
Abstract
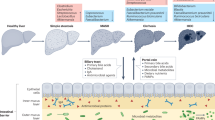
Immune dysregulation-induced inflammation serves as a driving force in the progression of metabolic dysfunction-associated steatohepatitis (MASH), while the underlying cellular and molecular mechanisms remain largely uncharted. A Western diet (WD) is employed to construct mouse models of metabolic dysfunction associated steatotic liver disease (MASLD) or MASH. Mass cytometry identifies a c-kit+ cDC1 subset whose frequency is reduced in the livers of mice and patients with MASH compared with healthy controls. Adoptive cell transfer of c-kit+ cDC1 protects the progression of MASH. Moreover, analysis of gut microbe sequence shows that WD-fed mice and MASLD/MASH patients exhibit gut microbiota dysbiosis, with an elevated abundance of H2S-producing Desulfovibrio_sp. Transplanting of MASH-derived fecal flora, Desulfovibrio_sp., or injecting H2S intraperitoneally into MASLD mice decreases the c-kit+cDC1 population and exacerbates liver inflammation. Mechanistically, H2S induces autophagic cell death of cDC1 in a c-kit-dependent manner in cDC-specific c-kit-/- and Atg5-/- mice. We thus uncover that microbiota-derived H2S triggers the autophagic cell death of c-kit+ cDC1 and ignites the liver inflammatory cascade in MASH.
Similar content being viewed by others

Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) is considered the hepatic manifestation of metabolic syndrome. Its global incidence rate is about 25%, of which 20–25% will progress to metabolic dysfunction-associated steatohepatitis (MASH)1. MASH differs from MASLD histologically, as it involves not only hepatic fat accumulation but also inflammation and fibrosis2. However, the mechanism of MASH development is still incompletely understood.
Immune cell-mediated inflammation drives MASLD to MASH. Recent advancements in single-cell transcriptomics have led to a better understanding of the refined landscape of immune cell subsets reshaped during MASH in mice3 and cirrhotic liver in humans4. Importantly, single-cell proteomics provides additional value as there is often a poor correlation between mRNA and protein expression levels for most genes5. Despite the constraint imposed by the limited number of markers, mass cytometry provides measurement of simultaneous protein parameters at a single-cell resolution, thereby having a unique advantage in fully explaining the genotype-phenotype relationships and understanding biological processes6. Unfortunately, this technology is seldom used in MASLD/MASH research at present.
Myeloid cells are believed to be closely associated with the development of inflammation in MASH progression7. Dendritic cells (DCs) link innate and adaptive immunity and represent a tolerogenic role within the steady-state liver8. Hepatic DCs consist of various subsets: monocyte-derived DCs (MoDCs), plasmacytoid DCs (pDCs), classical type-1 DCs (cDC1; CD103+CD11b-) and classical type-2 DCs (cDC2; CD103-CD11b+)9. DCs overall exhibit protective properties in limiting inflammatory and fibrotic injury in MASH mice models and patients10,11. However, different DC subsets have opposite abilities, with CD103+ cDC1 protecting against the progression of steatosis toward MASH12, while cDC2 and pDC mediate inflammation11,13. Here, we studied the immunopathological changes of MASH, with a specific focus on the cDC1 subset, aiming to elucidate the mechanisms underlying MASH.
The involvement of the communication between gut microbiota and liver immune cells has been extensively demonstrated in the pathogenesis of MASLD/MASH in mice models and patients14. Microbial-associated molecular patterns from gut microbiota directly impact the number, function, and maturation of hepatic Kupffer cells, DCs, and NKT cells, intensifying immunological liver injury15. The excessive activation of liver macrophages or hepatocytes/hepatic stellate is crucial in the development of MASLD or hepatocellular carcinoma16,17. Additionally, bacterial metabolites like secondary bile acids, deoxycholic acid, and lipoteichoic acid regulate liver immunity, modulating NKT cells, inflammatory secretome, and prostaglandin E2 expression, respectively18,19,20. Therefore, the gut-liver axis provides new directions for screening potential novel therapeutic targets.
In this study, we have discovered a previously unrecognized hepatic c-kit+ cDC1 subset in MASH patients and mice, further confirmed its protective function, and identified the mechanism underlying the reduction in its quantity, which triggered inflammation and contributed to the progression from MASLD to MASH.
Results
Landscape of hepatic immune cell lineages in MASH mice
Through flow cytometric analysis, we confirmed a significant increase in the infiltration of CD45+ immune cells in the liver of MASH mice compared to the control mice fed a normal chow diet (NCD) for 30 weeks (W) (Fig. 1a). The concentration of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF), C-C motif chemokine ligand 2 (CCL2), and C-X-C motif chemokine ligand 10 (CXCL10), was markedly elevated in MASH mice, while the anti-inflammatory cytokine IL-10 significantly decreased (Fig. 1b). Hepatic triglyceride (TG) content (Fig. 1c), liver coefficient and body weight (Fig. 1d and Supplementary Fig. 1a) were significantly higher in MASH mice compared to control mice. Glucose tolerance test (GTT) and insulin tolerance test (ITT) revealed impaired glucose metabolism in MASH mice and elevated area under the curve (AUC) value for glucose (Fig. 1e, f). In the liver of MASH mice, hepatocellular lipid droplets and ballooning, as well as inflammatory cell infiltration were observed through H&E staining, which were consistent with the elevated NAFLD Activity Score (NAS) (Fig. 1g). Additionally, the liver fibrosis of MASH mice was evidenced by Sirius red staining, as well as elevated hydroxyproline content and ACTA2 abundance (Fig. 1g–i). The serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity were significantly increased in MASH mice (Fig. 1j).

a Number of hepatic immune cells. n = 8/group. b Liver cytokines content. n = 8/group. c Liver TG content. n = 15/group. d Liver coefficient. n = 15/group. e GTT with corresponding AUC. n = 8/group. f ITT with corresponding AUC. n = 8/group. g H&E, Sirius red staining and NAS. Magnification ×40, scale bar = 100 µm. n = 15/group. h Liver hydroxyproline content. n = 15/group. i Liver ACTA2 mRNA level. n = 15/group. j Serum ALT and AST activity. n = 15/group. k H-SNE plots showing the relative protein expression. n = 5/group. l H-SNE plots showing immune cell lineages. n = 5/group. m Percentage of each lineage among hepatic immune cells. n = 5/group. Each dot symbol was an average of technical quadruplicates. Data were represented as mean ± SEM and analyzed using a two-tailed unpaired Student’s t-test (a–j). Significance levels were reported as *P < 0.05, **P < 0.01. TG triglyceride, AUC area under curve, GTT glucose tolerance test, ITT insulin tolerance test; H&E hematoxylin and eosin, NAS NAFLD activity score, ALT alanine aminotransferase, AST aspartate aminotransferase. Source data were provided as a Source Data file.
We then applied a mass cytometry panel comprising 38 antibodies (Supplementary Table 1) to capture the heterogeneity of the hepatic immune cells. We identified the single and live immune cells (Supplementary Fig. 1b) and mined the dataset via hierarchical stochastic neighbor embedding (hSNE) analysis21. All labeled antibodies demonstrated high specificity and fluorescence intensity (Supplementary Fig. 1c). At an overview level, hSNE landmarks depicted clear separation of the T cell (CD3+TCRβ+), B cell (CD3-CD19/TCRγδ+), innate lymphoid cell (ILC, CD3-CD19/TCRγδ-CD161c+/CD127+), and myeloid cell (CD3-CD19/TCRγδ-CD161c- - CD127-CD11b+/CD11c+) lineages (Fig. 1k, l). Compared to the control mice, MASH mice exhibited a significant increase in the proportion of myeloid cells and a decrease in T cells (Fig. 1m). A second hSNE analysis was performed to myeloid cells, thereby identified polymorphonuclear neutrophils (PMN, Ly6G+CD11b+), macrophage cells (Mφ, CD11b+Ly6C+F4-80low/MHCII+), monocyte cells (Mo, CD11blowCD11c-MHCII+), Kupffer cells (KCs, CD11b+CD11c-F4-80+/Ly6Clow), and dendritic cells (DCs, Ly6G-CD11c+MHCII+) (Supplementary Fig. 2a, b). Compared with the control mice, the proportion of DCs, Mo, and KCs decreased while Mφ and PMN increased markedly in MASH mice (Supplementary Fig. 2c).
Composition of hepatic myeloid cells: a redefined c-kit+ cDC1 subset reduced in MASH mice
We then subjected myeloid cells to t-distributed stochastic neighbor embedding (t-SNE) in Cytosplore. A total of 64 myeloid cell clusters, forming 29 subsets, were identified (Fig. 2a). Among them, 18 clusters of DCs (Supplementary Fig. 2d) were further divided into pDC (Ly6C+CD64-CD206-), MoDC (CD64+CD206+), cDC1 (CD64-CD206-CD11blow), and cDC2 (CD64-CD206-CD11b+) based on their marker expression22. Interestingly, the cDC1 subset included not only the traditionally defined CD11b-CD103+ subset (6.8 % of myeloid cells) but also a group of unrecognized novel CD11blowCD103- cDC1 (10.6 % of myeloid cells) (Fig. 2a, b). Because both of the two subsets expressed low levels of c-kit, they were renamed as c-kit+CD103+ cDC1 and c-kit+CD103- cDC1, respectively, or collectively referred to as the c-kit+ cDC1 subset in the following description (Fig. 2a, b and Supplementary Fig. 2d). Within the myeloid cell subsets, the proportion of pDC, c-kit+CD103- cDC1, c-kit+CD103+ cDC1, and most KCs subsets decreased, while all PMN subsets and several Mφ subsets (such as CD86+CD14+ Mφ) increased (Fig. 2b and Supplementary Fig. 2e, f) significantly. Pearson correlation analysis revealed that the quantity of c-kit+ cDC1 was positively correlated with Ly6G+ KCs and Ly6C+MHCII+ KCs while negatively with NAS and all PMN subsets (Supplementary Fig. 2g).

