
Abstract
Human lifespan is shaped by genetic and environmental factors. To enable precision health, understanding how genetic variants influence mortality is essential. We conducted a survival analysis in European ancestry participants of the UK Biobank, using age-at-death (N=35,551) and last-known-age (N=358,282). The associations identified were predominantly driven by cancer. We found lifespan-associated loci (APOE, ZSCAN23) for common variants and six genes where burden of loss-of-function variants were linked to reduced lifespan (TET2, ATM, BRCA2, CKMT1B, BRCA1, ASXL1). Additionally, eight genes with pathogenic missense variants were associated with reduced lifespan (DNMT3A, SF3B1, TET2, PTEN, SOX21, TP53, SRSF2, RLIM). Many of these genes are involved in oncogenic pathways and clonal hematopoiesis. Our findings highlight the importance of understanding genetic factors driving the most prevalent causes of mortality at a population level, highlighting the potential of early genetic testing to identify germline and somatic variants increasing one’s susceptibility to cancer and/or early death.
Similar content being viewed by others

Introduction
Human lifespan is a complex trait influenced by both genetic and environmental factors and their interactions1. According to previous studies, genetics accounts for less than 10%2 or up to 25% of the heritability of longevity3. Identifying the genetic variants that contribute to earlier death or prolonged survival can highlight key biological pathways linked to lifespan and inform genetic testing for general health and screening and enabling precision health. Previous genome-wide association studies (GWAS) have identified over 20 associated loci including APOE4,5, CHRNA3/56, HLA-DQA1 and LPA7. Recently, a burden analysis of protein-truncating variants from whole-exome sequencing (WES) data identified four additional genes (BRCA2, BRCA1, ATM, and TET2) linked to reduced lifespan8. However, most previous research on lifespan genetics has predominantly used proxy data, such as parents’ age at death, due to a lack of proband lifespan data. While proxy-based GWAS have been useful to gain genomic insights into age-related diseases in cohorts primarily composed of middle-aged individuals, and show some consistency with associations related to lifespan8, they may fail to fully capture the genetic influences that directly impact individual lifespan, particularly CHIP-related somatic variants9. On the other hand, some studies have employed logistic regression models on cases of extreme longevity and younger controls10,11,12. This approach may offer new insights by focusing on exceptionally long-lived individuals, yet they can be limited and costly. Moreover, replication of borderline significant variants remains an issue due to varying case definitions across studies, with some defining cases as individuals who survive to ages beyond 90 or 100 years or using the 90th or 99th survival percentiles as the age cutoff.
In this study, we carried out a genetic analysis of direct mortality data in the UK Biobank (UKB), the genetic database with the largest number of reported deaths (35,551 subjects) and aged individuals (344,237 subjects over 60 years old). To assess the association of genetic variants with lifespan in a survival analysis, we performed GWAS of common variants imputed from microarray data as well as burden/sequence kernel association test-optimized (SKAT-O) association of rare non-synonymous variants from WES data.
Results
Genome-wide association analyses in imputed array data
Our GWAS assessed 10,104,569 common variants (minor allele frequency (MAF) \(\ge\) 0.1%) using Martingale residuals on 393,833 individuals including 35,551 deceased subjects (mean age at death: 71.2 years) and 358,282 living subjects (mean current age: 70.7) from UKB (Supplementary Table 1)13. Two loci reached genome-wide significance (GWS) (\(p\) < \(5.0\times {10}^{-8}\)) on chromosomes 19 and 6 (Fig. 1A). On chromosome 19, rs429358 was the lead variant at the APOE locus (β = 0.013, p = 3.9 × 10−45), MAF = 15.6%). We tested whether the presence of APOE-ε4 was enriched in certain primary causes of death. Among the top four causes of death, each representing over 5% of total deaths (Fig. 1B), only those due to “Diseases of the circulatory system” (Chi-square \(p\) = 1.6 × 10−16) and “Diseases of the nervous system” (\(p\) = 1.1 × 10−71) showed a significant enrichment in the proportion of ε4 carriers compared to the prevalence of ε4 carriers among all subjects (Fig. 1C). On chromosome 6 locus overlapping ZSCAN23, the top genome-wide significant variant was rs6902687, located 2.2 kb upstream of the transcription start site (TSS) (rs6902687_C: β = 0.004, p = 2.7 × 10⁻⁸, MAF = 36.6%). This variant is in almost perfect linkage disequilibrium (R² > 0.99) with three other significant variants in this region, including rs13215804_G (located 4.2 kb upstream of the TSS), rs111859903_G (located in an intron) and rs13190937_A (situated in the 5’ untranslated region) (Fig. 1D).

A Manhattan plot. B The proportion of cause of death for the top 4 categories, each accounting for more than 5% of total deaths. C Association of causes of death with APOE-ε4 genotype. D LocusZoom and E colocalization plots at the ZSCAN23 locus, colocalized with ZSCAN23 eQTL in pancreatic tissue in GTEx. PP4 posterior probability of colocalization.
To explore a potential regulatory function for variants at the ZSCAN23 locus, we investigated whether the lead SNPs were expression quantitative trait loci (eQTLs) in the Genotype-Tissue Expression Project (GTEx) v8 database. rs13190937 was significantly associated with increased ZSCAN23 expression in pancreatic tissue and the GWAS on Martingale residuals signal colocalized with the ZSCAN23 expression quantitative trait loci (eQTL) (posterior probability of colocalization (PP4) = 0.934; Fig. 1E). Phenome-wide association study analysis (PheWAS) using PheWeb14 based on UKB Neale v1 dataset shows that the main associations of rs13190937 are with celiac disease and intestinal malabsorption (\(p\) = 1.8\(\times {10}^{-57}\), OR = 1.003) (Supplementary Fig. 1).
