
Abstract
Variation in malaria infection risk, a product of disease exposure and immunity, is poorly understood. We genotypically profiled over 13,000 blood samples from a six-year longitudinal cohort in Mali to characterize malaria infection dynamics with detail. We generated Plasmodium falciparum amplicon sequencing data from 464 participants (aged 3 months – 25 years) across the six-month 2011 transmission season and profiled a subset of 120 participants across the subsequent five annual transmission seasons. We measured infection rate as the molecular force of infection (molFOI, number of genetically distinct parasites acquired over time). We found that molFOI varied extensively among individuals (0–55 in 2011) but was independent of age and consistent within individuals over multiple seasons. Reported bednet usage was nearly universal. The HbS allele was associated with lower molFOI, and functional antibody signatures for the CSP C-term and RH5 antigens were correlated with low molFOI participants, identifying candidate immune correlates of protection. The large inter-individual variability in molFOI and consistency of intra-individual infection rate over time exhibits much greater dynamic range than malaria case incidence, and is most likely due to heterogeneous exposure to infectious mosquito bites. This and other factors contributing to variable infection risk should be considered in future clinical trials and implementation of malaria interventions.
Similar content being viewed by others


Introduction
Disease risk varies widely among hosts for many communicable diseases1,2,3. Risk of disease is a function of both the risk of infection by the pathogen and individual susceptibility to the development of symptomatic disease once infected. For pathogens that can cause asymptomatic infections, characterization of both symptomatic disease and asymptomatic infection is necessary to characterize the infectious reservoir, disease risk among the infected, and transmission dynamics4. Understanding variability in infection risk is important for designing and interpreting clinical trial data and evaluations of other potential interventions; if not accounted for, host variability in disease exposure and/or immunity could confound interpretations of intervention efficacy1,5,6. More importantly, a better understanding of what contributes to the heterogeneity in risk of infection could allow interventions to be targeted towards those at highest risk, or towards those with less naturally acquired immunity (NAI)7.
Despite the importance of heterogeneous infection risk, it can be difficult to study for diseases like malaria, where the gradual development of NAI through successive infections reduces risk of symptomatic disease without affecting infection risk, masking heterogeneity in infection incidence5. Consequently, an area of high transmission is a favorable setting in which to study infection risk, as low infection incidence may be interpreted as a consequence of low risk, rather than random chance. However, a disadvantage to high-transmission settings is that polyclonal infections resulting from newly incident infections in already-infected people will be more common8, preventing estimates of infection incidence through binary infection detection methods such as blood smear microscopy or polymerase chain reaction (PCR)9. To better understand complex infection dynamics, a high-resolution methodology is required to detect new infections among already-infected individuals.
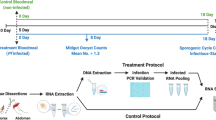
In this study, we used Illumina-based amplicon sequencing of four highly polymorphic malaria parasite antigens to provide an unparalleled portrait of polyclonal infection dynamics in a high transmission setting over 6 years for a large cohort of participants. We sequenced Plasmodium falciparum DNA from 13,152 dried blood spot samples from a longitudinal cohort in Kalifabougou, Mali (Fig. 1)9. Kalifabougou is a small, rural community with strong, seasonal malaria transmission during approximately half the year from July to December10. Approximately half (n = 6432) of the samples we genotyped are from the 2011 season, and represent 464 participants, ages 3 months−25 years, each of whom provided a finger prick blood sample every 2 weeks during the 6-month transmission season. The remaining samples (n = 6720) represent a subset of 120 participants (ages 3 months−8 years at enrollment), studied over the subsequent five annual transmission seasons, during which participants provided a blood sample every month. Participants also provided blood samples at any self-referred clinic visits when malaria was suspected by study clinicians. Prior studies of this cohort, using PCR to detect the first infection of the 2011 transmission season in all participants irrespective of symptoms, found that the majority of participants became infected with P. falciparum during a single transmission season, establishing Kalifabougou as an appropriately high transmission setting for evaluating infection rate heterogeneity9.

As described in Tran et al., the Kalifabougou cohort began in May 2011. We sequenced samples from 464 participants from the 2011 transmission season, as well as 120 participants who continued in the study from 2012 to 2016.
We performed genotyping of participant specimens to characterize infection rate heterogeneity and to look for associations between infection rate and features that might drive its variation. We estimated the molecular force of infection (molFOI, the number of genetically distinct strains that infect a participant over a set period of time—Supplemental Fig. 1)11,12. molFOI, originally estimated for malaria with capillary electrophoresis genotyping data12, has recently been extended to a variety of molecular data types13,14. Alternative metrics for estimating variation in infection rate, such as time to measuring the time to first infection via microscopy or PCR detection of parasitemia, provide information on a single infection event per person, and they cannot distinguish new infections in individuals who are infected at the start of a study. In comparison, molFOI represents new infection incidence at the level of strain over the full duration of a longitudinal study, with the potential to include many more infection events in the infection rate estimate15,16. We use molFOI to summarize infection history in 464 participants across one or more transmission seasons. We explored diverse factors that may contribute to the consistent patterns of heterogeneous infection rate that we observe among participants, including hemoglobin types, demography, geospatial data17, and immunological features18.
