
Abstract
Large-scale cancer genetic/genomic studies demonstrated that papillary renal cell carcinoma (pRCC) is featured with a frequent shallow deletion of the upstream tumor suppressors of the Hippo/YAP signaling pathway, suggesting that this signaling pathway may play a role in pRCC development. Here we develop a transgenic mouse model with a renal epithelial cell-specific hyperactivation of YAP1 and find that hyperactivation of YAP1 can induce dedifferentiation and transformation of renal tubular epithelial cells leading to the development of pRCC. We analyze at the single-cell resolution the cellular landscape alterations during cancer initiation and progression. Our data indicate that the hyperactivated YAP1, via manipulating multiple signaling pathways, induces epithelial cell transformation, MDSC (Myeloid-derived suppressor cells) accumulation, and pRCC development. Interestingly, we find that depletion of MDSC blocks YAP1-induced kidney overgrowth and tumorigenesis. Inhibiting YAP1 activity with MGH-CP1, a recently developed TEAD inhibitor, impedes MDSC accumulation and suppresses tumor development. Our results identify the disrupted Hippo/YAP signaling as a major contributor to pRCC and suggest that targeting the disrupted Hippo pathway represents a plausible strategy to prevent and treat pRCC.
Similar content being viewed by others


Introduction
The Hippo/YAP signaling pathway is an evolutionarily conserved regulator of organ size in many species1,2. It regulates organ size and maintains tissue homeostasis via integrating multiple upstream inputs to control cell proliferation, differentiation, and apoptosis during development and normal physiological function3. The core of the classical Hippo pathway in mammals is a kinase cascade consisting of STK4/3 (Serine/Threonine Kinase 4/3, also known as MST1/2) and LATS1/2 (Large Tumor Suppressor Kinase 1/2), their adapter proteins SAV1 (Salvador homolog 1) and MOB kinase activators (MOB1A/1B), and the downstream effectors YAP1(Yes-associated protein 1) and WWTR1 (WW domain containing transcription regulator 1, also known as TAZ)1,4 (Suppl Fig. S1). Upon activation, the kinase cascade of the Hippo pathway culminates in the phosphorylation/activation of LATS1/2 leading to cytoplasmic retention and degradation of YAP1 and WWTR1, two effectors of the Hippo signaling pathway. Inactivation of the Hippo pathway results in the activation and nuclear translocation of YAP1 and WWTR1. In the nucleus, YAP1 and WWTR1 act as co-activators of transcription factors such as TEAD1/2/3/4 to promote the expression of genes that drive cell proliferation and maintain cell viability1,4. Disruption of the Hippo signaling pathway results in constitutive activation of YAP1 and WWTR1 leading to developmental dysgenesis and/or tumorigenesis3,5,6,7,8.
The contribution of the disrupted Hippo pathway in tumorigenesis has been well documented6,9. Correspondingly, many translational studies are ongoing to discover Hippo pathway-based targeted therapies for different types of cancers10,11. However, large-scale cancer genome sequencing efforts revealed that the major components of the Hippo pathway are only deleted and/or mutated in select human cancers7,12. This suggests that precision medicine targeting the disrupted Hippo pathway may be effective in select cancers. By mining the TCGA database13,14, we found that instead of deep deletions and mutations, in high-grade serous ovarian cystadenocarcinoma (HGSOC) and papillary renal cell carcinoma (pRCC) the major components of the Hippo signaling pathway are featured by shallow deletions. pRCC is the second most prevalent renal cell carcinoma15,16. The cellular and mechanisms underlying pRCC development are understudied and, as a result, there are no effective therapeutic options for this deadly disease15,17. The frequent shallow deletion of its major components in pRCC implies that the Hippo signaling pathway plays a role in the initiation and/or progression this relatively rare cancer. In this study, we generated a pRCC transgenic mouse model and combined this model with Single-cell RNAseq technology to uncover the cellular and molecular mechanisms underlying pRCC initiation and progression. Our results demonstrate that the disrupted Hippo/YAP pathway alone is sufficient to induce renal epithelial neoplasia and pRCC development. Our mechanistic studies indicate that YAP1-induced accumulation of myeloid-derived suppressor cells (MDSCs) in renal tissues plays a critical role in neoplastic cell immunosurveillance evasion. Our preclinical studies suggested that targeting the disrupted Hippo signaling with a YAP/TEAD inhibitor MGH-CP1 provides a strategy for pRCC prevention and treatment.
Results
The Hippo signaling pathway is disrupted in human pRCC
The large-scale, multi-dimension genetic/genomic sequencing data in The Cancer Genome Atlas (TCGA) database allows for a comprehensive examination of the involvement of the disrupted Hippo/YAP signaling in pRCC. Data extracted from TCGA datasets demonstrated that alterations of the Hippo pathway are not common in renal clear cell carcinoma (ccRCC). However, The Hippo pathway is frequently disrupted in papillary renal cell carcinoma (pRCC) (Fig. 1A). The major upstream tumor suppressors of the Hippo signaling cascade, such as FAT1/2/3/4, AJUBA, NF2, SAV1, TJP1/2, YWHAH, and CSNK1E, are frequently deleted or mutated (Fig. 1A). Interestingly, the genetic alterations of these genes are featured by shallow deletions, not mutations or deep deletions (Fig. 1A). For example, NF2, a major upstream tumor suppressor of the hippo pathway, is deleted in 30% of pRCC patients, with only 4% deep deletion but 27% shallow deletion. YWHAH, another major YAP1 inactivator, has only 3% deep deletions but 30% shallow deletions in pRCC (Fig. 1A). Of note, we found that the deletion (shallow and deep deletions) of these upstream tumor suppressors of the Hippo signaling was consistently associated with poorer overall and disease-free survival rates (Fig. 1B, Suppl. Fig. S2). Consistent with these observations, we found that the deletion of upstream tumor suppressors was associated with upregulation of pRCC tumor-promoting factors (e.g., CCNE1, BIRC5, MYC) and downregulation of tumor-suppressive genes such as VHL18,19 (Fig. 1C, Suppl. Fig. S3). The abnormal expression of CCNE1 and VHL predicts poor survival of pRCC (Fig, 1D). In addition, we observed that the shallow deletions of the Hippo pathway upstream tumor suppressors and the subsequent alteration of downstream target genes are correlated with pRCC progression (Fig. 1E & 1F). These pieces of evidence suggest that the shallow deletion of the major components of the Hippo/YAP pathway is a critical contributor to pRCC development.

pRCC patient clinical data and gene expression profiling data were extracted from the TCGA datasets (TCGA firehose study) and analyzed using the GraphPad Prism 9 software. A Copy number variation (CNV) of genes encoding the key components of the Hippo signaling pathway (n = 293). B Deletion (both deep & shallow deletions) of genes encoding the major components of the Hippo signaling cascade predicts poor overall survival in pRCC patients. del: deletion; dip: diploid. Data were analyzed using the Kaplan-Meier simple survival analysis with the Mantel-Cox test and Gehan-Breslow-Wilcoxon test. The P value of each comparison is presented on the top of the corresponding graph. C Deletion of genes encoding the key components of the Hippo signaling pathway is associated with dysregulation of known regulators of the pRCC development (e.g., CCNE1 and VHL, etc.). Data were analyzed with unpaired t-tests (two-tailed). The P value of each comparison is presented on the top of the corresponding graph (FAT1-dip: n = 174; FAT1-Del: n = 23; LATS2-Dip: n = 170; LATS2-Del: n = 20; NF2-Dip: n = 159; NF2-Del: n = 51). D Dysregulated CCNE1 and VHL is associated with poor patient survival rate. Data were analyzed using the Kaplan-Meier simple survival analysis with the Mantel-Cox test and Gehan-Breslow-Wilcoxon test. The P value of each comparison is presented on the top of the corresponding graph (CCNE1-high: n = 42, CCNE1-low: n = 153; VHL-high: n = 132, VHL-low: n = 28). E, F Deletion of the upstream suppressor of the Hippo signaling pathway and dysregulation of their downstream genes (e.g., CCNE1, VHL, etc.) are associated with pRCC progression. Source data are provided as a Source Data file.
Hyperactivated YAP1 promotes the proliferation and transformation of renal epithelial cells
The most direct impact of the disrupted Hippo signaling is the hyperactivation of YAP1, a tumorigenic transcription coactivator6. To clarify the effect of the disrupted Hippo pathway on renal cell function, we transfect HK2 cell (an immortalized renal epithelial cell line) with lentiviral vectors expressing wild-type YAP1 (YAP) or constitutively active YAP1 (YAPS127A, with a serine to alanine mutation that results in the constitutive activation of YAP1) (Fig. 2A, B). Ectopic expression of YAP1 or YAPS127A significantly stimulated HK2 cell proliferation (Fig. 2C), leading to multiple-layer growth of HK-2 cells in the traditional 2D culture (Fig. 2D). Consistently, soft agar colony formation assays showed that HK2-YAPS127A cells grew in an anchorage-free manner (Fig. 2E), suggesting that these cells are transformed. Renal capsule implantation of these cells resulted in the growth of tumor-like tissue expressing high levels of Ki67 and vimentin (Fig. 2F–H) in the implant compared to adjacent control renal tissue. Subcutaneous injection of HK2-YAPS127A cells into athymic nude mice resulted in metastatic renal cell carcinoma in mice. All tumor-carrying mice died within 545 days post cell inoculation (Fig. 2I). No tumors developed in control HK2-MX cells (HK2 cells transfected with control vectors) 570 days after cell inoculation (Fig. 2I). Tumor cells expressed high levels of YAP1 and vimentin and are very proliferative, which is indicated by the high expression of Ki67 (Fig. 2J). These studies suggest that hyperactivated YAP1 can drive the malignant transformation of renal epithelial cells.