In sex-stratified GWAS (180,970 males and 212,863 females), the APOE locus was again linked to reduced lifespan in both males and females (Supplementary Table 1 and Supplementary Fig. 2A, B). In males, a significant association with reduced lifespan was observed for rs577106756_A located in intron of PRKD3 on chromosome 2 (β = 0.09, p = 3.2\(\times {10}^{-8}\), MAF = 0.1%). PheWAS analysis, based on the UKB Neale v1 dataset, revealed that rs577106756_A was associated with ICD10 code C10.9, Malignant neoplasm of oropharynx, unspecified, as the primary cause of death as the primary cause of death (\(p\) = 4.5\(\times {10}^{-8}\), OR = 1.04), and self-reported “Stomach Cancer” (\(p\) = 6.1\(\times {10}^{-7}\), OR = 1.003) (Supplementary Fig. 2C). Additionally, a borderline significant association with reduced lifespan was observed at rs35705950_T, located between MUC5AC and MUC5B on chromosome 11 (β = 0.01, p = 6.6\(\times {10}^{-8}\), MAF = 11.2%) (Supplementary Fig. 2D). This variant was notably linked to increased MUC5B expression in lung tissue with the GWAS on Martingale residuals signal colocalizing with a MUC5B eQTL (PP4 = 0.99; Supplementary Fig. 2E). PheWAS analysis showed associations with a diagnosis of pulmonary fibrosis (\(p\) = 4.4 × 10−13), OR = 1.002), “Other interstitial pulmonary diseases with fibrosis” as the primary cause of death (\(p\) = 1.7\(\times {10}^{-5}\), OR = 1.001), and paternal history of lung cancer (\(p\) = 2.1\(\times {10}^{-4}\), OR = 1.004), but no association with maternal history of lung cancer (\(p\) = 0.07) (Supplementary Fig. 2F). In female, a significant association with reduced lifespan was observed for rs547541271_T, located in the intron of CELF2 on chromosome 10 (β = 0.04, p = 3.1\(\times {10}^{-8}\), MAF = 0.3%). PheWAS analysis indicated rs547541271_T was associated with a diagnosis of “Myositis” (\(p\) = 1.0\(\times {10}^{-6}\), OR = 1.002), and self-reported Polycystic Ovaries/Polycystic Ovarian Syndrome (\(p\) = 2.5\(\times {10}^{-5}\), OR = 1.003) (Supplementary Fig. 2G).
Further validation of these significant variants was carried out using data from the FinnGen and LifeGen cohorts. Specifically, for common variants, we queried FinnGen (https://r11.finngen.fi/) and obtained summary statistics15 from the LifeGen consortium via the GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics) to assess their association in independent datasets. In FinnGen, rs13190937 was not significantly associated with the “Death” phenotype (p = 0.2), while it was significantly associated with a decrease in “Parental age at death” in the UKB and LifeGen consortium (\(p\) = 1.4\(\times {10}^{-4}\), β = −0.015). Similarly, rs35705950 showed a significant association with increased death in FinnGen (\(p\) = 6.0\(\times {10}^{-3}\), β = 0.034) and with decrease in “Parental age at death” in the UKB and LifeGen consortium (\(p\) = 6.6\(\times {10}^{-3}\), β = −0.023). However, neither rs577106756 nor rs547541271 showed a significant association with the “Death” phenotype in FinnGen (p = 0.55 and 0.93, respectively) (Supplementary Table 2).
Gene-based rare variant association analyses in whole-exome data
Among 26,230,624 variants with MAF < 1%, a total of 1,830,070 variants (17,174 genes) were annotated as loss-of-function (LoF) or missense variants. We excluded 199 genes with fewer than 10 total variant carriers, resulting in 476,447 predicted LoF variants (15,908 genes with a median of 23 variants per gene), 751,523 missense variants predicted as damaging by AlphaMissense (15,212 genes with a median of 37 variants per gene), and 262,866 missense variants predicted as damaging by rare exome variant ensemble learner (REVEL) (9231 genes with a median of 15 variants per gene). Of variants classified by each, 23.4% of AlphaMissense and 66.8% of REVEL variants were also pathogenic by the other classifier. A list of SNPs list used for the gene-based analyses is provided in Supplementary Data 1.
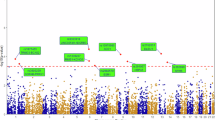
We identified six genes whose burden of LoF variants is significantly associated with reduced lifespan: TET2 (\(p\) = 2.6\(\times {10}^{-34}\)), ATM (\(p\) = 6.4\(\times {10}^{-10}\)), BRCA2 (\(p\) = 1.2\(\times {10}^{-33}\)), CKMT1B (\(p\) = 4.3\(\times {10}^{-7}\)), BRCA1 (\(p\) = 5.6\(\times {10}^{-12}\)) and ASXL1 \(\left(\right.p\) = 1.3 × 10−51) (Fig. 2A and Table 1). All of these but CKMT1B also showed gene-wide significance in a direction-agnostic (SKAT-O) approach (Supplementary Fig. 3A). Additionally, in eight genes, the burden of missense variants predicted as pathogenic by AlphaMissense was associated with reduced lifespan: DNMT3A (\(p\) = 6.9\(\times {10}^{-12}\)), SF3B1 (\(p\) = 1.9\(\times {10}^{-13}\)), TET2 (\(p\) = 9.2\(\times {10}^{-8}\)), PTEN (\(p\) = 1.6\(\times {10}^{-8}\)), SOX21 (\(p\) = 2.2\(\times {10}^{-8}\)), TP53 (p = 8.6 × 10–17), SRSF2 (\(p\) = 1.8 × 10−94) and RLIM (\(p\) = 6.0\(\times {10}^{-7}\)) (Fig. 2B). Lastly, three genes showed gene-wide significance for burden of missense variants predicted by REVEL: DNMT3A (\(p\) = 5.2 × 10−11), PTEN (\(p\) = 1.2\(\times {10}^{-7}\)), and TP53 (\(p\) = 2.2 × 10−9) (Supplementary Fig. 4 and Supplementary Table 3). SKAT-O identified additional associations with pathogenic missense variants predicted by AlphaMissense in C1orf52 (\(p\) = 7.2\(\times {10}^{-8}\)) and IDH2 (p = 5.4 × 10−42) (Supplementary Fig. 3B), and by REVEL in NMNAT2 (\(p\) = 6.7\(\times {10}^{-7}\)) and TERT (\(p\) = 3.3\(\times {10}^{-10}\)) (Supplementary Fig. 3C and Supplementary Table 3).

Rare variant burden association with lifespan, considering loss-of-functions (A) and AlphaMissense pathogenic variants (B). Genes highlighted in red represent those not previously identified as significant in ref. 8. A gene-wide significance threshold of \(p\) = 7.4\(\times {10}^{-7}\) was applied.