Results
molFOI is not associated with age
We define the molFOI as the number of genetically distinct strains that infected a given participant over a period of time12, where a newly incident infection by a genetically distinct strain was defined as observation of a novel amplicon haplotype (not seen in the previous two samples from the participant) at one or more of the four genotyped loci (Supplemental Fig. 1)12. We calculated the molFOI for each participant over the entirety of the 2011 season (Fig. 2a). The molFOI range was 0–55 (mean: 11, median: 10, standard deviation: 9). We found no overall correlation between participant age and molFOI (quasi-Poisson regression, age coded as a continuous variable, P = 0.077), though infection rate may be lower in the youngest and oldest participants We also examined the number of clinical disease episodes per participant in the same 2011 cohort, using the number of malaria treatments as a proxy for clinical disease diagnoses (Fig. 2b). The range in number of treatments per participant is smaller than molFOI, ranging from 0 to 6 (mean: 2, median: 2, standard deviation: 1). We found a negative correlation between participant age and number of treatments (quasi-Poisson regression, P = 0.00073, β = −0.02). We directly compared molFOI and number of treatments per participant (Supplemental Fig. 3a). The number of treatments was significantly associated with molFOI (quasi-Poisson model, t(462) = 3.07, P = 0.0023), with inclusion of treatments accounting for 2.02% of deviance in 2011 molFOI values when compared to the base model, implying that disease rate explains only a small fraction of the observed variation in infection rate (deviance explained calculated as the difference between the deviance observed by this model and the intercept-only model, divided by the intercept-only model deviance). These observations suggest that the NAI that accumulates with age in this population9 reduces the risk of clinical disease but does not provide measurable protection against infection.

a Each point represents a molFOI value for each of the 464 participants over the 2011 season. The x-axis groups participants by age, aiming for similar sized age groups for comparisons. We found no significant association between age and molFOI (quasi-Poisson regression, two-sided t test; t(462) = 1.78, P = 0.077, β = 0.014 [95% CI: −0.0014 to 0.029]). b Each point represents the number of treatments a participant received for symptomatic disease over the 2011 season for each of the 464 participants. The x-axis matches that in part a. We found a significant negative association between age and number of treatments (quasi-Poisson regression, two-sided t test; t(462) = −3.40, P = 0.00073, β = −0.02 [95% CI: −0.036 to −0.0096]). In both plots, violin plots represent the density distributions, with horizontal lines at the 25th, 50th, and 75th percentiles. CI confidence interval, molFOI molecular force of infection.
Intra-individual molFOI is consistent across seasons
We next compared individual molFOI across transmission seasons to determine whether molFOI variation in 2011 was driven by stochastic factors, or whether differences in infection rate among participants are consistent over time. We hypothesized that while heterogeneity in infection rate might obscure age-associated differences when comparing age bins cross-sectionally, these differences might be evident when profiling individuals longitudinally. We estimated the cumulative molFOI over the subsequent five seasons (2012–2016) and compared that value to the value from 2011 for individual participants (Fig. 3a). We rejected the null hypothesis that variation in 2011 molFOI is stochastic; participants with high molFOI in 2011 generally had high molFOI in 2012-2016 as well (Spearman’s rank correlation, ρ(111) = 0.53 (95% CI = [0.38, 0.66]), P = 1.51e−9; linear regression, P = 3.4e−13, R2 = 0.38). We performed the same analysis using symptomatic disease incidence (as estimated by number of malaria treatments) in place of molFOI in Fig. 3b. In contrast to molFOI, while the linear regression (adjusted R2 = 0.03, P = 0.041) between number of treatments in 2011 and 2012–2016 was significant, Spearman’s rank correlation (ρ(111) = 0.17 (95% CI = [−0.02, 0.35]), P = 0.073) did not indicate the presence a significant correlation in symptomatic disease incidence across time periods. This contrast between infection and treatment rates suggests that molFOI may be a more durable estimator of individual infection risk, as it does not appear to be modified by age-dependent NAI.

