
Abstract
Beyond its well-known role in orofacial recurrent infections, HSV-1 has garnered significant attention in neuroscience for contrasting reasons. On one hand, it has been found to be involved in neurodegenerative processes; on the other, it may represent a versatile platform for gene therapy of brain diseases, due to its large genome that enables the delivery of sizable or multiple genes. These opposite features underscore the importance of understanding HSV-1 interactions with neural tissues in view of its employment as a gene therapy platform. We recently developed a new generation of highly defective backbones that proved very efficient and safe after direct injection in the brain parenchyma. Here we aimed at probing in depth the safety of viral batches that lack obvious unwanted (specifically, fusogenic) activities during production and, therefore, may escape negative selection. We employed whole-genome sequencing, electrophysiology, and viral engineering to compare different viral batches. We identified mutations (in particular A to I at position 549 in the UL27 gene) that confer fusogenic capacity to the envelop glycoprotein gB, inducing a hyperexcitable phenotype in transduced neurons. Such syncytial variants should be identified and avoided for any application of HSV-1 vectors implicating their direct injection in the nervous system.
Similar content being viewed by others
Introduction
Herpes simplex virus type 1 (HSV-1) is a DNA virus with a neuronal tropism and a high worldwide prevalence. HSV-1 can have both a lytic (productive) and a latent (dormant) stage of infection in cells. HSV-1 DNA can persist for life in a latent state in sensory (mostly trigeminal) and central nervous system (CNS) neurons [1]. While the most typical manifestation of HSV-1 infection occurs in the face or mouth, where it results in small groups of blisters, HSV-1 is also the leading cause of sporadic viral encephalitis in adults, often presenting with acute, necrotizing inflammation of the temporal lobes [2]. In addition, HSV-1 is a predominant cause of acute retinal necrosis [3]. Its lifelong presence in the CNS may also be associated with neurodegenerative diseases, like Alzheimer’s disease [4,5,6,7,8,9]. HSV-1 infection may lead to impairment of synaptic function and autophagic processes, as well as alterations in intracellular calcium dynamics [10] and mitochondrial activity [11]. Moreover, syncytial strains of HSV-1 infecting rat sensory neuronal cultures can cause fusion and electrical coupling between cells, leading to uncontrolled spike frequency and synchronous, spontaneous network activity [12].
Beyond its implications in disease, HSV-1 has garnered significant attention in neuroscience as a potentially versatile platform for the delivery of therapeutic genes [13]. Gene therapy offers the possibility of specifically transferring therapeutic genes into diseased cells even in a highly heterogeneous tissue like the brain, and is therefore viewed as a promising alternative to currently available treatments of neurological disorders [14]. In this respect, the most developed approaches are based on viral vectors, in particular those derived from adeno-associated virus (AAV) or lentivirus (LV). Indeed, an AAV-based gene therapy is already available clinically for the treatment of spinal muscular dystrophy [15]. However, one important hurdle for the application of AAV or LV gene therapies to other neurological diseases is their limited cargo capacity. In many instances, an essential requirement for an effective therapeutic approach is the delivery of large genes or complex multigene cassettes. Therefore, the development of vectors able to accommodate large payloads is crucial and, in this respect, HSV-1 represents an optimal option [16].
Multiple HSV-1-based vector types have been developed: replication-competent, replication-defective, and amplicons [17]. All these platforms have been obtained by modifying the viral genome, which consists of a linear molecule of double-stranded DNA, about 152 kb long. While this extraordinarily large genome offers the opportunity to transfer incomparably greater amounts of DNA than AAV or LV (theoretically, up to 40-50 kb with replication-defective, up to almost 150 kb with amplicon vectors), genetic engineering and manufacturing of HSV-1 vectors remains challenging [17, 18]. Thus, their use for neurological diseases is not yet common. However, HSV-1-based vectors are already clinically tested in other areas, e.g., a replication-defective HSV-1 vector, beremagene geperpavec, has been approved in the US for the treatment of wounds [19], and replication-competent HSV-1 vectors proved successful for oncolytic therapies [20].
Considering the toxicity of wild-type HSV-1 infection in vivo in the CNS, modeling and characterization of the impact of HSV-derived vectors on cellular physiology is essential. In the present study, we tackled this issue by using the most recently developed vector backbone, namely the Joint deleted No Immediate 8 (J∆NI8) vector [17, 21]. This vector is deleted for the joint region and for the ICP0, ICP4 and ICP27 immediate-early (IE) genes. The ICP22 IE gene is converted to early-expression kinetics, such that no HSV IE gene gets expressed. In addition, the virion host shutoff gene (vhs, UL41) is also deleted, and bacterial sequences are removed [22]. After stereotactic injection in the brain, this vector backbone proved very safe (absence of detectable toxicity or inflammation) and ensured robust and long-lasting transgene expression [23]. Even if effective and reproducible protocols have been developed for the purification and storage of HSV vectors [18, 24], we noticed in the phase of clonal isolation that a few vector clones can display propensity to cause fusion of the transduced cells. These vector clones are usually negatively selected for further monoclonal expansion.
Here, we explored the effects on neuronal physiology of some of these variants. We provide evidence that infection by replication-defective HSV-1-based vectors with mutations in UL27, the gene encoding for glycoprotein B, recapitulates many of the negative effects observed with wild-type HSV-1, namely alterations in basal electrophysiological properties of neurons and calcium homeostasis that ultimately led to hyperexcitability. These findings provide novel insights for monitoring and controlling genetic variations in HSV-1-based gene therapy vectors.
Materials and methods
Materials
Cell culture media and reagents, if not otherwise stated, were from ThermoFisher Scientific (Carlsbad, CA, USA). Plates and flasks were from Nalgene Nunc (ThermoFisher Scientific). Petri dishes were from Falcon BD (Franklin Lakes, NJ, USA). Chemicals were from Tocris (Bristol, UK) or Merck-Sigma (Darmstadt, Germany).
Antibodies
Primary antibodies used for immunocytochemistry were: mouse anti-β-III-Tubulin - (Biolegend #801201, previously Covance #MMS-435P, dilution 1:1000) and mouse anti-Glial fibrillary acidic protein (GFAP) – (Sigma; G3893, dilution 1:250). Secondary antibody: Alexa Fluor 594 Goat anti-mouse (Invitrogen; A-11005, dilution of 1:250).
Cell lines
U2OS-ICP4/ICP27 complementing cell line has been generated (and kindly donated) by Yoshitaka Miyagawa through lentiviral transduction of the ICP4 and ICP27 HSV-1 genes into U2OS cells, allowing the stable expression of the transgene following infection. The ICP4 product is stably expressed but ICP27 must be induced by the viral function VP16. All these cells were maintained at 37 °C with 5% CO2 and grown in Dulbecco’s Modified Minimum Essential Medium (DMEM, Lonza) supplemented with 10% Fetal Bovine Serum (Gibco, Thermofisher), 2 mM L-glutamine (Gibco, Thermofisher), 100 U/mL Penicillin (Merck, Millipore), and 100 μg/ml Streptomycin (Merck, Millipore). Cells were routinely tested for mycoplasma contamination.
Primary culture of rat hippocampal neurons
The Institutional Animal Care and Use Committee of the San Raffaele Scientific Institute approved the animal use procedures. Primary cultures of hippocampal neurons were prepared according to [25] from 2 to 3-day-old Sprague–Dawley rats. Briefly, after brain removal from the skull, and quick subdivision of hippocampi into small pieces, the tissue was incubated into Hank’s solution containing 3.5 mg/mL trypsin type IX and 0.5 mg/mL DNase type IV for 5 min. The pieces were then mechanically dissociated in Hank’s solution supplemented with 12 mM MgSO4 and 0.5 mg/mL DNase IV. After centrifugation, cells were plated onto poly-ornithine coated coverslips and maintained in Neurobasal medium (ThermoFisher) supplemented with B27, 2 mM glutamax and 5 μM 1-β-D-cytosine-arabinofuranoside (Ara-C). Cultures were maintained at 37°C in a 5% CO2 humidified incubator.