A Western blots showing successful overexpression of the wild-type YAP1 (YAP) and constitutively active YAP1 (YAPS127A) in HK2 cells. Each experiment was repeated at least three time and a representative image was presented to show the relative level of YAP1 protein. B Fluorescent immunocytochemistry showing the expression and subcellular location of YAP (red) in HK2-MX, HK2-YAP, and HK2-YAPS127A cells; Each experiment was repeated independently at least three time and a representative image was presented to show the location and relative level of YAP1 protein. Nuclei were stained with DAPI (blue); scale bar: 10 μm. C Cell growth curves showing that YAP activity promotes the proliferation of HK2 cells. Each time point represents the mean ± SEM (n = 4~6 biological replicates). The cell numbers on day 12 were analyzed with One-Way ANOVA followed by the Tukey multiple comparisons test. MX v.s YAP: p = 0.2869 (ns); MX vs. YAPS127A: P = 0.0002 (***); YAP vs. YAPS127A: P = 0.0039. D Overexpression of YAP & YAPS127A stimulates multilayer growth of HK2 cells. Scale Bar: 100 μm. E The anchorage-free growth ability of modified HK2 cells. Left panel: Representative images showing colony formation in HK2 control cells (MX) and HK2 cells expressing YAPS127A, scale bar: 500 μm. Right panel: A violin plot (with minima, maxima, median, and percentile etc.) showing colony numbers formed by HK2-MX, HK2-YAP, and HK2-YAPS127A cells (n = 5 biological replicates). Data were analyzed with One-Way ANOVA followed by the Tukey multiple comparisons test. MX vs YAP: P = 0.0085 (**); MX vs. YAPS127A: P < 0.0001 (****); YAP vs. YAPS127A: P < 0.0001. F A representative image showing the orthotopic inoculation of modified HK2 cells under mouse kidney capsules. G A representative image showing the orthotopic tumor formed by HK2-YAPS127A cells implanted under mouse kidney capsules. H Representative images of H-E staining and immunohistochemistry (IHC) from six biological repeats showing the histological morphology of orthotopic tumors and expression of Ki67, Vimentin, and YAP proteins in orthotopic tumors. IPC: implanted cells, R: renal tissue, scale bar: 50 μm. I Survival rate of mice injected with HK2-MX cells (n = 5, no tumor) and HK2-YAPS127A cells (n = 10, all with metastatic tumor). J Representative H-E and IHC images showing the diverse histology of tumor (two H–E staining images) and expression of Ki67, Vimentin, and YAP1 proteins in tumors (three IHC staining images) derived from mice injected with HK2-YAPS127A cells. Scale bar: 50 μm. Source data for panels (A, C, E, and I) are provided as a Source Data file.
Hyperactivation of YAP1 in renal epithelial cells induces pRCC in vivo
We then generated transgenic mouse models to examine whether the disrupted Hippo pathway induces renal cell carcinoma in vivo. Since PAX8 is predominantly expressed in the renal epithelial cells20,21, we generated a Pax8-rtTA;tet-on-YAPS127A mice with doxycycline (Dox)-inducible, Pax8 promoter-driven expression of YAPS127A. Dox was added to the drinking water (1 mg/ml) to induce transgene expression when the transgenic mice were eight weeks old. After induction for 7–10 days, we observed that the kidney size of the Dox-induced transgenic mice increased 2–3 fold, suggesting that the expression of YAPS127A was successfully induced in renal tissues of these transgenic mice leading to overgrowth of the kidney (Fig. 3A, B). The Dox-induced Pax8-rtTA;tet-on-YAPS127A mice are referred to as Pax8-YAPS127A mice hereinafter. Compared to the control group, the color of the kidney in the Pax8-YAPS127A mice was lighter, potentially due to overgrowth-induced destruction of renal tissues. These mice had to be euthanized within 3 weeks of dox-induction due to poor health. Generally, we found that these transgenic mice were very sensitive to the doxycycline dosage. Mice die within three weeks if the doxycycline concentration in drinking water reaches 100ug/ml. If induced with a low concentration of doxycycline (0.03 mg/ml), these mice could be maintained for several months. By employing an intermittent doxycycline induction, we could successfully maintain mice induced with a relatively higher concentration of Dox (50ug/ml in drinking water) for up to 5–7 months. The color of the enlarged kidney collected from mice with long-term dox induction changed from dark red to grayish brown, with some irregular white intervals observed in the renal tissues (Fig. 3A). Histological analysis confirmed a neoplastic growth of renal epithelial cells and hyperactivation of YAP1 in these cells (Fig. 3C–D). The typical tubular structure is disrupted due to the depolarization and overgrowth of epithelial cells (Fig. 3C). The transformed epithelial cells expressed high levels of cell proliferation marker Ki67 (Fig. 3D). The hyperproliferative cells in the abnormal tissues lack clear cell features, indicated that the neoplasia developed in these transgenic mice are not typical clear cell carcinoma (Fig. 3A, C, D). The high expression of pRCC biomarkers such as Pax8, Vim, Ck7, Birc5, and Mdm2, as well as negative/low expression of Ca9 and Cd10 in the neoplastic/cancer tissues demonstrated that tumors derived from these mice are pRCC (Fig. 3D, Suppl. Fig. S4). This is further confirmed by the development of discrete renal tumors with clear papillary histology in transgenic mice induced with intermittent Dox for more than five months (Fig. 3E). To ensure that the observed neoplastic tissues/tumors originated from YAPS127A-expressing renal epithelium, we implanted kidney tissues from Pax8-rtTA;tet-on-YAPS127A mice to the athymic nude mice. As expected, the addition of Dox to the drinking water of implant-carrying nude mice also induced pRCC (Suppl. Fig. S5).

A Development of neoplasia and cancer in Pax8-YAPS127A transgenic mice. Left panel: Kidney size and morphology of mice with a Pax8 promoter-driven expression of YAPS127A (Pax8-YAPS127A) after inducing with doxycycline for ten days and five months. Age-matched wild-type mice and non-induced transgenic mice, which have similar phenotypes, were used as controls (CTL). Middle and right panels: Longitudinal (middle) and horizontal (right) section images (H-E stained) showing the size of kidneys from CTL and Pax8-YAPS127A mice after inducing gene expression with doxycycline for ten days and five months. Scale bar =1 mm. B Violin plots (with minima, maxima, median, and percentile information) showing changes in kidney weight, body weight, and kidney/body ratio in control (CTL) and Dox-induced (for 10 days or 5 months) Pax8-YAPS127A mice (n = 5 biological replicates for the groups of 10-days study; n = 7~9 biological replicates for the groups of 5-months study). Data were analyzed with unpaired t-tests (two-tailed). The P value of each comparison is presented on the top of the corresponding plot. C Representative H-E staining images from showing the histological morphology of kidneys from control (CTL) and Pax8-YAPS127A mice induced with doxycycline for 10 days and 5 months. The experiments with the similar observations have been independently repeated for more than ten times. Scale bar: 50 μm. D Immunohistochemical images showing expression of YAP1, Ki67, and vimentin in kidney tissues from control (CTL) and Pax8-YAPS127A mice induced with doxycycline for 10 days and 5 months. E H–E staining showing papillary structures derived from the acute Dox-induced neoplastic tissue (E1) and chronic intermittent Dox-induced cancerous tissues (E2–E4). Pax8-rtTA;Tet-on-YAPS127A mice were induced with doxycycline for two weeks (acute) or five months (chronic) before histological analyzes. Red arrows point to papillae observed in several Dox-induced transgenic mice. The experiments with the similar results have been independently repeated for more than ten times. Scale bar: 50 μm. Scale bar: 50 µm. Source data for (B) are provided in the Source Data file.
Single-cell RNA sequencing (SC-RNAseq) reveals the cellular landscape alterations during pRCC development
The above data demonstrated that the Pax8-rtTA;tet-on-YAPS127A mouse model is a powerful tool to uncover cellular and molecular mechanisms underlying pRCC development. Here we combine this unique animal model with SC-RNAseq to determine the alterations in the cellular landscape in the renal epithelium during pRCC initiation and progression. Single cells were isolated from renal tissues of non-induced mice (Control or CTR), mice induced with Dox for seven days (D7), and mice induced with Dox for four months (M4). Altogether, 20,008 cells were analyzed. Based on their molecular features and reported biomarker genes22,23, these cells were classified and annotated into 18 major cell clusters, including proximal convoluted tubule cells (PCT, clusters C1, C3, C4, C6, C15), neoplastic/cancerous cells (cluster C5, C11), distal convoluted tubule cells (DCT, cluster C7), collecting tube cells (CT, cluster C14), endothelial cells (cluster C9), fibroblast (cluster C17), myeloid-derived cells (MDCs, clusters C0, C8, C13, C2), NK cells (cluster C10), B cells (cluster C16), and T cells (cluster C12) (Fig. 4A–B, Suppl. Fig. S6). SC-RNAseq data showed that although Pax8 is expressed in almost all renal epithelial cell sub-populations, Pax8 promoter-driven expression of YAPS127A only induced drastic expansion in clusters C5 and C11. No significant change was observed in C1, C3, C4, C6, and C15 subsets of epithelial cells (Fig. 4C–D, Suppl. Fig. S6). High expression of pRCC biomarker genes (e.g., Krt7, Tert, Vim, Birc5, Pax8, Myc, etc.) in C5 and C11 subpopulations indicated that these are neoplastic/cancerous cells induced by hyperactivated YAP1 and should be the cell-of-origin of pRCC (Fig. 4B, Suppl. Fig. S7). Another major alteration in the cellular landscape of the renal tissue of Pax8-YAPS127A mice is the drastic accumulation of myeloid-derived immune cells (Figs. 4A, C, and Suppl. Fig. S6), which is identified by the high expression of Ptprc, Itgam, Arg1, Vsir, Csf1r, Cx3cr1, and Cd84 genes (Fig. 4B), as well as the high expression of S100a9, Arginase, F4/80, and VISTA proteins (Suppl. Fig. S8, detected by IHC) in these cells. The significant increase in endothelial cell components in the M4 tumors suggested that angiogenesis might be involved in the pRCC progression (Fig. 4C–D).

A UMAP plot showing the overview of 18 major cell types identified in renal tissues in control (CTL, non-induced mice) and Pax8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7) and four months (M4). B Violin plots of representative marker genes across the identified cell clusters. C UMAP plots showing the cellular components of renal tissues in Pax8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7) and four months (M4). D Dynamic alteration of major cell components in renal tissues in control and doxycycline-induced Pax8-rtTA;tet-on-YAPS127A mice during hyperactivated YAP1-induced tumorigenesis. *: significantly different when compared to CTR. Statistical difference were performed with scCODA with FDR = 0.05. Please note that mice in the M4 group were generated using a low dose/chronic induction protocol in order to avoid acute neoplasia-associated kidney failure.