a Each point represents a single participant, showing molFOI from the 2011 season (x-axis) and cumulative molFOI across the 2012–2016 seasons (y-axis). The axis scales vary due to the difference in time periods and sampling strategy between the two datasets (see Fig. 1). The line represents a linear regression (adjusted R2 = 0.38, P = 3.4e−13). We also computed the two-sided Spearman’s rank correlation between molFOI in 2011 and 2012–2016; we found a highly significant, positive correlation (⍴(111) = 0.53 (95% CI = [0.38, 0.66]), P = 1.51e−9). b Replicates the figure in (a), but with number of treatments for symptomatic disease in 2011 on the x-axis and number of treatments for symptomatic disease in 2012–2016 on the y-axis. The line represents a linear regression (adjusted R2 = 0.03, P = 0.04), but a two-sided Spearman’s rank correlation did not identify a significant association between number of treatments in 2011 and 2012–2016 (⍴(111) = 0.17 (95% CI = [−0.02, 0.35]), P = 0.073). c Average molFOI of all participants of a given age (on the x-axis) during a given season (on the y-axis). d Average number of treatments of all participants of a given age (on the x-axis) during a given season (on the y-axis). e molFOI from the 2011 season, stratified by infection status at enrollment in May 2011. molFOI for participants who were infected at enrollment is significantly higher than for those not infected at enrollment (Kruskal–Wallis test, χ2(1) = 78.42, P = 8.33e−19). f Replicates the analysis in (c), but with number of treatments for symptomatic disease in 2011 on the y-axis. Participants who were infected at enrollment had significantly more treatments than those who were not infected at enrollment (Kruskal–Wallis test, χ2(1) = 7.85, P = 0.0051). Violin plots in (e, f) represent the density of the distributions, with horizontal lines at the 25th, 50th, and 75th percentiles. CI confidence interval, molFOI molecular force of infection.
We also stratified molFOI and treatment data for each participant by season, resulting in five molFOI values per participant (Supplemental Fig. 2). The individual molFOI-per-season values ranged from 0 to 106 (mean: 10, median: 6, standard deviation: 12). We found no significant differences in the overall distribution of molFOI values stratified by season, suggesting no major temporal or weather factors impacted any one season (Kruskal–Wallis test, χ2(4) = 4.36, P = 0.36). We visualized the patterns of average molFOI per age in each season (Fig. 3c). We found that a specific sub-cohort of participants that were age nine in 2012 consistently exhibited high average molFOI values over time, despite their increasing age and cumulative parasite exposure. We similarly visualized average treatment counts per age (Fig. 3d), but did not observe the same sub-cohort trend as with molFOI.
We further explored consistency in individual infection rate over time by fitting a linear mixed-effects model (Table 1) to these data, with participant age as a fixed effect and participant identity as a random effect, aiming to capture intra-individual changes in molFOI. In this model, fixed effects (participant age) had a pseudo-R2 value of 0.00, while the pseudo-R2 value for all effects was 0.50, further demonstrating the lack of relationship between age and infection rate. The mixed-effects model fit the data better than an age-only linear model (linear regression, adjusted R2 = 0.076, P = 4.4e−12; models compared via likelihood rank test, χ2(1) = 170.69, P = 5.24e−39), suggesting that participant identity is a more significant driver of variation in infection rate than age. We also fit linear mixed-effects models to subsets of this cohort by age (Table 1). In all of these models, age is not significantly associated with molFOI, and the pseudo-R2 value for all effects is notably larger than the pseudo-R2 value for age alone (models for participants of all ages, 0–4 years, 4–7 years, and 8+ years, respectively; pseudo-R2 for age as a fixed effect: 0.00, 0.00, 0.01, 0.00 in each model, respectively, and pseudo-R2 for all effects, including participant identity as a random effect: 0.50, 0.71, 0.37, 0.52, respectively).
We also examined infection status in May 2011 (at study enrollment) as a predictor of infection rate; 44% of participants (n = 202) were asymptomatically infected at enrollment. We found that molFOI from the 2011 season was significantly greater for participants who were infected at enrollment (Fig. 3e, mean molFOI for infected = 15.9; uninfected = 7.6; Kruskal–Wallis test, χ2(1) = 78.42, P = 8.33e−19). Again, we replicated this analysis with symptomatic disease incidence (as estimated by treatment counts) in place of molFOI (Fig. 3f). We found a significant difference for disease incidence as well (mean number of treatments for infected = 2.2, for uninfected = 1.9; Kruskal–Wallis test, χ2(1) = 7.85, P = 0.0051), though the range of this metric (0–6) is narrower than that of molFOI (0–55).
We compared infection rate to rates of symptomatic disease by evaluating molFOI with respect to the number of treatments per participant within a transmission season or the parasitemia of individual samples (Supplemental Fig. 3). We also hypothesized that lower density infections would correlate with lower molFOI, if participants had parasitemia near the level of detection of the genotyping assay. However, participants grouped by low, middle, or high molFOI in 2011 exhibited no significant differences in parasite densities (Kruskal–Wallis test, χ2(2) = 4.68, P = 0.096). In fact, participants with high molFOI in 2012-2016 had slightly lower densities (mean densities: 2.5e4 (low molFOI), 2.1e4 (mid molFOI), 2.4e4 (high molFOI); Kruskal–Wallis test, χ2(2) = 10.52, P = 0.0052; Dunn test with Holm correction, P = 0.0037 (low vs. high)), rejecting our null hypothesis. We speculate that individuals with more NAI may have lower parasite densities, and may be more likely to exhibit high molFOI due to reduced incidence of symptomatic disease and antimalarial treatment.
