
Abstract
RNA can fold into structures that mediate diverse cellular functions. Understanding how RNA primary sequence directs the formation of functional structures requires methods that can comprehensively assess how changes in an RNA sequence affect its structure and function. Here we have developed a platform for performing high-throughput cotranscriptional RNA biochemical assays, called Transcription Elongation Complex display (TECdisplay). TECdisplay measures RNA function by fractionating a TEC library based on the activity of cotranscriptionally displayed nascent RNA. In this way, RNA function is measured as the distribution of template DNA molecules between fractions of the transcription reaction. This approach circumvents typical RNA sequencing library preparation steps that can cause technical bias. We used TECdisplay to characterize the transcription antitermination activity of >1 million variants of the Clostridium beijerinckii pfl ZTP riboswitch designed to perturb steps within its cotranscriptional folding pathway. Our findings establish TECdisplay as an accessible platform for high-throughput RNA biochemical assays.
Similar content being viewed by others

Introduction
RNA folding is dynamic and begins during transcription1,2,3. Despite the biological importance of structured RNAs, our understanding of how RNA folds into functional structures is limited because the interplay between RNA sequence, structure, and function is complex. Linking RNA sequence, folding, and function, therefore requires methods that can assess the structural and functional consequences of thousands of RNA sequence perturbations simultaneously4,5. The state-of-the-art approach for high-throughput cotranscriptional RNA biochemistry couples high-throughput DNA sequencing with in situ transcription on an Illumina sequencer flow cell so that fluorescence-based RNA assays can be performed for tens of thousands of RNA sequence variants in parallel4,5. This strategy was pioneered by the High-Throughput Sequencing-RNA Affinity Profiling (HiTS-RAP)6 and Quantitative Analysis of RNA on a Massively Parallel Array (RNA-MaP)7 methods, which were initially developed to quantify RNA-protein interactions and have been collectively referred to as RNA array on a High-Throughput Sequencer (RNA-HiTS) methods. RNA-MaP assays for diverse RNA-mediated functions have since been developed8,9,10,11,12,13,14,15,16,17,18.
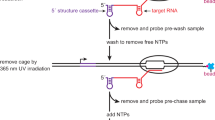
The primary limitation of RNA-HiTS experiments is that they require custom instrumentation. Although detailed procedures are available for building the requisite instrument8, many laboratories lack the expertise needed to assemble and maintain a custom instrument. To address this, we have developed a modular platform for cotranscriptional RNA biochemical assays, called Transcription Elongation Complex display (TECdisplay) that quantifies the function of at least one million RNA sequence variants simultaneously using an RNA-dependent DNA fractionation strategy that can be performed at the laboratory bench (Fig. 1a). In a TECdisplay experiment, an RNA variant library is cotranscriptionally displayed from E. coli RNA polymerase (RNAP) using quantitative in vitro transcription. This TEC library is then fractionated by the function of the nascent RNA. Because template DNA is physically coupled to nascent RNA in the TEC, the distribution of template DNA molecules for a given variant between fractions reflects the function of the RNA that it encodes. Consequently, template DNA can be recovered, quantitatively tagged with fraction-specific barcodes, and sequenced to determine the activity of each RNA variant in the library.

a General TECdisplay workflow. A library of RNA variants is encoded in template DNA that contains a promoter that is compatible with dU-excision tagging and a transcription stall site that prevents RNAP from running off the template. Single-round transcription is performed under conditions that yield TECs with a 1:1:1 DNA:RNAP:RNA composition. The TEC library is then fractionated by RNA function. This fractionation step can vary depending on the RNA function being measured. The protein and RNA components of the transcription reaction are then degraded and fraction-specific barcodes are quantitatively appended to template DNA using the one-pot dU-excision tagging procedure. Barcoded fractions are pooled together, the non-transcribed DNA strand is degraded by lambda exonuclease, and the resulting ssDNA is amplified for ~7 cycles of PCR and sequenced to determine the distribution of template DNA across fractions for each variant in the library. Because DNA was fractionated by RNA function, the distribution of DNA between fractions reflects RNA function. b Strategy for measuring riboswitch-mediated transcription termination/antitermination activity using TECdisplay. TECs are synchronized using the C3-SC1 leader sequence and immobilized on streptavidin-coated magnetic beads by a biotinylated capture oligonucleotide. An etheno-dA transcription stall site is positioned downstream of the riboswitch intrinsic terminator to retain RNAP on DNA. When transcription is resumed, template DNA molecules on which a termination event happened are released into the supernatant. Conversely, template DNA molecules on which a terminator readthrough event happened are retained in the bead pellet. c Quantification of C. beijerinckii pfl ZTP riboswitch activity in various conditions using the fractionation strategy shown in panel b. Fraction terminator readthrough was calculated using the distribution of template DNA between supernatant and pellet fractions and using the distribution of terminated and full-length RNA for each sample. The gel image is representative of n = 2 replicates. Uncropped source gels are available in Supplementary Fig. 9. Source data are provided as a Source Data file. RNAP RNA polymerase, C3-SC1 Cap3-structure cassette 1.
Here we describe the first TECdisplay assay, which measures transcription termination efficiency. We used TECdisplay to systematically assess how sequence changes affect riboswitch folding and antitermination activity. Riboswitches are cis-acting RNA chemical sensors that regulate gene expression in response to diverse ligands19,20 and have been used to develop biomolecular sensors21,22 and chemical biology tools23,24. We used TECdisplay to quantify the transcription termination and antitermination activity of over 1 million variants of the Clostridium beijerinckii (C. beijerinckii, Cbe) pfl ZTP riboswitch designed to perturb ZTP aptamer and transcription terminator folding. The ZTP riboswitch, which regulates genes associated with purine biosynthesis in response to the metabolites ZTP and ZMP25, is a potential antibiotic target26 and has been used as a reporter in a screen for folate biosynthesis inhibitors27. Our analysis of ZTP riboswitch folding produced expected outcomes that validate the accuracy of TECdisplay, identified sequence combinations that would naively be predicted to be functional based on sequence but are non-functional due to misfolding, and uncovered how stabilization of a non-canonical base pair increases ZMP-responsiveness. Our findings establish TECdisplay as an accessible platform for high-throughput cotranscriptional RNA functional assays that can likely be used to characterize diverse RNA-mediated biochemical activities.
Results
The cotranscriptional RNA-dependent DNA fractionation strategy
TECdisplay is a modular platform for performing high-throughput cotranscriptional RNA biochemical assays. TECdisplay experiments comprise three steps: First, an RNA variant library is synthesized using a quantitative E. coli single-round in vitro transcription reaction in which virtually all DNA template molecules yield a single RNA product28 (Fig. 1a). This is accomplished by using the C3-SC1 leader sequence to purify synchronized C3-SC1TECs, which are >98% active28. The template DNA used for this reaction invariably contains one of several modifications that halt E. coli RNAP transcription, which prevents run-off transcription so that nascent RNA molecules are physically coupled to the DNA template from which they were transcribed29. Second, the TEC library is fractionated by the biochemical activity of the nascent RNA (Fig. 1a). For example, in this work, we describe a TECdisplay assay for measuring transcription termination efficiency. In this assay, RNAP is tethered to a magnetic bead by the Cap3 oligonucleotide, which anneals to the C3 hybridization site in the C3-SC1 leader transcript28, and an etheno-dA transcription stall site29,30 is positioned downstream of the transcription termination site (Fig. 1b). If transcription termination occurs, nascent RNA is released from the TEC and template DNA is released into the supernatant. Conversely, if RNAP bypasses the termination site, transcription halts at the etheno-dA stall site, and the template DNA is retained in the bead pellet. In this way, DNA molecules are separated based on whether transcription termination occurred so that the efficiency of terminator readthrough can be calculated using the distribution of template DNA across supernatant and pellet fractions (Fig. 1c). This fractionation step is modular and can be changed depending on the RNA biochemical activity that will be measured. Third, the template DNA is converted into an Illumina sequencing library. In this step, RNA and protein are degraded and fraction- and molecule-specific barcodes are quantitatively appended to the template DNA in a one-pot deoxyUridine eXcision tagging (dUX-tagging) reaction31 (Fig. 1a and Supplementary Fig. 1a). Following the dUX-tagging reaction, the pellet and supernatant fractions are pooled together and excess tagging oligonucleotide is depleted by solid-phase reversible immobilization (SPRI). The non-transcribed DNA strand is then degraded using lambda exonuclease so that only the transcribed DNA strand, which was used to generate the nascent RNA, will be sequenced (Supplementary Fig. 1). This step safeguards the experiment against the potential presence of heteroduplexes in the DNA template preparation, which would confound the experimental measurement. The barcoded template DNA is then amplified by ~7 PCR cycles. The resulting library is then sequenced using the Illumina platform to determine the distribution of template DNA between pellet and supernatant fractions for each variant, which reflects the functional properties of the nascent RNA.
The RNA-dependent DNA fractionation strategy used by TECdisplay is advantageous in two ways. First, TECdisplay circumvents typical RNA-sequencing library preparation steps like reverse transcription and single-stranded nucleic acid ligations, which can potentially introduce technical bias into the measurement32,33,34,35,36,37,38,39. Second, TECdisplay is compatible with destructive RNA functions in which RNA sequence information is lost during the assay because sequence information is recovered directly from the DNA.
Validation: pfl ZTP aptamer pseudoknot folding
Strobel et al., previously performed a mid-throughput analysis of several hundred variants of the C. beijerinckii pfl ZTP riboswitch40 using an RNA-sequencing-based assay inspired by Sequencing-based Mutational Analysis of RNA Transcription Termination (SMARTT)41, which distinguishes terminated and terminator readthrough transcripts by their 3’ end. This analysis included 64 variants in which conserved base pairs within the pfl ZTP aptamer pseudoknot and terminator were randomized according to the ZTP riboswitch consensus sequence (Fig. 2a). This library contains four possible intact pseudoknot/terminator configurations, which are referred to as wild type (WT), flip (FL), strong (ST), and weak (WK) throughout the text below (Fig. 2a). We assessed the accuracy of TECdisplay by repeating this analysis and measured the termination and antitermination activity of the pfl riboswitch pseudoknot variants at several ZMP concentrations that were not originally assessed. As in the original analysis, transcription was performed at 500 μM NTPs. Comparison of replicates collected for all ZMP concentrations (R2 = 0.997) shows that TECdisplay measurements are reproducible (Fig. 2b). To assess the accuracy of TECdisplay, we first compared solid-phase fraction terminator readthrough measurements made for the pfl riboswitch using TECdisplay to previous solution-phase measurements made with and without the C3-SC1 leader28. This comparison assesses how the use of a solid-phase transcription configuration in which TECs are tethered to a surface by the nascent transcript in the TECdisplay assay affects ZTP riboswitch activity. The ZMP-mediated transcription antitermination response measured by TECdisplay approximates solution-phase measurements for the leaderless pfl riboswitch (Fig. 2c). However, the basal terminator readthrough measured for the C3-SC1-containing pfl riboswitch by TECdisplay is ~10 percentage points higher than was observed in the solution-phase assay (Fig. 2c). To assess how sequencing library preparation affects the accuracy of TECdisplay, we compared TECdisplay fraction terminator readthrough measurements made for the wild type pfl riboswitch with 0 and 1 mM ZMP to gel-based measurements made using TECdisplay in vitro transcription conditions. Gel-based fraction terminator readthrough measurements are ~7.5 percentage points higher than TECdisplay measurements (Fig. 2d). This is comparable to the difference between fraction terminator readthrough measurements that were made using an RNA-sequencing-based assay and a gel-based assay previously40 (Fig. 2d).

a Sequence of the C. beijerinckii pfl ZTP riboswitch pseudoknot variant library. b Correlation of replicate TECdisplay fraction terminator readthrough measurements for all ZMP concentrations that were assessed. c Wild-type C. beijerinckii pfl ZTP riboswitch dose-response curves measured by TECdisplay and by gel-based assays (ref. 28). d, e Sequencing and gel-based fraction terminator readthrough measurements for the wild type (d) and flip (e) pseudoknot variants. f, g Correlation of fraction terminator readthrough measurements made by TECdisplay and by an RNA-sequencing-based assay (ref. 40) for the pseudoknot variant library in panel a. h Dose-response curves for the wild type C. beijerinckii pfl ZTP riboswitch and variants with pseudoknot and terminator perturbations, measured by TECdisplay. i–k Dose-response curves for the wild type, flip, strong, and weak pseudoknot variants in the context of U44 (i), A44 (j), and G44 (k), measured by TECdisplay. A44 and G44 data are from the library shown in Fig. 3a. TECdisplay data for the library shown in panel a are from n = 2 replicates. In f, g, data from the two replicates were combined. In j, k, TECdisplay data for the library shown in Fig. 3a are from n = 2 replicates for the 0 mM and 1 mM ZMP conditions and are n = 1 for the 31.6 μM, 100 μM, 316 μM, and 3.16 mM conditions. Gel quantification data are from n = 2 replicates; the TECdisplay gel quantification data in panel d are from Fig. 1c. Uncropped source gels are available in Supplementary Fig. 9. The gel-based dose-response curves in panel c are from Kelly, Szyjka, and Strobel, Purification of synchronized Escherichia coli transcription elongation complexes by reversible immobilization on magnetic beads, J Biol Chem., 2022 Apr; 298(4):10178928. The RNA-sequencing-based measurements of pseudoknot variant library function in panels f, g are from Strobel et al. A ligand-gated strand displacement mechanism for ZTP riboswitch transcription control, Nat. Chem. Biol. 2019 Nov; 15(11):1067–107640. Source data are provided as a Source Data file.