Viral titration in plaques forming units (p.f.u/ml)
Titration of viral supernatant by plaque assay was performed on 48-well plates of U2OS-ICP4/ICP27 cells [22] at a density of 120,000 cells per well to achieve 80% confluency at the time of infection. Cells were infected with serial 10-fold dilutions of viral supernatant in 120 μl of DMEM (1% P/S, serum-free) media and incubated at 37 °C in 5% CO2 for approximately 3 hours. Subsequently, 100 μl of DMEM (1% P/S, serum-free) per well was added and the cells were incubated at 33 °C with 5% CO2 until lysis plaques formed (3-4 days). Plaque counts were performed by light or fluorescence microscopy and the result expressed as p.f.u./ml (plaque forming units per ml of viral preparation).
Vector engineering
The gB D285N and A549T mutations were introduced by scarless Red recombination, as described [22]. The necessary pBAD-IsceI and pEPkan-S2 plasmids were kindly provided by Nikolaus Osterrieder (Free University of Berlin, Berlin, Germany). The pgB1:D285N/A549T-kan plasmid described previously [21] was used as a template for amplification with primers gB-HA F (seq: GTTCCACCGGTACGGGACGACGGTAAACTGCATCGTCGAGGAGGTGGACG) and gB-HA R (seq: GGAGACGGCCATCACGTCGCCGAGCATCCGCGCGCTCACCCGCCGGCCCA), and the product was recombined with the native gB gene of KOS-37 BAC, followed by I-SceI-enhanced deletion of the aphAI gene in pBAD-I-sceI plasmid-transformed bacteria. Recombinants were confirmed by field inversion gel electrophoresis of restriction enzyme digested BAC DNAs and DNA sequencing across the gB genomic region.
Virus growth curves
Cells were plated at a density of 4 × 104 cell/well in 48-well plates (Corning) and infected the following day at MOI of 0.0005 genome copies (gc)/cell; MOI was determined by the average cell count of a monolayer of cells at the time of infection. Supernatants were collected every 24 h for up to 6 dpi. DNA was collected using the DNeasy blood and tissue kit (Qiagen) and total genome copies per sample were determined by real-time quantitative (q)PCR as previously described [22].
High-scale viral production
Viruses were propagated on U2OS-ICP4/ICP27 cells plated in T182 tissue culture flasks. In order to get high titer stocks, about 20 T182 flasks per virus have been used. Twenty-four hours before infection, U2OS-ICP4/ICP27 were plated as a 50% confluent monolayer in order to have about 90–100% confluent cells the day after. The amount of virus for infection was established by calibrating the multiplicity of infection (MOI, between 0.01 and 0.05), in serum-free media; the infected cells were incubated at 33 °C in 5% CO2.
Four to five days after infection, supernatant was collected and separated from cellular debris by centrifugation at 3000 revolutions per minute (rpm) for 10 min, followed by filtration through a 0.8 μm Versapor filtering membrane (PALL Corporation). The virus was then concentrated by 19500 rpm centrifugation for 45’ and the viral pellet was resuspended in about 250 μl Phosphate-Buffered-Saline (PBS) 1X supplemented with 10% glycerol by slow overnight rotation at 4 °C. Table 1 summarizes the different viral batches used in this manuscript, the experiment in which they have been used, and the mutations detected in the gB gene.
DNA extraction from J∆NI8 viral preparation
Twenty uL of pure viral preparation were used to extract DNA for sequencing. DNA was extracted with DNeasy Blood & Tissue Kits (Qiagen) according to manufacturer’s instruction. Purified DNA was delivered to the Center for Omics Science at San Raffaele Scientific Institute.
Classification analysis and variant calling
The classification analysis was performed with Kraken2 (version 2.1.1). Kraken is a taxonomic classification tool that uses exact k-mer matches to find the lowest common ancestor of a given sequence (Kraken2 is the newest version of Kraken). To classify the genomics reads, we use the standard Kraken database built with the “kraken-built” command.
The sequencing reads were demultiplexed with Illumina bcl2fastq (version 2.20) and aligned to the HSV viral reference, KOS strain (https://www.ncbi.nlm.nih.gov/nuccore/JQ673480.1) by bwa-mem. SNP and small indel variants were detected by Freebayes and filtered with bcftools, according to the snippy (https://github.com/tseemann/snippy) pipeline (version 4.6).
The key parameters we use in this analysis are (1) the minimum number of reads covering a site to be considered (set to 10); (2) the minimum proportion of those reads that must differ from the reference (set to 0.9); (3) the minimum VCF variant call “quality” used to filter variants (set to 100).
J∆NI8 transduction of primary neurons
To prevent osmotic shock at the time of infection, 1/3 (~400 μL) of the culture media of primary neurons, seeded on glass slides in a 24-well plate, was collected every three days and replaced with fresh medium. The conditioned medium was stored at 4 °C and used to resuspend the viral preparation. Eight days after seeding, the entire medium was collected and approximately 300 μL/well of conditioned medium were supplemented with the proper amount of viral volume (MOI 1; 200.000 neurons per slide) and added to the cells. Neurons were incubated for 1 h at 37 °C in 5% CO2. Subsequently, cells were washed with 300 μL of conditioned medium, and cells were then cultured with 2/3 of conditioned media and 1/3 of fresh one.
Immunofluorescence
Neurons were grown on glass coverslips in vitro, fixed with 4% paraformaldehyde solution, containing 4% sucrose in PBS. Primary and secondary antibodies were incubated for 1 h at RT, diluted in blocking solution (1% normal goat serum/0.1% Triton in PBS). Afterwards, coverslips were incubated with DAPI for 5’ at RT, deepened in water, and mounted on microscope slides with Fluorescence Mounting Medium (DAKO, Agilent).
Imaging and analysis
Confocal images have been acquired in the Advanced Light and Electron Microscopy BioImaging Center (Alembic). Images were acquired in an 8-bit format at a resolution of 1024 × 1024 pixels. For all images, we used a Leica SP8 confocal system equipped with an Acousto-Optical Beam Splitter which allows the selection of a specific frequency range for every fluorophore.
Image composition
Image compositions were made with Adobe Illustrator cc 2017 (Adobe System, San Jose, CA, USA). Drawings were created with BioRender.com under a license granted to M.S.
Electrophysiology
Primary culture slides were submerged in a recording chamber mounted on the stage of an upright BX51WI microscope (Olympus, Japan) equipped with differential interference contrast optics (DIC). Slides were continuously perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 25 NaHCO3, 1 MgCl2 and 11 D- glucose saturated with 95% O2, 5% CO2 (pH 7.3) flowing at a rate of 2–3 ml/min at room temperature. Whole-cell patch-clamp recordings were performed using glass pipettes filled with a solution containing the following (in mM): 30 KH2PO4, 100 KCl, 2 MgCl2, 10 NaCl, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, 0.02 Na-GTP, (pH 7.2, adjusted with KOH; tip resistance: 6–8 MΩ). For the dye-coupling experiments internal solution was modified by additional loading of 0.1% of Neuriobiotin-488 (LabVector).
All recordings were performed using a MultiClamp 700B amplifier interfaced with a PC through a Digidata 1440 A (Molecular Devices, Sunnyvale, CA, USA). Data were acquired and analyzed using pClamp10 software (Molecular Devices). Voltage- and current-clamp traces were sampled at a frequency of 30 kHz and low-pass filtered at 2 kHz.
Analysis of bursting activity was calculated by measuring the median instantaneous frequency with Clampfit Software (Molecular Devices). Instantaneous frequency is a measure of the inter-event intervals, which is the time between peaks, i.e. the time from the previous peak to the current peak. The final measure is calculated by first converting each inter-event interval into a frequency, and then averaging these frequencies together.
