
Abstract
The CRISPR-Cas9 system has frequently been used for genome editing in Streptomyces; however, cytotoxicity, caused by off-target cleavage, limits its application. In this study, we implement innovative modification to Cas9, strategically addressing challenges encountered during gene manipulation using Cas9 within strains possessing high GC content genome. The Cas9-BD, a modified Cas9 with the addition of polyaspartate to its N- and C-termini, is developed with decreased off-target binding and cytotoxicity compared with wild-type Cas9. Cas9-BD and similarly modified dCas9-BD have been successfully employed for simultaneous biosynthetic gene cluster (BGC) refactoring, multiple BGC deletions, or multiplexed gene expression modulations in Streptomyces. We also demonstrate improved secondary metabolite production using multiplexed genome editing with multiple single guide RNA libraries in several Streptomyces strains. Cas9-BD is also used to capture large BGCs using a developed in vivo cloning method. The modified CRISPR-Cas9 system is successfully applied to many Streptomyces sp., providing versatile and efficient genome editing tools for strain engineering of actinomycetes with high GC content genome.
Similar content being viewed by others


Introduction
Streptomyces is a Gram-positive bacterial species that produces valuable secondary metabolites, such as anticancer agents, antibiotics, and immunosuppressive compounds1,2,3,4. Considering that many of the metabolites are clinically active, Streptomyces has been recognized as an industrially important species. Efforts have been made to improve the production of valuable secondary metabolites from various Streptomyces species by focusing on biosynthetic gene cluster (BGC) refactoring to enhance gene expression or through host strain engineering to increase the supply of precursor metabolites4,5,6. In addition, to resolve obstacles such as slow growth rates or complicated regulation of native Streptomyces strains, BGCs have been captured and expressed in a heterologous strain that is easier to cultivate and engineer7,8,9,10. However, genetic engineering of Streptomyces is still a challenge because of the high GC content genome, resulting in slow progress in the manipulation of BGCs to produce secondary metabolites8.
The clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein (Cas) system is a leading genome editing tool used in prokaryotic and eukaryotic cells11,12,13. High substrate specificity, recognizing 20 bp of the target site and the protospacer adjacent motif (PAM) sequence in the genome, makes the CRISPR-Cas system the most powerful genome-editing tool. Moreover, it has resolved several difficulties in Streptomyces genome engineering, such as introducing mutation(s) or BGC refactoring14,15. In addition, dCas9-mediated tools such as CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) for gene expression control, or in vitro CRISPR-Cas tools such as BGC capturing are other examples used in engineering Streptomyces16,17,18. Despite its high specificity, the CRISPR-Cas9 system has a drawback in that it cleaves at sites with sequence similar to the target, called off-target cleavage19. This causes unexpected problems during genome engineering, including the generation of undesired mutations or detrimental effects on cell growth20,21. This problem is particularly fatal to Streptomyces genome editing because similar sequences are commonly present in the modular enzymes of BGC with similar functions, generating many off-target cleavages22,23. Additionally, the widely-used genome editing enzyme SpCas9 from Streptococcus pyogenes recognizes the ‘-NGG’ as a PAM sequence, which is often found in Streptomyces genome due to its high GC content. To address the issue of off-target cleavage in Streptomyces, previous studies have reduced Cas9 activity by either decreasing expression levels or expressing cognate inhibitors24,25,26,27. However, these methods reduce the efficiency of genome editing. Another strategy to reduce off-target cleavage efficiency is to use Cas12a from Francisella novicida U112 (FnCas12a), which recognizes the ‘-NTTT sequence’ as a PAM sequence. FnCas12a has been successfully demonstrated to capture large BGCs and manipulate genomic DNA7,28. Although FnCas12a helps increase target specificity for BGC capture or genome editing, it presents difficulties in selecting target sites because its PAM sequence ‘-NTTT’ is not widely distributed in the Streptomyces genome, attributed to the high GC content of the latter. Base editor systems (BEST), such as adenine base editors (ABE) or cytosine base editors (CBE), are another method that has been used to reduce the toxic effects of Cas9 by avoiding double-strand breaks. These methods have been used to successfully manipulate multiple sites of the genome in Streptomyces simultaneously29, but a limitation in editing target sequences of promoters or genes exists because they can only be used for A-to-G or C-to-T switches in short regions near the binding site of a single guide RNA (sgRNA).
In this study, the objective is to enhance the utility of Cas9 by introducing innovative modification to reduce off-target cleavage efficiency, overcoming previous challenges in utilizing Cas9 within various research. To achieve this, Cas9 originating from S. pyogenes is modified to reduce off-target cleavage efficiency and is utilized to engineer the genome of Streptomyces. Specifically, polyaspartate (DDDDD) is added to the N- and/or C-termini of Cas9 using a flexible glycine-serine linker. The off-target cleavage efficiency of the modified Cas9, named Cas9-BD, is dramatically decreased compared to that of wild-type Cas9, whereas it does not affect the on-target cleavage efficiency. This reduced off-target cleavage decreases the cellular toxicity when Cas9 is expressed in Streptomyces at high levels, allowing for versatile applications of Cas9-BD and dCas9-BD in genome engineering. Multiplexed promoter refactoring, simultaneous BGC deletion, and the inhibition of multiple metabolic pathway genes have been successfully demonstrated using modified proteins in various species of Streptomyces. In addition, we develop an in vivo BGC capturing method for the Streptomyces genome with large sizes ( > 100 kb) using Cas9-BD. The tools developed in this study are expected to accelerate the genetic manipulation and strain development of various species genomes with high GC content.
Results
Modification of Cas9 to reduce off-target cleavage by polyaspartate linking
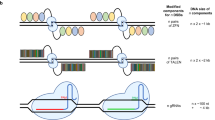
To reduce off-target cleavage during editing a genome with a high GC content, five aspartate residues were linked to the C- and/or N-termini of the Cas9 protein from S. pyogenes using a flexible glycine-serine linker (Fig. 1a)30. The addition of acidic residues to the Cas9 protein was expected to reduce the charge-charge interaction between the binding site of Cas9, which has several basic side chains, and the phosphate backbone of the target DNA (Supplementary Fig. 1a, b). Owing to the differences in the structure and affinity of Cas9 protein for on- and off-target DNA31,32, the linked polyaspartate can inhibit the binding of Cas9 to off-target DNA by interfering with basic residues without inhibiting the strong binding of Cas9 to on-target DNA. Modified Cas9 with polyaspartate at the N- and/or C-termini, or both ends of the polypeptide, was developed (Figs. 1b, Supplementary Fig. 1b) and named Cas9-ND, -CD, and -BD, respectively. Each constructed Cas9 was expressed and purified in Escherichia coli and their secondary structural conformations were examined using circular dichroism spectroscopy (Supplementary Fig. 1c, d). The analysis indicated that the addition of polyaspartate to Cas9 did not affect protein folding or its ability to bind with sgRNA. The modified Cas9s mixed with sgRNA and 1.3 kb linear double-stranded DNAs for the in vitro reaction31,33. DNAs containing various sequences were tested for the on- and off-target effects compatible with sgRNA or PAM (Figs. 1c, Supplementary Data 1, Supplementary Fig. 2). The results clearly showed that the modified Cas9s, especially Cas9-ND and Cas9-BD, exhibited significantly reduced cleavage efficiencies of off-target DNAs to sgRNA compared to wild-type Cas9 (Figs. 1d, Supplementary Fig. 3). Furthermore, the modified Cas9s showed reduced off-target cleavage of DNA with a non-PAM sequence, especially those containing -NGA or -NGT. For DNA with -NGC as a non-PAM sequence, all the Cas9s demonstrated very low cleavage efficiencies. Interestingly, despite a notable reduction in off-target cleavage efficiency, the on-target DNA cleavage efficiency of the modified Cas9s was decreased by less than 20% compared to wild-type Cas9. Modifications of Cas9s with negatively charged residues at the terminals significantly reduced off-target cleavage, whereas they did not trigger a significant change in cleavage efficiencies of the on-target DNA.

a Cas9 modification scheme and proposed differential binding effects on the on- and off-target DNAs. b Representation of the wild-type and various modified Cas9s. c The 1.3 kb double stranded DNAs showing the specified target sequences used for in vitro cleavage reactions. d Cleavage efficiencies of various modified Cas9s on target DNAs. The efficiency of reactions was measured after 30 min of reaction and visualized using agarose gel electrophoresis. All reactions were performed in triplicates (n = 3). The mean was plotted with error bar representing standard deviation. P values were determined by unpaired two-tailed Student’s t-test based on cleavage efficiency of each protein with on-target linear DNA (ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Source data are provided as a Source Data file.