In addition, we validated these findings within the UKB dataset using two approaches: an independent sample separate from our discovery data and a five-fold cross-validation (CV) within the discovery cohort. This independent validation included 73,281 subjects who were not categorized as having European ancestry based on genetic ethnic grouping. These participants were classified into five groups based on their self-reported ethnicity (Field: 21000): White (66.3%), Asian (14.4%), Black (9.9%), Other (5.7%), and Mixed (3.7%). Among the 21 novel genes identified in the discovery, four achieved significance under the Bonferroni correction threshold 1.1\(\times {10}^{-3}\) (0.05/42) in this validation cohort: BRCA2 (\(p\) = 1.1\(\times {10}^{-3}\), burden), ASXL1 (\(p\) = 1.2\(\times {10}^{-5}\), burden; \(p\) = 6.7\(\times {10}^{-6}\), SKAT-O) with LoF variants and (IDH2 \(p\) = 2.0\(\times {10}^{-7}\), SKAT-O) and SRSF2 (\(p\) = 9.2\(\times {10}^{-9}\), burden; \(p\) = 2.2 × 10−10), SKAT-O) with pathogenic missense variants predicted by AlphaMissense (Supplementary Table 4). To further validate our findings, we performed five-fold CV within the discovery dataset of 393,833 individuals, dividing it into five folds. For each fold, 80% of the data (315,066 individuals) was used for analysis. The results across folds were highly consistent. For example, TET2, BRCA2, BRCA1, ASXL1 (LoF), SF3B1, DNMT3A, IDH2, TP53, SRSF2 (AlphaMissense) and DNMT3A (REVEL) achieved gene-wide significance across all five folds. Except for CKMT1B (LoF), C1orf52, TET2, RLIM (AlphaMissense) and NMNAT2 (REVEL), all other genes that showed significance in the main analysis were significant in at least 3 out of 5 folds. This consistency across folds confirms the robustness of the associations identified in our study. Fold-specific results for each gene and variant category are provided in Supplementary Data 2.
For sex-specific gene-based analysis, an additional four genes not identified in the whole-cohort analysis showed gene-wide significance in males by either burden or SKAT-O: CDKN1A and PTPRK (LoF); COA7 and TG (AlphaMissense) (Supplementary Figs. 5A, 6A and Supplementary Table 5). In females, we identified five additional genes associated with reduced lifespan: PORCN (AlphaMissense); UGT1A8, CBX3, IFITM10, and OLIG1 (REVEL) (Supplementary Figs. 5B, 6B and Supplementary Table 6).
Gene-burden survival analysis
For the 14 gene-wide significant genes in the burden analyses, we assessed the association of variant carrier status with lifespan using Cox proportional hazards regression. Carriers of LoF variants in six genes were associated with decreased survival compared to non-carriers: CKMT1B (HR = 3.9, \(p\) = 2.6\(\times {10}^{-6}\)), ASXL1 (HR = 2.5, \(p\) = 6.2 × 10−33) (Fig. 3A), TET2 (HR = 2.3, \(p\) = 1.9 × 10−22), ATM (HR = 1.7, \(p\) = 3.0\(\times {10}^{-10}\)), BRCA2 (HR = 2.4, \(p\) = 9.7 × 10−39), and BRCA1 (HR = 2.2, \(p\) = 1.3 \(\times {10}^{-12}\)) (Supplementary Fig. 7A). Similarly, carriers of AlphaMissense-predicted pathogenic variants exhibited significantly earlier mortality compared to non-carriers on the following genes: DNMT3A (HR = 1.5, \(p\) = 1.4\(\times {10}^{-9}\)), SF3B1 (HR = 2.3, \(p\) = 4.4\(\times {10}^{-10}\)), PTEN (HR = 4.0, \(p\) = 1.1\(\times {10}^{-9}\)), SOX21 (HR = 1.9, \(p\) = 3.3\(\times {10}^{-8}\)), TP53 (HR = 3.9, \(p\) = 1.9\(\times {10}^{-14}\)), SRSF2 (HR = 5.8, \(p\) = 3.3 × 10−61), RLIM (HR = 3.1, \(p\) = 2.9\(\times {10}^{-4}\)) (Fig. 3B) and TET2 (HR = 1.5, \(p\) = 8.4\(\times {10}^{-7}\)) (Supplementary Fig. 7B). Carriers of pathogenic variants predicted by REVEL showed similar trends: DNMT3A (HR = 1.6, \(p\) = 2.4\(\times {10}^{-8}\)), PTEN (HR = 4.8, \(p\) = 1.0\(\times {10}^{-9}\)), and TP53 (HR = 2.5, \(p\) = 1.5\(\times {10}^{-8}\)) (Supplementary Fig. 7C).

Survival curves comparing carriers and non-carriers of variants on genes with a significant burden of loss-of-function (A) and AlphaMissense pathogenic (B) variants. For each gene, the survival curve includes Cox regression hazard ratio (HR), p value, the number of carriers, and their proportion within the total sample. A gene-wide significance threshold of \(p\) = 7.4\(\times {10}^{-7}\) was applied.
To explore the contribution of individual rare variants to mortality in each gene-wide significant gene in the burden and SKAT-O tests, we conducted Cox proportional hazards regression for each variant with a minor allele count (MAC) of three or more (Table 2). In total, 587 variants including LoF, AlphaMissense, and REVEL variants were examined. After applying a Bonferroni correction for multiple testing, setting the significance threshold at 8.3\(\times {10}^{-5}\) (0.05/599), we identified significant associations with reduced lifespan for four LoF variants: rs370735654 in TET2 (MAC = 17, HR = 7.9, \(p\) = 6.1\(\times {10}^{-10}\)), rs587779834 in ATM (MAC = 113, HR = 2.5, \(p\) = 3.1\(\times {10}^{-5}\)), rs80359705 in BRCA2 (MAC = 13, HR = 11.4, \(p\) = 2.5\(\times {10}^{-9}\)), and rs750318549 in ASXL1 (MAC = 201, HR = 2.8, \(p\) = 3.5\(\times {10}^{-19}\)). Additionally, significant associations with AlphaMissense variants were noted in seven genes, impacting lifespan: rs769009649 in C1orf52 (MAC = 62, HR = 3.3, \(p\) = 3.4\(\times {10}^{-7}\)), rs147001633 in DNMT3A (MAC = 269, HR = 1.8, \(p\) = 8.8\(\times {10}^{-6}\)), rs377023736 in SF3B1 (MAC = 12, HR = 7.3, \(p\) = 2.5 \(\times {10}^{-9}\)), rs121913502 in IDH2 (MAC = 45, HR = 6.8, \(p\) = 9.2\(\times {10}^{-25}\)), rs11540652 in TP53 (MAC = 5, HR = 11.5, \(p\) = 2.3\(\times {10}^{-5}\)), rs751713049 in SRSF2 (MAC = 51, HR = 6.7, \(p\) = 9.3\(\times {10}^{-31}\)) and rs75871009 in RLIM (MAC = 6, HR = 6.2, \(p\) = 6.0\(\times {10}^{-7}\)). For missense variants predicted by REVEL, rs201746612 in NMNAT2 (MAC = 5, HR = 11.0, \(p\) = 1.7\(\times {10}^{-6}\)), rs1043358053 in TERT (MAC = 5, HR = 16.8, \(p\) = 1.7\(\times {10}^{-8}\)), and rs11540652 in TP53 (MAC = 5, HR = 11.5, \(p\) = 2.3\(\times {10}^{-5}\)) were significantly linked to reduced lifespan (Supplementary Table 7).
