cause its inhibition [71]. Glud1 converts glutamate to α-ketoglutarate, a key molecule in the Krebs cycle [72]. Inhibiting the Glud1 activity causes increased levels of glutamate in the presynaptic neurons. The accumulating glutamate can be converted to GABA by GAD1 [71], shifting the glutamate/GABA balance. The levels of SNOed synaptophysin (Syp) were diminished in 5xFAD mice. Syp is a presynaptic protein essential for proper synaptic functioning [73]. Previously, upregulation of Syp was found in the hippocampal dentate gyrus of the kainic acid-induced mouse model of epilepsy, suggesting increased synaptogenesis. This effect was alleviated by a selective nNOS inhibitor in an epilepsy model [74]. These results are consistent with the finding that the Syp levels are decreased in 3–6 month-old P301S mice [23]. Since the levels of the SNO-proteins depend on the extent of their S-nitrosylation, it can be speculated that Syp denitrosylation may negatively affect synaptogenesis, contributing to synaptic dysfunction in AD.
Collectively, our results point to the important role of SNO in regulating the activity of several proteins and signaling pathways contributing to AD progression and suggest future experimental plans to validate it.
The increased 3-Ntyr levels found in the cortex and hippocampus of P301S mice and the hippocampus of 5xFAD mice indicate elevated nitrosative stress, which results from excessive NO levels [75, 76] and oxidative stress [75] observed in AD. We tested the levels of iNOS and nNOS to understand the possible source of excessive NO in these mice. iNOS levels appeared to be significantly increased in both regions only in the P301S brains. However, in the brain regions of 5xFAD mice, this protein insignificantly differed from the brains of the WT mice. The increased NO production by iNOS is expected in AD because iNOS produces NO• upon inflammation [77], characteristic of AD [78]. Still, iNOS activity may be altered, without changes in its levels. Lastly, nNOS levels did not change either in P301S or 5xFAD mice. AD mice show neuron loss in later stages of the disease [23, 79]. Since nNOS is mainly found in neurons [76], it is logical to suggest that nNOS activity is elevated in these mice, yet its levels remain unchanged due to neuron loss. The nNOS activity measurements in AD mice could be performed in future experiments.
Our systems biology analysis showed that the processes, such as mTORC1 signlaing, glutamatergic, and GABAergic systems regulation, were affected in the two transgenic mouse models. The shared processes indicate that along with protein phosphorylation, SNO may govern glutamatergic and GABAergic neurotransmission and the mTOR signaling pathway. Therefore, we chose to assess and quantify the levels of some critical players in these systems to provide initial information about the alterations in the brains of diseased mice.
We tested the levels of the glutamatergic synapse marker NR1 (the obligatory subunit of NMDA receptor), the GABAergic marker VGAT, which loads GABA into synaptic vesicles to be released to the postsynaptic cleft, and GAD1, the enzyme that catalyzes the production of gamma-aminobutyric acid from L-glutamic acid) proteins. The WB analysis showed alterations in the GABAergic and glutamatergic neurotransmission. In the P301S model of AD, we found a significant increase in the levels of NR1 in the cortex region and a marked decrease in NR1 levels in the hippocampus compared to the WT mice. In contrast, in the 5xFAD mice, the levels of NR1 were significantly decreased in both regions. The decrease in NR-1 levels could be caused by the accumulation of amyloid-beta plaques that can reduce the surface expression of NMDA receptor subunits, including NR1 [80]. It has been found that in P301S mice, Tau protein decreases NMDA receptor expression in the postsynaptic density [81]. It can be suggested that the increased levels of NR1 found in the cortex of these mice is a compensatory mechanism in response to amyloid-beta toxicity or NMDA receptor-related neuronal hyperactivity that precedes amyloid-beta accumulation in the AD brain [82].
In P301S brains, VGAT levels were significantly decreased only in the hippocampus, while in the 5xFAD mice, this change was observed in both the cortex and hippocampus compared to the WT counterparts. The reduced levels of VGAT were also found in previous studies in several AD mouse models [83]. In our experiments, no significant changes in GAD1 levels were detected in the brains of either AD mouse model. This observation, however, does not exclude changes in GAD1 expression in these mice, and further validation at different disease stages is required. Thus, in experiments on 6–10 month-old 5xFAD mice, Wang et al. [84], found a significant decrease in the GAD1 (GAD67) levels, suggesting that GAD1 depletion plays a role in AD pathology. It could be speculated that the GAD1 levels are age-dependent, and we may observe the depletion of this protein in older mice of these models. Several studies have shown that the hippocampus and other areas, such as the cingulate cortex and amygdala, have reduced GABA levels in the brains of AD patients [85].