Calcium measurement with Fura-2 calcium indicator
Ca2+ measurements were performed in Krebs-Ringer-Hepes buffer (KRH - 5 mM KCl, 125 mM NaCl, 2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 6 mM glucose and 20 mM Hepes, pH 7.4).
Cells were loaded with 4 µM fura-2 acetoxymethyl ester (Thermo Fisher Scientific) for 40 min at 37°C. After dye loading, cells were washed twice with KRH and kept in the same buffer for the entire duration of the experiments.
The single-cell experiments were performed with a video imaging setup consisting of an Axioskope 2 microscope (Zeiss, Oberkochen, Germany) and a Polychrome IV (Till Photonics, GmbH, Martinsried, Germany) light source. Fluorescence images were collected by a cooled CCD video camera (PCO Computer Optics GmbH, Kelheim, Germany). The ‘Vision’ software (Till Photonics) was used to control the acquisition protocol and to perform data analysis [26].
Statistical analysis
Statistical analysis was performed with GraphPad Prism software version 9.0 (GraphPad Software Inc., La Jolla, CA, USA). Results are given as dot plots or histograms with mean ± SEM as specified in the figure legends. The number of experiments and p-value are indicated in the figure legends, while biological replicates are shown as individual points in graphs. To assess for normal distribution of the data we applied the normality test (D’Agostino-Pearson) and equal variance test (Brown-Forsythe). Generally, in the case of normally distributed data, statistical significance was evaluated (with 95% confidence intervals) by one-way ANOVA followed by Dunnett post hoc test (for multiple comparisons against a single reference group), Newman–Keuls or Tukey’s post hoc test (for multiple comparisons between groups), two-tailed Student t-test (for comparisons between two average values); for samples with non-normal distributions, the nonparametric Mann–Whitney U test (for significant differences between two experimental groups) and the Kruskal–Wallis one-way analysis of variance followed by Dunn’s post hoc test (for the analysis of multiple experimental groups) were used. A value of P < 0.05 was considered statistically significant. Sample size was calculated using G Power software (version 3.1), based on effect sizes calculated from our preliminary experiments, with a power of 0.8 and alpha= 0.05.
Results
Some J∆NI8 vector batches can alter neuronal physiology
In order to exploit HSV-1 replication-defective vectors as gene therapy tools, we recently generated a JΔNI8 recombinant vector (J∆NI8-1) expressing the red fluorescent protein tdTomato (tdT) under the Ubiquitin C (UbC) promoter and used rat primary hippocampal neuronal cultures to characterize the capability of these vectors to induce expression of transgenes and to assess the effects of viral transduction on neuronal physiology (Fig. 1A, B). Primary hippocampal neurons were transduced at 8 days in vitro (DIV) and analyzed between 10 and 12 DIV (Fig. 1B). At a multiplicity of infection (MOI) of 1, J∆NI8-1 vectors displayed a high efficiency of transduction in neurons, as shown by the expression of tdTomato in β-III tubulin+ cells (Fig. 1C, D). No obvious cytotoxic effect or morphological change was observed in these neurons. Although HSV-1 has a natural tropism for neurons, it has been reported that it can also infect glial cells [27]. Under our experimental conditions, however, no red fluorescent signal was detected in the few GFAP+ astrocytes that are present in the preparation (Fig. S1).

A Schematic representation of the J∆NI8-1 recombinant vector genome, in which the Ubiquitin (Ub) promoter driving tdTomato (tdT) expression has been inserted in the ICP4 locus. B Schematic representation of the experimental set-up used for primary neuron transduction with HSV-1-based vectors. C Representative immunofluorescence images showing rat primary hippocampal neurons transduced with J∆NI8-1 at a multiplicity of infection of 1. Representative immunofluorescence images for β-III-Tubulin (C, green); DAPI (C’, blue), tdTomato (C”, red). D Histogram summarizing the average percentage of tdT-positive cells over DAPI in rat primary hippocampal neurons transduced with J∆NI8-1 at a multiplicity of infection of 1, E, F Voltage response of control and J∆NI8-1 transduced neurons to suprathreshold depolarizing ramp currents. Sequential sweeps are represented in grey. The one with greater amplitude current injection is shown in black. Note that J∆NI8-1 transduced neurons require injection of greater amplitude currents to elicit action potentials. G, H Voltage-clamp recordings of control and J∆NI8-1 transfected neurons (Vhold = −60 mV). I–L Histograms summarizing average Resting Membrane Potential, Input Resistance, spontaneous event Frequency, and Instantaneous Frequency of control and J∆NI8-1 transfected neurons. Data are shown as Mean ± SEM. Every dot represents a single cell. I CTRL = −55.7 ± 1.6 mV, J∆NI8-1 = -66 ± 1.4 mV. J CTRL = 296 ± 30 MΩ, J∆NI8-1 = 63 ± 7 MΩ. K CTRL = 0.28 ± 0.21 Hz, J∆NI8-1 = 2.47 ± 0.44 Hz. L CTRL = 0.78 ± 0.47 Hz, J∆NI8-1 = 14.42 ± 2.07 Hz. Statistical significance in (I–L) was calculated using an unpaired Student’s t-test. ***p < 0.001. M Examples of spontaneous activity in control (upper trace) and J∆NI8-1 transduced neurons (lower trace). Note the bursting activity of transduced cells which arises from a highly hyperpolarized resting membrane potential compared to controls. N, O Histograms summarizing average AHP amplitude and AP amplitude of control and J∆NI8-1 transduced neurons. Data are shown as Mean ± SEM. Every dot represents a single cell. N CTRL = −5,1 ± 1,3 mV; J∆NI8-1 = +9,6 ± 1.0 mV. O CTRL = +73 ± 4 mV; J∆NI8-1 = +68 ± 2 mV. Statistical significance in (N, O) was calculated using an unpaired Student’s t-test. ***p < 0.001. P Average voltage traces (V), and their first derivative (V’), of the spontaneous action potential of control (black) and transduced (red) neurons. Below, phase plot highlighting the slope trajectory during the entire AP cycle. Data are the mean of three cells per condition.
Because it has been previously reported that certain strains of wild-type HSV-1 are capable of causing electrophysiological alterations in dorsal root neurons [12], we decided to check our vectors in this respect. Surprisingly, we found variability between different batches of vectors, with some able to induce severe electrophysiological alterations. We characterized these defects in detail in a particular batch that was clearly displaying them (J∆NI8-1), as specified in Table 1. Transduced, tdTomato-positive neurons were able to fire action potentials (AP) when suprathreshold depolarizing ramp currents were applied (Fig. 1E, F). However, the amount of current needed to evoke an AP was greatly higher in transduced compared to control cells. Voltage clamp recordings of transduced cells indicated the presence of rhythmic single inward currents or bursts of currents, coherent with the APs and the excitatory post-synaptic potentials (EPSPs) observed in current clamp experiments (Fig. 1G, H). We analyzed the basic electrophysiological properties of J∆NI8-1 transduced or control neurons and observed a strong reduction in input resistance (Rin) and a more negative resting membrane potential (RMP) in transduced neurons (Fig. 1I, J). Furthermore, we quantified the spontaneous firing of neurons by measuring both action potential frequency and instantaneous frequency, which has been used as a measure of bursting activity. Both these measurements indicated a significantly higher firing frequency of transduced cells compared to controls (Fig. 1K, L). Moreover, J∆NI8-1 transduced neurons were spontaneously firing AP (Fig. 1M), which were replaced by EPSP-like depolarizing events when they were too hyperpolarized to reach the AP threshold.