We next compared fraction terminator readthrough measurements made for the library in Fig. 2a using TECdisplay to measurements made for the same variants using an RNA-sequencing-based assay previously40. The agreement of fraction terminator readthrough measurements made by TECdisplay and RNA-sequencing for the 0 and 1 mM ZMP conditions is bimodal and depends on the identity of position 24. Fraction terminator readthrough measurements for A24 variants agree strongly (0 mM ZMP: R2 = 0.962; 1 mM ZMP: R2 = 0.924) (Fig. 2f, g). Fraction terminator readthrough measurements for G24 variants are well-correlated (0 mM ZMP: R2 = 0.934; 1 mM ZMP: R2 = 0.853) but are higher in the TECdisplay assay than in the RNA-sequencing assay (Fig. 2f, g). Notably, fraction terminator readthrough in the absence of ZMP is higher for the flip pseudoknot variant, which contains G24, than for the wild-type riboswitch, which contains A24, in all sequencing and gel-based assays that were assessed (Fig. 2d, e). This indicates that the severity of the termination defect caused by G24 may be exacerbated or reduced depending on the conditions used for in vitro transcription.
Perturbations to base pairs in the pfl ZTP aptamer pseudoknot and terminator match several expectations (Fig. 2h): First, variants with a disrupted pseudoknot and intact terminator terminate transcription efficiently but do not respond to ZMP. Second, variants with an intact pseudoknot and disrupted terminator fail to terminate transcription efficiently regardless of ZMP concentration. Third, variants in which both the pseudoknot and the terminator are disrupted do not respond to ZMP and terminate transcription more efficiently than when only the terminator is disrupted, which agrees with previous findings that the pseudoknot is a barrier to terminator base pair propagation40,42. Each of the four variants in which both pseudoknot and terminator base pairs are intact exhibit a distinct ZMP-mediated antitermination response (Fig. 2i). These trends are similar to those observed for the same pseudoknot configurations in the context of A44 and G44 as part of a 32,768-variant library (Fig. 2j, k). In all cases, weak pseudoknot variants are less sensitive to ZMP, presumably because the weaker pseudoknot folds less efficiently. The increased basal terminator readthrough that is observed for the G24-containing flip and strong pseudoknot variants is examined in the sections below. Importantly, the ZMP-mediated transcription antitermination response measured by TECdisplay for the intact pseudoknot and terminator variants could be fit to an equation that describes the transcription antitermination response of a glycine riboswitch, which was derived by Torgerson et al.41 (Supplementary Fig. 2, see Methods).
Systematic perturbation of pfl ZTP aptamer folding
To further explore how sequence variation affects riboswitch folding, we characterized the activity of 32,768 C. beijerinckii pfl ZTP riboswitch variants in which conserved purine and pyrimidine nucleotides within the ZTP aptamer P1 stem, pseudoknot (PK), and terminator are randomized (Fig. 3a). Like the 64-variant library described above, this library perturbs the competition between pseudoknot and terminator folding and transcription was performed at 500 μM NTPs. However, this expanded data set, which contains 512 times more variants, enabled us to confidently dissect how sequence variation in pseudoknot and terminator base pairs dictates RNA folding outcome because the same competition between pseudoknot and terminator folding was observed repeatedly for different P1 stem variants. Comparison of replicates collected with 0 mM (R2 = 0.987) or 1 mM ZMP (R2 = 0.989) shows that TECdisplay measurements remain reproducible at increased scale (Fig. 3b). Bulk visualization of ZMP-mediated antitermination efficiency in response to increasing ZMP concentration revealed that the variant library contains discrete clusters of non-functional variants with variable terminator efficiency and functional variants with broadly variable terminator efficiency and responsiveness to ZMP (Fig. 3c). Filtering the 0 mM and 1 mM ZMP data for variants in which variable P1, pseudoknot, and terminator nucleotides form Watson–Crick or GU pairs enriches for functional variants, although many non-functional variants remain (Fig. 3d, e). Filtering these data for variants in which variable nucleotides in the pseudoknot and terminator form Watson–Crick pairs isolates a set of functional riboswitches in which the increase in fraction terminator readthrough in the presence of 1 mM ZMP is at least 0.2 for 94.5% of the variants (Fig. 3e, f). In agreement with this observation and observations made for the 64-variant library above, the ZMP dose-response for select variants matches four expectations: First, variants with intact pseudoknot and terminator base pairs exhibit ZMP-mediated transcription antitermination (Fig. 3g). Second, disrupting pseudoknot base pairs eliminates ZMP-mediated antitermination (Fig. 3h). Third, disrupting terminator base pairs causes a termination defect (Fig. 3i). Fourth, disrupting pseudoknot and terminator base pairs eliminates ZMP-mediated antitermination and restores some terminator activity, which agrees with previous findings that the pseudoknot is a barrier to terminator base pair propagation40,42 (Fig. 3j). Furthermore, the discrete clusters of non-functional variants are attributable to distinct classes of variants defined by the identity of pseudoknot and terminator base pairs (Supplementary Fig. 3). Together, these data indicate that the TECdisplay assay for transcription termination efficiency can be scaled to assess at least tens of thousands of variants simultaneously.

a Sequence of the C. beijerinckii pfl ZTP riboswitch consensus variant library. b Correlation of replicate TECdisplay fraction terminator readthrough measurements for the 0 and 1 mM ZMP conditions. c Plots showing fraction terminator readthrough for the sequence variants shown in panel a as measured by TECdisplay with variable ZMP concentration. The ZMP concentration that was used for each data set is indicated above each plot. Variants are plotted along the x-axis by the difference in fraction terminator readthrough measured for the 1 mM ZMP and 0 mM ZMP conditions so that each variant is located at the same x-axis position in all plots. d–f Plots showing fraction terminator readthrough for the 0 mM ZMP (black) and 1 mM ZMP (red) conditions for all variants (d), variants in which P1, the pseudoknot, and the terminator are fully paired and can contain Watson–Crick or GU pairs (e), and variants in which P1, the pseudoknot, and the terminator are fully paired but the pseudoknot and terminator contain only Watson–Crick base pairs (f). g–j Dose-response curves for wt-like variants with strong pseudoknot and terminator pairs (g), pseudoknot mismatches and strong terminator pairs (h), strong pseudoknot pairs and terminator mismatches (i), and pseudoknot and terminator mismatches (j). TECdisplay data for the 0 and 1 mM ZMP conditions are from n = 2 replicates that were combined. TECdisplay data for the 31.6 μM, 100 μM, 316 μM, and 3.16 mM conditions are n = 1. Source data are provided as a Source Data file. PK pseudoknot, term terminator, WC Watson–Crick, MM mismatch.
To determine whether the throughput of TECdisplay experiments could be increased further, we characterized the activity of a pfl ZTP riboswitch library that contained 1,048,576 variants at 100 μM NTPs with and without 1 mM ZMP. This library expands the randomization scheme described above for the 32,768-variant library by including N35, N37, and ΔU113/ΔC116 (Fig. 4a). This additional randomization is relevant to several observations made about the 32,768-variant library shown in Fig. 3a, and a subset of these variants were also assessed in separate lower-throughput experiments at 500 μM NTPs. The findings from these data sets are described in the sections below. Fraction terminator readthrough measurements from individual replicates were in general agreement but exhibit more variability than is observed in lower-throughput experiments (Fig. 4b). Unsurprisingly, variants that are better represented in the library exhibit less variability (Fig. 4c). Replicate data were merged to increase read depth and fraction terminator readthrough measurements for a 32,768-variant subset of the 1,048,576-variant library were compared to measurements made for the same 32,768 variants as separate library (Fig. 4d). The agreement of both 0 mM (R2 = 0.977) and 1 mM ZMP (R2 = 0.985) data indicates that TECdisplay can be applied to libraries of at least 1 million variants. This comparison is also shown for wild type, flip, strong, and weak pseudoknot variants in Fig. 4e. Importantly, the agreement of measurements made using the 1,048,576-variant and 32,768-variant libraries demonstrates that increasing library complexity does not meaningfully affect the accuracy of TECdisplay.

a Sequence of the C. beijerinckii pfl ZTP riboswitch 1,048,576-variant library. b Histograms of the difference in fraction terminator readthrough observed in replicate experiments (replicate 1–replicate 2) for the 0 and 1 mM ZMP conditions. c Plots of the difference in fraction terminator readthrough observed in replicate experiments (replicate 1–replicate 2) against the combined sequencing depth of the replicates. d Correlation of fraction terminator readthrough measurements for a 32,768-variant subset of the 1,048,576-variant library with measurements made for the same variants as a standalone 32,768-variant library. e Correlation of fraction terminator readthrough measurements for the wild type, flip, strong, and weak pseudoknot variants made using the 1,048,576-variant and 32,768-variant libraries. All TECdisplay data are from n = 2 replicates. In panels d, e, replicate data were combined. Source data are provided as a Source Data file.
Differential terminator activity of pfl riboswitch pseudoknot variants is caused by aptamer misfolding
The analysis of the 64-variant pfl riboswitch library above shows that pseudoknot identity has a substantial effect on the ZMP-mediated transcription antitermination response (Fig. 2i). As above, the pfl riboswitch consensus variant library contains four possible pseudoknot/terminator classes, which are referred to as wild type (WT), flip (FL), strong (ST), and weak (WK) (Fig. 5a). In the text below, sub-elements of these classes are referred to as “sub-elementclass”. For example, the wild-type J1-2 and terminator sub-elements are referred to as J1-2WT and TermWT, respectively. Filtering the 0 mM and 1 mM pfl riboswitch consensus variant data sets for variants in which the pseudoknot and terminator comprise Watson–Crick base pairs and P1 is intact isolates a set of variants that appears to contain at least two subpopulations (Fig. 3f). Filtering the variants identified in Fig. 3f by pseudoknot identity shows that each pseudoknot configuration forms a distinct cluster, but that there is a relationship between the terminator efficiency of wild type and weak pseudoknot variants and between flip and strong pseudoknot variants (Fig. 5b). 97% of wild type and weak pseudoknot variants exhibit terminator efficiencies between ~75 and ~90%, and the efficiency of termination is independent of the ZMP-mediated antitermination response (Fig. 5b). In contrast, flip and strong pseudoknot variants exhibit terminator efficiencies ranging from ~50 to ~90% and the amplitude of the ZMP-mediated antitermination response correlates with terminator efficiency (Fig. 5b). Whereas flip and strong pseudoknot variants contain non-native G24:C92 and C92:G112 base pairs, wild type and weak pseudoknot variants contain the native A24:U92 and U92:A112 base pairs (Fig. 5a). To identify how these nucleotides contribute to the termination defects observed for flip and strong pseudoknot variants, we assessed how each possible J1-2 and terminator hairpin combination affects terminator efficiency in the absence of ZMP (Fig. 5c). In the presence of J1-2WT (A24, G27) or J1-2WEAK (A24, A27), TermFLIP and TermSTRONG terminate transcription comparably to the cognate terminators (Fig. 5c, groups 1–3 and 14–16). This shows that neither TermFLIP nor TermSTRONG is intrinsically defective. This indicates that the identity of position 24, which distinguishes J1-2WT and J1-2WEAK (A24) from J1-2FLIP and J1-2STRONG (G24), contributes to the termination defect observed for flip and strong pseudoknot variants while the identity of the 92:112 base pair does not.

a Sequence of the C. beijerinckii pfl ZTP riboswitch consensus variant library annotated to show all pseudoknot and terminator configurations in which variable nucleotides form Watson–Crick base pairs. b Plots showing fraction terminator readthrough for the four variant classes in which variable nucleotides in the pseudoknot and terminator form Watson–Crick base pairs. Variants are plotted along the x-axis by the difference in fraction terminator readthrough measured for the 1 mM ZMP and 0 mM ZMP conditions so that each variant is located at the same x-axis position in both plots. c, d Plots showing the efficiency of each terminator from panel a in the presence of each J1-2 sequence for U113, C116, and ΔU113, ΔC116 variants in the presence of 500 μM NTPs (c) and 100 μM NTPs (d). The median fraction terminator readthrough for each variant group was calculated using the n = 288 variants that comprise the group. e Illustration of an alternative pseudoknot that is favored if J1-2 contains G24. f Illustration of a model for how the identity of the 89:117 terminator base pair affects the propagation of terminator base pairs through the alternative pseudoknot depicted in panel e. Data are from the experiments shown in Figs. 3, 4. Source data are provided as a Source Data file. WT wild type, FL flip, ST strong, WK weak, PK pseudoknot, term terminator, alt. PK alternative pseudoknot.
The distribution of terminator efficiencies is broader when J1-2 contains G24 regardless of which terminator hairpin is present (Fig. 5c, groups 5–12). However, variants that contain G24 terminate transcription more efficiently in the presence of TermWT or TermSTRONG than in the presence of TermFLIP or TermWEAK (Fig. 5c, groups 5–12). This indicates that the identity of the 89:117 pair, which distinguishes TermWT and TermSTRONG (C89:G117) from TermFLIP and TermWEAK (U89:A117), also contributes to the termination deficiencies observed for flip and strong pseudoknot variants.
