
Abstract
Hybrid necrosis, a century-old mystery in wheat, is caused by complementary genes Ne1 and Ne2. Ne2, encoding a nucleotide-binding leucine-rich repeat (NLR) immune receptor, has been cloned, yet Ne1 remains elusive. Here, we report that Ne1, which encodes an alpha/beta hydrolase (ABH) protein generated by structural variation, triggers hybrid necrosis with Ne2 by activating autoimmune responses. We further verify that not only allelic variation but also copy number variation (CNV) of Ne1 are pivotal for hybrid necrosis diversity in wheat. Ne1 likely originates from wild emmer wheat, potentially through duplication and ectopic recombination events. Unlike Ne2, which is frequently selected for rust resistance in wheat breeding, the lower prevalence of Ne1 in modern wheat cultivars is attributed to its association with hybrid necrosis. Altogether, these findings illuminate the co-evolution of the NLR/ABH gene pair in plant development and innate immunity, offering potential benefits for wheat breeding.
Similar content being viewed by others
Introduction
Hybrid necrosis represents a prevalent phenomenon observed in plant hybrids and is recognized as a common form of genetic incompatibility contributing to gene-flow barriers1,2. It arises from epistatic interactions between divergent alleles contributed by different parents in certain hybrids, resulting in autoimmune-like symptoms in the absence of pathogens, including leaf necrosis, crinkling, dwarfism, stunted growth, and reduced fertility1,3. Hybrid necrosis has been documented across various species, including Arabidopsis thaliana4, Capsella5, wheat6, rye7, lettuce8, rice9, and cotton10. The genetic basis of hybrid necrosis aligns with the principles of the Bateson-Dobzhansky-Muller (BDM) model, generally involving two-locus interactions11,12. The BDM model asserts that a genetic change at locus A in one population and a genetic change at locus B in another population may be incompatible when residing in the same genome upon the hybridization between individuals of the two populations, which could result in postzygotic incompatibility and lead to infertility or inferiority13.
Emerging genetic and biochemical evidence indicates that hybrid necrosis is closely linked to plant immune responses. Numerous causal genes associated with hybrid necrosis have been identified, the majority of which encode proteins related to immunity. The Arabidopsis hybrid necrosis genes, Dangerous Mix 1 (DM1) and DM3d, and the cotton hybrid lethality gene Le4 are known to encode nucleotide-binding leucine-rich repeat (NLR) immune receptor proteins4,10. The Arabidopsis DM3 protein, a member of the ABH family, interacts with the NLR protein DM2 is associated with hybrid necrosis3. In lettuce, hybrid necrosis is governed by specific isoforms of Rin4, which is recognized for its interactions with various resistance (R) genes8. Epistatic interactions between NPR1 and RPP5 orthologues result in genetic incompatibility within Capsella species5. Rice Hwi1 and Hwi2, which encode an LRR-RLK immune receptor and a subtilisin-like protease, respectively, activate the autoimmune response in interspecific hybrids9.
Structural variations, including copy number variations (CNVs) and chromosomal rearrangements, exert substantial influence on the genomic landscape of plants14. In particular, CNVs have been recognized as potent drivers of genetic diversity by instigating alterations in gene dosage and expression levels15. For example, CNVs contribute to grain size diversity in rice16 and enhance nematode resistance in soybean17. In addition, genomic structural changes and copy number variation at the Sc locus confer japonica–indica hybrid male sterility in rice18.
Hybrid necrosis in wheat has been initially documented by Sax19. Hybrid necrosis has been reported in both intraspecific crosses of common wheat6 and interspecific crosses between tetraploid wheat and Aegilops tauschii20. Hybrid necrosis impedes the genetic improvement of wheat, acting as a barrier to both the integration of desirable traits from diverse common wheat genotypes and the introgression of genes from related species into commercial cultivars6. Hybrid necrosis in common wheat is controlled by the complementary dominant genes Ne1 and Ne2, which are located on chromosome arms 5BL and 2BS, respectively21. In 2021, three independent groups successfully cloned and characterized Ne2, which encodes an NLR protein22,23,24. Ne2 is the same gene as wheat leaf rust resistance gene Lr13 and allelic to wheat stripe rust resistance gene Yr27, exhibits pleiotropic effects against rust and hybrid necrosis23,25. However, the causal gene for Ne1 has yet to be determined, despite the development of high-density genetic maps by three separate teams26,27,28.
In this study, we report the map-based cloning of Ne1 using two separate genetic populations. Ne1 is validated by mutagenesis and transgenic approaches. We also trace the ancestral lineage of Ne1 and highlight the influence of allelic and copy number variations of Ne1 on the phenotypic diversity of hybrid necrosis in wheat. Furthermore, we investigate the prevalence of Ne1 in worldwide tetraploid and hexaploid wheat.
Results
Phenotypic characterization of hybrid necrosis in wheat
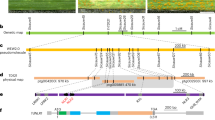
Previously, we cloned Ne2 from wheat line M114 (Ne1Ne1Ne2Ne2), which is identical to wheat leaf rust resistance gene LrZH22/Lr13 from Zhoumai 22 (ZM, ne1ne1Ne2Ne2)23 (Supplementary Data 1). M114 showed necrosis symptoms starting at the leaf tips, which spread with plant development, while ZM remained healthy (Fig. 1a and Supplementary Fig. 1a, b). The F1 hybrids from M114 × ZM displayed necrosis confined to the leaf tips without base involvement at the grain filling stage (Fig. 1a). The 356 F2:3 progenies of the M114 × ZM population segregated as 79 homozygous necrotic, 182 segregating, and 95 normal families, conforming to a 1:2:1 single Mendelian locus ratio (χ21:2:1 = 1.617, P < 0.05) (Supplementary Data 2).

a Phenotypes of necrotic line M114 (Ne1Ne1Ne2Ne2), Zhoumai22 (ZM, ne1ne1Ne2Ne2), and F1 plant derived from M114 × ZM at the grain filling stage in the field. Scale bar in plant, 8 cm; Scale bar in leaf, 2 cm. b Fine genetic map of Ne1 constructed using the F2 populations derived from M114 × ZM. c Physical map of Ne1 region according to Chinese Spring (CS) reference genome sequence IWGSC RefSeq v2.1. The blue oval indicates Ne1. d Fine genetic map of Ne1 constructed using segregation populations derived from Zhengnong17 (ZN17, ne1ne1Ne2Ne2) and Yangbaimai (YBM, Ne1Ne1ne2ne2). Ne1 is highlighted in blue. e Gene structure and sequence variation of ABH.1 gene. Black boxes and gray lines indicate exons and introns, respectively. The variation of coding sequence (c) and amino acid (p) sequences between CS, YBM, and M114 are indicated. f Protein structure prediction of ABH.1. Hydrolase domain, AAA ATPase with vWA domain, and membrane protease YdiL domain are represented in orange, green, and purple, respectively. The signal peptide is indicated in red. Source data are provided as a Source Data file.
We also identified hybrid necrosis in a segregating population derived from wheat accessions Zhengnong 17 (ZN17, ne1ne1Ne2Ne2) and Yangbaimai (YBM, Ne1Ne1ne2ne2), with Ne2 cloned from ZN1722. To evaluate the necrotic syndrome, we generated a pair of near-isogenic lines, NIL-ne1 (ne1ne1Ne2Ne2) and NIL-Ne1 (Ne1Ne1Ne2Ne2), at the NE1 locus. Necrosis was characterized by premature leaf death and impaired growth potential (Supplementary Fig. 1c), initially appearing on the tip of basal leaves (Supplementary Fig. 1d). The 3,3’-diaminobenzidine hydrochloride (DAB) staining indicated that excessive localized hydrogen peroxide (H2O2) accumulation may trigger leaf lesions (Supplementary Fig. 1e), similar to the hypersensitive response (HR) induced by pathogens.
Map-based cloning of Ne1
Bulked segregant RNA-Seq (BSR-Seq) experiments were performed to map Ne1 on chromosome arm 5BL using M114 × ZM segregating population (Supplementary Fig. 2). We then used 7,659 F2 individuals from M114 × ZM to narrow down Ne1 within a 0.07 cM genetic interval flanked by markers XM14 and XM6 (Fig. 1b and Supplementary Data 3), corresponding to a 3.61 Mb genomic region according to Chinese Spring (CS) genome sequence IWGSC RefSeq v2.129 (Fig. 1c and Supplementary Data 4), overlapping with genetic interval of Ne1 using ZN17 and YBM population26 (Fig. 1d). Due to previous report of genomic structural variations at the NE1 locus26, we developed specific markers based on the fifteen high-confidence genes in the Ne1 genomic interval to investigate M114 and ZM. The results show that M114 has a long genomic fragment insertion at the NE1 locus compared to ZM (Supplementary Fig. 3).