SC-RNAseq uncovers genes and pathways involved in renal epithelial cell carcinogenesis
The gene expression profiling results showed that YAP1 is predominantly expressed in the epithelial cells (Fig. 5A), which is consistent with the GSEA assay demonstrating the enrichment of YAP1 conserved signature in proximal tubal epithelial cells of both D7 and M4 renal tissue (Fig. 5B). Further analyzes demonstrated that among all epithelial cell clusters, conserved YAP1 signature is mainly enriched in C5 and C11 clusters (Fig. 5C). Correspondingly, we found that rapid cell proliferation also predominantly occurred in these two subpopulations (Fig. 5D), leading to the observed neoplasia and subsequent tumorigenesis. Since C5 and C11 subpopulations are present in the normal renal tissue, these cells could be the tumor-initiating cells (TIC) in the kidney. To uncover genes and pathways involved in the YAP1-induced transformation of these critical sub-populations, we examined the differentially expressed genes (DEGs) in control (non-induced) and doxycycline-induced C5 and C11 cells. Compared to the non-induced control, induction of YAP1 expression in C5 epithelial cells with Dox for seven days or four months upregulated genes associated with cell dedifferentiation, extracellular matrix remodeling, tumorigenesis, and myeloid cell recruitment, while down-regulated expression of genes associated with cell differentiation and cell death (Fig. 5E). This observation is further confirmed by the GSEA analyzes which showed that hyperactivation of YAP1 in C5 cells activates signaling pathways that contribute to increased cell stemness, metabolic acidosis, and the development of malignant genitourinary tract tumors (Fig. 5F). A similar pattern of gene expression regulation was observed in C11 cluster of renal epithelial cells (Fig. 5G, H). When compared to other subtypes of epithelial cells, the C5 subtype is featured by signaling pathways associated with immunosuppression, while the C11 subtype is primarily featured by signaling pathways related to cell division. Notably, Our results demonstrated that hyper-activation of YAP1 in C5 and C11 clusters induced enrichment of genes and pathways that are associated with myeloid cell development and migration (Fig. 5E–H), suggesting that hyperactivated YAP1 in epithelial cells may facilitate cancer development by remodeling the extracellular matrix to target the anti-tumor immunity. This speculation was confirmed by the upregulation of many growth factors, cytokines, and chemokines reportedly associated with the remodeling of the extracellular matrix and suppression of anti-tumor immunity in both C5 and C11 sub-populations expressing YAPS127A (Fig. 5I).

A UMAP plots showing the expression of YAP1 in different types of cells in the renal tissues. Please note that YAP1 is primarily expressed in epithelial cells. B GSEA analyzes showing the enrichment of genes associated with YAP1 conserved signature in renal epithelial cells induced with Dox for seven days (D7) or four months (M4). C UMAP plots showing the changes of epithelial cell subpopulations in control (CTR) and YAPS127A-induced (for 7 days and 4 months) renal tissues. Please note that the numbers of cells in cluster 5 (C5) and cluster 11 (C11) are significantly increased after doxycycline induction of transgene expression. D GSEA analyzes showing the enrichment of genes associated with YAP1 conserved signature in C5 and C11 subpopulations after inducing transgene expression with doxycycline for 7 days (D7) or 4 months (M4). E Heatmap demonstrates significantly altered genes in the C5 cluster of proximal tubal epithelial cells after inducing transgene with doxycycline for 7 days (D7) or 4 months (M4). Renal epithelial cells from the non-induced mice were used as control (CTR). The significantly altered genes involved in extracellular matrix reprogramming are listed on the left side of the heatmap. F Gene ontology enrichment analysis showing alteration of signaling pathways involved in the transformation of cluster 5 (C5) cells in D7 early lesions and M4 pRCC tumor tissues. P.adjust is Benjamini-Hochberg adjusted p values. And P values were derived from GSEA permutation based on an adaptive multi-level split Monte-Carlo scheme implemented in fgsea R package. G Heatmap demonstrates significantly altered genes in the C11 cluster of proximal tubal epithelial cells after inducing transgene with doxycycline for 7 days (D7) or 4 months (M4). Renal epithelial cells from the non-induced mice were used as control (CTR). The significantly altered genes involved in extracellular matrix reprogramming are listed on the left side of the heatmap. H Gene ontology enrichment analysis showing alteration of signaling pathways involved in the transformation of cluster 11 (C11) cells in D7 early lesions and M4 pRCC tumor tissues. P.adjust is Benjamini-Hochberg adjusted p values. And P values were derived from GSEA permutation based on an adaptive multi-level split Monte-Carlo scheme implemented in fgsea R package. I Heatmap showing expression of genes encoding for chemokines, cytokines, and growth factors that are associated with MDSC accumulation and immune suppression in C5 and C11 epithelial cells. Please note that mice in the M4 group were generated using a low dose/chronic induction protocol in order to avoid acute neoplasia-associated kidney failure.
SC-RNAseq reveals genes and pathways involved in MDSC accumulation and function
Consistent with the upregulated genes associated with extracellular matrix remodeling and immunosuppression in epithelial cells, we observed a drastic accumulation of myeloid-derived immune cells in YAPS127A-induced neoplastic/cancerous renal tissues (Fig. 4C). DEG analyzes demonstrated that hyperactivation of YAP1 in epithelial cells not only increased myeloid cell population size in renal tissues of dox-induced transgenic mice but also altered their molecular signature. Compared to the control, myeloid cells in renal tissues of the D7 and M4 groups had upregulation of genes associated with extracellular matrix remodeling, immune suppressive cytokines, and tumorigenesis (Fig. 6A, B). These observations were supported by the GSEA analyzes showing that hyperactivation of YAP1 in renal epithelial cells also induced the enrichment of genes and signaling pathways associated with immune suppression and tumorigenesis in myeloid cells of the D7 and M4 groups (Fig. 6C, D). Based on the transcriptomics of individual cells, we performed the graph-based clustering analysis and annotated myeloid cells into M-MDSC (monocytic MDSC, clusters 1, 4, & 5), TRM (tissue-resident macrophage, cluster 2), PMN-MDSC (polymorphonuclear MDSC, cluster 3), Cd209high DC (cDC2, cluster 6), Clec9ahigh DC (cDC1, cluster 8), and Mono-M (monocytic macrophage, cluster 7) (Fig. 6E, F). As shown in the UMAP plots, hyperactivation of YAP1 in renal epithelial cells induced accumulation of immunosuppressive M-MDSC and PMN-MDSCs in the renal neoplastic/cancerous tissues (Figs. 6G, H, and Suppl. Fig. S6), especially in the early stage of tumorigenesis (D7). The drastic accumulation of MDSCs in the early lesions suggests that MDSCs may play a critical role in tumor initiation, potentially by suppressing anti-tumor immunity. This hypothesis is supported by the significantly increased expression of immunosuppressive genes, including Arg1, S100a8, Csf1r, Stat3, Csf2rb, Vsir, Cx3cr1, Wfdc17, and CD84 in MDSCs in the neoplastic/cancerous tissues (Fig. 6I).

A, B Heatmap demonstrates significantly altered genes in myeloid cells in renal tissues of Pax8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7, A) or four months (M4, B). Myeloid cells in renal tissues of non-induced mice were used as control (CTR). Significant genes involved in renal tissue microenvironment reprogramming are listed on the left side of the heatmap. C, D Gene set ontology enrichment analysis showing significantly altered signaling pathways in myeloid cells in renal tissues of Pax8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7, C) or four months (M4, D). Myeloid cells in renal tissues of non-induced mice were used as control (CTR). Representative pathways involved in renal tissue microenvironment reprogramming are listed in the plots. P.adjust is Benjamini-Hochberg adjusted p values. And P values were derived from GSEA permutation based on an adaptive multi-level split Monte-Carlo scheme implemented in fgsea R package. E UMAP projection of eight clusters identified in the myeloid cells (clusters are labeled in UMAP). F Violin plots of representative marker genes across identified myeloid cell clusters. G UMAP plots showing changes in myeloid cell subpopulations in renal tissues of Pax8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7) and four months (M4). Renal tissues from non-induced mice were used as a control (CTR). Please note the drastic accumulation of myeloid-derived suppressor cells (MDSCs) in the renal tissue of Pax8-YAPS127A mice. H Dynamic change of myeloid cell subpopulations in renal tissue of PAX8-rtTA;Tet-on-YAPS127A mice induced with doxycycline for seven days (D7) and four months (M4). Renal tissues from non-induced mice were used as a control (CTR). *: significantly different when compared to CTR. Statistical difference were performed with scCODA with FDR = 0.05. Please note that the vast majority of increased myeloid cells are MDSCs. I) UMAP plots showing high expression of key genes associated with MDSC functionality in renal tissues of Pax8-YAPS127A mice induced with doxycycline for seven days (D7) and four months (M4). Renal tissues from non-induced mice were used as a control (CTR). Please note that mice in the M4 group were generated using a low dose/chronic induction protocol in order to avoid acute neoplasia-associated kidney failure.
To verify the clinical relevance of findings from our animal models, we downloaded and reanalyzed the human pRCC single-cell RNA sequencing data [GSE152938]. Intriguingly, we found that YAP1 signaling is highly activated in human pRCC patient samples (Suppl. Figure S9) and MDSC was also accumulated in human pRCC tissues (Suppl. Figure S10). These findings verified the clinical relevance of our animal models.
To further confirm the contribution of MDSC to pRCC initiation, we depleted MDSC in Pax8-YAPS127A mice using a clodronate-based cell depletion protocol24,25,26. We found that clodronate treatment can efficiently delete MDSC, which was indicated by the significant reduction in YAPS127A-induced accumulation of Cd11b + ;Gr1+ cells in renal tissues of Pax8-YAPS127A mice (Fig. 7A). Importantly, we found that the decrease of MDSC population was associated with the restoration of kidney size and renal tubal structure (Fig. 7B, C). Clodronate treatment had minimal impact on the body weight of treated mice, suggesting that the dosage of clodronate was well tolerated (Fig. 7C). The IHC analyzes demonstrated that clodronate treatment significantly reduced YAPS127A-induced expression of CD45 and F4/80 in neoplastic renal tissues (Fig. 7D). Depletion of MDSC eradicated YAPS127A-induced neoplasia, suggesting that YAPS127A-associated MDSC accumulation is essential for pRCC initiation.