The observation that G24 inhibits both transcription termination and ZMP-mediated antitermination suggests that it can cause the pfl aptamer to misfold. One possible mechanism for misfolding is the formation of an alternative pseudoknot between nucleotides 22–25 and 87–84, which is stabilized by G24 and interrupted by A24 and is adjacent to the native pseudoknot (Fig. 5e). This model is consistent with the observation that a U89:A117 terminator base pair exacerbates the termination defect caused by G24. Displacement of the alternative pseudoknot during terminator base pair propagation is anchored by two contiguous base pairs due to a bulge at C116 (Fig. 5f). The presence of a strong base pair within the anchoring dinucleotide may favor displacement of the alternative pseudoknot. To assess this model, we used TECdisplay to characterize a second pfl ZTP riboswitch consensus variant library in which both terminator hairpin bulges were removed by deleting U113 and C116. Deletion of the terminator hairpin bulges improves terminator efficiency for all J1-2/terminator configurations except wild type (Fig. 5c, groups 17–32). The improvement in terminator efficiency is greater for G24 variants than for A24 variants, indicating that removing the terminator hairpin bulges partially compensates for the G24-associated termination defect (Fig. 5c, groups 17–32). Slowing transcription by reducing the concentration of NTPs from 500 μM to 100 μM has a similar effect to deleting the terminator hairpin bulges, except that terminator efficiency is also improved for variants with wild type J1-2 and terminator sequences (Fig. 5d, groups 33–48). This indicates that the G24-associated termination defect can be overcome by allowing additional time for terminator folding to occur. Deleting the terminator hairpin bulges and reducing NTP concentration further improves terminator efficiency (Fig. 5d, groups 49–64). Together, these observations support a model in which G24 variants interfere with terminator base pair propagation.
pfl ZTP aptamer variants are prone to pseudoknot misfolding
To assess the model that G24 stabilizes an alternative pseudoknot, we designed a 16,384-variant library that varies two alternative pseudoknot base pairs as N22:G87 and N24:N85 and varies several other nucleotides to allow for complementarity in P1, P3, and the terminator hairpin (Fig. 6a). As expected, mismatches in either or both of the terminator base pairs that were randomized causes a termination defect (Fig. 6b). Variants with intact terminators exhibit a broad range of terminator efficiencies that result from several classes of folding defects (Fig. 6b). The first class of defective terminators contains U97, which likely stabilizes a competitor helix that competes with P3 folding and inhibits terminator hairpin folding by blocking formation of the first terminator base pair (Fig. 6a, c). The second class of defective terminators contains C24, which increases terminator readthrough in some sequence contexts (Fig. 6d). The remaining classes of defective terminators are associated with pseudoknot misfolding and are described in detail below.

a Sequence of the C. beijerinckii pfl ZTP riboswitch alternative pseudoknot perturbation library. b Plot showing fraction terminator readthrough in the absence of ZMP for variants with intact or disrupted terminator base pairs before and after variants that are prone to misfolding have been filtered. c, d Plots showing fraction terminator readthrough for variants that contain each of the sixteen intact terminator hairpins that occur in the library shown in panel a before (c) and after (d) variants that are prone to misfolding have been filtered. In panel d, C24 variants are separated from A24 and G24 variants because they exhibit context-dependent termination defects. TECdisplay data are from n = 2 replicates that were combined. Source data are provided as a Source Data file.
Each of the 16 variant classes in which terminator base pairing is intact contains a group of three variants for which fraction terminator readthrough is ~90% in the absence of ZMP (Fig. 6c). These variants all contain G22 and U24, which enables a non-native pseudoknot to form by pairing with C90 and A88, respectively (Fig. 7a). Disruption of these base pairs restores terminator efficiency to a variable degree that depends on the identity of the 85:121 and 97:107 terminator base pairs. (Fig. 7b and Supplementary Fig. 4). Notably, C44 partially restores terminator function by pairing with G22, which competes with pseudoknot misfolding by stabilizing P1 (Fig. 7b and Supplementary Fig. 4).

a, c, e Secondary structure of pseudoknot configurations that were identified in the library shown in Fig. 6a, which cause transcription termination defects. b, d, f Plots showing fraction terminator readthrough in the absence of ZMP for variants that can form the alternative pseudoknots shown in panels a, c, e and for variants in which alternative pseudoknot base pairing is perturbed. Data are from the experiment shown in Fig. 6. Source data are provided as a Source Data file.
Variants that contain A22 can extend the native pseudoknot by forming A22:U94 and C21:G95 base pairs (Fig. 7c). For most intact terminator classes, extending the native pseudoknot increases the amount of terminator readthrough that occurs in the absence of ZMP, presumably by stabilizing the pseudoknot (Fig. 7d and Supplementary Fig. 5). In these cases, disrupting either or both of the A22:U94 and R24:U92 pairs restores terminator efficiency. U44 partially restores terminator function by pairing with A22 in P1, which competes with an extension of the native pseudoknot (Fig. 7d and Supplementary Fig. 5). However, this effect is small in comparison to the effect of stabilizing P1 with a G22:C44 pair described above.
Once the sources of terminator dysfunction described above had been identified, it became possible to assess whether G24 causes a terminator defect by stabilizing an alternative pseudoknot between nucleotides 22–25 and 87–84 (Fig. 7e). In combination, the C22:G87 and G24:C85 pairs reduce the efficiency of all terminator classes except for variants that contain U97, which are predicted to form a competitor helix that is mutually exclusive with one alternative pseudoknot base pair (Fig. 7f and Supplementary Fig. 6). As expected, weakening or disrupting alternative pseudoknot base pairs restores terminator efficiency. Notably, the presence of a C22:G44 pair restores terminator efficiency, presumably by stabilizing P1 so that the alternative pseudoknot cannot fold. This is consistent with the observation that the G24-containing flip and strong pseudoknot variants exhibit higher basal terminator readthrough in the presence of a U22:U44 pair than in the presence of U22:A44 or U22:G44 (Fig. 2i–k).
P1 destabilization causes a transcription termination defect
In the initial analysis of the 32,768-variant library depicted in Fig. 3a, we identified a class of variants in which the efficiency transcription termination and ZMP-mediated antitermination were both reduced43. These variants share a sequence that is predicted to form two competitor helices, termed CH1 and CH2, that compete with the native P1 subdomain structure (Supplementary Fig. 7a, b). The apparent disruption of either competitor helix restores termination efficiency (Supplementary Fig. 7c). Similarly, disrupting the pseudoknot restores termination efficiency except for variants that contain G43 and G44 (Supplementary Fig. 7d, e). To assess the competitor helix model, we performed TECdisplay on a library of 32,768 variants that contain G35N and U37N, which perturbs base pairs within CH1 in the context of the wild-type pseudoknot and terminator (Supplementary Fig. 7a, b). If the competitor helix model is correct, disrupting the 21:35 and 19:37 base pairs should resolve P1 subdomain misfolding and restore the efficiency of termination. Variants that contain the native G35 and U37 nucleotides behave identically to the same variants when they were measured in the library depicted in Fig. 3a (Supplementary Fig. 7c, f). However, disrupting either or both of the 21:35 and 19:37 base pairs does not resolve the termination defect that was originally attributed to competitor helix folding (Supplementary Fig. 7g). Furthermore, the termination defect is more severe in the presence of an A19/C37 mismatch, which disrupts CH1, than in the presence of a G19:C37 pair, which stabilizes CH1 (Supplementary Fig. 7g, first row). Notably, variants in which CH1 is stabilized by both C21:G35 and G19:C37 pairs exhibit a more severe termination defect in the presence of G43 and G44 (Supplementary Fig. 7g, C21:G35, R19/C37 plot). This suggests that CH1 and CH2 can form given sufficiently strong base pairing and that the termination defect caused by the competitor helices only occurs in the presence of G43 and G44, although the role of these nucleotides is not clear.
The observations described above show that the competitor helix model does not explain the termination defects that are associated with P1 sequence variation. Upon further examination, we determined that the sequence combinations that are associated with the termination defect all contain G17:U49 and U18:G48 base pairs in P1. This suggests that the termination defects described above may be caused by P1 destabilization. In support of this model, Y16:R50, R17:Y49, Y18:G48, and R19:A47 base pair variants that further destabilize P1 exacerbate the termination defect and variants that stabilize P1 reduce the termination defect (Supplementary Fig. 8a). Slowing transcription by reducing NTP concentration from 500 to 100 μM partially resolves the termination defect (Supplementary Fig. 8b). Slowing transcription and deleting the U113 and C116 bulges, which interrupt pseudoknot base pair displacement during terminator base pair propagation, fully resolves the termination defect (Supplementary Fig. 8c). This indicates that the termination defect that is associated with P1 destabilization is caused by decreasing the efficiency of terminator folding but does not cause the terminator to misfold catastrophically. Disrupting the pseudoknot by combining the wild-type J1-2 sequence with TermSTRONG restores terminator efficiency except for variants that contain G43 and G44 (Supplementary Fig. 8d). This indicates that, for most variants, the termination defect that is caused by P1 destabilization requires pseudoknot formation. The observation that this termination defect can be overcome by slowing transcription and deleting nucleotides that interrupt the displacement of pseudoknot base pairs by terminator base pairs suggests that destabilizing P1 increases the stability of the pseudoknot.
P1 and J2-1 nucleotides modulate the ZMP-mediated transcription antitermination response
In addition to the variability in terminator activity between the four pseudoknot/terminator classes, variants within each class exhibit a variable ZMP-mediated antitermination response (Fig. 5b). This variability is primarily attributable to the identity of nucleotides 43 and 46, which are present in J2-1 and in a region of P1 that contains several non-canonical base pairs, respectively (Fig. 5a). To determine how the identity of nucleotides 43 and 46 affects ZMP-mediated antitermination, we filtered wild type pseudoknot variants for each of the four possible sequence configurations of positions 43 and 46 and generated aggregate dose-response curves (Fig. 8a). Variants that exhibit the P1 destabilization-associated termination defects shown in Supplementary Fig. 8 were excluded from this analysis. The EC50 of G43, A46 variants (677 μM) is 2.7X that of A43, G46 variants (250 μM), whereas the EC50 of G43, G46 (374 μM) and A43, A46 (426 μM) variants fall between these values (Fig. 8b). This indicates a sequence preference of [A43, G46] > [G43, G46] ≈ [A43, A46] > [G43, A46]. Notably, the sub-optimality of the wild-type sequence (A43, A46, EC50 = 426 μM) may enable discrimination between basal and folate stress-induced Z nucleotide concentrations, which in Salmonella typhimurium are ~100 μM and >1 mM, respectively44. The preference for G46, which is non-native and pairs with A20 in P1, is presumably attributable to P1 stabilization, since G46 forms three hydrogen bonds with A20, whereas A46 only forms two hydrogen bonds with A20 (Fig. 8c). The preference for A43, which is native and stacks with the U22: R44 base pair, is less clear. The structure of the Actinomyces odontolyticus ZTP aptamer determined by Trausch et al. identified a base pair between the P2-proximal adenine base in J1-2 and a guanine base at the position equivalent to nucleotide 43 in the Cbe pfl ZTP aptamer45 (Fig. 8d). In the Cbe pfl aptamer, the DMS reactivity of the P2-proximal adenine at position 29 increases upon ZMP binding38 (Fig. 8e). This indicates that ZMP binding causes the Cbe pfl aptamer to undergo a conformational change in which the Watson–Crick face of A29 becomes unpaired. An interaction between A29 and G43 might interfere with the ZMP-mediated conformational change that causes A29 to become DMS reactive. However, this speculation cannot be assessed rigorously by TECdisplay or cotranscriptional RNA chemical probing. Nonetheless, it is clear that the nucleotide composition of P1 can modulate the ZMP-mediated transcription antitermination response.

a Plots showing the ZMP-mediated transcription antitermination response for C. beijerinckii pfl ZTP riboswitch variants with intact P1 base pairs and wild-type pseudoknot and terminator base pairs filtered by the identity of nucleotides 43 and 46. The shaded region shows the mean fraction terminator readthrough ± two standard deviations of each variant group (n = 60) for each ZMP concentration. Data are from the experiment in Fig. 3. b Plot showing aggregate dose-response curves for the variant groups shown in panel a. Plotted data are the mean ± one standard deviation of each variant group (n = 60) for each ZMP concentration. c Structure of the Thermosinus carboxydivorans (PDB:4ZNP81) and Fusobacterium ulcerans (PDB:5BTP82) ZTP aptamer base pairs that correspond to the Cbe A20:G46 and A20:A46 base pairs, respectively. d Structure of the Actinomyces odontolyticus ZTP aptamer (PDB:4XW745) base pair between nucleotides that correspond to nucleotides A29 and G43 in the pfl riboswitch aptamer. Crystal structures were visualized using UCSF Chimera83 and are labeled using Cbe numbering. e Plot of transcript length-dependent DMS reactivity changes at A29 measured using TECprobe-VL38. Z indicates when ZMP can bind to the aptamer, and T indicates when the terminator hairpin can begin to fold. Decreased A29 DMS reactivity at transcript ~130 is caused by the presence of terminated transcripts in the probing reaction. TECprobe-VL data are from Szyjka and Strobel, Observation of coordinated RNA folding events by systematic cotranscriptional RNA structure probing, Nat. Commun., 2023 Nov 29;14(1)783938. Source data are provided as a Source Data file. WT wild type, frac. fraction.