The long insertion fragment specifically encompasses six high-confidence genes, designated as TraesCS5B03G0561600-TraesCS5B03G0564000 according to IWGSC RefSeq v2.129 (Fig. 1c and Supplementary Data 4). Notably, TraesCS5B03G0561800 (ABH.1), encodes an alpha/beta hydrolase (ABH) family protein, which is recognized for its roles in plant immunity, such as ABH family proteins EDS1 and PAD430,31, as well as Arabidopsis hybrid necrosis protein DM3 (At3g61540, ABH family protein) as an interactor of NLR protein DM23. In addition, TraesCS5B03G0565000 (ABH.2), which also encodes an ABH family protein, is located within the Ne1 genetic interval but not on the long insertion fragment. However, sequence analysis revealed no sequence differences within ABH.2 between ZN17 and YBM. Therefore, ABH.1 is considered the likely candidate for Ne1.
We then cloned the complete genomic sequences of ABH.1 from M114 and YBM using specific primers (Supplementary Data 3). ABH.1 comprises eleven exons (Fig. 1e) and encodes an ABH family protein that contains a hydrolase domain, an AAA ATPase with vWA domain, and a membrane protease YdiL domain (Fig. 1f). Sequence comparison between ABH.1CS and ABH.1M114 revealed one T/C SNP at the first intron and another C/G SNP at the sixth exon, resulting in a proline to alanine (P691A) amino acid substitution in the ABH.1 protein (Fig. 1e). Compared to ABH.1CS, a 9 bp insertion and a 3 bp deletion (AGCTCAGTT/CAA) was observed at the fifth exon of ABHYBM, resulting in codon insertions/deletions (DN423-424EAQF) (Fig. 1e).
Mutagenesis and transgenic assay validation of the ABH.1 as Ne1
To verify the role of ABH.1, seeds of necrotic lines NIL-Ne1 and M114 were mutagenized with ethyl methanesulfonate (EMS). A total of 4500 and 2360 M2 families from NIL-Ne1 and M114, respectively, were screened for non-necrotic leaf phenotype in the field. Finally, 11 and 14 M3 non-necrotic lines were identified from the NIL-Ne1 and M114 EMS-mutagenized populations, respectively. The complete genomic sequences of ABH.1 and Ne2 were amplified and sequenced from the non-necrotic mutants. Five NIL-Ne1-derived non-necrotic mutants (N-m45, N-m61, N-m70, N-m81, and N-m119) and six M114-derived non-necrotic mutants (M114-m1 to M114-m6) displayed a distinct EMS-induced single nucleotide change (G/C to A/T) in ABH.1 (Fig. 2a, b). Moreover, three NIL-Ne1-derived non-necrotic mutants (N-m04, N-m28, and N-m34) and six M114-derived non-necrotic mutants (M114-m7 to M114-m12) exhibited missense mutations in Ne2. Two mutants (M114-m13 and M114-m14) harbored nonsense mutations resulting in a premature stop codon in Ne2 (Fig. 2c, d). The identification of multiple independent EMS non-necrotic mutants from two necrotic backgrounds indicated that ABH.1 is most likely Ne1. Furthermore, three NIL-Ne1-derived non-necrotic mutants (N-m40, N-m51, and N-m328) did not demonstrate any SNPs in either Ne2 or ABH.1, as well as other genes within the Ne1 mapping interval, suggesting the presence of mutations in alternative unknown regulators of the necrosis pathway associated with Ne1-Ne2.

a, c Phenotypes of non-necrotic mutants generated from the necrotic NIL-Ne1 (Ne1Ne1Ne2Ne2) and M114 (Ne1Ne1Ne2Ne2) background by ethyl methanesulfonate (EMS) treatment. Gene structure and EMS mutant analysis of ABH.1 (b) and Ne2 (d). The positions of the EMS-derived loss-of-function mutations are indicated by black lines. Black boxes and gray straight lines represent exons and introns, respectively. The coding sequence (c) changes and their predicted effects on the amino acid (p) are indicated. Scale bar in (a and c), 1 cm. Mutation names in red indicate nonsense mutations. Source data are provided as a Source Data file.
To further validate the function of ABH.1, we performed multiple gene complementation tests with the ABH.1YBM and ABH.1M114 alleles, respectively. We amplified the full-length coding region of ABH.1 from YBM and constructed an overexpression construct, proUbi::ABH.1YBM (Fig. 3a). Then, this construct was introduced into the hexaploid wheat cultivar Fielder (ne1ne1ne2ne2) using Agrobacterium-mediated transformation. All four positive transgenic lines, OEABH.1YBM#1-#4, carrying only ABH.1YBM and not Ne2, exhibited normal growth without leaf necrosis (Fig. 3b). To further confirm that ABH.1YBM can induce leaf necrosis in the presence of Ne2, we conducted a complementation test by crossing line ZZ6903 (ne1ne1Ne2Ne2) with the four homozygous ABH.1YBM transgenic lines and Fielder, respectively. The F1 hybrids derived from Fielder × ZZ6903 displayed normal growth, while all the F1 hybrids from the crosses between ZZ6903 and ABH.1YBM transgenic lines exhibited leaf necrosis (Fig. 3b).

a Structure of proUbi::ABH.1YBM used for transformation of wheat cultivar Fielder. The construct contains the ABH.1YBM coding sequence (CDS) and maize ubiquitin promoter region (proUbi). LB left border; RB, right border; Nos Ter, termination sequence of the Agrobacterium tumefaciens nopaline synthase gene. b Genetic complementation of ABH.1 and Ne2 was performed by crossing independent transformants (OEABH.1YBM#1-#4) carrying proUbi::ABH.1YBM and wheat cultivar ZZ6903 with the ne1ne1Ne2Ne2 genotype, which induced the expression of hybrid necrosis. Representative leaves of four F1 hybrids and their corresponding parental lines are presented. Scale bar, 1 cm. c Structure of proABH.1M114::ABH.1M114 used for transformation of the Ne2 overexpression transgenic line OE-T1−1-1 (ne1ne1Ne2Ne2, OENe2) in the Fielder genetic background and the F1 plants (ne1ne1Ne2ne2) of ZM × Fielder, respectively, by Agrobacterium-mediated transformation. The construct proABH.1M114::ABH.1M114 consisted of a 13,118 bp genomic fragment of ABHM114 from M114, comprising 8157 bp the entire gene body, 2406 bp upstream native promoter and 2,556 bp downstream regulatory sequences, respectively. d Phenotypes of OENe2, positive transgenic lines (OENe2 + COMABH.1M114#1-#3) in OENe2 background, ZM, positive transgenic lines (Ne2Ne2 + COMABH.1M114#1-#3, and ne2ne2 + COMABH.1M114#1) in ZM × Fielder background. Scale bar, 1 cm. Source data are provided as a Source Data file.
We also generated a construct, proABH.1M114::ABH.1M114, consisting of a 13,118 bp genomic fragment of ABH.1M114 from M114, which includes 8157 bp of the entire gene body, 2406 bp of the upstream native promoter, and 2556 bp of the downstream regulatory sequence (Fig. 3c). This construct was transformed into the Ne2 overexpression transgenic line OE-T1−1-1 (ne1ne1ne2ne2Ne2ZMNe2ZM, hereafter named OENe2), in the Fielder genetic background23 and the F1 plants (ne1ne1Ne2ne2) of the ZM × Fielder cross, respectively, using Agrobacterium-mediated transformation. We obtained 10 and 8 positive T0 transgenic individuals with the confirmed ABH.1M114 transgene sequence from OENe2 and ZM × Fielder transformation events, respectively. The positive T0 individuals were advanced to produce T1 and T2 generations. T2-positive transgenic plants carrying ABH.1M114 in the OENe2 transformation event exhibited pronounced necrosis at the grain filling stage, while OENe2 exhibits slight chlorosis at the leaf tip (Fig. 3d). Similarly, in the ZM × Fielder transformation events, T2-positive transgenic plants carrying ABH.1M114 exhibited leaf necrosis when homozygous for Ne2Ne2 (Fig. 3d), while displaying normal leaves when homozygous for ne2ne2 (Fig. 3d). The results from multiple independent EMS non-necrotic mutants and transgenic experiments confirm that ABH.1 is indeed Ne1.