A FACS showing the effect of clodronate (CLD) on the accumulation of MDSCs in the renal tissues of doxycycline-induced Pax8-rtTA;tet-YAPS127A mice. Mice were treated with Dox ± CLD for 2 weeks before FACS analysis. Please note that the administration of CLD significantly reduced MDSCs in the neoplastic renal tissues. The violin plot (with minima, maxima, center, and percentile, etc.) on the right shows changes in MDSC ratios (n = 4-5 biological replicates). Data were analyzed with One-Way ANOVA followed by the Tukey multiple comparisons test. PBS + H2O vs. PBS + DOX: P < 0.0001 (****); PBS + DOX vs. CLD + DOX: P = 0.0081 (**). B Morphology and histology of kidneys in Pax8-rtTA;tet-YAPS127A mice treated with or without doxycycline (Dox) and clodronate (CLD) for 2 weeks. Scale bar: 25 µm. C Violin plots (with minima, maxima, median, and percentile, etc.) showing the effect of CLD on the body, kidney, and spleen weights of the Pax8-rtTA;tet-YAPS127A mice treated with or without Dox for 2 weeks (n = 5 biological replicates). Data were analyzed with One-Way ANOVA followed by the Tukey multiple comparisons test. Statistics for the kidney weight: PBS + H2O vs. PBS + DOX: P < 0.0001 (****); PBS + DOX vs. CLD + DOX: P = 0.0001 (***). Statistics for the kidney somatic index: PBS + H2O vs. PBS + DOX: P < 0.0001 (****); PBS + DOX vs. CLD + DOX: P = 0.0148 (*).D) Representative images (from six biological repeats) showing the effect of CLD on the expression of CD45 (pan-immune cell marker) and F4/80 (macrophage marker) in renal tissues of the Pax8-rtTA;tet-YAPS127A mice treated with or without Dox for 2 weeks. Scale bar: 25 µm. Source data for (A and C) are provided in the Source Data file.
The availability of differentially-expressed genes encoding ligands and receptors in different types of cells provided an opportunity to understand the context-dependent communication between neoplastic/cancerous cells and MDSCs using CellPhoneDB, a repository of ligands, receptors and their interactions27. As expected, the inferred results demonstrated that the ligands derived from neoplastic/cancer cells and the receptors expressed in MDSCs contributed to the majority of cell-cell communications, which predominately occurred in the D7 and M4 groups (suppl. Figure S11). This observation is further confirmed by data visualized using LIANA, another ligand-receptor analysis framework28, which also showed that crosstalk involving Cxcl and Ccl family and Csf cytokines between C5/C11 clusters and MDSCs predominantly occurred in neoplastic/cancerous tissues (suppl. Figure S12). Consistently, we found that inferred interactions between MDSCs-derived ligands (chemokines of Cxcl and Ccl subfamilies and Csf cytokines) and neoplastic/cancer cells-expressed receptors are less active in neoplastic/cancerous tissues during pRCC development (suppl. Figure S13). It seems that interactions between ligands derived from neoplastic cells and receptors expressed in MDSCs predominates the communications between neoplastic cells and MDSCs during pRCC initiation and progression.
YAP1-induced MDSCs impair T cell activities during pRCC development
T cells play a central role in adaptive immunity and immune surveillance against cancer. MDSCs facilitate tumorigenesis by creating an immunosuppressive microenvironment capable of interrupting the anti-tumor immunity of T cells29,30,31. We reasoned that YAP1-induced MDSC accumulation may impact T cell function, so we examined markers of T cell differentiation and function. Based on transcriptomics of individual cells, T cells in the control and doxycycline-induced renal tissues could be classified into CD8+ effector/memory T cells (CD8_Teff/mem), CD8+ exhaust T cells (Tex, including pro-exhaust T cells), CD8+ Early active T cells (CD8_Early Active), CD8+ Naïve like T cells (CD8_Naivelike), CD4+ Naïve T cells (CD4_Naivelike), follicular Helper T cells (Tfh), type 1 helper T cells (Th1), and Regulatory T cells (Treg) (Fig. 8A). Consistent with YAP1-induced accumulation of immunosuppressive MDSCs, hyperactivation of YAP1 in renal epithelial cells resulted in a significant decrease in CD8_Teff/mem population but drastic increases in the number of CD8_Tex, CD8_Tpex (pro-exhaust T cells), and Treg cells (Fig. 8B). Hyperactivated YAP1 in renal cells also induced increases in CD4_Naivelike, Th1 and Tfh populations and reduced the number of CD8_Naive-like cells in neoplastic and cancerous tissues (Fig. 8B). Moreover, hyperactivated YAP1 in renal epithelial cells induced significant increase in the expression of immunosuppressive receptors such as Ctla4, Pd-1 (Pdcd1), and Il10rb (Fig. 7C), and upregulated cell cycle arrest-associated genes such as Trp53, cdkn1a, and Cdkn2a in T cells (Fig. 8D). Consistent with the upregulated expression of immunosuppressive receptor genes, transcription factors controlling the expression of these receptor genes such as Nr4a1, Nr4a2, Nr4a3, Nfat1, Nfat2, Nfat5, Tox, Eomes, and Batf in neoplastic/cancerous tissues were significantly higher than that of the non-induced control tissues (Fig. 8E). In addition, the hyperactivated YAP1 in renal epithelial cells induced upregulation of inhibitory receptor (e.g., Ctla4, Pdcd1), enzymes (e.g., Gzmb), and cytokines (e.g., Tgfb1, Il10) in Treg and Tfh cells (Fig. 8F, G). The clinical relevance of these findings is supported by the observation that YAP1 expression is significantly correlated with the expression of T cell suppressive genes and exhaustion markers such as PD-L1 (CD274), AGR2, TOX, TOX2, and VISTA (VISR) in human pRCC32 (suppl. Figure S14).

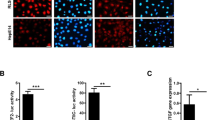
A Radar plots showing the functional clusters of T cells in renal tissue with or without induction with doxycycline. The molecular features of reference cells are labeled in black. Molecular features of T cell clusters in our control and transgenic animal models are labeled in red. CD8_Teff/mem: CD8+ effector/memory T cells; Tex: CD8+ exhaust T cells; Tpex: CD8+ pro-exhaust T cells; CD8_Early Activ: CD8+ Early active T cells; CD8_Naivelike: CD8+ Naïve like T cells; CD4_Naivelike: CD4+ Naïve-like T cells; Tfh: follicular Helper T cells; Th1: type 1 helper T cells; Treg: Regulatory T cells. B Dynamic alternation of T cell subpopulations in control (CTR), early renal epithelial neoplastic lesions (D7), and pRCC (M4). *: significantly different when compared to CTR. Statistical difference were performed with scCODA with FDR = 0.05. a: p = 0.064818. C–G Notched Box plots (with minima, maxima, center, and percentile, etc.) were generated at single-cell level to demonstrate the upregulation of cell surface immune suppressive molecules in Tex cells (C), apoptosis-associated molecules in tumor-infiltrating T lymphocytes (D), alterations of the intrinsic transcription factors that drive the expression of cell surface immune suppressive molecules (E), upregulation of cell surface immune suppressive molecules in Tfh cells (F), and upregulation of molecules associated with Treg suppression of T effectors (G). CTR: n = 95; D7: n = 199; M4: n = 260. Please note that mice in the M4 group were generated using a low dose/chronic induction protocol in order to avoid acute neoplasia-associated kidney failure. H FACS analyzes showing the impact of MDSC on T cell expansion. T cells were activated with a combination of IL2 (10 ng/ml), anti-CD3 (α-CD3, 1 ug/ml), and anti-CD28 (α-CD28, 1 ug/ml). T Cell expansion in the co-culture was determined by a carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution assay. Representative FACS graphs showing the expansion of Cd4+ T cells (left panel) and Cd8+ T cells (right panel) (both isolated from mouse spleen) co-cultured with or without 1:1 MDSCs (isolated/purified from Pax8-YAPS127A mice) for 72 h. The same amount of bone marrow cells (BMC) was used as negative control. “Non-stimulated”: T cells without stimulation/activation (negative control). “MDSC (or BMC): T = 1: 1”: MDSCs (or BMCs) and activated T cells were cocultured with a ratio of 1:1. Please note that addition of MDSCs, not BMCs, blocked the expansion of activated Cd4+ and Cd8+ T cells in the co-culture system.
An MDSC and T cell co-culture system was established to provide direct evidence that the accumulated MDSCs contribute to the immune-suppressive microenvironment. MDSCs were isolated from the neoplastic renal tissues of Pax8-YAPS127A mice using an EasySep™ Mouse MDSC (CD11b + Gr1 + ) Isolation Kit (suppl. Figure S15). Cd4+ and Cd8+ T cells were isolated from spleens of normal control mice and activated using a well-established IL2/α-CD3/α-CD28 protocol33,34. Myeloid cells isolated from bone marrow of wild-type mice (BMC) were also used as negative control cells. We found MDSC, not BMC, could block the expansion of stimulated Cd4+ and Cd8+ T cells in the co-culture system (Fig. 8H). The suppressive effect of the isolated MDSC on T cell expansion was repeated using an Incucyte® Live Cell imaging and Analysis system (data no shown). These observations clearly suggested that YAPS127A-induced accumulation of MDSC in renal tissue contributes significantly to the suppression of anti-tumor immunity during pRCC initiation.