Phenome-wide association studies
For the nine novel genes identified in the burden test (CKMT1B, ASXL1, DNMT3A, SF3B1, PTEN, SOX21, TP53, SRSF2 and RLIM), we examined the burden of LoF or pathogenic missense variants through PheWASs across 1670 UKB phenotypes including disease occurrences derived from electronic health record, self-reported family history, and physical measures (Supplementary Fig. 8). The burden of LoF variants in ASXL1 and AlphaMissense variants in DNMT3A, SF3B1, PTEN, TP53 and SRSF2 were strongly linked to an increased risk of leukemia: acute myeloid leukemia (ASXL1: Odds Ratio (OR) = 1.05; \(p\) = 8.6\(\times {10}^{-170}\); DNMT3A: OR = 1.03, \(p\) = 2.1\(\times {10}^{-150}\); SRSF2: OR = 1.3, \(p\) = 1.2\(\times {10}^{-195}\); TP53: OR = 1.05, \(p\) = 4.7\(\times {10}^{-35}\)), monocytic leukemia (DNMT3A: OR = 1.01, \(p\) = 2.5 × 10−9), chronic lymphoid leukemia (SF3B1: OR = 1.07, \(p\) = 4.1\(\times {10}^{-68}\)) and acute lymphoid leukemia (PTEN: OR = 1.01, \(p\) = 2.1\(\times {10}^{-14}\)). Additionally, the burden of LoF in CKMT1B was associated with hypopharynx cancer (OR = 1.03, \(p\) = 3.9\(\times {10}^{-26}\)), vertiginous syndromes (OR = 1.03, \(p\) = 3.0\(\times {10}^{-17}\)) and salivary glands cancer (OR = 1.03; \(p\) = 3.2\(\times {10}^{-12}\)). SOX21 burden was associated with increased acne (OR = 1.01, \(p\) = 6.9\(\times {10}^{-7}\)) and spinocerebellar disease (OR = 1.01, \(p\) = 2.3\(\times {10}^{-6}\)). Lastly, the burden of AlphaMissense variants in RLIM were associated with chromosomal anomalies and genetic disorders (OR = 1.02, \(p\) = 4.2\(\times {10}^{-16}\)), other and unspecified congenital anomalies (OR = 1.02, \(p\) = 8.2\(\times {10}^{-11}\)), malignant neoplasm of small intestine, including duodenum (OR = 1.02, \(p\) = 3.5\(\times {10}^{-6}\)) and cancer of oropharynx (OR = 1.02, \(p\) = 1.3\(\times {10}^{-5}\)).
Somatic mutation and clonal hematopoiesis of indeterminate potential
We computed the variant allelic fraction (VAF) per carrier for each variant included in the analysis. Generally, germline variants have a mean VAF close to 50%, while somatic variants’ mean VAF will be lower16. Thus, when an association is linked to clonal hematopoiesis of indeterminate potential (CHIP), we expect the distribution of VAF to be left-shifted compared to a normal distribution centered at VAF = 50%. Considering LoF variants, TET2 (mean VAF across variants [95% bootstrap confidence interval for the mean VAF] = 0.33 [0.31,0.34]) and ASXL1 (mean VAF = 0.32 [0.31,0.33]) burden test associations are supported by variants with a left-shifted VAF distribution (Supplementary Table 6 and Supplementary Fig. 9A). Similarly, considering pathogenic AlphaMissense variants, in DNMT3A (mean VAF = 0.24 [0.23–0.24]), TET2 (mean VAF = 0.36 [0.34,0.38]), TP53 (mean VAF = 0.28 [0.24,0.34]), SRSF2 (mean VAF = 0.30 [0.28,0.31]) and SF3B1 (mean VAF = 0.31 [0.26,0.37]) are also left-shifted and the observed associations may be linked to CHIP (Supplementary Table 8 and Supplementary Fig. 9B).
Discussion
In this study, we report several known and novel findings related to genetic risks associated with lifespan analyzing 393,833 European participants from the UKB. In the common variant GWAS, five independent loci associated with increased mortality risk were identified. In the gene-based analysis of rare non-synonymous variants, 16 genes were associated with lifespan via burden or SKAT-O tests.
Consistent with previous reports, rs429358, determining the APOE-ε4 allele dosage, was associated with decreased lifespan across both sexes. APOE-ε4 is well known for its associations with Alzheimer’s Disease17 and cardiovascular disease18. In our dataset, the proportion of ε4 carriers was significantly higher for deaths caused by “Disease of the circulatory system” and “Diseases of the nervous system” compared to the general prevalence of ε4 carriers, which could explain the effect of ε4 on lifespan. Examining the subcategories of these ICD-10 chapters, “Disease of the circulatory system” includes cardiovascular disease (I51.6), while “Diseases of the nervous system” covers Alzheimer’s disease (G30). We also identified a genome-wide association at the ZSCAN23 locus, which, while not previously reported in human lifespan studies, has been associated with rheumatoid arthritis19,20, multiple sclerosis19, and COVID-1920 in other studies. Our colocalization analysis revealed that the lifespan-associated signal colocalizes with a ZSCAN23 eQTL in pancreatic tissue with increased expression observed in minor allele carriers. Although the role of ZSCAN23 remains unclear, recent studies have linked its expression to pancreatic tumors, supporting our colocalization findings21. Sex-specific GWAS in males identified a significant association in PRKD3, which PheWAS linked to neoplasms and stomach cancer. PRKD3 has been highlighted as a potential oncogene in various cancer types22,23. Additionally, a borderline significant association was found in males between MUC5AC and MUC5B, which highly colocalizes with a MUC5B eQTL in lung tissue, and several studies have linked this variant to pulmonary disease like idiopathic pulmonary fibrosis24,25 and COVID-1926,27. In females, sex-stratified GWAS identified a variant in CELF2 associated with reduced lifespan. CELF2, an RNA-binding protein, is a candidate gene for certain neurological disorders28,29,30, and its activity has also been implicated in the development of ovarian and breast cancers31,32. Previously reported SNP associations with lifespan were concordant in our dataset but none of these passed the GWAS suggestive threshold (\(p\) = 1.0\(\times {10}^{-5}\)) except for those at the APOE locus (Supplementary Table 9). This phenomenon likely resulted from previous studies relying on proxy data such as parental age at death, which may capture a different set of genetic factors than direct proband mortality data.