Finally, the spontaneous action potential of transduced and control neurons displayed an increase of the afterhyperpolarization (AHP) amplitude (i.e., the difference in voltage between RMP and the voltage after the AP peak; Fig. 1N). No difference in the AP amplitude was instead detected between control and transduced cells (Fig. 1O). In addition, the AP shape at threshold values for the initiation of the AP showed a sharper inflection as represented by the higher slope value at the peak in transduced neurons compared to controls (Fig. 1P).
Monoclonal expansion of J∆NI8 vectors can result in genetic mutations
Because HSV naturally mutates at the rate of about 1 nucleotide change every 3000 bp [28], we hypothesized that, in spite of the absence of detectable signs of cytotoxicity or morphological defects, mutations might have occurred in J∆NI8-1 that cause the electrophysiological alterations described above. We therefore extracted the genomic DNA and performed whole-genome sequencing of J∆NI8-1 to clarify whether the mutations were already present in the original Bacterial Artificial Chromosome (BAC) or arose during vector production. We also included a second independent clone derived from the same BAC, here referred to as J∆NI8-Syn, which exhibited a clear syncytial phenotype during vector production and served as proof that mutations can accumulate over production cycles. Because of its obvious syncytial phenotype, this clone would be routinely discarded.
These viral batches (Fig. 2A) were sequenced using the short-reads Illumina NGS technology (read length = 250 bp) and the wild-type HSV-1-KOS strain (NCBI Accession N. JQ673480) was used as a reference for detecting variations. Before proceeding with the variant calling analysis, we performed a quality control assay to check the correct mapping of the sequenced DNA on the HSV-1 DNA. We used Kraken2, a taxonomic classification tool used to find the lowest common ancestor of a given sequence, to conduct a read classification analysis and check where the reads map (Fig. 2B). The percentage of reads mapping to the host cell genome (human) reached over 80%, while the percentage of reads mapping on the viral genome spanned between 4 and 22%. These data were expected, because eukaryotic DNA contamination is known to be present in the final viral batch, likely resulting from the viral replication process during vector production, even if cellular DNA contamination can be reduced by benzonase treatment. A representation in Integrative Genome Viewer (IGV) of the mapped reads against the reference genome is presented in Fig. 2C.

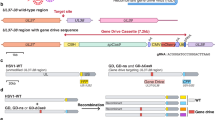
A The upper part represents a schematic representation of the HSV-1 genome (KOS strain) and J∆NI8 recombinant vector in which a Gene of Interest (GOI, e.g., tdTomato) can be inserted through Gateway cloning. The bottom part illustrates the two clones of J∆NI8 viral vectors. B Kraken 2 classifications summary, showing the percentage of reads falling into the top 5 taxa across different ranks. C IGV snapshot of BigWig file showing the DNA coverage across the HSV-1 Kos genome strain. Each sample auto-scaled individually. D Variant calling summary with different detection sensitivity parameters. E Identified mutation in the UL27 gene (glycoprotein B; gB) in the monoclonal expanded J∆NI8 batches. Mutations common to the two batches are boxed in pink, those identified in a single one (J∆NI8-Syn) are in violet.
We then filtered analysis only on the DNA aligned with the HSV viral reference. We performed a variant calling analysis on filtered reads consistently mapping on the viral HSV genome. Most of the variants we detected were single nucleotide polymorphisms (SNPs; Fig. 2D). To estimate the sensitivity of the variant detection we applied the “minfrac” parameter (minimum proportion of reads which differ from the reference) in a range of 0.85 to 0.95. The number of detected variables slightly decreases by increasing the minfrac parameter. Ultimately, a minfrac value of 0.90 was chosen for further evaluation.
Nearly all detected SNPs either fell in intergenic regions or caused silent synonymous mutations. Of note, most mutations were observed in both batches, suggesting that they may have been present in the parental plasmid employed to prepare the vector. We thus sequenced the parental BAC employed in viral vector production. Sequencing results showed that some SNPs were indeed already present in the ancestral BAC, while others accumulated during the early stages of production (Supplemental Table S1). Missense mutations observed in the two viral batches affected the genes UL27, UL46, US7 and US10. UL46 codes for a tegument protein that can anchor the viral particle to the cell membrane [29], but its function is believed to be more closely associated with inhibition of the immune response [30]. US7 encodes for glycoprotein gI, which, along with gE, is involved in the cell-to-cell spreading of the virus [31]. US10 encodes a protein associated with the capsid/tegument, whose function is still unclear [32].
We decided to focus on UL27, because this gene, which encodes glycoprotein B (gB), one of the most important fusogenic proteins of the Herpesviridae family [33], hosted multiple missense mutations, namely D285N, A315T, A549I (Fig. 2E). The D285N mutation was expected as we previously described and introduced it in the parental plasmid [21]. The A549I (Alanine converted into Isoleucine) mutation was on a residue previously described to increase gB transduction efficiency when Alanine was converted into Threonine (A549T). The A315T mutation was not reported previously. J∆NI8-Syn, which showed a marked syncytial phenotype during propagation, displayed two additional mutations in the gB sequence (R858H and S869N), that arose during vector production (Fig. 2E, Supplemental Table S1). The presence of these mutations is coherent with the observed phenotype, because R858H was already annotated as a fusogenic mutation, and S869N localizes in a domain relevant for the fusogenic activity of gB [34, 35].
It is known that mutations in the gB coding sequence may lead to a range of different phenotypes. On one extreme, loss of function mutations may cause the inability to fuse lipid membranes [36]; on the other, gain of function mutations may lead to the generation of aggressively fusogenic strains [34]. During vector production, viral clones carrying gain of function mutations acquire a growth advantage over those carrying wild-type gB. As mentioned above, such highly mutated clones (like J∆NI8-Syn) are generally identified and excluded, as multinucleated competent cells are observed during the production process. Table 1 summarizes the different viral batches we used in the manuscript and their respective mutations in the gB gene.
To confirm that the mutations (D285N, A315T, A549I) in the gB gene of J∆NI8-1 were already present in the BAC and did not arise during vector production, DNA was extracted from multiple BACs produced over the past few years, and the gB gene was sequenced. As expected, these mutations were consistently detected across all samples, indicating that they were present in the original BACs. Nevertheless, we cannot rule out that the genomic instability of the viral DNA during vector propagation, even if not leading to a recognizable syncytial phenotype, may still result in subtle yet potentially harmful syncytial phenotypes. Thus, we decided to investigate this possibility in our J∆NI8-1 vector.
J∆NI8-1 vector-induced electrophysiological alterations are associated with fusogenic events
Despite the absence of obvious morphological changes, we asked if the J∆NI8-1 vector carrying, among others, the gB NTI variant (D285N, A315T, A549I) might be able to trigger subtle events of hemifusion and pore formation among neuronal cells. To this aim, a green fluorescent dye (Neurobiotin) was loaded in the patch clamp pipette, enabling its diffusion in the cytoplasm of the patched cell during voltage-clamp recordings. The green dye filled the patched neuron first and then labeled neighboring tdTomato-positive (but not tdTomato-negative) neurons (Fig. 3A, B). Figure 3C shows an example of the bursting activity of the patched cell recorded throughout the whole dye-filling experiment in voltage-clamp configuration. These findings suggest that, coherent with our hypothesis, J∆NI8-1 may induce fusion between neuronal branches and thereby allow diffusion of the fluorescent dye. To further test this hypothesis, we also performed paired patch-clamp recordings of tdTomato-positive neurons. Pairs of tdTomato positive cells displayed robust electrical coupling (n = 5/5 pairs recorded) with a mean coupling coefficient of 0.65 ± 0.11 (Fig. 3D). These results suggest that viral vector-induced membrane fusion can produce electrical coupling between hippocampal neurons, supporting and increasing spontaneous activity.