Cas9-BD for improved genome editing efficiency in Streptomyces species
To verify the in vivo effect of modified Cas9 on the genome editing of Streptomyces sp., the pCRISPomyces-2 plasmid containing codon-optimized Cas9 with a rpsL promoter34 was used. The Cas9 gene in the plasmid was modified to express Cas9-BD instead of Cas9, and the resulting plasmid was named pCRISPomyces-2BD. We hypothesized that a reduction in the off-target cleavage tendency of Cas9-BD would decrease cytotoxicity in Streptomyces strains. When pCRISPomyces-2BD was transformed into S. coelicolor M1146, growth of several colonies was observed (Figs. 2, Supplementary Fig. 4b). In contrast, the strain conjugated with pCRISPomyces-2 formed much less colonies. This result is in accordance with a previous report that expressed Cas9 using a strong promoter, such as rpsLp, caused significant cytotoxicity in Streptomyces strains25. To confirm the on-target cleavage ability of Cas9-BD, sgRNA with a sequence targeting the desferrioxamine B synthesis gene in the S. coelicolor M1146 genome was inserted into a pCRISPomyces-2BD plasmid (pCRISPomyces-2BD-dB in Supplementary Data 2). As expected, when the pCRISPomyces-2BD-dB plasmid was transformed into S. coelicolor M1146, no exconjugant was observed owing to genome cleavage. In this experiment, Cas9-BD showed substantially lower toxicity than wild-type Cas9, possibly attributable to reduced off-target cleavage in the genome while maintaining high on-target cleavage efficiency, as expected from the in vitro experiments. For quantitative comparison of the editing efficiency of Cas9 and Cas9-BD in Streptomyces, a deletion of matAB genes, responsible for forming aggregates into a pellet, was carried out in the genome of S. coelicolor M1146 (Supplementary Fig. 4a). Using Cas9-BD resulted in a 77-fold increase in the number of exconjugants compared to Cas9, along with high editing efficiency, 98.1 ± 1.40% (Supplementary Fig. 4b–d). Whole-genome sequencing of the edited strains revealed a higher incidence of off-target mutations with wild-type Cas9 (Supplementary Fig. 4e, f, Supplementary Data 3). Since double-strand breaks of the genome lead to cell death in this strain35, it is likely that off-target cleavages are significantly more frequent when using Cas9.

S. coelicolor M1146 colonies on agar plates after conjugation with various types of plasmids to compare toxicity between Cas9 and Cas9-BD.
Given the low toxicity of Cas9-BD, genome editing experiments were designed to improve secondary metabolite production in Streptomyces sp. First, a silent BGC for the production of oviedomycin, a compound with antitumor activity, was targeted for activation in S. antibioticus NRRL 323836. In a previous report, promoters of the ovmOI, ovmOII, and ovmF genes in the BGC were activated to produce oviedomycin36 (Fig. 3a). First, Cas9 and Cas9-BD were used to replace the promoter of ovmOI gene with a strong one (Supplementary Fig. 5a). Experiments with Cas9-BD successfully produced exconjugants in S. antibioticus NRRL 3238 with high editing efficiency (Supplementary Fig. 5c, e), while no conjugates were observed with Cas9. Additionally, most of the edited strains demonstrated increased oviedomycin production compared to the wild-type S. antibioticus NRRL 3228 strain during flask cultivation in MS media (Supplementary Fig. 5g).

a The design of the promoter for the exchange of genes in the oviedomycin BGC in S. antibioticus NRRL 3238. The target genes, ovmOI, ovmOII, and ovmF, are colored red, purple, and green, respectively. The inserted promoters, ermE*p, kasO*p, and sp44p, are also represented. b Results of flask cultivation of the wild-type and engineered S. antibioticus NRRL 3238. Engineered strains with activated oviedomycin BGCs exhibited a dark brown color. c Exchanged promoters of each strain are listed. Blank fields in the table indicate that the promoter was not exchanged. d The design of the promoter for the exchange of genes in rapamycin BGC in S. rapamycinicus NRRL 5491. The genes of polyketide synthase, rapA, rapB, and rapC, are colored green, and the BGC activator gene and rapH are colored red. The inserted promoters, ermE*p, kasO*p, and sp44p, are also presented. e Results of flask cultivation with the wild-type and engineered S. rapamycinicus NRRL 5491 are shown. f Exchanged promoters of each strain are listed. Blank fields in the table indicate that the promoter was not exchanged. All cultivations were performed in triplicates (n = 3). The mean was plotted with error bar representing standard deviation. P values were determined by unpaired two-tailed Student’s t-test (ns, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001). Source data are provided as a Source Data file.
Then, sgRNAs targeting promoters of ovmOI, ovmOII and ovmF genes were assembled into pCRISPomyces-2BD. In addition, for recombination after cleavage at each site, three heterologous promoters, ermE*p, kasO*p, and sp44p, and their respective homologous regions on both sides of the endogenous promoters were inserted into the pCRISPomyces-2BD plasmid harboring sgRNA (Supplementary Data 1). In total, nine plasmids that could replace the promoters of ovmOI, ovmOII, and ovmF with three strong promoters, ermE*p, kasO*p, and sp44p, respectively, were constructed. Subsequently, the DNA fragments of the nine homologous regions and promoter sequences, as well as each sgRNA targeting the three promoter regions, were amplified using PCR and inserted into the pCRISPomyces-2BD plasmid using the Golden Gate assembly to create a plasmid library (Supplementary Fig. 6). The library was designed to target one to three promoters (ovmOI, ovmOII, and ovmF) and replace them with one of the three strong promoters (ermE*p, kasO*p, and sp44p). Finally, transformation was performed by conjugation of the plasmids with S. antibioticus NRRL 3238. A total of five exconjugant colonies appeared on the agar plates of triplicated experiments, which were then inoculated into tryptic soy broth (TSB) media containing apramycin and cultured in round-bottom tubes for 2 d. The activation of BGC was confirmed by the appearance of brown-colored culture, indicating oviedomycin production, which was not observed in the culture of wild-type S. antibioticus NRRL 3238 (Fig. 3b). The strains were subjected to additional cultivation in fresh TSB medium and then transferred to MS medium for flask cultivation. Four of the five edited strains produced oviedomycin and were confirmed to have activated the BGC. Specifically, the S. antibioticus-1 strain produced 83.4 mg/mL of oviedomycin. Sequencing of the extracted genomic DNAs revealed that all three targeted promoters in the oviedomycin BGC were refactored in the S. antibioticus-1 strain, and the exchange of ovmOI promoter with either kasO*p or ermE*p was commonly identified in the strains producing oviedomycin (Fig. 3c).
Rapamycin is a valuable compound produced by Streptomyces rapamycinicus NRRL 549137. The BGC contains three type I polyketide synthase (PKS) genes, rapA, rapB, and rapC, which are activated by a regulator encoded in the rapH gene (Fig. 3d). Similarly to oviedomycin, we aimed to enhance rapamycin production by refactoring the promoter (Supplementary Fig. 5b). The promoter of the rapH gene was replaced with kasO*p using the modified Cas9-BD (Supplementary Fig. 5d, f). The engineered strains exhibited increased rapamycin production compared to the wild-type strain during flask cultivation (Supplementary Fig. 5h). Additionally, Cas9-BD was utilized to replace the promoters of the rapA, rapB, rapC, and rapH genes. Similar to the oviedomycin BGC activation, three strong promoters, ermE*p, kasO*p, and sp44p, were combined with the homologous sequences of the promoter regions of each gene, and a library of plasmids was constructed using sgRNAs and transformed into S. rapamycinicus NRRL 5491 by conjugation. Three exconjugants were obtained in triplicated experiments, which were individually cultured in TSB media twice, followed by transfer to fermentation media for flask cultivation. Two of the three engineered strains, S. rapamycinicus-1 and -3 produced 87.7 mg/L, and 75.1 mg/L of rapamycin, respectively, which were higher than those produced by the wild-type S. rapamycinicus NRRL 5491 (58.2 mg/L) (Fig. 3e). While confirming the successful insertion of the promoters, we found that up to three promoters were engineered in S. rapamycinicus-1 and -3 strains, and the exchange of the rapH promoter with kasO*p was effective in improving the production of rapamycin (Fig. 3f). These results showed that multiplexed genome editing in a single experiment was enabled by modified Cas9 in Streptomyces strains, which allowed the simultaneous refactoring of several promoters in a BGC for higher metabolite production.
Multiplexed BGC deletion in Streptomyces genome using Cas9-BD
To improve metabolite production in Streptomyces, other BGCs are frequently deleted to increase precursors for target metabolites. Because multiple BGCs exist in Streptomyces sp., the simultaneous deletion of many BGCs is desirable. Therefore, in this study, Cas9-BD was used for the simultaneous deletion of multiple BGCs in Streptomyces coelicolor M114610,36,38 (Fig. 4a). The BGCs targeted in S. coelicolor M1146 were desferrioxamine B (7.4 kb), SapB (5.6 kb), and SCO-2138 (22 kb). sgRNAs binding to the middle of the three BGCs and two homologous regions outside of the BGC for DNA recombination were designed and inserted into pCRISPomyces-2BD (Supplementary Data 1). The BGCs were effectively deleted individually using the constructed plasmids for each BGC deletion with 2.0 × 10−5, 1.8 × 10−5, and 1.2 × 10−5 of conjugation efficiency, respectively (Fig. 4b, c, d). BGC deletion plasmids for both desferrioxamine B and SapB were constructed and conjugated to M1146, and all six exconjugants contained deletions in both BGCs with 6.0 × 10−6 efficiency (Fig. 4e). Finally, triple BGC deletion was successfully performed in a single experiment using modified Cas9-BD in S. coelicolor M1146 with 1.0 × 10−6 efficiency (Fig. 4f). No unintended mutations were found when the predicted off-target sites in the genome were sequenced following the deletion experiments. (Supplementary Fig. 7).

a Representation of the plasmids constructed for BGC deletions in S. coelicolor using Cas9-BD. Insertion of sgRNA to create a cleavage of BGC, and two homologous regions for recombination of ~1 kb were inserted after cleavage into a plasmid containing the gene Cas9-BD. To perform multiple deletion, sets of sgRNA and homologous regions were inserted in the plasmids. b–d Scheme of BGC deletion of desferrioxamine B, SapB, and SCO-2138 and confirmation via nested PCR. e Result of double deletion of desferrioxamine B and SapB. f Triple deletion, including desferrioxamine B, SapB, and SCO-2138 BGC. All conjugations were performed in a single replicate (n = 1). Source data are provided as a Source Data file. Ctl, product of PCR amplification using the genomic DNA of S. coelicolor M1146 as the template. Mar, DNA ladder marker; Des. B, desferrioxamine B.