Discussion
We have described how TECdisplay quantifies riboswitch-mediated transcription antitermination using a cotranscriptional RNA-dependent DNA fractionation strategy. While the current study focuses on the measurement of transcription termination efficiency, TECdisplay is a modular platform for building assays that measure diverse RNA-mediated functions. As outlined in Fig. 1, the general TECdisplay procedure records RNA function in an Illumina sequencing library in three steps: First, RNA is cotranscriptionally displayed from E. coli RNAP using a quantitative single-round in vitro transcription reaction in which RNAP is halted at a chemically-encoded transcription roadblock. Second, TECs are fractionated by the activity of the cotranscriptionally displayed transcript. Third, template DNA molecules are quantitatively tagged with fraction-specific barcodes. The fractions are then pooled back together, excess tagging oligo is depleted, the non-transcribed DNA strand is selectively degraded, and the transcribed DNA strand is amplified for Illumina sequencing by limited-cycle PCR. This modular organization is designed to facilitate the rapid development of new TECdisplay assays. Whereas the procedures for quantitatively barcoding template DNA and preparing an Illumina library are universal elements of all TECdisplay assays, the procedures for cotranscriptionally displaying RNA and fractionating the TEC library are designed to be tailorable to the application at hand. Towards this end, we have already established a high-performance tool kit for building cotranscriptional RNA assays. While the current procedure uses an etheno-dA modification to halt RNAP, we previously showed that the biotin-triethylene glycol and desthiobiotin-triethylene glycol affinity tags quantitatively halt E. coli RNAP transcription, when placed in the transcribed DNA strand29. Similarly, Nadon et al. showed that the NPOM-caged-dT modification can be used as a photoreversible transcription roadblock46. The biotin-triethylene glycol modification is likely to be particularly useful for TECdisplay because it can be used to isolate roadblocked TECs with a 1:1:1 DNA:RNAP:RNA composition without using the non-native C3-SC1 leader sequence47,48. We envision that these tools will facilitate the development of TECdisplay assays for diverse RNA functions.
TECdisplay is directly inspired by the HiTS-RAP6 and RNA-MaP7 (RNA-HiTS) methods, in which RNA is cotranscriptionally displayed in situ on an Illumina flow cell for use in high-throughput, fluorescence-based RNA functional assays. Like HiTS-RAP and RNA-MaP, TECdisplay enables the activity of RNA variants to be characterized systematically in a high-throughput experiment. The primary advantage of TECdisplay is that it is broadly accessible because it does not require custom instrumentation and can be performed using commercially available reagents with the exception of GreB, which is readily purified49. The primary disadvantage of TECdisplay relative to RNA-HiTS methods is that TECdisplay is not a direct biophysical measurement. Consequently, RNA-HiTS experiments have superior temporal resolution and maybe more quantitatively accurate in some cases. While there is substantial overlap in the potential applications of TECdisplay and RNA-HiTS assays, these strategies are also likely to be complementary due to the technical nuances of implementing fractionation-based and imaging-based assays for RNA function.
Torgerson et al., previously developed a high-throughput transcription termination assay called Sequencing-based Mutational Analysis of RNA Transcription Termination (SMARTT)41, which has been used to characterize mechanisms of riboswitch-mediated transcription control50 and was integrated with a RelE cleavage assay51 to measure riboswitch-mediated translation control52. SMARTT quantifies transcription termination efficiency using an RNA-sequencing approach in which sequencing reads that originate from termination products are distinguished from full-length reads by the identity of their 3’ end. The TECdisplay assay presented here is also designed to systematically quantify transcription termination efficiency, but accomplishes this measurement using an orthogonal fractionation-based approach. TECdisplay is a more complex procedure than SMARTT because it is designed to be a modular platform for developing cotranscriptional RNA assays rather than a dedicated assay for transcription termination efficiency. For some RNA targets, the TECdisplay assay for quantifying transcription termination efficiency described here will be advantageous because it does not require an RNA 3’ ligation which can be a source of technical bias32,33,36,38. In cases where the target RNA cannot accommodate the C3-SC1 leader sequence, which is required for the TECdisplay assay, SMARTT will be advantageous because it does not require a non-native leader sequence.
RNA-sequencing-based strategies for systematically quantifying RNA function have also been developed for other RNA-mediated biochemical activities, such as self-cleavage53,54,55, protein binding56,57, ligation58, and pri-miRNA processing59. While the initial application of TECdisplay described here measures transcription termination efficiency, it is likely that TECdisplay assays can be developed for other systems. Whether TECdisplay is advantageous is application-specific and depends on several factors: First, in order for the development of a TECdisplay assay for a given RNA-mediated function to be possible, it must be possible to fractionate a TEC library using the function of interest. RNA-mediated functions that cannot be used to fractionate a TEC library are not compatible with TECdisplay. Second, RNA targets that must fold or function cotranscriptionally will likely benefit from TECdisplay because it is an intrinsically cotranscriptional assay. RNA assays that require thermal refolding are not compatible with TECdisplay. Third, assays in which RNA sequence information is destroyed will likely benefit from TECdisplay because sequence information is recovered from DNA in TECdisplay experiments. Fourth, TECdisplay can only be performed in vitro. Therefore, while the development of a TECdisplay assay will be advantageous for some applications, established RNA-sequencing-based approaches will remain preferable in other cases.
There are several potential sources of error that must be avoided to ensure that cotranscriptional RNA-dependent DNA fractionation is robust during TECdisplay experiments. First, because sequence information for each variant is recovered from DNA, heteroduplexes in the DNA template preparation will confound TECdisplay measurements. Strategies such as performing reconditioning PCR cycles can minimize the presence of heteroduplexes60. To further safeguard against the potential presence of heteroduplexes in the DNA template preparation, the non-transcribed DNA strand is degraded by lambda exonuclease after template DNA fractions are barcoded and pooled back together during the TECdisplay sequencing library preparation. This guarantees that all sequencing reads are derived from the transcribed DNA strand, which directly corresponds to the RNA sequence. In later DNA template preparations, we also implemented a procedure in which the template DNA strand is degraded by lambda exonuclease after PCR and subsequently resynthesized using a primer that contains the internal etheno-dA modification (see Methods). This approach provides an additional safeguard against heteroduplexes on the front end of the procedure. Second, it is essential that virtually every DNA template molecule in the transcription reaction yields a single, complete RNA product because DNA templates that do not contain a single TEC that has completed transcription will partition into one reaction fraction by default and cause noise in the TECdisplay measurement. Quantitative in vitro transcription is accomplished by isolating C3-SC1TECs28,61, which are synchronized and >98% active, and by including the transcript cleavage factor GreB62, which rescues TECs that have arrested due to RNAP backtracking63,64,65, in the transcription reaction. It is also possible for long-lived pauses that are not backtracked66,67,68,69 to prevent RNAP from completing transcription, however this can typically be resolved by increasing NTP concentration or the duration of the transcription reaction. Finally, it is critical that template DNA barcoding is quantitative for both fractions of the transcription reaction. This is accomplished by the dUX-tagging procedure31, which is structured as a one-pot reaction to minimize sample handling variability and appends the fraction barcode to the constant PRA1 promoter sequence to eliminate the possibility of variant-dependent variability in barcoding efficiency. Together, the procedures described above enable the activity of RNA variants to be accurately measured using the cotranscriptional RNA-dependent DNA fractionation strategy.
Characterization of the C. beijerinckii pfl ZTP riboswitch consensus variant library revealed several pathways by which the ZTP aptamer can misfold and identified how stabilization of P1 base pairs can improve ZMP-responsiveness. The identification of these characteristics of pfl riboswitch folding required an approach in which classes of variants were isolated hierarchically. Base pair variation within the ZTP aptamer pseudoknot and terminator hairpin caused the most substantial effect on ZTP riboswitch function (Fig. 5b and Supplementary Fig. 3). This is unsurprising because ZTP riboswitches control transcription termination using a mechanism that requires helical switching between pseudoknot and terminator base pairs25,40,42. Once the effect of pseudoknot and terminator hairpin variability was isolated, it became clear that the composition of nucleotides 43 and 46 in P1 contribute to ZMP-responsiveness (Fig. 8). However, rigorous evaluation of how nucleotides 43 and 46 contribute to ZMP-mediated transcription antitermination was only possible once a sub-population of variants that exhibit a termination defect due to P1 destabilization was isolated (Supplementary Fig. 8).
In the context of the C. beijerinckii pfl ZTP riboswitch scaffold, G24 variants exhibit a broad range of terminator efficiencies due to the formation of an alternative pseudoknot (Fig. 5c). In the P1 subdomain, nucleotides that participate in the alternative pseudoknot partially overlap with nucleotides that participate in the native pseudoknot (Fig. 5a, e). In the P3 subdomain, nucleotides that participate in the alternative pseudoknot are adjacent to nucleotides that participate in the native pseudoknot, but emerge from RNAP first (Fig. 5a, e). In combination, the proximity of alternative pseudoknot base pairs to native pseudoknot base pairs and the earlier emergence of downstream alternative pseudoknot nucleotides from RNAP may cause G24 variants to be prone to aptamer misfolding. It is notable that the severity of the G24-associated termination defect depends on the identity of the 89:117 terminator base pair, which affects how efficiently terminator base pairs are able to displace alternative pseudoknot base pairs (Fig. 5f). This demonstrates that minor differences in expression platform sequence can have substantial context-dependent effects on riboswitch function. When validating the model that G24 variants are prone to pseudoknot misfolding, we identified two additional variant classes in which the formation of a non-native pseudoknot caused a transcription termination defect (Fig. 7). The first class is similar to the G24 class in that pseudoknot formation is shifted upstream in the ZTP riboswitch folding pathway but also increases the number of pseudoknot base pairs, causing a severe termination defect (Fig. 7a). The second class extends the native pseudoknot by two base pairs (Fig. 7c). The observation that relatively minor changes in J1-2 and P3 sequence readily give rise to misfolded pseudoknot structures suggests that the architecture of the ZTP riboswitch aptamer may promote long-range base pairing between these regions in a way that accommodates diverse pseudoknot pairing configurations. Therefore, one possible mode by which cotranscriptional RNA misfolding may arise is the mutation-induced formation of a non-native alternative structure that approximates a native structure but is non-functional.
Transcription termination defects were also observed when P1 base pairs are weakened or disrupted (Supplementary Fig. 8). These termination defects depend on pseudoknot formation in nearly all cases and can be overcome by slowing transcription and deleting bulges within the terminator hairpin that would otherwise interrupt displacement of pseudoknot base pairs. Therefore, P1 destabilization most likely causes a transcription termination defect by increasing the stability of the pseudoknot, which then reduces the efficiency of terminator base pair propagation. Destabilization of P1 base pairs likely increases the flexibility of J1-2. Therefore, one possible explanation for how P1 destabilization interferes with terminator base pair propagation is that increased flexibility in J1-2 places the pseudoknot in a less strained state than when P1 base pairs are intact.
The alternative pseudoknot and disrupted P1 misfolded states are described in the model for ZTP riboswitch folding shown in Fig. 9, which expands upon models that were proposed by ref. 40 and ref. 42 previously. The current model expands the previous models by including a pathway in which the ZTP aptamer misfolds into a terminator-resistant state that favors transcription terminator readthrough. Despite structural differences, both classes of terminator-resistant misfolded aptamers disfavor terminator base pair propagation and can be rescued by slowing transcription or deleting bulged nucleotides that interrupt terminator folding. This supports a model in which both misfolded aptamer classes favor terminator readthrough by enabling RNAP to bypass the termination site before the terminator hairpin can fold. These observations are consistent with findings from a systematic analysis of the Clostridium tetani glycine riboswitch type 1 singlet, which found that helical switching requires competing helices to be close in energy41.

Diagram of on-pathway and off-pathway ZTP riboswitch folding states. Secondary structures are adapted from ref. 40 and were updated using structure probing data from ref. 38. Purple nucleotides indicate structures within the apo ZTP aptamer that are stabilized by ZMP binding. Green nucleotides indicate the stabilization of these structures and other nucleotides that exhibit ZMP-dependent changes in benzoyl cyanide and/or dimethyl sulfate reactivity in TECprobe-VL40 experiments.
The RNA-dependent DNA fractionation strategy used by TECdisplay experiments is conceptually similar to the mRNA display70 and ribosome display71 strategies for peptide evolution. In mRNA and ribosome display experiments, a nascent polypeptide is tethered to its mRNA template by covalent fusion or the ribosome complex, respectively. In this way, mRNA can be enriched based on the function of its encoded polypeptide. In TECdisplay experiments, template DNA is fractionated by the activity of its encoded RNA, which is physically coupled to its DNA template by the TEC. RNA-dependent DNA fractionation was previously used for in vitro selection by the DNA display method in which a run-off RNA transcript is tethered to the DNA molecule from which it was transcribed by a capture oligonucleotide that is covalently linked to the template DNA72. In principle, the procedures we have developed for TECdisplay could be used to perform cotranscriptional in vitro selection. For example, the strategy for separating DNA molecules on which a transcription termination event occurred from DNA molecules on which a terminator readthrough event occurred could potentially be applied to isolate riboswitches that regulate transcription. This approach would likely be synergistic with recent advances in methods for isolating synthetic riboswitches in which natural riboswitch aptamers are used as scaffolds for in vitro evolution22,73.
Methods
An inventory of all reagents used in this study, including the manufacturer and catalog number of each reagent, is provided in Supplementary Data 1.
Oligonucleotides
All oligonucleotides were purchased from Integrated DNA Technologies. A detailed description of all oligonucleotides, including sequence, modifications, and purifications, is presented in Supplementary Table 1. The sequences of the Ultramer oligonucleotides used in this study are presented in Supplementary Table 2.
Proteins
Q5 DNA Polymerase, Q5U DNA Polymerase, Vent (exo-) DNA polymerase, Sulfolobus DNA Polymerase IV, E. coli RNA Polymerase Holoenzyme, RNase If, Thermolabile Proteinase K, Thermolabile USER II Enzyme, T4 DNA Ligase, ET SSB, and Lambda Exonuclease were purchased from New England Biolabs (NEB). UltraPure BSA was purchased from Invitrogen. GreB was a gift from R. Landick (University of Wisconsin, Madison).
