
Abstract
The mammalian pattern-recognition receptor TLR4/MD-2 (Toll-like receptor 4/myeloid differentiation factor-2) can be activated by a wide variety of pathogen-associated and endogenous molecules, with Gram-negative bacterial lipopolysaccharide (LPS) being the primary natural TLR4 agonist. Activation of TLR4 triggers cellular signaling that enables the beneficial innate immune responses and enhances adaptive immunity, thereby emphasizing the potential of TLR4 agonists for the management of diseases with an immunopathological background and for use as vaccine adjuvants. Given the challenges associated with LPS-derived products, including structural complexity, heterogeneity, toxicity, and species specificity, synthetic molecules targeting TLR4/MD-2 offer a promising alternative. Here, we elucidate the structural basis for the recognition of synthetic LPS-mimicking glycolipids, Disaccharide Lipid A Mimetics (DLAMs), by human and mouse TLR4/MD-2 through cryo-EM structures of six dimeric [TLR4/MD-2/ligand]2 complexes resolved at 2.2-3.1 Å. We reveal that the specific binding modes of DLAMs, distinct from those of LPS, are essential for the species-independent TLR4 agonistic activity. DLAMs function as a molecular bridge, effectively induce the dimerization of TLR4/MD-2 complexes through specific carbohydrate structure-relevant ligand-protein interactions. Our findings reveal the distinct molecular modes of TLR4 activation, and provide a structural basis for the rationale design and development of innovative, highly potent TLR4-targeting immunotherapeutics and adjuvants.
Similar content being viewed by others
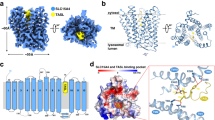

Introduction
TLR4/MD-2 is a pattern recognition receptor (PRR) responsible for the innate immune responses against infectious diseases and plays an important role in immune homeostasis. TLR4 is widely expressed on the cell surface of primary immune cells such as dendritic cells, macrophages, and monocytes, where it senses and responds to circulating pathogen-associated and endogenous danger-associated molecular patterns (PAMPs and DAMPs), as well as on epithelial cells, primarily of lung/bronchial and intestinal epithelia, in the heart, and other tissues. Activation of TLR4 by various PAMPs and DAMPs leads to the induction of intracellular pro-inflammatory signaling pathways that contribute to infection clearance and healing1,2,3,4,5,6,7. Systemic dysregulation of TLR4-mediated signaling has been implicated in a variety of chronic diseases with an immunopathological background favouring the use of TLR4 agonists as therapeutics8. Targeted activation of TLR4 by lipopolysaccharide (LPS) or lipid A variants has been shown to ameliorate the Alzheimer’s disease-related pathology9,10, to have beneficial therapeutic effects in asthma11,12 and to contribute to the management of bacterial and viral infections13,14,15,16. Activation of TLR4-mediated signaling promotes antigen processing and presentation by APCs (Antigen-Presenting Cells), thereby bridging innate and adaptive immunity, making the TLR4 complex an ideal target for the discovery of leads to act as adjuvants17,18. Mounting evidence suggests that TLR4 activation on tumor cells and in the cells of the tumor microenvironment can enhance antitumor immunity and the application of LPS-derived TLR4 agonists or synthetic lipid A has shown potential in treating solid tumors and reducing cancer metastasis19,20,21,22,23,24,25,26. The most relevant TLR4 agonists with therapeutic potential include isolated and/or genetically engineered and biochemically modified LPS/lipid A variants27,28. While monophosphoryl lipid A (MPLA) derived from the partial hydrolysis of S. Minnesota Re595, is approved as an adjuvant in several vaccines, significant heterogeneity and the presence of TLR4-antagonizing impurities in clinical-grade MPLA highlight the need for the development of molecularly defined vaccine adjuvants29,30.
LPS is a micro-heterogeneous glycolipid, which is the major component of outer membrane of Gram-negative bacteria. LPS with a molecular weight of 10-20 kDa, consists of a membrane-bound immunomodulatory portion lipid A and an extracellular polysaccharide composed of a relatively conserved ‘core’ region, and a variable O-antigen (Fig. 1a)31. The structure of lipid A is based on a β(1→6)-linked diglucosamine backbone, with (R)-3-alkanoyloxyalkanoyl and/or (R)-3-hydroxyalkanoyl lipid chains attached at positions 2, 3, and 2´, 3´ in symmetric or asymmetric distribution and two phosphate groups in positions 1 and 4´ (Fig. 1a). Many factors such as the number (four to seven), chemical structure, length, and attachment sites of the lipid chains as well as the number and position of the phosphate groups (at C1-OH and/or C4′-OH) determine whether an LPS variant can be recognized by the TLR4 complex and induce the pro-inflammatory intracellular signaling32. The structural basis for the recognition of the most efficient TLR4 agonist, hexaacylated bisphosphorylated Escherichia coli LPS (Fig. 1a), has been elucidated by co-crystal structures of human (h) and mouse (m) TLR4 complexes with E. coli Ra-LPS (PDB code 3FXI) and Re-LPS (PDB code 3VQ2), respectively, revealing that LPS-binding induces the formation of an m-shaped [TLR4/MD-2/LPS]2 complexes (Fig. 1b)33,34. The juxtaposition of two TLR4/MD-2/LPS complexes leads to the formation of a close contact area (homodimerization interface), which is a crucial receptor-activating event leading to the proximity-induced recruitment of intracellular adapter proteins and the assembly of cytosolic multiprotein signaling hubs3,7,35. The ligand-induced dimerization of the extracellular domains of TLR4, which are physically associated with the co-receptor protein MD-2, is driven by numerous hydrophobic and ionic interactions between LPS, the parent TLR4/MD-2 complex and the second TLR4*, as shown by crystal structures, molecular dynamics (MD)-simulations and mutagenesis studies33,34,36,37,38,39,40,41,42, with significant contributions from both the acyl chains of lipid A and the sugar residues of the Core region (Fig. 1b), emphasizing the structural requirements for potent TLR4 activation. However, the use of wild-type or genetically engineered LPS variants as potential therapeutics has several limitations, such as their enormous structural heterogeneity and cellular toxicity, making synthetic molecularly defined TLR4 ligands with a simplified structure and safer biological profile a more feasible option. Although synthetic lipid A or its derivatives are a valuable alternative, lipid A is known to be at least 10-fold less active than Re-LPS (Kdo2-lipid A)43, and its biological activity is difficult to predict due to the influence of numerous complex structural factors32,44. The inherent flexibility of the GlcNβ(1 → 6)GlcN backbone of lipid A (Fig. 1a), which readily adapts its molecular shape to fit into the hydrophobic, capacious binding pocket of MD-2, contributes to the complexity of predicting the immunological outcome of TLR4/MD-2-lipid A interactions45. Despite these shortcomings, synthetic and genetically engineered lipid A variants are most widely used as TLR4 agonists27,32,46,47. Atomic resolution structures of agonist-bound dimeric TLR4/MD2 are 3FXI (hTLR4/MD-2/Ra-LPS), 3VQ2 (mTLR4/MD-2/Re-LPS), 3VQ1 (mTLR4/MD-2/lipid IVa), 5IJD (mTLR4/MD-2/lipid A), and 7MLM (mTLR4/MD-2/Sulfatides), which serve as a critical foundation for the rational design of TLR4 agonists33,34,36,37.

a Schematic representations of LPS, structure of Re/Ra-LPS (stick model from PDB: 3FXI), and chemical structure of E. coli LPS. b Crystal structure of the hTLR4/MD-2/Ra-LPS complex (PDB code: 3FXI). TLR4 and MD-2 are shown as cartoons, with Ra-LPS displayed as spheres. c Design of Disaccharide Lipid A Mimetics (DLAMs) based on α,α-1,1′-disaccharide scaffold.
In order to develop LPS-mimicking TLR4 agonists with a simplified structure lacking core sugars, while retaining the full immunostimulatory activity characteristic of LPS, we employed a strategy of restricting the conformational flexibility of the native βGlcN(1→6)GlcN backbone of LPS by replacing it with a synthetic conformationally confined nonreducing disaccharide reflecting the molecular shape of the TLR4/MD-2 bound diglucosamine backbone of lipid A (Fig. 1c)48,49. Using chemical synthesis, we have created a class of carbohydrate-based TLR4 agonists featuring hexa-lipidated and di- or mono-phosphorylated nonreducing disaccharide backbone: the Disaccharide Lipid A Mimetics (DLAMs)50,51. We have chemically synthesized a library of hexaacylated DLAMs based on the αGlcN(1↔1)αMan scaffold (Fig. 2a) and demonstrated that these glycolipids can induce potent (pico- to nanomolar range) TLR4-mediated cellular responses in a variety of primary immune cells of human and mouse origin as well as airway epithelial cells48,52. By varying the acylation and phosphorylation patterns on the Man moiety, the induction and release of cytokines could be fine-tuned, allowing tailored species-independent modulation of cellular responses52. Given the importance of understanding the molecular basis for the specific TLR4-mediated agonistic activity of DLAMs, we used DLAMs as versatile probes to determine atomic resolution cryogenic electron microscopy (cryo-EM) structures of [TLR4/MD-2/DLAM]2 complexes.