Targeting YAP1 blocked MDSC accumulation and improved the pRCC survival rate
The above evidence suggests that the disrupted Hippo signaling pathway in epithelial cells drives pRCC development by inducing malignant transformation of epithelial cells and recruiting MDSCs, which may enable the transformed cells to evade anti-tumor immunosurveillance. Since TEA domain transcription factors (TEADs) are the key transcription factors that mediate YAP1 activity, we hypothesized that targeting the YAP1-TEADs interaction would prevent pRCC initiation and progression. To test our hypothesis, we examined whether MGH-CP1, a potent and orally active TEAD2/4 inhibitor developed recently by one of our research labs35, could prevent pRCC development. In the cultured pRCC ACHN cells, MGH-CP1 suppressed cell proliferation in a concentration-dependent manner (Suppl. Fig. S16). In the transgenic mice, administration of MGH-CP1 successfully blocked YAP1-induced renal tissue neoplasia, as indicated by the normalization of kidney size and weight ten days post doxycycline induction (Fig. 9A–D). MGH-CP1 treatment also significantly increased the survival rate of transgenic mice (Fig. 9E, P < 0.05, compared to vehicle-treated Pax8-YAPS127A mice), and partially recovered hyperactivated YAP1-induced damage of renal tissues (Fig. 9F). Intriguingly, MGH-CP1 treatment successfully blocked YAP1-induced accumulation of MDSCs in doxycycline-induced transgenic mice (Fig. 9G). Of note, treatment with MGH-CP1 at a dosage of 75 mg/kg did not cause a significant change in animal body weight (Suppl. Fig. S17A) and had no adverse effects on the histological morphology of major organs such as heart, lung, liver, pancreas, spleen, intestine, and testis (Suppl. Fig. S17B).

A Structure of the MGH-CP1 molecule. B Kidney size and morphology of control mice (Ctrl), Pax8-YAPS127A mice (YAP, Dox-induced Pax8-rtTA;Tet-on-YAPS127A mice), and Pax8-YAPS127A mice treated with MGH-CP1 (YAP + MGH-CP1). Ruler scale: 1 mm. C Changes of body weight in control mice (Ctrl), Pax8-YAPS127A mice (YAP), and MGH-CP1 treated Pax8-YAPS127A mice (YAP + MGH-CP1). Data were represented as the mean ± SEM (Ctrl: n = 10; YAP: n = 12; YAP + MGH-CP1: n = 18 biological replicates). Data were analyzed with One-Way ANOVA followed by the Tukey multiple comparisons test. Statistics for the kidney weight: CTL vs. YAP: P < 0.0001 (****); YAP vs. YAP + MGH-CP1: P < 0.0001 (****). Statistics for the kidney/body ratio (%): CTL vs. YAP: P < 0.0001 (****); YAP vs. YAP + MGH-CP1: P = 0.0001 (***).D Representative H-E staining images showing changes in kidney size and histological morphology in control mice (Ctrl), Pax8-YAPS127A mice (YAP), and MGH-CP1 treated Pax8-YAPS127A mice (YAP + MGH-CP1). E Survival curves of control mice (Ctrl), Dox-induced Pax8-YAPS127A mice (YAP), and Pax8-YAPS127A mice treated with MGH-CP1 (YAP + MGH-CP1). F representative H-E staining images showing renal tissue histology of control mice (Ctrl), Pax8-YAPS127A mice (YAP), and MGH-CP1 treated Pax8-YAPS127A mice (YAP + MGH-CP1). The experiments with the similar results have been independently repeated for more than ten times. Scale bar: 50μm. G IHC images showing the expression of YAP1, CD45, and MDSC biomarkers (Arginase 1, F4/80, and VISTA) in renal tissues from control mice (Ctrl, n = 6), Pax8-YAPS127A mice (YAP, n = 6), and MGH-CP1 treated Pax8-YAPS127A mice (YAP + MGH-CP1, n = 6). Source data for panels C and E are provided in the Source Data file.
Discussion
Previous studies, including ours, provided convincing evidence that disrupting the Hippo/YAP signaling pathway contributes to the carcinogenesis of fallopian tube epithelial cells and the development of high-grade serous ovarian carcinoma (HGSOC)36,37,38,39,40. Since the fallopian tube epithelial cell (oviduct epithelial cell in mice) is recognized as a cell-of-origin of HGSOC, and Pax8 promoter-driven deletion of Pten and Brca1 genes in mouse oviduct epithelial cells results in HGSOC41,42, we anticipated that Pax8 promoter-driven hyperactivation of YAP1 in mouse oviduct epithelial cells would induce HGSOC. Unexpectedly, we found Pax8 promoter-driven hyperactivation of YAP1 in mice induced neoplasia of renal epithelium within seven days. Our findings clearly demonstrate that constitutive activation of YAP1 in renal epithelial cells resulted in the carcinogenesis of renal epithelium and the development of pRCC.
pRCC is a malignant renal parenchymal tumor and accounts for ~15% of renal cell carcinoma, which is among the few cancers whose incidence consistently increases in all ages and genders43,44. Since most renal cancer research focuses on renal clear cell carcinoma (ccRCC), the mechanisms underlying pRCC initiation and progression remain largely unidentified. Previous studies demonstrated that mutations in MET, FH (fumarate hydratase), and TP53 are associated with pRCC15. However, these mutations are relatively rare in pRCC (8% for MET, 0.7% for FH, and 2.5% for TP53). The exact contribution of these mutations to pRCC initiation and progression is unclear. Although increased oxidative stress and activation of the NRF2-antioxidant response element (ARE) pathway are frequently observed in more aggressive type 2 tumors, their contributions to pRCC development still need to be confirmed15,45. The results derived from our transgenic mouse model demonstrated that hyperactivation of YAP1 in renal epithelial cells induced renal neoplasia within 1-2 weeks and pRCC within four months, providing convincing evidence that the disrupted Hippo/YAP signaling pathway plays a key role in the pRCC initiation and progression. Consistent with findings from our animal model, several previous studies using high-throughput analyzes have demonstrated that the Hippo pathway may be involved in human pRCC development46,47. These findings didn’t attract attention because they only identified low-frequent deep deletion and mutation of genes encoding several key components of the Hippo signaling pathway in pRCC patient samples. Data presented in the present study demonstrated that deletion of the Hippo pathway tumor suppressors, which results in hyperactivation of YAP1, is a high-frequency event in human pRCC. For example, NF2, a major upstream tumor suppressor of the hippo pathway and recently recognized pRCC tumor suppressor48,49, is deleted in 30% of pRCC patients (4% deep deletion, 27% shallow deletion). Similarly, YWHAH, a major YAP1 oncogene inactivator, is deleted in 33% of pRCC patients (3% deep deletion, 30% shallow deletion). It seems that human pRCC is featured by shallow deletions of the tumor suppressors of the Hippo signaling pathway (Fig. 1A). Although shallow deletions may not cause a complete loss of an upstream tumor suppressor of the Hippo pathway, the observed concurrent shallow deletions may result in additive effects leading to YAP/TAZ hyperactivation. Our findings in this study identify the shallow deletion-induced disruption of the Hippo signaling as a potential key contributor to pRCC development. Nevertheless, the factor(s) leading to the deletion of the Hippo tumor suppressors need further investigation.
Currently, the cell-of-origin of pRCC is not fully understood. Previous studies indicated that pRCC might originate from proximal tube epithelial cells50,51. Evidence from the acute kidney injury (AKI) model demonstrated that pRCC developed from progenitor cells of the tubular epithelium52. In addition, a more recent study based on the genome-wide chromatin accessibility profiling data indicated that a small portion of pRCC may be derived from collecting duct principal cells53. Our inducible pRCC transgenic animal model provides a powerful tool for monitoring the initiation of pRCC from the very beginning. Results from this unique model demonstrate that although YAP1 was highly expressed in almost all renal epithelial cells, hyperactivated YAP1 only induced overgrowth in C5 and C11 subpopulations, suggesting that these cells are potential cell-of-origin of pRCC. Supporting this notion, we observed that these two subpopulations are featured by high expression of Krt7, Tert, Vim, and Birc5, which are known pRCC biomarkers. The SC-RNAseq analyzes also demonstrated that these two subpopulations possess the molecular signature of malignant cells. Identifying cellular orthologs in the benign tissues that share transcriptional signatures with the tumor cells indicated that these cells are potential cell-of-origins of this subtype of pRCC. Further research is warranted to confirm the clinical significance of these findings and their potential applications in pRCC prevention and early detection.
Under physiological conditions, the immune system can effectively identify and eliminate pre-cancerous and cancerous cells before they cause harm54,55. However, cancer cells developed strategies to evade immune surveillance54,55,56. Understanding the cellular and molecular mechanisms by which cancer cells evade immune surveillance is a prerequisite for improving cancer prevention, early detection, and therapy. Results from our preclinical animal model demonstrated that the Hippo pathway disruption and the subsequent YAP1 hyperactivation contribute significantly to pRCC immunosurveillance evasion. Data derived from our SC-RNAseq analyzes demonstrated that hyperactivated YAP1 not only induced hyperproliferation of C5 and C11 epithelial subpopulations but also stimulated upregulation of genes encoding growth factors, cytokines, and chemokines associated with immunosuppression. Of note, hyperactivated YAP1 in C5 and C11 epithelial cells induced overexpression of CSF1, CSF2, and CSF3 and many other genes associated with the recruitment, expansion, and activation of MDSCs (e.g., Cxcl1, Ccl2, Il6, S100a6, S100a11, Fst, Il34, Vegf)29,30. Correspondingly, MDSCs in the YAP1-induced neoplastic/cancerous tissues expressed high levels of cytokine receptor molecules, interacting proteins, and many immunosuppressive molecules (e.g., Agr1, Stat3, S100a8, Vsir, Apoe, Cd84, and Wfdc17, etc.) to mediate the immunosuppressing signals from the neoplastic/cancerous epithelial cells. MDSC depletion blocked renal neoplasia and pRCC development, suggesting that MDSC accumulation is an essential step for pRCC initiation.
Previous studies also indicated that MDSCs serve as a competent accomplice to drive tumorigenesis in many different tissues29,31,57,58. T cells play a central role in tumor immunosurveillance and are the primary target of MDSCs29,59. This is confirmed by our coculture study which demonstrated that MDSCs isolated from YAP1-induced neoplastic renal tissues strongly suppressed T cell expansion. Activated MDSCs employ multiple cellular and molecular mechanisms to inhibit proliferation, promote apoptosis, block trafficking, and induce dysfunction of T cells29,59,60. In our preclinical model, we found that compared to that of the control group, the population size of effector and memory T cells significantly decreased, whereas the number of exhaust T cells significantly increased in the neoplastic/cancerous renal tissues. Increased expression of Trp53, Cdkn1a, and cdkn2a in the exhaust T cells demonstrated the activation of apoptotic signaling pathways in these cells. Overexpression of transcription factors (Nr4a1/2/3, Nfat1/2/5, Tox, and Batf, etc.) that govern the expression of immune checkpoint molecules and the consistent upregulation of Ctla4 and Pdcd1 in exhaust T cells of the YAP1-hyperactivated renal tissues suggested that YAP1-induced neoplastic/cancer cells interact with MDSCs to induce imbalanced immune checkpoint system causing dysfunction of cytotoxic T cells and thereby facilitating neoplastic/cancer cells to evade T cell-mediated immune surveillance. Co-overexpression of Arg1, Stat3, and S100a8 in MDSCs suggests that multiple molecular mechanisms are involved in MDSC-induced T cell dysfunction. Therefore, recruiting MDSC is one of the most important mechanisms by which YAP1-induced neoplastic/cancerous cells evade immunosurveillance. These findings uncover previously unprecedented cellular mechanisms underlying pRCC development and identify MDSCs as a cellular target for pRCC prevention and treatment.