DNA template preparation
Supplementary Table 3 provides details for the oligonucleotides and processing steps used for every DNA template preparation used in this work. Supplementary Table 4 provides DNA template sequences. Initial experiments with the Ultramer 004 variant library used DNA templates that were prepared by PCR amplification and translesion synthesis. Later experiments with the Ultramer 015, 016, 017, 018, 019, and 020 variant libraries used an improved procedure in which PCR products are treated with lambda exonuclease to generate ssDNA, which is then used as a template for primer extension to generate internally modified dsDNA. Both procedures are described below.
Preparation of internally modified DNA templates by PCR amplification and translesion synthesis
Because the maximum Ultramer length is 200 nt, it was necessary to amplify DNA templates for TECdisplay in two steps. The first PCR, which appended sequence from the PRA1 promoter, was performed as a 100 μl reaction containing 1X Q5 Reaction Buffer (NEB), 1X Q5 High GC Enhancer (NEB), 200 μM dNTPs (NEB), 250 nM TECD020.PRA1_C3SC1.F (Supplementary Table 1), 250 nM TECD017.dRP1_NoMod.R (Supplementary Table 1), 0.025 nM ULT004.C3SC1_pflCONSv1_VRA3 template (Supplementary Table 2), and 0.02 U/μl Q5 DNA polymerase (NEB). Amplification was performed using the program: 95 °C for 3 min, [95 °C for 20 s, 65 °C for 30 s, 72 °C for 20 s] x 12 to 15 cycles, 72 °C for 5 min. PCR products were mixed with an equal volume of SPRI beads, purified as described below in the section Purification of DNA by solid-phase reversible immobilization (SPRI), and eluted into 20 μl of 10 mM Tris-HCl (pH 8.0). DNA was quantified using the Qubit dsDNA HS Assay Kit (Invitrogen) with a Qubit 4 Fluorometer (Invitrogen). Molarity was determined using the length of the DNA.
DNA templates that contained an internal etheno-dA transcription stall site were PCR amplified from the purified dsDNA prepared above. This second PCR was performed as four 100 μl reactions containing 1X Q5U Reaction Buffer (NEB), 1X Q5 High GC Enhancer, 200 μM dNTPs, 500 nM TECD007.pRA1_2dU.F (Supplementary Table 1), 500 nM TECD002.dRP1iEthDA.R (Supplementary Table 1), 1 μl of 1 pM DNA template 4 (Supplementary Table 3), and 0.02 U/μl Q5U DNA polymerase (NEB). Amplification was performed as a 2-step PCR using the program: 98 °C for 30 s, [98 °C for 10 s, 72 °C for 2 min] x 30 cycles, 72 °C for 1 min. After the reaction was complete, the thermal cycler was held at 72 C, 33 μl of the original PCR was diluted into 66 μl of fresh PCR master mix that contained 1X Q5U Reaction Buffer, 1X Q5 High GC Enhancer, 200 μM dNTPs, 750 nM TECD007.pRA1_2dU.F (Supplementary Table 1), 750 nM TECD002.dRP1iEthDA.R (Supplementary Table 1), and 0.02 U/μl Q5U DNA polymerase. These diluted reactions were then amplified for two additional cycles.
PCR products were ethanol precipitated by mixing with 3 volumes of 100% ice-cold ethanol, 0.1 volumes of 3 M sodium acetate (pH 5.5), and 1.5 μl of GlycoBlue Coprecipitant (Invitrogen) and chilling at −70 °C for at least 30 min. The samples were centrifuged at 18,500×g and 4 °C for 30 min, the supernatant was removed, the samples were centrifuged again briefly to pull down residual ethanol, and residual ethanol was removed. The pellets were washed by adding 500 μl of 70% ethanol and centrifuged at 18,500×g and 4 °C for 2 min. The supernatant was removed, the samples were centrifuged again briefly to pull down residual ethanol, and residual ethanol was removed. The pellets were dissolved in 100 μl of 10 mM Tris-HCl (pH 8.0) and mixed with 20 μl of 6X DNA Loading Dye [10 mM Tris-HCl (pH 8.0), 30% (v/v) glycerol, 0.48% (w/v) SDS, 0.05% (w/v) Bromophenol Blue]. The sample was run on an 8% native TBE-polyacrylamide gel at 120 V for 75 min. Gels were stained with SYBR Gold Nucleic Acid Stain (Invitrogen) for 10 min. The modified DNA template of interest was visualized using a blue light transilluminator and cut out of the gel using a clean razor blade. The gel slice was placed inside a 600 μl microcentrifuge tube with a needle hole in the bottom, which was placed in a 2 ml collection tube and centrifuged at 18,500×g and 4 °C for 5 min to crush the gel. The gel was resuspended in 3 μl of Polyacrylamide Gel Extraction Buffer (300 mM sodium acetate [pH 5.5], 1 mM EDTA) per mg of gel and incubated at room temperature with rotation (~15 rpm) overnight. The sample was centrifuged at 18,500×g, and 4 °C for 5 min, and the supernatant was transferred to a Spin-X 0.22-μm cellulose acetate centrifuge tube filter. The gel was resuspended in 1 μl of Polyacrylamide Gel Extraction Buffer per mg of gel, mixed by vortexing, centrifuged at 18,500×g and 4 °C for 5 min, and the supernatant was transferred to the filter column. Samples were centrifuged at 2000×g and 4 °C for 3 min, and the filtrate was collected. The filtrate was mixed with an equal volume of Tris (pH 8)-buffered phenol:chloroform:isoamyl alcohol (25:24:1, v/v), mixed by vortexing and inversion, and centrifuged at 18,500×g and 4 °C for 5 min. The aqueous phase was collected and transferred to a new tube. The DNA was ethanol precipitated as described above and resuspended in 100 μl of 10 mM Tris-HCl (pH 8.0).
The purified DNA template was then completed using the translesion primer extension, which extends the non-transcribed DNA strand beyond the etheno-dA modification in the template DNA strand29. Translesion DNA synthesis using Sulfolobus DNA polymerase IV (NEB) was performed by incubating two 100 μl reactions containing the purified PCR product, 1X ThermoPol Buffer, 200 μM dNTPs, 0.02 U/μl Vent (exo-) DNA Polymerase, and 0.02 U/μl Sulfolobus DNA Polymerase IV at 55 °C for 1 h in a thermal cycler with a heated lid set to 105 °C29,74. The resulting dsDNA was mixed with an equal volume of SPRI beads and purified as described below in the section Purification of DNA by solid-phase reversible immobilization (SPRI). Following purification, the resulting DNA template was quantified using the Qubit dsDNA HS Assay Kit (Invitrogen) with a Qubit 4 Fluorometer, and molarity was determined using the length of the DNA.
Preparation of internally modified DNA by PCR amplification, lambda exonuclease treatment, and primer extension
PCR was performed as eight 100 μl reactions containing 1X Q5U Reaction Buffer, 1X Q5 High GC Enhancer, 200 μM dNTPs, 250 nM TECD029.PRA1C3SC1p22.F (Supplementary Table 1), 250 nM TECD026.dRP1_5P_iA.R (Supplementary Table 1), 0.025 nM ultramer oligonucleotide template (Supplementary Table 2), and 0.02 U/μl Q5U DNA polymerase. TECD029.PRA1C3SC1p22.F contains both the 2x dU modified PRA1 promoter and part of the C3-SC1 leader, which eliminates the need for performing multiple rounds of PCR to extend the ultramer sequence, as was done above. TECD026.dRP1_5P_iA.R contains a 5’ phosphate so that the template DNA strand can be selectively degraded by lambda exonuclease. Amplification was performed as a 2-step PCR using the program: 98 °C for 30 s, [98 °C for 10 s, 72 °C for 2 min] x 12–17 cycles, 72 °C for 2 min. The optimal number of PCR cycles was determined for each Ultramer by performing a test amplification in which PCR products were assessed by native TBE-PAGE. Optimal cycle numbers had the following characteristics: (i) excess primers were present and (ii) no aberrant PCR products were present.
The eight PCRs were combined into two 400 μl pools, mixed with two sample volumes (800 μl) of SPRI beads, and purified as described below in the section Purification of DNA by solid-phase reversible immobilization (SPRI). DNA was eluted in 25 μl of 10 mM Tris (pH 8.0) per 100 μl PCR (100 μl per pooled sample). Lambda exonuclease reactions were performed as eight 50 μl reactions that contained 25 μl of purified PCR product, 1X Lambda Exonuclease Reaction Buffer (NEB), and 0.1 U/μl Lambda Exonuclease (NEB). The lambda exonuclease reactions were incubated on a thermal cycler at 37 °C for 30 min and then at 75 °C for 10 min to heat inactivate lambda exonuclease. The reactions were transferred to separate 1.7 ml microcentrifuge tubes and mixed with 100 μl of 10 mM Tris (pH 8.0). Samples were then mixed with an equal volume (150 μl) of Tris (pH 8)-buffered phenol:chloroform:isoamyl alcohol (25:24:1). The samples were centrifuged at 18,500×g and 4 °C for 5 min. The aqueous phase was collected, mixed with 3 volumes of ice-cold 100% ethanol, 0.1 volumes of 3 M sodium acetate (pH 5.5), and 1.5 μl of GlycoBlue Coprecipitant, and chilled at −20 °C overnight or −70 °C for 30–60 min to precipitate the ssDNA. The samples were centrifuged at 18,500×g and 4 °C for 30 min, the supernatant was aspirated and discarded, the samples were briefly spun in a mini centrifuge, and the residual supernatant was aspirated and discarded. Samples were then washed by adding 1 ml of ice-cold 70% ethanol and gently inverting the tube. The samples were centrifuged at 18,500×g and 4 °C for 5 min, the supernatant was aspirated and discarded, the samples were briefly spun in a mini centrifuge, and the residual supernatant was aspirated and discarded. Each pellet was resuspended in 50 μl of 10 mM Tris (pH 8.0).
100 μl primer extension reactions contained 50 μl of purified ssDNA, 1X Q5U Reaction Buffer, 1X Q5 High GC Enhancer, 200 μM dNTPs, 300 nM TECD002.dRP1iEthDA.R (Supplementary Table 1), and 0.02 U/μl Q5U DNA polymerase. Primer extension was performed by incubating the samples at 95 °C for 3 min, 65 °C for 10 min, and 72 °C for 10 min. The eight primer extension reactions were then combined into two 400 μl pools, mixed with two volumes (800 μl) of SPRI beads, and purified as described in the section Purification of DNA by solid-phase reversible immobilization (SPRI) below. DNA was eluted in 50 μl of 10 mM Tris (pH 8.0) per pooled sample, and purified by SPRI a second time to remove residual primer. DNA was then eluted in 50 μl of 10 mM Tris (pH 8.0) per pooled sample. DNA concentration was quantified using a Qubit dsDNA HS Assay Kit and Qubit 4 fluorometer, and the resulting dsDNA product was assessed by native TBE-PAGE with SYBR Gold staining.
Purification of DNA by solid-phase reversible immobilization (SPRI)
SPRI beads were prepared in-house using the ‘DNA Buffer’ variation of the procedure by Jolivet and Foley75. Samples were mixed with a variable amount of SPRI beads (depending on the procedure, details are in each relevant section), incubated at room temperature for 5 min, and placed on a magnetic stand for 3 min so that the beads were collected on the tube wall. The supernatant was aspirated and discarded, and the beads were washed twice by adding a volume of 70% ethanol at least 200 μl greater than the combined volume of the sample and SPRI beads to the tube without disturbing the bead pellet while it remained on the magnetic stand. The samples were incubated at room temperature for 1 min before aspirating and discarding the supernatant. Residual ethanol was evaporated by placing the open microcentrifuge tube in a 37 °C dry bath for ∼15 s with care taken to ensure that the beads did not dry out. Purified DNA was eluted by resuspending the beads in a variable amount of 10 mM Tris-HCl (pH 8.0) (depending on the procedure, details are in each relevant section), allowing the samples to sit undisturbed for 3 min, placing the sample on a magnetic stand for 1 min so that the beads collected on the tube wall, and transferring the supernatant, which contained purified DNA, into a screw-cap tube with an O-ring.
TECdisplay
Preparation of streptavidin-coated magnetic beads
5 μl of 10 mg/ml Dynabeads MyOne Streptavidin C1 beads (Invitrogen) per 25 μl sample volume were equilibrated in Buffer TX (1X Transcription Buffer (defined below), 0.1% (v/v) Triton X-100) in bulk exactly as described previously28,38,61. The original method is presented below: After placing the beads on a magnetic stand and removing the storage buffer, the beads were resuspended in 500 μl of hydrolysis buffer (100 mM NaOH, 50 mM NaCl) and incubated at room temperature for 10 min with rotation. Hydrolysis buffer was removed, and the beads were resuspended in 1 ml of high salt wash buffer (50 mM Tris-HCl (pH 7.5), 2 M NaCl, 0.5% (v/v) Triton X-100), transferred to a new tube, and washed by rotating for 5 min at room temperature. High salt wash buffer was removed, and the beads were resuspended in 1 ml of binding buffer (10 mM Tris-HCl (pH 7.5), 300 mM NaCl, 0.1% (v/v) Triton X-100), transferred to a new tube, and washed by rotating for 5 min at room temperature. After removing the binding buffer, the beads were washed twice with 500 μl of Buffer TX by resuspending the beads, transferring them to a new tube, washing with rotation for 5 min at room temperature, and removing the supernatant. After washing the second time with Buffer TX, the beads were resuspended at a concentration of ~2 μg/μl in Buffer TX (25 μl per sample volume), split into 25 μl aliquots, and stored on ice until use.