Development of an in vivo BGC capturing method mediated by Cas9-BD
Improving the production of secondary metabolites in Streptomyces is often challenging owing to difficulties in optimizing the cultivation conditions and genetic manipulation. In various studies, in vitro capture of BGCs and production of metabolites in heterologous hosts, such as S. avemitilis, S. lividans, S. coelicolor, and S. albus have been utilized to overcome these difficulties7,9,10,36. However, the in vitro capture method requires high concentrations and purity of Streptomyces genomic DNA, thereby increasing the time and labor required. Therefore, in this study, an in vivo capture method was developed to clone large BGCs using Cas9-BD, without the need for purifying genomic DNA. This was applied to the production of the clinically promising immunosuppressive compound FK5064, which is produced by S. tsukubaensis NRRL 18488. The pCRISPomyces and pCRISPomyces-2BD plasmids, with or without sgRNAs, were transformed into S. tsukubaensis NRRL 18488 (Supplementary Fig. 8a). Exconjugants were only formed when using plasmids containing Cas9-BD. Initially, two sgRNAs were designed to cleave both end regions of the FK506 BGC and cloned into pCRISPomyces-2BD together with two homologous regions that could recombine with DNA after cleavage (Figs. 5a, Supplementary Data 1). The resulting plasmid for BGC capture, pCap-00 (Supplementary Data 2), was transformed into S. tsukubaensis NRRL 18488 by conjugation. Four exconjugants were obtained (Supplementary Fig. 8a) and cultured in 50 mL of TSB medium containing apramycin. The plasmids extracted from all four strains successfully captured the FK506 BGC, as confirmed by PCR on eight loci within the BGC (Supplementary Fig. 8b, c). To produce FK506 in the heterologous host strain, the BGC region of the cloning vectors was transferred to a pExp-00 plasmid containing an integrase (Supplementary Data 2), and the resulting plasmid was designated pFK (Figs. 5a, Supplementary Fig. 9b, c). The entire sequence of the pFK plasmid harboring FK506 BGC was validated through PCR amplification and subsequent sequence analysis (Supplementary Figs. 10a, b, Supplementary Data 4). To improve the production of FK506, we performed refactoring of the fkbN promoter4 in the BGC to kasO*p and ermE*p using homologous recombination with λ red recombinase in E. coli39 incorporated into the pFK plasmid. The resulting plasmids were named pFK-KN and pFK-EN, respectively. The resulting plasmids were transformed into the heterologous host strain S. coelicolor M1146 by conjugation. The resulting strains, S. coelicolor-FKKN and -FKEN, produced up to 33.9 and 12.0 mg/mL of FK506, respectively, when cultivated in fermentative medium (Fig. 5b, c). The in vivo BGC capture and transfer to a heterologous host proved to be effective, resulting in a substantial higher level of FK506 production in S. coelicolor M1146 containing refactored BGCs compared to the wild-type S. tsukubaensis NRRL 18488.

a The scheme for capturing, cloning, and refactoring large BGC to produce FK506 in S. coelicolor M1146. b HPLC plots of different strains harvested 8 d after cultivation. Metabolite peaks shown in both S. tsukubaensis NRRL 18488 and S. coelicolor-FKKN strains are pointed by dotted gray lines. FK506 peak is highlighted by a star. c FK506 production during flask cultivation of different strains was presented. All cultivations were performed in triplicates (n = 3). The mean was plotted with error bar representing standard deviation. P values were determined by unpaired two-tailed Student’s t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Source data are provided as a Source Data file.
Development of dCas9-BD with reduced toxicity
dCas9 was modified with a polyaspartate linkage using the same method as in Cas9-BD to reduce off-target binding. The pCRISPomyces-2 plasmid was used to construct dCas9 by introducing the mutations D10A and H840A, and the resulting plasmid was named pdCRISPomyces-2. Next, dCas9-BD with polyaspartate introduced into the C- and N-termini of dCas9 was constructed, generating plasmid pdCRISPomyces2-BD. To measure the inhibition efficiency, gusA as a reporter gene10,40 was inserted into both plasmids, pdCRISPomyces-2 and pdCRISPomyces-2-BD; the plasmids were transformed into S. coelicolor M1146 to produce strains SCO0G and SCO0G-BD, respectively. Finally, three respective sgRNAs were designed to bind the promoter (1), 5’ (2), and middle (3) regions of the gusA gene and added to both plasmids (Supplementary Data 1). Another plasmid containing all three sgRNAs was constructed (Supplementary Fig. 11a). All constructed plasmids were transformed into the S. coelicolor M1146 strain. In flask cultivation, the growth rates of the strains with wild-type dCas9 were significantly reduced compared to the control strain, S. coelicolor M1146, with the gusA gene and without dCas9 in the plasmid (SCOXG). Interestingly, the strains with dCas9-BD showed similar or slightly reduced growth compared with the control (Figs. 6a, Supplementary Fig. 11b). This demonstrated the lower toxicity of dCas9-BD compared to dCas9. When comparing gusA expression levels, the inhibitory effects of dCas9-BD were generally greater than those of dCas9 (Fig. 6b; Supplementary Figs. 11e, g, i, k). The strain with dCas9-BD showed higher inhibition of the target gusA gene without leaky expression than strain with dCas9 (Figs. 6b, Supplementary Fig. 11i). In addition, the strain containing all three sgRNAs showed little growth reduction, with strong inhibition of gusA gene expression when dCas9-BD was used (Supplementary Figs. 11j, k). To access the transcriptional effects of dCas9 and dCas9-BD, transcriptomic analysis was performed on SCOXG, SCO0G, SCO0G*, SCO3G, and SCO3G* strains using RNA-seq (Supplementary Fig. 12a). Consistent with the GusA assay results, dCas9-BD with sgRNAs (SCO3G) presented a similar level of gusA gene expression inhibition as dCas9 (SCO3G*) in the transcriptomic data (Supplementary Fig. 12d, e). The results showed that the modified dCas9-BD was less toxic to the strain and produced similar inhibitory effects than dCas9.

a Cell growth measured by phenylamine staining and (b) Expression of the gusA gene measured by GUS assay at 48 h after cultivation. The results of the strain without dCas9 and sgRNA are represented in black columns. Strains expressing wild-type dCas9 and dCas9-BD are represented in red and blue, respectively. Expressed sgRNA listed as C describes the absence of both dCas9 and sgRNA; 0 represents dCas9 without sgRNA; 1 describes the sgRNA targeted promoter region of the gusA gene; 2 describes the sgRNA targeted 5’ region of a gusA gene; 3 describes the sgRNA targeting the middle of a gusA gene; and 3 g describes expressing all three sgRNAs (1-3). All cultivations were performed in triplicates (n = 3). The mean was plotted with an error bar representing the standard deviation. P values were determined by unpaired two-tailed Student’s t-test with results obtained from strain lacking both dCas9 and sgRNA (ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001). Source data are provided as a Source Data file.
Multiple gene expression inhibition by dCas9-BD for improved secondary metabolite production
To improve the production of secondary metabolites in Streptomyces, it is necessary to simultaneously inhibit the expression of multiple genes to increase precursor levels for secondary metabolite synthesis40. Because multiple sgRNAs did not cause serious growth reduction, dCas9-BD with multiplexed inhibition was employed to improve the production of actinorhodin, undecylprodigiosin, and coelimycin p2 in Streptomyces coelicolor A3(2)41,42. sgRNAs targeting five genes, pyc, gltA, adhE, fabD, and fabH, were designed to increase the levels of the precursors acetyl-CoA and malonyl-CoA40,43,44,45 (Fig. 7a). The five sgRNAs were separately cloned into pCRISPomyces-2BD using the Golden Gate assembly (Supplementary Fig. 13). Next, the sgRNAs were amplified using PCR and cloned into pdCRISPomyces-2BD. The transformation was performed via conjugation of the plasmids from the library into S. coelicolor A3(2). Strains showing faster coloration on MS plates compared to the wild-type S. coelicolor A3(2) strain were selected (Supplementary Fig. 14). A total of 23 selected strains were cultured in TSB containing apramycin. Most of the selected strains exhibited more coloration than the wild type within 2 d. The strains were harvested, and secondary metabolites were extracted with methanol (Fig. 7b). Among them, five strains showed significantly increased secondary metabolite content, which might be caused by the accumulations of acetyl-CoA and malonyl-CoA in all five strains (Supplementary Fig. 15). The selected five strains were cultivated in flasks in MS medium for 7 d. Plasmids harbored by the five strains were extracted, and their sgRNA regions were sequenced. Most of the strains contained sgRNAs targeting the adhE gene, which encodes acetaldehyde dehydrogenase, and the fabH gene, which encodes 3-oxoacyl-ACP-synthase III (Fig. 7f). S. coelicolor A3(2)-1 demonstrated a significantly improved secondary metabolite content with an eight-fold increase in actinorhodin, a four-fold increase in undecylprodigiosin, and a two-fold increase in coelimycin P2 production (Fig. 7c, d, e). This strain harbored sgRNAs targeting gltA, adhE, and fabH. These results confirmed that the multiplexed repression of a few genes was very efficient in isolating a high producer of a secondary metabolite, which was easily achieved using dCas9-BD in Streptomyces sp.