A Representative sequential images showing green fluorescence monitoring at different time points during voltage-clamp recording of a J∆NI8-1 transduced neuron using a Neurobiotin-488 loaded pipette. Note the dye diffusing through the cytoplasm of the patched cell; asterisks represent the non-specific labeling of dead cells prior to patching. As shown in (B) Neurobiotin-488 can diffuse in neighboring tdTomato+ cells (arrows), but not within tdTomato- neurons (indicated by arrowheads in the bright field image in (A)). C Voltage clamp recording during the dye loading of the patched cell. Insert represents a higher magnification of the bursting activity. D Dual patch clamp recording of two J∆NI8-1 neurons demonstrating the electrical coupling of putatively fused cells. Note that only one neuron (blue) has been injected with suprathreshold depolarizing current steps, while the second (green) displays a coherent and synchronous response.
Blocking voltage-gated calcium channels reduces hyperactivity of J∆NI8-1 transduced neurons
To characterize alterations in ion homeostasis that may occur in J∆NI8-1 transduced neurons, intracellular calcium levels were measured with Fura-2 dye. Following the same protocol used for patch-clamp experiments, primary cultures were transduced at 8 DIV and calcium was measured at 11-12 DIV under resting conditions (Fig. 4A). Neurons transduced with J∆NI8-1 displayed increased intracellular Ca2+ basal levels compared to control cells (Fig. 4B). Moreover, patch clamp experiments showed that treatments with either TTX (a blocker of voltage-gated sodium channels) or NNC-55-0396 (a blocker of T-type calcium channels) were able to reduce the spiking frequency of transduced neurons (Fig. 4C–G). Conversely, bath application of NBQX (an AMPA receptor blocker) did not stop nor reduce the higher spiking activity induced by viral vector transduction (Fig. 4E–G). These data further support the idea that the predominant driving force of the increased firing frequency induced by the mutated vector is electrical coupling, rather than synaptically-mediated hyperexcitation and synchronization.

A Pseudocolored images representative of calcium level measured with the Fura-2 Ca2+ dye in control and J∆NI8-1 transduced neurons at resting state. B Histograms summarizing mean intracellular calcium levels under resting conditions. Data are shown as Median with interquartile range; each dot represents a single cell. CTRL = 190, n = 258 cells of 5 independent experiments, J∆NI8-1 = 269, n = 202 cells of 5 independent experiments. Statistical significance in (B) was calculated using the Mann-Whitney U test. ***p < 0.001. C Voltage clamp recording of transduced neurons during bath application of the voltage-gated sodium channels blocker TTX (1 µM). D–E Voltage clamp recording of transduced neurons during bath application of either the T-type calcium channel blocker NNC 55-0396 (10 µM, D) or the AMPA receptor antagonist, NBQX (5 µM, E). F, G Histograms summarizing average bursting frequency and instantaneous frequency. Data are shown as Mean ± SEM. F ACSF = 5.61 ± 1.13 Hz (black), NBQX = 6.52 ± 2.46 Hz (orange), NNC = 0.20 ± 0.12 Hz (light blue) and TTX = 0 Hz (magenta). G ACSF = 17.67 ± 3.35 Hz (black), NBQX = 16.13 ± 1.95 Hz (orange), NNC = 6.63 ± 3.5 Hz (light blue) and TTX = 0 Hz (magenta). Statistical significance in F and G was calculated using the Kruskal–Wallis one-way analysis of variance followed by the Dunn’s post hoc test. *p < 0.05; *** p < 0.001.
The A549I mutation in the gB gene is sufficient to increase the fusogenic properties of J∆NI8
As noted (Supplemental Table 1), the J∆NI8-1 vector consistently carries mutations in other genes besides UL27, like US7, which may also contribute to the fusogenic phenotype [31]. In addition, the consequences of gB NTI mutations on viral biological properties are essentially unknown. Therefore, we decided to test if the gB NTI mutations alone were sufficient to produce a fusogenic phenotype. We thus generated two new viral batches starting from a new J∆NI8 BAC, whose clones carry mutations only in gB. We named these batches J∆NI8_A549I and J∆NI8_A549T. In J∆NI8_A549I, we introduced our newly discovered mutations in the gB NTI gene (D285N, A315T, A549I); in J∆NI8_A549T, we introduced a gB NTT mutation (D285N, A315T, A549T), i.e., Alanine converted into Threonine in position 549, a previously characterized mutation [21] (Fig. 5J).

A–D Histograms summarizing average Resting Membrane Potential, Input Resistance, spontaneous event Frequency, and Instantaneous Frequency of neurons transduced with different viral batches at a multiplicity of infection of 1 (MOI = 1). Data are shown as Mean ± SEM. Every dot represents a single cell of 3 different biological replicates. A NT = -57 ± 1.3 mV, J∆NI8_A549I = −58 ± 1.5 mV, J∆NI8_A549T = -56 ± 1.9 mV, J∆NI8-1 = -65 ± 2.7 mV. B NT = 368 ± 28 MΩ, J∆NI8_A549I = 441 ± 31 MΩ, J∆NI8_A549T = 382 ± 27 MΩ, J∆NI8-1 = 97 ± 11 MΩ. C NT = 0.1 ± 0.04 Hz, J∆NI8_A549I = 0.54 ± 0.2 Hz, J∆NI8_A549T = 0.19 ± 0.07 Hz, J∆NI8-1 = 3.1 ± 0.5 Hz. D NT = 0.4 ± 0.27 Hz, J∆NI8_A549I = 1.79 ± 0.61 Hz, J∆NI8_A549T = 0.28 ± 0.1 Hz, J∆NI8-1 = 17.57 ± 2.78 Hz. Statistical significance in (A–D) was calculated using one-way ANOVA followed by Tukey’s post-test for multiple comparisons. *p < 0.05; **p < 0.01, ***p < 0.001. E–G. Histograms summarizing average Resting Membrane Potential, Input Resistance, spontaneous event Frequency, and Instantaneous Frequency of neurons transduced with vectors carrying either J∆NI8_A549I or J∆NI8_A549T at MOI 1 or 3. E NT = −53 ± 1.7 mV, J∆NI8_A549I (MOI 1) = −55 ± 3 mV, J∆NI8_A549I (MOI 3) = -64 ± 2 mV, J∆NI8_A549T (MOI 1)= −57 ± 2.6 mV, J∆NI8_A549T (MOI 3)= -58 ± 1.8 mV. F NT = 430 ± 54 MΩ, J∆NI8_A549I (MOI 1) = 231 ± 19 MΩ, J∆NI8_A549I (MOI 3) = 280 ± 27 MΩ, J∆NI8_A549T (MOI 1) = 424 ± 83 MΩ, J∆NI8_A549T (MOI 3) = 458 ± 52 MΩ. G: NT = 0.28 ± 0.11 Hz, J∆NI8_A549I (MOI 1) = 1.24 ± 0.3 Hz, J∆NI8_A549I (MOI 3) = 2.1 ± 0.5 Hz, J∆NI8_A549T (MOI 1) = 0.35 ± 0.15 Hz, J∆NI8_A549T (MOI 3) = 0.32 ± 0.08 Hz. H NT = 0.55 ± 0.27 Hz, J∆NI8_A549I (MOI 1) = 3.76 ± 1.11 Hz, J∆NI8_A549I (MOI 3) = 7.86 ± 1.7 Hz, J∆NI8_A549T (MOI 1) = 0.84 ± 0.56 Hz, J∆NI8_A549T (MOI 3) = 1.06 ± 0.31 Hz. Statistical significance in (E–G) was calculated using one-way ANOVA followed by Tukey’s post-test for multiple comparisons. *p < 0.05; **p < 0.01, ***p < 0.001. I Examples of spontaneous activity in control (black trace) and J∆NI8_A549I or J∆NI8_A549T transduced neurons at different MOI. J Schematic representation of the two gB variants analyzed.