Preparation of C3-SC1TECs for quantitative in vitro transcription
Quantitative in vitro transcription was performed by isolating magnetic bead-immobilized, synchronized TECs using the C3-SC1 leader (C3-SC1TECs) essentially as described previously28. A detailed protocol for the preparation of C3-SC1TECs is available61. The original method for C3-SC1TEC purification with application-specific modifications is presented below. One sample volume is defined as 25 μl.
In vitro transcription reactions containing 1X Transcription Buffer (20 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol (DTT), and 0.1 mM EDTA), 0.1 mg/ml BSA, 100 μM ApU dinucleotide, 1X UGC Start NTPs (25 μM UTP, 25 μM GTP, 25 μM CTP [Cytiva, cat. no. 27202501]), 25 nM TECD018.Cap3_3BioTEG oligonucleotide (Supplementary Table 1), 20 nM template DNA, and 0.032 U/μl E. coli RNAP holoenzyme (NEB) were prepared in bulk on ice; at this point, the total reaction volume per sample was 20 μl due to the omission of 10X (200 μg/ml) heparin (Millipore Sigma, catalog no. H5515) and 10X Start Solution (100 mM MgCl2, 100 μg/ml rifampicin). The bulk transcription reaction was placed in a dry bath set to 37 °C for 20 min to form open promoter complexes. After 20 min, 2.5 μl of 200 μg/ml heparin per sample volume was added to the reaction, and the sample was mixed by pipetting and incubated at 37 °C for 5 min to sequester free RNAP and enrich for heparin-resistant open promoter complexes; the final concentration of heparin was 20 μg/ml. After 5 min, 2.5 μl of room temperature 10X Start Solution per sample volume was added to the transcription reaction for a final concentration of 10 mM MgCl2 and 10 μg/ml rifampicin. The transcription reaction was mixed by pipetting and incubated at 37 °C for 20 min to walk RNAP to the C+31 synchronization site in the C3-SC1 leader and hybridize the TECD018.Cap3_3BioTEG oligonucleotide (Supplementary Table 1) to nascent RNA. At this time, 25 μl of ~2 μg/μl pre-equilibrated streptavidin-coated magnetic beads per sample volume, Wash Buffer (1X Transcription Buffer, 10 mM MgCl2, and 0.05% Tween-20), Elution Buffer 1 (1.28X Transcription Buffer, 0.1 mg/ml BSA, 12.82 mM MgCl2, 10 μg/ml Rifampicin, and 100 nM GreB [a gift from R. Landick, UW Madison]), and Elution Buffer 2 (1X Transcription Buffer, 0.1 mg/ml BSA, 10 mM MgCl2, and 10 μg/ml Rifampicin) were placed at room temperature.
After ~18 min, the magnetic beads were placed on a magnetic stand, and the storage buffer was removed. After 20 min, the pre-equilibrated streptavidin-coated magnetic beads were resuspended with the bulk transcription reaction by pipetting and incubated at room temperature with rotation (~15 rpm) for 15 min to immobilize C3-SC1TECs. After 15 min the bead binding mixture was spun briefly in a Labnet Prism mini centrifuge by quickly flicking the switch on and off so that liquid was removed from the tube cap, but the speed of the mini centrifuge remained as low as possible. The sample was placed on a magnetic stand for 1 min, and the supernatant was discarded. The 1.7 ml tube containing the sample was removed from the magnetic stand and the beads were gently resuspended in 1 ml of room temperature Wash Buffer and incubated at room temperature with rotation for 5 min. The sample was placed on a magnet stand for 1 min, and the supernatant was discarded. Immobilized C3-SC1TECs were gently resuspended in 22 μl of room temperature Elution Buffer 1 per sample volume.
Termination-dependent fractionation of template DNA
The C3-SC1TECs and 1.7 ml microcentrifuge tubes containing ZMP Chase Mix were placed in a dry bath set to 37 °C for 2 min to prewarm. For samples that contained ≤1 mM ZMP, ZMP Chase Mix contained 2.5 μl of 5 mM NTPs (Cytiva, cat. no. 27202501) and 0.5 μl of 50 mM ZMP solution in DMSO or, in the case of 0 mM ZMP samples, 0.5 μl of DMSO. For the 3.16 mM ZMP sample, ZMP Chase Mix contained 2.5 μl of 5 mM NTPs and 1.58 μl of 50 mM ZMP. The final NTP concentration for all samples was 500 μM. Because the 50 mM ZMP stock was dissolved in DMSO, samples that contained ≤1 mM ZMP also contained 2% DMSO; the 3.16 mM sample contained 6.32% DMSO. After 2 min, 22 μl (≤1 mM ZMP samples) or 20.9 μl (3.16 mM sample) of immobilized C3-SC1TECs were mixed with ZMP Chase Mix and incubated at 37 °C for 3 min. The transcription elongation factor GreB was included in this reaction (as part of Elution Buffer 1) to rescue backtracked elongation complexes, which could potentially cause experimental noise because DNA that contains a backtracked TEC will always partition into the pellet fraction. After 3 min of transcription, samples were placed on a magnetic stand for 1 min, and the supernatant was transferred to a 200 μl thin-walled PCR tube; this is the unbound (UNB) fraction. The remaining beads were gently resuspended in 25 μl of Elution Buffer 2 and transferred to a 200 μl thin-walled PCR tube; this is the bound (BND) fraction. Samples remained on ice until RNA degradation. We previously showed that, in the absence of a biotin tag, >97% of DNA (either free DNA or DNA that contains a TEC) partitions into the supernatant, which indicates that there is no non-specific bead binding28.
RNA degradation
0.5 μl of RNase If (NEB) was added to each 25 μl transcription reaction fraction and mixed by pipetting. Samples were incubated on a thermal cycler set to 37 °C for 15 min. Samples were then incubated at 70 °C for 20 min to heat-inactivate RNase If. The bound fraction was placed on a magnetic stand for 1 min, and the supernatant, which contains eluted template DNA, was transferred to a new 200 μl thin-walled PCR tube.
Protein degradation
1 μl of Thermolabile Proteinase K (NEB) was added to each 25.5 μl sample and mixed by pipetting. Samples were incubated on a thermal cycler set to 37 °C for 30 min. Samples were then incubated at 65 °C for 20 min to heat-inactivate Thermolabile Proteinase K. At this point samples were either barcoded as described below or stored at −20 °C overnight before barcoding.
Fraction barcoding by deoxyuridine excision tagging (dUX-tagging)
Template DNA was barcoded using a modified version of the dUX-tagging procedure31, which quantitatively appends a barcode and Illumina adapter to dsDNA in a one-pot reaction. USER digestions were prepared on ice by adding 3 μl of 10X T4 DNA Ligase Buffer (NEB) to each 26.5 μl sample, pipetting to mix, then adding 0.5 μl of Thermolabile USER II Enzyme (NEB) and pipetting to mix again. Samples were incubated at 37 °C for 30 min on a pre-warmed thermal cycler block with a heated lid set to 45 °C and held at 12 °C after the incubation was complete. Following USER digestion, 0.4 μl of 500 ng/μl (200 ng) ET SSB (NEB), and 0.6 μl of 2.5 μM (1.5 pmol) of the TECD021.5pBND_pRA1m12_VRA5 or TECD022.5pUNB_pRA1m12_VRA5 oligonucleotides (for bound and unbound fractions, respectively; Supplementary Table 1) were added to each sample and mixed by pipetting. To anneal the tagging oligo and inactivate Thermolabile USER II Enzyme, the master mix was placed on a thermal cycler block set to 70 °C with a heated lid set to 105 °C and slowly cooled using the protocol: 70 °C for 5 min, ramp to 65 °C at 0.1 °C/s, 65 °C for 5 min, ramp to 60 °C at 0.1 °C/s, 60 °C for 2 min, ramp to 37 °C at 0.1 °C/s, hold at 37 °C. After annealing the tagging oligo, 1 μl of T4 DNA ligase (NEB) was added to each sample, and the samples were mixed by pipetting while the reaction remained at 37 °C. The ligation reactions were incubated at 37 °C for 5 min and then at 65 °C for 10 min to heat-inactivate T4 DNA ligase. A 100 μl primer extension reaction containing the 32 μl tagged DNA sample (which at this point contains a 5’ overhang), 1X ThermoPol Buffer (NEB), 0.2 mM dNTPs, 2.5% formamide, and 0.02 U/μl Vent (exo-) DNA Polymerase (NEB) was prepared on ice for each sample. These primer extension reactions were placed on a preheated 72 °C thermal cycler block for 5 min and then moved to an ice-cold aluminum PCR tube block to snap cool. At this point the template DNA is fully barcoded, and the bound and unbound fractions of each sample were combined. The barcoded DNA was then mixed with 200 μl of SPRI beads and purified as described above in the section Purification of DNA by solid-phase reversible immobilization (SPRI) to deplete excess tagging oligonucleotide. Purified DNA was eluted into 50 μl of 10 mM Tris-HCl (pH 8.0).
Non-transcribed DNA strand degradation
Following template DNA barcoding and purification, the non-transcribed DNA strand was selectively degraded using lambda exonuclease so that sequencing libraries are amplified exclusively from the transcribed DNA strand, which is guaranteed to correspond to the transcribed RNA sequence. This ensures that TECdisplay measurements are not confounded by heteroduplexes, which may be present in the DNA template preparation. Lambda exonuclease reactions were prepared on ice in 200 μl thin-walled PCR tubes by adding 5.56 μl of 10X lambda exonuclease reaction buffer (NEB) to the 50 μl purified DNA samples, pipetting to mix, adding 0.5 μl of lambda exonuclease (NEB), and pipetting to mix again. Samples were incubated at 37 °C on a thermal cycler block for 5 min, and then at 75 °C for 10 min to heat-inactivate lambda exonuclease.
Purification of ssDNA by phenol-chloroform extraction
Following non-transcribed DNA strand degradation, the volume of each sample was raised to 150 μl by adding 94 μl of 10 mM Tris-HCl (pH 8.0). Samples were mixed with an equal volume (150 μl) of Tris (pH 8)-buffered phenol:chloroform:isoamyl alcohol (25:24:1, v/v), mixed by vortexing and inversion, and centrifuged at 18,500×g and 4 °C for 5 min. The aqueous phase was collected and transferred to a new tube. ssDNA was precipitated by mixing the aqueous phase with 450 μl of 100% ice-cold ethanol, 15 μl of 3 M sodium acetate (pH 5.5), and 1.5 μl of GlycoBlue Coprecipitant and chilling at −70 °C for at least 30 min. The samples were centrifuged at 18,500×g and 4 °C for 30 min, the supernatant was removed, the samples were centrifuged again briefly to pull down residual ethanol, and residual ethanol was removed. The pellets were washed by adding 500 μl of 70% ice-cold ethanol and centrifuged at 18,500×g and 4 °C for 2 min. The supernatant was removed, the samples were centrifuged again briefly to pull down residual ethanol, and residual ethanol was removed. The pellets were dissolved in 100 μl of 10 mM Tris-HCl (pH 8.0).
Test amplification of TECdisplay libraries
The number of PCR amplification cycles needed for TECdisplay libraries was determined by performing a test amplification adapted from ref. 76. 20 μl PCRs contained 14 μl of Q5 PCR master mix and 6 μl of a 16-fold or 64-fold dilution of the ssDNA libraries, such that the final concentration of the reaction components were 1X Q5 Reaction Buffer, 1X Q5 High GC Enhancer, 200 μM dNTPs, 250 nM RPIX (Supplementary Table 1), 250 nM TECD017.dRP1_NoMod.R (Supplementary Table 1), and 0.02 U/μl Q5 DNA Polymerase. One PCR for each ssDNA dilution was amplified for 11 or 12 cycles using the program: 98 °C for 30 s, [98 °C for 10 s, 62 °C for 20 s, 72 °C for 20 s] x 11 or 12 cycles, 72 °C for 2 min. This format yields test amplifications that correspond to 4, 5, 6, and 7 cycles in the final library amplification described below. Following amplification, each PCR was mixed with 4 μl of 6X DNA Loading Dye and run on a native TBE-polyacrylamide gel to determine the number of cycles needed to amplify the libraries for high-throughput sequencing.
Preparation of DNA libraries for sequencing
Indexed dsDNA libraries were prepared for Illumina sequencing by amplifying each sample in a 50 μl PCR that contained 1X Q5 Reaction Buffer, 1X Q5 High GC Enhancer, 200 μM dNTPs, 500 nM RPI Indexing Primer (Supplementary Table 1), 500 nM TECD017.dRP1_NoMod.R (Supplementary Table 1), 12 μl of ssDNA library, and 0.02 U/μl Q5 DNA Polymerase. Amplification was performed using the program 98 °C for 30 s, [98 °C for 10 s, 62 °C for 20 s, 72 °C for 20 s] x [# of cycles], 72 °C for 2 min, where “# of cycles” corresponds the optimal number of cycles determined by test amplification. To minimize the sequencing of PCR duplicates, TECdisplay libraries that contained 1,048,576 variants were amplified in eight separate PCRs. Following amplification, 50 μl PCRs were mixed with 100 μl of SPRI beads and purified as described in the section Purification of DNA by solid-phase reversible immobilization (SPRI). DNA was eluted into 20 μl of 10 mM Tris-HCl (pH 8.0), mixed with 40 μl of SPRI beads, and purified a second time. Twice-purified DNA was eluted into 10 μl of 10 mM Tris-HCl (pH 8.0) and quantified using the Qubit dsDNA HS Assay Kit (Invitrogen) with a Qubit 4 Fluorometer.