a The central metabolic pathway involved in the secondary metabolite production of S. coelicolor A3(2). The malonyl-CoA and acetyl-CoA highlighted in blue are precursors of target metabolites, actinorhodin, undecylprodigiosin, and coelimycin P2. The genes highlighted in red, pyc, gltA, adhE, fabD, and fabH, present the inhibition targets to achieve precursor accumulation. b The secondary metabolites actinorhodin, prodigiosin, and coelimycin P2, were produced by the 23 strains engineered using dCas9-BD. The five strains showing a dramatic improvement in secondary metabolite production are marked with blue arrows. c ACT, actinorhodin; d RED, undecylprodigiosin; e CPK P2, coelimycin P2 productions of the five strains in a flask cultivation; WT, S. coelicolor A3(2); WT*, S. coelicolor A3(2) harboring pdCIRSPomyces-2BD without sgRNA; S. coelicolor A3(2)*, S. coelicolor A3(2) harboring pdCRISPomyces-2BD without sgRNA. f The harbored sgRNAs in five high-producing strains. Flask cultivations were performed in triplicates (n = 3). The mean was plotted with an error bar representing the standard deviation. Source data are provided as a Source Data file.
Discussion
Although many Streptomyces sp. are promising industrial strains that produce clinically important natural products, the high GC content in their genomes makes genome editing Cas9 challenging8,22,23. In particular, many repeated or similar PAM sequences increase the chances of off-target binding and cleavage by Cas931. To reduce Cas9-mediated off-target cleavage, decreasing the expression of Cas9, expressing inhibitory proteins, and introducing mutations in the binding site of Cas9 to the target DNA have been attempted. In this study, we proposed an additional strategy of attaching polyaspartate residues to Cas9. The added negatively charged residues were expected to function as an anti-CRISPR protein, AcrIIA4, which inhibits binding between the Cas9 protein and the target DNA by interacting with positively charged residues within the binding site of Cas946. AcrIIA4 was utilized in the genome editing of human K562 erythroleukemia cells by introducing it 6 h after Cas9 treatment, which successfully reduced off-target cleavage47. On that note, it has been revealed that aspartyl residues in AcrIIA4 mediate the interaction with the DNA binding region of Cas9. We expected that the addition of aspartyl residues to Cas9 would have a similar effect, eliminating the inconvenience of the time-delayed expression of the protein. Owing to the different binding affinities of Cas9 to on- and off-target DNA, it was expected that the introduction of aspartyl residues would more effectively inhibit the weak binding of Cas9 to off-target DNA than its strong binding to on-target DNA. As expected, the Cas9 protein linked with five aspartyl residues to the C- and/or N-termini (Cas9-BD) showed much lower off-target cleavage than wild-type Cas9 (Figs. 1d, Supplementary Fig. 3). More specifically, Cas9-ND presented a much lower off-target cleavage efficiency than Cas9-CD, which may be because the DNA-binding region is located near the N-terminus of Cas948,49 (Supplementary Fig. 1b). Overall, Cas9-BD presented the lowest off-target cleavage efficiency, whereas the on-target cleavage efficiency was comparable to that of wild-type Cas9. This modification is expected to be applicable to various types of Cas9-mediated tools, such as BEST and prime editing, to reduce off-target cleavage29,50,51.
With reduced toxicity when expressed in the strain, Cas9-BD was employed for genome editing of Streptomyces sp. to enhance secondary metabolite production in various ways. For example, native promoters in BGCs are often exchanged with constitutively strong promoters to increase secondary metabolite production13,23,26. However, it is difficult to simultaneously engineer several promoters in a BGC with optimal expression strength. Using Cas9-BD, we successfully constructed a combinatorial plasmid library with multiple sgRNAs that could target and exchange promoters in a BGC. This method was used to activate silent BGC-producing oviedomycin in S. antibioticus and increase rapamycin production in S. rapamicinicus in a single round of experiments. Considering that the time to manipulate a promoter in Streptomyces sp. typically takes 7–10 d and additional time is required for plasmid removal, spore collection, and validation through sequence analysis in each round of the genome editing experiment, a considerable reduction in experiment time is expected with this method. Multiple editing or deletion using Cas9-BD could more conveniently increase the efficiency of the activation of silent BGC expression or improve target metabolite production via the deletion of competitive BGCs. This knowledge can also be applied for the identification and deletion of unnecessary regions within the genome or even for the development of a minimal Streptomyces sp. genome.
Cas9-BD has also been used to develop an in vivo capture method for large BGCs. Our results were remarkable regarding large BGC capture with a high probability, without the time-consuming process of preparing high concentrations of linear pure genomic DNA to obtain BGCs7,10,52. In addition, when Cas9-mediated linearization and recombination of large DNA fragments occur simultaneously within the cell, double recombination at both ends of the BGC by Cas9 can be utilized for the selection of a circular plasmid, contributing to high engineering efficiency. Furthermore, recombination of the target BGC triggered the removal of DNA fragments in pCas-00 coded sgRNAs, thereby avoiding cleavage mediated by Cas9 after capture53. Moreover, this method is expected to ensure the stability of large DNA owing to intracellular cleavage and recombination processes. Using this approach, various large BGCs up to 230 kb in size were successfully captured (Supplementary Fig. 16). Consequently, the developed in vivo capture method is expected to reduce the challenges of manipulating large-sized DNA, facilitating functional studies of large BGCs in Streptomyces.
Although dCas9 is a highly valuable tool for controlling the expression of genes or localizing proteins at desired sites in the genome, it has been shown to be toxic to Streptomyces because of the high possibility of off-target binding24,27. To avoid this, an additional process is required to reduce off-target binding, such as reducing the expression of dCas9 or expressing various sgRNAs, to find an appropriate approach, as shown by previous research40. In this study, the strains expressing dCas9-BD did not show significant growth inhibition, even with several sgRNAs (Fig. 6b, Supplementary Fig. 11j). Therefore, dCas9-BD allowed multiplexed CRISPRi in the Streptomyces strain. This enabled us to construct a combinatorial sgRNA library, which was transformed together with dCas9-BD into the S. coelicolor A3(2) strain to reduce the expression of genes involved in competitive metabolic pathways with target molecules. This facilitated the screening of target genes in host metabolic engineering by randomly controlling the expression of various genes via CRISPRi or CRISPRa at various sites of genomic DNA using multiple sgRNAs.
In summary, we developed a Cas9 modification to reduce off-target cleavage efficiency. It was confirmed via in vitro reactions that the modified Cas9-BD presented a lower off-target cleavage efficiency than wild-type Cas9 without forfeiting on-target cleavage efficiency. The modified proteins exhibited low toxicity to Streptomyces when expressed at high levels. However, genome sequencing revealed that off-target editing still occurred with Cas9-BD, indicating the need to work with multiple exconjugants following the editing experiment. Using Cas9-BD and similarly constructed dCas9-BD, the production of secondary metabolites in Streptomyces was dramatically improved in a single experiment using a library containing multiple sgRNAs. In addition to engineering genomic DNA, Cas9-BD was utilized for capturing large FK506 BGC, resulting in successful FK506 production in a heterologous strain, S. coelicolor M1146, through promoter refactoring. The Cas9 modification method developed in this study was confirmed to be effective for Cas9 derived from S. pyogenes, particularly in microorganisms with high GC content. This includes not only Streptomyces but also a range of other high GC content organisms (Supplementary Fig. 17). This modification can also be employed for the genetic engineering of various cells with Cas9–based tools, such as BEST or prime editing, and Cas9 variants, such as those derived from Staphylococcus aureus, Neisseria meningitidis, or Streptococcus thermophilus, which share a similar crystal structure with SpCas9. Furthermore, multiplexed promoter refactoring for optimizing BGC expression and in vivo capture of large BGCs was demonstrated in Streptomyces using modified Cas9. This tool is expected to reduce the time and labor required to study secondary metabolites encoded by BGCs in Streptomyces.
Methods
Development of E. coli BL21 strains expressing various Cas9s
The plasmids used in this study were cloned in E. coli DH5α (Real Biotech Co., Taipei, Taiwan, RH619), and the strains used in this study are described in Supplementary Data 2. Cas9 modification was performed following PCR amplification using primers including the polyaspartate sequence, listed in Supplementary Data 2, with a synthesized gene encoding Cas9 as a template (Supplementary Data 1, Bionics Co., Seoul, Korea, 20210110-0001). Nested PCR was performed for all the amplifications to obtain the desired products. The polyaspartate sequence was added to the N- and/or C-termini of Cas9. The original and modified Cas9 sequences were subcloned into the pETduet-1 plasmid, downstream of the pBAD promoter containing a His6-tag sequence. For insertion, ligation was performed on the Cas9s sequence digested with the BsaI enzyme and the pETduet-1 plasmid digested with BamHI and HindIII enzymes (New England Biolabs Inc., Massachusetts, USA, R3136L; R0104L). All the plasmids developed in this study were confirmed using colony PCR and sequence analysis (Bionics Co., ABI-3730XL). The verified plasmids were named pET_Cas, expressing wild-type Cas9; pET_Cas-CD, Cas9 linked with polyaspartate at the C-terminus; pET_Cas-ND, Cas9 linked with polyaspartate at the N-terminus; and pET_Cas-BD, Cas9 linked with polyaspartate on both sides of the C- and N- termini. Then plasmids were transformed into the BL21 DE3 strain using electroporation, and the transformed cells were selected on Luria Bertini (LB) agar plates supplemented with 50 µg/mL of carbenicillin. E. coli BL21 strain harboring pET_Cas were named BL21_C, pET_Cas-CD as BL21_C-CD, pET_Cas-ND as BL21_C-ND, and pET_Cas-BD as BL21_C-BD, respectively.