ABH.1 triggers hybrid necrosis with Ne2 by activating immune responses
To better understand the function of ABH.1, we investigated its expression patterns by reverse transcription quantitative PCR (RT-qPCR). The results indicate that ABH.1 is expressed across all tissues examined, with the highest expression level in the leaf blade and comparatively lower level in the young stem and spike (Supplementary Fig. 4a). The necrotic syndrome, which closely resemble a hypersensitivity response (Supplementary Fig. 1), along with the identity of Ne2 that is the high-temperature leaf rust resistance gene Lr1323, suggest a possible link between hybrid necrosis and immune response. Expression analysis of pathogen-related genes, including PR1, PR2, PR3, PR5, and LOX2, revealed significant differential expression between near-isogenic lines NIL-ne1 and NIL-Ne1 (Supplementary Fig. 4b). Furthermore, the levels of salicylic acid (SA) and jasmonic acid (JA), two well-known phytohormones involved in plant immune responses, were significantly higher in NIL-Ne1 compared to NIL-ne1 (Supplementary Fig. 4c). These findings imply a role for autoimmunity activation in the initiation of hybrid necrosis.
The ABH.1 gene encodes alpha/beta-hydrolases comprising 1671 amino acids, featuring a predicted signal peptide spanning initial residues 1 to 17 (Supplementary Fig. 5a). Both the yeast signal peptide secretion assay and the 2,3,5-triphenyltetrazolium chloride (TTC) staining assay suggest that the predicted signal peptide within ABH.1 may have the potential secretory capability (Supplementary Fig. 5b). To investigate the subcellular localization of ABH.1, we performed co-transfection of Nicotiana benthamiana leaves with the pro35S::ABH.1-GFP construct and the plasma membrane-specific marker pro35S::PIP2-mCherry32. The co-localization of ABH.1-GFP with the PIP2-mCherry marker indicates that ABH.1 is mainly localized to the plasma membrane (Supplementary Fig. 6a, b). In addition, Ne2 is predominantly localized to the cytoplasm and nucleus (Supplementary Fig. 6c).
Allelic and copy number variation effects of ABH.1 on hybrid necrosis
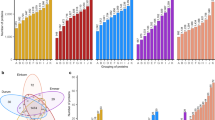
To investigate the sequence variation of ABH.1, we conducted a haplotypic analysis using whole-genome re-sequencing data from 540 hexaploid33,34,35 (Supplementary Data 5) and 157 tetraploid35,36,37 (Supplementary Data 6) wheat accessions. Seven haplotypes (Hap1-Hap7) were identified (Fig. 4a), and 366 wheat accessions lacked ABH.1 (Hap0) (Supplementary Data 5 and 6). Notably, although classified as haplotype Hap5, Ripper and Hallam displayed multiple heterozygous SNPs in the intronic regions of ABH.1 when compared to Mania (Supplementary Fig. 7a). Thus, we speculate that there may be additional copy number variations or structural variations within the NE1 locus. We then investigated structural variations at the NE1 locus using whole-genome re-sequencing data from 540 hexaploid wheat33,34,35 and identified four structural variations, Zhengnong17-like (ZN17-like, 310 accessions), Yangbaimai-like (YBM-like, 192 accessions), Youzimai-like (YZM-like, 29 accessions), and Wuyuanmai-like (WYM-like, 9 accessions) (Fig. 4b and Supplementary Data 5). Compared to the ZN17-like, YBM-like, YZM-like, and WYM-like materials harbored 1, 2 and 3 copies of the 2.89 Mb-fragment, respectively (Fig. 4b). Moreover, we also found the multiple 2.89 Mb-fragments in the YZM-like or WYM-like subtype materials are not identical, suggesting the potential presence of complex structural variation within Ne1 genetic interval (Supplementary Fig. 7b).

a Haplotype analysis of ABH.1 using whole-genome re-sequencing data from 540 hexaploid and 157 tetraploid wheat accessions. Variants at the coding sequence of ABH.1 were clustered into seven major haplotypes (Hap1-Hap7), while 366 accessions lacked ABH.1 (Hap0). Introns and exons are represented by lines and black boxes, respectively. The coding nucleotide and amino acid sequences of Chinese Spring (CS) are shown in black font, and the bracketed numbers represent the relative positions of nucleotide or amino acid sequences concerning ATG or Met, respectively. Deletions are indicated by the symbol ‘-’. YM14, Yumai14; YBM, Yangbaimai. b Four major representative structural variations of the NE1 locus. Reads from these wheat samples were aligned to the Chinese Spring reference genome sequence IWGSC RefSeq v2.1, and coverage depth for each 5 kb window was computed. The coverage depth for each 5 kb window was then normalized based on the total coverage depth. Finally, the normalized depth for the target region (Chr5B:385.5-390.5 Mb) was presented in the form of a scatter plot, with each data point representing a 5 kb window. c Coverage depth of the ABH.1 gene in the published re-sequence dataset for 540 hexaploidy wheat accessions. The ABH.2 (TraesCS5B03G0565000) gene is situated downstream of ABH.1 (TraesCS5B03G0561800), with the sequencing coverage depth of ABH.2 used as an indicator of normal resequencing outcomes within this genomic region. Reads from wheat samples were aligned to the Chinese Spring RefSeq v2.1, and coverage depth was determined for each gene. The coverage depth for each gene was subsequently normalized by the total coverage depth, and the resulting normalized depth for the target gene was depicted in a scatter plot, with each data point representing a specific wheat accession. The n number is the sample size used to derive statistics. d The expression of ABH.1 was compared among the four CNV (copy number variation) groups (two-sided Student’s t test; centerline, median). RNA-seq data from 305 common wheat genomes (one biological replicate) were used to quantify ABH.1 expression for the four CNV groups. P-values are shown at the top of data points, and n, the sample size used to derive statistics, is listed at the bottom of the figure. TPM, transcripts per million. Source data are provided as a Source Data file.
Furthermore, we conducted an investigation to determine if ABH.1 is present in different copies. Utilizing the coverage depth as a criterion, four copy number variations (CNVs) of ABH.1, CNV-0, CNV-1, CNV-2, and CNV-3, were identified using whole-genome re-sequencing data from 540 hexaploid wheat33,34,35 (Supplementary Data 5). These CNVs correspond to the presence of zero, one, two, or three copies of ABH.1, respectively, and they also aligned with four structural variations ZN17-like, YBM-like, YZM-like, and WYM-like, respectively (Fig. 4c). The copy number analysis of ABH.1 in the eight representative materials confirmed the CNVs of NE1 locus (Supplementary Fig. 8a and Supplementary Data 7). CNVs play a pivotal role in modulating gene expression and influencing phenotypic diversity14. RNA-sequencing data from 305 hexaploid wheat33 also demonstrated that the expression levels of ABH.1 were significantly higher in the CNV-2 and CNV-3 accessions relative to the CNV-1 accessions (Fig. 4d and Supplementary Data 8).
Previous reports revealed that there are at least three Ne1 alleles, Ne1w-weak, Ne1m-moderate, and Ne1s-strong, with diverse effects based on the severity of necrosis observed in F1 progenies38. To illuminate the effects of ABH.1 alleles, we chose wheat accessions representing different haplotypes of ABH.1, including CS (Hap1), YM14 (Hap2), YBM (Hap3), PI 171028 (Hap4), and Mania (Hap5) (Fig. 5a), all of which belong to CNV-1 (Fig. 5b), and crossed them with ZZ6903 (Hap0, CNV-0) with the ne1ne1Ne2Ne2 genotype. The F1 hybrids from the crosses of YM14 × ZZ6903 and YBM × ZZ6903 exhibited necrotic symptoms that were similar to, or slightly more severe than, those observed in the CS × ZZ6903 cross (Fig. 5c, d). CS is previously believed to carry Ne1w allele6. Therefore, Hap1, Hap2, and Hap3 of ABH.1 could correspond to Ne1w allele. The F1 hybrids from the crosses PI 171028 × ZZ6903 and Mania × ZZ6903 displayed an extremely severe necrotic symptom, leading to complete leaf desiccation and death at the flowering stage (Fig. 5d) and resulting in very small seeds. These findings suggest that Hap4 and Hap5 of ABH.1 could correspond Ne1s allele.

a Phenotypes of a series of materials carrying different ABH.1 alleles. Scale bar, 10 cm. b Structural variations of the ABH.1 locus from materials carrying different ABH.1 alleles. c–e Phenotypes of ZZ6903, and the representative F1 plants derived from crosses between wheat accessions (carrying ne2ne2 and different ABH.1 haplotypes) × ZZ6903 (ne1ne1Ne2Ne2 genotype). ZZ6903, Zhengzhou 6903; YM14, Yumai 14; YBM, Yangbaimai; CS, Chinese Spring. Scale bar in plant, 10 cm. Source data are provided as a Source Data file.
To further investigate whether CNVs of ABH.1 also affect necrosis in addition to haplotypes of ABH.1, we utilized PI 182704, Ripper, and Hallam, which are belong to Hap5 and the CNV-3, cross with ZZ6903, respectively (Fig. 5a, b). The F1 plants from the crosses PI 182704 × ZZ6903, Ripper × ZZ6903, and Hallam × ZZ6903 exhibited exacerbated necrosis and significantly reduced seed production compared to those from Mania × ZZ6903 (Fig. 5d, e). RT-qPCR analysis confirmed that the transcript levels of ABH.1 are significantly higher in the leaves of PI 182704, Ripper, and Hallam relative to Mania (Supplementary Fig. 8b). These findings suggest that both allelic and copy number variations of ABH.1 contribute to hybrid necrosis.