a Heatmap showing the median expression values and hierarchical clustering of the identified subpopulations. n = 5/group. b Composition of the myeloid cells. Color as shown in A. n = 5/group. c, d Gating strategy and representative flow cytometry dot plots of c-kit+CD103+ and c-kit+CD103- cDC1 in the liver. Lin includes CD3, CD19, NK1.1. n = 5/group. e Representative flow cytometry dot plots of hepatic c-kit+CD103+ cDC1 and c-kit+CD103- cDC1, as well as their proportion in XCR1+cDC1. n = 5/group. f Liver mIHC images stained with CD103 (red), c-kit (green), and XCR1 (white). Cell nuclei were stained with DAPI (blue). Magnification ×40, scale bar = 100 µm. Arrows indicate c-kit+CD103+ cDC1, and arrowheads indicate c-kit+CD103- cDC1. n = 5/group and two sections were stained from each liver sample. Data were represented as mean ± SEM and analyzed using a two-tailed unpaired Student’s t-test (b, d). Significance levels were reported as *P < 0.05, **P < 0.01. Source data were provided as a Source Data file.
Flow cytometry with specific antibodies for c-kit+ cDC1 (single live CD45+Lin-CD11c+CD11blow/-CD103-/+ c-kit+) (Supplementary Table 2) confirmed a significant reduction in their number in the liver of MASH mice (Fig. 2c, d). It was believed that liver cDC1 expressed CD10312. Given the emergence of the new CD103-c-kit+ cDC, in order to determine the type of DC subgroup it belongs to, two recognized marker XCR1 and CD172a were introduced to help to identify cDC1 and cDC2, respectively. Notably, this subset of cells highly expresses XCR1 but does not express CD172a, indicating that it also belongs to cDC1. In addition, all c-kit expressing cells (CD103-/+c-kit+) account for over 95% of cDC1, indicating the co-expression of XCR1 and c-kit (Fig. 2e). Furthermore, multiplex immunohistochemistry (mIHC) analysis confirmed the presence of the c-kit+ cDC1 subset and their decrease in quantity in the liver of MASH mice (Fig. 2f). Interestingly, a comparison of the transcriptome between cDC1 (c-kit+) and cDC2 (c-kit-) revealed no significant change in c-kit mRNA levels (Supplementary Fig. 2h), suggesting that c-kit expression may distinguish cDC1 from other myeloid subsets only at the translation (protein) level. This also explains why previous single-cell sequencing studies failed to detect the specific expression of c-kit in cDC1.
The hepatic c-kit+ cDC1 subset inhibits liver inflammation and injury in MASH mice
To investigate the role of the c-kit+ cDC1, we used a liver- and spleen-derived c-kit+CD103- cDC1 and c-kit+CD103+ cDC1 to adoptively transfer to MASH mice lasting for 4 W, respectively (Fig. 3a). The cells transferred successfully reached the liver (Supplementary Fig. 3a). Both subsets significantly reduced the inflammatory response in the liver of MASH mice, evidenced by decreased content of liver pro-inflammatory TNF, CCL2, and CXCL10 while increased IL-10 (Fig. 3b), as well as reduced CD45+ immune cell infiltration (Fig. 3c). Compared to the control mice, the MASH mice exhibited a higher liver coefficient, which was attenuated by the adoptive transfer of each subset (Fig. 3d). Moreover, cell transfer improved the glucose tolerance (Fig. 3e, f), decreased hepatic TG accumulation (Fig. 3g), and alleviated liver injury (Fig. 3h) of MASH mice. Additionally, cell transfer improved the hepatocellular lipid droplets, ballooning, and inflammatory cell infiltration, resulting in decreased NAS (Fig. 3i). Data of Sirius staining, hydroxyproline content, and ACTA2 abundance showed that cell transfer also alleviated the fibrosis level (Fig. 3i–k). Cell transfer also reduced the weight of mice, but the difference was not significant (Supplementary Fig. 3b). These data suggested an essential protective role of c-kit+ cDC1 in the development of inflammation in MASH. Interestingly, the above effects of the two subsets were similar in the aforementioned aspects, indicating that the expression of c-kit rather than CD103 may be important for their survival or function.

a A schematic of the adoptive transfer experiment. b Liver cytokine content. n = 8/group. c Quantity of liver immune cells. n = 8/group. d Liver coefficient. n = 8/group. e GTT with corresponding AUC. n = 8/group. f ITT with corresponding AUC. n = 8/group. g Liver TG content. n = 8/group. h Serum ALT and AST activity. n = 8/group. i H&E, Sirius red staining, and NAS. Magnification ×40, scale bar = 100 µm. n = 8/group. j Liver hydroxyproline content. n = 8/group. k Liver ACTA2 mRNA level. n = 8/group. Data were represented as mean ± SEM and analyzed using one-way ANOVA with Tukey’s post hoc test (b–k). Significance levels were reported as *P < 0.05, **P < 0.01. AUC area under curve, TG triglyceride, GTT glucose tolerance test, ITT insulin tolerance test, H&E hematoxylin and eosin, NAS NAFLD activity score, ALT alanine aminotransferase, AST aspartate aminotransferase. Source data were provided as a Source Data file.
The reduction of the hepatic cDC1 subset is c-kit-dependent during the transition from MASLD toward MASH
To explore the function of c-kit in cDC1, we introduced a cDC-specific c-kit knockout mouse model (zDC, c-kit-/-). According to the flow cytometry data, over 98% of c-kit expression was removed in cDC1 (marked by XCR1+) from different tissues including the liver, colon, adipose, and spleen in c-kit-/- mice (Supplementary Fig. 4a, b). Notably, c-kit knockout had no obvious effect on the cDC1 number without WD feeding (Supplementary Fig. 4c). We separately selected naïve T cells derived from OT-I (CD8+) and OT-II (CD4+) mice that target specific OVA peptide. By co-culturing them with cDC1 in vitro, we detected whether c-kit is involved in regulating the function of cDC1. The purity of naïve T cells (CD3+CD44-CD62L+) obtained through magnetic bead sorting was as high as 97.5% (Supplementary Fig. 5a). Surprisingly, the in vitro co-culture experiment discovered that c-kit knockout in cDC1 had no significant impact on the number and the expression of CD107a and IFN-γ of naïve T cells (Supplementary Fig. 5b, c), suggesting that c-kit is not related to the antigen presentation function of cDC1. Additionally, adoptively transfer of cDC1 from control or c-kit-/- mice to MASH mice showed similar effects on CD45+ immune cell infiltration (Supplementary Fig. 5d), liver coefficient (Supplementary Fig. 5e), glucose tolerance (Supplementary Fig. 5f, g), hepatic TG accumulation (Supplementary Fig. 5h), liver injury (Supplementary Fig. 5i), NAS (Supplementary Fig. 5j), and fibrosis (Supplementary Fig. 5j–l). These pieces of evidence indicate that c-kit does not participate in the regulation of cDC1 function.
Mice were fed with a WD of 15 W or 30 W, corresponding to the stage of MASLD or MASH, respectively23. In wild-type (WT) mice, the number of hepatic cDC1 significantly increased at the MASLD stage, but sharply decreased at the MASH stage (Fig. 4a). Interestingly, after WD feeding, c-kit knockout in cDC1 reversed their number decrease at the MASH stage while having no significant effect at the MASLD stage (Fig. 4a). This evidence suggests that c-kit may be necessary for the reduction of cDC1 number in the liver of MASH mice. Furthermore, we observed that MASH mice exhibited approximately two-fold higher levels of inflammatory cell infiltration and NAS compared to MASLD mice (Fig. 4b, f). Remarkably, c-kit knockout in cDC1 reversed these effects, as demonstrated by reduced inflammatory cell infiltration and NAS (Fig. 4b, f). Consistently, the levels of pro-inflammatory cytokines (TNF, CCL2, and CXCL10) in the liver gradually increased with the prolongation of WD feeding, while c-kit knockout significantly retarded their elevation at the MASH stage (Fig. 4c). Along with the chance of hepatic c-kit+ cDC1, the level of IL-10, an anti-inflammatory cytokine can be produced by c-kit+ cDC1 (Supplementary Fig. 5m), showed an increased trend in the liver at the MASLD stage and a markedly decrease in the MASH stage (Fig. 4c). However, in the c-kit-/- MASH mice, an upward persisted during WD feeding (Fig. 4c). Additionally, compared to the MASLD mice, MASH mice did not show further exacerbation in glucose tolerance (Fig. 4d), but exhibited increased liver fat accumulation (Fig. 4f, g) and liver injury (Fig. 4h). However, c-kit knockout in cDC1 slightly improved glucose tolerance (Fig. 4d) and significantly reduced liver coefficient, liver TG content, and serum ALT levels in MASH mice (Fig. 4e–h). Furthermore, knockout of c-kit in cDC1 also alleviated weight gain in mice fed with WD at 30 W (Supplementary Fig. 5n). Compared to the control and MASLD mice, MASH mice showed increased liver fibrosis, which was improved by c-kit knockout in cDC1 (Supplementary Fig. 6a–d). These results suggest that c-kit protein is essential for the reduction of the hepatic protective cDC1 in MASH mice, which contributes to the imbalance between pro- and anti-inflammatory actions, as well as subsequent metabolic changes and fibrosis.

a Quantity of hepatic XCR1+ cDC1. n = 5/group. b Quantity of liver immune cells. n = 8/group. c Liver cytokines content. n = 8/group. d AUC of GTT and ITT. n = 8/group. e Liver coefficient. n = 8/group. f H&E staining and NAS. Magnification ×40, scale bar = 100 µm. n = 8/group. g Liver TG content. n = 8/group. h Serum ALT and AST activity. n = 8/group. Data were represented as mean ± SEM and analyzed using the two-way ANOVA with Tukey’s post hoc test (a–h). a–d, groups sharing no common letters indicate statistically significant differences (P < 0.05), groups sharing partial letters indicate no significant difference (P > 0.05). AUC area under curve, GTT glucose tolerance test, ITT insulin tolerance test, H&E hematoxylin and eosin, NAS NAFLD activity score, TG triglyceride, ALT alanine aminotransferase, AST aspartate aminotransferase. Source data were provided as a Source Data file.
Interestingly, except in the liver, cDC1 number also decreased in the colon, but not in the adipose and spleen in MASH mice (Supplementary Fig. 6e). Similarly, their decrease could be reversed by c-kit knockout (Supplementary Fig. 6e), suggesting that the factor reducing hepatic c-kit+ cDC1 in MASH mice may originate from the intestine.