In vitro Cas9 reaction and calculation of cleavage efficiency
E. coli BL21 strains harboring plasmids containing Cas9 and modified Cas9s were cultivated in a flask with 50 mL of LB media containing 50 µg/mL of carbenicillin at 30 °C. After 4 h of cultivation, during the early exponential phase, 20 µ M of IPTG was added to induce the expression of each type of Cas9. After 20 h of cultivation, the cells were collected by centrifugation at 20,000 g for 10 min. The collected cells were washed twice with distilled water, followed by centrifugation, and redissolved in 5 mL of lysis buffer (pH 8.0) containing 20 mM Tris and 250 mM NaCl. The resolved cells were disrupted using sonication for 10 min, followed by centrifugation at 20,000 g for 10 min. After centrifugation, the supernatant was collected, and the Cas9 dissolved in the supernatant was purified using Nickel Agarose Beads and GoldBio Plastic Columns (Gold Biotechnology Inc., Olivette, MO., USA, H-320; P-301). The concentration of purified Cas9s was measured using the Quick Start Bradford Dye Reagent (Bio-Rad Lab. Inc., California, USA, 5000205).
For the in vitro reaction, 200 nM of each Cas9 and 600 nM of sgRNA synthesized (Macrogen Inc., Seoul, Korea, ACR210619-0101) as listed in Supplementary Data 2 were mixed in a 25 µL reaction solution containing 10% of 3.1 buffer (New England Biolabs Inc., B7203) for each reaction. The mixed solutions were pre-incubated for 30 min at 37 °C and 700 ng of various types of linear DNA substrates were added to the prepared solutions (Fig. 1c). To prepare linear DNA, plasmids containing target sequences were constructed by ligating pETduet-1 and annealing the oligomer-coded target sequences after digestion with the restriction enzymes XbaI and PacI. The resulting nine plasmids were named aspET_target_1 to pET_target_9 (Supplementary Data 2), and linear DNAs were amplified by PCR using the constructed plasmids. The in vitro reactions were performed for 2 h at 37 °C, and 5 µL of samples were collected at 10, 30, 60, and 120 min of reaction. To stop the reaction, EDTA and protease K were added to the collected samples at final concentrations of 50 mM and 1 µg/µL, respectively. Samples were incubated at room temperature for 30 min and resolved using electrophoresis on 0.8 % of agarose gel stained with RedSafe (iNtRon Biotech. Inc., Gyeonggi-do, Korea, 21141; Supplementary Fig. 2). After electrophoresis, the samples were visualized using a WUV-L10 transilluminator (Daihan Scientific Co., Seoul, Korea, DH.WUV00010) and photographed using a cell phone. The intensities of each fragment were measured using the ImageJ program33,54. The cleavage efficiencies were calculated as cleavage fragment intensity/total fragment intensity (Supplementary Fig. 3). The in vitro reactions of each Cas9 were performed in triplicate.
Measurement of protein circular dichroism
The Cas9 proteins were purified above-described method. After measuring concentration using the Quick Start Bradford Dye Reagent, each protein was loaded in Vivaspin Turbo 4 Ultrafiltration Unit (Sartorius Inc., Göttingen, Germany, VS04T91) and centrifugated at 10,000 g for 10 min. After centrifugation, 2 mL of distilled water was added in unit and one more centrifugation at 10,000 g was performed for 10 min. Then buffer solution, containing 100 mM NaCl, 10 mM Tris-Hcl, 0.1 mM EDTA, 1 mM DTT, and 50% glycerol, was added for dilution of proteins to concentration of 1-3 mg/mL. To measure circular dichroism, proteins were diluted in nuclease free water to concentration of 1 µM. For preparing samples of sgRNA-Cas9 complexes, 1 µM of sgRNA was added in solution including proteins and samples were rested at room temperature at least 30 min. Prepared samples were loaded in 21/10/Q/10 quartz cuvette (Starna Sientific Ltd., London, UK, 21-Q-1) and measured by JASCO J-1000 CD Spectrometer (JASCO Co., Yokyo, Japna, J-1000) with scanning speed of 100 nm/min for 190-350 nm spectra range with 1 nm bandwidth and 10 scans of accumulation. Samples were analyzed several times, up to 10 times, and means were calculated for each sample.
Development of plasmids expressing modified Cas9 and dCas9
To develop modified Cas9-BD plasmids, the wild-type Cas9 sequence from the pCRISPomyces-2 plasmid34 was amplified using PCR with primers containing the polyaspartate sequence. The PCR products were then cloned into pCRISPomyces-2 using Gibson Assembly at the Cas9 site, resulting in pCRISPomyces-2BD. To construct plasmids expressing dCas9, DNA fragments of dCas9 containing mutations D10A and H840A were prepared by PCR amplification using the pCRISPomyces-2 plasmid as a template. These fragments were then assembled using Gibson Assembly and cloned into the same pCRISPomyces-2 plasmid to generate pdcCRISPomyces-2 containing dCas9. Similar to Cas9-BD, dCas9-BD was developed using PCR and cloned into pdCRISPomyces-2 via Gibson assembly to contain the dCas9-BD gene instead of dCas9, resulting in pdCRISPomyces-2BD. All the cloning was performed using E. coli DH5α and transformed by heat shock. Strains were selected on LB agar plates supplemented with a 50 µg/mL concentration of apramycin. Developing plasmids were confirmed using colony PCR and sequence analysis. The plasmids with the correct sequences were transformed into Streptomyces sp. strain via conjugation mediated by E. coli strain ET12567/pUZ800210.
For conjugation, E. coli ET12567/pUZ8002 harboring plasmids were inoculated in 15 mL conical tube with 1 mL of LB media containing 20 µg/mL of apramycin, chloramphenicol, and kanamycin. After overnight, broth was transferred to 250 mL flask with 50 mL fresh LB media containing the same antibiotics. Flask cultivation was performed until optical density reached at 0.4 with 600 nm wavelength. Grown cell was washed and collected using distilled water and centrifugation. The 10 µL of spore stock, concentrated to 1.0 × 108 CFU/mL, were transferred into 100 µL of fresh LB media and spore in LB media was germinated at 50°C for 10 min. Germinated spore was mixed with collected E. coli pellet and spread on MS agar media, containing 20 g mannitol, 20 g soybean meal, and 20 g agar powder per 1 L containing 10 mM MgCl2. After cultivation for 16 h, 1 mL of distilled water containing 0.5 mg nalidixic acid and 1.25 mg apramycin was overlayed on MS agar media. 7 d after conjugation, formed exconjugants were cultivated into round bottom tube with 2 mL of TSB media for 3 d. Cultivated cells were collected by centrifugation and genomic DNAs of each strain were extracted. To confirm genome editing, PCR amplification was performed using extracted genomic DNA as templates. The PCR products were resolved by gel electrophoresis on a 0.8% agarose gel stained with RedSafe, visualized using a WUV-L10 transilluminator, and confirmed by sequence analysis.
Gene manipulations in Streptomyces sp. using Cas9 and Cas9-BD
To develop plasmid for gene editing, annealed conserved sequence oligomers were inserted into pCRISPomyces-2 and pCRISPomyces-2BD plasmid to prepare sgRNA, targeting gene of interest, via Golden Gate assembly using T4 ligase and BbsI restriction enzyme34. sgRNAs for gene manipulation in this study were designed using CRISPR RGEN tool (http://www.rgenome.net/)55 To predict off-target sites of sgRNAs, BLAST searches with NCBI database and RGEN OFFinder Tool56 were used. For deletion experiments, the homologous sequence regions flanking target sites were amplified using PCR with the genomic DNA of S. coelicolor Sp. For exchanging promoters, flaking homologous sequence regions of target promoters and sequence of kasO* promoter57 were prepared by PCR amplification using genomic DNA of each strain and synthesized genes (Supplementary Data 1). The prepared PCR products were digested with the BbsI restriction enzyme and inserted into digested plasmids containing sgRNA using an XbaI restriction enzyme and T4 ligase. The developed plasmid was also confirmed by PCR amplification and sequence analysis. The confirmed plasmids were transformed into S. coelicolor M1146 strain via conjugation mediated by E. coli ET12567/pUZ8002.
For engineered S. antiboticus NRRL 3238, 50 mL of MS media, containing 20 g mannitol and 20 g soybean meal per 1 L distilled water, was used for flaks cultivation and cultivation was performed for 7 d. For engineered S. rapamycinicus NRRL 5491, 50 mL of fermentative media used in previous research38 was used and cultivation was performed for 14 d. Samples were harvested by centrifugation of 50 mL culture broth and analyzed by reverse phase HPLC with a C18 reverse phase column36,37. Flask cultivation of each strain was performed in triplicate.
Combinatorial promoter refactoring and multiple gene deletions using Cas9-BD and multiple sgRNA
sgRNAs targeting the promoter regions of the target genes in the BGC were prepared similarly as described above. Three promoter sequences57,58, ermE*p, kasO*p, and sp44p, were prepared (Supplementary Datas 1 and 2) and inserted into the homologous sequence regions and cloned into sgRNA-containing pCRSIPomyces-2BD plasmids. After cloning and confirmation, DNA fragments of the plasmids, including sgRNA and homologous regions containing the promoter, were obtained by PCR amplification using a mixture of primer pairs, lib_cond_1_FW and lib_cond_1_RV, and another primer pair, lib_cond_2_FW and lib_cond_2_RV, in a 1:4 ratio (Supplementary Data 2). The PCR products were purified after digestion with the BsaI restriction enzyme and randomly inserted into the pCRISPomyces-2BD plasmid using Golden Gate assembly with the AvrII restriction enzyme and T4 ligase. The resulting library was transformed into an E. coli DH5α strain via heat shock, and the transformed colonies were inoculated in LB media containing 50 µg/mL of apramycin, to develop the library. The library, capable of randomly exchanging the promoters of the target genes with three different promoters, was transformed into the ER12567/pUZ8002 strain via electroporation, followed by conjugation with the S. antibioticus NRRL 3238 strain and the S. rapamycinicus NRRL 5491 strain. For conjugation, 10 µL of spore stock for S. antibioticus NRRL 3238 and 30 µL of spore stock for S. rapamycinicus NRRL 5491 were used. The conjugation of each strain was performed triplicate. All exconjugants were cultured and the metabolites were analyzed as described above.