Host factors explain some heterogeneity in molFOI
We next hypothesized that host-specific factors could affect infection rate. We found that participant sex was not associated with molFOI in 2011 (Supplemental Fig. 4a, mean molFOI for females = 11.6, males = 11.1; Kruskal–Wallis test, χ2(1) = 2.47, P = 0.12) or 2012–2016 (Supplemental Fig. 4c, mean cumulative molFOI for females = 47.7, males = 54.0; Kruskal–Wallis test, χ2(1) = 0.77, P = 0.38). Similarly, we stratified symptomatic disease incidence by participant sex, finding that participant sex was also not associated with symptomatic disease counts in either 2011 (Supplemental Fig. 4b, mean cumulative number of treatments for females = 2.1, males = 1.9; Kruskal–Wallis test, χ2(1) = 1.17, P = 0.28) or 2012–2016 (Supplemental Fig. 4d, mean cumulative number of treatments for females = 4.4, males = 4.7; Kruskal–Wallis test, χ2(1) = 0.15, P = 0.70).
We also considered participant ethnicity, bednet usage, and socioeconomic factors19 as potential explanations for molFOI heterogeneity and temporal consistency20. However, neither ethnicity nor bednet usage varied significantly among participants. The majority of participants in both the 2011 and 2012–2016 cohorts are of the Bambara ethnic group (418/464 participants or 90% in 2011 and 105/120 participants in 2012–2016, or 87.5%). Participants nearly universally reported daily bednet usage when surveyed in 2013 (561/563 households owned bednets and 559/563 households reported daily usage). We used roof type as a proxy for socioeconomic status, as in prior studies of this cohort17, where metal roofs are a proxy for relative wealth. In the 2011 cohort, we found a significant difference in molFOI between participants living in homes with flat roofs (n = 143, mean molFOI = 14.1) vs. those from homes with metal roofs (n = 269, mean molFOI = 10.5; Kruskal–Wallis test, χ2(1) = 8.12, P = 0.004). We observed a similar trend in the 2012-2016 data between participants from homes with flat roofs (n = 37, mean molFOI = 58.6) vs. participants from homes with metal roofs (n = 76, mean molFOI = 49.8), but the difference was not statistically significant (Kruskal–Wallis test, χ2(1) = 1.64, P = 0.20).
Given that many individuals in this population carry malaria-protective variant alleles at the hemoglobin subunit beta (HBB) locus17, we examined molFOI stratified by HBB genotype (Fig. 4, stratifications of cohorts by HBB genotype and age or sex in Supplemental Table 1). Of the 464 participants in our 2011 cohort, 372 were homozygous for the ancestral allele, A (HbAA genotype). A total of 43 participants were heterozygous for the S allele (HbAS genotype), which confers the sickle cell trait and has been associated with lower risk of both uncomplicated and severe malaria disease21,22,23. A total of 47 participants were heterozygous for the C allele (HbAC), a genotype that has not been as clearly associated with protection from uncomplicated malaria as the HbAS genotype21 Due to the low sample size, we excluded two participants with HbCC and HbSC genotypes from the analysis in Fig. 4a only. We found significant differences in the molFOI in 2011 for participants with HbAS compared to both HbAA and HbAC genotypes (Fig. 4a, mean molFOI for HbAA (11.8), for HbAC (13.1), for HbAS (5.3). Kruskal–Wallis test, χ2(2) = 23.53, P = 7.8e−6, ε2 = 0.05. Dunn test with Holm adjustment, P = 7.6e−6 (HbAA vs. HbAS), 9.1e−5 (HbAC vs. HbAS), 0.51 (HbAA vs. HbAC)). In the 2012–2016 cohort, which had fewer participants (n = 120) and thus lower statistical power compared to the 2011 cohort, fewer participants had variant genotypes (6 HbAC and 17 HbAS); molFOI values were lower in HbAC (mean molFOI: 45.5) and HbAS (mean molFOI: 34.3) participants than in HbAA participants (mean molFOI: 54.3), but they did not reach the threshold of significant difference (Fig. 4c; Kruskal–Wallis test, χ2(2) = 0.96, P = 0.62). We also examined symptomatic disease stratified by HBB genotype (Fig. 4). We found significant differences in the number of treatments for individuals with HbAS genotype compared to HbAA in both the 2011 cohort (Fig. 4b, Kruskal-Wallis test, χ2(2) = 6.55, P = 0.038, ε2 = 0.014. Dunn test with Holm adjustment, P = 0.032 (HbAA vs. HbAS), 0.12 (HbAC vs. HbAS), 0.93 (HbAA vs. HbAC) and the 2012-2016 cohort (Fig. 4d, (Kruskal–Wallis test, χ2(2) = 12.58, P = 0.0019, ε2 = 0.11. Dunn test with Holm adjustment, P = 0.0016 (HbAA vs. HbAS), 0.36 (HbAC vs. HbAS), 0.53 (HbAA vs. HbAC)).