In our gene-based rare variant analysis, 16 genes achieved gene-wide significance (\(p\) < 7.4\(\times {10}^{-7}\)) in either the burden or SKAT-O test. Four of these, TET2, ATM, BRCA2, and BRCA1, were reported in a previous rare-variant analysis of lifespan in UKB8. We identified 13 novel genes associated with lifespan—CKMT1B, ASXL1, C1orf52, DNMT3A, SF3B1, PTEN, SOX21, IDH2, TP53, SRSF2, RLIM, NMNAT2 and TERT—when assessing variants causing genetic LoF or missense variants classified as pathogenic by REVEL or AlphaMissense. ASXL1 was missed by a previous study considering protein truncating variants8, as they excluded variants annotated as end-truncation, notably the main ASXL1 variant driving CHIP (rs750318549 in Table 2). Of note, LoF and missense variant analyses identified mostly separate genes with only one overlap (TET2). This supports the use of both categories in rare variant analyses and may indicate that missense variants as classified by AlphaMissense capture a wider range of variation missed when only assessing LoF variants, which are generally interpreted as resulting in haploinsufficiency. Importantly, missense variants may lead to increased or decreased protein function. In our analyses, IDH2 was not gene-wide significant with the burden test (\(p\) = 1.4\(\times {10}^{-4}\), Table 1) but was highly significant with SKAT-O (\(p\) = 5.4\(\times {10}^{-42}\), Table 1). Since SKAT-O does not lose power when variants have differing directions of effect, this suggests that different mutations in IDH2 can lead to either increased or decreased lifespan. These results underline the gain in information achieved when studying rare missense variants as well as LoF using appropriate statistical techniques.
Strikingly, most of the genes we identified carrying lifespan-associated rare variants have been previously linked to cancer. TET2, ASXL1, DNMT3A, and SF3B1 are all known to harbor causal leukemia variants33,34,35,36, and somatic variants in SRSF2 have been described in myelodysplastic syndrome37. ATM, BRCA2, and BRCA1 mutations have been well characterized in breast, ovarian, and other cancers38,39,40. RLIM appears to be a regulator of estrogen-dependent transcription, an important pathway in breast cancer41, and has been recently described as a potential tumor suppressor42. PTEN and TP53 are well studied due to their critical role in genomic stability and are the two most mutated genes in human cancer43. IDH2 is also frequently mutated in many kinds of cancer44. The antisense long noncoding RNA SOX21-AS1, but not SOX21, has been linked to oral, cervical, and breast cancer45,46,47. A recent study found potential for CKMT1B expression as a prognostic biomarker in glioma48. NMNAT2 expression has been found to be upregulated in colorectal cancer49. Finally, variation in both the coding and promoter sequences of TERT has been associated with a variety of cancer types50,51.
Our PheWAS results also suggest that most of these genes are associated with cancer, specifically blood-based tumors such as myeloid leukemia. Combined with the common ZSCAN23 locus we identified, associated with pancreatic tumors, this points to cancer being the major genetic factor currently affecting lifespan in UKB. This is consistent with a previous study of health span that found cancer to be the first emerging disease in over half of disease cases in UKB52. These results likely reflect the characteristics of the cohort, comprised of predominantly middle-aged individuals, with age-at-death ranging from 40.9 to 85.2 years and last-known ages between 52.6 and 88.7 years.
For sex-specific rare variant analyses, we identified four novel genes (CDKN1A, PTPRK, COA7, TG) in males and five genes (PORCN, UGT1A8, CBX3, IFITM10 and OLIG1) in females. Some of these genes have been found to be associated with sex-specific diseases. In one study, advanced prostate cancer patients had a higher frequency of a variant on the 3’UTR of CDKN1A53 and the gene has received attention as a potential therapeutic target for prostate cancer54. PORCN is located on the X chromosome and mutations on it can cause Goltz-Gorlin Syndrome55, but it has also been found to regulate a signaling pathway that controls cancer cell growth56. UGT1A8 expression is altered in endometrial cancer57 and amino acid substitutions in it may modulate estradiol metabolism leading to an increased risk of breast and endometrial cancer58.
Since UKB collected DNA from peripheral blood mononuclear cell samples, we explored whether the variants were potentially of somatic origin, picked up by WES genotyping due to CHIP. The VAF distribution of variants included in our analysis emphasizes that several associations are likely linked to CHIP and notably include the well-established CHIP-related genes TET2, ASLX1, DNMT3A, SF3B1, TP53 and SRSF2. While WES heterozygote genotypes for these variants will not include all variants with some degree of CHIP within these genes, it does capture CHIP-related somatic variants sufficiently to establish robust associations with lifespan. In UKB the mean duration between the primary visit (blood draw date) and death is currently 9.2 years (\(\pm\) 3.8) and suggests that WES screening for CHIP variants may be used as a precision health tool to contribute to earlier cancer detection by assessing individuals with higher susceptibility risks59. In addition to known cancer variants, such as breast cancer-related BRCA1/BRCA2, our study highlights novel associations that should be considered in cancer susceptibility screenings.
While our study lacks an independent external replication and only a small number of genes formally replicated in the independent test set within UKB, the stability of the burden test association to five-fold CV suggests that these results are not due to outliers and are robust within the UKB.
By combining large-scale GWAS with rare variant analysis, this study enhances our understanding of the genetic basis of human lifespan. Our results emphasize the importance of understanding the genetic factors driving the most prevalent causes of mortality on a population level, highlighting the potential for early genetic testing to identify germline and somatic variants that place some individuals at risk of early death. Understanding the biological pathways through which these genes influence cancer and aging, as well as the environmental factors interacting with these pathways, will be essential for developing therapeutic targets aimed at extending a healthy lifespan. Our study’s implications thus extend beyond genetics, as they touch on the broader aspects of health care, public health policy, and preventive strategies against age-related diseases.
In conclusion, this study enhances our understanding of the genetic basis of human lifespan by combining large-scale GWAS with detailed rare variant analysis. The novel loci identified warrant further exploration to understand their biological roles and interactions with environmental factors, which will be crucial for unraveling the complex nature of aging and developing strategies to mitigate its adverse effects.
Methods
Study participants
The UKB is a large population-based longitudinal cohort study with recruitment from 2006 to 2010 in the United Kingdom60. In total, 502,664 participants aged 40–69 years were recruited and underwent extensive phenotyping including health and demographic questionnaires, clinic measurements, and blood draw at one of 22 assessment centers, of whom 468,541 subjects have been genotyped by both SNP array and WES.
We restricted our analysis to 393,833 individuals who self-reported their ethnic background as “white British” and were categorized as European ancestry based on genetic ethnic grouping (Field: 21000). Among them, 35,551 subjects were reported deceased, and their ages at death were recorded from the UK Death Registry (Field: 40007). For the other 358,282 subjects without death records, we assumed they were still alive by the latest censoring date (November 30, 2022). We determined their last known ages by subtracting their year and month of birth (Field: 33) from the censoring date.
All participants provided written informed consent, as outlined in the UK Biobank ethics framework (https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/governance/ethics-advisory-committee).