Next, we tested the levels of critical components of the mTOR signaling pathway. In the P301S mice, the analysis showed significantly increased levels of p-AKT at Ser473 (a positive regulator of mTOR). The levels of TSC2 (the master negative regulator of the mTORC1 activity [86] were decreased. p-mTOR was upregulated in both regions, indicating its activation. Finally, we found a significant increase in the p-RPS6 levels in the cortex, but not the hippocampus, reflecting the increased mTORC1 activity in the cortex of mice of P301S. Thus, our WB results indicate an overactivation of the mTOR pathway in the P301S tauopathy mouse model.
In the 5xFAD mouse model of AD, no significant changes in the levels of the components of the mTOR signaling pathway was found in either region, except for a significant increase in the levels of p-RPS6 at Ser235 in the cortex region. Ser235 is one of four main phosphorylation sites for RPS6, which can be phosphorylated not only by mTOR but also by other signaling pathways and kinases [87]. Thus, RPS6 can be phosphorylated by the RAS/ERK (extracellular signal-regulated kinase) pathway via the activation of p90 ribosomal S6 kinase (RSK) [88, 89] and casein kinase [90]. Still, we cannot rule out the RPS6 phosphorylation by mTOR signaling. Overactivation of the mTOR pathway is one of the underlying mechanisms in AD. It leads to several downstream effects, such as inhibition of autophagy, resulting in amyloid-beta and hyperphosphorylated tau tangles accumulation [42].
In conclusion, our study highlights the importance of S-nitrosylation and nitric oxide in AD. It suggests a link between SNO and the glutamatergic/GABAergic systems and the mTOR signaling pathway at the early stages of AD. The similarities and differences between the two mouse models of AD, the P301S and 5xFAD transgenic mice, are summarized in Table 1. Our findings uncovered critical molecular mechanisms in the early stages of two AD mouse models, which may serve as potential biomarkers and therapeutic targets for early-stage AD.
Data availability
The proteomics data is available upon request from the corresponding author.
References
Mencer S, Kartawy M, Lendenfeld F, Soluh H, Tripathi MK, Khaliulin I, et al. Proteomics of autism and Alzheimer’s mouse models reveal common alterations in mTOR signaling pathway. Transl Psychiatry. 2021;11:1–10.
Tripathi MK, Kartawy M, Amal H. The role of nitric oxide in brain disorders: autism spectrum disorder and other psychiatric, neurological, and neurodegenerative disorders. Redox Biol. 2020;34:101567.
Zvěřová M. Clinical aspects of Alzheimer’s disease. Clin Biochem. 2019;72:3–6.
Kuslansky G, Buschke H, Katz M, Sliwinski M, Lipton RB. Screening for Alzheimer’s disease: the memory impairment screen versus the conventional three‐word memory test. J Am Geriatrics Soc. 2002;50:1086–91.
Morshed N, Ralvenius WT, Nott A, Watson LA, Rodriguez FH, Akay LA, et al. Phosphoproteomics identifies microglial Siglec‐F inflammatory response during neurodegeneration. Mol Syst Biol. 2020;16:e9819.
Jahn H. Memory loss in Alzheimer’s disease. Dialogues Clin Neurosci 2022.
Palop JJ, Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: two faces of the same coin? Neuromolecular Med. 2010;12:48–55.
Mann DM. Pyramidal nerve cell loss in Alzheimer’s disease. Neurodegeneration. 1996;5:423–7.
Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–81.
Chen G, Xu T, Yan Y, Zhou Y-R, Jiang Y, Melcher K, Xu HE. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacologica Sin. 2017;38:1205–35.
Norfray JF, Provenzale JM. Alzheimer’s disease: neuropathologic findings and recent advances in imaging. Am J Roentgenology. 2004;182:3–13.
Korolev IO. Alzheimer’s disease: a clinical and basic science review. Med Stud Res J. 2014;4:24–33.
Bertram L, Tanzi RE. The genetics of Alzheimer’s disease. Prog Mol Biol Transl Sci. 2012;107:79–100.
Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88:3–12.
Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 2017;14:e1002270.
Shapira R, Gdalyahu A, Gottfried I, Sasson E, Hadanny A, et al. Hyperbaric oxygen therapy alleviates vascular dysfunction and amyloid burden in an Alzheimer’s disease mouse model and in elderly patients. Aging (Albany NY). 2021;13:20935–61.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–40.
Ju IG, Son SY, Lee S, Im H, Huh E, Eo H, et al. Protective effects of CCL01 against Aβ-induced neurotoxicity in 5xFAD transgenic mouse model of Alzheimer’s disease. Biomedicine Pharmacotherapy. 2023;158:114105.
Shin J, Park S, Lee H, Kim Y. Thioflavin-positive tau aggregates complicating quantification of amyloid plaques in the brain of 5XFAD transgenic mouse model. Sci Rep. 2021;11:1617.
Kourti M, Metaxas A. A systematic review and meta-analysis of tau phosphorylation in mouse models of familial Alzheimer’s disease. Neurobiol Dis. 2024;192:106427.
Morshed NF. Phosphoproteomics analysis of Alzheimer’s disease: Massachusetts Institute of Technology; 2021.
Amal H, Gong G, Gjoneska E, Lewis SM, Wishnok JS, Tsai LH, et al. S-nitrosylation of E3 ubiquitin-protein ligase RNF213 alters non-canonical Wnt/Ca+ 2 signaling in the P301S mouse model of tauopathy. Transl Psychiatry. 2019;9:1–12.
Yoshiyama Y, Higuchi M, Zhang B, Huang S, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51.
Yuste JE, Tarragon E, Campuzano CM, Ros-Bernal F. Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci. 2015;9:322.
Schulz JB, Matthews RT, Beal MF. Role of nitric oxide in neurodegenerative diseases. Curr Opin Neurol. 1995;8:480–6.
Zhao B. Nitric oxide in neurodegenerative diseases. Front Bioscience-Landmark. 2005;10:454–61.
Abdel-Haq M, Ojha SK, Hamoudi W, Kumar A, Tripathi MK, Khaliulin I, et al. Effects of extended-release 7-nitroindazole gel formulation treatment on the behavior of Shank3 mouse model of autism. Nitric Oxide. 2023;140:41–9.
Kruglyakov D, Ojha SK, Kartawy M, Tripathi MK, Hamoudi W, Bazbaz W, et al. Nitric Oxide Synthase Inhibition Prevents Cell Proliferation in Glioblastoma. J Mol Neurosci. 2023;73:875–83.
Steinert JR, Amal H. The contribution of an imbalanced redox signalling to neurological and neurodegenerative conditions. Free Radic Biol Med. 2023;194:71–83.
Tripathi MK, Kartawy M, Ginzburg S, Amal H. Arsenic alters nitric oxide signaling similar to autism spectrum disorder and Alzheimer’s disease-associated mutations. Transl Psychiatry. 2022;12:127.
Tripathi MK, Ojha SK, Kartawy M, Hamoudi W, Choudhary A, Stern S, et al. The NO answer for autism spectrum disorder. Adv Sci. 2023;10:2205783.
Tripathi MK, Ojha SK, Kartawy M, Khaliulin I, Hamoudi W, Amal H. Mutations associated with autism lead to similar synaptic and behavioral alterations in both sexes of male and female mouse brain. Sci Rep. 2024;14:10.
Yang H, Oh C, Amal H, Wishnok JS, Lewis S, Schahrer E, et al. Mechanistic insight into female predominance in Alzheimer’s disease based on aberrant protein S-nitrosylation of C3. Sci Adv. 2022;8:eade0764.
Hamoudi W, Tripathi MK, Ojha SK, Amal H. A cross-talk between nitric oxide and the glutamatergic system in a Shank3 mouse model of autism. Free Radic Biol Med. 2022;188:83–91.
Bazbaz W, Kartawy M, Hamoudi W, Ojha SK, Khaliulin I, Amal H. The Role of Thioredoxin System in Shank3 Mouse Model of Autism. J Mol Neurosci. 2024;74:90.
Ojha SK, Amal H. Air pollution: an emerging risk factor for autism spectrum disorder. Brain. 2024;1:4.
Oh C, Nakamura T, Zhang X, Lipton SA. Redox regulation, protein S-nitrosylation, and synapse loss in Alzheimer’s and related dementias. Neuron. 2024;112:3823–50.