Infection of U2OS-ICP4/27 at 0.0005 gc/cell resulted in comparable levels of viral DNA at all time points analyzed (Fig. S2), indicating that these mutations did not alter virus entry or spread. We analyzed the electrophysiological profiles of neurons untransduced or transduced with either J∆NI8_A549I, J∆NI8_A549T, or the fusogenic J∆NI8-1. Confirming the data described above, at MOI 1 J∆NI8-1 transduction reduced Rin and resulted in a more hyperpolarized RMP, while increasing both spiking frequency and instantaneous frequency (Fig. 5A–D). Conversely, both J∆NI8_A549I and J∆NI8_A549T did not display any statistically significant difference compared to control neurons in any of the analyzed parameters, even if a tendency to increased frequency was observed in neurons transduced with J∆NI8_A549I.
When using a higher viral particle load (MOI of 3), however, neurons transduced with J∆NI8_A549I (but not those transduced with J∆NI8_A549T) displayed a significantly more negative RMP and a clear tendency toward a reduced Rin (Fig. 5E, F), as well as an increased firing frequency and bursting activity (Fig. 5G–I). In other words, J∆NI8_A549I but not J∆NI8_A549T partially mimicked the effects of J∆NI8-1. Although these electrophysiological alterations were more prominent in the J∆NI8-1, these data argue that the gB NTI mutations, and specifically the conversion of Alanine into Isoleucine at residue 549 (A549I), are (is) sufficient to generate the fusogenic phenotype.
Discussion
The results from this study demonstrate that monoclonal expansion of J∆NI8 HSV-1 vectors can result in genetic mutations, some of which can significantly alter the functional properties of the vector, in particular its fusogenic potential. These findings align with the known genomic instability of herpesviruses during replication, underscoring the importance of rigorous quality control in vector production to ensure therapeutic safety.
Some batches of HSV-1 replication-defective vectors may alter neuronal physiology
Whole-cell patch-clamp experiments revealed several electrophysiological alterations in neurons transduced with some J∆NI8 vector batches that were not leading to obvious morphological alterations. We examined in detail the effects of one of these batches (J∆NI8-1), and observed increased spiking frequency and bursting activity, reduced Rin, a more hyperpolarized RMP, spontaneous spiking activity (single spike discharges or bursts of action potentials), and altered action potential properties (alterations in the AHP). These alterations are reminiscent of those reported in primary cultures of dissociated sensory neurons infected with syncytial HSV-1 strains, which are thought to be responsible for certain types of neuralgia [12]. Interestingly, despite the authors’ use of a wild-type HSV-1 replication-competent syncytial strain, no marked morphological changes were observed in neuronal cultures in their experimental conditions [12]. Similarly, in our hands neurons appeared viable and displayed no sign of cytotoxicity even at the latest time-point that we employed for patch-clamp experiments (i.e., 96 h after transduction). These data led us to hypothesize that, similar to syncytial HSV-1 strains, some J∆NI8 batches might exert fusogenic effects even if highly deficient and non-replicative. It is likely that the viral particles used to transduce neurons can fuse cells together, rather than induce de novo expression of gB. This hypothesis is supported by previous data on our replication-defective vectors, which show low, if not null, expression of viral genes [22]. Thus, we decided to focus our attention on potentially harmful mutations in the gB gene rather than the de novo expression of the protein.
Genetic mutations and their functional consequences
We then sequenced the DNA of two distinct batches of J∆NI8, one causing the electrophysiological alterations described above (J∆NI8-1) and another (that we named J∆NI8-Syn) that caused clearly detectable fusion in complementing cells during vector preparation and would therefore be routinely discarded. We identified several SNP mutations in these batches, in most of the cases synonymous or intergenic. However, we also identified missense mutations in the genes UL27, UL46, US7, and US10. Of particular interest were the missense mutations in UL27, because this gene codes for gB, one of the most important fusogenic proteins of the Herpesviridae family [33]. The R858H and S869N mutations in gB, observed only in the highly fusogenic J∆NI8-Syn, have established associations with increased membrane fusion activity [34, 35] and are therefore in line with the obvious phenotype of this vector batch. We hypothesized that the other mutations (D285N, A315T and A549I) that are also observed in the J∆NI8-1 batch may not only cause electrophysiological alterations but also favor the fusion of membranes between neurons. In fact, fusion may cause the spreading of excitation and, thereby, hyperexcitability.
We directly addressed this hypothesis in multiple manners. First, we performed a dye coupling experiment and found that Neurobiotin was able to diffuse from the patch pipette and label not only the patched cell but also neighboring tdTomato-positive neurons. Second, we performed dual cell patch-clamp experiments of tdTomato-positive neurons and found that J∆NI8-1 transduction of hippocampal neurons led to electrical coupling, facilitating an increased spontaneous activity. Taken together, these findings strongly support the view that transduced neurons were directly interconnected by membrane fusion.
Fusogenic events may explain the electrophysiological alterations observed in transduced neurons. One plausible interpretation of Rin reduction is an increased membrane surface. Based on the Ohm law, electrical resistance is given by:
where R is the electrical resistance, ρ is the resistivity of the material, L is the length of the material, and A is the cross-sectional area of the material through which the current passes. Because electrical resistance (R) is inversely proportional to the area (A), i.e., to the membrane surface, it is expected to decrease when cells fuse. In presence of a lower Rin, a stronger input would be necessary to trigger an AP in transduced neurons, but, at the same time, after the first AP, fused membranes would facilitate the spreading of AP between cells.
Other electrophysiological alterations cannot be explained directly by a fusogenic effect. For example, the hyperpolarized RMP may be interpreted as a homeostatic response of transduced neurons to increased rates of depolarizing inputs or as a direct consequence of viral envelope fusion with the cell membrane [37]. Therefore, we speculate that other mechanisms capable of modifying host cell biology might take place. For example, alterations in Ca2+ homeostasis have been reported to occur following HSV-1 infection of neuronal cultures [38,39,40]. Indeed, because synaptic activity, in particular excitatory glutamatergic transmission, leads to increases in intracellular calcium concentrations [Ca2+]i, we hypothesized that the hyperactivity of transduced neurons could increase the levels of [Ca2+]i. In Fura-2 experiments, in fact, hippocampal neurons transduced with the J∆NI8-1 vector displayed significantly higher [Ca2+]i compared to controls. In principle, prolonged increases of [Ca2+]i should be cytotoxic but, surprisingly, transduced neurons did not appear to be suffering.
In addition, we found that TTX completely blocked the excessive firing of J∆NI8-1 transduced neurons, indicating sodium dependence of this event. Interestingly, blocking T-type calcium channels mirrored the effect of TTX, suggesting a key role also for T-type calcium channels. Conversely, NBQX, a potent AMPA receptor antagonist, only partially blocked spontaneous spiking of transduced cells, indicating that this event is not dependent on excitatory synaptic activity, and further supporting the view that electrical coupling resulting from fusogenic events is the main driving force for the alterations that we detected.
Altogether, these results demonstrate that fusogenic events might arise after the transduction of neurons with the J∆NI8-1 vector, leading to increased firing frequency, alterations of basic membrane parameters, and increase in basal intracellular calcium levels.
Glycoprotein B plays a key role in membrane fusion processes
As noted, multiple genetic alterations were observed in the J∆NI8-1 vector. Thus, one final question was if any of those could be sufficient to cause the observed phenotypes. As described above, we hypothesize a key role of UL27, the gene coding for gB. Reports in the literature support the view that mutations in gB can enhance cell-cell fusion [41], while others demonstrate the impact of gB on neuronal physiology [37]. For instance, three different strains of replication-competent pseudorabies virus (PRV), which belong to the Herpesviridae family, exhibit distinct effects on neuronal physiology. These strains include the virulent Becker strain (PRV-151), an avirulent Becker strain that does not express gB (PRV-223 gB null), and the attenuated Bartha strain (PRV-152). PRV-151 caused several electrophysiological alterations (high firing frequency, spikelet-like events during hyperpolarization) and the formation of pores between neurons (as demonstrated by both dye and electrical coupling). Similar but delayed effects were observed with the attenuated virus PRV-152. Conversely, no alteration was observed with the gB null mutant PRV-233 [37].