SNP array genotyping and QC
A total of 488,000 UKB participants were genotyped using one of two closely related Affymetrix microarrays (UKB Axiom Array or UK BiLEVE Axiom Array) for approximately 850,000 variants. The genotyped dataset was phased using SHAPEIT3 and imputed with IMPUTE4, leveraging reference panels from UK10K, 1000 Genomes Project phase 3, and Haplotype Reference Consortium reference panels, resulting in approximately 97 million variants61. Additionally, we removed SNPs with imputation quality score <0.3, genotype missing rate >0.05, minor allele frequency (MAF) < 0.1%, and Hardy-Weinberg equilibrium \(p\) < \(1.0\times {10}^{-6}\).
Genome-wide association studies
We performed linear regression analyses using BOLT-LMM (v.2.3.4)62, which employs a linear mixed-effects model to test the association of common variants (MAF \(\ge\) 0.1%) with lifespan for the entire cohort, as well as stratified by sex. For all three analyses, we used Martingale residuals calculated using the Cox proportional hazards model as the outcome variable. The procedure for calculating Martingale residuals was as follows. First, a Cox proportional hazards model63 was fitted without genotype:
where \({H}_{0}(t)\) is the baseline hazard function at time point \(t\) given the age at enrollment (Field: 54), last-known age and dead/alive status, \({{{\rm{Z}}}}=[{{{{\rm{Z}}}}}_{1},\,\ldots,{{{{\rm{Z}}}}}_{k}]\) is a covariate matrix, and \({{{\rm{\beta }}}}=[{{{{\rm{\beta }}}}}_{1},\,\ldots,{{{{\rm{\beta }}}}}_{k}]\) is a coefficient matrix for Z. Here, we included sex and the first 40 principal components (PC) and geographic covariates (Field: 20118) under the Geographical and Location category (100113) as covariates, but for sex-specific analyses, sex was excluded. Then, Martingale residuals were calculated as:
where \({\delta }_{i}\) is the dead/alive status (0 = alive, 1 = dead) of the \(i\) th subject and \(\hat{{{{\rm{\beta }}}}}\) is the estimated coefficient matrix. We adapted the coxph function from the survival (v.3.2.13)64 in R (v.4.1.3) package to compute the Martingale residuals. Genome-wide significance threshold was set at the standard GWAS level (\(p\) = \(5.0\times {10}^{-8}\)). We used LocusZoom65 to generate regional plots and Python v.3.7 to create Manhattan plots.
Gene expression and colocalization analysis
To evaluate the effect of the significant loci identified in our GWAS, we examined expression quantitative trait loci (eQTLs) across 49 tissues having at least 73 samples from the Genotype-Tissue Expression Project (GTEx) version 866. Bayesian colocalization analysis was employed using the COLOC package (v.5.2.3)67 in R and the posterior probability of colocalization (PP4) was calculated between GWAS findings and eQTL associations within a 1 megabase (Mb) window. Additionally, colocalization was visualized using the locuscompareR package68.
Whole-exome sequencing and QC
Whole-exome sequencing (WES) data was available for 469,835 UKB participants. The dataset was generated by the Regeneron Genetics Center69. Details about the production and QC for the WES data was previously described69. We restricted the WES analysis to rare variants (MAF < 1%).
Rare variant annotation
Rare variants in WES data were annotated using Variant Effect Predictor (v. 112) provided by Ensembl70. We defined LoF variants as those with predicted consequences: splice acceptor, splice donor, stop gained, frameshift, start loss, stop loss, transcript ablation, feature elongation, or feature truncation. Missense variants were annotated using AlphaMissense71 and REVEL72 plugins and included if they had an AlphaMissense score \(\ge\) 0.7 or REVEL score \(\ge\) 0.75. All annotation was conducted based on GRCh38 genome coordinates.
Gene-based rare variant association studies
For testing groups of rare variants, genotype matrices were first transformed into a binary variable describing whether samples carry a variant of a given class as follows:
Where \({g}_{{ij}}\) is the minor allele count observed for subject \(i\) at variant \(j\) in the gene and \(k\) is the number of variants in the gene.
To account for relatedness and population structure, Martingale residuals were first adjusted using a linear mixed model approach implemented in fastGWA (--save-fastGWA-mlm-residual)73. The adjusted residuals were then used as the phenotype for the rare variant analyses, ensuring the robustness of the results in the presence of related individuals and population stratification.
We carried out two gene-based tests: the burden test and sequence kernel association test-optimized (SKAT-O)74. The burden test is a mean-based test that assumes the same direction of effects for all variants within a gene. On the other hand, SKAT-O employs a weighted average of the burden test and SKAT75, the latter a variance-based test that does not lose power when variants have opposing directions of effect.
Association tests were performed for each gene and rare variant class separately, including LoF variants, missense variants with an AlphaMissense score \(\ge\) 0.7, and missense variants with a REVEL score \(\ge\) 0.75, using Martingale residuals as the phenotype as in the common variant analyses. We excluded genes with fewer than 10 variant carriers to ensure the reliability of our analyses. A gene-wide significance threshold was established at \(p\) = 7.4\(\times {10}^{-7}\) based on the Bonferroni method accounting for the number of genes, variant classes, and statistical methods. Gene-based analyses were carried out using the SKAT package (v.2.2.5) in R.
To characterize the impacts of gene burden in significant genes, we compared lifespan survival depending on gene burden using Kaplan–Meier survival curves, and Cox proportional hazard regression analyses. Additionally, we performed Cox proportional hazards regression to assess the effect of each rare variant in a gene. The survival (v.3.2.13) package in R (v.4.1.3) was utilized for the survival analysis and the lifelines package (v.0.28.0) in Python v.3.7 was used for generating Kaplan–Meier survival curves.
Phenome-wide association studies
For gene-wide significant genes, we conducted phenome-wide association studies (PheWAS) of variant carrier status across 1670 phenotypes in the UKB derived from binary, categorical, and continuous traits using the PHEnome Scan ANalysis Tool76. Phenotypes included the International Classification of Disease 10 (ICD-10) codes, family history (e.g., father’s illness, father’s age at death), blood count (e.g., white blood cell count), blood biochemistry (e.g., Glucose levels), infectious diseases (e.g., pp 52 antigen for Human Cytomegalovirus), physical measures (e.g., BMI), cognitive test (e.g., pairs matching) and brain measurements (e.g., subcortical volume of hippocampus). For ICD-10 codes, we excluded phenotypes from the following ICD-10 chapters: “Injuries, poisonings, and certain other consequences of external causes” (Chapter XIX), “External causes of morbidity and mortality” (Chapter XX), “Factors influencing health status and contacts with health services” (Chapter XXI), and “Codes for special purposes” (Chapter XXII). The ICD-10 codes were then converted into Phecodes (v.1.2)77 which combine correlated ICD codes into a distinct code and improve alignment with diseases commonly used in clinical practice.