The reduction of hepatic c-kit+ cDC1 is caused by gut microbiota disorder
In our feces 16S rRNA sequencing data, the rarefaction curve indicated high quality and reasonable sample size (Fig. 5a). The α diversity, as measured by the Chao1 index, was not affected by a WD feeding (Fig. 5a). Principal component analysis (PCA) indicated that the gut microbiome of WD and NCD clustered separately with the prolonged feeding time, and WD feeding decreased the β diversity in Jaccard Anosim analysis (Fig. 5b). Compared to the Control and WD 15 W groups, the abundance of Desulfovibrio_sp. increased in the WD 30W group, while Lactobacillus_Murinus, Lactobacillus_johnsonii, Akkermansia_muciniphila, and Firmicutes_bacterium decreased (Fig. 5c and Supplementary Fig. 7a). Compared to the NCD 30W, the increased Desulfovibrio abundance was confirmed in WD 30W group, with Desulfovibrio_sp. increased drastically (Fig. 5c). Interestingly, among the above species, the abundance of Desulfovibrio_sp. was negatively correlated with the hepatic c-kit+ cDC1 number and positively correlated with the NAS value (Fig. 5d). On the other hand, Porphyromonadaceae_Bacterium, Lactobacillus_Murinus, Lactobacillus_Johansonii, and Lactobacillus_Reuteri showed the opposite correlation (Fig. 5d).

a–c 16S rRNA gene sequencing of the fecal microbiome of WT mice fed with NCD or WD for 0, 15, and 30W. n = 6 in the NCD group, n = 10 in the WD group. a Rarefaction curve and violin plot. Detailed information is presented in Source data. b PCA and boxplot of Jaccard distance. Detailed information is presented in Source data. c Quantity of data between NCD 30 W and WD 30 W in top 10 abundance at the phylum level, genus level, and species level in mice fed with NCD or WD for different durations. d Correlation analysis among hepatic c-kit+ cDC1 number, NAS, and fecal flora abundance in MASH mice. Spearman’s rank correlation coefficient test is performed. e Schematic of the FMT experiment. f Quantity of liver immune cells. n = 8/group. g Liver cytokine content. n = 8/group. h AUC of GTT and ITT. n = 8/group. i Liver TG content. n = 8/group. j Serum ALT and AST activity. n = 8/group. k H&E staining and NAS. n = 8/group. l Liver coefficient. n = 8/group. m Liver hydroxyproline content. n = 8/group. n Liver ACTA2 mRNA level. n = 8/group. o Quantity of hepatic c-kit+ cDC1. n = 5/group. Data were represented as mean ± SEM. P values calculated by a two-tailed unpaired Student’s t-test (c), one-way ANOVA with Tukey’s post hoc test (b, f–n) or two-way ANOVA with Tukey’s post hoc test (o). Significance levels were reported as *P < 0.05, **P < 0.01. a–d groups sharing no common letters indicate statistically significant differences (P < 0.05), groups sharing partial letters indicate no significant difference (P > 0.05). FMT fecal microbiota transplantation, AUC area under curve, GTT glucose tolerance test, ITT insulin tolerance test, H&E hematoxylin and eosin, NAS NAFLD activity score, TG triglyceride, ALT alanine aminotransferase, AST aspartate aminotransferase. Source data were provided as a Source Data file.
We transplanted gut microbiota from MASH mice (30 W) and commercialized Desulfovibrio_sp. to WD-feeding mice for 15 W (Fig. 5e). Following both fecal microbiota transplantation (FMT) and Desulfovibrio_sp. addition, the MASLD mice not only developed enhanced hepatic inflammatory state with increased infiltration of inflammatory cells (Fig. 5f, g), and aggravated metabolic disorders indicated by impaired glucose tolerance and increased liver TG (Fig. 5h, i), but also exhibited heightened liver injury (Fig. 5j) and NAS value (Fig. 5k). Additionally, in MASLD mice, the FMT and addition of desulfovibrio_sp. had a significant exacerbating effect on the liver coefficient (Fig. 5l) and fibrosis indexes, including the increased content of hydroxyproline and ACTA2 abundance (Fig. 5m, n). In MASLD mice, the supplementary FMT intervention remarkably mitigated the increase in body weight. On the other hand, despite the fact that the additional Desulfovibrio_sp. treatment led to an elevation in the average body weight, this change did not reach statistical significance (Supplementary Fig. 7c). Importantly, with the feeding of WD, the number of hepatic c-kit+ cDC1 showed an increasing trend and significantly increased at 15 W (Fig. 5o, Supplementary Fig. 7b). However, when FMT and Desulfovibrio_sp. addition were present, it began to decrease significantly at 5 W and reached the lowest level during 10-15 W (Fig. 5o and Supplementary Fig. 7b).
In the human study, consistent with the findings in mice, the presence of c-kit+ cDC1 and their reduction were confirmed in MASH patients (Fig. 6a). Additionally, data from the human gut microbiota database showed that the abundance of Desulfovibrio increased while Lactobacillus decreased significantly in the feces of MASLD patients (Fig. 6b).

a Human liver mIHC images stained with CD103 (red), c-kit (green), and XCR1 (white). Cell nuclei were stained with DAPI (blue). Magnification ×40, scale bar = 100 µm. Arrows indicate c-kit+CD103+ cDC1, and arrowheads indicate c-kit+CD103- cDC1. n = 5/group. b The relative abundance of Desulfovibrio (Normal, n = 4; MASLD, n = 5) and Lactobacillus (Normal, n = 11; MASLD, n = 24) in the intestine of normal individuals and MASLD patients. Data was obtained from the online human microbiota database (gmrepo). Samples that did not identify the target microbiota were removed. Each data point represents an individual sample. Data were shown as mean ± SEM and analyzed using a two-tailed unpaired student’s t-test. Significance levels were reported as **P < 0.01. The exact p values are shown in Source data. Source data were provided as a Source Data file.
Gut-derived H2S reduces the number of hepatic c-kit+ cDC1 and triggers the development of MASH
Desulfovibrio is the main source of H2S in the intestine. The H2S concentration in the liver and feces of MASH mice was markedly higher than that of MASLD and control mice (Fig. 7a and Supplementary Fig. 8a). In addition, both FMT and Desulfovibrio_sp. addition significantly increased the H2S levels in the liver of WD mice (Fig. 7b and Supplementary Fig. 8b). GYY4137, which sustained the release of H2S, was intraperitoneally injected into MASLD mice (Fig. 7c). The H2S concentration in the liver and serum increased in a dose-dependent manner after GYY4137 administration (Supplementary Fig. 8c, d). H2S treatment further increased the number of hepatic CD45+ cells and the levels of inflammatory cytokines (TNF, CCL2, and CXCL10), while decreased IL-10 in MASLD mice, suggesting an expansion of inflammation (Fig. 7d, e). Importantly, H2S treatment decreased the number of hepatic c-kit+ cDC1 by half (Fig. 7f). H2S treatment further aggravated metabolic disorder and liver injury in MASLD mice, as evidenced by increased AUC of GTT and ITT (Fig. 7g), hepatic TG accumulation (Fig. 7h), elevated serum ALT and AST activity (Fig. 7i), as well as NAS (Fig. 7k). Although the liver coefficient slightly increased, it was not significant (Fig. 7j). Furthermore, in MASLD mice, H2S treatment had a significant effect on the liver fibrosis indexes, including the increased liver hydroxyproline content and ACTA2 abundance (Fig. 7l, m). In MASLD mice, although the additional H2S treatment increased the average body weight, no significance was found (Supplementary Fig. 8e).

a Liver H2S concentration in NCD and WD mice. n = 8/group. b Liver H2S concentration in mice after microbiota transfer. n = 8/group. c Schematic of the H2S delivery. d Quantity of liver immune cells. n = 8/group. e Liver cytokines content. n = 8/group. f Quantity of hepatic c-kit+ cDC1. n = 8/group. g AUC of GTT and ITT analysis. n = 8/group. h Liver TG content. n = 8/group. i Serum ALT and AST activity. n = 8/group. j Liver coefficient. n = 8/group. k H&E staining and NAS. n = 8/group. l Liver hydroxyproline level. n = 8/group. m Liver ACTA2 mRNA level. n = 8/group. Data were represented as mean ± SEM. P values calculated by a two-tailed unpaired Student’s t-test (d–m), one-way ANOVA with Tukey’s post hoc test (b) or two-way ANOVA with Tukey’s post hoc test (a). Significance levels were reported as *P < 0.05, **P < 0.01. a–d the same letters indicate no significant differences (P > 0.05), different letters mean a significant difference (P < 0.05). FMT fecal microbiota transplantation, AUC area under curve, GTT glucose tolerance test, ITT insulin tolerance test, H&E hematoxylin and eosin, NAS NAFLD activity score, TG triglyceride, ALT alanine aminotransferase, AST aspartate aminotransferase. Source data were provided as a Source Data file.
H2S induces ACD of cDC1 in a c-kit-dependent manner
To gain better insight into the molecular pathological alterations in hepatic c-kit+ cDC1 during MASH development, we sorted the target subsets by flow cytometry and performed transcriptome sequencing analysis. Hepatic c-kit+ cDC1 from control and MASH mice were separated clearly (Supplementary Fig. 9a), with 585 upregulated and 2411 downregulated genes (Supplementary Fig. 9b, c). Among the cytokines secreted by DCs, IL-10 was upregulated, while Tnf, IL-6, Ccl2, Cxcl10, and IL-23a were downregulated, indicating that c-kit+ cDC1 may play an anti-inflammatory role in the development of MASH (Supplementary Fig. 9b, c). KEGG analysis revealed that differential genes were mainly enriched in signaling pathways such as autophagy and PPAR signaling pathway, etc. (Fig. 8a). GSEA analysis also confirmed the upregulated autophagy signaling (Fig. 8b). By labeling lysosome and autophagosome with CYTO-ID in flow cytometry analysis, we found that the autophagic flux of hepatic cDC1 was significantly enhanced in control MASH mice, but not in the c-kit-/- MASH mice (Fig. 8c), suggesting that c-kit plays an indispensable role in regulating the autophagy of hepatic cDC1 induced by WD feeding. To further verify whether H2S was involved, we administered GYY4137 intraperitoneally to control and c-kit-/- MASLD mice (Fig. 8d). H2S significantly increased autophagy in hepatic cDC1 in control but not in c-kit-/- mice in vivo (Fig. 8e). Importantly, WD feeding and H2S administration had no significant effect on the apoptosis of cDC1 (Supplementary Fig. 9d, e). This was consistent with the findings from mass cytometry, where no significant change in the proportion of apoptotic cDC1 was found between MASH and control mice (Supplementary Fig. 2e). Moreover, we found a positive correlation between hepatic H2S concentration and the autophagy of cDC1 (Fig. 8f).

