
Abstract
Fear memory is crucial for animals to effectively respond to dynamic environments and survive dangerous stimuli. However, aberrant fear memory contributes to various psychiatric disorders, such as post-traumatic stress disorder (PTSD). Despite its importance, the precise molecular mechanisms underlying fear memory remain insufficiently understood. In this study, we highlight the pivotal role of the epigenetic factor chromodomain Y-like protein (CDYL) in the regulation of fear memory. We discovered that ablation of CDYL in CaMKIIα+ excitatory neurons in the forebrain or hippocampus leads to increased fear memory in mice. CDYL is phosphorylated by cyclin-dependent kinase 5 (CDK5) at Ser147, which facilitates tripartite motif containing 32 (TRIM32)-mediated ubiquitination and degradation of CDYL in response to neural activity. Additionally, we developed an interfering peptide that specifically targets the phosphorylation of CDYL at Ser147, resulting in a decrease in contextual fear memory in mice. Collectively, our findings underscore the essential role of CDYL in fear memory and illustrate the modulatory function of CDK5 and TRIM32 on CDYL, positioning CDYL as a promising target for the modulation of fear memory.
Similar content being viewed by others


Introduction
Memory is one of the most vital brain functions, and among its forms, fear memory is critical for survival and is extensively studies [1]. It is essential for animals to appropriately respond to dynamic environments and survive dangerous stimuli [1, 2]. While adaptive fear memory aids survival, dysfunctions can lead to fear-related disorders [3], significantly impacting nearly every aspect of patients’ lives, such as in post-traumatic stress disorder (PTSD). PTSD is characterized by excessively strong and persistent fear memories, which impose substantial psychological and physical burdens on individuals [4, 5]. The lifetime prevalence of PTSD is estimated to range from 1.3% to 12.2% in the general population, escalating beyond 50% in those exposed to severe psychological stress [4]. Therefore, understanding the molecular mechanisms underlying fear memory is crucial for developing new therapeutic strategies for memory modulation.
During memory formation, neurons undergo lasting changes that encode new information and create distinct gene expression patterns in response to neural activity [6,7,8,9,10]. Epigenetic mechanisms, which integrate environmental stimuli to regulate memory-related gene transcription, thereby influencing synaptic plasticity, neuronal excitability, and other essential signaling pathways, are important in the regulation of fear memory [11,12,13]. However, these molecular processes are not yet fully understood.
The epigenetic factor chromodomain Y-like protein (CDYL) comprises an amino-terminal chromodomain and a carboxy-terminal enoyl-coenzyme A hydratase-isomerase catalytic domain [14, 15]; CDYL has been shown to repress gene transcription by acting as a crotonyl-coenzyme A hydratase and a histone H3K9/27 methyllysine reader [16,17,18]. Initially identified as a key regulator of mammalian spermatogenesis [16]. CDYL has since been recognized for its regulatory role in the development and function of the nervous system [17]. Further research demonstrated that CDYL represses the transcription of genes critical for brain function, such as Bdnf, Vgf, and Scn8a, negatively affecting the structural plasticity, intrinsic excitability, and migration of neurons. These mechanisms illustrate CDYL’s significant roles in neurodevelopment, epilepsy, and depression [17, 19,20,21]. Previously, our results indicated that CDYL levels decrease in response to neural activity, suggesting an activity-dependent role in gene expression regulation [17]. Although much is known about CDYL’s downstream mechanisms and functions in neurons, its role in regulating memory and how it is itself regulated remains uncertain.
Cyclin-dependent kinase 5 (CDK5) has been linked to the regulation of numerous neural functions and may connect extracellular stimuli to CDYL and the transcriptional program [22,23,24,25]. CDK5 translocates into the nucleus during neural activity and regulates Bdnf expression and dendrite development, both aligning with CDYL’s functions [17, 22]. In this study, we report that CDYL levels in forebrain or hippocampal excitatory neurons negatively influence fear memory in mice. We demonstrate that CDYL is phosphorylated at Ser147 by CDK5, promoting tripartite motif containing 32 (TRIM32)-mediated ubiquitination and degradation of CDYL in response to neural activity. Additionally, we developed an interfering peptide specifically targeting the phosphorylation of CDYL at Ser147, which increased CDYL levels and decreased contextual fear memory in mice. Our findings offer novel insights into CDYL’s role in fear memory modulation and the upstream regulatory mechanisms of CDYL, positioning CDYL as a potential target for treating fear-related disorders.
Materials and methods
Animals
ICR and C57BL/6 mice were obtained from Charles River Laboratories in Beijing, China. Cdyl-loxp mice were generated on a C57BL/6 J background by Biocytogen, also in Beijing, China. CaMKIIα-CREERT2 mice were provided by the Jackson Laboratory in Maine, USA. All mice were housed with their littermates (fewer than six per cage) under a 12:12 h light/dark cycle, with ad libitum access to food and water. Biochemical and behavioral experiments were performed in 8-weeks-old male mice. Efforts were made to minimize animal suffering and to reduce the number of animals used. In biochemical and histological experiments, at least 3 animals or 3 cultures were included in each group, and at least 7 in each group in behavioral tests. The exact number of animals used in experiments was indicated in the related figures and legends.
Ethics approval and consent to participate
The animal studies were approved by the Peking University Institutional Animal Care and Use Committee (Protocol Approval No. LA2014152) and performed in compliance with institutional guidelines. As no human participants were involved, informed consent was not required.