For binary traits, we removed phenotypes with fewer than 100 cases, and for continuous traits, those with fewer than 100 participants were excluded. Depending on the phenotype, we employed various regression models including binary logistic regression, ordinal logistic regression, multinomial logistic regression, and linear regression. All analyses included age and sex as covariates. Phenome-wide significance threshold was set at \(p\) = \(2.9\times {10}^{-5}\) based on the number of phenotypes.
Variant allelic fraction
To investigate whether some gene-level associations are enriched for somatic variants, we computed the variant allele frequency (VAF) for each heterozygous sample, reporting the mean VAF and VAF distribution per gene per variant class. VAF is defined as the number of reads with an alternate allele divided by the read depth at a given variant position. We also calculated the confidence interval for the mean VAF per gene using 10,000 bootstrap samples to ensure robust statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
GWAS summary statistics and rare variant results from SKAT-O and burden tests are available in the GWAS Catalog database under accession codes GCST90551884–GCST90551889. All phenotypic and genotypic data supporting the findings of this study are available from the UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research/register). Access to these data is available from the authors with UKB permission.
Code availability
The codes used for analyses in the present study are available at the following link: https://github.com/Junkkkk/Lifespan-studies. The code for PheWAS analysis was utilized from https://github.com/MRCIEU/PHESANT.
References
Passarino, G., De Rango, F. & Montesanto, A. Human longevity: genetics or lifestyle? It takes two to tango. Immun. Ageing 13, 1–6 (2016).
Ruby, J. G. et al. Estimates of the heritability of human longevity are substantially inflated due to assortative mating. Genetics 210, 1109–1124 (2018).
van den Berg, N., Beekman, M., Smith, K. R., Janssens, A. & Slagboom, P. E. Historical demography and longevity genetics: back to the future. Ageing Res. Rev. 38, 28–39 (2017).
Ryu, S., Atzmon, G., Barzilai, N., Raghavachari, N. & Suh, Y. Genetic landscape of APOE in human longevity revealed by high-throughput sequencing. Mech. Ageing Dev. 155, 7–9 (2016).
Sebastiani, P. et al. APOE alleles and extreme human longevity. J. Gerontol. A Biol. Sci. Med. Sci. 74, 44–51 (2019).
Joshi, P. K. et al. Variants near CHRNA3/5 and APOE have age- and sex-related effects on human lifespan. Nat. Commun. 7, 11174 (2016).
Joshi, P. K. et al. Genome-wide meta-analysis associates HLA-DQA1/DRB1 and LPA and lifestyle factors with human longevity. Nat. Commun. 8, 910 (2017).
Liu, J. Z. et al. The burden of rare protein-truncating genetic variants on human lifespan. Nat. Aging 2, 289–294 (2022).
Kessler, M. D. et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022).
Bae, H. et al. A genome-wide association study of 2304 extreme longevity cases identifies novel longevity variants. Int. J. Mol. Sci. 24, 116 (2022).
Sebastiani, P. et al. Four genome-wide association studies identify new extreme longevity variants. J. Gerontol. Ser. A 72, 1453–1464 (2017).
Deelen, J. et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 10, 3669 (2019).
Therneau, T. M., Grambsch, P. M. & Fleming, T. R. Martingale-based residuals for survival models. Biometrika 77, 147–160 (1990).
Gagliano Taliun, S. A. et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat. Genet. 52, 550–552 (2020).
Timmers, P. R. et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. elife 8, e39856 (2019).
Baer, C. et al. Somatic” and “pathogenic”—Is the classification strategy applicable in times of large-scale sequencing? Haematologica 104, 1515–1520 (2019).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Mahley, R. W. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 94, 739–746 (2016).
Wen, Y.-P. & Yu, Z.-G. Identifying shared genetic loci and common risk genes of rheumatoid arthritis associated with three autoimmune diseases based on large-scale cross-trait genome-wide association studies. Front. Immunol. 14, 1160397 (2023).
Yao, M., Huang, X., Guo, Y., Zhao, J. V. & Liu, Z. Disentangling the common genetic architecture and causality of rheumatoid arthritis and systemic lupus erythematosus with COVID-19 outcomes: genome-wide cross trait analysis and bidirectional Mendelian randomization study. J. Med. Virol. 95, e28570 (2023).
Du, Q., Zhang, M., Gao, A., He, T. & Guo, M. Epigenetic silencing ZSCAN23 promotes pancreatic cancer growth by activating Wnt signaling. Cancer Biol. Ther. 25, 2302924 (2024).
Liu, Y. et al. Oncogenic protein kinase D3 regulating networks in invasive breast cancer. Int. J. Biol. Sci. 13, 748–758 (2017).
Chen, Z. et al. PRKD3 promotes malignant progression of OSCC by downregulating KLF16 expression. Eur. Rev. Med. Pharm. Sci. 24, 12709–12716 (2020).
Lee, M.-G. & Lee, Y. H. A meta-analysis examining the association between the MUC5B rs35705950 T/G polymorphism and susceptibility to idiopathic pulmonary fibrosis. Inflamm. Res. 64, 463–470 (2015).
Nakano, Y. et al. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 193, 464–466 (2016).
van Moorsel, C. H. et al. The MUC5B promoter polymorphism associates with severe COVID-19 in the European population. Front. Med. 8, 668024 (2021).
Verma, A. et al. A MUC5B gene polymorphism, rs35705950-T, confers protective effects against COVID-19 hospitalization but not severe disease or mortality. Am. J. Respir. Crit. Care Med. 206, 1220–1229 (2022).
Ladd, A. N. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol. Cell. Neurosci. 56, 456–464 (2013).
Itai, T. et al. De novo variants in CELF2 that disrupt the nuclear localization signal cause developmental and epileptic encephalopathy. Hum. Mutat. 42, 66–76 (2021).
Duan, X. et al. CELF2 deficiency demonstrates autism-like behaviors and interferes with late development of cortical neurons in mice. Mol. Neurobiol. 62, 156–168 (2025).
Guo, Q. et al. The RNA-binding protein CELF2 inhibits ovarian cancer progression by stabilizing FAM198B. Mol. Ther. Nucleic Acids 23, 169–184 (2021).
Piqué, L. et al. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene 38, 7106–7112 (2019).
Quivoron, C. et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 (2011).
Abdel-Wahab, O. et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193 (2012).
Yang, L., Rau, R. & Goodell, M. A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 15, 152–165 (2015).
Wang, L. et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 (2011).
Kim, E. et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27, 617–630 (2015).