a Top 10 KEGG enrichment of hepatic c-kit+ cDC1 from WT mice fed with NCD and WD for 30 W. n = 3/group. b GSEA analysis of Autophagy signaling. c Flow cytometry histograms of CYTO-ID (autophagic flux). n = 5/group. d Schematic of the H2S delivery. e Autophagic flux analysis of hepatic XCR1+ cDC1. n = 5/group. f Correlation analysis between the concentration of H2S and the autophagic flux of XCR1+ cDC1 in the liver. Spearman’s rank correlation coefficient test was performed. 95% confidence interval (0.7132 to 0.9658), P < 0.0001. g TEM showing the autophagosome and autolysosome in c-kit+ DC induced in vitro. n = 3/group. h Relative expression of p62 and LC3. n = 3/group. i MTT data showing the cell survival rate of c-kit+ DC induced in vitro. j MTT data showing the cell survival rate of c-kit+ DC in Atg5flox/flox and Atg5-/- mice. n = 5/group. Data were represented as mean ± SEM. P values calculated by a two-tailed unpaired Student’s t-test (g, j), one-way ANOVA with Tukey’s post hoc test (c, e, i) or two-way ANOVA with Tukey’s post hoc test (h). The screening criteria for KEGG were adjusted p < 0.05 and FDR value (q value) < 0.25, and the p value correction method was Benjamini-Hochberg (a). Significance levels were reported as *P < 0.05, **P < 0.01. a–c, groups sharing no common letters indicate statistically significant differences (P < 0.05), groups sharing partial letters indicate no significant difference (P > 0.05). 3MA 3-Methyladenine, FDR False Discovery Rate, MTT methylthiazolyl diphenyl-tetrazolium bromide. Source data were provided as a Source Data file.
To obtain a sufficient cell number for in vitro experiments, we induced c-kit+ DCs24 using bone marrow (BM) stem cells in vitro, with a positive rate of up to 93% (Supplementary Fig. 10a, b). GYY4137 treatment enhanced the autophagy flux of c-kit+ DCs, which was manifested by the existence of a large number of autolysosomes containing different organelle (Fig. 8g), as well as increased LC3II and decreased p62 protein abundance (Fig. 8h). Importantly, GYY4137 treatment significantly decreased the cell survival rate of c-kit+ DCs (Fig. 8i). However, inhibition of autophagy by 3-MA treatment abolished the above adverse effects (Fig. 8i), indicating that high concentrations of H2S induce ACD of c-kit+ DCs. Additionally, we introduced myeloid-specific Atg5 knockout mice (Supplementary Fig. 10c), sorted c-kit+ cDC1, and stimulated them with GYY4137 in vitro. We found that autophagy is necessary for H2S induced ACD of c-kit+ cDC1 (Fig. 8j). Moreover, the inhibitors of apoptosis or necrosis, z-vad-fm or necrostatin-1, respectively, did not have a noticeable effect on the cell survival rate (Fig. 8i). These in vivo and in vitro findings indicate that H2S produced by gut-derived Desulfovibrio_sp. induces ACD of hepatic c-kit+ cDC1 in MASH mice.
Discussion
Innate immunity, in particular the myeloid compartment, plays a pivotal role in driving the progression of MASH7,25. Through mass cytometry, we defined a c-kit+ cDC1 subset, which consists of a definite CD103+ cDC1 subset and an unrecognized CD103- cDC1 subset. These cells have been confirmed to exist in the liver of both humans and mice, and play a regulatory role in the progression of MASH.
DCs have been demonstrated to play a protective role in the hepatic injury10, but some contradictory conclusions exist at the subset level, especially for the cDC111,12,13. Joel et al. examined human MASH livers using microarrays and reported that MASH induction in patients and the WD mice led to a decrease of hepatic cDC1 number, which displayed a negative association with liver pathology, but failed to detect the regulatory role of cDC111. Heier et al. had reported the existence of a protective CD103+ DC subset in MASH using Batf3-deficient mouse model with MCD12, but failed to detect markers of a rare cDC1 subset. On the contrary, in MCD-induced MASH, Deczkowska performed scRNAseq analysis to evidence cDC1 exacerbating liver inflammation through promoting inflammatory T cell reprogramming using XCR1DTA mice26, but inconsistent changes exist in the number of hepatic cDC1 in single-cell data from MASH mice and patients, as well as the lack of adoptive transfer experiments. In the MASH research, there is a significant difference in metabolic and immune stress caused by different models27,28. Although the MCD model imitates the histopathological characteristics of MASH, its shortcomings such as weight loss, lack of metabolic characteristics of human MASH, and inability to establish a transition from MASLD to MASH have limited its use in immunopathological studies29,30,31. Thus, through using the WD-induced MASH mice model, the present study found that the number of the hepatic c-kit+ cDC1 (CD103+ cDC1 and CD103- cDC1) firstly increased during the MASLD stage, but reduced drastically when developing into MASH. This reduction was also confirmed in MASH patients in our mIHC data. Importantly, adoptive transfer of c-kit+ cDC1 cells (CD103+ cDC1 and CD103- cDC1, respectively) to mice with MASH significantly reduced hepatic inflammation, steatosis, NAS, and liver injury, suggesting that c-kit+ cDC1 plays a protective role to against the pathological injury of MASH, which is supported by the finding of Joel T11 and Heier12. Among all the current evidence, there exist inconsistent results in studying MASH immunopathology in different models, and the reasons for this phenomenon may include factors such as modeling diet, modeling time, animal age, etc. Therefore, the research field must determine the most suitable animal model for studying human MASH immunopathology and treatment.
Inflammation is a key driver of pathological injury in MASH. Data from MCD-induced MASH mice demonstrated that cDC1 depletion induced the overproduction of CCL2 or TNF after LPS stimulation in the liver, while cDC1 supplementation reduced hepatic TNF and CCL2 levels10,12. In the WD-induced MASH model, we found a negative correlation between the number of hepatic c-kit+ cDC1 and the NAS. As the decrease of hepatic c-kit+ cDC1, the anti-inflammatory cytokine IL-10 was significantly decreased, while the pro-inflammatory cytokines TNF, CCL2, and CXCL10 were markedly increased. Importantly, we found that hepatic c-kit+ cDC1 express a high level of IL-10, which is consistent with a previous report where an IL-10-producing DC subset was found to have immune tolerance roles32,33. Previous studies have also shown a significant decrease in hepatic XCR1+ cDC1 and tolerogenic IL-10-producing CD141+ DC in MASH patients, and both of the above cell subpopulations have been confirmed to counteract inflammation during MASH development11,26,34. Additionally, hepatic DC were shown to limit inflammation through inhibiting CD8+ T cell expansion and restricting cytokine production in innate immune effector cells in MASH10. These evidences indicate that c-kit+ cDC1 alleviates the pathological damage of MASH through its anti-inflammatory effects.
A link between liver immunopathology and gut microbiome has been well-proven in MASLD/MASH patients and mice35,36. Abnormal microbiota metabolites mediate hepatic inflammation by affecting the function and survival of liver immune cells36. We revealed a worsening dysbiosis of the gut microbiota during the transition from MASLD to MASH in WD-feeding mice. Furthermore, our FMT data provided further evidence supporting the causal relationship between gut microbiota disorder and the reduction of hepatic c-kit+ cDC1, inflammation, and liver injury. Importantly, Desulfovibrio had garnered our attention due to its drastic increase in MASH mice and MASLD patients. Several human studies have also shown an elevated presence of gut Desulfovibrio in MASH patients37,38,39, which is believed to have potential clinical value in distinguishing between MASH and MASLD diseases40. By directly administering Desulfovibrio_sp., we confirmed that it promotes the occurrence of MASH by reducing the number of liver protective c-kit+ cDC1. In addition, hepatocellular carcinoma (HCC), a severe manifestation of MASH, is intimately tied to gut microbiota17. Research has linked the increased abundance of Desulfovibrio in the gut to a range of HFD-related cancers and tumors, including HCC41,42. Intriguingly, cDC1 has emerged as a protective force against HCC, facilitating the activation and proliferation of CD8+ T cells43. Although there is a lack of direct evidence, these collective findings hint at a potential role for the gut Desulfovibrio-liver cDC1 axis in accelerating the progression from NASH to HCC.
Desulfovibrio is a key player responsible for H2S production. We found an increased concentration of H2S in the liver of both MASH mice or WD mice following FMT and Desulfovibrio_sp. addition. Importantly, the accumulation of H2S in the liver can cause a significant decrease in the number of hepatic c-kit+ cDC1 and a deterioration of MASH, indicating that the intestinal H2S-induced reduction of hepatic anti-inflammatory immune cells is an important factor triggering MASH. Exogenous H2S can promote autophagy in various animal cells, which has been widely recognized44. However, excessive levels of intracellular autophagy can lead to ACD, a programmed death type depending on autophagy but not apoptosis and necrosis45. Through in vivo and in vitro experiments, we discovered that excessive H2S enhances autophagy and decreases the survival rate of c-kit+ cDC1. These effects can be protected by autophagy inhibitors, but not by apoptosis and necrosis inhibitors. Additionally, through introducing myeloid-specific Atg5 knockout mice, we found that autophagy is necessary for H2S-induced ACD of c-kit+ cDC1. These findings further confirming that ACD is a key process mediating the reduction of c-kit+ cDC1 caused by excessive H2S.