Evolutionary trajectory of ABH.1
Phylogenetic analysis revealed that orthologous genes of ABH.1 are widely present across gramineous plants (Supplementary Fig. 9). To investigate the evolutionary origins of ABH.1, we conducted a homologous analysis of ABH.1 in nine Sitopsis genomes39,40 and 157 re-sequenced tetraploid genomes35,36,37. Our results indicated that ABH.1 is absent in nine Sitopsis species (Supplementary Fig. 10). However, multiple haplotypes of ABH.1 and the four CNVs of ABH.1 were observed in tetraploid wheat species, including Triticum durum and Triticum dicoccoides (Supplementary Fig. 11 and Supplementary Data 6). These findings suggest that ABH.1 likely originated from wild emmer wheat.
ABH.1 has homologs in the A, B, and D sub-genomes of CS (Supplementary Fig. 9). Comparative genomic analysis demonstrated significant structural variation at NE1 locus, in contrast to the corresponding homologous regions in the A and D sub-genomes (Supplementary Fig. 12a). However, the NE1 locus exhibits good micro-collinearity within the homologous regions of the A, B and D sub-genome in hexaploid wheat Fielder, carrying the ne1 allele (Supplementary Fig. 12b). Genomic sequence comparison of Ne1 genetic interval between CS and Fielder indicate that the 2.89 Mb insertion fragment shares a strong similarity with the adjacent genomic region spanning from IF4F.2 to SP.8 (Supplementary Fig. 12c), suggesting this region may have resulted from duplication and ectopic recombination events.
The ABH.2 gene, which is located downstream of the 2.89 Mb insertion fragment within Ne1 genetic interval, shares an 92.83% identity in amino acid sequences with ABH.1 (Fig. 1c). ABH.1 differs from ABH.2 by lacking the initial 105 N-terminal amino acids and instead possesses an additional signal peptide of 17 amino acids (Supplementary Figs. 5, 13 and 14), which is unique to the orthologs of ABH.1 in cereal species (Supplementary Fig. 15). Sequence analysis identified multiple transposable elements (TEs) within the promoter region of ABH.1 (Supplementary Fig. 16). Interestingly, a 377 bp sequence of the 5’-end upstream region and 57 bp sequence of the first exon of ABH.1 show a 93.58% identity to the 3’-downstream region of SP.5 (Supplementary Fig. 17), which is the homolog of SP.1. Further analysis of the 3’-downstream sequence of SP.5 revealed that two adjacent fragments, designated as Fragment A and Fragment B, align with the 5’-end of ABH.1 and the 3’-downstream part of SP.1, respectively. Two fragments are separated by TEs (Supplementary Fig. 17).
Consequently, we propose a hypothetical evolutionary trajectory for ABH.1 (Fig. 6a). Following the segmental duplication containing genes ABH.2, SP.4, and SP.5, the entire gene SP.4, along with the promoter region and the first exon (315 bp) of ABH.2, was lost from the duplicated segment. Subsequently, TEs were inserted into the 3’-downstream region of gene SP.5. These transposons were then fused with a part of the downstream sequence of gene SP.5 and the truncated gene ABH.2 to create a new functional gene ABH.1.

a A proposed model for the evolutionary trajectory of ABH.1. Following a segmental duplication event encompassing the genomic loci of ABH.2, SP.4, and SP.5, designated as CNV-0 type, there was a subsequent deletional event within the duplicated segment. This resulted in the loss of the entire SP.4 gene sequence, as well as the promoter region and the initial 315 base pairs of the first exon of the ABH.2 gene. Subsequently, transposable elements (TEs) were inserted into the 3’ downstream region of the SP.5 gene, situated between Fragment A and Fragment B. The integration of these TEs with a segment of the downstream sequence of the SP.5 gene, in conjunction with the truncated remnant of the ABH.2 gene, catalyzed the genesis of a novel functional gene entity, characterized as the ABH.1 gene (CNV-1 type). As a hotspot for genomic recombination, this region has also undergone events resulting from one (CNV-2 type) and two (CNV-3 type) duplications. b Global distribution of Ne1/ABH.1 and ne1 alleles in a worldwide wheat collection (n = 853). Samples were categorized into Ne1/ABH.1 allele type and ne1 allele type based on their genetic type in the NE1 locus, and the proportions of samples in different regions were represented using pie charts. Initially, the geographic coordinates of the samples were imported as point layers and overlaid with corresponding national and continental boundary data. Subsequently, pie charts were added at the sampling locations in each region, with different colors representing distinct haplotype categories and the size of the pie charts reflecting the total number of samples in that region. c Ne1/ABH.1 allele was negatively selected in Chinese wheat breeding. A total of 89 landrace wheat and 449 cultivar wheat accessions were used to investigate the distribution of Ne1/ABH.1 and ne1 alleles. Source data are provided as a Source Data file.
Distribution of ABH.1
We utilized the diagnostic markers Ne1-Select2128 for Ne1 and Ne2-Select91 for Ne2 to screen a globally sourced diverse panel of tetraploid and hexaploid wheat (Supplementary Fig. 18). The results show that the presence proportion of Ne1 is 62.5% (120/192) in wild emmer wheat, 26.0% (52/200) in cultivated emmer wheat, 81.3% (156/192) in durum wheat, and 38.7% (330/853) in hexaploid wheat. However, Ne2 is not present in any of the tetraploid wheat tested, and its presence proportion in hexaploid wheat is 5.9% (50/853) (Supplementary Data 9 and 10). The frequency of ABH.1 in North American common wheat is significantly lower than that observed in Europe and Asia (Fig. 6b). The geographic distribution of ABH.1 in the central wheat region of China shows the highest frequency (51.76%) in Henan province, and the lowest (11.27%) in Shandong province (Fig. 6b). Geographic distribution analysis of Chinese common wheat revealed the frequency of ABH.1 is significantly lower in modern cultivars and breeding lines than in landrace (Fig. 6c), indicating that ABH.1 was negatively selected in Chinese wheat breeding.
Discussion
Wheat hybrid necrosis, mediated by the complementary genes Ne1 and Ne2, was first reported in 192119. The Ne2 gene was characterized as an NLR immune receptor22,23,24. Here, we successfully isolated Ne1, encoding an ABH protein, through map-based cloning, mutagenesis, and transgenic approaches. Furthermore, we found that the Ne1 allele likely originated from wild emmer wheat, potentially arising from segmental duplication and ectopic recombination. Notably, haplotypic and structural variations that altered the Ne1 gene expression are driving forces of hybrid necrosis diversification in wheat. This study sheds light on the long-standing mystery of Ne1-Ne2 interaction-induced hybrid necrosis in wheat. The Ne1-Ne2 (ABH.1-NLR) mediated necrosis in wheat reveals a parallel conserved function to the interaction of DM2-DM3 (NLR-ABH), which are involved in innate immunity and hybrid necrosis in Arabidopsis3,30,31.
Hybrid necrosis is often associated with an autoimmune response1. The genetic combination of Ne1 and Ne2 results in necrosis. However, when only Ne2 is present, it does not exhibit necrotic symptoms but does confer resistance to leaf rust23. Previous evidence suggests that the presence of Ne1 may lead to an increase in the expression of Ne222. In this study, we found that compared to NIL-ne1, the levels of pathogenesis-related genes expression and phytohormones, salicylic acid, and jasmonic acid in NIL-Ne1 are higher (Supplementary Fig. 4b, c), suggesting that Ne1 may be involved in autoimmunity activation. Similar results have been also observed in rice hybrid weakness mediated by Hwi1 and Hwi1. The activation of some PRs, such as PBZ1, was always associated with the occurrence of weakness syndrome under various conditions. Salicylic acid and jasmonic acid were highly accumulated in NIL(Hwi1) basal nodes9.
Gene duplication drives genome and genetic system evolution, facilitating functional diversification14. Our findings suggest that the 2.89 Mb insertion fragment containing ABH.1 likely formed through duplication and ectopic recombination events (Supplementary Fig. 12c). Multiple segmental duplications among hexaploid and tetraploid wheat indicated a duplication hotspot at NE1 locus (Fig. 6a). Notably, the YZM-like subtype materials harbors two 2.89 Mb insertion fragments; however, a segregation in heterozygosity is observed, which implies the potential presence of complex structural alterations within Ne1 genetic interval (Supplementary Fig. 7b). The similar situation was detected in the WYM-like subtype materials, which carries three 2.89 Mb insertion fragments (Supplementary Fig. 7b). Variations in gene copy number can alter the level of gene expression, thereby affecting traits14. Our results demonstrated that the expression level of ABH.1 was significantly higher in the CNV-2 and CNV-3 accessions relative to the CNV-1 accessions (Fig. 4d). Genetic experiments demonstrate that both allelic and copy number variation of ABH.1 contributes to hybrid necrosis.