We tested if the mutations that we observed in UL27 were sufficient to produce a fusogenic and hyperexcitable phenotype. Indeed, a newly generated vector batch in which gB NTI was engineered in an otherwise safe J∆NI8 backbone (J∆NI8_A549I; no mutations in other HSV-1 genes) recapitulated the electrophysiological alterations observed in spontaneously mutant batches. Another newly generated vector in which we engineered a gB carrying an NTT mutation, i.e., identical to the above but Alanine converted into Threonine in position 549 (J∆NI8_A549T), did not cause any electrophysiological alteration. Thus, the single most relevant mutation seems to be Alanine converted into Isoleucine at residue 549 (A549I). These results suggest that even subtle mutations in critical regions of gB can dramatically influence viral vector properties, emphasizing the need for a thorough genetic screening during vector development and production.
Implications for gene therapy
Gene therapy has enjoyed success in the past few years. Originally devoted to the treatment of rare monogenic diseases, it seems now to have reached a maturity level for broader approaches to treat diseases affecting millions of people worldwide. Both LV and AAV vectors have been already brought to clinical trials for treating CNS disorders [42,43,44,45]. However, while these vectors are proving effective for some applications, future gene therapies might require the delivery of large genes, complex multigene cassettes, or sophisticated genome editing approaches [46,47,48,49] that greatly exceed their payload capacity. Although several strategies either to miniaturize the expression cassette [50] or to split it in multiple vectors [51] are under development in many laboratories, it remains highly uncertain if these approaches will ultimately result in satisfactory efficacy.
In the past few years, our and other laboratories have made significant advancements in the field of viral vectors based on HSV-1 [23, 49, 52,53,54]. The most attractive feature of these vectors is that they can accommodate very large payloads, amplifying the potential impact of future gene therapy approaches. While last-generation backbones proved very safe in vivo, not expressing any viral protein after direct injection in the brain tissue [22], one prerequisite for applicability to human CNS diseases is the development of quality control measures that ensure the stability of the gene therapy product over multiple rounds of manufacturing.
The possible emergence during propagation of fusogenic mutations in gB that alter neuronal physiology has critical implications for the safety of HSV-1-based vectors, particularly in therapeutic contexts involving the CNS. Syncitial phenotypes have been described in HSV-infected culture, for example, in presence of mutations or truncations in glycoproteins (such as gB, gD, gH/gL, gE, gM, gK) involved in cell fusion [55]. Notably, the emergence of syncytial clones in replication-defective vectors (which, unlike wild-type HSV, have impaired growth) may be explained by the selective pressure for mutations that confer growth advantages. Mutations in the gB gene indeed enhance cell-to-cell spread and this may result in a growth advantage. Moreover, during vector production, there is a tendency to select clones that display strong reporter expression, as these often form more visible plaques. However, such clones may harbor syncytial mutations that become apparent only later. Since plaque selection is typically performed visually, this process is inherently operator-dependent and may not reliably exclude syncytial variants. Moreover, different mutations in the gB gene can confer varying degrees of syncytial activity, making these phenotypes difficult to detect and increasing the likelihood that syncytial clones persist and expand over the following passages.
To mitigate these risks, vector production protocols must incorporate stringent measures to identify and exclude clones harboring these deleterious mutations. High-throughput sequencing and functional assays, as performed in this study, represent essential tools for ensuring vector quality.
This study highlights the crucial need to monitor and control genetic variations in HSV-1–based vectors to ensure their safety for clinical applications. Although rare, DNA recombination may occur during BAC replication or repetitive cycles of cell infection during vector production.
One strategy to mitigate the risk of DNA recombination in specific genes could be to engineer cell lines suitable for viral production to stably express those genes. For instance, the insertion of UL27, providing gB in trans, could greatly reduce the risk of mutations during vector production. Ultimately, future efforts should focus on developing strategies to stabilize the viral genome.
Data availability
All data that support the key findings of this study are available within the article or from the corresponding author, upon reasonable request. This study includes genomic data accessible at NCBI SRA with the access code: PRJNA1305684.
References
Duarte LF, Farías MA, Álvarez DM, Bueno SM, Riedel CA, González PA. Herpes simplex virus type 1 infection of the central nervous system: insights into proposed interrelationships with neurodegenerative disorders. Front Cell Neurosci. 2019;13:46.
Zhu S, Viejo-Borbolla A. Pathogenesis and virulence of herpes simplex virus. Virulence. 2021;12:2670–702.
Van Gelder RN, Willig JL, Holland GN, Kaplan HJ. Herpes simplex virus type 2 as a cause of acute retinal necrosis syndrome in young patients. Ophthalmology. 2001;108:869–76.
Chauhan P, Begum MY, Narapureddy BR, Gupta S, Wadhwa K, Singh G et al. Unveiling the involvement of Herpes simplex virus-1 in Alzheimer’s disease: possible mechanisms and therapeutic implications. Mol Neurobiol. https://doi.org/10.1007/s12035-024-04535-4. 2024.
Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol. 1991;33:224–7.
Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349:241–4.
Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429:95–100.
Hyde VR, Zhou C, Fernandez JR, Chatterjee K, Ramakrishna P, Lin A, et al. Anti-herpetic tau preserves neurons via the cGAS-STING-TBK1 pathway in Alzheimer’s disease. Cell Rep. 2025;44:115109.
Álvarez G, Aldudo J, Alonso M, Santana S, Valdivieso F. Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J Neurosci Res. 2012;90:1020–9.
Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, et al. HSV-1 promotes Ca2+-mediated APP phosphorylation and Aβ accumulation in rat cortical neurons. Neurobiol Aging. 2011;32:2323.e13–2323.e26.
Zhao Y, Xu K, Shu F, Zhang F. Neurotropic virus infection and neurodegenerative diseases: Potential roles of autophagy pathway. CNS Neurosci Ther. 2024;30:e14548.
Mayer ML, James MH, Russell RJ, Kelly JS, Wise JCM, Pasternak CA. Spontaneous electrical activity induced by herpes virus infection in rat sensory neuron cultures. Brain Res. 1985;341:360–4.
Le Hars M, Joussain C, Jégu T, Epstein AL. Non-replicative herpes simplex virus genomic and amplicon vectors for gene therapy—an update. Gene Ther. 2024:1–11.
Bettegazzi B, Cattaneo S, Simonato M, Zucchini S, Soukupova M. Viral vector-based gene therapy for epilepsy: What does the future hold?. Mol Diagn Ther. 2024;28:5–13.
Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. 2021;29:464–88.
Nectow AR, Nestler EJ. Viral tools for neuroscience. Nat Rev Neurosci. 2020;21:669–81.
Artusi S, Miyagawa Y, Goins W, Cohen J, Glorioso J. Herpes Simplex virus vectors for gene transfer to the central nervous system. Diseases. 2018;6:74.
Kuroda S, Miyagawa Y, Sukegawa M, Tomono T, Yamamoto M, Adachi K, et al. Evaluation of parameters for efficient purification and long-term storage of herpes simplex virus-based vectors. Mol Ther Methods Clin Dev. 2022;26:132–43.
Dhillon S. Beremagene Geperpavec: first approval. Drugs. 2023;83:1131–5.
Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20:160–77.
Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, et al. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of Herpes simplex virus type 1 infection. J Virol. 2010;84:12200–9.
Miyagawa Y, Marino P, Verlengia G, Uchida H, Goins WF, Yokota S, et al. Herpes simplex viral-vector design for efficient transduction of nonneuronal cells without cytotoxicity. Proc Natl Acad Sci USA. 2015;112:E1632–E1641.