a Chemical structures of α,α-DLAMs and their EC50 values52 for the TLR4-mediated release of specific cytokines. b Size exclusion chromatography (SEC) (left) and native gel electrophoresis (right) of DLAMs or LPS-bound TLR4/MD-2 complex. The selected fractions for cryo-EM analysis (P2) are marked with the red square. SEC was performed three times for cryo-EM analysis (n = 3). Samples used for native gel electrophoresis were derived from the SEC experiment, and the gels were processed in parallel. The native gel electrophoresis experiment was repeated independently 2 times with similar results. Source data are provided as a Source Data file. c The cryo-EM maps of the h- and mTLR4/MD-2 in complex with DLAM5, DLAM3, or DLAM1. The colors of each component are indicated. d Side view of h- and mMD-2 (surface model) with bound DLAMs (spheres). GlcN portion of the DLAM and GlcN-linked lipid chains are colored blue; Man and Man-linked lipid chains are colored salmon.
Results
Cyro-EM structures of h- and m-TLR4/MD-2/DLAM complexes
To elucidate the structural basis for the recognition of DLAMs by the TLR4/MD-2 complex and to gain insight into the molecular interactions behind their tailored pico- to nanomolar potency, we determined the cryo-EM structures of h- and mTLR4/MD-2 in complex with three structurally different lipid A mimetics (DLAM5, DLAM3 and DLAM1), which exhibit different biological activities (Fig. 2a, Table 1, Supplementary Fig. 1)52. Whereas DLAM5 with the Man/C6-OH linked branched acyloxy lipid chain (R6/R6´) and a phosphate group at C4-OH is the most potent picomolar inducer of TLR4-mediated pro-inflammatory signaling in a variety of human and mouse primary immune cells, DLAM1 with a reversed acylation and phosphorylation pattern (acyloxy lipid chain at C4-OH and the phosphate group at C6-OH of the Man moiety) is slightly less efficient in inducing cytokine release (Supplementary Fig. 1c–e)52. DLAM3 - the monophosphorylated counterpart of DLAM1 lacking the phosphate group on the Man moiety – shows a significant loss of activity on both h- and mTLR4 (a drop from the picomolar to the nanomolar range) (Supplementary Figs. 1 and 2).
Initially, we examined whether DLAMs could induce the dimerization of h- and mTLR4/MD-2 complexes under in vitro conditions. Upon incubation of each DLAM with the TLR4 ectodomain/MD-2 complex (human or mouse), and subsequent gel filtration chromatography, three distinct peaks in the elution profiles were observed. The second peak (P2) corresponded to the elution volume of the LPS-bound TLR4/MD-2 heterotetramer ([TLR4/MD-2/LPS]2), suggesting that DLAMs can also induce TLR4/MD-2 dimerization (Fig. 2b and Table 2). The variability in the intensity of the P2 peak among different DLAMs likely reflects differences in their agonistic potencies. Among the tested DLAMs, DLAM5 was the most potent TLR4 agonist, inducing efficient dimerization of mTLR4/MD-2 in vitro, as evidenced by both SEC and native gel analysis (Fig. 2b, yellow, lane 10). In contrast, DLAM3 exhibited the weakest dimerization efficiency (Fig. 2b, green, lane 9), consistent with its reduced agonistic potency in mouse cell assays. For hTLR4/MD-2, agonist-induced dimerization was less efficient than that observed for mTLR4/MD-2. Even in the presence of LPS, less than half of the hTLR4/MD-2 complex formed dimers in both SEC and native gel electrophoresis (Fig. 2b, black, lane 2). Furthermore, for DLAM1 and DLAM5, the dimerized bands of hTLR4/MD-2 were not clearly observed on the native gel (Fig. 2b, purple and pink, lanes 3 and 5, respectively), likely due to the dissociation of hTLR4/MD-2 dimers during electrophoresis.
Despite this limitation, we selected the P2 fraction for cryo-EM structural studies because it represents the dimerized complex required for analysis, regardless of its relative abundance in SEC. This approach allowed us to successfully determine the cryo-EM structure of the TLR4/MD-2 in complex with DLAMs at atomic resolution (2.24 Å for [hTLR4/MD-2/DLAM5]2, 2.9 Å for [hTLR4/MD-2/DLAM3]2, 2.7 Å for [hTLR4/MD-2/DLAM1]2, 3.1 Å for [mTLR4/MD-2/DLAM5]2, 2.6 Å for [mTLR4/MD-2/DLAM3]2, and 2.6 Å for [mTLR4/MD-2/DLAM1]2) (Fig. 2c, Supplementary Figs. 3–8 and Supplementary Table 1). Atomic models for TLR4/MD-2 were adjusted after fitting the crystal structures of monomeric TLR4/MD-2 (PDB code: 3FXI (hTLR4/MD-2/Ra-LPS) and 3VQ2 (mTLR4/MD-2/Re-LPS))33,34. X-ray structural and quantum mechanics data for trehalose and other non-reducing disaccharides were used to construct the αGlcN(1↔1)αMan disaccharide backbone of DLAMs53,54,55,56, where both pyranose rings were presented in a 4C1 conformation with (R)-3-alkanoyloxyalkanoyl lipid residues linked via acyl- or amide bonds. The geometry of the entire glycolipid was precisely controlled using CIF files and then modeled within the well-defined cryo-EM density map. The overall structures of [TLR4/MD-2/DLAM]2 complexes closely resembled the crystal structure of LPS-bound [TLR4/MD-2]2 complexes, but appeared slightly larger overall when aligned by Cα atoms. This subtle increase in size is likely due to the absence of crystal lattice contacts, which can slightly compress adjacent TLR4/MD-2/LPS complexes in the X-ray structure. (Supplementary Fig. 9; Supplementary Table 2). Five out of the six lipid chains of the DLAMs were buried deep inside the MD-2 pocket, while the remaining lipid tail was exposed at the opening of the β-sandwich in MD-2 and participated in the formation of the secondary dimerization interface with TLR4* (Fig. 2d).
Species-Independent Binding of DLAM5 to h- and m-TLR4/MD-2
The Cryo-EM structure of h- and mTLR4/MD-2 in complex with picomolar TLR4 agonist DLAM5 showed that the glycolipid is accommodated in the binding pockets of h- and mMD-2 in very similar poses with GlcNα(1↔1)αMan backbone aligned along the secondary dimerization interface (Fig. 3a, b). Five out of six acyl chains of DLAM5 are deeply embedded in the hydrophobic pocket of MD-2 while the Man-linked 3-hydroxyalkanoyl chain R6´ is exposed on the surface of MD-2 where it participates in the formation of the secondary dimerization interface by interacting with conserved Phe residues of TLR4* and MD-2 (F440*hTLR4* or F438*mTLR4* and F126MD-2) (Fig. 3a, b). According to previously reported structural data, only the closed form of MD-2, which is characterized by the inward shift of the conserved side chain F126MD-2 to form a hydrophobic interaction with the exposed lipid chain of the ligand, is capable of dimerization33,34,36. In perfect agreement with this observation, our results confirm that DLAM5 binding induces a similar shift in F126MD-2, forming short-range hydrophobic contacts with the exposed lipid chain. (Supplementary Fig. 10).

The detailed interactions of DLAM5 with hTLR4/MD-2 (a) and mTLR4/MD-2 (b) complexes. The interactions of DLAM5 are marked by red (electrostatic interaction) and black (hydrophobic interaction) dashed lines. c Superimposition of DLAM5 bound to h- and mTLR4/MD-2. The distances between lipid chain tips are indicated as a–c. d Comparison of the distribution of hydrophobic residues in human and mouse MD-2 pockets, highlighting their contribution to the conformational variability and lipid chain orientation in DLAM5. The disaccharide moiety and overlapped chains of DLAM5 (R2′, and R6) were omitted. e The detailed interactions between hTLR4/MD-2 and lipid A portion of LPS in hTLR4/MD-2/Ra-LPS complex (PDB: 3FXI) f The detailed interactions between mTLR4/MD-2 and lipid A portion of LPS in mTLR4/MD-2/Re-LPS complex (PDB: 3VQ2). Only the lipid A portion is shown for clarity. Superimposition of DLAM5 and E. coli lipid A bound to hTLR4/MD-2 (g) and to mTLR4/MD-2 (h). The colors of DLAM5 and TLR4/MD-2 are consistent with Fig. 2c.
The whole DLAM5 molecule is tightly coordinated within the h- and mTLR4 complex through multiple electrostatic and H-bond interactions. The specific binding pose of the GlcNα(1↔1)αMan backbone of DLAM5 brings the GlcN-linked phosphate group P4´ in a favorable orientation where it is tethered by ionic bond to K362hTLR4 of the hTLR4 concave region. In mTLR4/MD-2/DLAM5 complex, this interaction is replaced by R337mTLR4/K360mTLR4 which coordinate the phosphate group P4´, while the Q339mTLR4 forms electrostatic interaction with the hydroxyl group Man-O3. The Man-linked phosphate group P4 forms ionic bond with the positively charged side chain K122hMD-2, which is replaced by E122mMD-2 in the mouse complex (Fig. 3a, b, Supplementary Fig. 11, and Supplementary Tables 3 and 4). Ionic interactions also contribute significantly to the maintenance of the dimerization interface between TLR4/MD-2/DLAM5 and TLR4*: the positively charged side chain of a conserved R90MD-2, which is stabilized by interaction with E92MD-2, forms a tight ionic bridge with E439*hTLR4* (E437*mTLR4* in the mouse complex) at the dimerization interface. In addition, the side chains of Q436*hTLR4* at the convex area of hTLR4* form electrostatic interactions with the carbonyl oxygen of R6′ (Fig. 3a, b). All these molecular forces appear to be critical for the high affinity of DLAM5 and the efficient ligand-induced dimerization of TLR4/MD-2 complex, leading to the potent species-independent agonistic activity.