Except for the MDSCs, hyperactivation of YAP1 in epithelial cells also induced a significant increase in the regulatory T cells, which promote tumorigenesis by hindering protective immunosurveillance of neoplasia and hampering anti-tumor immune response61,62. Moreover, the direct interaction between neoplastic/cancerous cells with MDSC or T cells may also play a critical role in suppressing anti-tumor immunity. For example, both C5 and C11 subpopulations have high expression of cytokines and chemokines such as Csf1/2/3, Ccl2, Cxcl1, Apoe, Lgals3, S100a8, S100a9, Cx3cl1, Tnf, Il10, allowing for a direct interaction between neoplastic/cancerous cells and cytotoxic T cells29,30,63. In addition, the high expression of Glb1, Trp53, and Cdkn1a in the exhaust T cells suggests that cancer cells may also utilize cellular senescence as a mechanism to suppress anti-tumor immunity during YAP1-induced pRCC development. Clearly, the disrupted Hippo signaling in renal epithelial cells employs multiple mechanisms to suppress anti-tumor immunity during pRCC development. More investigations are needed to uncover the exact role of disrupted Hippo signaling in tumorigenesis and immunosuppression.
Due to the lack of an agreed-upon treatment regimen for this disease, pRCC treatment is still a challenge to clinicians. Current targeted therapies for metastatic RCCs include mTOR and VEGF inhibitors. However, the response rate of pRCC to these modalities is lower than that of ccRCC. Based on the finding that a mutation in MET is associated with pRCC progression15, MET inhibitors were used to treat pRCC. The clinical trials showed that these inhibitors could improve the prognosis of metastatic pRCC17,64. Nevertheless, the response rate of pRCC to MET inhibitor is relatively low, possibly due to the relatively low mutation rate of MET in this type of cancer. In fact, the targeted therapy for pRCC in the past has been unsuccessful, mainly due to the fact that we do not have a clear understanding of the molecular basis of pRCC. In this study, evidence derived from our in vitro and in vivo models indicates that the disrupted Hippo/YAP signaling pathway drives pRCC initiation and progression via remodeling the cellular landscape of renal epithelium. Based on these findings, we used a recently developed drug, MGH-CP135, to target the constitutive activation of YAP1. As expected, MGH-CP1 almost eradicated YAP1-induced accumulation of MDSC and successfully blocked hyperactivated YAP1-induced renal cell epithelial neoplasia in our preclinical transgenic animal model. Importantly, our preliminary toxicity studies demonstrated that MGH-CP1 is well tolerated. These results suggested that targeting the disrupted Hippo/YAP signaling pathway represents a plausible strategy to prevent and treat pRCC, and MGH-CP1 could be used as a potential drug for pRCC prevention and therapy.
A limitation of the present study is that we do not have human pRCC early lesion samples to verify the relationship between the disrupted Hippo/YAP signaling pathway and pRCC initiation in human kidneys. However, data extracted from human pRCC datasets support our conclusion that YAP1-induced MDSC accumulation plays a critical role in pRCC initiation and progression. Future clinical trials based on findings derived from our animal models are expected to provide direct evidence that targeting the disrupted Hippo/YAP signaling has the potential to improve pRCC prevention and treatment.
In summary, we established a pRCC transgenic animal model which is expected to significantly contribute to pRCC etiology study and drug screening. Results derived from this preclinical model identify the disrupted Hippo/YAP signaling as a key driver of pRCC, uncover the cellular and molecular mechanisms underlying pRCC immunosurveillance evasion, and confirm the prevention and therapeutic potential of targeting the disrupted Hippo pathway in pRCC.
Methods
All experimental procedures were performed in accordance with the regulatory requirements, sponsor and institutional policies. No human subjects were involved in this study. Animal-associated studies (procedures for mouse handling, breeding, and treatment) were approved by the Institutional Animal Care and Use Committee (IACUC) of the University at Nebraska Medical Center (UNMC) and Massachusetts General Hospital (MGH).
Human renal cancer genetic/genomic data and analyzes
All patient genetic/genomic data were extracted from the Cancer Genome Atlas (TCGA) RCC datasets14. The deep sequencing data were retrieved from TCGA Kidney Renal Papillary Cell Carcinoma (firehose Legacy) studies (n = 293). The genetic/genomic alternations of the Hippo/YAP signaling pathway in renal cell carcinoma (RCC) were analyzed using the cBioPortal for Cancer Genomics online tools (available at http://www.cbioportal.org). Kaplan-Meier plot analysis was performed to examine the relationships between genetic/genomic alterations of the Hippo signaling and pRCC patient survival rate.
Establishing cell lines with distinct YAP expression and activation
Three cell lines derived from HK2 cells with differential YAP expression and activation were established to determine the effect of hyperactivated YAP1 in renal cancer development using a previously described protocol65. HK2 is an immortalized, non-tumorigenic renal epithelial cell line. Briefly, HK2 was cultured to 40% confluence in 6-well plates and then infected with retroviral vectors expressing wild-type YAP1 (YAP) or constitutively active form of YAP1 (YAPS127A, the serine to alanine mutation at residue 127 prevents phosphorylation of YAP1 at serine 127 leading to its constitutive activation). HK2 cells transfected with an empty MXIV vector (MX) were used as a negative control. Transfected cells were selected using G418 for positive infection.
Cell proliferation and soft agar colony formation
Cell proliferation was examined by counting cell numbers using an Invitrogen Countess Automated cell counter (Invitrogen, Carlsbad, CA) and MTT assay66. The anchorage-free cell growth of established cell lines was determined by the soft agar cell colony formation assay using a protocol previously established in our lab67.
Establishing xenograft and orthotopic tumor mouse models
Xenograft mouse tumor models were used to examine the tumorigenic potential of the established cell lines. Animal-associated studies (procedures for mouse handling, breeding, and treatment) were approved by the Institutional Animal Care and Use Committees (IACUC) at UNMC and MGH. Mice were housed in the comparative medicine core facilities. The room temperature was 23 ± 2 °C and the relative humidity varied from 65% to 70%. The professional staff of specially trained veterinarians, animal care managers, technicians, and husbandry personnel provide care for these animals. Each cage houses less than five adult mice. Mice received a 12 hr light/dark cycle and were provided water and mice chow ad libitum. To establish xenograft and orthotopic tumor models, HK2-MX or HK2-YAPS127A cells (5×106 cells in 0.1 mL sterile PBS) were injected subcutaneously into the left (HK2-MX) or right (HK2-YAPS127A) dorsal flank of 6-week-old athymic nude mice (The Jackson Laboratory, ME). Tumor formation was monitored daily. Based on the approved IACUC protocol, mice will be euthanized if the diameter of tumor rich 1.5 cm in any direction. Mice were humanely euthanized and recorded as dead once facility veterinarians provided evidence of serious illnesses (dysuria, wasting, or sluggishness) and incurableness. Tumor tissues were harvested and processed for molecular and pathohistological analyzes39.
To establish an orthotopic kidney tumor model, HK2-MX and HK2-YAPS127A cells (1x106 cells in 20 µl sterile PBS mixed with 20 µl in vivo high concentration Matrigel) were injected into the kidney capsule of 6-week-old athymic nude mice. Mice were monitored daily and euthanized every three months to examine tumorigenesis. Kidneys from each group were harvested for pathohistological analyzes.
Genetically modified mouse models
Since PAX8 is a nephric-lineage transcription factor required for the establishment of renal lineage cells20,21, the Pax8-rtTA mouse strain (The Jackson laboratory, Strain #:007176), which expresses the reverse tetracycline-controlled trans-activator (rtTA) protein in kidney tubule epithelial cells under the control of Pax8 promoter, was used to generate a renal epithelial cell-specific expression of the transgene68. The TRE-YAPS127A mouse strain, which expresses a constitutively active form of YAP1 protein (YAPS127A) under the control of a tetracycline regulatory element (TRE)69, was from Dr. Fernando Camargo’s lab at Boston Children’s Hospital. Pax8-rtTA mice were bred to the TRE-YAPS127A mice to produce Pax8-rtTA;TRE-YAPS127A mice (also referred to as Pax8-rtTA;tet-on-YAPS127A mice), which specifically express YAPS127A in renal epithelial cells upon tetracycline (e.g., doxycycline) induction. Genotyping was performed via RT-PCR using mouse tail tissue. Primers for genotyping and the size of PCR products are listed in Table S1. Based on our preliminary studies, the regular dose of doxycycline (1.0 mg/mL) was used to induce YAPS127A in short-term studies (7 days or 10 days, abbreviated as D7 or D10 in figures), whereas the low dose of doxycycline (0.03 - 0.05 mg/mL) was used for long-term treatment (4 months or 7 months, abbreviated as M4 or M7 in figures). Doxycycline was generally added to the drinking water of Pax8-rtTA;tet-on-YAPS127A mice when they were eight weeks or older. Doxycycline-induced Pax8-rtTA;tet-on-YAPS127A mice have Pax8 promoter-driven expression of YAPS127A (hereinafter referred to as Pax8-YAPS127A mice). Age-matched non-induced Pax8-rtTA;tet-on-YAPS127A mice and Doxycycline-induced Pax8-rtTA mice were used as controls (CTR in figures). Kidneys in each group were collected, weighed, and processed for histological and biochemical analyzes. Since neoplasia developed in 100% testing mice (regardless of sex) after doxycycline induction, male and female mice were randomly allocated into experimental groups.