Gene rearrangement, duplication, and fusion have the potential to create new genes. ABH.1 and ABH.2 are both located in the genetic interval of Ne1 and share 92.83% protein sequence similarity. Compared to ABH.2, ABH.1 misses the first 105 N-terminal amino acids and contains an additional 17-amino-acid signal peptide that is specific to ABH.1 orthologs in cereal species (Supplementary Figs. 5, 13–15). Sequence analysis reveals that ABH.1 is likely from gene duplication and ectopic recombination (Fig. 6a). Since the available wheat genome sequence data predominantly belong to the ZN17-like and YBM-like types (Supplementary Fig. 19), no published resequencing data exists for the materials of YZM-like and WYM-like type. Therefore, the direction of the additional 2.89 Mb fragment in YZM-like and WYM-like materials remains uncertain. With advancements in third-generation sequencing technology, this ambiguity may be resolved in the future. In our genetic experiments, the F1 hybrids derived from the cross PI 171028 × ZZ6903 exhibited necrosis symptoms (Fig. 5d). However, PI 171028, which carries the ABH.1, lacks the ABH.2 (Supplementary Fig. 20). In contrast, the F1 hybrids of ZZ6903 and ZN17, an accession lacked ABH.1 and shared the identical ABH.2 gene sequence of YBM (Supplementary Fig. 20), showed normal growth. Therefore, despite the high protein sequence identity between ABH.1 and ABH.2 (Supplementary Fig. 14), ABH.2 is not involved in wheat hybrid necrosis, suggesting that ABH.1 is a neofunctionalization gene.
In this study, among 58 T. dicoccoides wheat accessions primarily from Israel (36), Lebanon (6), Syria (8), and Turkey (6), CNV-0 and CNV-1 types were detected. However, CNV-2 and CNV-3 types were prevalent in cultivated emmer wheat subspecies (Supplementary Data 6). Further investigation is needed to determine the presence or absence of CNV-2 and CNV-3 in wild emmer wheat using more accessions.
Moreover, Ne2 was absent in all tested 584 tetraploid wheat accessions analyzed (Supplementary Data 9). The results suggest that Ne1 likely originated in wild emmer and integrated into hexaploid wheat through polyploidization, while Ne2 might be emerged post-hexaploidization or from other wheat relatives.
Wheat hybrid necrosis can hinder gene flow, impacting wheat genetic improvement38. The Ne1 functional marker Ne1-Select2128, combined with Ne2-Select91, a diagnostic marker for Ne222, can detect both Ne1 and Ne2 in a single PCR reaction (Supplementary Fig. 18b). Our findings indicate that none of the tested materials simultaneously carry both Ne1 and Ne2 (Supplementary Data 9 and 10), likely due to their detrimental interaction. The Ne2, which is identical to the leaf rust resistance gene LrZH22/Lr13 in the elite cultivar Zhoumai 2223, has experienced positive selection, while negative selection was observed for Ne1 (Fig. 6c). The positive selection for Ne2/LrZH22/Lr13 during breeding has significantly increased the frequency of the ne1 allele in modern breeding varieties. Cultivars like Andes, Chris, Ciano 67, and Pavon 76, which carry Lr13, were extensively used as leaf rust resistance sources in CIMMYT’s wheat breeding program41,42. The pleiotropic nature of the Lr13 and Ne2 genes may have contributed to the increased prevalence of Ne2 in US wheats (Fig. 6b), crowding out Ne1 and sharply reducing its proportion. The presence of Ne2/Lr13 in elite wheat varieties like Liangxing 99, Yannong 15, and Jimai 22 possibly explains the high frequency of the ne1 allele in wheat varieties of Shandong province23 (Fig. 6b).
Methods
Plant materials and growth conditions
The common wheat line M114 carries both Ne1 and Ne2, while Zhoumai 22 (ZM) is a commercially elite cultivar with Ne223. We utilized an F2 population derived from M114 × ZM for fine mapping Ne1, with subsequent phenotypic verification conducted on F3 progenies of key F2 recombinants within the Ne1 genetic interval. In another fine mapping population for Ne1, we constructed recombinant inbred lines from common wheat Zhengnong 17 (ZN17, ne1ne1Ne2Ne2) × Yangbaimai (YBM, Ne1Ne1ne2ne2) and Zhengzhou 6903 (ZZ6903, ne1ne1Ne2Ne2) × Yumai 14 (YM14, Ne1Ne1ne2ne2)26. RIL-368 (Ne1ne1Ne2Ne2) is a residual heterozygous line at the NE1 locus derived from the F8 recombinant inbred lines of the ZN17 × YBM cross. Aside from exhibiting differences in normal and necrotic phenotypes, RIL-368 does not display any significant visible variations. Therefore, we self-pollinated RIL-368 to generate nearly isogenic lines: NIL-ne1 and NIL-Ne1. To genetically verify the effect of the Ne1 allele, ZZ6903 was crossed with different wheat accessions (Mania, Chinese Spring, PI 171028, PI 182704, Hallam, and Ripper). A collection of 584 accessions of tetraploid wheat (Supplementary Data 9), and 853 accessions of hexaploid wheat (Supplementary Data 10) was used to determine Ne1 alleles. All these plant materials were planted in 1.5 m rows with 30 cm row spacing, leaving ~ 14 cm between each plant, at the experimental fields of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, in Shijiazhuang, Hebei province, China.
The transgenic experiments involved the use of common wheat cultivar Fielder, homozygous Ne2 overexpression transgenic line OE-T1−1-1 (OENe2)23, and the F1 hybrid plants from Fielder × ZM. All plants for transgenic experiments were cultivated in pots (20 cm × 20 cm) in a greenhouse with a 16 h-light/8 h-dark cycle (25 °C/18 °C, 65% relative humidity) at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China. Supplementary Data 1 presents the genotypes at the NE1 and NE2 locus for the wheat materials mentioned in this study.
Phenotyping
Wheat plants, including the parents, F1, F2, F2:3 families, EMS mutants, and transgenic plants, were evaluated for leaf necrosis at the grain-filling stage under field conditions or controlled greenhouse. Plants with complete necrotic death of flag leaf and leaves below it were scored as necrosis and those with all normal flag leaves and leaves below it were scored as non-necrotic. The Pearson’s Chi-squared (χ2) tests were performed to determine the fitness of segregation ratios to theoretical Mendelian ratios using the SAS 8.0 statistical analysis package (SAS Institute, Cary, NC, the United States).
Histochemistry
Accumulation of H2O2 at the site of lesion formation in NIL-Ne1 was visualized by DAB staining as previously described with minor modifications43. Specifically, at the flowering stage, when the necrotic area of the flag leaves of NIL-Ne1 plants was approximately 5–10%, whole flag leaf blades of both NIL-Ne1 and NIL-ne1 plants were collected. The entire leaves were then immersed in 1 mg/mL DAB staining solution for 24 h, followed by decolorization in a chloral hydrate solution for another 24 h, and rinsed twice with distilled water. Photographs were taken using a digital camera, and the staining intensities were compared. The experiment was conducted three times, each time using flag leaves from six NIL-Ne1 plants and six NIL-ne1 plants.
BSR-Seq analysis
In the bulked segregant RNA-sequencing (BSR-Seq) experiment44, the flag leaves from 50 homozygous necrotic and 50 homozygous normal F3 families from M114 × ZM at the flowering stage, represented by an equal amount of leaf tissues from single plants of each line, were collected randomly to compose the necrotic and normal RNA bulks for RNA-Seq. RNA samples were extracted using the TRIzol protocol (Tiangen, Beijing, China, Catalog No. DP424) and sequenced on an Illumina HISeq 4000 platform by Beijing Novogene Bioinformatics Technology Co. Ltd, Beijing, China, following the manufacturer’s standard protocol. The clean reads were aligned against the Chinese Spring reference genome sequence IWGSC RefSeq v2.129 using STAR software with default parameters45. SNP calling was conducted using GATK software46, with a filtration criterion of allele frequency difference (AFD) > 0.8 and Fisher’s Exact Test P-value < 1e-10.