To delete the BGCs, sgRNAs of the three BGCs of SapB, desferrioxamine B, and SCO-2138 and homologous sequence regions of ~1 kb, located on both sides of each BGC, were prepared and inserted into the pCRSPomyces-2BD ho To develop plasmids for multiple deletions, DNA fragments containing two homologous sequence regions and sgRNA for BGC deletion in SapB and SCO-2138 BGC were inserted into the plasmid containing homologous regions and sgRNA for the deletion of the desferrioxamine B BGC via Golden Gate assembly using the restriction enzymes, BbsI and AvrII, and T4 ligase (Supplementary Data 1). The confirmed plasmids were transformed into E. coli ET12567/pUZ8002 strains, and strains harboring the plasmids were conjugated to S. coelicolor M1146.BGC deletions were confirmed by sequence analysis. After BGC deletion, the genomic DNAs of engineered strains were extracted and used as templates for PCR amplification of predicted off-target sites. The PCR bands were identified by electrophoresis on a 0.8% agarose gel and subsequently used for sequencing analysis (Supplementary Fig. 7).
Measuring toxicity and inhibition efficiency of modified dCas9 in S. coelicolor M1146
To repress the gusA gene, three sgRNA sequences were designed to bind to the promoter, 5’ region, and middle of the gusA gene and inserted into plasmids pdCRISpomyces-2 and pdCRISPomyces-2BD, using Golden Gate assembly. The resulting plasmids were transformed into E. coli DH5α, and the transformed strains were confirmed by colony PCR and sequencing. For the utilization of multiple sgRNAs, the constructed sgRNA sequences that targeted the 5’ region and middle of the gusA gene were amplified using PCR from the developed plasmids and inserted into plasmids pdCRIPSomyces-2 and pdCRIPSomyces-2BD, containing sgRNA targeting the promoter region of the gusA gene, via Golden Gate assembly using restriction enzyme, AvrII and BbsI, and T4 ligase. After confirmation using colony PCR and sequence analysis, the sequences of the kasO*p and gusA genes amplified by PCR using the synthesized gene (Supplementary Data 1, Bionics Co., GS230628-019) were inserted into the developed plasmids encoding dCas9 or dCas9-BD with each sgRNA using Golden Gate assembly with restriction enzymes BbsI and XbaI and T4 ligase (Supplementary Data 1). To develop pdCRSISPomyces-2, which lacks dCas9 and contains only the gusA gene, the BamHI restriction enzyme was used for Golden Gate assembly. The constructed plasmids were transformed into E. coli ET12567/pUZ8002 and conjugated with S. coelicolor M1146.
The exconjugants were inoculated into round bottom tubes with 2 mL of TSB media containing 50 µg/mL of apramycin. After 3 d of cultivation at 30 °C, 100 µL of the broth with grown cells were transferred into 2 mL of fresh TSB media containing apramycin. The strains were grown for another 2 d, and 1 mL of the broth was transferred to a flask with 50 mL of TSB media containing 50 µg/mL of apramycin. At each 12-h interval, 500 µL of samples were collected and then centrifuged and the supernatant was removed. To dissolve the collected cells, 500 µL of distilled water was added, and the samples were divided into two 200 µL samples. Each sample was washed with distilled water and collected by centrifugation to perform the GUS assay10 and measure cell growth40. Flask cultivation for each strain was performed thrice.
Production of metabolites using dCas9-BD and multiplexed sgRNA
The sgRNAs targeting the 5’-UTR regions of pta, gltA, adhE, fabH, and fabD genes of S. coelicolor A3(2) were prepared (Supplementary Data 2) using Golden Gate assembly in the pdCRISPomyces-2BD plasmid. The constructed sgRNA sequences were amplified using nested PCR with a mixture of the primer pairs sgRNA_cond_1_FW and sgRNA_cond_2_RV, and sgRNA_cond_2_FW and sgRNA_cond_1_RV in a 1:5 ratio (Supplementary Data 2). The resulting PCR products were randomly inserted into pdCRISPomyces-2BD plasmids containing sgRNAs using Golden Gate assembly with the restriction enzymes XbaI and BbsI and T4 ligase. The developed library was transformed into an E. coli DH5 strain by heat shock and spread onto LB agar plates containing 50 µg/mL of apramycin. All formed colonies were transferred to 50 mL LB media containing 50 µg/mL of apramycin and cultivated. Then, the developed plasmids were extracted through plasmid preparation, and these plasmids were transformed into E. coli ET12567/pUZ8002 by electroporation and subsequently conjugated into S. coelicolor A3(2). Strains with rapid pigmentation on MS plates were selected (Supplementary Fig. 14), and these strains were cultured in 2 mL of TSB media containing 50 µg/mL of apramycin for two days in a round-bottom tube.
To measure the production levels of actinorhodin, undecylprodigiosin, and coelimycin P2, 500 μL of culture were harvested and mixed with an equal volume of methanol. The samples were vortexed for one hour and centrifuged at 20,000 g for 10 min. The 200 μL of supernatants were transferred to a 96-well plate, and absorbance was measured at 633 nm, 530 nm, and 460 nm using a SYNERGY H1 plate reader (BioTek Instruments, Winooski, VT, USA, 11-120-533) to measure the production of actinorhodin, undecylprodigiosin, and coelimycin P2, respectively. The absorbance of the fresh TSB medium was used as the baseline, and the production levels of each strain were measured and normalized to the absorbance of the wild-type S. coelicolor A3(2) strain.
The selected strains, which presented high metabolites production, and the wild-type S. coelicolor A3(2) strain were cultured in TSB media containing 50 µg/mL of apramycin for 3 d in a round-bottom tube. Then, 100 μL of culture containing the strains was transferred to 2 mL of fresh TSB media containing 50 µg/mL of apramycin and cultured for an additional 2 d. The broth was then transferred to 50 mL of MS media containing 50 µg/mL of apramycin in a flask and cultivated for 96 h. Every 12 h, 500 μL of samples were harvested and mixed with 500 μL of methanol, vortexing for one hour. After centrifugation at 20,000 g for 10 min, the 200 μL of supernatants were transferred to a microplate, and absorbance was measured to quantify the production levels of metabolites. The absorbance of the fresh TSB medium was used as the baseline, and the production levels of each strain were measured and normalized to the absorbance of the wild-type S. coelicolor A3(2) strain. Flask cultivation was independently performed in triplicate. To confirm the sequences of expressed sgRNAs in the engineered strains, the plasmids were subsequently extracted using the ZymoPure II Plasmid Midiprep Kit (Zymo Research Co., Tustin, CA., USA, D4201), and sequences of sgRNAs were confirmed via sequence analysis.
Quantifications of malonyl-CoA and acetyl-CoA
The engineered S. coelicolor A3(2) strains and the wild-type S. coelicolor A3(2) strain were cultured in a round bottom tube with 2 mL of TSB media containing 50 µg/mL of apramycin for 3 d. Then, 100 μL of culture containing the strains was transferred to 2 mL of fresh TSB media containing 50 µg/mL of apramycin and cultured for an additional 2 d. The 1 mL of cultured broths were transferred to 50 mL of MS media containing 50 µg/mL of apramycin in a flask and cultivated for 24 h. 1 mL of wet cells were harvested and pelleted using centrifugation at 15,000 g for 10 min. After removing supernatant, collected cells were resuspended in 1 mL of 6.0 % perchloric acid and 300 µL of 3 M potassium carbonate was added into each sample for neutralization. Each sample was vortexed at 4°C for 30 min and centrifugated at 15,000 g for 1 min at 4°C. Each supernatant was passed through a 0.22 nm syringe filter and 200 µL of samples were used for analysis by reverse phase HPLC with a C18 reverse phase column59. Flask cultivation of each strain was performed in triplicate.
Construction of pCap-00 plasmid and in vivo capture of FK506 BGC
To capture BGCs, two sgRNAs capable of cleaving both sides of the BGC were inserted into the pCRISPomyces-2BD plasmid using the Golden Gate assembly. The sgRNA sequences were then prepared using PCR amplification with the constructed plasmid as a template. The regions on both sides of the BGC, which were ~1 kb long and capable of recombination after cleavage, were prepared using PCR with the genomic DNA of S. tsukubaensis NRRL 18488 as a template. To create sequences of TTAATTAA and TCTAGA, the cleavage sites of the PacI and XbaI restriction enzymes, additional sequences were added at the 5’ sites of the primers FK506_HR_1_FW and FK506_HR_2_RV. Each prepared fragment was digested with the BsaI restriction enzyme and ligated with the digested pCRISPomyces-2BD plasmid using XbaI and EcoRI restriction enzymes by combining two sgRNAs between the two homologous regions (Supplementary Data 1). The constructed plasmid was verified using colony PCR and sequencing. The confirmed plasmid, pCap-00, was transformed into E. coli ET12567/pUZ8002 via electroporation and subsequently transferred into S. tsukubaensis NRRL 18488 by conjugation. For conjugation, 100 µL of spore stock, concentrated to 1.0 × 108 CFU/mL, were used. After conjugation, the plate was incubated at 30oC for 14 d and the formed exconjugants were inoculated into 50 mL TSB media containing 50 µg of apramycin. The strains were cultured in flasks for 7 d, and the plasmids were subsequently extracted using the ZymoPure II Plasmid Midiprep Kit (Zymo Research Co., Tustin, CA., USA, D4201). To confirm the capture of the BGC, eight sites in the BGC were amplified by PCR using the extracted plasmids and S. tsukubaensis NRRL 18488 genomic DNA as a template. The PCR bands were identified by electrophoresis on a 0.8% agarose gel and subsequently used for sequencing analysis (Supplementary Fig. 8c).