The orthotopic kidney tumor model derived from Pax8-rtTA;tet-on-YAPS127A strain was established by transplanting a small piece of kidney tissue (1 mm3) from Pax8-rtTA;tet-on-YAPS127A model into the kidney capsule of athymic nude mice under anesthetization. This design can ensure that tumors were derived from PAX8-induced transgene in the renal tissues. Doxycycline was added to the drinking water of implant-carrying nude mice to induce the expression of transgene. Mice were monitored daily and euthanized every month to examine tumorigenesis. Animal procedures in the in vivo studies have been approved by the Institutional Animal Care and Use Committees (IACUC) at UNMC and MGH.
Immunohistochemistry, Western blot, and quantitative PCR
Immunohistochemistry was used to examine the expression of YAP1, pRCC biomarkers, and immune cell signature molecules in normal and neoplastic/cancerous renal tissues using an established protocol70,71,72. Antibodies used in immunohistochemistry are listed in Table S2. The immunosignals were visualized using a polymer-based immunohistochemistry kit and a DAB assay kit (Vector Laboratories, Burlingame, CA)70,71,72. Stained slides were imaged with an Iscan Coreo Slide Scanner (Ventana Medical Systems, Inc., AZ).
Immunofluorescent staining was used to detect YAP1 expression in established cell lines using a protocol reported previously39,73. Briefly, cells cultured on chamber slides (70% confluent) were fixed in 4% paraformaldehyde, blocked with 10% normal donkey serum, and probed with YAP1 antibody at 4 °C for 16 hours, visualized with fluorescent 2nd antibody and DAPI (for nuclear staining), and imaged under a Zeiss 710 Meta Confocal Laser Scanning Microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY).
Western blot was used to determine the relative expression or phosphorylation levels of proteins. Protein extraction, quantification, and denaturation were performed using a standard protocol established in our lab36,37,71. Denatured protein samples were fractioned by 10% SDS-PAGE gels, transferred onto PVDF membranes, blocked with 5% BSA-TBST, and probed with appropriate primary and secondary antibodies. The immunosignal was detected using a Thermo Scientific SuperSignal West Femto Chemiluminescent Substrate Kit. The images were captured by a UVP gel documentation system (UVP, Upland, CA).
Quantitative PCR was used to determine the relative expression levels of mRNAs. Briefly, RNA was extracted using the TRIzol reagent (Invitrogen, CA) and a QIAGEN RNeasy mini kit (QIAGEN, CA) and quantified with a Nanodrop-based protocol. cDNA was prepared using an iScript™ Reverse Transcription Supermix protocol (BIO-RAD, Hercules, CA). PCR analysis was performed in a BIO-RAD CFX96 Real-time fast PCR system (BIO-RAD, Hercules, CA) using an iTaq™ Universal SYBR® Green Supermix kit (BIO-RAD, Hercules, CA).
Evaluating the role of MDSCs on pRCC initiation
The role of MDSC in pRCC initiation was evaluated by in vivo MDSC depletion. Clodronate-liposome, which induces macrophage apoptosis25,26, was injected into the Dox-induced Pax8-YAPS127A mice to deplete macrophages (including MDSC) following a well-established protocol24. Kidneys were harvested from control and treated groups two weeks after vehicle or chlodronate injection, and processed for morphology, histology, and FACS analyzes.
Isolated MDSCs from the neoplastic renal tissues were also co-cultured with T cells to evaluate their roles in creating an immune suppressive environment. MDSC were isolated with a mouse tumor cell isolation kit (Miltenyi Biotec, Auburn, CA) and purified using an EasySep™ Mouse MDSC (CD11b+Gr1+) Isolation Kit (STEMCELL Technologies Inc, Cambridge, MA). T cells were isolated using an EasySep™ Mouse T Cell Isolation Kit (STEMCELL Technologies Inc, Cambridge, MA), and stimulated with a combination of IL2 (10 ng/mL), anti-CD3 (1.0 µg/mL), and anti-Cd28 (1.0 µg/mL)34. Myeloid cells isolated from bone marrow (BMC) were used as negative control cells. The alteration of cell population sizes was evaluated using a carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution assay.
Targeting hyperactivated YAP1 to prevent MDSC accumulation and pRCC development
We use MGH-CP1, a potent orally active TEAD2/4 inhibitor developed recently by one of our research labs35, to determine whether targeting hyperactivated YAP1 has the potential to block MDSC accumulation and pRCC development. Eight-week-old Pax8-rtTA;tet-on-YAPS127A female mice were treated with or without Doxycycline (1.0 mg/mL, in drinking water) to induce the expression of YAPS127A in renal epithelial cells as described above. Age-matched Pax8-rtTA mice were used as a negative control. Doxycycline-induced mice were randomly distributed to treatment and vehicle groups (n = 8/group). MGH-CP1 (75 mg/kg BW in sterile saline with 5 % PEG-400, 5% Tween-80, and 5% DMSO) or vehicle (control) were injected IP twice/day for seven days or until the experiment endpoint. All mice were euthanized 48 hours after the last MGH-CP1 injection. Kidneys from each group were harvested, weighed, and processed for pathohistological analyzes.
Single-cell transcriptomics and Bioinformatic analyzes
Followed previous studies22,74, we pooled tissues from 3–5 mice to prepare one sample. Therefore, cells ( ~ 20,000) in each sample were originally from 3–5 mice. A minimum of 9 mice (3–5 mice/sample, n = 3 samples/group) were used in each group for cell preparation. Cells were collected from random regions of each kidney, but each sample included the cortex and medulla. Three samples in each group (CTR, D7, and M4) were run separately to ensure the reproducibility of our findings. Single cells isolated from kidneys of control and transgenic mice were used for Single-cell RNA sequencing. In brief, kidney tissues from control or transgenic mice were cut into small pieces ( ~ 1mm2) on ice, washed with cold PBS, and digested at 37 °C with a shaking speed of 150 rpm for 40 min in an air bath shaker. The digestion medium containing 450 units/ml collagenase1, 150 units/ml collagenase 2, 450 units/ml collagenase 3, 450 units/ml collagenase 4, 0.8 units/ml Elastase, 300 units/ml Hyaluronidase, 250 units/ml DNase I, and 1 unit/ml Dispase. The digested cells were mixed with cold RPMI 1640 with 2% FBS, filtered through a prewetted 70-μm & 40 µm strainer, and centrifuged at 1500 rpm at 4 °C for 5 minutes. The cell pellets were then treated for ACK lysis for 10 min, washed with ice-cold PBS + 0.04%BSA, and resuspended in cold PBS + 0.04%BSA. The prepared cells were then resuspended for evaluating cell viability, adjusting cell number, and preparing the library.
Single-cell library was prepared using the 10x Genomics Chromium Single Cell 5′ Library & Gel Bead Kit v2 (PN-1000014). Cell suspensions were loaded onto a Chromium Single-Cell Chip with the reverse transcription master mix and single cell 5′ gel beads, aiming for 10,000 cells per channel. Reverse transcription was performed using a C1000 Touch Thermal Cycler (Bio-Rad Laboratories). Quality control and quantification were performed using the Agilent 2100 Bioanalyzer with a DNA high-sensitivity chip. For input into the gene-expression library construction, 50 ng cDNA and 14 cycles were used. Libraries were sequenced using an Illumina HiSeq 2500 NexGen Sequencer.
Single-cell RNAseq Fastq files were processed using 10x Genomics Cell Ranger for 3’ v3 chemistry (version 3.0.2) with standard parameters. The resultant raw counts of each sample were individually assessed for quality control. The quality control was carried out using Seurat version 4.2.0 to generate Seurat Objects. Each Seurat Object then was normalized using SCTransform algorithm (version 2) and integrated accordingly in Seurat. Differential gene analysis was achieved by Wilcoxon tests. Clustering was based on the Jaccard similarity of the KNN graph-refined Euclidean distance in PCA space. These tasks were realized and visualized using Seurat and SCpubr version 1.0.4. Specifically, isolated cells were carefully filtered and only those with <35% mitochondrial genes, >500 RNA counts, and >200 RNA features were kept for analyzing. The cells passed to downstream have less than 2.6% RPL-type ribosomal genes and 1.7% RPS-type ribosomal genes suggesting high purity of the library.
Cell types were identified by determining differentially expressed genes (DEGs) in all isolated kidney cells at single cell level and comparing their signature scores calculated based on published renal cell biomarkers and marker panels22,23,53,75,76. Subtypes of myeloid cells and T cells were annotated based on the biomarkers and molecular profiles published recently30,31. Identification and visualization of T cell subpopulations were further confirmed using ProjecTILs version 3.0. Gene set enrichment analysis was carried out against the Molecular Signatures Database (MSigDB) and visualized using clusterProfiler version 3.16. Heatmaps were plotted using ComplexHeatmap 3.16. All the above packages, datasets, or libraries were implemented with R 4.2.
Statistics and Reproducibility
All experiments were repeated at least three times unless otherwise noted. Sample size (in both cellular and animal models) were determined based on our previous studies39,40,71,72. Samples were randomly allocated into experimental groups. Data are presented as mean ± SEM. Statistical analysis was conducted using GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA). Data were analyzed for significance using Student T-tests or one-way ANOVA unless otherwise noted. A value of p < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw and processed sc-RNAseq data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the accession number/code GSE230364. The human pRCC sc-RNAseq data used in this study are available in the NCBI GEO database under accession code GSE183276 and GSE152938. Other data supporting the findings of this study are available within the article and its supplementary information files. Source data are provided with a source data file. Source data are provided with this paper.
Code availability
The code that supports the findings within this paper is publicly available at the GitHub repositories [https://github.com/jliu678/PRCC-Nature_Communications & https://github.com/jliu678/SeqWins] and are available on Zenodo with https://doi.org/10.5281/zenodo.14728978 and https://doi.org/10.5281/zenodo.14728996.
Change history
30 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-61430-9
References
Yu, F. X. & Guan, K. L. The Hippo pathway: regulators and regulations. Genes Dev. 27, 355–371 (2013).
Zhao, B., Li, L., Lei, Q. & Guan, K. L. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 24, 862–874 (2010).
Zheng, Y. & Pan, D. The hippo signaling pathway in development and disease. Dev. Cell 50, 264–282 (2019).
Meng, Z., Moroishi, T. & Guan, K. L. Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 (2016).