a molFOI values from 2011, stratified by participant HBB genotype. molFOI is significantly different between some groups (Kruskal–Wallis test, χ2(2) = 23.53, p value = 7.8e−6, ε2 = 0.05. Dunn test with Holm adjustment, p values = 7.6e−6 (HbAA vs. HbAS), 9.1e−5 (HbAC vs. HbAS), 0.51 (HbAA vs. HbAC)). b Number of treatments for symptomatic disease in 2011, stratified by HBB genotype. Number of treatments is significantly different between some groups (Kruskal–Wallis test, χ2(2) = 6.55, p value = 0.038, ε2 = 0.014. Dunn test with Holm adjustment, p values = 0.032 (HbAA vs. HbAS), 0.12 (HbAC vs. HbAS), 0.93 (HbAA vs. HbAC)). c molFOI values from 2012 to 2016, stratified by participant HBB genotype. No significant differences between groups (Kruskal–Wallis test, χ2(2) = 0.96, p value = 0.62). d Number of treatments for symptomatic disease in 2012–2016 stratified by participant HBB genotype. Number of treatments is significantly different between some groups (Kruskal–Wallis test, χ2(2) = 12.58, p value = 0.0019, ε2 = 0.11. Dunn test with Holm adjustment, p values = 0.0016 (HbAA vs. HbAS), 0.36 (HbAC vs. HbAS), 0.53 (HbAA vs. HbAC)). In all panels, horizontal lines represent significant differences between groups, and colors represent the same categories as the x-axis. Violin plots represent the density of the distribution, with horizontal lines representing the 25th, 50th, and 75th percentiles. HBB hemoglobin subunit beta locus, molFOI molecular force of infection.
Additionally, we considered other potential effects that could be associated with HBB genotype (Supplemental Fig. 5). Heterozygous HBB genotypes did not cluster into a particular region of Kalifabougou (Supplemental Fig. 5a), nor did they have a different range in participant ages than HbAA (Supplemental Fig. 5b, Kruskal–Wallis test, χ2(2) = 1.84, P = 0.40). We also did not find a difference in parasite density for HbAS individuals, which could have potentially biased our ability to detect molFOI (Supplemental Fig. 5c, Kruskal–Wallis test, χ2(2) = 2.45, P = 0.29), though the sample size of available parasitemia data after stratification by HBB genotype is small.
We fit an additional linear mixed-effects model to the 2012–2016 molFOI data incorporating these additional host factors in addition to age, with participant identity as a fixed effect (Table 1). In this model, none of the fixed effects have a significant effect on molFOI on their own (P = 0.26, 0.47, 0.60, 0.09, 0.41 for age, roof type, HbAC genotype, HbAS genotype, and sex, respectively). As a whole, the fixed effects contributed a small amount to the model’s total explanatory power; pseudo-R2 for fixed effects alone was 0.03, and pseudo-R2 for all effects was 0.52.
Geographic features may explain some heterogeneity in molFOI
We next considered whether geographic factors could affect infection rate. First, we analyzed the spatial autocorrelation of the 2011 molFOI values (Fig. 5a), to look for general associations between household location and molFOI. We found a very small negative dispersion of molFOI (Moran’s I = −0.023, pseudo-P = 0.002, two-sided hypothesis, compared to a Monte-Carlo simulation of permutations of the data), suggesting that micro-geographic features do not drive locally similar molFOI values. We found a positive correlation between the distance from participants’ homes to the Kalifabougou study clinic and their molFOI in 2011 (Fig. 5b, linear regression, adjusted R2 = 0.089, P = 4.3e−8; quasi-Poisson regression, t(312) = 5.71, P = 2.68e−8). We also found a positive correlation between participants’ distance to the closest part of the river system and their 2011 molFOI (Fig. 5c, linear regression, adjusted R2 = 0.025, P = 0.0031; quasi-Poisson regression, t(312) = 3.00, P = 0.003). Taken together, these spatial analyses suggest that geographic differences may have a slight effect, but they do not explain the majority of the observed heterogeneity in infection rate in this cohort (2.45% and 8.30% of deviance explained by distance to clinic and rivers, respectively).

a Visualization of homes and molFOI in Kalifabougou, Mali. Each dot represents the geographic coordinates of one participant’s home, colored by their molFOI in 2011. Points are slightly jittered, for visibility. Black lines represent local rivers, geospatial data from Global Map of Mali © ISCGM/IGM. b Scatter plot of each participant’s molFOI in 2011 (x-axis) vs. the distance from their home to the Kalifabougou clinic (y-axis, labeled in kilometers) (quasi-Poisson regression, two-sided t test; t(312) = 5.71, P = 2.68e−8, β = 0.00021 [95% CI: 0.00014–0.00028]). The line shows the linear regression (adjusted R2 = 0.089, P = 4.3e−8). c A similar scatter plot to (b), except that the y-axis shows the distance from each participant’s home to the nearest river (quasi-Poisson regression, two-sided t test; t(312) = 3.00, P = 0.003, β = 0.19 [95% CI: 0.066–0.32]). Linear regression shown as described above (adjusted R2 = 0.025, P = 0.0031). CI confidence interval, molFOI molecular force of infection.