Marker development and linkage analysis
Based on the BSR-Seq results, SNPs and InDels associated with Ne1 were selected for marker development and validation. The flanking sequences, approximately 3 kb, of the associated SNPs and InDels were identified from the Chinese Spring reference genome IWGSC RefSeq v2.129 and used as templates for designing PCR primers using the web-based program available at the GSP website (http://probes.pw.usda.gov/GSP/) to amplify 800-1000 bp sequences containing the variant. Genomic DNA was extracted from young leaves of M114, ZM, 5 homozygous necrotic, and 5 homozygous normal F3 families using the cetyl trimethyl ammonium bromide (CTAB) method47. PCR was performed in a Biometra T3000 Thermocycler (Applied Biosystems, New York, NY, USA). Each 10 µL reaction mixture comprised 5 µL of PCR mixture (including Taq polymerase, dNTPs, and 10× PCR buffer with Mg2+), 2 µL ddH2O, 1 µL DNA (50 ng µL−1), and 1 µL of 10 µmol L−1 each of the forward and reverse primers. The PCR profile included initial denaturation at 95 °C for 3 min; 95 °C for 15 s, annealing at 56–61 °C (depending on the primers used) for 10 s, and 72 °C for 1 min, 35 cycles; and extension at 72 °C for 10 min. The sequences of the primers used are detailed in Supplementary Data 3. Sanger sequencing of the amplicons was performed to validate true polymorphisms (Tsingke, China). The closest SNP markers were used as queries for BLAST against the Chinese Spring whole genome assembly sequences. The genomic sequences in the target genomic region of Ne1 were used as templates to design SSR primers using BATCHPRIMER3 software (http://batchprimer3.bioinformatics.ucdavis.edu/)48. Polymorphic SSR markers between M114 and ZM and the contrasting DNA bulks were used to construct the genetic linkage map of Ne1 after genotyping the mapping population. Linkage analysis of molecular markers and the NE1 locus was conducted using the software MAPMAKER 3.0 with a LOD score threshold of 3.049.
Mutagenesis and identification of non-necrotic mutants
Seeds of the necrotic line NIL-Ne1 were subjected to mutagenesis with EMS. A total of 10,000 seeds were incubated in deionized water for seven hours at room temperature, followed by gentle shaking for 12 hours at ambient temperature using 2000 mL of 0.5% EMS (v/v) solution. Following treatment, the seeds underwent an eight-hour rinse under running water and were left to air-dry in the fume hood overnight (~ 16 h). Subsequently, the EMS-treated M1 seeds were artificially sown in the field (Hebei, China). Approximately 170 spikes exhibiting a potential reduction of necrotic symptoms were collected from about 6000 M1 plants based on preliminary visual inspection. A total of 156 independent M2 families, comprising ~ 30 plants per plot, were visually screened for non-necrotic mutants.
Non-necrotic mutants were also generated in the necrotic M114 background. Approximately 10,000 seeds of M114 were soaked in water for eight hours, then treated with 0.5% (v/v) EMS solution at room temperature for 16 h followed by washing with running water for 4 h, covered with Whatman paper, and air-dried at 4 °C50. EMS-treated M1 plants were generated at the Gaoyi Experimental Station (Hebei, China). M2 families (25 seeds per family) were planted in the field for phenotypic investigation. All M3 seeds obtained from M2 non-necrotic mutants were planted in the field to confirm the homozygous mutations.
Total RNA was extracted from the flag leaves of homozygous non-necrotic mutants at the flowering stage in the field using a TRIzol reagent (Invitrogen, USA). First-strand cDNA was synthesized utilizing the PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Japan). The full-length cDNA sequences for Ne1 candidate genes, as well as Ne2 in all M3 homozygous non-necrotic mutants, were amplified using specific primer pairs (Supplementary Data 3) designed using IWGSC RefSeq v2.129 reference sequence with BATCHPRIMER3 software48. PCR was performed in a Biometra T3000 Thermocycler (Applied Biosystems, New York, NY, USA) in 20 μL total volumes, consisting of 10 µL of PCR mixture (including Taq polymerase, dNTPs, and 10× PCR buffer with Mg2+), 5 µL ddH2O, 1 µL cDNA (50 ng µL−1), and 2 µL of 10 µmol L−1 each of the forward and reverse primers. The thermal cycling profile is denaturation for 5 min at 95 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 59–62 °C, 2 min at 72 °C and a final extension for 10 min at 72 °C. The PCR products were sequenced by Sanger sequencing service (Beijing Tsingke Biotech Co., Ltd., Beijing, China) following the manufacturer’s standard protocol. After the Sanger sequencing data is assembled, candidate genes for Ne1 or Ne2 genes obtained from the homozygous non-necrotic mutants and the wild type (M114 or NIL-Ne1) are subjected to multiple sequence alignment using the ClustalW (http://www.clustal.org/omega/) to confirm mutation sites.
Wheat transformation
To create Ne1-transgenic wheat plants, the coding sequence of Ne1 was amplified from YBM and inserted into the pTA1037 vector using the EZ-HiFi Seamless Cloning Kit (GenStar, China). The resulting recombinant construct, proUbi::Ne1YBM, containing the Ne1YBM gene regulated by the maize ubiquitin promoter51, was transferred into the EHA105 strain and subsequently introduced into immature embryos of common wheat Fielder with the ne1ne1ne2ne2 genotype via Agrobacterium-mediated transformation with the assistance of WeiMi Biotechnology Co., LTD, Jiangsu province, China. The Ne1YBM positive-transgenic plants was confirmed by the specific primer OE-Ne1-L/R, while the expression of Ne1 was analyzed by primer qNe1 (Supplementary Data 3). To conduct genetic complementation tests, line ZZ6903 (ne1ne1Ne2Ne2) was crossed with OENe1YBM#1-#4 (Ne1 homozygous transgenic lines, i.e., Ne1YBMNe1YBMne1ne1ne2ne2). In addition, a 13,118 bp genomic fragment of Ne1M114, including the 2406 bp upstream of the start codon, the 8157 bp complete coding region and introns, and the 2555 bp downstream of the stop codon, was cloned into the vector pCAMBIA-1300. The recombinant construct was introduced into the homozygous Ne2 overexpression transgenic line OE-T1−1-1 (OENe2)23 and Fielder × ZM F1 hybrid plants, respectively, via Agrobacterium - mediated transformation. Specific PCR primer Ne1HB-jc was designed to validate the presence of Ne1 in the transgenic plants. Primer qNe1 was used to analyze the expression of Ne1 in the transgenic plants. All transgenic plants for each transgenic event were planted in the greenhouse for phenotyping.
Quantitative real-time PCR analysis
Total RNA samples were extracted from leaves of NIL-ne1 and NIL-Ne1 using the TRIzol reagent (Invitrogen, USA), and expression analysis were performed according to the protocol established by Zhang et al. 52. Five representative individual plants were selected, and equivalent quantities of leaf tissue were collected for RNA extraction. The first-strand cDNA from total RNA was synthesized using PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Kyoto, Japan), and RT-qPCR was performed with SYBR green kit (TaKaRa, Kyoto, Japan) in a Roche 480 light cycler (Roche, Switzerland). The thermocycling conditions for the RT-qPCR included an initial denaturation of 3 min at 95 °C; 40 cycles of 30 s at 95 °C, 10 s at 58 °C, and 20 s at 68 °C, followed by a final extension of 5 min at 68 °C. The relative expression level of the gene was calculated using the 2−ΔΔCt method53. RT-qPCR analysis was conducted with three biological replicates. Primers used to evaluate the transcript levels of ABH.1 and pathogen-related marker genes, including PR1, PR2, PR3, PR5, and LOX2 are listed in Supplementary Data 3. TaActin was used as an endogenous control (Supplementary Data 3). The student’s t test was utilized to evaluate statistical significance.
Measurement of free SA and JA levels
The quantification of free SA and JA in NIL-ne1 and NIL-Ne1 plants was performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS), as modified from the method described by Xiang et al.54. Fresh leaves were collected from 45-day-old NIL-ne1 and NIL-Ne1 plants and immediately frozen in liquid nitrogen. Approximately 50 mg of ground leaf tissue was weighed and transferred into a 1.5 mL tube. The samples were spiked with 10 µL of an internal standard mixture (100 ng/mL) and 1 mL of extraction solvent (methanol/water/formic acid, 15:4:1, v/v/v). The mixture was vortexed for 10 min and then centrifuged at 12,000 × g for 5 min at 4 °C. The supernatant was transferred to a new centrifuge tube and concentrated under vacuum. The residue was reconstituted in 100 µL of 80% methanol/water solution, filtered through a 0.22 µm syringe filter, and transferred to an autosampler vial for LC-MS/MS analysis. The LC-MS/MS system consisted of an ExionLCTM AD ultra-performance liquid chromatography (UPLC) system coupled to a QTRAP® 6500 + tandem mass spectrometer (SCIEX, China). The chromatographic separation was achieved on a Waters ACQUITY UPLC HSS T3 C18 column (1.8 µm, 100 mm × 2.1 mm i.d.). The mobile phase consisted of (A) ultrapure water with 0.04% acetic acid and (B) acetonitrile with 0.04% acetic acid. The gradient elution program was as follows: 0 min, 95:5 (A/B, v/v); 1.0 min, 95:5 (A/B, v/v); 8.0 min, 5:95 (A/B, v/v); 9.0 min, 5:95 (A/B, v/v); 9.1 min, 95:5 (A/B, v/v); and 12.0 min, 95:5 (A/B, v/v). The flow rate was set at 0.35 mL/min, with a column temperature of 40 °C and an injection volume of 2 µL. The electrospray ionization (ESI) source was operated at 550 °C. The ionization voltage was set at + 5500 V in positive ion mode and − 4500 V in negative ion mode, with a curtain gas (CUR) pressure of 35 psi. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode, and each ion pair was scanned with optimized declustering potential (DP) and collision energy (CE) values. Data acquisition and processing were performed using Analyst 1.6.3 and MultiQuant 3.0.3 software (SCIEX, China). The chromatographic peaks detected in the samples were integrated and corrected based on the retention time and peak shape information of the reference standards to ensure accurate qualitative and quantitative analysis. A total of six samples were analyzed, comprising three biological replicates each from NIL-ne1 and NIL-Ne1.