Nakamura T, Oh C, Liao L, Zhang X, Lopez KM, Gibbs D, et al. Noncanonical transnitrosylation network contributes to synapse loss in Alzheimer’s disease. Science. 2021;371:eaaw0843.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84.
Pei JJ, Hugon J. mTOR‐dependent signalling in Alzheimer’s disease. J Cell Mol Med. 2008;12:2525–32.
Perluigi M, Di Domenico F, Barone E, Butterfield D. mTOR in Alzheimer disease and its earlier stages: links to oxidative damage in the progression of this dementing disorder. Free Radic Biol Med. 2021;169:382–96.
Davoody S, Asgari Taei A, Khodabakhsh P, Dargahi L. mTOR signaling and Alzheimer’s disease: what we know and where we are? CNS Neurosci Therapeutics. 2024;30:e14463.
Xie PL, Zheng MY, Han R, Chen WX, Mao JH. Pharmacological mTOR inhibitors in ameliorating Alzheimer’s disease: current review and perspectives. Front Pharmacology. 2024;15:1366061.
An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 2003;163:591–607.
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, et al. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochemistry. 2005;93:105–17.
Amal H, Barak B, Bhat V, Gong G, Joughin BA, Wang X, et al. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol Psychiatry. 2020;25:1835–48.
Seneviratne U, Nott A, Bhat VB, Ravindra KC, Wishnok JS, Tsai LH, et al. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc Natl Acad Sci. 2016;113:4152–7.
Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–906.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Society: Ser B (Methodol). 1995;57:289–300.
Parameshwaran K, Dhanasekaran M, Suppiramaniam V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol. 2008;210:7–13.
Bandookwala M, Sengupta P. 3-Nitrotyrosine: a versatile oxidative stress biomarker for major neurodegenerative diseases. Int J Neurosci. 2020;130:1047–62.
Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, et al. Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer’s disease. Exp Neurol. 1999;160:194–204.
Guix F, Uribesalgo I, Coma M, Munoz F. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–52.
Picón-Pagès P, Garcia-Buendia J, Munoz FJ. Functions and dysfunctions of nitric oxide in brain. Biochimica et Biophysica Acta -Molecular Basis Dis. 2019;1865:1949–67.
Zhang X, Chen Z, Xiong Y, Zhou Q, Zhu LQ, Liu D. The emerging role of nitric oxide in the synaptic dysfunction of vascular dementia. Neural Regeneration Res. 2025;20:402–15.
Kartawy M, Khaliulin I, Amal H. Systems biology reveals S-Nitrosylation-dependent regulation of mitochondrial functions in mice with Shank3 mutation associated with autism spectrum disorder. Brain Sci. 2021;11:677.
Steinert JR, Chernova T, Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist. 2010;16:435–52.
Shahani N, Sawa A. Protein S-nitrosylation: role for nitric oxide signaling in neuronal death. Biochimica et Biophysica Acta -General Subj. 2012;1820:736–42.
Okamoto S, Lipton SA. S-Nitrosylation in neurogenesis and neuronal development. Biochimica et Biophysica Acta -General Subj. 2015;1850:1588–93.
Sun Y, Chen G, He J, Huang ZG, Li SH, Yang YP, et al. Clinical significance and potential molecular mechanism of miRNA-222-3p in metastatic prostate cancer. Bioengineered. 2021;12:325–40.
Teerlink CC, Miller JB, Vance EL, Staley LA, Stevens J, Tavana JP, et al. Analysis of high‐risk pedigrees identifies 11 candidate variants for Alzheimer’s disease. Alzheimer’s Dement. 2022;18:307–17.
Vanlandingham PA, Barmchi MP, Royer S, Green R, Bao H, Reist N, et al. AP180 couples protein retrieval to clathrin‐mediated endocytosis of synaptic vesicles. Traffic. 2014;15:433–50.
Vigneault É, Poirel O, Riad M, Prud’Homme J, Dumas S, Turecki G, et al. Distribution of vesicular glutamate transporters in the human brain. Front Neuroanat. 2015;9:23.
Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614.
Gerendasy D. Homeostatic tuning of Ca2+ signal transduction by members of the calpacitin protein family. J Neurosci Res. 1999;58:107–19.
Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer’s disease. Int Psychogeriatr. 1998;10:11–23.
Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010;1362:13–22.
Hamoudi W, von Lendenfeld F, Kartawy M, Mencer S, Suloh H, Khaliulin I, et al. Regional differences in S-nitrosylation in the cortex, striatum, and hippocampus of juvenile male mice. J Mol Neurosci. 2021;71:2383–92.
Represa A, Deloulme JC, Sensenbrenner M, Ben-Ari Y, Baudier J. Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci. 1990;10:3782–92.
Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–32.
Raju K, Doulias PT, Evans P, Krizman EN, Jackson JG, Horyn O, et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal. 2015;8:ra68.
Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D. Alpha-ketoglutarate: physiological functions and applications. Biomolecules Ther. 2016;24:1.
Sobrevia L, Ooi L, Ryan S, Steinert JR. Nitric oxide: a regulator of cellular function in health and disease. Oxid Med Cell Longev. 2016;2016:9782346.
Beamer E, Otahal J, Sills GJ, Thippeswamy T. N w‐Propyl‐l‐arginine (L‐NPA) reduces status epilepticus and early epileptogenic events in a mouse model of epilepsy: behavioural, EEG and immunohistochemical analyses. Eur J Neurosci. 2012;36:3194–203.
Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56:1449–52.
Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–30.
Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–53.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421.
Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8:1–12.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–8.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–97.
Stargardt A, Swaab DF, Bossers K. The storm before the quiet: neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer’s disease. Neurobiol Aging. 2015;36:1–11.
Ghatak S, Dolatabadi N, Trudler D, Zhang X, Wu Y, Mohata M, et al. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. eLife. 2019;8:e50333.
Wang Y, Wu Z, Bai YT, Wu GY, Chen G. Gad67 haploinsufficiency reduces amyloid pathology and rescues olfactory memory deficits in a mouse model of Alzheimer’s disease. Mol Neurodegener. 2017;12:1–15.
Jiménez-Balado J, Eich TS, editors. GABAergic dysfunction, neural network hyperactivity and memory impairments in human aging and Alzheimer’s disease. Seminars in cell & developmental biology; 2021: Elsevier.
Carroll B, Maetzel D, Maddocks OD, Otten G, Ratcliff M, Smith GR, et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife. 2016;5:e11058.
Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochemical Sci. 2006;31:342–8.
Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1−/−/S6K2−/− mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24.
Pirbhoy PS, Farris S, Steward O. Synaptically driven phosphorylation of ribosomal protein S6 is differentially regulated at active synapses versus dendrites and cell bodies by MAPK and PI3K/mTOR signaling pathways. Learn Mem. 2017;24:341–57.
Hutchinson JA, Shanware NP, Chang H, Tibbetts RS. Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1. J Biol Chem. 2011;286:8688–96.
Acknowledgements
This work was funded by a US Department of Defense (DoD) grant, a DFG – German Research Foundation Grant, an Israeli Science Foundation (ISF, 669/22) grant, an Eagles Autism Foundation grant, a National Institute of Psychobiology in Israel (NIPI) grant, an Israeli Council for Higher Education Maof Grant, and a Berettler Centre for Research in Molecular Pharmacology and Therapeutics Grant. We also thank the Satell Family Foundation and the Neubauer Family Foundation for their support. This research was also supported by the ISF (2141/20), BrightFucos grant (A2022029S), NIH grant 1R21AG074846-01A1 (to U.A). We would like to thank Alex Smolar for his technical assistance.
Author information
Authors and Affiliations
Contributions
HS – Proteomics and biochemical analysis, and writing the draft, SKO – Biochemical analysis and writing the draft, MK – Systems biology analysis, WH – Systems biology analysis, MKT – Biochemical analysis, WB – Biochemical analysis, NS – Animals’ handling, UA – Provided the 5XFAD model and contributed to the discussion, IK – Data analysis and contributed to the discussion, HA – Conceived the idea, executed the study, and supervised the research.
Corresponding author
Ethics declarations
Competing interests
HA is a CSO of Point6 Bio and Neuro-NOS. No funds from both companies were received for this study. All other authors do not hold any competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Suloh, H., Ojha, S.K., Kartawy, M. et al. Shared early molecular mechanisms revealed in P301S and 5xFAD Alzheimer’s disease mouse models. Transl Psychiatry 15, 97 (2025). https://doi.org/10.1038/s41398-025-03321-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-025-03321-7