Several serological features distinguish between low and high infection rate participants
Systems serological profiling was previously performed on serum from 201 participants included in our sequencing study18. These data were generated using samples from May 2011, at enrollment into the study and before the transmission season began that year, with the objective of defining potential correlates of protection. Antigen-specific IgG1, IgG2, IgG3, IgG4, IgA1, and IgM to AMA1, full-length CSP, MSP1, RH5 and the N- and C-terminal domains of CSP were quantified. The functional potential of these antigen-specific antibodies was also captured, including the ability of the antigen-specific antibodies to bind to Fc receptors (FcRn, FcγRIIAH, FcγRIIAR, FcγRIIB, FcγRIIIAF, FcγRIIIAV, and FcγRIIIB) and recruit antibody-dependent complement deposition, antibody-dependent cellular phagocytosis, and antibody-dependent neutrophil phagocytosis. While the evolution of antigen-specific neutrophil activation was previously found to be correlated with rates of disease severity in this cohort18, here we aimed to define whether malaria-specific serological markers were positively or negatively associated with infection rate, as estimated by molFOI.
We set thresholds for molFOI in 2011 to create “low molFOI” (<4) and “high molFOI” (>13) groups, which correspond to the lowest and highest 12% of the molFOI distribution (Fig. 6a). To explore the sensitivity of the analysis to threshold used, we defined two additional different sets of thresholds, to consider the upper and lower 33% and 25% of the molFOI distribution (Supplemental Table 2). We primarily discuss results from the most stringent set of thresholds here; the others are highly concordant and are described in Supplemental Figs. 6 and 7. For each set of molFOI cutoffs, we compared each serological feature between low and high molFOI groups using Mann–Whitney tests with Benjamini–Hochberg corrections. In all three approaches to molFOI stratification, Fcγ receptor binding to the highly polymorphic c-terminal region of CSP (specifically, Fcγ receptor IIIAV, or FcγRIIIAV), as well as Fcγ receptor binding to RH5 (FcγRIIIAV), were enriched in participants with low molFOI (Mann–Whitney test with Benjamini–Hochberg correction, P = 0.015 and 0.030, respectively; Fig. 6c, d), suggesting that these may be protective against infection. While only these two features were significant in all three molFOI stratifications, 13 additional features were enriched in individuals with high molFOI in two of the three stratifications, including IgG1 specific to AMA1, similar to previous studies suggesting that it may be a marker of infection history24,25,26.

a Distribution of molFOI in the 2011 season for the 201 participants for whom we had both genetic and serology data. We compared low and high molFOI participants in the following analyses, who are colored in orange and teal, respectively (see Supplemental Table 2 for details on molFOI ranges used here and in other versions of this analysis). b We identified serological features with significant differences between high and low molFOI groups, using two-sided Mann–Whitney tests with Benjamini–Hochberg corrections for multiple comparisons. Each point here represents a serological feature in the analysis. The x-axis shows the difference between the average value of that feature in the high molFOI vs. low molFOI groups; points greater than zero were expressed more in the high molFOI group and points less than zero were expressed more in the low molFOI group. The y-axis shows the −log10 of the adjusted p values; points above the dashed line were significant. Significant features are labeled. c Raw data from FcγRIIIAV binding to the c-terminal of CSP, one of the features that was significant in this analysis (two-sided Mann–Whitney test with Benjamini–Hochberg correction for multiple comparisons; W = 112, uncorrected P = 0.00018, corrected P = 0.015; confidence interval not applicable). d Raw data from FcγRIIIAV binding to RH5, the other significant feature in this analysis (two-sided Mann–Whitney test with Benjamini–Hochberg correction for multiple comparisons; W = 127, uncorrected P = 0.00068, corrected P = 0.030; confidence interval not applicable). In (c, d), each point represents a single participant. Violin plots represent the density of the distribution, with horizontal lines representing the 25th, 50th, and 75th percentiles. CSP circumsporozoite protein, molFOI molecular force of infection, RH5 reticulocyte-binding protein homolog 5.
Discussion
In this study, we identified an unexpectedly large degree of malaria infection rate heterogeneity among individuals in a small community. This heterogeneity in infection rate was consistent over time, and was only partially explained by measured geographic, demographic, behavioral, and host genetic factors. Heterogeneity in infection risk has likely been previously under characterized in most studies using microscopy or PCR positivity to detect infection7,27,28, and was measured with precision in this study through multiplexed amplicon genotyping of a very densely sampled longitudinal sample collection from 464 participants.
Our data indicate that host genotype may be one modulator of infection rate. Participants from this cohort were previously genotyped at the HBB locus, given the known protective effects of the sickle (HbS) and HbC alleles against severe disease21. Similar to a previous study that estimated force of infection via capillary electrophoresis genotyping16, our finding of lower molFOI in HbAS participants than HbAA or HbAC participants (Fig. 4) may indicate that HbAS participants are protected against blood-stage infection, and/or that blood stage infection duration was shorter and less likely to be detected. Recent studies have identified four genomic loci within P. falciparum associated with individuals with HbS alleles29,30, and three of these four loci are found at >10% frequency in genomes from Mali in the MalariaGEN Pf7 data resource30,31. This association suggests that the pool of parasites commonly infecting HbAS individuals may be a subset of the larger parasite population, which could lead to lower molFOI. However, HbAS participants only represent 10% of the cohort participants, and the effect size of this association was small (ε2 = 0.05). Despite the role of HBB as the most important locus conferring protection against clinical malaria21, it only explains a small portion of variation in infection rate in this cohort. Other unmeasured host genetic factors may also influence infection rate, but are likely to exhibit smaller effect sizes than HbS and HbC.