Subcellular localization
A. tumefaciens-mediated transient gene expression in N. benthamiana assays was performed with minor modifications32. In short, the coding region of Ne1 was cloned into the pMDC43 vector and subsequently transformed into Agrobacterium tumefaciens GV3101. Then, the construct pro35S::Ne1-GFP was co-expressed with PIP2-mCherry, plasma membrane marker32 in 4-week-old N. benthamiana leaves through A. tumefaciens-mediated infiltration. Following infiltration, the plants were incubated in a greenhouse at 25 °C for 48 h, and the fluorescence signals were assessed and captured using a laser confocal scanning microscope (Zeiss LSM780) in accordance with the manufacturer’s guidelines.
Yeast signal sequence trap system
To assess the secretory capability of the Ne1 signal peptide (SP), we conducted a yeast secretion assay to measure yeast growth on sucrose or raffinose media55. Yeast transformation was carried out following the protocol of the Yeast maker™ Yeast Transformation System 2 (Clontech, USA). The invertase-deficient yeast strain (YTK12), negative (pSUC2-Mg87), and positive (pSUC2-Avr1b) control plasmids were generously provided by Professor Qianhua Shen from the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences. The signal sequence of Ne1 was predicted with the SignalP 4.1 server56. The predicted SP sequence of Ne1 was amplified using specific primer pSUC2-Ne1-SP-F/R (Supplementary Data 3) and fused with a truncated SUC2 gene that encodes an invertase lacking an SP via the EZ-HiFi Seamless Cloning Kit (GenStar, China). The resulting construct, pSUC2-Ne1-SP, was subsequently transformed into the yeast strain YTK12. After transformation, yeast cells were selected on yeast minimal tryptophan dropout plates (CMD-W) containing 2% sucrose, 2% agar, 0.67% yeast nitrogen base without amino acids, 0.1% glucose, and 0.075% tryptophan dropout supplement. Transformed colonies were transferred to fresh CMD-W medium and incubated at 30 °C for three days. For the invertase secretion assay, transformed yeast colonies were replica plated onto YPRAA plates (2% raffinose, 2% peptone, 2% agar, 1% yeast extract, and 2 µg/mL antimycin A). After three days of incubation at 30 °C, the plates were evaluated for growth and imaged. In addition, 2, 3, 5-triphenyltetrazolium chloride (TTC) dye conversion to the insoluble red-colored triphenylformazan was used to verify invertase secretion.
Structural and allelic variation analysis for Ne1
For the structural and allelic variation analysis of Ne1, 540 hexaploid wheat33,34,35 (Supplementary Data 5), and 157 tetraploid wheat genome re-sequencing data35,36,37 (Supplementary Data 6) were analyzed. Reads from these samples were mapped to IWGSC RefSeq v2.129 with bwa software (https://sourceforge.net/projects/bio-bwa/files/) with default parameter settings. The ensuing coverage depth for every 5 kilobase (kb) window was ascertained with mosdepth software (https://github.com/brentp/mosdepth), incorporating parameter settings (-n –fast-mode –by 5000 -F 1796 -Q 40). The coverage depth for every 5 kb window was later normalized based on each sample’s total coverage depth. Furthermore, 157 tetraploid wheat genome re-sequencing data35,36,37 and nine Sitopsis genome data39,40 were utilized to conduct an origin analysis of Ne1.
Copy number variation validation of Ne1
In this study, we employed RT-qPCR technology to statistically analyze the relative copy number of Ne1 (Supplementary Data 3 and 8). Building upon previous research, we identified that Ne2 lacks homologs in the common wheat genome, existing as a single copy22, making it an endogenous control gene. We designed specific markers, qCNV-Ne1-F/R and qCNV-Ne2-F/R, targeting the unique sequences of Ne1 and Ne2, respectively. Genomic DNA was extracted from plant leaves using the cetyltrimethylammonium bromide (CTAB) method47. The relative copy number of Ne1 was determined following the experimental procedures and data analysis methods described above for gene expression level detection, with the modification that genomic DNA was utilized in place of cDNA in the reaction mixture.
Distribution of Ne1
We designed and validated a dominant marker, Ne1-Select2128 (Supplementary Data 3), targeting the specific sequence of Ne1. This marker can effectively detect the presence or absence of Ne1 alleles in both tetraploid and hexaploid wheat accessions. For Ne1-Select2128, each PCR reaction (10 µL) consisted of 5 µL 2 × Taq PCR Starmix (GenStar, China), 0.5 µL of 10 µM sense and antisense primers, 2 µL DNA template (approximately 50-90 ng), and 2 µL ddH2O. The PCR thermocycling involved an initial denaturation at 95 °C for 5 min, followed by 8 cycles of 94 °C for 30 s, 64 °C with a 0.7 °C reduction per cycle for 30 s, 72 °C for 30 s; and 25 cycles of 30 s at 94 °C, 30 s at 57 °C, 30 s at 72 °C, culminating with a final extension of 2 min at 72 °C. The PCR products were then assessed and visualized by electrophoresis on 1% agarose gels. Materials carrying Ne1 can be amplified using Ne1-Select2128, producing a distinct 1,110 bp band. A functional marker Ne2-Select91 for the detection of Ne2 was also developed22. In addition, to visualize the geographic distribution of the Ne1 allele, we generated proportional symbol maps using ArcMap 10.8.157. The maps were constructed using both national and global base maps, with underlying data sourced from Shapefile files57.
Statistics and reproducibility
In the study, we conducted most experiments with three biological replicates, except the RNA-seq data from 305 common wheat samples used to quantify ABH.1 expression for the four CNV groups represents a single biological replicate. Sample sizes were chosen based on our prior experience and the standard practices in the field. No statistical method was used to predetermine the sample size. All experiments were designed and conducted with randomization in the selection of samples to minimize potential biases. The investigators were blinded to the allocation during experiments and outcome assessment to ensure unbiased data collection and analysis. No data were excluded from the analyses. To further ensure the reproducibility of our experiments, we followed standardized protocols and used the same reagents and equipment across all replicates. Statistical analyses were conducted (two-sided Student’s t test), and charts were created using GraphPad Prism 8 (GraphPad Software). The sequence alignment charts were produced using DNAMAN version 8.
Primers
All the primers used in this study are listed in Supplementary Data 3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited in the Genome Sequence Archive in the National Genomics Data Center (NGDC), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences database under accession code CRA005878. The Ne2 and multiple Ne1 alleles sequence data generated in this study have been deposited in the GenBank database under accession code: Ne2 (MW756036), Ne1-M114 (PP781974), Ne1-CS (PP760338), Ne1-YM14 (PP760339), Ne1-YBM (PP760340), Ne1-PI182704 (PP760341), Ne1-Mania (PP760342). Source data are provided in this paper.
References
Bomblies, K. & Weigel, D. Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 8, 382–393 (2007).
Li, L. & Weigel, D. One hundred years of hybrid necrosis: hybrid autoimmunity as a window into the mechanisms and evolution of plant-pathogen interactions. Annu. Rev. Phytopathol. 59, 213–237 (2021).
Chae, E. et al. Species-wide genetic incompatibility analysis identifies immune genes as hot spots of deleterious epistasis. Cell 159, 1341–1351 (2014).
Bomblies, K. et al. Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 5, e236 (2007).
Sicard, A. et al. Divergent sorting of a balanced ancestral polymorphism underlies the establishment of gene-flow barriers in Capsella. Nat. Commun. 6, 7960 (2015).
Hermsen, J. G. T. The genetic basis of hybrid necrosis in wheat. Genetica 33, 245–287 (1963).
Ren, Z. L. & Lelley, T. Genetics of hybrid necrosis in Rye. Plant Breed. 100, 173–180 (1988).