Verlengia G, Miyagawa Y, Ingusci S, Cohen JB, Simonato M, Glorioso JC. Engineered HSV vector achieves safe long-term transgene expression in the central nervous system. Sci Rep. 2017;7:1507.
Kuroda S, Miyagawa Y, Sato Y, Yamamoto M, Adachi K, Kinoh H, et al. Protocol optimization for the production of the non-cytotoxic JΔNI5 HSV vector deficient in expression of immediately early genes. Mol Ther Methods Clin Dev. 2020;17:612–21.
Bettegazzi B, Sebastian Monasor L, Bellani S, Codazzi F, Restelli LM, Colombo AV, et al. Casein Kinase 2 dependent phosphorylation of eIF4B regulates BACE1 expression in Alzheimer’s disease. Cell Death Dis. 2021;12:1–14.
Rosato C, Bettegazzi B, Intagliata P, Balbontin Arenas M, Zacchetti D, Lanati A, et al. Redox and calcium alterations of a müller cell line exposed to diabetic retinopathy-like environment. Front Cell Neurosci. 2022;16:862325.
Hensel N, Raker V, Förthmann B, Detering NT, Kubinski S, Buch A, et al. HSV-1 triggers paracrine fibroblast growth factor response from cortical brain cells via immediate-early protein ICP0. J Neuroinflammation. 2019;16:248.
Drake JW, Hwang CBC. On the mutation rate of Herpes simplex virus type 1. Genetics. 2005;170:969–70.
Murphy MA, Bucks MA, O’Regan KJ, Courtney RJ. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology. 2008;376:279–89.
You H, Zheng S, Huang Z, Lin Y, Shen Q, Zheng C Herpes Simplex Virus 1 Tegument Protein UL46 Inhibits TANK-Binding Kinase 1-Mediated Signaling. mBio 2019;10: https://doi.org/10.1128/mbio.00919-19.
Johnson DC, Webb M, Wisner TW, Brunetti C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol. 2001;75:821–33.
Nishiyama Y, Yamada H, Yamashita Y, Daikoku T, Tsurumi T, Jiang YM. The product of the US10 gene of herpes simplex virus type 1 is a capsid/tegument-associated phosphoprotein which copurifies with the nuclear matrix. J Gen Virol. 1997;78:2923–31.
Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci USA. 2007;104:2903–8.
Okubo Y, Uchida H, Wakata A, Suzuki T, Shibata T, Ikeda H, et al. Syncytial mutations do not impair the specificity of entry and spread of a glycoprotein D receptor-retargeted herpes simplex virus. J Virol. 2016;90:11096–105.
Ambrosini AE, Enquist LW. Cell-fusion events induced by α-herpesviruses. Future Virol. 2015;10:185–200.
Connolly SA, Longnecker R. Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry. J Virol. 2012;86:6386–93.
McCarthy KM, Tank DW, Enquist LW. Pseudorabies virus infection alters neuronal activity and connectivity in vitro. PLoS Pathog. 2009;5:e1000640.
Mayer ML. Selective block of inward but not outward rectification in rat sensory neurones infected with herpes simplex virus. J Physiol. 1986;375:327–38.
Zhang Q, Hsia S-C, Martin-Caraballo M. Regulation of T-type Ca2+ channel expression by herpes simplex virus-1 infection in sensory-like ND7 cells. J Neurovirol. 2017;23:657–70.
Koyuncu OO, Enquist LW, Engel EA. Invasion of the nervous system. Current Issues Mol Biol. 2021:1–62.
Fan Q, Longnecker R, Connolly SA. Herpes simplex virus glycoprotein B mutations define structural sites in domain I, the membrane proximal region, and the cytodomain that regulate entry. J Virol. 2021;95: https://doi.org/10.1128/jvi.01050-21.
Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, et al. Epilepsy gene therapy using an engineered potassium channel. J Neurosci. 2019;39:3159–69.
Szczygieł JA, Danielsen KI, Melin E, Rosenkranz SH, Pankratova S, Ericsson A, et al. Gene Therapy Vector Encoding Neuropeptide Y and Its Receptor Y2 for Future Treatment of Epilepsy: Preclinical Data in Rats. Front Mol Neurosci. 2020;13:232.
Jensen TL, Gøtzsche CR, Woldbye DPD. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021;14. https://www.frontiersin.org/article/10.3389/fnmol.2021.695937 (accessed 14 Jan2022).
Jablonka S, Hennlein L, Sendtner M. Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders. Neurol Res Pr. 2022;4:2.
Terzic B, Melin E, Fagergren P, Dobry D, Cattaneo S, Giupponi I, et al. AAV-mediated gene therapy for focal epilepsy by expressing neuropeptide Y and Y2 receptor in rodent and non-human primate hippocampus. Mol Therapy. 2025;33:4239–58.
Colasante G, Lignani G, Brusco S, Berardino CD, Carpenter J, Giannelli S, et al. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther. 2020;28:235–53.
Lubroth P, Colasante G, Lignani G. In vivo genome editing therapeutic approaches for neurological disorders: Where are we in the translational pipeline?. Front Neurosci. 2021;15:632522.
Ingusci S, Cattaneo S, Verlengia G, Zucchini S, Simonato M. A matter of genes: the hurdles of gene therapy for epilepsy. Epilepsy Curr. 2019;19:38–43.
Xu X, Chemparathy A, Zeng L, Kempton HR, Shang S, Nakamura M, et al. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol Cell. 2021;81:4333–4345.e4.
Tornabene P, Trapani I, Minopoli R, Centrulo M, Lupo M, Simone S, et al. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci Transl Med. 2019. https://doi.org/10.1126/scitranslmed.aav4523.
Ingusci S, Verlengia G, Soukupova M, Zucchini S, Simonato M. Gene therapy tools for brain diseases. Front Pharmacol. 2019;10:724.
Soukupová M, Zucchini S, Trempat P, Ingusci S, Perrier-Biollay C, Barbieri M, et al. Improvement of HSV-1 based amplicon vectors for a safe and long-lasting gene therapy in non-replicating cells. Mol Ther Methods Clin Dev. 2021;21:399–412.
Miyagawa Y, Verlengia G, Reinhart B, Han F, Uchida H, Zucchini S, et al. Deletion of the virion host shut-off gene enhances neuronal-selective transgene expression from an HSV vector lacking functional IE genes. Mol Ther - Methods Clin Dev. 2017;6:79–90.
Weed DJ, Nicola AV. Herpes simplex virus membrane fusion. Adv Anat Embryol Cell Biol. 2017;223:29–47.
Acknowledgements
The authors thank the ALEMBIC imaging facility at the San Raffaele Scientific Institute.
Author information
Authors and Affiliations
Contributions
S.C., B.B., G.V., and M.S. designed the research. S.I., M.M., and J.C.G. contributed to the conception and design. A.S.T., M.J.M., and S.Z. contributed to the acquisition and analysis of data. S.C., B.B., G.V., F.C., and S.I. prepared the vectors and performed the experiments. S.C., B.B., and M.S. wrote the paper with the input of all co-authors.
Corresponding author
Ethics declarations
Competing interests
J.C.G. is founder and CSO of Adolore Biotherapeutics, Inc., Senior Advisor of Replay Bio, and consultant of EG247. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. This work was supported by a grant from the European Community (FP7- HEALTH Project 602102 [EPITARGET]).
Ethical approval
The Institutional Animal Care and Use Committee of the San Raffaele Scientific Institute approved the animal use procedures. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cattaneo, S., Bettegazzi, B., Ingusci, S. et al. Genetic mutations in HSV-1 replication-defective vectors: Implications for their safety in gene therapy applications. Gene Ther 32, 581–593 (2025). https://doi.org/10.1038/s41434-025-00566-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41434-025-00566-1