As hydrophobic interactions are known to play an important role in the ligand binding by MD-2, we compared the binding characteristics of DLAM5 within the deep hydrophobic pockets of h- and mMD-2. Lipid chains R2′ and R6 occupy essentially the same space (Fig. 3c, d), whereas the R6′, R2″, and R3′ chains of DLAM5 fold back into the pocket of hMD-2, resulting in tip end displacements of 5.2, 7.6, and 7.0 Å, respectively, compared to mMD-2 (Fig. 3c). The tip of the exposed R6′ chain in the hTLR4/MD-2 complex orients toward V135 within the hMD-2 pocket to avoid steric hindrance to the hydrophilic side chain of S127hMD-2. In contrast, the tip of the R6′ chain in the mTLR4/MD-2 complex is positioned closer to the dimerization interface due to favorable hydrophobic interactions with P127mMD-2 (Fig. 3d). The species-specific variations in the hydrophobic lining the MD-2 pocket (e.g., V135hMD-2 vs. A135mMD-2 or I63hMD-2 vs. V63mMD-2) cause a slight shift in the positioning of the lipid chains (Fig. 3d), while maintaining high ligand affinity in both hTLR4 and mTLR4 complexes. Additionally, the binding orientation of the αGlcN(1↔1)αMan backbone of DLAM5 showed a slight shift in h- or mMD-2 (Fig. 3c), since the strong ionic interactions between P4 and K122hMD-2 are replaced by the hydrogen bond with E122mMD-2. Moreover, the elongated R434*mTLR4* side chain, unlike the shorter Q436*hTLR4*, allows the disaccharide moiety to be pushed further away from the mMD-2/mTLR4* dimerization interface, displacing GlcN-O1 of DLAM5 by approximately 1.1 Å. Q339mTLR4 interacts with the Man-O3 and the strong interaction between P4′ and R337mTLR4 exerts more pulling force on DLAM5. These combined effects contribute to a slight shift of DLAM5 in the mMD-2 pocket, compared to hMD-2.
Since the lipid A mimetic DLAM5 is able to induce the intracellular signaling pathway with LPS-like potency, we compared its binding interactions with those of the lipid A portion of E.coli Ra- or Re-LPS (Fig. 3e, f). In general, two possible binding orientations (usually associated with opposite biological activities) have been previously identified for LPS ligands: a ‘primary’ binding pose characteristic of the TLR4 agonist LPS in complex with h- and mTLR433,34 and an ‘inverted’ binding orientation (rotation by 180°) typically adopted by TLR4 antagonists (although only in complex with hTLR4/MD-2)57,58. A ‘primary’ binding pose, where the phosphate group P1 (attached to the proximal GlcN residue of lipid A) faces the dimerization interface and the 2N-linked lipid chain of the proximal GlcN moiety is exposed on the surface of MD-2, is characteristic of agonistic LPS ligands independently of their acylation pattern [e.g., E. coli LPS in complex with hTLR4/MD-2 (PDB: 3FXI) and with mTLR4/MD-2 (PDB: 3VQ2) and lipid IVa in complex with mTLR4/MD-2 (PDB: 3VQ1) (Fig. 3e, f)33,34. A 180° inverted binding orientation was shown for two lipid A-like TLR4 antagonists: tetraacylated lipid IVa (PDB: 2E59)58 and the synthetic compound Eritoran (PDB: 2Z65)57, both in complex with hMD-2. In these co-crystal structures, the diglucosamine backbone of lipid IVa is rotated by 180°, so that the P1 group faces the concave region of TLR4 (the primary interface between TLR4 and MD-2), while neither lipid chain is exposed, consistent with ligand’s antagonistic activity57,58.
The most striking difference in the binding of the lipid A portion of LPS and DLAM5 lies in the orientation of their disaccharide backbones. Unlike the primary binding pose of the βGlcN(1→6)GlcN backbone in E. coli lipid A, the GlcNα(1↔1)αMan backbone of DLAM5 is rotated by 90° and aligns along the secondary dimerization interface (Fig. 3g, h). This specific binding orientation leads to repositioning of the phosphate groups P4 and P4′ of DLAM5 relative to the those of LPS (P1 and P4′) within the complex resulting in the appearance of new short-range electrostatic interactions. The most prominent are the ionic bond tethering P4′ via side chains of K362hTLR4 and the interactions between P4 and K122hMD-2. In addition, the hydrogen bonds formed between the edge residues (S118hMD-2/S120hMD-2/K122hMD-2) and LPS (P4′/R2-N/R3-O/GlcN-O4) are absent in DLAM5. Superimposition of TLR4/MD-2-bound DLAM5 and E. coli LPS reveals that all lipid chains of both molecules are substantially overlapped except for the tip of the exposed lipid tail (R6′ for DLAM5 and R2 for LPS) (Fig. 3g, h). When bound to the mTLR4 complex, DLAM5 forms strong ionic interactions between P4′ and R337mTLR4/K360mTLR4 driven by a conformational change in the N359-S362 loop of mTLR4 (Fig. 3b). However, in the mTLR4/MD-2/Re-LPS complex, the ionic bond between K360mTLR4 and P1 of the Re-LPS keeps the N359-S362mTLR4 loop close to the proximal GlcN (Fig. 3f). These conformational changes highlight the conformational adaptability of the mTLR4/MD-2 to accommodate structurally different ligands and explain the specific species-independent picomolar potency of DLAM5. Additionally, the binding orientation of LPS is influenced by its bulky oligosaccharide core, which significantly contributes to defining the primary binding pose of GlcNβ(1→6)GlcN backbone of LPS in the TLR4/MD-2/LPS complex. In contrast, the binding mode of the small lipid A-mimicking molecule DLAM5 to the TLR4/MD-2 complex is determined solely by the bisphosphorylated GlcNα(1↔1)αMan backbone and its six acyl chains.
Species-Specific Binding of DLAM3 to h- and m-TLR4/MD-2
A weak TLR4 agonist DLAM3 (Supplementary Figs. 1 and 2) lacking the second phosphate group on the Man moiety and featuring a branched lipid chain attached at C4-OH of the Man (Fig. 2a) – was found to adopt deviating binding poses in complex with h- and mTLR4/MD-2 (Fig. 4a, b). When complexed with hTLR4/MD-2, the αGlcN(1↔1)αMan backbone is aligned along the heterodimerisation interface and the single phosphate group P4′ on the GlcN moiety of DLAM3 forms tight ionic bonds with hTLR4 (R264hTLR4, K341 hTLR4, and K362hTLR4) (Fig. 4a, and Supplementary Fig. 11; Supplementary Table 5). The most prominent ligand-protein interactions are observed between hMD-2 and the acyloxy groups linking the lipid chains to the disaccharide backbone of DLAM3, whereas only a few long-range contacts are found at the dimerization site with hTLR4*. The oxygens of sugar hydroxyl group Man-O3 and acyl group Man-R4-O are anchored by the main chain of S120hMD-2 and K122hMD-2 through hydrogen bonds. In addition, the charged side chains of E92/R90hMD-2 and E439*hTLR4* form long-range electrostatic interactions with the oxygens of lipid ester groups, while the Man-linked R4´ chain of DALM3 is exposed on the surface of hMD-2 and undergoes hydrophobic contacts with F440*hTLR4* at the heterodimerization interface. Compared to the primary binding pose of the βGlcN(1→6)GlcN backbone in E. coli LPS, the GlcNα(1↔1)αMan backbone of DLAM3 in human MD-2 pocket is rotated by 60° (Fig. 4d).

a and b The detailed interactions of DLAM3 with hTLR4/MD-2 (a) and mTLR4/MD-2 (b). The direct interactions with DLAM3 are indicated by dashed lines as in Fig. 3. The colors of DLAMs and TLR4/MD-2 are consistent with Fig. 2c. c Superimposition of DLAM3 bound to h- and mTLR4/MD-2 complexes demonstrating species-specific binding poses. Superimposition of DLAM3 and E.coli lipid A part of LPS bound to hTLR4/MD-2 (d) and mTLR4/MD-2 (e). The colors of DLAMs and TLR4/MD-2 are consistent with Fig. 2c.
When bound to mTLR4/MD-2, the αGlcN(1↔1)αMan backbone of DLAM3 is rotated by 90°, bringing its single phosphate group P4′ into close apposition to the secondary dimerization interface, while the distribution of all lipid chains is completely reversed compared to the hTLR4-bound ligand, placing the GlcN-linked lipid chain R3″ close to the MD-2 cavity mouth, which leads to its exposure on the surface of MD-2 (Fig. 4b, c). In this way, a single phosphate group P4′ of DLAM3 forms an ionic bridge with R434*mTLR4* and the carbonyl oxygen of R3″ makes a hydrogen bond with R434*mTLR4*, thus contributing to the ligand-induced dimerization (Fig. 4b and Supplementary Fig. 11; Supplementary Table 6). Hydrogen bonds tightly coordinate the molecule through the interactions of hydroxyl groups Man-O2/Man-O6 with and K360mTLR4 and S120mMD-2, while the acyl groups R4-O/R4′-O are tethered by hydrogen bonds with K263mTLR4 and Y102mMD-2, respectively. Despite the different binding pose of DLAM3 in hTLR4/MD-2 and mTLR4/MD-2, the conserved E92 mMD-2, R90 mMD-2, and E437* mTLR4* facilitate long-range electrostatic interactions with the carbonyl oxygen of R4′/R2″/R3″ on DLAM3 (Fig. 4b). Superimposition of DLAM3 and LPS in mTLR4/MD-2 complex reveals a 4 Å shift in the disaccharide αGlcN(1↔1)αMan backbone of DLAM3 compared to the diglucosamine backbone of LPS (Fig. 4e). This displacement repositions the phosphate group P4′ on DLAM3, enabling its interaction with the charged residue R434*mTLR4*.