Previous studies have linked infection risk heterogeneity with geographic factors32,33. We found significant but small associations between molFOI heterogeneity and distance to the central clinic in Kalifabougou, as well as distance to the nearest river (Fig. 5), suggesting that geographic factors may contribute a small amount to the heterogeneity of infection rate we observed. Studies with a wider range in geographic and ecological factors34 than this one may benefit from considering geographic factors. Behavioral factors, including bednet usage, have also been indicated in previous studies of disease rate heterogeneity12; however, participants in this cohort almost universally reported daily bednet usage. Despite the reported consistency of bednet usage among participants, the most likely explanation for heterogeneous infection rate in this study remains variable exposure to infectious mosquito bites, a parameter for which validated markers are lacking for measurement at an individual level35.
We conducted a systems serology analysis to identify candidate immune correlates of infection rate vs. infection protection, using molFOI as a composite measure of disease risk/protection. Analysis of systems serology data from a subset of participants profiled in this study identifies several potential correlates of infection rate or protective immunity18. We found that participants with low molFOI in 2011 (low rate, and/or high protection; the majority of participants in this group were also uninfected at study enrollment) had enriched Fcγ receptor IIIA binding antibody responses to both the C-terminus of CSP (Fig. 6c) and RH5 (Fig. 6d). While previous studies have found low antibody titers for RH5 in individuals in malaria-endemic regions36,37, RH5 antibodies have been associated with protection against malaria36,38,39. Because RH5 is expressed in the blood stages of the parasite life cycle40, protective antibodies cannot prevent blood-stage infection, but may contribute to reduced molFOI estimates by shortening the duration of infections and/or lowering parasitemia below the limit of detection of the genotyping assay used in this study. However, given that CSP is expressed in sporozoites, an immune response to CSP could potentially prevent progression to blood-stage infection and reduce molFOI. This finding accords with recent studies of serological responses to the CSP-based RTS,S/AS01 vaccine that suggested a protective role for Fc-effector functions, particularly those specific to CSP41,42, as well as previous work with this cohort that found an association between functional CSP-specific IgG and protection from clinical disease and decreased parasite density18. Further work examining responses to different vaccine dose regimens found an increased response to CSP c-terminal-specific responses in participants who received the dose regimen that had higher vaccine efficacy43. Finally, a recent study examining responses to the radiation-attenuated whole sporozoite PfSPZ vaccine found increased expression of genes related to Fcγ receptor-mediated phagocytosis correlated with protective outcomes, regardless of vaccination status44. Taken together, these results suggest that increased response to CSP with antibodies able to bind FcγRIIIA and activate peripheral or liver-resident NK cells, phagocytic cells, and memory CD8+ T cells may be an important mechanism of immune protection for both natural and vaccine-induced immunity to infection, even if the implicated regions of CSP differ (C-terminus and NANP central repeat42, respectively).
This work has some limitations. Amplicon sequencing data recovery was variable across samples, with dropout of one or more amplicons in 35% (n = 1540) of 4383 parasite-positive samples. We mitigated this by requiring conservative approaches to define new infections. The age of some of the dried blood spots from which we extracted DNA may have impacted our sensitivity to detect new infections in a systematic manner, but we observed consistent distributions of molFOI from samples from 2012 to 2016, despite a 5-year difference in original sample age. Additionally, heterogeneity in the number of treatments a participant received over a period of time and the density of parasites present within samples have the potential to bias these analyses, by creating variable periods of time during which participants were refractory to new infections. We did find expected correlations between both parasite density and number of treatments with participant age (Supplemental Fig. 3); older participants tended to have fewer treatments and lower density infections. However, if these factors were to bias our analyses, we expect that molFOI would decrease in participants with lower density infections and/or fewer treatments. Instead, we found slightly positive correlations—participants who were treated more often tended to have higher molFOI than those treated less. We found no difference in parasite density data for high vs low molFOI participants in 2011, though we did find a slightly lower density for participants with high molFOI in 2012–2016 than those with low molFOI. Overall, these differences trend in the opposite direction than what we hypothesized could bias our analysis, though the differences are small.
We were also unable to measure exposure to infectious mosquito bites among the participants in this study. While we examined distance to the main river, we were unable to capture other ecological variables that could play an important role in the observed heterogeneous infection rate. Although smaller mosquito breeding sites (ponds, puddles) were not captured by these analyses, the overall negative geographic dispersion of molFOI suggests that microgeographic factors are not significant in this setting. We were able to include household roof type as a proxy for socioeconomic status, but further details on housing variability could serve as better surrogates for mosquito exposure.