Plasmids
Complementary DNAs (cDNAs) for mouse wild-type (WT) CDYL, CDK5, and TRIM32 were amplified using polymerase chain reaction (PCR) and ligated into pEGFP-N1, pET-28(+), and pGEX-5X-1 vectors, respectively. CDYL mutants, including S147A, S147D, and S162A, were produced using QuikChange Site-Directed Mutagenesis Kits (Stratagene, #200517-5, WA, USA), with site mutants of CDYL cloned into the pEGFP-N1 vector. Serial deletion mutants of CDYL were generated and cloned into the pGEX-5X-1 plasmid. His-tagged TRIM32 was cloned into the pET-28(+) vector. Short hairpin RNA (shRNA) specific for mouse TRIM32 was utilized from a previous study [26]. and cloned into the pLKO.1 vector. The shRNA sequences were: sense, 5’-CCGGTGAAGTTGAGAAGTCCAATAGCTCGAGCTATTGGACTTCTCAACTTCATTTTTG-3’; antisense, 5’-AATTCAAAAATGAAGTTGAGAAGTCCAATAGCTCGAGCTATTGGACTTCTCAACTTCA-3’. All the primers used for constructing these plasmids are listed in Supplementary Table 1.
Primary cultures of cortical neurons and transfection
Cortical tissues from embryonic day 16 mouse embryos were isolated and digested with 0.25% trypsin and DNase (Sigma–Aldrich, MA, USA) for 30 min at 37 °C. The cells were then triturated with a pipette in plating medium (DMEM with 10% fetal bovine serum) and plated onto poly-D-lysine-coated dishes (Sigma–Aldrich). After 4 h of culture, the medium was replaced with Neurobasal medium supplemented with 2% B27 and 0.5 mM GlutaMAX-I (Invitrogen, CA, USA). Transfections were performed on Day 5 in vitro (DIV 5) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and cells were harvested at DIV 7. For drug-induced experiments, KCl (final concentration of 60 mM), roscovitine (20 μM, Sigma–Aldrich), and the interfering peptide (10 μg) were added at DIV 7, and cultures were harvested at the designated times. In experiments focused on ubiquitin detection, MG132 (20 μM, Sigma–Aldrich) was added to the medium 6 h before harvesting the neurons. At least 3 cultures were included in each group in experiments using primary culture of cortical neurons for biological replicates.
Pull-down assay
Recombinant plasmids containing deletion mutants of CDYL (GST-CDYL 1-60, GST-CDYL 61-180, GST-CDYL 181-309, and GST-CDYL 310-544) and His6-CDK5 were transformed into DH5α cells and induced with IPTG (Sangon, Shanghai, China). The fusion proteins were purified using GST and His beads (Sigma–Aldrich). For the pull-down assay, the His6-CDK5 fusion protein was incubated with CDYL deletion mutants or a GST control at 4 °C with shaking overnight, followed by incubation with GST beads for 2 h. The beads were washed six times with TBST and boiled in SDS loading buffer for Western blot analysis.
In vitro phosphorylation
Fusion proteins of His6-CDK5 and mutants of CDYL were incubated in reactive buffer containing 32P and 1,4-dithiothreitol at 30 °C for 30 min. Then, the reaction buffers were boiled in SDS loading buffer and subjected to SDS–PAGE, followed by Coomassie blue staining and radioautography.
Western blotting and coimmunoprecipitation
Western blotting and coimmunoprecipitation experiments were performed as described previously [27, 28]. The tissues and the harvested primary neurons were lysed in RIPA buffer with protease inhibitors and ready for western blotting. The bands were quantified by ImageJ (NIH). The primary neurons were transfected with plasmids and prepared for coimmunoprecipitation. The commercial antibodies were anti-β-actin (ZSGB-BIO, #TA09, Beijing, China), anti-CDK5 (Santa Cruz, #sc-6247, CA, USA), anti-CDYL (ABclonal, #WG-03686, Wuhan, China), anti-CDYL (Proteintech, #17763-1-AP, IL, USA), anti-pS147-CDYL (customized by MBL Beijing Biotech, Beijing, China, Peptide antigen: CLVPK(pS)PVKSR), anti-GFP (Santa Cruz, #sc-8334), anti-GST (Proteintech, #66001-2-Ig), anti-His (PPLYGEN, #C1301, Beijing, China), anti-P35 (Santa Cruz, #sc-293184), anti-Ubiquitin (CST, #43124S, MA, USA), anti-TRIM32 (Proteintech, #10326-1-AP). The expected molecular weight of CDYL was between 55 kDa and 70 kDa, the CDYL antibody that was used in this study identified two to three distinct bands within the range, all bands were consider specific and quantified together as the total level of CDYL. The main band of p-CDYL was above 70 kDa, one or two bands could be detected depending on the source and the extraction process of samples.
Histology and immunofluorescence staining
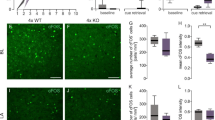
3 knock out mice and 3 wild type mice were used to examine the ablation of CDYL in CaMKIIα+ neurons. Mice were perfused with 20 mL of PBS and then with 20 mL of 4% paraformaldehyde (PFA). The perfused brains were post-fixed in 4% paraformaldehyde. Fixed brain were dehydrated in 20–30% (w/v) sucrose for cryoprotection and embedded in Tissue‐Tek O.C.T. compound (#4583, SAKURA). Brains were sectioned with a cryostat at 30‐µm thickness. Sections were washed with phosphate‐buffered saline (PBS) once for 20 min and blocked for 1 h at room temperature with PBS containing 0.1% Triton X‐100 and 3% bovine serum albumin (BSA). Then sections were incubated with primary antibodies at 4 °C overnight. Primary antibodies were used in a 1:1,000 concentration: CaMKIIα (Cell Signaling Technology, #3357) and CDYL (Sigma-Aldrich, SAB2103346). Next, sections were washed with PBS three times for 10 min each and incubated with appropriate secondary antibodies (Alexa Fluor 488‐conjugated donkey anti‐mouse, Invitrogen, 1: 1000, A‐21202; Alexa Fluor 594‐conjugated donkey anti‐rabbit, Invitrogen, 1: 1000, A‐2120) at 4 °C overnight. Finally, sections were stained with Hoechst (1:500, Solarbio, C0031) and coverslips were applied. Images were acquired using an inverted fluorescence microscope and a confocal laser‐scanning microscope (Leica TCS SP8 STED), at least 3 images were captured in each slice and one image was shown as representative.