In terms of molecular mechanisms, the function of H2S is often achieved through the persulfidation of the receptor tyrosine kinases (RTKs) family, which influences cell proliferation, survival, and differentiation. C-kit is a member of the RTKs family. For most cells, as they mature, the expression of the c-kit decreases until disappears. However, a few DC subsets retain the expression of c-kit, which is necessary for their corresponding functions46. In our mass cytometric data, c-kit was demonstrated expressed in all hepatic cDC1 subsets (CD103+ cDC1 and CD103- cDC1). Functionally, we found that c-kit knockout in cDCs had no significant effect on the hepatic cDC1 quantity at the MASLD stage, but reversed their quantity reduction and liver injury at the MASH stage. Mechanically, through employing cDC-specific c-kit-knockout mice, we discovered in in vitro co-culture and in vivo adoptive transfer experiments that the antigen presentation and protective function of cDC1 in MASH is not reliant on c-kit. Thus, further research is necessary to delve into the molecular mechanisms underlying how they exert their protective functions. Importantly, c-kit knockout abolished the ACD of hepatic cDC1 and subsequent pathological injury of MASH induced by H2S administration, indicating that excessive H2S-induced ACD in cDC1 and their reduction is c-kit-dependent. However, the direct combination of H2S and c-kit needs further evidence. Notable, regarding the selection of cDC1-specific c-kit-knockout mice, considering that Batf3 is involved in the functional regulation of regulatory T cells and type 9 helper T47,48, and XCR1 also targets a small fraction of CD4+ T cells49, we ultimately chose the Zbtb46 mouse model without direct impact on the immune system, although there have been reports that Zbtb46 is expressed not only on cDC but also on endothelial cells50.
Apart from DCs, other myeloid cell types also contribute to the progression of MASH. The emergence of TREM-2+ Mφ has garnered attention as an inflammatory mediator in MASH, underscoring its inflammatory contribution3. The function of hepatic KCs exhibits a paradoxical nature, akin to a “double-edged sword” in response to low or high LPS exposure51,52. Our current investigation echoes these findings, demonstrating a near-disappearance of hepatic Mo, a decrease in KCs, and a surge in inflammatory Mφ. Moreover, PMN accumulation exacerbate inflammation by facilitating the recruitment of other pro-inflammatory immune cells. In rodent models, PMN depletion mitigated hepatic lipid accumulation and inflammation in HFD-induced MASH53. Our data also indicate an expansion of the PMN proportion in the livers of MASH mice. Additionally, we observed inverse correlations between hepatic PMN/Mo-Mφ counts and c-kit+ cDC1 populations, as well as between KCs and c-kit+ cDC1, suggesting a potential complex interplay among distinct myeloid cell subsets.
In conclusion, we discovered an unrecognized hepatic c-kit+ cDC1 and confirmed their protective role in the development of MASH. The accumulation of hepatic H2S derived from gut Desulfovibrio_sp. further results in a c-kit-dependent ACD of cDC1, reducing their quantity and triggering the development of MASH. These findings may provide insights into the therapeutic strategies in MASH management.
Methods
Animals and treatments
The protocol was approved by the Ethics Committee on the Use and Care of Animals of Jilin University (No. SY202012022 and SY202302019) and performed in accordance with the institutional guidelines. In the present study, the male mice utilized for the FMT experiment were at the age of 4 weeks. Conversely, for the other experiments conducted, all the male mice involved were 6 weeks old. This study adhered rigorously to the following endpoint determination, formulated by our institutional ethical board for this research: A 20% reduction in body weight relative to the pre-study value, or a body condition score <2/5; Persistent anorexia, diarrhea, or vomiting for 4 consecutive days, accompanied by substantial body-weight loss; Abnormal behaviors: decreased mobility, loss of consciousness, spontaneous vocalizations, self-harm, continuous convulsions, incessant circling, paresis/paralysis; Severe depression or abnormal/exaggerated responses to external stimuli.
C57BL/6 mice weighing 18-20 g were purchased from Vital River Laboratories (Cat. 219). T-cell receptor (TCR)-transgenic OT-I mice (harboring OVA257-264-specific CD8+ T cells, Cat. 003831) and OT-II (harboring OVA323-339-specific CD4+ T cells, Cat. 004194) mice were purchased from Jackson Laboratories. zDC-Cre (B6. Cg-Zbtb46tm3.1(cre)Mnz/J, Cat. 028538) mice carrying the Zbtb46-driven Cre recombinase transgene were obtained from The Jackson Laboratory. The control c-kitflox/flox (C57BL/6J-Kitem1 flox Cya, Cat. S-CKO-03726) mice and CD45.1 (B6 Cd45.1, Cat. C001195) mice were obtained from Cyagen biological company. Conditional zDC (c-kit-/-) mice (c-kit knockout in cDCs) were generated by commercial breeding zDC-Cre mice with c-kitflox/flox mice. Lyz2‑Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J, Cat. 004781) expressing the Cre recombinase within the lysozyme Mlocus were purchased from the Jackson Laboratory. Atg5flox/flox mice (C57BL/6Smoc-Atg5em2(flox)Smoc, Cat. NM-CKO-00131) were obtained from Shanghai Model Organisms Center. Conditional myeloid Atg5-/- (Atg5flox/flox-Lyz2-Cre) mice were generated by breeding commercial Lyz2‑Cre mice and Atg5flox/flox mice. We utilized the Gubra Amylin MASH (GAN), a Western diet (WD)-induced MASH model that has been proven to successfully simulate the development of human MASH23. Mice were fed with a NCD (5053, Lab diet) and provided with normal water, or a WD containing 40 kcal% fat (Palm Oil), 20 kcal% fructose, and 2% cholesterol (D09100310, Research diets), along with a high sugar solution consisting of fructose 23.1 g/L (F8100, Solarbio Science & Technology) and 18.9 g/L glucose (G8150, Solarbio Science & Technology) for a duration of 30 W. Five mice were housed per cage in a specific pathogen-free (SPF) environment with a 12-h light/dark cycle at a temperature of 22.5 ± 2.5 °C and humidity of 50 ± 5%. Both experimental and control animals were housed in the same SPF environment, but placed separately. A series of experiments were conducted, including control and MASH modeling with WT mice fed with NCD and WD; adoption transfer experiments with WT mice fed with NCD and WD, C-kit-/- mouse modeling with c-kitflox/flox and c-kit-/- mice fed with NCD and WD, FMT experiments with WT mice fed WD, H2S treatment experiments with WT mice fed NCD and WD, and autophagy-related experiments with c-kitflox/flox and c-kit-/- mice fed with NCD and WD.
Patients
Liver sections were obtained from patients at The First Bethune Hospital of Jilin University. The human studies were approved by the Ethical Committee of The First Bethune Hospital of Jilin University (No. 2023-573) and were performed in accordance with the ethical guidelines on the participation of human subjects in the Declaration of Helsinki. For mIHC stains, a series of n = 5 Normal participants (n = 3 males and n = 2 females) and n = 5 MASH patients was included. All patients signed the informed consent. The sex factor has no effect on the number of c-kit+ cDC1 cells (Supplementary Fig. 10d). The inclusion and exclusion criteria for selecting the subjects who provided the liver slices were as follows: (1) adults aged between 18 and 65, (2) without end-stage liver disease, (3) without congenital liver disease, (4) without autoimmune liver disease, drug-induced liver injury, viral liver disease, or alcoholic liver disease, (5) without malignant tumors, (6) without mentally ill, pregnant or lactation, (7) a weekly ethanol consumption <140 g in females and <210 g in males, and (8) without use of antibiotics within a month.
GTT and ITT
For the intraperitoneal glucose tolerance tests (GTT), the mice underwent an overnight fast (16 h) and were then administered a sterile glucose solution via intraperitoneal injection at a dosage of 2 g/kg body. For the insulin tolerance tests (ITT), the mice were fasted for 5 h and received an intraperitoneal injection of 0.5 U/kg insulin. Blood samples were meticulously collected from the tail vein at 30, 60, 90, and 120 min post-injection, respectively. The blood glucose levels were measured using a glucose meter (Freestyle Lite, Abbot Diabetes Care Inc.)
Hydroxyproline assays
To determine the amount of collagen deposited in the liver, the hydroxyproline levels in fresh liver were detected with hydroxyproline assay kit (BC0255, Solarbio Science & Technology). The absorbance was read on a Synergy LX Multimode reader. The concentration of hydroxyproline was calculated by standard curve according to the manufacturer’s instructions.
Biochemical analysis
The activity of ALT and AST, as well as the hepatic TG content, were detected using commercial kits from Applygen Technologies (ALT, E2022; AST, E2024; TG, E1013) following the protocols provided by the manufacturer. Protein concentration in liver homogenization was quantified using a bicinchoninic acid (BCA) protein assay kit. Absorbance readings were taken using a Synergy LX Multimode reader (BioTek Instruments).
Histological analysis
Samples from the right lobe of the mouse livers were fixed in 4% paraformaldehyde at room temperature for over 24 h and embedded in paraffin. Thin sections of 4 μm thickness were cut and stained with hematoxylin and eosin (H&E; G1120, Solarbio Science & Technology). Liver histopathology was evaluated by three experienced liver pathologists without knowing the patient’s information based on the NASH Clinical Research Network. The degree of steatosis (percentage of hepatocytes containing fat droplets) was graded as 0 (<5%), 1 (5–33%), 2 (>33–66%), and 3 (>66%). Hepatocyte ballooning was categorized as 0 (none), 1 (few), or 2 (many cells/prominent ballooning). Foci of lobular inflammation were scored as 0 (no foci), 1 (<2 foci per 200× field), 2 (2–4 foci per 200× field), and 3 (>4 foci per 200× field). The three components were added together to determine the NAFLD activity score (NAS) which ranges from 0 to 8. Three liver sections from each liver sample were assessed blindly to preliminarily determine the NAS. Patients with NAS ≥ 5 were defined as MASH and those with macrovesicular steatosis <5% were not considered as MASH. The basic data of the patients were shown in Supplementary Table 1.
Mice liver tissue sections were also stained with Sirius red (GL472, Solarbio Science & Technology) to evaluate the liver fibrosis degree. The Image J was used to measure the positive areas.