Cremona, C. A. & Behrens, A. ATM signalling and cancer. Oncogene 33, 3351–3360 (2014).
Wooster, R. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792 (1995).
Ford, D., Easton, D. F., Bishop, D. T., Narod, S. A. & Goldgar, D. E. Risks of cancer in BRCA1-mutation carriers. Lancet 343, 692–695 (1994).
Johnsen, S. A. et al. Regulation of estrogen-dependent transcription by the LIM cofactors CLIM and RLIM in breast cancer. Cancer Res. 69, 128–136 (2008).
Gao, R., Wang, L., Cai, H., Zhu, J. & Yu, L. E3 ubiquitin ligase RLIM negatively regulates c-Myc transcriptional activity and restrains cell proliferation. PLoS ONE 11, e0164086 (2016).
Yin, Y. & Shen, W. H. PTEN: a new guardian of the genome. Oncogene 27, 5443–5453 (2008).
Yang, H., Ye, D., Guan, K.-L. & Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin. Cancer Res. 18, 5562–5571 (2012).
Yang, C.-M. et al. Aberrant DNA hypermethylation-silenced SOX21-AS1 gene expression and its clinical importance in oral cancer. Clin. Epigenetics 8, 129 (2016).
Zhang, X., Zhao, X., Li, Y., Zhou, Y. & Zhang, Z. Long noncoding RNA SOX21-AS1 promotes cervical cancer progression by competitively sponging miR-7/VDAC1. J. Cell. Physiol. 234, 17494–17504 (2019).
Sheng, X.-Y., Wang, C.-H., Wang, C.-F. & Xu, H.-Y. Long-chain non-coding SOX21-AS1 promotes proliferation and migration of breast cancer cells through the PI3K/AKT signaling pathway. Cancer Manag. Res. 12, 11005–11014 (2020).
Shi, H., Song, Y., Song, Z. & Huang, C. CKMT1B is a potential prognostic biomarker and associated with immune infiltration in Lower-grade glioma. PLoS ONE 16, e0245524 (2021).
Qi, J. et al. Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncol. Lett. 16, 5829–5837 (2018).
Baird, D. M. Variation at the TERT locus and predisposition for cancer. Expert Rev. Mol. Med. 12, e16 (2010).
Vinagre, J. et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 4, 2185 (2013).
Zenin, A. et al. Identification of 12 genetic loci associated with human healthspan. Commun. Biol. 2, 41 (2019).
Kibel, A. S. et al. CDKN1A and CDKN1B polymorphisms and risk of advanced prostate carcinoma1. Cancer Res. 63, 2033–2036 (2003).
Li, S. et al. miR-3619-5p inhibits prostate cancer cell growth by activating CDKN1A expression. Oncol. Rep. 37, 241–248 (2017).
Lombardi, M. P. et al. Mutation update for the PORCN gene. Hum. Mutat. 32, 723–728 (2011).
Covey, T. M. et al. PORCN moonlights in a Wnt-independent pathway that regulates cancer cell proliferation. PLoS ONE 7, e34532 (2012).
Zhao, F. et al. The function of uterine UDP-glucuronosyltransferase 1A8 (UGT1A8) and UDP-glucuronosyltransferase 2B7 (UGT2B7) is involved in endometrial cancer based on estrogen metabolism regulation. Hormones 19, 403–412 (2020).
Thibaudeau, J. et al. Characterization of common UGT1A8, UGT1A9, and UGT2B7 variants with different capacities to inactivate mutagenic 4-hydroxylated metabolites of estradiol and estrone. Cancer Res. 66, 125–133 (2006).
Koch, Z., Li, A., Evans, D. S., Cummings, S. & Ideker, T. Somatic mutation as an explanation for epigenetic aging. Nat. Aging, https://doi.org/10.1038/s43587-024-00794-x (2025).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Loh, P.-R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Cox, D. R. Regression models and life‐tables. J. R. Stat. Soc. Ser. B Methodol. 34, 187–202 (1972).
Therneau, T. A package for survival analysis in S. R Package version 2, 2014 (2015).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Liu, B., Gloudemans, M. J., Rao, A. S., Ingelsson, E. & Montgomery, S. B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 51, 768–769 (2019).
Szustakowski, J. D. et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 53, 942–948 (2021).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Millard, L. A. C., Davies, N. M., Gaunt, T. R., Davey Smith, G. & Tilling, K. Software application profile: PHESANT: a tool for performing automated phenome scans in UK Biobank. Int. J. Epidemiol. 47, 29–35 (2018).
Wu, P. et al. Mapping ICD-10 and ICD-10-CM codes to phecodes: workflow development and initial evaluation. JMIR Med. Inform. 7, e14325 (2019).
Karlsson, Q. et al. Rare germline variants in ATM predispose to prostate cancer: a PRACTICAL Consortium Study. Eur. Urol. Oncol. 4, 570–579 (2021).
Dorling, L. et al. Breast cancer risk genes—association analysis in more than 113,000 women. N. Engl. J. Med. 384, 428–439 (2021).
Easton, D. F. et al. A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am. J. Hum. Genet. 81, 873–883 (2007).
Papaemmanuil, E. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016).
Ley, T. J. et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 (2010).
Usui, Y. et al. Helicobacter pylori, homologous-recombination genes, and gastric cancer. N. Engl. J. Med. 388, 1181–1190 (2023).
Andersson, A. K. et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25, 1570–1577 (2011).
Jordan, J. J. et al. Altered-function p53 missense mutations identified in breast cancers can have subtle effects on transactivation. Mol. Cancer Res. 8, 701–716 (2010).
Acknowledgements
This research has been conducted using the UK Biobank Resource under application number 45420. We thank all the participants and researchers of UK Biobank for making these data open and accessible to the research community. This research was supported by the Dean’s Postdoctoral Fellowship at the School of Medicine, Stanford University. Additionally, this research was partially supported by the Biostatistics Shared Resource (B-SR) of the NCI-sponsored Stanford Cancer Institute: P30CA124435 and by the following NIH funding source of Stanford’s Center for Clinical and Translational Education and Research award, under the Biostatistics, Epidemiology and Research Design (BERD) Program: 1UM1TR004921-01.
Author information
Authors and Affiliations
Contributions
J.P. conducted all analyses, prepared all figures and drafted the manuscript. A.P.T. contributed to refining the manuscript. L.T. provided critical comment on the manuscript. M.D.G. and Y.L.G. planned, organized and supervised the entire study and revised the manuscript. All authors have approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Luke Pilling, Paul Timmers and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J., Peña-Tauber, A., Talozzi, L. et al. Rare genetic associations with human lifespan in UK Biobank are enriched for oncogenic genes. Nat Commun 16, 2064 (2025). https://doi.org/10.1038/s41467-025-57315-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57315-6