High-throughput DNA sequencing
Sequencing of TECdisplay libraries was performed by Novogene Co. on an Illumina HiSeq X Ten System using 2 × 150 PE reads with 35% PhiX spike in. 32,768-variant TECdisplay libraries were sequenced at a depth of ~200 to ~250 million PE reads, and libraries that contained fewer variants were sequenced at a proportionally lower depth. 1,048,576-variant libraries were sequenced at a depth of ~1 billion PE reads per replicate. Initial sequencing libraries contained constant sequence at the head of read 1 and were pooled with libraries of a different type (e.g., TECprobe-VL38 libraries) to circumvent issues with low library complexity. In later experiments, the inclusion of five random bases at the head of read 1 enabled TECdisplay libraries to be sequenced without the need for pooling with other library types.
TECdisplay data analysis
Source code and documentation for custom data analysis tools is available at https://github.com/e-strobel-lab/TECtools. Target sequences used to map sequencing reads were generated by variant_maker. Sequencing read preprocessing and alignment was performed by TECdisplay_mapper, which manages sequencing read preprocessing using fastp77 and maps reads to targets that were generated by variant_maker. During preprocessing, fastp trims adapter sequences (which are rarely present in the sequencing reads), performs error correction in overlapping regions of read pairs, merges read pairs, and extracts the fraction barcode from the head of read 2. TECdisplay_mapper then maps merged sequencing reads to targets by identifying the location of the SC1 hairpin in the sequencing read, generating a key from bases at locations within the read that are expected to contain variable bases, using the key to search a hash table of all target sequences for a potential match, and verifying the match by comparing the target RNA sequence from the sequencing read to the potential target match. If a match to a reference target is identified, the read is assigned to the bound or unbound fraction based on its fraction barcode. After read mapping is complete, the fraction of bound reads for each variant is calculated by dividing the number of bound reads by the sum of bound and unbound reads. During the interpretation of the data, variants within each data set were filtered by nucleotide and base pair identity using TECdisplay_navigator or TECdisplay_Hnav, which coordinates hierarchical TECdisplay_navigator analyses. Detailed instructions for using all of the software described above are available at https://github.com/e-strobel-lab/TECtools78.
Dose-response curve fitting
For variants that exhibited a ZMP-mediated transcription antitermination response, Ymin, Ymax, and EC50 were determined using a three-parameter nonlinear regression in GraphPad Prism 10 (GraphPad Software). Variants that did not respond to ZMP were fit using a simple linear regression. The resulting dose-response curves are shown in Figs. 2,8. In Supplementary Fig. 2, Ymin, Ymax, and EC50 were used to fit TECdisplay data to the following equation, which was described by ref. 41:
where X is the ligand concentration, Ymax is the fraction terminator readthrough observed (or predicted by the three-parameter logistic fit) at saturating ligand concentration, Ymin is the fraction terminator readthrough in the absence of ligand, and EC50 is the concentration at which half-maximal fraction terminator readthrough was observed (or predicted by the three-parameter logistic fit). Fitting was performed using the script CT2018fit, which can be accessed at https://github.com/e-strobel-lab/Publications/.
Characterization of RNA-dependent DNA fractionation accuracy
To assess the accuracy of RNA-dependent DNA fractionation, C3-SC1TECs were prepared as described above in the section Preparation of C3-SC1TECs for quantitative in vitro transcription using the PRA1_2dU_C3SC1_CbePfl_dRP1_iEthdA and PRA1_2dU_C3SC1_CbePfl_FlipPK_dRP1_iEthdA DNA templates (Supplementary Table 4), which encode the wild-type and flip Cbe pfl ZTP riboswitches, respectively. After C3-SC1TEC purification, transcription was resumed by adding NTPs to a final concentration of either 100 or 500 μM in the presence and absence of 1 mM ZMP, and template DNA was fractionated as described above in the section Termination-dependent fractionation of template DNA. Upon collection, the 25 μl supernatant of each sample was mixed with 125 μl of Stop Solution (0.6 M Tris-HCl [pH 8.0], 12 mM EDTA). Each bead pellet was resuspended in 25 μl of formamide elution solution (95% formamide (v/v), 10 mM EDTA) and incubated at 100 °C for 5 min. The pellet samples were placed on a magnetic stand, and each 25 μl supernatant was mixed with 125 μl of Stop Solution. The 150 μl samples were mixed with an equal volume of Tris (pH 8)-buffered phenol:chloroform:isoamyl alcohol (25:24:1, v/v) and purified by phenol/chloroform extraction and ethanol precipitation as described above in the section Purification of ssDNA by phenol-chloroform extraction except that the resulting pellets were resuspended in 15 μl of formamide loading dye (90% (v/v) deionized formamide, 1X transcription buffer, 0.01% (w/v) bromophenol blue) and assessed by denaturing PAGE as described in the section Denaturing Urea-PAGE. Quantification of band intensity was performed using ImageJ 1.53k by plotting each lane, drawing a line at the base of each peak to subtract the background, and determining the area of the closed peak47. For DNA bands, fraction terminator readthrough was calculated by dividing the intensity of the pellet band by the sum of the intensities of the supernatant and pellet bands. For RNA bands, band intensity was normalized by RNA length, and fraction terminator readthrough was calculated by dividing the normalized intensity of the readthrough band by the sum of the normalized intensities of the terminated and readthrough bands40.
Intermediate fraction analysis of the dUX-tagging procedure
To visualize intermediate products of the optimized dUX-tagging procedure, the PRA1_2dU_C3SC1_CbePfl_dRP1 DNA template (Supplementary Table 4) was barcoded in bulk using the procedure described above in the section Fraction barcoding by deoxyuridine excision tagging (dUX-tagging) and intermediate fractions were collected after key processing steps. Note that the volume of each fraction changes to account for the increase in sample volume as reagents are added throughout the dUX-tagging procedure. The input DNA fraction was collected by transferring 24.5 μl of the reaction to 125 μl of Stop Solution before the USER enzyme was added. The USER-digested fraction was collected by transferring 25 μl of the reaction to 125 μl of Stop Solution after USER digestion. For experiments in which the tagging oligo ligation was performed at 37 °C for 5 min, which contained ET SSB during the ligation, the post-ligation fraction was collected by transferring 26.9 μl of the reaction to 125 μl of Stop Solution. For experiments in which the tagging oligo ligation was performed at 25 °C for 60 min, which did not contain ET SSB during the ligation, the post-ligation fraction was collected by transferring 26.5 μl of the reaction to 125 μl of Stop Solution. The post-primer extension fraction was collected by mixing the 100 μl primer extension reaction with 50 μl of Stop Solution supplemented with 1.8 μl of 500 mM EDTA (pH 8.0) so that the final EDTA concentration was 10 mM. Primer extension reactions were mixed with 200 μl of SPRI beads and purified as described in above in the section Purification of DNA by solid-phase reversible immobilization (SPRI). Purified DNA was eluted into 50 μl of 10 mM Tris-HCl (pH 8.0), and one sample was collected as the post-purification fraction by mixing with 100 μl of Stop Solution. The final sample was treated with lambda exonuclease as described in the section Non-transcribed DNA strand degradation, and the ~56 μl ssDNA product fraction was mixed with 94 μl 10 mM Tris-HCl (pH 8). Fractions were mixed with an equal volume (150 μl) of Tris (pH 8)-buffered phenol:chloroform:isoamyl alcohol (25:24:1, v/v) and processed by phenol/chloroform extraction and ethanol precipitation as described above in the section Purification of ssDNA by phenol-chloroform extraction. Following ethanol precipitation, each pellet was resuspended in 15 μl of formamide loading dye. The samples were analyzed by denaturing PAGE as described in the section Denaturing Urea-PAGE.
Optimization of dUX-tagging ligation time
Eight 25 μl dUX-tagging reactions containing 1X T4 DNA Ligase Buffer, 5 nM PRA1_2dU_C3SC1_CbePfl_dRP1 template DNA (Supplementary Table 4), and 0.02 U/μl Thermolabile USER II Enzyme were prepared in bulk in thin-walled 200 μl PCR tubes on ice; One 24.5 μl reaction volume was transferred to 125 μl of Stop Solution for collection as an input DNA fraction before USER enzyme was added. USER digestion was performed as described in the section Fraction barcoding by deoxyuridine excision tagging (dUX-tagging) and one reaction volume (25 μl) was transferred to 125 μl of Stop Solution for collection as the USER-digested sample and kept on ice. 0.6 μl of 2.5 μM TECD021.5pBND_pRA1m12_VRA5 or TECD022.5pUNB_pRA1m12_VRA5 (Supplementary Table 1) per reaction volume and 0.4 μl of ET SSB per reaction volume were added to the master mix. The tagging oligo was annealed as described in the section Fraction barcoding by deoxyuridine excision tagging (dUX-tagging), and the sample was held at 37 °C. One 25.9 μl fraction was collected as a zero time point before T4 DNA ligase was added. The master mix was aliquoted into five separate thin-walled 200 μl PCR tubes. Each sample was mixed with 1 μl of T4 DNA ligase and incubated at 37 °C for the indicated amount of time (30 s, 1 min, 2 min, 5 min, or 10 min) before being incubated at 65 °C for 10 min to heat-inactivate T4 DNA ligase. After heat inactivation, samples were mixed with 125 μl of Stop Solution and kept on ice. The samples were phenol/chloroform extracted and ethanol precipitated as described in the section Purification of ssDNA by phenol-chloroform extraction, and each pellet was resuspended in 15 μl of formamide loading dye. The samples were analyzed by denaturing PAGE as described in the section Denaturing urea-PAGE.
Optimization of lambda exonuclease digestion time
Seven 25 μl dUX-tagging reactions were prepared in bulk as described in the section Optimization of dUX-tagging ligation time and processed as described in the section Fraction barcoding by deoxyuridine excision tagging (dUX-tagging) through SPRI bead purification; One 24.5 μl reaction volume was transferred to 125 μl of Stop Solution for collection as an input DNA fraction before USER enzyme was added to the initial master mix, and a second 50 μl sample was transferred to 100 μl of Stop Solution after SPRI bead purification for collection as the pre-lambda exonuclease digestion fraction. Each of the remaining five samples was mixed with 5.56 μl of lambda exonuclease reaction buffer and 0.5 μl of lambda exonuclease and incubated at 37 °C for the indicated amount of time (30 s, 1 min, 2 min, 5 min, or 10 min) before being mixed with 1.1 μl of 0.5 M EDTA and incubated at 75 °C for 10 min to heat inactivate lambda exonuclease. Samples were phenol/chloroform extracted and ethanol precipitated as described in the section Purification of ssDNA by phenol-chloroform extraction, and each pellet was resuspended in 15 μl of formamide loading dye. The samples were analyzed by denaturing PAGE as described in the section Denaturing urea-PAGE.
Denaturing urea-PAGE
Denaturing urea-PAGE was performed exactly as described previously47. Briefly, samples in formamide loading dye were heated at 95 °C for 5 min, and snap-cooled on ice for 2 min. Denaturing urea-PAGE was performed using 8 or 10% gels prepared using the SequaGel UreaGel 19:1 Denaturing Gel System (National Diagnostics) for a Mini-PROTEAN Tetra Vertical Electrophoresis Cell as follows: The gel apparatus was assembled such that buffer in the outer chamber covered ~1 cm of the gel plates. The gel was then pre-run at 480 V for 30 min, the wells were flushed by pipetting, the denatured samples were loaded, and the gel was run at 480 V for ~11 min47. Gels were stained with SYBR Gold Nucleic Acid Stain (Invitrogen) and scanned using a Typhoon RGB biomolecular imager or an Azure Sapphire biomolecular imager.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data generated in this study have been deposited in the Sequencing Read Archive (https://www.ncbi.nlm.nih.gov/sra) with the BioProject accession code PRJNA976257. Individual BioSample accession codes are available in Supplementary Table 5. The fully processed sequencing data have been deposited in Zenodo (10.5281/zenodo.10412935)79. The TIFF images of all gels generated in this study have been deposited in Zenodo (10.5281/zenodo.10425136)80. Source data are provided with this paper.
Code availability
TECtools can be accessed at https://github.com/e-strobel-lab/TECtools/releases/tag/v1.1.078. The script used for fitting TECdisplay data to Equation 1 can be accessed at https://github.com/e-strobel-lab/Publications/.
References
Pan, T. & Sosnick, T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 35, 161–175 (2006).
Lai, D., Proctor, J. R. & Meyer, I. M. On the importance of cotranscriptional RNA structure formation. RNA 19, 1461–1473 (2013).
Scharfen, L. & Neugebauer, K. M. Transcription regulation through nascent RNA folding. J. Mol. Biol. 433, 166975 (2021).
Denny, S. K. & Greenleaf, W. J. Linking RNA sequence, structure, and function on massively parallel high-throughput sequencers. Cold Spring Harb. Perspect. Biol. 11, a032300 (2019).
Marklund, E., Ke, Y. & Greenleaf, W. J. High-throughput biochemistry in RNA sequence space: predicting structure and function. Nat. Rev. Genet. 24, 401–414 (2023).