Behavioral tests
All behavioral experiments were conducted blinded to the treatment and genotype of the mice. Only 2- to 4-month-old male mice were used for behavioral testing.
Open field test
Mice were allowed to individually explore in a plastic open-field chamber (50 × 50 × 50 cm) for 30 min and recorded by a Smart 3.0 video tracking system (Panlab, DC, USA). The total distance traveled in 30 min, time spent in the center region (25 cm × 25 cm) and the number of entries into the center in the first 5 min were recorded.
Elevated zero maze test
The maze was comprised of a platform (25 cm inner diameter, 5 cm width), elevated 60 cm above ground level and divided into four equal quadrants. Two opposing quadrants were open, and the remaining two were enclosed by 20 cm-high walls. Mice were free to individually explore the apparatus for 5 min while being recorded by Smart 3.0 video tracking system.
Y maze test
The Y maze was comprised of three arms (50 × 10 × 30 cm) at angles of 120° from each other. Mice were free to individually explore the maze for 8 min, as recorded by Smart 3.0 video tracking system. The travel distance and spontaneous alternation in arm selection were recorded.
Morris water maze test
The water maze was comprised of a circular tank (with a diameter of 120 cm and a depth of 60 cm) filled with water and equipped with a platform hidden just beneath the surface; visual clues were placed around the tank. The water maze was divided into four quadrants: the target quadrant (containing the platform), and three nontarget quadrants (Fig. 1I). The test was performed as previously described [29] and recorded by SuperMaze V2.0 (Softmaze Information Technology Co. Ltd., Shanghai, China). Briefly, on Day 0, mice were trained to find the platform with a visible flag. On Days 1-5, mice were placed into the maze to search for the platform without flag for 60 s. Their escape latency was recorded. The probe test was performed on Day 6, with the platform removed. Occupancy time (%) in all quadrants, number of platform crossings, and the latency to first cross the target platform position (s) were recorded.
Fear conditioning test
The test had 3 phases: training, contextual test, and cued test. On the training day (Day 0), mice were placed into a Skinner box (Softmaze Information Technology, Shanghai, China) and allowed to explore for 1 min before the onset of a tone stimulus (85 dB, 2.8 kHz), followed by a 0.5 mA electric foot shock for 2 s; this pair of stimuli was repeated once more. For Western blot experiments to detect CDYL and p-CDYL, mice were subjected to an additional round of training, the amplitude of electric shock was 1.0 mA. The contextual test was conducted on Day 1, during which mice were placed into the same box to explore for 6 min without tone or foot shock stimulus. For the cued test (Day 2), mice were placed into a box with a different context (changes in the color of walls, smell, background noise and illumination), allowed to explore for 3 min and then presented with the same sound used in the training phase over the following 3 min. During each phase, the percentage of time exhibiting freezing behavior was recorded by SuperFcs (Softmaze Information Technology).
Surgery and injection
Tamoxifen injection
For tamoxifen-induced conditional knockout of CDYL in forebrain excitatory neurons, offspring mice of Cdyl-loxp mice and CaMKIIα-CREERT2 mice were intraperitoneally injected with tamoxifen (100 mg/kg, diluted in corn oil) daily for 5 consecutive days when they were 8 weeks old, and all experiments were conducted 2 weeks after the last injection.
Virus microinjection
Wild type male C57 mice were randomly divided into two groups (GFP or CRE, blinded to the experimenters) when they were 8 weeks old. AAV viruses were bilaterally injected into the dCA1 region (AP: −2.06 mm, ML: ± 1.50 mm, DV: −1.30 mm) at a volume of 300 nL at 60 nL/min by a Hamilton microsyringe with a stereotaxic apparatus (RWD Life Science, Shenzhen, China). Three weeks after virus injection, mice were subjected to the following experiments. AAV9-CaMKIIα-CRE-GFP and control AAV9-CaMKIIα-GFP, derived from Obio Technology (Shanghai, China).
In cannula administration of interfering peptides
All the interfering peptides were synthesized by GL Biochem (Shanghai, China). Cannulas for administration of the peptide were implanted and targeted the bilateral dCA1 regions of wild type male C57mice when they were 8 weeks old. One week after the surgery, the mice were randomly divided into control group or experimental group and then injected with 500 nL peptides per site at a speed of 100 nL/min, and the experiments were performed at the indicated times.
Statistical analysis
All of the statistics were analyzed using GraphPad Prism 8.0 (GraphPad Software, CA, USA). Normality and lognormality tests were performed prior to statistical comparisons between two groups, two-tailed t tests were performed if the data passed normality and lognormality tests, otherwise, Mann-Whitney test was performed. For multiple comparisons, one-way ANOVA with Tukey’s multiple comparisons or two-way ANOVA with Sidak’s multiple comparison was used. Outliers were detected using ROUT method in GraphPad Prism 8.0 software, with the parameter Q = 5% in a pre-established manner. All data were presented as means ± S.E.M. *p < 0.05, **p < 0.01 and ***p <0.001 were considered significant; n.s. indicates not significance.