Harvey, K. & Tapon, N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat. Rev. Cancer 7, 182–191 (2007).
Harvey, K. F., Zhang, X. & Thomas, D. M. The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257 (2013).
Kulkarni, A., Chang, M. T., Vissers, J. H. A., Dey, A. & Harvey, K. F. The Hippo pathway as a driver of select human cancers. Trends Cancer 6, 781–796 (2020).
Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 (2010).
Calses, P. C., Crawford, J. J., Lill, J. R. & Dey, A. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer 5, 297–307 (2019).
Pobbati, A. V. & Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 10, 3622–3635 (2020).
Zagiel B., Melnyk P., Cotelle P., Progress with YAP/TAZ-TEAD inhibitors: a patent review (2018-present). Expert Opin. Ther. Pat., 32, 899–912 (2022).
Wang, Y. et al. Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 25, 1304–1317 e1305 (2018).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
N. Cancer Genome Atlas Research. et al.Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med 374, 135–145 (2016).
Mendhiratta, N., Muraki, P., Sisk, A. E. Jr & Shuch, B. Papillary renal cell carcinoma: review. Urol. Oncol. 39, 327–337 (2021).
Martinez Chanza, N. et al. Cabozantinib in advanced non-clear-cell renal cell carcinoma: a multicentre, retrospective, cohort study. Lancet Oncol. 20, 581–590 (2019).
Velickovic, M., Delahunt, B., Storkel, S. & Grebem, S. K. VHL and FHIT locus loss of heterozygosity is common in all renal cancer morphotypes but differs in pattern and prognostic significance. Cancer Res 61, 4815–4819 (2001).
Buscheck, F. et al. Prevalence and clinical significance of VHL mutations and 3p25 deletions in renal tumor subtypes. Oncotarget 11, 237–249 (2020).
Blake, J. A. & Ziman, M. R. Pax genes: regulators of lineage specification and progenitor cell maintenance. Development 141, 737–751 (2014).
Tong, G. X. et al. Expression of PAX8 in normal and neoplastic renal tissues: an immunohistochemical study. Mod. Pathol. 22, 1218–1227 (2009).
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
Miao, Z. et al. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat. Commun. 12, 2277 (2021).
Nguyen, T., Du, J. & Li, Y. C. A protocol for macrophage depletion and reconstitution in a mouse model of sepsis. STAR Protoc. 2, 101004 (2021).
van Rooijen, N., Sanders, A. & van den Berg, T. K. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods 193, 93–99 (1996).
Naito, M. et al. Liposome-encapsulated dichloromethylene diphosphonate induces macrophage apoptosis in vivo and in vitro. J. Leukoc. Biol. 60, 337–344 (1996).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Dimitrov, D. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 13, 3224 (2022).
Yang, Y., Li, C., Liu, T., Dai, X. & Bazhin, A. V. Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front Immunol. 11, 1371 (2020).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: facilitators of cancer and obesity-induced cancer. Annu. Rev. Cancer Biol. 5, 17–38 (2021).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 48, W509–W514 (2020).
Nijhuis, E. W., vd Wiel-van Kemenade, E., Figdor, C. G. & van Lier, R. A. Activation and expansion of tumour-infiltrating lymphocytes by anti-CD3 and anti-CD28 monoclonal antibodies. Cancer Immunol. Immunother. 32, 245–250 (1990).
Kongkaew, T. et al. TIL expansion with high dose IL-2 or low dose IL-2 with anti-CD3/anti-CD28 stimulation provides different quality of TIL-expanded T cell clones. J. Immunol. Methods 503, 113229 (2022).
Li, Q. et al. Lats1/2 sustain intestinal stem cells and Wnt activation through TEAD-dependent and independent transcription. Cell Stem Cell 26, 675–692.e678 (2020).
He, C. et al. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression. Oncogene 34, 6040–6054 (2015).
Hua, G. et al. YAP induces high-grade serous carcinoma in fallopian tube secretory epithelial cells. Oncogene 35, 2247–2265 (2016).
He C. et al., YAP1-LATS2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO Rep. 20, e44948 (2019).
LV, X. et al. Reprogramming of ovarian granulosa cells by YAP1 leads to development of high-grade cancer with mesenchymal lineage and serous features. Sci. Bull. (Beijing) 65, 1281–1296 (2020).
He, C. et al. HPV-YAP1 oncogenic alliance drives malignant transformation of fallopian tube epithelial cells. EMBO Rep. 25, 4542–4569 (2024).
Perets, R. et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24, 751–765 (2013).
Russo, A. et al. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 37, 1976–1990 (2018).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Xia, C. et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin. Med J. (Engl.) 135, 584–590 (2022).
Farber, N. J. et al. Renal cell carcinoma: the search for a reliable biomarker. Transl. Cancer Res. 6, 620–632 (2017).
Ricketts, C. J. et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 23, 313–326.e315 (2018).
Chen, F. et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep. 14, 2476–2489 (2016).
Sourbier, C. et al. Targeting loss of the Hippo signaling pathway in NF2-deficient papillary kidney cancers. Oncotarget 9, 10723–10733 (2018).
White, S. M. et al. YAP/TAZ Inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2-deficient tumor cells. Dev. Cell 49, 425–443 (2019).
Lindgren, D. et al. Cell-type-specific gene programs of the normal human nephron define kidney cancer subtypes. Cell Rep. 20, 1476–1489 (2017).
Zhang Y. et al., Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 118, e2103240118 (2021).
Peired A. J. et al., Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells. Sci Transl Med. 12, eaaw6003 (2020).
Wang, Q. et al. Single-cell chromatin accessibility landscape in kidney identifies additional cell-of-origin in heterogenous papillary renal cell carcinoma. Nat. Commun. 13, 31 (2022).
Galon, J. & Bruni, D. Tumor immunology and tumor evolution: intertwined histories. Immunity 52, 55–81 (2020).
Gonzalez, H., Hagerling, C. & Werb, Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018).
Spranger, S. & Gajewski, T. F. Mechanisms of tumor cell–intrinsic immune evasion. Annu. Rev. Cancer Biol. 2, 213–228 (2018).
Ding, A. S., Routkevitch, D., Jackson, C. & Lim, M. Targeting myeloid cells in combination treatments for glioma and other tumors. Front Immunol. 10, 1715 (2019).
Law A. M. K., Valdes-Mora F., Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 9, 561 (2020).
Groth, C. et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 120, 16–25 (2019).
Raskov, H., Orhan, A., Christensen, J. P. & Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 124, 359–367 (2021).
Togashi, Y., Shitara, K. & Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 16, 356–371 (2019).
Saleh, R. & Elkord, E. FoxP3(+) T regulatory cells in cancer: prognostic biomarkers and therapeutic targets. Cancer Lett. 490, 174–185 (2020).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Choueiri, T. K. et al. Biomarker-based phase ii trial of savolitinib in patients with advanced papillary renal cell cancer. J. Clin. Oncol. 35, 2993–3001 (2017).
Fu, D. et al. YAP regulates cell proliferation, migration, and steroidogenesis in adult granulosa cell tumors. Endocr. Relat. Cancer 21, 297–310 (2014).
Wang, C. et al. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell death Dis. 4, e869 (2013).
Hua, G. et al. The four and a half LIM domains 2 (FHL2) regulates ovarian granulosa cell tumor progression via controlling AKT1 transcription. Cell death Dis. 7, e2297 (2016).
Traykova-Brauch, M. et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 14, 979–984 (2008).
Camargo, F. D. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 (2007).
Huang C. et al., Human papillomavirus targets the YAP1-LATS2 feedback loop to drive cervical cancer development. Oncogene, 41, 3761–3777 (2022).
He, C. et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 7, 1426–1449 (2015).
He, C. et al. A human papillomavirus-independent cervical cancer animal model reveals unconventional mechanisms of cervical carcinogenesis. Cell Rep. 26, 2636–2650 e2635 (2019).
LV, X. et al. G-1 inhibits breast cancer cell growth via targeting colchicine-binding site of tubulin to interfere with microtubule assembly. Mol. cancer therapeutics 16, 1080–1091 (2017).
Hochane, M. et al. Single-cell transcriptomics reveals gene expression dynamics of human fetal kidney development. PLoS Biol. 17, e3000152 (2019).
Han, X. et al. Mapping the mouse cell atlas by microwell-Seq. Cell 172, 1091–1107 (2018).
Ransick, A. et al. Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev. Cell 51, 399–413 (2019).
Acknowledgments
This work was partially supported by the National Cancer Institute/National Institute of Health (2R01CA197976, 1R01CA201500, 5R01CA279385, CW), the Colleen’s Dream Foundation (no number, CW), the Ruggles Family Foundation (CW), the Vincent Memorial Hospital Foundation (CW), and the Vincent Department of Obstetrics and Gynecology, Massachusetts General Hospital (No number). We thank Laiping Wong for her contribution to the sequencing data analyzes.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.W., X.L., J.L. Funding acquisition, project administration, supervision: C.W. Methodology – in vivo studies: X.L., J.R., J.L., C.W., C.He., P.C., C.H., G.H., L.H., X.W. Methodology – in vitro studies: X.L., J.R., C.He., J.L., P.C., C.H., G.H., X.Z., L.C., H.W., M.M., D.S., I.M., A.D., X.C., R.K., O.Y., Bioinformatics and mega data visualization: J.L., R.S., S.Y., Manuscript Writing: C.W., X.L., J.L., J.R., Review & Editing: C.W., X.L., J.L., J.R., P.C., J.S.D., X.W., A.D., M.M., C.He., G.H., R.S., O.Y.
Corresponding author
Ethics declarations
Competing interests
Dr. Xu Wu has a financial interest in Tasca Therapeutics, which is developing small molecule modulators of TEAD palmitoylation and transcription factors. Dr. Wu’s interests were reviewed and are managed by Mass General Hospital, and Mass General Brigham in accordance with their conflict-of-interest policies. The rest authors have no competing interests to declare.
Peer review
Peer review information
Nature Communications thanks Dean Yimlamai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lv, X., Liu, J., Ruan, J. et al. Targeting the disrupted Hippo signaling to prevent neoplastic renal epithelial cell immune evasion. Nat Commun 16, 2858 (2025). https://doi.org/10.1038/s41467-025-57697-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57697-7