Tome, J. M. et al. Comprehensive analysis of RNA-protein interactions by high-throughput sequencing-RNA affinity profiling. Nat. Methods 11, 683–688 (2014).
Buenrostro, J. D. et al. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol. 32, 562–568 (2014).
She, R. et al. Comprehensive and quantitative mapping of RNA-protein interactions across a transcribed eukaryotic genome. Proc. Natl Acad. Sci. USA 114, 3619–3624 (2017).
Denny, S. K. et al. High-throughput investigation of diverse junction elements in RNA tertiary folding. Cell 174, 377–390 e320 (2018).
Becker, W. R. et al. High-throughput analysis reveals rules for target RNA binding and cleavage by AGO2. Mol. Cell 75, 741–755.e711 (2019).
Jarmoskaite, I. et al. A quantitative and predictive model for RNA binding by human pumilio proteins. Mol. Cell 74, 966–981.e918 (2019).
Wu, M. J., Andreasson, J. O. L., Kladwang, W., Greenleaf, W. & Das, R. Automated design of diverse stand-alone riboswitches. ACS Synth. Biol. 8, 1838–1846 (2019).
Yesselman, J. D. et al. Sequence-dependent RNA helix conformational preferences predictably impact tertiary structure formation. Proc. Natl Acad. Sci. USA 116, 16847–16855 (2019).
Andreasson, J. O. L., Savinov, A., Block, S. M. & Greenleaf, W. J. Comprehensive sequence-to-function mapping of cofactor-dependent RNA catalysis in the glmS ribozyme. Nat. Commun. 11, 1663 (2020).
Bonilla, S. L. et al. High-throughput dissection of the thermodynamic and conformational properties of a ubiquitous class of RNA tertiary contact motifs. Proc. Natl Acad. Sci. USA 118, e2109085118 (2021).
Ober-Reynolds, B. et al. High-throughput biochemical profiling reveals functional adaptation of a bacterial Argonaute. Mol. Cell 82, 1329–1342.e1328 (2022).
Sadee, C. et al. A comprehensive thermodynamic model for RNA binding by the Saccharomyces cerevisiae pumilio protein PUF4. Nat. Commun. 13, 4522 (2022).
Shin, J. H., Bonilla, S. L., Denny, S. K., Greenleaf, W. J. & Herschlag, D. Dissecting the energetic architecture within an RNA tertiary structural motif via high-throughput thermodynamic measurements. Proc. Natl Acad. Sci. USA 120, e2220485120 (2023).
Serganov, A. & Nudler, E. A decade of riboswitches. Cell 152, 17–24 (2013).
McCown, P. J., Corbino, K. A., Stav, S., Sherlock, M. E. & Breaker, R. R. Riboswitch diversity and distribution. RNA 23, 995–1011 (2017).
Hallberg, Z. F., Su, Y., Kitto, R. Z. & Hammond, M. C. Engineering and in vivo applications of riboswitches. Annu. Rev. Biochem. 86, 515–539 (2017).
Porter, E. B., Polaski, J. T., Morck, M. M. & Batey, R. T. Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 13, 295–301 (2017).
Topp, S. & Gallivan, J. P. Emerging applications of riboswitches in chemical biology. ACS Chem. Biol. 5, 139–148 (2010).
Braselmann, E. et al. A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nat. Chem. Biol. 14, 964–971 (2018).
Kim, P. B., Nelson, J. W. & Breaker, R. R. An ancient riboswitch class in bacteria regulates purine biosynthesis and one-carbon metabolism. Mol. Cell 57, 317–328 (2015).
Tran, B. et al. Parallel discovery strategies provide a basis for riboswitch ligand design. Cell Chem. Biol. 27, 1241–1249.e1244 (2020).
Perkins, K. R., Atilho, R. M., Moon, M. H. & Breaker, R. R. Employing a ZTP riboswitch to detect bacterial folate biosynthesis inhibitors in a small molecule high-throughput screen. ACS Chem. Biol. 14, 2841–2850 (2019).
Kelly, S. L., Szyjka, C. E. & Strobel, E. J. Purification of synchronized Escherichia coli transcription elongation complexes by reversible immobilization on magnetic beads. J. Biol. Chem. 298, 101789 (2022).
Strobel, E. J., Lis, J. T. & Lucks, J. B. Chemical roadblocking of DNA transcription for nascent RNA display. J. Biol. Chem. 295, 6401–6412 (2020).
Pupov, D., Ignatov, A., Agapov, A. & Kulbachinskiy, A. Distinct effects of DNA lesions on RNA synthesis by Escherichia coli RNA polymerase. Biochem. Biophys. Res. Commun. 510, 122–127 (2019).
Strobel, E. J. Efficient linear dsDNA tagging using deoxyuridine excision. Chembiochem 22, 3214–3224 (2021).
Alon, S. et al. Barcoding bias in high-throughput multiplex sequencing of miRNA. Genome Res. 21, 1506–1511 (2011).
Hafner, M. et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17, 1697–1712 (2011).
Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A. & Weeks, K. M. Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 10, 1643–1669 (2015).
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015).
Strobel, E. J., Watters, K. E., Nedialkov, Y., Artsimovitch, I. & Lucks, J. B. Distributed biotin-streptavidin transcription roadblocks for mapping cotranscriptional RNA folding. Nucleic Acids Res. 45, e109 (2017).
Lucas, J. K., Gruenke, P. R. & Burke, D. H. Minimizing amplification bias during reverse transcription for in vitro selections. RNA 29, 1301–1315 (2023).
Szyjka, C. E. & Strobel, E. J. Observation of coordinated RNA folding events by systematic cotranscriptional RNA structure probing. Nat. Commun. 14, 7839 (2023).
Verwilt, J., Mestdagh, P. & Vandesompele, J. Artifacts and biases of the reverse transcription reaction in RNA sequencing. RNA 29, 889–897 (2023).
Strobel, E. J., Cheng, L., Berman, K. E., Carlson, P. D. & Lucks, J. B. A ligand-gated strand displacement mechanism for ZTP riboswitch transcription control. Nat. Chem. Biol. 15, 1067–1076 (2019).
Torgerson, C. D., Hiller, D. A., Stav, S. & Strobel, S. A. Gene regulation by a glycine riboswitch singlet uses a finely tuned energetic landscape for helical switching. RNA 24, 1813–1827 (2018).
Binas, O., Schamber, T. & Schwalbe, H. The conformational landscape of transcription intermediates involved in the regulation of the ZMP-sensing riboswitch from Thermosinus carboxydivorans. Nucleic Acids Res. 48, 6970–6979 (2020).
Kelly, S. L. & Strobel, E. J. Systematic analysis of cotranscriptional RNA folding using transcription elongation complex display. Preprint at bioRxiv https://doi.org/10.1101/2023.12.22.573115 (2023).
Bochner, B. R. & Ames, B. N. ZTP (5-amino 4-imidazole carboxamide riboside 5’-triphosphate): a proposed alarmone for 10-formyl-tetrahydrofolate deficiency. Cell 29, 929–937 (1982).
Trausch, J. J., Marcano-Velazquez, J. G., Matyjasik, M. M. & Batey, R. T. Metal ion-mediated nucleobase recognition by the ZTP riboswitch. Chem. Biol. 22, 829–837 (2015).
Nadon, J. F. et al. Site-specific photolabile roadblocks for the study of transcription elongation in biologically complex systems. Commun. Biol. 5, 457 (2022).
Strobel, E. J. Preparation of E. coli RNA polymerase transcription elongation complexes by selective photoelution from magnetic beads. J. Biol. Chem. 297, 100812 (2021).
Strobel, E. J. Isolation of E. coli RNA polymerase transcription elongation complexes by selective solid-phase photoreversible immobilization. Methods Enzymol. 691, 223–250 (2023).
Perederina, A. A. et al. Cloning, expression, purification, crystallization and initial crystallographic analysis of transcription elongation factors GreB from Escherichia coli and Gfh1 from Thermus thermophilus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 44–46 (2006).
Torgerson, C. D., Hiller, D. A. & Strobel, S. A. The asymmetry and cooperativity of tandem glycine riboswitch aptamers. RNA 26, 564–580 (2020).
Focht, C. M. & Strobel, S. A. Efficient quantitative monitoring of translational initiation by RelE cleavage. Nucleic Acids Res. 50, e105 (2022).
Focht, C. M., Hiller, D. A., Grunseich, S. G. & Strobel, S. A. Translation regulation by a guanidine-II riboswitch is highly tunable in sensitivity, dynamic range, and apparent cooperativity. RNA 29, 1126–1139 (2023).
Kobori, S. & Yokobayashi, Y. High-throughput mutational analysis of a twister ribozyme. Angew. Chem. Int. Ed. Engl. 55, 10354–10357 (2016).
Zhang, Z. et al. Accurate inference of the full base-pairing structure of RNA by deep mutational scanning and covariation-induced deviation of activity. Nucleic Acids Res. 48, 1451–1465 (2020).
Sumi, S., Hamada, M. & Saito, H. Deep generative design of RNA family sequences. Nat. Methods 21, 435–443 (2024).
Lambert, N. et al. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell 54, 887–900 (2014).
Smyth, R. P. et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods 12, 866–872 (2015).
Pitt, J. N. & Ferre-D’Amare, A. R. Rapid construction of empirical RNA fitness landscapes. Science 330, 376–379 (2010).
Auyeung, V. C., Ulitsky, I., McGeary, S. E. & Bartel, D. P. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell 152, 844–858 (2013).
Thompson, J. R., Marcelino, L. A. & Polz, M. F. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR. Nucleic Acids Res. 30, 2083–2088 (2002).
Strobel, E. J., Kelly, S. L. & Szyjka, C. E. Isolation of synchronized E. coli elongation complexes for solid-phase and solution-based in vitro transcription assays. Methods Enzymol. 675, 159–192 (2022).
Borukhov, S., Sagitov, V. & Goldfarb, A. Transcript cleavage factors from E. coli. Cell 72, 459–466 (1993).
Komissarova, N. & Kashlev, M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3’ end of the RNA intact and extruded. Proc. Natl Acad. Sci. USA 94, 1755–1760 (1997).
Nudler, E., Mustaev, A., Lukhtanov, E. & Goldfarb, A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell 89, 33–41 (1997).
Laptenko, O., Lee, J., Lomakin, I. & Borukhov, S. Transcript cleavage factors GreA and GreB act as transient catalytic components of RNA polymerase. EMBO J. 22, 6322–6334 (2003).
Artsimovitch, I. & Landick, R. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl Acad. Sci. USA 97, 7090–7095 (2000).
Larson, M. H. et al. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344, 1042–1047 (2014).
Vvedenskaya, I. O. et al. Interactions between RNA polymerase and the “core recognition element” counteract pausing. Science 344, 1285–1289 (2014).
Saba, J. et al. The elemental mechanism of transcriptional pausing. Elife 8, e40981 (2019).
Roberts, R. W. & Szostak, J. W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl Acad. Sci. USA 94, 12297–12302 (1997).
Hanes, J. & Pluckthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad. Sci. USA 94, 4937–4942 (1997).
MacPherson, I. S., Temme, J. S. & Krauss, I. J. DNA display of folded RNA libraries enabling RNA-SELEX without reverse transcription. Chem. Commun. 53, 2878–2881 (2017).
Mohsen, M. G., Midy, M. K., Balaji, A. & Breaker, R. R. Exploiting natural riboswitches for aptamer engineering and validation. Nucleic Acids Res. 51, 966–981 (2023).
Strobel, E. J. Preparation and characterization of internally modified DNA templates for chemical transcription roadblocking. Bio. Protoc. 11, e4141 (2021).
Jolivet, P. & Foley, J. SPRI bead mix. protocols.io. https://doi.org/10.17504/protocols.io.bnz4mf8w (2020).
Mahat, D. B. et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 11, 1455–1476 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kelly, S.L. & Strobel, E.J. Systematic analysis of cotranscriptional RNA folding using transcription elongation complex display, e-strobel-lab/TECtools: TECtools v1.2.0, https://zenodo.org/records/10413339 (2023)
Kelly, S. L. & Strobel, E. J. Systematic analysis of cotranscriptional RNA folding using transcription elongation complex display, TECdisplay data for Clostridium beijerinckii pfl ZTP riboswitch variant libraries. Zenodo https://doi.org/10.5281/zenodo.13829757 (2023).
Kelly, S. L. & Strobel, E. J. Systematic analysis of cotranscriptional RNA folding using transcription elongation complex display, TECdisplay validation gel images. https://zenodo.org/records/10425136 (2023).
Ren, A., Rajashankar, K. R. & Patel, D. J. Global RNA fold and molecular recognition for a pfl riboswitch bound to ZMP, a master regulator of one-carbon metabolism. Structure 23, 1375–1381 (2015).
Jones, C. P. & Ferre-D’Amare, A. R. Recognition of the bacterial alarmone ZMP through long-distance association of two RNA subdomains. Nat. Struct. Mol. Biol. 22, 679–685 (2015).
Pettersen, E. F. et al. UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank R. Landick (University of Wisconsin—Madison) for providing GreB. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM147137 (to E.J.S.) and by start-up funding from the University at Buffalo (to E.J.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
E.J.S., conceptualization, methodology, software, supervision, and funding acquisition; S.L.K., investigation; S.L.K. and E.J.S, Validation, formal analysis, writing—original draft, and writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rhiju Das and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kelly, S.L., Strobel, E.J. Systematic analysis of cotranscriptional RNA folding using transcription elongation complex display. Nat Commun 16, 2350 (2025). https://doi.org/10.1038/s41467-025-57415-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57415-3