Results
Conditional knockout of CDYL in CaMKIIα+ neurons increases fear memory but does not affect spatial or working memory in mice
Initially, we examined the baseline expression pattern of CDYL in the adult mouse brain. Consistent with previous studies, CDYL was found to be widely expressed throughout the brain (Supplementary Figure S1A). To explore CDYL’s role in regulating fear memory, we generated Cdyl-loxP transgenic mice (Supplementary Figure S2A) and crossed homozygous Cdyl-loxp mice with CaMKIIα-CREERT2 mice, which express Cre recombinase upon tamoxifen induction under the CaMKIIα promoter. CDYL was ablated in excitatory neurons in the forebrain by tamoxifen injection (Fig. 1A). We confirmed the reduced CDYL levels in conditional knockout (cKO) mice and verified the specificity of CDYL knockout in CaMKIIα+ neurons using Western blot and immunofluorescence staining, respectively (Fig. 1B, Supplementary Figure S1B-C). We then assessed the impact of CDYL cKO on fear memory using a fear conditioning test (Fig. 1C). Results indicated that cKO mice exhibited significantly increased freezing times in both contextual and cued fear tests compared to WT littermates (Fig. 1D), suggesting enhanced fear memory in cKO mice. To determine whether CDYL knockout affects the response to an unconditioned stimulus (electric shock), we analyzed the proportion of freezing time following exposure to the unconditioned stimulus during the first round of the contextual fear-conditioning procedure. Our findings suggest that reducing CDYL via tamoxifen-induced knockout does not impact unconditioned freezing behavior, indicating that changes in CDYL levels are specifically related to associative learning rather than the innate freezing response to the unconditioned stimulus (Supplementary Figure S2H).
To investigate whether CDYL influences working memory, we subjected the mice to the Y maze test and found no differences between cKO mice and WT littermates (Fig. 1E, F), indicating that spatial working memory was unaffected. Further examination of spatial memory using the Morris water maze test (Fig. 1G) similarly revealed no significant differences between cKO and WT mice (Fig. 1H–J, Supplementary Figure 2B). These results demonstrate that cKO of CDYL in CaMKIIα+ neurons does not affect spatial or working memory in mice.
Additionally, we evaluated locomotion and anxiety-like behaviors in cKO mice using the open field test (Fig. 1K) and the elevated zero maze test (Fig. 1M). No differences in ambulatory distance or total distance were observed between cKO mice and WT littermates (Supplementary Figure S2C, D, F), indicating unaffected locomotion. Anxiety-like behaviors were also unchanged in cKO mice, as shown by the results (Fig. 1K, N, Supplementary Figure 2E, G). Collectively, these findings demonstrate that the ablation of CDYL in forebrain excitatory neurons specifically enhances fear memory.

A Schematic of tamoxifen-induced conditional ablation of CDYL in CaMKIIα+ neurons in mice. B Western blot analysis of CDYL protein levels in the hippocampus of wild-type (WT) and conditional knockout (cKO) mice. (Unpaired t-test, n = 5 WT, 4 cKO). C Diagram of the fear conditioning protocol. D Percentage of time mice exhibited freezing behavior in contextual (left) and cued (right) fear memory tests. (Unpaired t-test; contextual: n = 15 WT, 15 cKO; cued: n = 16 WT, 15 cKO; one outlier excluded). E, F Y-maze test diagram (E) and percentage of spontaneous alternation triplets (F). (Unpaired t-test, n = 9 WT, 8 cKO). G-J Morris water maze. G Diagram of the Morris water maze. H Escape latency time during the training period. Two-way ANOVA with Sidak’s multiple comparisons test. I Time distribution in the four quadrants during the test. Two-way ANOVA with Sidak’s multiple comparisons test. J Latency to first cross the target platform position (left), unpaired t test. One sample was identified as an outlier and thus excluded, n (WT, cKO) = 14, 14. The number of platform area crossings during the test period (right), Mann–Whitney test, n (WT, cKO) = 15, 14. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ns, no significance. K, L Open field test diagram (K) and time spent in the center area during the first 5 min (L). Unpaired t test, n (WT, cKO) = 15, 14. M, N Elevated zero maze diagram (M) and the time spent in open arms during the test (N). Unpaired t test, n (WT, cKO) = 15, 14.
Conditional knockout of CDYL in dorsal hippocampal excitatory neurons leads to an increased fear memory
The hippocampus is a critical brain region for fear memory regulation [30,31,32,33]. We hypothesized that CDYL in the hippocampus is essential in this process. To test this, we specifically ablated CDYL in excitatory neurons of the dorsal hippocampus by bilaterally injecting AAV-CaMKIIα-CRE-GFP into the dorsal CA1 region (dCA1) of Cdyl-loxP mice (Fig. 2A, B). This led to a significant reduction of CDYL levels in the hippocampus of mice receiving AAV-CRE compared to controls (Fig. 2C). Behavioral tests, including the open field and elevated zero maze tests, showed no differences between the two groups, indicating unaffected locomotion and anxiety-like behaviors (Fig. 2D, Supplementary Figure S3A-E). However, in the fear conditioning test, mice that received AAV-CRE exhibited significantly increased freezing times in the contextual fear memory test, with no differences observed in the cued fear memory test (Fig. 2E). These findings suggest that CDYL knockout in dorsal hippocampal excitatory neurons specifically enhances contextual fear memory without affecting locomotion or anxiety-like behaviors.