Heterologous expression of FK506 BGC in S. coelicolor M1146 strain
The plasmid for expressing BGC was prepared using a combination of pSET-BAC plasmid10 and pJKR-H-mphR plasmid54. Both plasmids were used as templates for PCR amplification, and each PCR product was digested using a restriction enzyme. The product of the pSET-BAC plasmid was digested with Nsil and SwaI, and that of pJKR-H-mphR was digested with BaegI and NaeI. After purification, digested products were combined by ligation, resulting in plasmid pExp-00. pCap-00 harboring the FK506 BGC was linearized using restriction enzymes PacI and XbaI, and pExp-00 was digested using restriction enzymes PacI and AvrII after linearization via PCR amplification. To remove the restriction enzyme after digestion, the linearized BGC was heated at 60oC for 20 min, and the linearized pExp-00 was purified using a DNA purification kit (Promega, WI, USA). After preparation, the two linear fragments were ligated together and transferred to E. coli EPI400 using heat shock. The strain containing the desired plasmid was selected on LB agar plates containing 50 µg/mL of apramycin and carbenicillin. To eliminate template contamination, only white colonies were selected for confirmation (Supplementary Fig. 9a). The plasmid confirmed using colony PCR and sequence analysis, was designated pFK (Supplementary Figs. 9b, c). To improve FK506 production, the promoter of fkbN was replaced with ermE*p and kasO*p by homologous recombination39. The gene cassettes, containing chloramphenicol resistance gene and promoter sequences, were prepared by PCR amplification using pZA_ermE and pZA_kasO as templates, and transformed into E. coli EPI400 harboring pFK and pKD4 plasmids, expressing λ red recombinase. The resulting plasmids were confirmed using PCR amplification and sequence analysis and named pFK-EN and pFK-KN, respectively. The constructed plasmids were transformed into the S. coelciolor M1146 strain by conjugation. The exconjugants were cultured in TSB media containing 50 µg/mL of apramycin for 3 d. Then, 100 µL of inoculum was transferred to fresh TSB media containing 50 µg/mL of apramycin. One milliliter of the inoculum was transferred to 50 mL of fermentative media in flasks, similar to the media used for rapamycin production, and the strain was cultivated for 14 d in a flask. Samples were harvested daily and analyzed using reverse-phase HPLC with a C18 reverse-phase column4. The flask cultivations were performed in triplicates.
Sequencing library preparation
For genome sequencing, the genomic DNA was extracted as described previously60. The cell pellet was washed twice with 1 mL of 10 mM EDTA, and incubated for 45 min at 30 °C in 600 µL of lysozyme solution (2 mg/mL lysozyme and 40 mM EDTA). After treatment of lysozyme, cells were centrifuged down and the supernatant was removed. The genomic DNA was extracted using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA, A1120) according to the manufacturer’s instruction. The sequencing library was prepared by using Illumina DNA PCR-Free Prep kit (Illumina Inc., San Diego, CA, USA, 20041797) according to the manufacturer’s instruction.
For RNA-seq, RNA was extracted as described previously61 About 300 µL of wet cell pellet was resuspended in 1 mL of resuspension buffer (25 mM Tris-HCl pH 8.0, 10 mM EDTA, 50 mM glucose and 2 mg/mL lysozyme) and incubated at 30 °C for 10 min. After incubation, cells were centrifuged down and the supernatant was removed. The cell pellet was resuspended in 550 µL of lysis buffer (50 mM sodium acetate pH 5.2, 10 mM EDTA and 1% SDS). The cell suspension was mixed with 600 µL of phenol:chloroform = 5:1 solution and incubated at 65 °C for 5 min. After incubation, the mixture was centrifuged at 16,000 g for 20 min and upper aqueous phase was saved. The total RNA was extracted from the supernatant by using a Zymo RNA Clean & Concentrator-5 (Zymo Research, Irvine, CA, USA, R1014), and DNA contamination was removed by treating DNase I (New England Biolabs, Ipswich, MA, USA, M0303L). From the total RNA, ribosomal RNA was removed by using RiboRid method as described previously61,62,63. DNase I-treated RNA was hybridized with rRNA-specific oligo probes. A 33 µL hybridization reaction mixture containing 1 µg of total RNA, 1.5 µL of 10× DNase I Reaction Buffer (New England BioLabs, B0303S), 15 µL of hybridization buffer (90 mM Tris-HCl pH 7.5 and 200 mM KCl), 1 µL of oligo probe mixture (Supplementary Data 5), and 2 µL of 50 mM MgCl2 was heated to 90 °C for 1 sec and cooled to 65 °C in a thermal cycler. After hybridization, 10 U of Hybridase™ Thermostable RNase H (LGC Biosearch Technologies, Hoddesdon, UK, E0038-5D) was added to the reaction mixture, and the mixture was incubated at 65 °C for 20 min, 90 °C for 1 sec, and 65 °C for 10 min in a thermal cycler. After incubation, the reaction was cleaned using a Zymo RNA Clean & Concentrator-5 kit, and the oligo probes were removed by DNase I treatment. For DNase I treatment, 10 U of DNase I and 5 µL of 10× DNase I Reaction Buffer were used in a total reaction volume of 50 µL. The reaction was carried out through consecutive 5 min incubations at 25, 30, 35, 40, and 45 °C. Afterward, the reaction was cleaned using a Zymo RNA Clean & Concentrator-5 kit, and the rRNA-depleted RNA was subjected to sequencing library preparation using the TruSeq Stranded mRNA Library Prep Kit (Illumina, RS-122-2201) according to the manufacturer’s instructions.
High-throughput sequencing and data processing
The sequencing libraries were sequenced using the Illumina NextSeq 1000 system with 2 × 50 bp read length, and the raw reads were processed using the CLC Genomics Workbench (CLC Bio, Aarhus, Denmark, 832023). The reads were mapped to the reference genome (accession number: NC003888.3 [https://www.ncbi.nlm.nih.gov/nuccore/NC_003888.3?report=genbank]) or the plasmid sequence with the following parameters: mismatch cost = 2, insertion cost = 2, deletion cost = 3, and length fraction = 0.9, and only uniquely mapped reads were used for analysis. Due to the inherent challenges in sequencing high G-C genome, only mutations with high fidelity (frequency > 80% and minimum coverage of 5) were regarded as the genuine mutations60. To determine the differentially expressed genes (DEGs) across the conditions, gene expression was normalized by using DESeq2 package, and genes with |expression fold change | > 2 and p-value < 0.01 were regarded as DEGs64.
Statistics and reproducibility
To perform statistical analysis, GraphPad Prism 8 and Excel 365 were used. All experiments included in this study were performed three times at least. Results of experiments were presented as mean ± standard deviation, as indicated in legends of figures. To determine P values, an unpaired two-tailed Student t-test was performed. P values < 0.05 were considered statistically significant. No statistical method was used to predetermine sample size, and no data was excluded from the analysis. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to support the conclusion of the paper is provided in the Supplementary Information and Source Data file. Source data are provided with this paper. The sequencing raw data files were deposited in the European Nucleotide Archive (ENA) under the accession number https://www.ebi.ac.uk/ena/browser/view/PRJEB79564. Source data are provided with this paper.
References
Takahashi, K., Yoshihara, T. & Kurosawa, K. Ushikulides A and B, immunosuppressants produced by a strain of Streptomyces sp. J. Antibiot. (Tokyo) 58, 420–424 (2005).
Noomnual, S., Thasana, N., Sungkeeree, P., Mongkolsuk, S. & Loprasert, S. Streptanoate, a new anticancer butanoate from Streptomyces sp. DC3. J. Antibiot. (Tokyo) 69, 124–127 (2016).
Matselyukh, B. et al. N-methylphenylalanyl-dehydrobutyrine diketopiperazine, an A-factor mimic that restores antibiotic biosynthesis and morphogenesis in Streptomyces globisporus 1912-B2 and Streptomyces griseus 1439. J. Antibiot. (Tokyo) 68, 9–14 (2015).
Wu, Q. B., Zhang, X. Y., Chen, X. A. & Li, Y. Q. Improvement of FK506 production via metabolic engineering-guided combinational strategies in Streptomyces tsukubaensis. Microb. Cell. Fact. 20, 1–11 (2021).
Wang, R. et al. A biobricks metabolic engineering platform for the biosynthesis of anthracyclinones in streptomyces coelicolor. ACS Synth. Biol. 11, 4193–4209 (2022).
Qi, Y., Nepal, K. K. & Blodgett, J. A. V. A comparative metabologenomic approach reveals mechanistic insights into Streptomyces antibiotic crypticity. Proc. Natl. Acad. Sci. USA 118, 31 (2021).
Enghiad, B. et al. Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination. Nat. Commun. 12, 1171 (2021).
Gren, T. et al. Characterization and engineering of Streptomyces griseofuscus DSM 40191 as a potential host for heterologous expression of biosynthetic gene clusters. Sci. Rep. 11, 18301 (2021).
Chen, M. et al. Discovery of novel septacidin congeners from a high yield heterologous expression strain Streptomyces albus 1597. J. Antibiot. (Tokyo) 75, 172–175 (2022).
Kim, D. K., Gu, B., Kim, D. G. & Oh, M. K. Quorum sensing-based metabolic engineering of the precursor supply in Streptomyces coelicolor to improve heterologous production of neoaureothin. Biotechnol. Bioeng. 120, 2039–3044, (2023).