Species-specific binding of DLAM1 to h- and m-TLR4/MD-2
Species specificity is a well-recognized challenge in ligand recognition by TLR4/MD-2, complicating the translation of in vitro cellular experiments conducted on human primary immune or epithelial cells to in vivo studies in mice and, ultimately, to human clinical trials. To gain deeper insight into the structural features responsible for species-specific recognition of monophosphate DLAM3 - a mimetic of monophosphoryl lipid A (MPLA) - by h- and mTLR4/MD-2, we determined the cryo-EM structure of DLAM1 (a bisphosphorylated counterpart of DLAM3) in complex with h- and mTLR4/MD-2 (Figs. 2a and 5a, b). In complex with hTLR4, the αGlcN(1↔1)αMan backbone of DLAM1 adopts a binding pose resembling the primary binding orientation of the lipid A portion of E. coli LPS (Fig. 5a). As DLAM1 is a conformationally confined analog of E. coli LPS, the binding orientation of its αGlcN(1↔1)αMan backbone and the overall distribution of the lipid chains were found to closely resemble those of LPS. The only notable difference was a slightly altered orientation of the tip of the Man-linked R4′ lipid chain, which is exposed at the secondary dimerization interface (corresponding to the R2 lipid chain of LPS) (Fig. 5d). Upon superimposition of two structures, the phosphate groups P4′ of Ra-LPS and DLAM1 nearly align, both forming an H-bond with S118hMD-2 and an electrostatic interaction with R264hTLR4. Meanwhile, the primary phosphate group P6 of DLAM1 (which replaces the P1 of LPS) is anchored by K362hTLR4 and K341hTLR4 of the parent hTLR4 (Figs. 5a and 3e). Nearest to the dimerization interface, the carbonyl oxygen R4′-O forms hydrogen bond with S415*hTLR4* and Q436*hTLR4*, while F440*hTLR4* and F126hMD-2 form a hydrophobic region that secures the R′ lipid chain and keeps it exposed on the surface of MD-2 (Fig. 5a, Supplementary Fig. 11 and Supplementary Table 7). The molecule is further stabilized by the interaction of multiple ester groups with both the parent TLR4/MD-2 complex and the second TLR4*, i.e. the carbonyl oxygen R3′-O is tethered by main chain of S120hMD-2, while the charged residues E92hMD and R90hMD on the hDM-2 rim, along with E439*hTLR4*, stabilize the ester groups R2″-O and R4′-O through the long range electrostatic interactions (Fig. 5a, Supplementary Fig. 11; Supplementary Table 7).

a, b The detailed interactions of DLAM1 with hTLR4/MD-2 (a) and mTLR4/MD-2 (b) complexes. The direct interactions of DLAM1 with TLR4/MD-2 are marked by dashed lines as in Fig. 3. c Superimposition of DLAM1 in h- and mTLR4/MD-2 complexes. Two-color models of DLAM1 are present. Superimposition of DLAM1 and E.coli lipid A part of LPS bound to hTLR4/MD-2 (d) and mTLR4/MD-2 (e). The colors of DLAMs and TLR4/MD-2 are consistent with Fig. 2c.
Unexpectedly, the additional phosphate group attached to the Man moiety of DLAM1 (compared to DLAM3) did not prove to be the game changer in overcoming species-specific recognition by the mTLR4/MD-2 complex, as DLAM1 adopted a 180° rotated binding pose in complex with mTLR4/MD-2, with the GlcN-linked R3″ lipid chain exposed at the secondary dimerization interface, a feature previously observed for DLAM3 (Fig. 5b, c). The P6 group of DLAM1 forms direct ionic contacts with K263mTLR4, while the P4′ exposed at the heterodimerization interface interacts with R434*mTLR4* (Fig. 5b, Supplementary Fig. 11 and Supplementary Table 8). The electrostatic interaction formed between Man-O2/R3″-O and K360mTLR4/E437*mTLR4* also contribute to the DLAM1 stabilization. The five lipid chains of DLAM1 are buried within the deep hydrophobic mMD-2 pocket, while the secondary acyl chain R3″ extends towards the heterodimerization interface, forming hydrophobic contacts with F438*mTLR4* and F126mMD-2. Superimposition of mTLR4/MD-2-bound DLAM1 and LPS/mTLR4 reveals that the disaccharide αGlcN(1↔1)αMan backbone of DLAM1 is shifted upwards by 5 Å compared to diglucosamine backbone of LPS (Fig. 5e). This displacement repositions the phosphate groups of DLAM1, allowing them to efficiently interact with the charged residues of TLR4 and TLR4*. In this instance, DLAM1 acts as a bridge that effectively cross-links the two receptor proteins together.
Comparison of binding orientations of DLAMs
Superimposition of the carbohydrate backbone of DLAM3 and pico-molar agonist DLAM5 complexed with hTLR4/MD-2 shows a 30° shift between the two disaccharides, whereas the lipid chains are nearly overlapping including those exposed at the dimerization interface (R4′ for DLAM3 and R6′ for DLAM5) (Fig. 6a). Also, the inward reorientation of the F126MD-2, which is considered the key event preceding TLR4 complex dimerization, is identical for both ligands (Supplementary Fig. 10). Thus, the moderate TLR4-mediated activity of monophosphoryl DLAM3 can be justified by a specific binding pose of the monophosphorylated αGlcN(1 ↔ 1)αMan backbone, which determines the location of a single phosphate group within the complex. Since the GlcN-linked P4′ is positioned far from the contact area between the two complexes, the secondary dimerization interface between hTLR4/MD-2 and hTLR4* is weakened in the hTLR4/MD-2/DLAM3 complex. The electrostatic interactions observed between DLAM5 and hTLR4* Q436*hTLR4* are absent in the hTLR4/MD-2/DLAM3 complex. The loss of these interactions leads to reduced ligand affinity compared to DLAM5, ultimately resulting in altered kinetics and a low level of TLR4 activation.

Superimposition of a DLAM5 and DLAM3 in h- and mTLR4/MD-2 complex, b DLAM3 and DLAM1 in h- and mTLR4/MD-2 complex, c DLAM5 and DLAM1 in h- and mTLR4/MD-2 complex. In the bottom panel, only the αGlcN(1↔1)αMan backbone is shown for clarity and the lipid chains are omitted. The colors of DLAMs are consistent with Fig. 2c.
Given that bisphosphorylated DLAM1 is a conformationally constrained prototype of the lipid A of E. coli LPS, whereas monophosphate DLAM3 mimics MPLA, comparison of the binding modes of these two molecules could provide valuable insights into the role of phosphate groups in recognition, binding and ligand-induced dimerization of TLR4/MD2 by natural lipid A ligands. Superimposing hMD-2 with bound DLAM1 or DLAM3 shows that the six lipid chains of the glycolipids nearly align, while the αGlcN(1↔1)αMan backbone of DLAM1 is rotated by 30° relative to DLAM3, causing a significant repositioning of the P4′ phosphate group (Fig. 6b). As a result, these two lipid A mimetics, which differ solely by the presence or absence of a second phosphate group on the Man moiety, exhibit significant differences in the binding poses of their carbohydrate backbones when complexed with hTLR4/MD-2. However, comparing the binding poses of DLAM1 and DLAM3 in complex with mTLR4/MD-2 reveals a close overlap of the two ligands, both in terms of their disaccharide backbones and lipid chains (Fig. 6b).
Discussion
Despite the advancements in understanding ligand recognition in the TLR4/MD-2 system, there is limited knowledge of the molecular mechanisms that distinguish LPS/lipid A with different acylation and phosphorylation patterns. To complement the existing structural data with information on the binding of therapeutically relevant synthetic glycolipid molecules to TLR4/MD-2, we determined six atomic resolution cryo-EM structures of human and mouse TLR4/MD-2 in complex with three different DLAMs displaying pico- and nanomolar TLR4 agonistic activity. The disaccharide lipid A mimetics (α,α-DLAMs) are constructed on a synthetic, conformationally constrained nonreducing disaccharide αGlcN(1↔1)αMan, designed to mimic the 3D molecular shape of the native diglucosamine [GlcNβ(1→6)GlcN] backbone of TLR4/MD-2-bound hexaacylated E. coli Ra-LPS (Fig. 1c)48. By design, the GlcN moiety of the αα-DLAMs retains the same acylation and phosphorylation pattern as E. coli LPS, while the Man fragment features distinct attachment sites for the branched lipid chain and a phosphate group. Due to the exceptional rigidity of the non-reducing disaccharide backbone (imposed by specific carbohydrate-related effects), the molecular shape of the αGlcN(1↔1)αMan disaccharide remains unchanged upon protein binding54,55,56,59,60. This is in contrast to the flexible GlcNβ(1→6)GlcN diglucosamine backbone of natural lipid A, which undergoes significant conformational changes when bound by the TLR4/MD-2 complex. Despite their relatively small size, with a molecular weight of about 2 kDa, and the absence of core sugars, the bisphosphorylated DLAMs (DLAM1 and DLAM5) induce TLR4-mediated signaling with pico- to nanomolar potency, comparable to that of Re/Ra-LPS48,52. Through a comparative analysis of six [TLR4/MD-2/DLAM]2 complexes, we identified the binding modes of structurally diverse glycolipid molecules with varying TLR4-activating capacities, with a particular focus on species-specific recognition by h- and mTLR4/MD-2 and structural requirements for TLR4 agonists with therapeutic potential.