A Schematic of the conditional knockout of CDYL in hippocampal CaMKIIα+ neurons. B Injection site of the virus in the dCA1. Scale bar, 400 μm. C Western blot analysis of CDYL protein levels in the hippocampus. (Unpaired t-test, n = 4 GFP, 4 CRE). D Open field test, time spent in the center area during the first 5 min (left). (Unpaired t test. One outlier excluded, n = 10 WT, 12 cKO). Elevated zero maze, time spent in open arms during the test (right). (Unpaired t test, n = 11 WT, 12 cKO). E Fear conditioning test, contextual fear test (left) and cued fear test (right). The percentage of time spent freezing. (Mann-Whitney test. One outlier excluded, n = 10 GFP, 12 CRE for contextual; n = 11 GFP, 12 CRE for cued).
CDYL is phosphorylated at Ser147 by CDK5
CDYL plays a crucial role in the nervous system by regulating gene expression, yet its upstream regulatory mechanisms remain poorly understood. Previous studies indicated mismatched levels of CDYL protein and mRNA after neural activity [17], indicating posttranscriptional regulatory mechanisms of CDYL levels. By comparing the sequences of the CDYL protein among human, mouse and rat, we identified Ser147 and Ser162 of CDYL as two potential evolutionarily conserved phosphorylation sites of CDK5 (Supplementary Figure S4). CDK5 is involved in various neural processes, including synaptic plasticity and gene expression, aligning with CDYL’s functions [17, 22, 23, 34,35,36,37]. We hypothesized that CDK5 regulates CDYL in an activity-dependent manner.
To verify CDK5 interaction with CDYL, we constructed and analyzed GST-tagged CDYL deletion mutants, identifying CDYL 181-309 as a direct binding site for CDK5 (Fig. 3A). In vitro phosphorylation analysis revealed that CDYL 61-180, containing the predicted sites, was phosphorylated by CDK5 (Fig. 3B). Roscovitine, a CDK5 inhibitor, significantly reduced this phosphorylation. Mutation experiments showed that Ser147, but not Ser162, is the specific phosphorylation site for CDK5.

A In vitro pull-down assays examining interaction between CDK5 and CDYL deletion mutants. Poly-His tag fused to CDK5 and GST tag fused to CDYL mutants. Detection using anti-His for His6-CDK5 and anti-GST for GST-CDYL (red star). B In vitro 32P radioautography assays of CDYL mutants and CDYL mutants, showing phosphorylation. Top panel: 32P radiography; bottom panel: Coomassie blue staining. Ros, roscovitine treatment. C, D Protein levels of CDYL and p-CDYL in primary neurons after roscovitine treatment. (Unpaired t test, n = 3 DMSO, 3 Ros). E-H CDYL and p-CDYL levels in primary neurons post-KCl treatment. Duration of long-term (E, F) and short-term (G, H) KCl exposure as indicated. (One-way ANOVA with Dunnett’s test; all timepoints n = 3). I, J CDYL and p-CDYL levels in the mouse hippocampus 1 h (Unpaired t test, n = 4 No stress, 4 Fear conditioning) or 24 h (Unpaired t test, n = 5 No stress, 5 Fear conditioning) after fear conditioning (FC) training. All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ns not significance.
To explore this further in neurons, primary neurons were treated with roscovitine or DMSO, resulting in decreased phosphorylated CDYL (p-CDYL) levels (Fig. 3C). Upon 60 mM KCl-induced neural activity, CDYL levels decreased over 12 h, consistent with past findings, while p-CDYL levels dropped significantly within 1 h (Fig. 3E, F). Shortening the KCl stimulation showed stable CDYL levels at 1 h, but p-CDYL levels decreased within 30 min (Fig. 3G, H). In vivo, 1 h after fear conditioning training, p-CDYL in hippocampus showed a trend towards decrease compared to β-actin and a significant reduction of p-CDYL/CDYL proportion (Fig. 3I). Levels of CDYL and p-CDYL decreased significantly 24 h post-training (Fig. 3J). These data indicated that CDYL and p-CDYL in hippocampus underwent a dynamic process of degradation after fear conditioning training. However, levels of CDYL and p-CDYL in cortex were unaffected under the same experimental conditions (Supplementary Figure 5), highlighting the specific role of CDYL in hippocampus.
In summary, these results identify Ser147 of CDYL as a phosphorylation target of CDK5, elucidating the relationship between CDYL and p-CDYL levels in response to neural activity both in vitro, this mechanism may contribute to the down-regulation of CDYL and p-CDYL in hippocampus after fear conditioning training.
Phosphorylation at Ser147 promotes the ubiquitination and subsequent degradation of CDYL, a process mediated by TRIM32
Previous studies have shown that MG132, a proteasome inhibitor, prevents CDYL degradation induced by neural activity. When primary neurons were treated with MG132 for 6 h and KCl for 3 h, MG132 reversed the KCl-induced decrease in CDYL and p-CDYL levels (Fig. 4A), indicating that the ubiquitin–proteasome pathway mediates their degradation.

A Protein levels of CDYL and phosphorylated CDYL (p-CDYL) in neurons under various treatments. Analyzed by one-way ANOVA from three independent experiments with Tukey’s multiple comparisons test. B Immunoprecipitation analysis of CDYL ubiquitination in treated neurons. CDYL is fused to GFP, and ubiquitin to HA. Lysates were immunoprecipitated with GFP antibody and ubiquitination detected with anti-ubiquitin antibody. C Ubiquitination levels of CDYL in neurons transfected with GFP-tagged CDYL variants: wild-type, CDYL-S147A, or CDYL-S147D, alongside HA-tagged ubiquitin. D Ubiquitination levels of CDYL in neurons treated and transfected as follows: GFP-tagged CDYL, HA-tagged ubiquitin, vector, shTRIM32, or His-tagged TRIM32. Neurons were further treated with KCl in combination with DMSO or roscovitine. All data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ns not significance.