Jeuken, M. J. et al. Rin4 causes hybrid necrosis and race-specific resistance in an interspecific lettuce hybrid. Plant Cell 21, 3368–3378 (2009).
Chen, C. et al. A two-locus interaction causes interspecific hybrid weakness in rice. Nat. Commun. 5, 3357 (2014).
Deng, J., Fang, L., Zhu, X., Zhou, B. & Zhang, T. A CC-NBS-LRR gene induces hybrid lethality in cotton. J. Exp. Bot. 70, 5145–5156 (2019).
Dobzhansky T. Genetics and the origin of species. (New York: Columbia Univeristy Press, 1937).
Muller, H. J. Isolating mechanisms, evolution, and temperature. Biol. Symp. 6, 71–125 (1942).
Orr, H. A. Dobzhansky, Bateson, and the genetics of speciation. Genetics 144, 1331–1335 (1996).
Li, X. et al. Large-scale gene expression alterations introduced by structural variation drive morphotype diversification in Brassica oleracea. Nat. Genet. 56, 517–529 (2024).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 20, 38 (2019).
Wang, Y. et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 47, 944–948 (2015).
Lee, T. G., Diers, B. W. & Hudson, M. E. An efficient method for measuring copy number variation applied to improvement of nematode resistance in soybean. Plant J. 88, 143–153 (2016).
Shen, R. et al. Genomic structural variation-mediated allelic suppression causes hybrid male sterility in rice. Nat. Commun. 8, 1310 (2017).
Sax, K. Sterility in wheat hybrids. I. sterility relationships and endosperm development. Genetics 6, 399–416 (1921).
Mizuno, N., Shitsukawa, N., Hosogi, N., Park, P. & Takumi, S. Autoimmune response and repression of mitotic cell division occur in inter-specific crosses between tetraploid wheat and Aegilops tauschii Coss. that show low temperature-induced hybrid necrosis. Plant J. 68, 114–128 (2011).
Chu, C. G., Faris, J. D., Friesen, T. L. & Xu, S. S. Molecular mapping of hybrid necrosis genes Ne1 and Ne2 in hexaploid wheat using microsatellite markers. Theor. Appl. Genet. 112, 1374–1381 (2006).
Si, Y. et al. Ne2, a typical CC-NBS-LRR-type gene, is responsible for hybrid necrosis in wheat. New Phytol. 232, 279–289 (2021).
Yan, X. et al. High-temperature wheat leaf rust resistance gene Lr13 exhibits pleiotropic effects on hybrid necrosis. Mol. Plant 14, 1029–1032 (2021).
Hewitt, T. et al. Wheat leaf rust resistance gene Lr13 is a specific Ne2 allele for hybrid necrosis. Mol. Plant 14, 1025–1028 (2021).
Athiyannan, N. et al. Long-read genome sequencing of bread wheat facilitates disease resistance gene cloning. Nat. Genet. 54, 227–231 (2022).
Si, Y. et al. Fine mapping of hybrid necrosis gene Ne1 in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 134, 2603–2611 (2021b).
Li, N., Tan, Q., Ding, J., Pan, X. & Ma, Z. Fine mapping of Ne1, the hybrid necrosis gene complementary to Ne2 in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 134, 2813–2821 (2021).
Zhang, M. et al. Fine mapping and distribution analysis of hybrid necrosis genes Ne1 and Ne2 in wheat in China. Theor. Appl. Genet. 135, 1177–1189 (2022).
Zhu, T. et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 107, 303–314 (2021).
Jia, A. et al. TIR-catalyzed ADP-ribosylation reactions produce signaling molecules for plant immunity. Science 377, eabq8180 (2022).
Pruitt, R. N. et al. The EDS1-PAD4-ADR1 node mediates Arabidopsis pattern-triggered immunity. Nature 598, 495–499 (2021).
Lee, H. K. et al. Drought stress-induced Rma1H1, a RING membrane-anchor E3 ubiquitin ligase homolog, regulates aquaporin levels via ubiquitination in transgenic Arabidopsis plants. Plant Cell 21, 622–641 (2009).
Niu, J. et al. Whole-genome sequencing of diverse wheat accessions uncovers genetic changes during modern breeding in China and the United States. Plant Cell 35, 4199–4216 (2023).
Hao, C. et al. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol. Plant 13, 1733–1751 (2020).
Zhou, Y. et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 52, 1412–1422 (2020).
Wang, H. et al. Sympatric speciation of wild emmer wheat driven by ecology and chromosomal rearrangements. Proc. Natl. Acad. Sci. USA 117, 5955–5963 (2020).
Wang, Z. et al. Dispersed emergence and protracted domestication of polyploid wheat uncovered by mosaic ancestral haploblock inference. Nat. Commun. 13, 3891 (2022).
Vikas, V. K. et al. Hybrid necrosis in wheat: evolutionary significance or potential barrier for gene flow? Euphytica 194, 261–275 (2013).
Li, L. F. et al. Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Mol. Plant 15, 488–503 (2022).
Yang, Y. et al. Genome sequencing of Sitopsis species provides insights into their contribution to the B subgenome of bread wheat. Plant Commun. 4, 100567 (2023).
Anonymous (1973) CIMMYT annual report on maize and wheat improvement 1972.
Anonymous (1976) CIMMYT annual report on maize and wheat improvement 1974.
Fitzgerald, H. A., Chern, M. S., Navarre, R. & Ronald, P. C. Overexpression of (At)NPR1 in rice leads to a BTH- and environment-induced lesion-mimic/cell death phenotype. Mol. Plant Microbe . 17, 140–151 (2007).
Xie, J. et al. A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol 228, 1011–1026 (2020).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325 (2006).
You, F. M. et al. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinform. 9, 253 (2008).
Lincoln, S. E., Daly, M. J. & Lander, E. S. Constructing linkage maps with MAPMAKER/Exp Version 3.0. A tutorial reference manual, 3rd edn. (Whitehead Institute for Medical Res., Cambridge, MA 1993).
Saintenac, C. et al. Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341, 783–786 (2013).
Cornejo, M. J., Luth, D., Blankenship, K. M., Anderson, O. D. & Blechl, A. E. Activity of a maize ubiquitin promoter in transgenic rice. Plant Mol. Biol. 23, 567–581 (1993).
Zhang, Y. et al. Mediator subunit 16 functions in the regulation of iron uptake gene expression in Arabidopsis. New Phytol. 203, 770–783 (2014).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Xiang, Y. A jacalin-related lectin-like gene in wheat is a component of the plant defence system. J. Exp. Bot. 62, 5471–5483 (2011).
Oh, S. K. et al. In planta expression screens of RXLR effectors reveal diverse phenotypes, including activation of the disease resistance protein Rpi-blb2. Plant Cell 21, 2928–2947 (2009).
Nielsen, H. Predicting secretory proteins with SignalP. Methods Mol. Biol. 1611, 59–73 (2017).
ESRI. ArcGIS Desktop: Release 10.8.1. Environmental Systems Research Institute, Redlands. (2020).
Acknowledgements
This work was supported by the STI 2030–Major Projects (2023ZD04070 to M.M.L.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA24010204 to H.-Q.L.), the National Natural Science Foundation of China (31991211 to H.-Q.L.; 32101735 to M.M.L.; 32302364 to X.C.Y.; U21A20224 to Z.Y.L.), the National Key Research and Development Program of China (2023YFF1000600 to L.L.D. and 2023YFD1200402 to Z.Y.L.), the Major Project of Agricultural Biological Breeding (2023ZD0402501 to H.Z.Z.), the Major Basic Research Program of Shandong Natural Science Foundation (ZR2019ZD15 to H.-Q.L.). This project was also supported by grants from the Hainan Seed Industry Laboratory (B21HJ0111 to Z.Y.L.), China Postdoctoral Science Foundation (2022M710163 to Y.Q.S.), Key Research and Development Program of Hebei (22326305D to C.G.Y.) and Yazhouwan National Laboratory project (2310JM01 to S.W.M.).
Author information
Authors and Affiliations
Contributions
H.-Q.L., Z.L., M.L., and L.L.D. conceived and designed the study. Y.S., H.Z., S.Z., J.N., S.T., X.C., K.Z., Q.L., Z.Z., T.D., P.L., M.G., Y.C., and Q.W. carried out the experiments, conducted fieldwork and analyzed data. S.M., J.X., G.G., and L.L.D. performed bioinformatic analysis. Z.F.L., X.Y., C.Y., Y.L., and H.W. provided germplasm, scientific support, and advice. H.-Q.L., Z.L., M.L., and Y.S. wrote the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks André Laroche and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Si, Y., Zhang, H., Ma, S. et al. Genomic structural variation in an alpha/beta hydrolase triggers hybrid necrosis in wheat. Nat Commun 16, 2655 (2025). https://doi.org/10.1038/s41467-025-57750-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57750-5