Since our investigation focused on ligands with similar hydrophobic volumes (all hexaacylated lipid A mimetics with optimal C14-C12 length of lipid chains), the shape of the clamshell-like cavity of MD-2 remained consistent across all structures and the binding of DLAMs was reinforced by interactions of (R)-3-acyloxyacyl lipid tails with the hydrophobic lining of the binding cavity of MD-2. The DLAM binding induced local conformational changes in the co-receptor protein by shifting the F126 loop of MD-2 inward compared to the initial ligand-free state MD-2apo (PDB ID: 2E56 (hMD-2) and 2Z64 (mMD-2)) (Supplementary Fig. 10), thereby stabilizing the exposure of the 6th lipid chain at the opening of the β-sandwich in MD-2. The hydroxyl groups of the disaccharide backbone, along with the carbonyl groups of the lipid chains and the phosphate groups of DLAMs, were found to interact with hydrophilic and charged side chains at the TLR4 concave region, the rim of the binding pocket of MD-2 and the convex region of the second TLR4*.
The disaccharide backbone GlcNα(1↔1)αMan of structurally different DLAMs adopts different binding poses (±90° and ±180°) with either the Man- or GlcN-linked lipid chains located in close proximity to the MD-2 cavity mouth resulting in their exposure on the surface of MD-2 (Fig. 2d and Supplementary Fig. 10). Binding pose and binding orientation refer to the positioning of the glycolipid molecule at the interface formed by the binding cavity of MD-2, the concave region of the parent TLR4 and the convex region of the second TLR4*, the latter known as the secondary dimerization interface. The binding pose of DLAMs is primarily determined by a specific gauche-gauche conformation of the non-reducing disaccharide GlcNα(1↔1)αMan backbone53,54,55,56, and the site of attachment of the branched lipid chain to the Man moiety. The Man-linked phosphate group P6, which is absent in DLAM3, doesn’t play a significant role in determining the binding orientation. The αGlcN(1↔1)αMan backbone of DLAM1 is rotated by approximately 30° relative to DLAM3 in complex with hMD-2, thereby resulting in a significant relocation of the P4′ phosphate group (Fig. 6b). This shift primarily affects the orientation of the carbohydrate backbone, but not the overall arrangement of the lipid chains in the binding pocket. Consequently, both diphosphate DLAM1 and monophosphate DLAM3 (a monophosphorylated version of DLAM1 with the same acylation pattern) exhibit a comparable arrangement of lipid chains in complex with hTLR4/MD-2 and nearly overlapping arrangement in complex with mTLR4/MD-2 (Fig. 6b). It is noteworthy that the rim of mMD-2 pocket lacks most of the positively charged side chains found in hMD-2 (K122, K125, K128, and K58), with exception of the conserved R90 (Supplementary Fig. 13). As a result, hydrophobic interactions and hydrogen bonds dominate in the ligand-protein interactions in the mouse complex and also dictate the species-specific binding orientation of the DLAM1 and DLAM3 (Fig. 2d). In contrast, the disaccharide backbones of DLAM5 adopts the same binding pose in complex with both h- and mTLR4, which differs from the binding orientation of the lipid A portion of E. coli LPS by being rotated by 90° (Fig. 3g, h). A species-independent binding pose of DLAM5 places all important functional groups (phosphates, carbonyl groups, disaccharide oxygens) in a favorable orientation, allowing them to form multiple interactions with specific residues on MD-2, TLR4 and TLR4*, making binding and dimerization more efficient.
The key events driving ligand-induced dimerization—exposure of a lipid tail of LPS on the MD-2 surface and inward repositioning of the F126 aromatic side chain—were previously characterised through NMR studies using a chemical shift perturbation approach. It was proposed that the lipid chain closest to the mouth opening of MD-2 protrudes from the hydrophobic pocket, as judged by its higher susceptibility to paramagnetic attenuation, and that it is stabilised by the aromatic side chain of F12661,62. This hypothesis was further reinforced by co-crystal structures of TLR4/MD-2 in complex with potent TLR4 agonists Ra/Re-LPS33,34 and supported by molecular dynamics simulation studies40,41,42. However, information on the behavior of F126 with TLR4 agonists less potent than LPS, such as MPLA, is controversial. On the one hand, the low TLR4-activating potential of MPLA has been explained by a less efficient dimerization of the TLR4/MD-2 complex, as demonstrated by an antibody assay63, and proposed by molecular dynamics simulations, which suggest an intermediate level of F126 switching for TLR4/MD-2-bound MPLA42. On the other hand, recent data reveal that a weak mTLR4 agonist, diacylated 3-sulfogalactosyl ceramide (Gal-Cer) in which three Gal-Cer molecules mimic the binding of six lipid chains of E. coli lipid A, induces the same degree of F126 repositioning as other more potent agonistic LPS ligands (PDB: 7MLM)37. Likewise, a co-crystal structure of mTLR4 with a μM synthetic agonist Neoseptin (PDB: 5IJC) shows similar conformational properties36.
DLAM binding induces a local conformational rearrangement of the F126 in a β-cup fold of MD-2, with consistent RMSD values across all ligands/complexes, showing no correlation with the ligand potency in triggering downstream signaling. Our data suggest that both events - lipid chain exposure and inward repositioning of the aromatic side-chain of F126—are intrinsic features of the dimerized [TLR4/MD-2/ligand]2 complexes, regardless of the structural features of the disaccharide skeleton or the potency of the TLR4 activation. This hypothesis is consistent with the observation that about 20% of ligand-free dimeric TLR4/MD-2 complexes pre-exist in unstimulated cells, with their number remaining constant at low concentrations of TLR4 agonists, but increasing as the concentration of agonistic ligands rises35. The TLR4/MD-2 complex in cells may be in a balance between different accessible states (MD-2 in an ‘open’ or ‘closed’ conformation) that have a natural affinity for specific LPS variants, while potent agonists not only bind to a pre-existing dimeric complex but are also able to induce a local rearrangement in MD-2, bringing it into a dimerization-ready ‘closed’ state. This suggests that ligand affinity and the ability to stabilize and maintain [TLR4/MD-2/ligand]2 complexes are the major determinants of the ligand activation potency. Moreover, the kinetics of Myddosome formation downstream of TLR47, as determined by Western blot analysis of phosphorylation of key signaling molecules, including IRAK4, TBK and p38 (Supplementary Fig. 2), appear to align with the binding data. Stimulation of moDCs with DLAM5 and DLAM1 results in a rapid phosphorylation of TBK and p38 within 15 min, peaking at 30 min before decreasing by 60 min. In contrast, stimulation of moDCs with DLAM3 leads to delayed phosphorylation of both molecules, which continues to increase up to 60 min. Regarding IRAK4 phosphorylation, DLAM5 and DLAM1 induce the strongest response at 30 and 60 min, respectively, whereas DLAM3 induces only weak IRAK4 phosphorylation.
According to our data, the structure of the sugar portion of the lipid A mimetic and its acylation pattern define the binding orientation, influencing the number, type and strength of ligand-protein interactions, while the phosphorylation status is important for affinity and the efficiency of dimerization. The significance of ionic interactions provided by the phosphate groups of lipid A in the recognition of LPS by the TLR4 complex has also been confirmed in previous studies64,65. A specific binding pose of DLAM5, conserved across species, positions the phosphate groups favorably to promote high affinity and effective cross-linking between two TLR4/MD-2/DLAM5 complexes. In contrast, the species-specific binding orientation of DLAM3 in h- and mTLR4 complexes, along with its monophosphorylation, leads to reduced affinity, which translates into a lower potency and species-specific differences in cellular responses. DLAM3 fails to efficiently cross-link two TLR4/MD-2/DLAM complexes due to the absence of several important electrostatic interactions and hydrogen bonds both with the parent TLR4/MD-2 and at the dimerization interface, which is likely to result in a rather short-lived active TLR4 dimer and a low level of TLR4 activation. The diphosphate DLAM1 is particularly intriguing as it binds to h- and mTLR4 complexes in reversed by 180° orientations, yet triggers similar innate immune responses in both human and mouse cells, while the activation potency is strongly CD14-dependent (Supplementary Figs. 1 and 2)52. Our structural data can also be utilized to predict potential binding modes of lipid A and its derivatives. However, certain limitations remain due to the inherent flexibility of the GlcNβ(1→6)GlcN backbone and the consequent adaptability of natural lipid A molecules to the geometry of the binding pocket of TLR4/MD-2 complex.
Although the analysis of the protein-protein dimerization interfaces shows similar protein-protein interaction types across hTLR4/MD-2/DLAM complexes, except for DLAM5/hTLR4 where two additional amino acids T584 and E586 are involved (Supplementary Fig. 12, Supplementary Tables 3–8), the number of short-range interactions (<3.5 Å) at the dimerization interface is highest for DLAM5/hTLR4, followed by DLAM1/hTLR4 and lowest for DLAM3/hTLR4. Unlike the number of ‘contact’ residues involved in the protein-protein dimerization interface, which remains comparable across TLR4 complexes with different DLAMs (Supplementary Fig. 11), the glycolipid functional groups participating in the dimerization interface vary between complexes. In particular, the GlcNα(1↔1)αMan backbone of DLAM5 in [TLR4/MD-2/DLAM5]2 complex is positioned closer to the dimerization interface compared to DLAM1 and DLAM3 (Fig. 6), thereby enhancing ligand-protein interactions between DLAM5 and TLR4*, which is translated into enhanced TLR4 activation potency. In the case of DLAM1, the phosphate group (P6 in [hTLR4/MD-2/DLAM1]2 and P4′ in [mTLR4/MD-2/DLAM1]2) are situated close to the dimerization site, likely contributing to the nanomolar TLR4 activation potency of DLAM1.