To explore how this pathway and phosphorylation affect CDYL levels, primary neurons transfected with GFP-CDYL and HA-ubiquitin were exposed to KCl and roscovitine. Immunoprecipitation with an anti-GFP antibody revealed that KCl increased CDYL ubiquitination, whereas roscovitine inhibited it, regardless of KCl presence (Fig. 4B). To assess Ser147 phosphorylation effects on CDYL ubiquitination, neurons were transfected with HA-ubiquitin and either wild-type GFP-CDYL or mutants mimicking Ser147 phosphorylation (S147D) and dephosphorylation (S147A). S147D enhanced CDYL ubiquitination, while S147A reduced it (Fig. 4C), demonstrating that CDK5-mediated Ser147 phosphorylation promotes CDYL ubiquitination.
Additionally, we previously established that TRIM32 ubiquitinates and degrades CDYL in resting neurons. To confirm if phosphorylation-enhanced ubiquitination under neural activity is TRIM32-dependent, we modulated TRIM32 expression in neurons treated with KCl and either DMSO or roscovitine. Knocking down TRIM32 inhibited CDYL ubiquitination during neural activity, and roscovitine suppressed this ubiquitination in TRIM32-overexpressing neurons (Fig. 4D). These findings suggest that CDYL phosphorylation by CDK5 facilitates TRIM32-mediated ubiquitination in response to neural activity.
Disruption of CDYL phosphorylation at Ser147 using an interfering peptide leads to a reduction in contextual fear memory in mice
To explore the role of Ser147 phosphorylation on CDYL, we developed an interfering peptide, Tat-S147, mimicking CDYL residues 140-153, fused with the cell-penetrating HIV-1 transactivator protein (Tat). A control peptide, Tat-S147A, included the same sequence but with Ser147 mutated to alanine (Fig. 5A). Both peptides were tagged with FITC for visualization (Fig. 5B). In vitro phosphorylation analysis revealed that Tat-S147 significantly reduced phosphorylation of the CDYL 61-180 mutant compared to Tat-S147A (Fig. 5C).

A, B Construction of interfering peptides: a peptide mimicking the sequence near Ser147 was synthesized and fused to the Tat cell-penetrating peptide (Tat-S147). A control peptide with Ser147 mutated to alanine (Tat-S147A) was similarly constructed (A). Both peptides were additionally tagged with FITC for visualization (B). C In vitro 32P radioautography assay: purified His-CDK5 and GST-CDYL 61-180 were incubated in the presence of 32P-labeled ATP and treated with TAT-S147A or TAT-S147 as indicated. D-G Localization and relative levels of FITC-tagged interfering peptide in the hippocampus: FITC signal intensity assessed at 1 h (D), 3 h (E), 6 h (F), and 12 h (G) post-injection. The distribution of FITC signal was matched to coronal section of mouse brain at Bregma -2.06 mm. Scale bars: 800 μm (left), 50 μm (right). H, I Protein levels of CDYL and p-CDYL in mouse hippocampus: post-treatment with Tat-S147 and Tat-S147A (Unpaired t-test, n = 3 each for Tat-S147A, Tat-S147). J Diagram of peptide injection via cannula and fear conditioning test. K Contextual fear conditioning test results: mice treated with Tat-S147 or Tat-S147A (Unpaired t-test, n = 13 for Tat-S147A, 11 for Tat-S147). L Location and signals of peptides three hours after being injected into the hippocampus: Scale bar: 700 μm. All data are represented as mean ± SEM. *p < 0.05.
In vivo, peptide persistence was assessed, showing strong fluorescence in the hippocampus 3 h post-injection, but minimal signal after 6 or 12 h (Fig. 5D–G). Mice injected with FITC-Tat-S147 exhibited increased CDYL levels and a downward trend in p-CDYL levels in hippocampal extracts after 3 h (Fig. 5H, I).
To test the peptide’s impact on fear memory, we administered it bilaterally to the hippocampus via cannula 3 h before fear conditioning training (Fig. 5J). Peptide injections were verified in the dCA1 region of the hippocampus (Fig. 5L). Mice treated with FITC-Tat-S147 showed significantly reduced freezing time in the contextual fear test, though cues were unaffected compared to controls (Fig. 5L). These findings indicate that the interfering peptide successfully inhibited CDYL phosphorylation at Ser147, thereby increasing CDYL levels and reducing contextual fear memory in mice.
Discussion
In this study, we elucidated the crucial role of CDYL in managing fear memory and detailed its regulatory mechanisms. Conditional knockout of CDYL in hippocampal excitatory neurons resulted in enhanced contextual fear memory in mice. Our findings revealed that CDYL interacts directly with, and is phosphorylated at Ser147 by, CDK5 in response to neural activity, boosting TRIM32-mediated CDYL ubiquitination. We developed an interfering peptide to block CDYL phosphorylation at Ser147, thereby elevating CDYL levels and reducing contextual fear memory, emphasizing the critical nature of Ser147 phosphorylation in CDYL regulation and its impact on fear memory (Fig. 6).