Van de Kooij, B., Kruswick, A., van Attikum, H. & Yaffe, M. B. Multi-pathway DNA-repair reporters reveal competition between end-joining, single-strand annealing and homologous recombination at Cas9-induced DNA double-strand breaks. Nat. Commun. 13, 5295 (2022).
Qiao, J. et al. Co-expression of Cas9 and single-guided RNAs in Escherichia coli streamlines production of Cas9 ribonucleoproteins. Commun. Biol. 2, 161 (2019).
Zhang, Y. et al. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 10, 1053 (2019).
Zhang, M. M. et al. CRISPR-Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat. Chem. Biol. 13, 607–609 (2017).
Low, Z. J. et al. Identification of a biosynthetic gene cluster for the polyene macrolactam sceliphrolactam in a Streptomyces strain isolated from mangrove sediment. Sci. Rep. 8, 1–13 (2018).
Van Bergeijk, D. A. et al. The ubiquitous catechol moiety elicits siderophore and angucycline production in Streptomyces. Commun. Chem. 5, 14 (2022).
Ameruoso, A., Villegas Kcam, M. C., Cohen, K. P. & Chappell, J. Activating natural product synthesis using CRISPR interference and activation systems in Streptomyces. Nucleic Acids Res. 50, 7751–7760 (2022).
Kudo, K. et al. In vitro Cas9-assisted editing of modular polyketide synthase genes to produce desired natural product derivatives. Nat. Commun. 11, 4022 (2020).
Thomas, T. Off-target Cas9 crystallized. Nat. Struct. Mol. Biol. 29, 1–1 (2022).
Hennig, S. L. et al. Evaluation of mutation rates, mosaicism and off target mutations when injecting Cas9 mRNA or protein for genome editing of bovine embryos. Sci. Rep. 10, 1–9 (2020).
Hoijer, I. et al. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 13, 627 (2022).
Tong, Y., Weber, T. & Lee, S. Y. CRISPR/Cas-based genome engineering in natural product discovery. Nat. Prod. Rep. 36, 1262–1280 (2019).
Alberti, F. & Corre, C. Editing streptomycete genomes in the CRISPR/Cas9 age. Nat. Prod. Rep. 36, 1237–1248 (2019).
Wang, K. et al. Multi-layer controls of Cas9 activity coupled with ATP synthase over-expression for efficient genome editing in streptomyces. Front. Bioeng. Biotechnol. 7, 304 (2019).
Ye, S., Enghiad, B., Zhao, H. & Takano, E. Fine-tuning the regulation of Cas9 expression levels for efficient CRISPR-Cas9 mediated recombination in Streptomyces. J. Ind. Microbiol. Biotechnol. 47, 413–423 (2020).
Tong, Y., Charusanti, P., Zhang, L., Weber, T. & Lee, S. Y. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth. Biol. 4, 1020–1029 (2015).
Jiang, Y. H. et al. Fine-tuning Cas9 activity with a cognate inhibitor AcrIIA4 to improve genome editing in streptomyces. ACS Synth. Biol. 10, 2833–2841 (2021).
Li, L. et al. CRISPR-Cpf1-assisted multiplex genome editing and transcriptional repression in streptomyces. Appl. Environ. Microbiol. 84, e00827–18 (2018).
Zhao, Y. et al. Multiplex genome editing using a dCas9-cytidine deaminase fusion in Streptomyces. Sci China Life Sci 63, 1053–1062 (2020).
Van Rosmalen, M., Krom, M. & Merkx, M. Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins. Biochemistry 56, 6565–6574 (2017).
Chen, J. S. et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017).
Pacesa, M. et al. Structural basis for Cas9 off-target activity. Cell 185, 4067–4081–, (2022).
Kim, H. et al. Enhancement of target specificity of CRISPR-Cas12a by using a chimeric DNA-RNA guide. Nucleic Acids Res. 48, 8601–8616 (2020).
Cobb, R. E., Wang, Y. & Zhao, H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 4, 723–728 (2015).
Hoff, G. et al. Multiple and variable NHEJ-like genes are involved in resistance to DNA damage in Streptomyces ambofaciens. Frontiers in Microbiology 7, 1901 (2016).
Gu, B. et al. Heterologous overproduction of oviedomycin by refactoring biosynthetic gene cluster and metabolic engineering of host strain Streptomyces coelicolor. Microb. Cell Fact. 22, 212 (2023).
Jo, H. G. et al. Comparative genomic analysis of Streptomyces rapamycinicus NRRL 5491 and its mutant overproducing rapamycin. Sci. Rep. 12, 10302 (2022).
Gomez-Escribano, J. P. & Bibb, M. J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 4, 207–215 (2011).
Serra-Moreno, R., Acosta, S., Hernalsteens, J. P., Jofre, J. & Muniesa, M. Use of the lambda Red recombinase system to produce recombinant prophages carrying antibiotic resistance genes. BMC Mol. Biol 7, 31 (2006).
Tian, J. et al. Developing an endogenous quorum-sensing based CRISPRi circuit for autonomous and tunable dynamic regulation of multiple targets in Streptomyces. Nucleic Acids Res. 48, 8188–8202 (2020).
Yang, Y. H. et al. Novel method for detection of butanolides in Streptomyces coelicolor culture broth, using a His-tagged receptor (ScbR) and mass spectrometry. Appl. Environ. Microbiol. 71, 5050–5055 (2005).
Chen, X. et al. A new bacterial tRNA enhances antibiotic production in Streptomyces by circumventing inefficient wobble base-pairing. Nucleic Acids Res. 50, 7084–7096 (2022).
Florova, G., Kazanina, G. & Reynolds, K. A. Enzymes involved in fatty acid and polyketide biosynthesis in Streptomyces glaucescens: role of FabH and FabD and their acyl carrier protein specificity. Biochemistry 41, 10462–10471 (2002).
Lin, H., Vadali, R. V., Bennett, G. N. & San, K. Y. Increasing the acetyl-CoA pool in the presence of overexpressed phosphoenolpyruvate carboxylase or pyruvate carboxylase enhances succinate production in Escherichia coli. Biotechnol. Prog. 20, 1599–1604 (2004).
Krivoruchko, A., Zhang, Y., Siewers, V., Chen, Y. & Nielsen, J. Microbial acetyl-CoA metabolism and metabolic engineering. Metab. Eng. 28, 28–42 (2015).
Yang, H. & Patel, D. J. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol. Cell 67, 117–127 (2017).
Shin, J, et al. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 3, e1701620 https://www.science.org/doi/10.1126/sciadv.1701620 (2017).
Nishimasu, H. et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014).
Guo, M. et al. Structural insights into a high fidelity variant of SpCas9. Cell Res. 29, 183–192 (2019).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Tong, Y. et al. CRISPR-Cas9, CRISPRi and CRISPR-BEST-mediated genetic manipulation in streptomycetes. Nat. Protoc. 15, 2470–2502 (2020).
Jiang, W. J. et al. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun. 6, 8101 (2015).
Wang, K. H. et al. Defining synonymous codon compression schemes by genome recoding. Nature 539, 59–64 (2016).
Kim, D. G., Kim, M. & Oh, M. K. Enriching intracellular macrolides in Escherichia coli improved the sensitivity of bioluminescent sensing systems. Talanta 249, 123626 (2022).
Park, J., Bae, S. & Kim, J. S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016 (2015).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Dong, H. J. et al. Improved A40926 production from using the promoter engineering and the co-expression of crucial genes. J. Biotechnol. 324, 28–33 (2020).
Wang, W. S. et al. An Engineered Strong Promoter for Streptomycetes. Appl. Environ. Microb. 79, 4484–4492 (2013).
Tsuchiya, Y., Pham, U. & Gout, I. Methods for measuring CoA and CoA derivatives in biological samples. Biochem. Soc. T. 42, 1107–1111 (2014).
Lee, N. et al. Thirty complete Streptomyces genome sequences for mining novel secondary metabolite biosynthetic gene clusters. Sci. Data 7, 55 (2020).
Lee, Y. et al. Genome-scale determination of 5′ and 3′ boundaries of RNA transcripts in Streptomyces genomes. Sci. Data 7, 436 (2020).
Choe, D. et al. RiboRid: A low cost, advanced, and ultra-efficient method to remove ribosomal RNA for bacterial transcriptomics. Plos Genet. 17, e1009821 (2021).
Lee Y., et al. Genome-scale determination of 5′ and 3′ boundaries of RNA transcripts in Streptomyces genomes. figshare.https://doi.org/10.6084/m9.figshare.c.5044730 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Acknowledgements
This study was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean Government (MSIT) (No. RS-2024-00509338 to M.-K.O., No. RS-2024-00357320 to M.-K.O. and No. RS-2021-NR056596 to B.-K.C.).
Author information
Authors and Affiliations
Contributions
D.G.K., B.-K.C., and M.-K.O. conceived of and designed the study. D.G.K., B.G., Y.C., and J.H. developed the plasmids and strains used in this study. D.G.K. and J.H. cultivated all strains. B.G. and Y.C. performed metabolite analysis. The whole genome sequencing and RNA-sequencing were performed by Y.L. and G.K. D.G.K., B.-K.C., and M.-K.O. wrote and edited the manuscript. All the authors discussed the results of the study and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Christophe Corre, Shuangxia Jin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, D.G., Gu, B., Cha, Y. et al. Engineered CRISPR-Cas9 for Streptomyces sp. genome editing to improve specialized metabolite production. Nat Commun 16, 874 (2025). https://doi.org/10.1038/s41467-025-56278-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56278-y
This article is cited by
-
Exploring pigment-producing Streptomyces as an alternative source to synthetic pigments: diversity, biosynthesis, and biotechnological applications. A review
World Journal of Microbiology and Biotechnology (2025)