Cytokine measurement
Liver samples were homogenized in cold RIPA buffer containing 1% phenylmethylsulfonyl fluoride (PMSF) using the tissue homogenizer (D1000, Benchmark Scientific). After a brief centrifugation step (4 °C, 12,000 × g), protein concentration in the supernatants was quantified using a BCA protein assay kit. Concentrations of TNF, IL-10, IL-1β, IL-23, CCL2, CXCL10, and CXCL9 in liver were measured using commercial kits (TNF, MTA00B; IL-10, M1000B-1; IL-1β, MLB00C; IL-23, M2300; CCL2, MJE00B; CXCL9, MCX900, R&D Systems; and CXCL10, BMS6018, Thermo Fisher) according to the manufacturer’s instructions. The absorbance was read on a Synergy LX Multimode reader.
Quantitative Reverse-Transcription PCR Assay
Total RNA of tissue was isolated with Trizol reagent (15596026, Invitrogen). A PrimeScript Reverse Transcriptase Kit (6110B, TaKaRa Biotechnology Co. Ltd.) was used to reverse-transcribe total RNA into cDNA according to the supplier’s protocol. Real-time PCR was carried out using a 7500 Real-Time PCR System (Applied Biosystems Inc.). The PCR reactions were performed in triplicate for each of the 3 individual cell preps and determined by the 2−ΔΔCT method. The level of β-actin RNA expression was used to normalize the data. The primer sequences were shown in Supplementary Table 2.
Mass cytometry and data visualization
Antibodies used for mass cytometry are listed in Supplementary Table 3. Purified antibodies were conjugated with metal reporters by using a MaxPar X8 Antibody Labeling Kit (201165 A, Fluidigm). Procedures for antibody staining and data acquisition were described previously21. Briefly, liver cells were incubated with 5 μM Cell-ID Intercalator-103Rh (201103B, Fluidigm) for 15 min, followed with staining with a cocktail of metal-conjugated antibodies for 45 min at room temperature. After washing, cells were incubated with 125 nM Cell-ID Intercalator-Ir (201192 A, Fluidigm) in MaxPar Fix and Perm Buffer (201067, Fluidigm) overnight at 4 °C. Data were acquired using a Helios mass cytometer (Fluidigm) and normalized using EQ Four Element Calibration Beads with the reference EQ Passport P13H2302 (Fluidigm). Single, live, and CD45+ cells were gated individually using FlowJo, followed with hyperbolic arcsinh transforming with a cofactor of 5 using Cytosplore software54. The overview immune lineages were then identified by performing a hSNE analysis (perplexity: 30; iterations: 1,000). All Gaussian mean-shift clustering-derived cell clusters and t-SNE plots were generated in Cytosplore. The phenotype heat map was created in Cytosplore, and the vertical bar for cytometry data were generated in R (4.2.0).
Immune cell isolation
To isolate hepatic immune cells, the in-situ perfusion with enzymatic fluids method was employed, following the previously described procedure55. In brief, a catheter filled with saline was carefully inserted into the portal vein, and the inferior vena cava was cut to allow blood outflow. Until the liver turned pale, the normal saline was replaced by 20 mL dissociation buffer containing 1 × HBSS, 0.01% collagenase IV (C4-BIOC, Sigma), 0.02% BSA, 0.001% DNase I (10104159001, Roche). Subsequently, the liver was removed carefully, cut into small pieces, and incubated with 10 mL dissociation buffer at 37 °C for 30 min. The resulting mixture was then passed through a 70 µm nylon cell strainer (352350, Falcon) to obtain a single-cell suspension. The cell suspension was centrifuged at 50 × g for 5 min to collect the supernatant, which was subjected to density centrifugation using Percoll (30%/75%, 17089109, Cytiva) with acceleration and deceleration set at 3 and 2, respectively, at 1000 × g for 25 min. The hepatic immune cells were collected from the interface, washed twice with PBS, and red blood cells were lysed using RBC Lysis Buffer following the manufacturer’s guidelines. Finally, isolated immune cells were resuspended in RPMI 1640 for further analysis.
The epididymal white adipose tissue was excised and cut into tiny pieces56. The tissue was incubated with 10 mL dissociation buffer at 37 °C for 40 min and filtered through a 70 µm nylon cell strainer. After centrifugation at 500 × g for 5 min with acceleration 3, the cells were resuspended in RBC Lysis Buffer and incubated on ice for 5 min.
Splenic immune cells were harvested by mechanical dissociation and then filtered through a 70 µm nylon cell strainer. The red blood cells were lysed using RBC Lysis Buffer.
For the immune cells of colon lamina propria, the single-cell suspension was prepared as described previously57. Briefly, the colon was isolated and washed in cold PBS after the removal of Peyer’s patches and mesenteric fat. The tissue was opened longitudinally, cut into 1-2 cm segments, and then incubated in epithelial cell removal solution including 1 × HBSS, 2% fetal calf serum inactivated, 5 mM EDTA, and 10 mM HEPES for 20 min at 37 °C. The supernatant was discarded, and the remaining segments were washed three times with cold PBS, minced, and then digested in the dissociation buffer at 37 °C for 30 min. Immune cell enrichment was performed by Percoll gradient centrifugation as described above.
Flow cytometry
The antibodies used for flow cytometric analysis are from Biolegend, as shown in Supplementary Table 4. Isolated single cells were suspended at a concentration of 1 × 106 cells/mL and incubated with 7-AAD viability staining solution to evaluate cell viability. Fc Block (Biolegend) was added to block the non-specific bindings. The cells were then strained with fluorochrome-tagged antibodies as mentioned above, and incubated at 4 °C in the dark for 30 min in PBS containing 2% FBS and 5 mM EDTA to detect cell surface markers. After three washes, the strained cells were resuspended in 200 μL PBS and analyzed using a DxFLEX flow cytometer (B73415, Beckman Coulter, Inc.) or sorted using a BD FACSAria II (643180, Becton Dickinson). The flow cytometry standard (FCS) files were processed using flowjoVX10 (BD Biosciences). Debris (SSC vs FSC) and single cells (FSC vs FSC) were excluded. Immune cells were identified as the CD45+ population from live cells. Specific immune cells were classified based on cell surface markers. Cell apoptosis was determined using the Annexin V Apoptosis Detection Kit I (556547, BD Biosciences) according to the manufacturer’s instructions.
mIHC
The collected livers were fixed with 10% polymethyl methacrylate and embedded in paraffin. The paraffin blocks were then sliced into 4 μm sections and adhered to glass slides. The tissue slides were subjected to a dry oven at 60 °C for 1 h to facilitate deparaffinization. After deparaffinization with xylene, the slides were rehydrated in a series of ethanol solutions (100%, 95%, and 70%). Antigen retrieval was performed by incubating the slides in a citric acid solution at 95 °C for 20 min, followed by cooling to room temperature. To inhibit endogenous peroxidase activity, the slices were treated with 3% H2O2 for 15 min. Subsequently, the slides were incubated with 10% goat serum for 30 min to block non-specific binding sites. Primary antibodies, including Rabbit anti-c-kit (1:200, AF6153, Affinity Biosciences), Rabbit anti-CD103 (1:100, ab224202, Abcam), and Rabbit anti-XCR1 (1:50, NBP1-02343, Novus Biologicals), were applied to the slides and incubated at 37 °C for 1 h in a humidified chamber. After washing twice, the sections were incubated with HRP-coupled goat anti-rabbit IgG (1:1000, ab6721, Abcam) as secondary antibodies at room temperature for 10 min. Heat-induced epitope retrieval was performed after each staining cycle to remove all antibodies, including primary and secondary antibodies. For mIHC staining, the AlphaTSA Multiplex IHC kit (AXT34100011, AlphaX) was used according to the manufacturer’s instructions. Finally, the sample nuclei were stained with DAPI for 10 min, and multispectral images were scanned using Zeiss AxioScan7.
Adoptive transfer assay
Single-cell suspensions of livers and spleens from NCD mice were prepared as described above. C-kit+CD103+ and c-kit+CD103- cDC1 were sorted from the suspensions by flow cytometry using BD FACSAria II cell sorter with a purity of 98 to 99%. The sorted cells were then washed three times with sterile PBS. Subsequently, 2 × 105 cells were resuspended in 100 µL of sterile saline. Mice fed with a WD for 26 W received weekly injections of the c-kit+CD103+ and c-kit+CD103- cDC1 cells via the tail vein for a total of 4 W. The recipient WD mice were chosen with close body weights, with a median of 56.85 g, a maximum value of 59.1 g, a minimum value of 55.6 g, and a coefficient of variation (CV) of 1.44%. The control group was injected with sterile saline. One week after the final cell transfer, mice were euthanized by cervical dislocation by trained and experienced staff.
Gut microbiota profiling
Feces from mice fed with a WD for 0, 15, and 30 W were collected, immediately frozen, and stored at -80 °C. The DNA from fecal microbiota was extracted using the CTAB following the manufacturer’s instructions. The full-length 16S rRNA gene was amplified using primers 27 F (5’-AGRGTTTGATYNTGGCTCAG-3’) and 1492 R (5’- TASGGHTACCTTGTTASGACTT-3’), which were tagged with specific barcode for per sample. The PCR amplification was performed with an initial denaturation at 95 °C for 2 min, followed by 25 cycles of denaturation at 95 °C for 30 sec, annealing at 55 °C for 30 sec, and extension at 72 °C for 1 min. A final extension step was carried out at 72 °C for 5 min. The amplicons were extracted from a 2% agarose gel electrophoresis using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences) according to the manufacturer’s instructions. After quantified by QuantiFluor-ST (Promega), the amplicon pools were prepared for library construction. SMRTbell libraries were prepared using the Pacific Biosciences SMRTbell Template Prep kit 1.0 (PacBio) and sequenced on PacBio RS II (LC-Bio Technology Co., Ltd.). Circular Consensus Sequence (CCS) reads were generated from raw subreads by SMRT Link (v6.0) with the following parameters: minPasses = 5; minPredictedAccuracy = 0.9. Then lima (v1.7.1) was used to distinguish CCS reads from different samples, and cutadapt (v1.9) was applied to identify primers. The CCS reads between 1200 bp to 1650 bp remained after the length filtration. After dereplication and filtering chimeric sequences using DADA2, the feature table and feature sequence were obtained. Alpha and beta diversity were calculated by normalizing to the same sequences randomly. The ASVs were annotated by aligned feature sequences with the SILVA database (release 138). The method parameters are as follows classify-sklearn (confidence = 0.7), classify-consensus-vearsh (identity = 0.9, coverage = 0.9). Other diagrams were implemented using the R packages.