This model illustrates how CDYL contributes to fear memory formation. CDYL is phosphorylated by CDK5 in response to neural activity, which enhances TRIM32-mediated ubiquitination and degradation of CDYL in the hippocampus. This process contributes to the strengthening of fear memory. The introduction of an interfering peptide that specifically blocks CDYL phosphorylation at Ser147 effectively reduces fear memory in mice.
Previous studies indicated that CDYL targets various genes vital for neural functions and is linked to seizures and depression [19, 21]. However, its involvement in memory regulation was unclear. In this work, we position CDYL as an essential regulator of fear memory, consistent with its effects on neuronal plasticity and excitability. Our data showed transgenic KO of CDYL in the forebrain and hippocampus increased contextual memory, while cued fear memory increased only with forebrain CDYL ablation. This heightened phenotype from forebrain KO likely involves additional regions such as the cortex and thalamus. Although amygdala was widely recognized as an important brain region that regulates fear memory and CaMKIIα-cre-dependent knockout may have potential effect on amygdala, we observed that CDYL expression in amygdala was very limited and not influenced by CaMKIIα-cre-dependent knockout (data not shown), therefore amygdala was unlikely to involve in CDYL-regulated fear memory. Interestingly, tamoxifen-induced CDYL cKO mice showed unchanged spatial and working memory, which was assessed via Morris water maze and Y maze, indicating CDYL’s non-critical role in these memory types. The disparate impacts on fear and spatial memory underline CDYL’s complex role in memory regulation, suggesting different mechanisms for distinct types of memory, particularly those linked to hippocampal activities in contextual fear conditioning.
While many studies have explored CDYL’s downstream mechanisms, how CDYL itself is regulated hasn’t been fully grasped. We previously discovered TRIM32 as a promoter of CDYL degradation, a ubiquitin ligase essential for neuronal differentiation and associated with Alzheimer’s disease [26, 38,39,40]. This study clarifies CDYL’s regulation in response to neural activity, highlighting that its phosphorylation at Ser147 by CDK5 facilitates TRIM32 ubiquitination and degradation of CDYL. CDK5 is integral to numerous neuronal processes and known to enter the nucleus post-neuronal depolarization to regulate gene expression [22, 23, 34,35,36,37]. Our findings affirm that Ser147 is a critical CDK5 phosphorylation site on CDYL, with activity-driven CDYL degradation reliant on the ubiquitin–proteasome pathway. CDK5-mediated Ser147 phosphorylation enhances CDYL ubiquitination, as shown by TRIM32 response to neural activity. Furthermore, inhibiting CDK5 halted CDYL’s increased ubiquitination under TRIM32 overexpression, reinforcing the phosphorylation’s role. Our research highlights a novel mechanism for converting extracellular stimuli into transcriptional programs crucial for lasting neuronal changes during memory processes.
The significance of Ser147 phosphorylation for CDYL regulation and fear memory is further evidenced by the interfering peptide developed to obstruct Ser147 phosphorylation. Injections into the dCA1 led to diminished contextual fear memory without affecting cued fear memory, showcasing CDYL’s distinct involvement in different fear memory types. Our results underscore the pivotal role of Ser147 phosphorylation in CDYL and fear memory regulation, suggesting potential intervention points in CDYL or its Ser147 phosphorylation for fear memory modulation. However, other kinases could also potentially phosphorylate CDYL at Ser147. Generally, various modifications of epigenetic elements can affect their roles, necessitating further research to comprehensively understand CDYL’s regulation [41, 42].
This study provides multifaceted evidence of CDYL’s specific role in regulating fear memory. We demonstrated that CDYL undergoes phosphorylation and ubiquitination following neural activity, mediated by CDK5 and TRIM32, respectively. Furthermore, we developed an interfering peptide targeting CDYL’s regulatory mechanisms to influence fear memory. Our findings offer new insights into the mechanisms underlying fear memory and detail the molecular processes governing CDYL regulation, suggesting CDYL as a promising target for modulating fear memory.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. The plasmids that were used in this study are available from the corresponding author upon request.
References
Izquierdo I, Furini CR, Myskiw JC. Fear memory. Physiol Rev. 2016;96:695–750.
Raber J, Arzy S, Bertolus JB, Depue B, Haas HE, Hofmann SG, et al. Current understanding of fear learning and memory in humans and animal models and the value of a linguistic approach for analyzing fear learning and memory in humans. Neurosci Biobehav Rev. 2019;105:136–77.
Flores A, Fullana MA, Soriano-Mas C, Andero R. Lost in translation: how to upgrade fear memory research. Mol Psychiatry. 2018;23:2122–32.
Shalev A, Liberzon I, Marmar C. Post-traumatic stress disorder. N Engl J Med. 2017;376:2459–69.
Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–87.
Uddin LQ. Cognitive and behavioural flexibility: neural mechanisms and clinical considerations. Nat Rev Neurosci. 2021;22:167–79.
Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–86.
Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–24.
Lisman J, Cooper K, Sehgal M, Silva AJ. Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat Neurosci. 2018;21:309–14.
Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–52.
Kwapis JL, Wood MA. Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci. 2014;37:706–20.
Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102–10.
Campbell RR, Wood MA. How the epigenome integrates information and reshapes the synapse. Nat Rev Neurosci. 2019;20:133–47.
Caron C, Pivot-Pajot C, van Grunsven LA, Col E, Lestrat C, Rousseaux S, et al. Cdyl: a new transcriptional co-repressor. EMBO Rep. 2003;4:877–82.
Lahn BT, Page DC. Retroposition of autosomal mRNA yielded testis-specific gene family on human Y chromosome. Nat Genet. 1999;21:429–33.