The DLAM molecules function as a molecular bridge anchoring MD-2 through hydrophobic interactions of the aliphatic lipid chains. More importantly, the phosphate groups attached on either side of the non-reducing disaccharide backbone are key for cross-linking two TLR4/MD-2 complexes through ionic interactions and/or hydrogen bonds. The carbonyl oxygens of the acyloxyacyl chains mediate interactions with all three proteins, while the distinct binding poses of DLAMs play a crucial role in determining short-range contacts and ensure efficient dimerization, ultimately translating into differential potency in the induction of TLR4-mediated signaling. Whereas the binding orientation of LPS is strongly influenced by the need to accommodate the voluminous oligosaccharide core, which is likely to determine the primary binding pose of the GlcNβ(1→6)GlcN backbone of lipid A in the Ra-LPS/TLR4 complex, smaller glycolipids such as DLAMs are not subject to such limitations and the binding mode is determined solely by the chemical structure of the lipidated/phosphorylated disaccharide, opening up vast opportunities for the rational design of TLR4 agonist with tailored activating potencies. Given that activation of TLR4-mediated cellular signaling is considered a promising tool for the management of infections, autoimmune diseases and cancer, our findings provide a structural basis for the design and development of innovative, highly potent TLR4-targeting adjuvants and immunotherapeutics.
Methods
Ethics statement
Approval for these studies was obtained from the Institutional Ethics Committee at the University of Lübeck (Lübeck, Germany; Az. 12-202A) according to the Declaration of Helsinki. All donors gave written informed consent. As the blood donation was anonymized, no data on gender and age is available. Gender specificity was not considered in the study design. Donors were compensated with up to 35 € per 450 mL of blood.
Constructs and virus preparation of the TLR4/MD-2 complex
The ectodomains of hTLR4 (residues 27–631) and mTLR4 (residues 26–629) were inserted into BamHI and NotI sites of a pAcGP67 expression vector (BD Biosciences). Genes encoding hMD-2 (residues 19–160) and mMD-2 (residues 19–160) were connected to the protein A gene and inserted into the same BamHI and NotI sites of the pAcGP67 expression vector (BD Biosciences). A thrombin cleavage site was incorporated between the MD-2 and protein A genes to facilitate tag removal. The primer sequences used for TLR4 and MD-2 constructs were reported in a previous study33,57. Baculoviruses were generated by transfecting the cloned TLR4 or MD-2 vector along with the AB vector into Sf9 insect cells (Invitrogen). High-titer baculoviruses from passage 4 (P4) for each construct were subsequently produced and collected for protein expression in further experiments.
Expression and purification of the TLR4/MD-2 complex
For protein expression, High Five insect cells (Invitrogen, catalog no. B85502) were infected with recombinant P4 baculoviruses at a TLR4:MD-2 ratio of 1:4. Infected cells were incubated for 72 h at 28 °C with gentle agitation at 105 rpm. After cell pelleting through centrifugation, supernatants containing TLR4/protein A-fused MD-2 complexes were applied to IgG resins (GE Healthcare Life Sciences, catalog no. GE17-0969-01). The resin was washed with 10 column volumes of buffer [20 mM Tris-HCl (pH 8.0), 200 mM NaCl] and incubated with thrombin [0.5% (v/v) in wash buffer] at 4 °C overnight for protein A tag removal. Subsequently, eluted TLR4/MD-2 complexes were concentrated to ~0.5 mg/ml using an Amicon Ultra centrifugal filter (50 kDa MW cutoff; Millipore, catalog no. UFC9050). Further purification was carried out by subjecting concentrated proteins to size-exclusion chromatography on a Superose 6 Increase 10/300 GL column (GE Healthcare Life Sciences, catalog no. GE29-0915-96) in a buffer containing 20 mM Tris-HCl (pH 8.0) and 200 mM NaCl. The resulting peak fractions of each TLR4/MD-2 were subsequently concentrated to 0.5 mg/ml using an Amicon Ultra centrifugal filter (50 kDa MW cutoff; Millipore, catalog no. UFC9050). These purified proteins were employed for DLAM binding and cryo-EM experiments.
Generation of TLR4/MD-2/DLAM complexes
DLAMs and LPS-Ra were incubated with pre-purified TLR4/MD-2 protein to generate heterotetrameric complexes. LPS-Ra (catalog no. L9641; Sigma) was used as the positive control for TLR4/MD-2 dimerization. DLAMs (1 mg/ml, ~565 μM) dissolved in dimethyl sulfoxide and LPS-Ra (2 mg/ml, ~465 μM) dissolved in distilled water were sonicated for 10 min in a bath sonicator and mixed with prepurified TLR4/MD-2 protein at a molar ratio of 20:1. The TLR4/MD-2/DLAM mixture was incubated in a glass vial at 37 °C for 3 h with agitation at 199 rpm. After incubation, the mixture was further purified through size-exclusion chromatography using a Superose 6 Increase 10/300 GL column (GE Healthcare Life Sciences, catalog no. GE29-0915-96) in a buffer containing 20 mM Tris-HCl (pH 8.0) and 200 mM NaCl. DLAM or LPS-Ra binding was assessed using 7% basic native polyacrylamide gel electrophoresis. Heterotetrameric peak fractions of the TLR4/MD-2/DLAM complex (P2 fraction) were used to prepare cryo-EM samples on the grid.
Cryo-EM sample preparation and data collection
Graphene oxide-coated Quantifoil R1.2/1.3 300 mesh grids (Quantifoil Micro Tools, catalog no. Q350CR1.3) were glow discharged using a PELCO easiGlow Glow Discharge Cleaning system (Ted Pella, catalog no. 91000S) for 5 to 10 s at 10 mA. Approximately 2.5 to 4 μl of purified TLR4/MD-2/DLAM complexes were applied to the grids and incubated for 10 s in 100% humidity at 4 °C. After blotting for 2 to 4 s, the grids were rapidly vitrified by plunging them into liquid ethane using a Vitrobot MkIV (Thermo Fisher Scientific). hTLR4/MD-2/DLAM5 (#micrographs = 9930), hTLR4/MD-2/DLAM1 (#micrographs = 4407), mTLR4/MD-2/DLAM1 (#micrographs=8,759), mTLR4/MD-2/DLAM3 (#micrographs=18,733), and mTLR4/MD-2/DLAM5 (#micrographs=6,999) micrographs were acquired on a TFS Krios G4 TEM operated at 300 keV with a K3 direct electron detector (Gatan) at the Institute for Basic Science, utilizing a slit width of 20 eV on a GIF quantum energy filter. Micrographs of hTLR4/MD-2/DLAM3 (#micrographs=2,072) were obtained on a TFS Krios G3 TEM operating at 300 keV with a Falcon 3EC direct electron detector at the Korea Basic Science Institute. EPU software was employed for automated data collection under the single-electron counting mode and correlated double sampling (CDS) mode. Detailed image acquisition parameters for each TLR4/MD-2/DLAM complex is summarized in Supplementary Table 1.
Image processing, model building, and refinement
The detailed image processing workflow and statistics are shown in Supplementary Figs. 3–8 and Supplementary Table 1. Raw movies were corrected for motion using MotionCorr266, and contrast transfer function (CTF) parameters were estimated by CTFFIND467 in cryoSPARC version 468. Initially, particles were picked with a template picker employing TLR4/MD-2/LPS (PDB IDs 3FXI and 3VQ2) as templates. The resulting particle sets were binned two times, followed by 2D classification, ab initio modeling, and heterogeneous refinement for data cleanup within cryoSPARC. Final particles from 3D classes with good secondary structural features were selected and re-extracted into the original pixel size. Nonuniform refinement was performed on the resulting volume using per-particle defocus and per-group CTF optimization applied69. The final refinement yielded maps of TLR4/MD-2/DLAMs at an overall resolution ranging from 2.24 to 3.0 Å (with tight masks). The mask-corrected Fourier shell correlation (FSC) curves were calculated in cryoSPARC, and the reported resolutions were based on the gold-standard FSC = 0.143 criterion70. Local resolutions of density maps were estimated by Blocres71.