Fecal collection and FMT
Based on the previous study, fresh feces (20-30 pellets) were collected from mice that were fed a WD for 30 W using anal stimulation methods every morning at 9:00 am. The collected feces were then dissolved and mixed with sterile saline (1 pellet/mL saline), vigorously shaken, and swirled until no visible fecal particles remained. Subsequently, the samples were centrifuged at 800 × g, 4 °C for 10 min to discard the precipitate and collect the supernatant. The supernatant was further centrifuged at 12,000 × g, 4 °C for 5 min to obtain total bacteria, which were subsequently filtered twice in saline. Finally, the bacteria suspension was resuspended in saline to a concentration of 109 colony-forming units (CFU)/200 µL for oral gavage. Four-week-old C57BL/6 mice were administered with an antibiotic cocktail (containing 0.2 g/L ciprofloxacin, 0.5 g/L vancomycin, 1 g/L neomycin sulfate, 1 g/L metronidazole, 1 g/L ampicillin) in their drinking water ad libitum for 2 W to deplete the original gut microbiota. Then, they received daily oral gavage of bacteria suspension prepared above or sterile saline (200 µL/mouse) for 15 W. Given that both the recipient mice (MASLD) and donor mice (MASH) in the FMT experiment were WD-fed mice, we still opted to use oral saline after pretreatment with antibiotics as the control group, which can avert the uncertain influence of introducing new microbiota of NCD mouse on the microbiota of WD mouse.
Desulfovibrio_sp. transplantation
Desulfovibrio_sp. (ATCC-700045) was purchased from ATCC, and cultured overnight in anaerobically sterilized ATCC medium 1249 under strict anaerobic conditions. Four-week-old C57BL/6 mice were treated with antibiotics for 2 weeks to clear their gut microbiota, and then were gavaged daily with living Desulfovibrio_sp. in saline solution of 109 CFU/200uL for 15 weeks, and fed with WD diet at the same time.
H2S measurement
The H2S concentrations in the liver and serum were measured using the H2S concentration determining kit (BC2055, Solarbio Science & Technology) according to the manufacturer’s instructions. In brief, sulfide is initially trapped with a metal, and the resulting complex reacts with N, N-dimethyl-p-phenylenediamine to produce methylene blue in the presence of Fe3+ under acidic conditions. Subsequently, the absorbance of methylene blue at 665 nm was measured using a Synergy LX Multimode reader (BioTek Instruments).
Induction of c-kit+ DCs in vitro
According to previous studies64, mouse BM cells were isolated to induce c-kit+ DCs in vitro. The BM cells were obtained from the tibias, femurs, and pelvises of 8-week-old male C57BL/6 mice. They were cultured in a 24-well plate with RPMI medium containing granulocyte macrophage colony stimulating factor (GM-CSF, 20 ng/mL, 315-03, Peprotech) and interleukin-4 (IL-4, 20 ng/mL, 214-14, Peprotech) at a density of 2 × 106 cells/mL per well for ten days. Subsequently, non-adherent and loosely adherent cells were collected and cultured in RPMI medium containing tumor necrosis factor-α (TNF, 20 ng/mL, 315-01A, Peprotech) for 2 d before obtaining the c-kit+ DCs. The purity of c-kit+ DCs was analyzed using flow cytometry.
DCs and T cell co-culture
XCR1+ cDC1 were isolated from the liver of c-kitflox/flox mice and c-kit-/- mice, respectively. The spleens of OT-I and OT-II mice were collected and the naïve T cells were isolated using the EasySep Mouse Pan-Naïve T Cell Isolation Kit (19848, STEMCELL Technologies) according to manufacturer’s instructions. The sorted cDC1 were treated with OVA257-264 peptide (1 µg/mL, HY-P5400, MCE) and OVA323-339 peptide (1 µg/mL, HY-P0286, MCE), and co-cultured with isolated naïve T cells in a ratio of cDC1: T cell = 1:4 in a 96-well plate. After 72 h of co-culture, T cell proliferation was detected using the CellTrace CFSE cell proliferation kit (C34554, Thermo Fisher Scientific) according to manufacturer’s instructions. The cDC1 treated with OVA257-264 peptide were co-cultured with naïve T cells isolated from OT-I mice for 10 d. The ability of cDC1 to induce differentiation of CD8+ T cells was evaluated by detecting the level of CD107a on the surface of CD3+CD8+ T cells. The naïve T cells isolated from OT-II mice were treated with Brefeldin A (HY-16592; MCE) for 4 h before co-culture with cDC1 treated with OVA323-339 peptide. After co-culture for 10 d, the ability of cDC1 to induce differentiation of CD4+ T cells was evaluated by detecting the intracellular IFN-γ level of CD3+CD4+ T cells.
Detection of IL-10 in c-kit+ cDC1
C-kit+ cDC1 were isolated from the liver of mice fed with 30 W NCD or WD by flow cytometry (BD FACSAria II cell sorter). Cells were stimulated for 4 h with PMA (50 ng/mL, Sigma Aldrich), ionomycin (1 μg/mL, Sigma Aldrich) and Brefeldin A in a 37 °C incubator with a humidified atmosphere containing 95% air and 5% CO2. After washed with PBS, cells were fixed using medium A of FIX & PERM kit (GAS003, Thermo Fisher) in the dark for 15 min at room temperature. Then cells were incubated with medium B and IL-10-FITC antibody for 30 min. Fluorescence intensity was detected by flow cytometry.
GYY4137 treatment in vivo and in vitro
GYY4137 is a H2S donor that releases H2S slowly. In vivo study, 6-week-old male C57BL/6 mice fed a WD diet for 13 W were administrated with GYY4137 (150 mg/kg/day, HY-107632, MCE) or saline once daily for 2 W via intraperitoneal injection. The injection concentration of GYY4137 was determined through concentration gradient experiments. In vitro study, the induced c-kit+ DCs were treated with GYY4137 (1.6 mM) for 2 h according to a previous study65.
Autophagy detection
In vivo, autophagy flux of c-kit+ cDC1 in mice was detected by flow cytometry using the Cyto-ID Autophagy Detection Kit 2.0 (ENZ-KIT175-0050, Enzo Life science Inc) according to the manufacturer’s instructions. In vitro, induced c-kit+ DCs were treated with or without 3-methyladenine (5 mM, 3MA, HY-19312, MCE) for 24 h before GYY4137 stimulation. The protein abundance of p62 and LC3 in cells was detected by western blotting. The number of autophagosomes and autolysosome was evaluated using a transmission electron microscope (TEM, H-7650 electron microscope, Hitachi). The autophagosome and autophagic lysosome were counted from at least 15 random cell sections in each sample and expressed as the average number per cell section.
Western blotting
Total proteins were extracted using a commercial protein extraction kit and concentration was measured with the BCA assay kit. A total of 30 μg protein from each sample was separated by 8–15% SDS-PAGE with a known prestained protein ladder. The target protein on the gel was electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane. PVDF membranes were blocked in 5% skim milk in Tris-buffered saline with 0.1 % Tween-20 detergent (TBS-T) for 2 h at room temperature, followed by incubation overnight at 4 °C with primary antibodies against LC3 (1:1000, ab48394, Abcam), sequestosome-1 (p62, 1:1000, 23214, Cell Signaling Technology), Atg5 (1:1000, DF6010, Affinity) and β-actin (1:2,000, ab8226, Abcam), respectively. Subsequently, PVDF membranes were washed with TBS-T 3 times and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit (1:5000, SA00001-2, ProteinTech Group) or anti-mouse (1:5000, SA00001-1, ProteinTech Group) immunoglobulin at room temperature for 1 h. The membrane was washed with TBS-T, and the HRP activity was detected using an enhanced chemiluminescence solution (ECL). β-actin was used as a reference protein for protein. The image was scanned using the Protein Simple Imager (ProteinSimple, Santa Clara) and analyzed using the Image-pro Plus (Media Cybernetics; Rockville).
MTT assay
Cell viability was determined using the methylthiazolyl diphenyl-tetrazolium bromide (MTT) cell proliferation and cytotoxicity assay kit (C0009S, Beyotime) according to the manufacturer’s instructions. In brief, 100 µL of cell suspension (5 × 104 cells/mL) was seeded into a 96-well plate and incubated with 10 µL of MTT (5 mg/mL). After incubation at 37 °C for 4 h, 100 µL of formazan lysis solution was added to each well and incubated for another 4 h to fully dissolve the crystals. The absorbance was measured at 570 nm using a Synergy LX Multimode reader (BioTek Instruments). Cell viability was subsequently calculated by comparing the obtained optical value to that of the control group.
Transcriptome sequencing
Transcriptome sequence and data analysis were performed following standard procedures at Shanghai Genesky Biotechnology Company. Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions DNase I treatment was performed to remove DNA contamination. Subsequently, total RNA was quantified by the Qubit 3.0 Spectrophotometer (Thermo Fisher Scientific) and Agilent 2100 bioanalyzer system (Agilent Biotechnologies). The RNA library was sequenced on an Illumina Hiseq 4000 platform, generating 150 bp paired-end reads.
For the sequencing data analysis, clean reads were mapped to the reference genome using HISAT2 v2.0.4. The mapped reads of each sample were assembled using StringTie v1.3.1 in a reference-based approach66. The assembled transcripts were analyzed for coding potential using RNA_seQc (http://www.broadinstitute.org/cancer/cga/rna-seqc). Transcripts without coding potential were considered as candidate sets of lncRNAs. Fragments per kilobase of exon per million (FPKMs) reads for both lncRNAs and coding genes in each sample were calculated by using Cuffdiff v2.1.1. Gene FPKMs were calculated by summing the FPKMs of transcripts within each gene group. Differentially expressed mRNAs and lncRNAs were identified using the FPKM values for each gene. Additionally, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using the clusterProfiler R package to gain insights into the potential roles of differentially expressed genes.
Statistics & Reproducibility
Our study’s experimental design is shown in Figs. 3a, 5e, 7c, and 8d. Detailed information can be found in the corresponding section above. The sample size was determined based on similar studies and a reliable tool (https://www.bu.edu/researchsupport/compliance/animal-care/working-with-animals/research/sample-size-calculations-iacuc). Liver sections were randomly and blindly assessed for NAS determination. No data were excluded from the analyses.