Liu S, Yu H, Liu Y, Liu X, Zhang Y, Bu C, et al. Chromodomain protein CDYL acts as a Crotonyl-CoA hydratase to regulate histone crotonylation and spermatogenesis. Mol Cell. 2017;67:853–66 e855.
Qi C, Liu S, Qin R, Zhang Y, Wang G, Shang Y, et al. Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH2. J Neurosci. 2014;34:4494–508.
Zhang Y, Yang X, Gui B, Xie G, Zhang D, Shang Y, et al. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J Biol Chem. 2011;286:42414–25.
Qin R, Cao S, Lyu T, Qi C, Zhang W, Wang Y. CDYL deficiency disrupts neuronal migration and increases susceptibility to epilepsy. Cell Rep. 2017;18:380–90.
Liu Y, Lai S, Ma W, Ke W, Zhang C, Liu S, et al. CDYL suppresses epileptogenesis in mice through repression of axonal Nav1.6 sodium channel expression. Nat Commun. 2017;8:355.
Liu Y, Li M, Fan M, Song Y, Yu H, Zhi X, et al. Chromodomain Y-like protein-mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry. 2019;85:635–49.
Liang Z, Ye T, Zhou X, Lai KO, Fu AK, Ip NY. Cdk5 regulates activity-dependent gene expression and dendrite development. J Neurosci. 2015;35:15127–34.
Kawauchi T. Cdk5 regulates multiple cellular events in neural development, function and disease. Dev Growth Differ. 2014;56:335–48.
Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–91.
Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18.
Liu L, Liu TT, Xie GG, Zhu XQ, Wang Y. Ubiquitin ligase TRIM32 promotes dendrite arborization by mediating degradation of the epigenetic factor CDYL. FASEB J. 2022;36:e22087.
Veschi S, De Lellis L, Florio R, Lanuti P, Massucci A, Tinari N, et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J Exp Clin Cancer Res. 2018;37:236.
Li Y, Jia Y, Wang D, Zhuang X, Li Y, Guo C, et al. Programmed cell death 4 as an endogenous suppressor of BDNF translation is involved in stress-induced depression. Mol Psychiatry. 2021;26:2316–33.
Huang S, Zheng C, Xie G, Song Z, Wang P, Bai Y, et al. FAM19A5/TAFA5, a novel neurokine, plays a crucial role in depressive-like and spatial memory-related behaviors in mice. Mol Psychiatry. 2021;26:2363–79.
Bouton ME, Maren S, McNally GP. Behavioral and neurobiological mechanisms of pavlovian and instrumental extinction learning. Physiol Rev. 2021;101:611–81.
Maddox SA, Hartmann J, Ross RA, Ressler KJ. Deconstructing the gestalt: mechanisms of fear, threat, and trauma memory encoding. Neuron. 2019;102:60–74.
Sharma V, Sood R, Khlaifia A, Eslamizade MJ, Hung TY, Lou D, et al. eIF2alpha controls memory consolidation via excitatory and somatostatin neurons. Nature. 2020;586:412–6.
Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–99.
Brito V, Giralt A, Masana M, Royes A, Espina M, Sieiro E, et al. Cyclin-Dependent Kinase 5 dysfunction contributes to depressive-like behaviors in Huntington’s disease by altering the DARPP-32 phosphorylation status in the nucleus accumbens. Biol Psychiatry. 2019;86:196–207.
Zhou J, Chow HM, Liu Y, Wu D, Shi M, Li J, et al. Cyclin-Dependent Kinase 5-Dependent BAG3 degradation modulates synaptic protein turnover. Biol Psychiatry. 2020;87:756–69.
Plattner F, Hayashi K, Hernandez A, Benavides DR, Tassin TC, Tan C, et al. The role of ventral striatal cAMP signaling in stress-induced behaviors. Nat Neurosci. 2015;18:1094–1100.
Lai KO, Wong AS, Cheung MC, Xu P, Liang Z, Lok KC, et al. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15:1506–15.
Yokota T, Mishra M, Akatsu H, Tani Y, Miyauchi T, Yamamoto T, et al. Brain site-specific gene expression analysis in Alzheimer’s disease patients. Eur J Clin Invest. 2006;36:820–30.
Sato T, Okumura F, Kano S, Kondo T, Ariga T, Hatakeyama S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J Cell Sci. 2011;124:3492–502.
Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–25.
Oliveira AM, Hemstedt TJ, Freitag HE, Bading H. Dnmt3a2: a hub for enhancing cognitive functions. Mol Psychiatry. 2016;21:1130–6.
Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. 2013;110:2647–52.
Acknowledgements
This work was supported by grant from the Ministry of Science and Technology of China (2021ZD0203204 to Y.W.) and grant from the National Natural Science Foundation of China (32030052 to Y.W.).
Author information
Authors and Affiliations
Contributions
Conceptualization, N.Y.L., G.G.X., T.J.L. and Y.W.; Methodology, N.Y.L. G.G.X., and Z.W.H.; Investigation, N.Y.L., G.G.X., Z.W.H., and T.J.L.; Writing – Original Draft, N.Y.L. and G.G.X.; Writing – Review & Editing, N.Y.L., Z.W.H., G.G.X., R.Q., L.C. and Y.W.; Funding Acquisition, Y.W.; Resources, N.Y.L., Z.W.H., G.G.X., T.J.L. and Y.W.; Supervision, Y.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lyu, NY., Xie, GG., Hu, ZW. et al. Activity-dependent phosphorylation of CDYL by CDK5 regulates fear memory in mice. Transl Psychiatry 15, 334 (2025). https://doi.org/10.1038/s41398-025-03568-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03568-0