Model building for the TLR4/MD-2/DLAM complexes were initiated using TLR4/MD-2 subunits derived from the high-resolution structures of human and mouse TLR4/MD-2 complexes bound to LPS derivatives: PDB IDs 3FXI (human TLR4/MD-2/LPS-Ra) and 3VQ2 (mouse TLR4/MD-2/LPS-Re). To minimize potential biases introduced by the pre-dimerized X-ray structures, only monomeric structure of TLR4/MD-2 after excluding LPS molecule was used for initial fitting into the cryo-EM maps. The fitting process was performed using the Dock in Map module of the Phenix package (version 1.19.2)72, ensuring that the structural information was guided solely by the cryo-EM density rather than assumptions based on dimeric templates. Subsequently, the structures of dimerized TLR4/MD-2/DLAM complexes were reconstructed and manually adjusted in Coot (version 0.9.4.1)73 based on the cryo-EM density map, with particular attention given to the DLAM density within the MD-2 pocket. This step ensured that the final model accurately reflected the experimental cryo-EM data, independent of potential biases from the initial PDB structures. The PDB and restraint CIF files of DLAMs were generated using REEL in the Phenix package and each entire DLAM was generated as a single molecule. The overall geometry of DLAMs, including bond length, angle, dihedral, and plane, is strictly controlled during refinement. After several rounds of refinement against the cryo-EM map using the Real-Space Refinement module in the Phenix package, the TLR4/MD-2/DLAMs structures with the expected geometry for both protein and ligand were obtained. The refinement statistics from Phenix validation are summarized in Supplementary Table 1. All figures for cryo-EM maps and structure models were generated using UCSF ChimeraX (version 1.6.1)74 and Pymol (version 2.3.1)75.
Stimulation of TLR4/MD-2/CD14(+/-) transfected HEK293 cells with DLAMs
HEK293 cells ((#ATCC-CRL-1573, LGC standards, Wesel, Germany) were transfected for 24 h with plasmids coding for human TLR4 (kind gift of P. Nelson, USA), human MD-2 (kind gift of K. Miyake, Tokyo, Japan), and human CD14 (kind gift of D. Golenbock, Worcester, USA) using Lipofectamine 2000 (#11668027, Thermo Fisher, Darmstadt, Germany) according to the manufacturer’s instruction. DLAM1, DLAM3 and DLAM5 were dissolved in DMSO (1 mg/mL) and diluted with DMEM cell medium supplemented with 10% FCS (fetal calf serum, #FCS-62A, Capricorn Scientific, Ebsdorfergrund, Germany) under vortex to provide the indicated concentrations. Next, transiently transfected HEK293 cells were incubated with increasing concentrations of DLAMs (0.01–1000 nM) for 20 h while recombinant human TNF-α (kind gift of D. Männel, Regensburg, Germany) served as transfection-independent control. DLAMs-induced IL-8 production was measured by human IL-8 CytoSet ELISA (#CHC1303, Thermo Fisher, Darmstadt, Germany) according to the manufacture’s instruction. Data are from n = 2 independent experiments.
Cytokine release by DLAMs in monocyte-derived dendritic cells
Approval for these studies was obtained from the Institutional Ethics Committee at the University of Lübeck (Lübeck, Germany; Az. 12-202 A) according to the Declaration of Helsinki. All donors gave written informed consent. As the blood donation was anonymized, no data on gender and age is available. Gender specificity was not considered in the study design. Donors were compensated with up to 35 € per 450 mL of blood. Heparinized blood from healthy volunteers was used to prepare MNC (peripheral human blood mononuclear cells) by gradient centrifugation (#L6115, Biocoll, Merck, Darmstadt, Germany) followed by incubation in 96-well tissue culture plates at a volume of 150 µL and a concentration of 1 × 106/mL using RPMI-1640 as medium supplemented with 10.000 U/mL penicillin & 10 mg/mL streptomycin (#P06-07100 PAN-Biotech Aidenbach, Germany), and 10% FCS (#FCS-62A, Capricorn Scientific, Ebsdorfergrund, Germany). CD14+ monocytes were subsequently isolated by magnetic sorting (MojoSort, BioLegend). MoDCs were generated by addition of interleukin 4 and granulocyte-macrophage colony-stimulating factor (both 500U/ml, Immuno Tools, Germany) to monocytes in 6-well plates for 7 days, harvested and resuspended in complete RPMI medium. MoDCs were grown and incubated in a humidified atmosphere of 5% carbon dioxide at 37 °C. Solutions of DLAMs were prepared from stock solutions in DMSO (1 mg/mL) using RPMI cell medium supplemented with 10% FCS. The solutions of DLAMs were added to moDCs (1 × 106/ml) in the indicated concentrations and incubated for 20 h. Cytokine production by moDCs after 20 h stimulation was quantified using commercial ELISA Kits (ThermoFisher, Darmstadt, Germany) that are specific for IL-6 (#CHC1263), IL-10 (#CHC1323) and IL-12-p70 (#88-7126-76) in supernatants. Data are combined from n = 3 independent donors (n = 2 for IL-10); error bars indicate standard error of the mean (IL-6 and IL-12) or from 2 independent donors (IL-10).
Western blot
Human monocyte-derived dendritic cells have been stimulated with DLAMs and E. coli O111 LPS (estimated MW of 10 kD) at 10 nM for the indicated times. Subsequently, cells were lysed and subjected to SDS page western blot analysis. Early signal transduction events have been investigated by probing with specific antibodies to the phosphorylated forms (all from Cell Signaling) of TBK (#5483S), IRAK4 (#11927S) and p38 (#9216S) as well as the unphosphorylated forms (TBK (#3504T), Cell Signaling; IRAK4 (#NBP2-37575), Novus Biologicals; p38 (#9212S, Cell Signaling). Results shown are from the identical blot membranes (DLAM1, DLAM3, DLAM5 and LPS). One out of three independent results is shown.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates and cryo-EM maps for the TLR4/MD-2/DLAM complex have been deposited to the Protein Data Bank and Microscopy Data Bank, respectively. The accession numbers for each complex are as follows: PDB ID 8WO1 and EMD-37677 for hTLR4/MD-2/DLAM5; PDB ID 8WSA and EMD-37803 for mTLR4/MD-2/DLAM5; PDB ID 8WTA and EMD-37831 for hTLR4/MD-2/DLAM3; PDB ID 8WRY and EMD-37794 for mTLR4/MD-2/DLAM3; PDB ID 9J03 and EMD-61047 for hTLR4/MD-2/DLAM1, and PDB ID 8WQT and EMD-37753 for mTLR4/MD-2/DLAM1. The previously published accession codes (PDB ID) used in this study is listed below: 3FXI (hTLR4/MD-2/Ra-LPS), 3VQ2 (mTLR4/MD-2/Re-LPS), 3VQ1 (mTLR4/MD-2/lipid Iva), 5IJD (mTLR4/MD-2/lipid A), 7MLM (mTLR4/MD-2/Sulfatides), 2E59 (hMD-2/lipid Iva), 2Z65 (hMD-2/Eritoran), 2E56 (hMD-2), 2Z64 (mTLR4/MD-2) and 5IJC (mTLR4/MD-2/Neoseptin). All other data are available in the manuscript or the supplementary materials. Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding authors (hm_kim@kaist.ac.kr, alla.zamyatina@boku.ac.at). Source data are provided as a Source Data file. Source data are provided with this paper.
References
Taylor, K. R. et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 282, 18265–18275 (2007).
Bryant, C. E., Spring, D. R., Gangloff, M. & Gay, N. J. The molecular basis of the host response to lipopolysaccharide. Nat. Rev. Microbiol. 8, 8–14 (2010).
Gay, N. J., Symmons, M. F., Gangloff, M. & Bryant, C. E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 14, 546–558 (2014).
Fitzgerald, K. A. et al. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 (2003).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Freudenberg, M. A. et al. Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: benefits and hazards of LPS hypersensitivity. Immunobiology 213, 193–203 (2008).
Fisch, D. et al. Molecular definition of the endogenous Toll-like receptor signalling pathways. Nature 631, 635–644 (2024).
Hernandez, A. et al. Immunobiology and application of toll-like receptor 4 agonists to augment host resistance to infection. Pharmacol. Res. 150, 104502 (2019).
Qin, Y. et al. Stimulation of TLR4 attenuates Alzheimer’s disease–related symptoms and pathology in tau-transgenic mice. J. Immunol. 197, 3281–3292 (2016).
Michaud, J.-P. et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl Acad. Sci. USA 110, 1941–1946 (2013).
Hollingsworth, J. W. et al. TLR4 signaling attenuates ongoing allergic inflammation. J. Immunol. 176, 5856–5862 (2006).
Shalaby, K. H. et al. The TLR4–TRIF pathway can protect against the development of experimental allergic asthma. Immunology 152, 138–149 (2017).
Bohannon, J. K. et al. Treatment with TLR4 agonists protect against infection after severe burn injury. J. Immunol. 196, 200.217–200.217 (2016).
Gregg, K. A. et al. A lipid A-based TLR4 mimetic effectively adjuvants a Yersinia pestis rF-V1 subunit vaccine in a murine challenge model. Vaccine 36, 4023–4031 (2018).
Haupt, R. et al. BECC Adjuvanted Vaccine Provides Cross-Protection from Both Homologous and Heterologous Influenza A Infections. Vaccine 39, 5205 (2021).
Bohannon, J. K., Hernandez, A., Enkhbaatar, P., Adams, W. L. & Sherwood, E. R. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock 40, 451–462 (2013).
Pouliot, K. et al. Contribution of TLR4 and MyD88 for adjuvant monophosphoryl lipid A (MPLA) activity in a DNA prime–protein boost HIV-1 vaccine. Vaccine 32, 5049–5056 (2014).
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004).
Apetoh, L. et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Matzner, P. et al. Perioperative treatment with the new synthetic TLR‐4 agonist GLA‐SE reduces cancer metastasis without adverse effects. Int. J. cancer 138, 1754–1764 (2016).
Nagarsheth, N., Wicha, M. S. & Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572 (2017).
Han, S., Wang, C., Qin, X., Xia, J. & Wu, A. LPS alters the immuno-phenotype of glioma and glioma stem-like cells and induces in vivo antitumor immunity via TLR4. J. Exp. Clin. Cancer Res. 36, 1–11 (2017).