Statistical analysis was performed using Prism software version 8.3.0 (GraphPad Software). Data were first checked to be normally distributed using the Shapiro-Wilk test. In comparison between the 2 groups, the difference was determined using an unpaired two-tailed Student’s t-test with normal distribution or a Mann-Whitney U test without normal distribution. For three or more groups, data were statistically analyzed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test or Kruskal-Wallis nonparametric test. Two-way ANOVA was used to analyze experiments with multiple groups and two independent variables. Spearman’s rank correlation coefficient test was performed to analyze correlations. Data were presented as means ± SEM. *P < 0.05 was considered significant, **P < 0.01 was considered markedly significant. All error bars represent SEM and statistical details are included in the figure legends.
All statistics details including statistical test used, exact value of n and statistical significance are reported in figure legend and source data. Each data point represents genuine replication (biological replicates) and was obtained from a single measurement or from multiple measurements illustrated by the mean. For the reproducibility of the results, all the experiments were repeated independently with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The fecal microbiome data generated in this study have been deposited in the National Microbiology Data Center of China (https://data.mendeley.com/datasets/j8bw86mc5h/1, https://doi.org/10.17632/j8bw86mc5h.1). Mass cytometry data generated in this study have been deposited in the Flow Repository (https://data.mendeley.com/datasets/vm5rkkk96d/1, https://doi.org/10.17632/vm5rkkk96d.1). RNA-sequencing data generated in this study have been deposited in the Genome Sequence Archive under project PRJCA034212 (accession number CRA021908, https://ngdc.cncb.ac.cn/gsa/browse/CRA021908). The human gut microbiota data used in this study have been deposited in the GMrepo database under project Project PRJNA373901 (https://gmrepo.humangut.info/data/project/PRJNA3 73901). Source data are provided with this paper. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Estes, C., Razavi, H., Loomba, R., Younossi, Z. & Sanyal, A. J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018).
Brunt, E. M., Kleiner, D. E., Wilson, L. A., Belt, P. & Neuschwander-Tetri, B. A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 53, 810–820 (2011).
Xiong, X. et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e645 (2019).
Ramachandran, P. et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019).
Buccitelli, C. & Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644 (2020).
Spitzer, M. H. & Nolan, G. P. Mass cytometry: single cells, many features. Cell 165, 780–791 (2016).
Eckert, C., Klein, N., Kornek, M. & Lukacs-Kornek, V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 6, 179 (2015).
Audiger, C., Rahman, M. J., Yun, T. J., Tarbell, K. V. & Lesage, S. The importance of dendritic cells in maintaining immune tolerance. J. Immunol. 198, 2223–2231 (2017).
Dalod, M., Chelbi, R., Malissen, B. & Lawrence, T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J. 33, 1104–1116 (2014).
Henning, J. R. et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 58, 589–602 (2013).
Haas, J. T. et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 1, 604–614 (2019).
Heier, E. C. et al. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J. Hepatol. 66, 1241–1250 (2017).
Shin, J. Y., Wang, C. Y., Lin, C. C. & Chu, C. L. A recently described type 2 conventional dendritic cell (cDC2) subset mediates inflammation. Cell Mol. Immunol. 17, 1215–1217 (2020).
Schnabl, B. & Brenner, D. A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146, 1513–1524 (2014).
Chen, J. et al. Natural killer T cells play a necessary role in modulating of immune-mediated liver injury by gut microbiota. Sci. Rep. 4, 7259 (2014).
Vespasiani-Gentilucci, U. et al. Hepatic toll-like receptor 4 expression is associated with portal inflammation and fibrosis in patients with NAFLD. Liver Int. 35, 569–581 (2015).
Dapito, D. H. et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 (2012).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Loo, T. M. et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 7, 522–538 (2017).
van Unen, V. et al. Visual analysis of mass cytometry data by hierarchical stochastic neighbour embedding reveals rare cell types. Nat. Commun. 8, 1740 (2017).
Segura, E. Review of mouse and human dendritic cell subsets. Methods Mol. Biol. 1423, 3–15 (2016).
Hansen, H. H. et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 20, 210 (2020).
Barroeta Seijas, A. B. et al. GM-CSF inhibits c-Kit and SCF expression by bone marrow-derived dendritic cells. Front. Immunol. 8, 147 (2017).
Cai, J., Zhang, X. J. & Li, H. Role of innate immune signaling in non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 29, 712–722 (2018).
Deczkowska, A. et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 27, 1043–1054 (2021).
Alshawsh, M. A. et al. A comparison of the gene expression profiles of non-alcoholic fatty liver disease between animal models of a high-fat diet and methionine-choline-deficient diet. Molecules 27, 858 (2022).
Connolly, M. K. et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Invest. 119, 3213–3225 (2009).
Caballero, F. et al. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 285, 18528–18536 (2010).
Febbraio, M. A. et al. Preclinical models for studying NASH-driven HCC: How useful are they? Cell Metab. 29, 18–26 (2019).
Im, Y. R. et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 74, 1884–1901 (2021).
Schülke, S. Induction of Interleukin-10 producing dendritic cells as a tool to suppress allergen-specific T Helper 2 responses. Front. Immunol. 9, 455 (2018).
Goddard, S., Youster, J., Morgan, E. & Adams, D. H. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Am. J. Pathol. 164, 511–519 (2004).
Kelly, A. et al. CD141+ myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 60, 135–142 (2014).
Schwimmer, J. B. et al. Microbiome signatures associated with steatohepatitis and moderate to severe fibrosis in children with nonalcoholic fatty liver disease. Gastroenterology 157, 1109–1122 (2019).
Sharpton, S. R., Ajmera, V. & Loomba, R. Emerging role of the gut microbiome in nonalcoholic fatty liver disease: from composition to function. Clin. Gastroenterol. Hepatol. 17, 296–306 (2019).
Lee, G. et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 11, 4982 (2020).
Lin, Y. C., Lin, H. F., Wu, C. C., Chen, C. L. & Ni, Y. H. Pathogenic effects of Desulfovibrio in the gut on fatty liver in diet-induced obese mice and children with obesity. J. Gastroenterol. 57, 913–925 (2022).
Zhu, L. et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609 (2013).
Li, C. et al. Using intestinal flora to distinguish non-alcoholic steatohepatitis from non-alcoholic fatty liver. J. Int. Med. Res. 48, 300060520978122 (2020).
Chen, J. et al. A high-fat diet promotes cancer progression by inducing gut microbiota-mediated leucine production and PMN-MDSC differentiation. Proc. Natl Acad. Sci. USA 121, e2306776121 (2024).
Taglialegna, A. Fat, Desulfovibrio and cancer. Nat. Rev. Microbiol. 22, 388 (2024).
Tan, J. et al. Anti-PD-L1 antibody enhances curative effect of cryoablation via antibody-dependent cell-mediated cytotoxicity mediating PD-L1highCD11b+ cells elimination in hepatocellular carcinoma. Acta Pharm. Sin. B 13, 632–647 (2023).
Wang, S. S. et al. Hydrogen sulfide promotes autophagy of hepatocellular carcinoma cells through the PI3K/Akt/mTOR signaling pathway. Cell Death Dis. 8, e2688 (2017).
Jung, S., Jeong, H. & Yu, S. W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 52, 921–930 (2020).
Zhou, S. et al. CD117+ Dendritic and mast cells are dependent on RasGRP4 to function as accessory cells for optimal natural killer cell-mediated responses to lipopolysaccharide. PLoS One 11, e0151638 (2016).
Lee, W., Kim, H. S., Hwang, S. S. & Lee, G. R. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp. Mol. Med. 49, e393 (2017).
Li, J. et al. IL-9 and Th9 cells in health and diseases-From tolerance to immunopathology. Cytokine Growth Factor Rev. 37, 47–55 (2017).
Mattiuz, R. et al. Novel Cre-expressing mouse strains permitting to selectively track and edit Type 1 conventional dendritic cells facilitate disentangling their complexity in vivo. Front. Immunol. 9, 2805 (2018).
Satpathy, A. T. et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 209, 1135–1152 (2012).
Knolle, P. et al. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 22, 226–229 (1995).
Seki, E. et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor88 that is critically involved in induction of production of IL-12 and IL-1b. J. Immunol. 166, 2651–2657 (2001).
Ou, R. et al. Neutrophil depletion improves diet-induced non-alcoholic fatty liver disease in mice. Endocrine 57, 72–82 (2017).
Li, N. et al. Memory CD4(+) T cells are generated in the human fetal intestine. Nat. Immunol. 20, 301–312 (2019).
Shi, W. et al. Isolation and purification of immune cells from the liver. Int. Immunopharmacol. 85, 106632 (2020).
Jaitin, D. A. et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e614 (2019).
Ignacio, A. et al. Small intestinal resident eosinophils maintain gut homeostasis following microbial colonization. Immunity 55, 1250–1267.e1212 (2022).
Acknowledgements
This work was supported by the National Key R&D Program of China, grant number (2023YFD1801100 to X.L.), the National Natural Science Foundation of China (Beijing, China; grant no. U24A20454 to X.L., 32473104 to Y.S.), and the Fundamental Research Funds for the Central Universities (Jilin, China) to X.L.
Author information
Authors and Affiliations
Contributions
Y.S. and X.L. designed the experiments. Y.S., N.L., S.J., and K.W. performed the experiments. Y.S. and X.L. provided funding support. N.L. provided technical support in data visualization. Y.L. and Z.F. provided human liver samples. G.L., X.D., W.G., L.L, and Z.W. provided data analysis. Y.S. wrote the original draft, and X.L. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nobuhito Taniki and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Y., Li, N., Jiang, S. et al. Microbiota-derived H2S induces c-kit+ cDC1 autophagic cell death and liver inflammation in metabolic dysfunction-associated steatohepatitis. Nat Commun 16, 2222 (2025). https://doi.org/10.1038/s41467-025-57574-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57574-3
This article is cited by
-
Immunopathogenic mechanisms and immunoregulatory therapies in MASLD
Cellular & Molecular Immunology (2025)
-
Gut microbiota in hepatic encephalopathy: a 3PM-guided three-tier health strategy
EPMA Journal (2025)