Yahiro, K. et al. Activation of TLR4 signaling inhibits progression of osteosarcoma by stimulating CD8-positive cytotoxic lymphocytes. Cancer Immunol., Immunother. 69, 745–758 (2020).
Albershardt, T. C. et al. Intratumoral immune activation with TLR4 agonist synergizes with effector T cells to eradicate established murine tumors. npj Vaccines 5, 50 (2020).
Blazquez, R. et al. Intralesional TLR4 agonist treatment strengthens the organ defense against colonizing cancer cells in the brain. Oncogene 41, 5008–5019 (2022).
Bhatia, S. et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with Merkel cell carcinoma. Clin. Cancer Res. 25, 1185–1195 (2019).
Gregg, K. A. et al. Rationally designed TLR4 ligands for vaccine adjuvant discovery. MBio 8, 10.1128/mbio. 00492-00417 (2017).
Pupo, E., Hamstra, H.-J., Meiring, H. & van der Ley, P. Lipopolysaccharide engineering in Neisseria meningitidis: structural analysis of different pentaacyl lipid A mutants and comparison of their modified agonist properties. J. Biol. Chem. 289, 8668–8680 (2014).
Mata-Haro, V. N. et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316, 1628–1632 (2007).
Wang, Y.-Q., Bazin-Lee, H., Evans, J. T., Casella, C. R. & Mitchell, T. C. MPL adjuvant contains competitive antagonists of human TLR4. Front. Immunol. 11, 577823 (2020).
Trent, M. S., Stead, C. M., Tran, A. X. & Hankins, J. V. Invited review: diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 12, 205–223 (2006).
Needham, B. D. et al. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc. Natl Acad. Sci. 110, 1464–1469 (2013).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 458, 1191–1195 (2009).
Ohto, U., Fukase, K., Miyake, K. & Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl Acad. Sci. USA 109, 7421–7426 (2012).
Latty, S. L. et al. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. elife 7, e31377 (2018).
Wang, Y. et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl Acad. Sci. USA 113, E884–E893 (2016).
Su, L. et al. Sulfatides are endogenous ligands for the TLR4–MD-2 complex. Proc. Natl Acad. Sci. USA 118, e2105316118 (2021).
Martín-Santamaría, S. et al. Full-atom model of the agonist LPS-bound toll-like receptor 4 dimer in a membrane environment. Chemistry 27 15406–15425 (2021).
Vašl, J., Oblak, A., Gioannini, T. L., Weiss, J. P. & Jerala, R. Novel roles of lysines 122, 125, and 58 in functional differences between human and murine MD-2. J. Immunol. 183, 5138–5145 (2009).
DeMarco, M. L. & Woods, R. J. From agonist to antagonist: structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol. Immunol. 49, 124–133 (2011).
Garate, J. A. & Oostenbrink, C. Lipid A from lipopolysaccharide recognition: structure, dynamics and cooperativity by molecular dynamics simulations. Proteins Struct. Funct., Bioinform. 81, 658–674 (2013).
Paramo, T., Piggot, T. J., Bryant, C. E. & Bond, P. J. The structural basis for endotoxin-induced allosteric regulation of the Toll-like receptor 4 (TLR4) innate immune receptor. J. Biol. Chem. 288, 36215–36225 (2013).
Raetz, C. R. et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. lipid Res. 47, 1097–1111 (2006).
Simpson, B. W. & Trent, M. S. Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 17, 403–416 (2019).
Artner, D. et al. Conformationally constrained lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem. Biol. 8, 2423–2432 (2013).
Zhang, Y., Gaekwad, J., Wolfert, M. A. & Boons, G.-J. Modulation of Innate Immune Responses with Synthetic Lipid A Derivatives. J. Am. Chem. Soc. 129, 5200–5216 (2007).
Gaekwad, J. et al. Differential Induction of Innate Immune Responses by Synthetic Lipid A Derivatives*[S]. J. Biol. Chem. 285, 29375–29386 (2010).
Adanitsch, F. et al. Synthetic glycan-based TLR4 agonists targeting caspase-4/11 for the development of adjuvants and immunotherapeutics. Chem. Sci. 9, 3957–3963 (2018).
Strobl, S., Hofbauer, K., Heine, H. & Zamyatina, A. Lipid A mimetics based on unnatural disaccharide scaffold as potent TLR4 agonists for prospective immunotherapeutics and adjuvants. Chemistry 28, e202200547 (2022).
Heine, H. & Zamyatina, A. Therapeutic targeting of TLR4 for inflammation, infection, and cancer: a perspective for disaccharide lipid A mimetics. Pharmaceuticals 16, 23 (2023).
Strobl, S. et al. Nonreducing sugar scaffold enables the development of immunomodulatory TLR4‐specific LPS mimetics with picomolar potency. Angew. Chem. Int. Ed. 63, e202408421 (2024).
Heine, H. et al. Tailored modulation of cellular pro-inflammatory responses with disaccharide lipid A mimetics. Front. Immunol. 12, 621 (2021).
Bock, K., Defaye, J., Driguez, H. & Bar‐Guilloux, E. Conformations in solution of α, α‐trehalose, α‐d‐glucopyranosyl α‐d‐mannopyranoside, and their 1‐thioglycosyl analogs, and a tentative correlation of their behaviour with respect to the enzyme trehalase. Eur. J. Biochem. 131, 595–600 (1983).
Färnbäck, M., Eriksson, L. & Widmalm, G. Octa-O-acetyl-α, α-trehalose ethanol disolvate. Acta Crystallogr. Sect. E: Struct. Rep. Online 60, o1483–o1485 (2004).
French, A. D., Johnson, G. P., Kelterer, A.-M., Dowd, M. K. & Cramer, C. J. Quantum mechanics studies of the intrinsic conformation of trehalose. J. Phys. Chem. A 106, 4988–4997 (2002).
Brown, G. et al. The crystal structure of α, α-trehalose dihydrate from three independent X-ray determinations. Acta Crystallogr. Sect. B: Struct. Crystallogr. Cryst. Chem. 28, 3145–3158 (1972).
Kim, H. M. et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917 (2007).
Ohto, U., Fukase, K., Miyake, K. & Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634 (2007).
Batta, G., Kövér, K. E., Gervay, J., Hornyák, M. & Roberts, G. M. Temperature dependence of molecular conformation, dynamics, and chemical shift anisotropy of α, α-trehalose in D2O by NMR relaxation. J. Am. Chem. Soc. 119, 1336–1345 (1997).
Perić-Hassler, L., Hansen, H. S., Baron, R. & Hünenberger, P. H. Conformational properties of glucose-based disaccharides investigated using molecular dynamics simulations with local elevation umbrella sampling. Carbohydr. Res. 345, 1781–1801 (2010).
Yu, L. et al. NMR studies of hexaacylated endotoxin bound to wild-type and F126A mutant MD-2 and MD-2· TLR4 ectodomain complexes. J. Biol. Chem. 287, 16346–16355 (2012).
Teghanemt, A. et al. Novel roles in human MD-2 of phenylalanines 121 and 126 and tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J. Biol. Chem. 283, 1257–1266 (2008).
Casella, C. R. & Mitchell, T. C. Inefficient TLR4/MD-2 heterotetramerization by monophosphoryl lipid A. PLoS One 8, e62622 (2013).
Meng, J., Gong, M., Björkbacka, H. & Golenbock, D. T. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J. Immunol. 187, 3683–3693 (2011).
Meng, J., Lien, E. & Golenbock, D. T. MD-2-mediated ionic interactions between lipid A and TLR4 are essential for receptor activation. J. Biol. Chem. 285, 8695–8702 (2010).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. methods 11, 63–65 (2014).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Kagami, L. P., das Neves, G. M., Timmers, L. F. S. M., Caceres, R. A. & Eifler-Lima, V. L. Geo-measures: a PyMOL plugin for protein structure ensembles analysis. Comput. Biol. Chem. 87, 107322 (2020).
Acknowledgements
We thank the staff of the IBS Research Solutions Center and the Center for Electron Microscopy Research, Korea Basic Science Institute for their assistance with cryo-EM data collection. Computational work for this research was performed on the data analysis hub, Olaf, in the IBS Research Solution Center. We are also grateful to the Global Science experimental Data hub Center (GSDC) at Korea Institute of Science and Technology Information (KISTI) for computing resources and technical support. We gratefully acknowledge Katrin Böhnstedt and Ina Goroncy for excellent technical assistance. This research was supported by the Institute for Basic Science (IBS-R030-C1 to Y.F., H.K., A.H., D.S.L., and H.M.K.), the National Research Foundation of Korea (RS-2025-00523615 to H.M.K.) and the KAIST Convergence Research Institute Operation Program (to H.M.K.). Financial support from Austrian Science Fund FWF (grants FWF-P−32397-N28 and FWF-PAT2965423) is gratefully acknowledged (AZ).
Author information
Authors and Affiliations
Contributions
Y.F., H.K., H.H., A.Z., and H.M.K. designed the research; Y.F. and H.K. determined the cryo-EM structures; A.H. helped with data processing; D.S.L. purified proteins; Y.F., H.K., H.H., A.Z. and H.M.K. analyzed the data; Y.F., H.H., A.Z. and H.M.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Y., Kim, H., Lee, D.S. et al. Structural insight into TLR4/MD-2 activation by synthetic LPS mimetics with distinct binding modes. Nat Commun 16, 4164 (2025). https://doi.org/10.1038/s41467-025-59550-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59550-3