In summary, the significant inter-individual variation in infection rate we observed in this cohort study is only partially explained by the diverse measured variables we studied, motivating future studies to identify these factors. In particular, factors associated with risk of symptomatic disease in previous studies could play a role, particularly variability in mosquito biting rates among people20,45,46,47,48, or intrinsic immune factors like HLA genotype that may not be dependent on age and previous exposure49. Serological markers of exposure to mosquito bites have been developed for the population level but are thus far not accurate at the individual level50. Heterogeneity in infection rate could be informative in clinical trials for selective enrollment of high-risk individuals in small clinical trials, or where intervention efficacy may be affected by individual infection status or risk, as has been hypothesized for several malaria vaccine trials44,51,52. Individuals with a higher infection risk may exhibit a distinct pre-existing natural immunity profile, and/or a distinct response to new immune stimuli, which could help or hinder an intervention. For example, RTS,S/AS01 exhibits significant enhanced protection in individuals who are infected at first vaccination and/or experience high infection rate53. While it would be impractical to measure molFOI directly for trial participants before treatment group assignment, it can be assessed retrospectively, and this study as well as a recent phase 2b trial of RTS,S/AS01 observed a strong association between molFOI and infection status as enrollment53, which we suggest may serve as a useful proxy for conducting analyses of efficacy stratified by infection risk. Finally, an enhanced understanding of the drivers of infection risk heterogeneity could inform measures to alleviate risk and assist in targeting interventions to better protect those most vulnerable to infection, especially when deployment of interventions is constrained due to manufacturing limitations54 or shortfalls in local public health resources.
Methods
Samples from cohort
We processed samples from the Kalifabougou longitudinal cohort study previously described9 (Fig. 1). The Kalifabougou cohort study was approved by the Ethics Committee of the Faculty of Medicine, Pharmacy and Dentistry at the University of Sciences, Technique and Technology of Bamako, and the Institutional Review Board of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH IRB protocol number: 11IN126; https://clinicaltrials.gov/; trial number NCT01322581). The original approvals authorized ancillary genetic and epidemiological investigations, which covered the current study. The original study authors obtained written informed consent from parents or guardians of participating children before inclusion in the study, and they collected these samples between 2011 and 2016. The original study randomly sampled 20% of the population of Kalifabougou by age and invited them to enroll in the study (n = 857). Six hundred and ninety-five participants were enrolled in May 2011. We selected a subset of the participants from 2011—we excluded any individuals who did not have a PCR-positive result for P. falciparum at any point in the 2011 season in the original study9, and we required at least 10 samples per person over the 2011 season. For participants ages 7–10 years, we performed a random selection stratified by sex and age, and for other ages, we included all individuals. These stratifications resulted in 464 participants from 2011. For participants who continued in the study through the end of the 2016 season, we required at least 10 samples per person per season and restricted participant age to 10 years or younger at enrollment in May 2011. We included all participants ages 6 years and under meeting those criteria, and we again randomly selected participants ages 7–10 at enrollment meeting those criteria by age and sex.
DNA extraction and sequencing
We physically randomized all samples prior to DNA extraction, to minimize the impact of any batch effects. We extracted DNA from the clinical samples and sequenced them using the 4CAST amplicon panel, as previously described55. This multiplexed amplicon sequencing panel targets highly polymorphic regions within the csp (c-terminus), ama1, sera2, and trap genes of P. falciparum. The csp and trap genes are expressed during the mosquito and liver stages of the malaria parasite life cycle, whereas sera2 and ama1 are expressed during the blood stage. The high haplotypic diversity exhibited by these antigens enables detection of superinfections on top of polyclonal infections, a requirement for accurate molFOI estimation in this transmission setting. The primer sequences used to amplify these four loci are provided in Supplemental Table 3; the full protocol is unchanged from the original publication55.
We sequenced samples from 2011 on Illumina MiSeq instruments, with 96–384 samples within each run. We sequenced samples from 2012 to 2016 on Illumina NovaSeq instruments, with 768 samples per lane. Data from these samples were submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession PRJNA1129562.
Data processing
We processed the paired-end sequencing data through a custom pipeline55, based on the Divisive Amplicon Denoising Algorithm (DADA2)56, which produces pseudo-CIGAR strings summarizing the observed polymorphisms in each haplotype. In the 2011 dataset, where the number of samples varied per sequencing batch, we divided the read-pairs at this stage by 2 or 4, for batches of 192 or 96 samples, respectively. Next, we required a haplotype to have a minimum of 10 reads per sample, as well as a minimum of 1% of the within-sample reads per locus. In these data after filtering, we observed a median of 631 total reads per sample, and a mean of 7947 reads per sample. In the 2012–2016 dataset (sequenced on NovaSeq instruments), we required a haplotype to have a minimum of 200 reads per sample, with the same within-sample frequency minimum as before. In these data, we observed a median of 27,600 reads per sample and a mean of 169,886 reads per sample.
Additionally, for each dataset, we assessed the number of samples in which each haplotype appeared (Supplemental Fig. 8). Despite the difference in read depth between the two datasets, we found similar numbers of haplotypes present in more than one sample within each dataset; we found 552 haplotypes present in at least two samples in the 2011 dataset, and 597 in the 2012–2016 dataset. We did find more “singleton” or unique haplotypes (found in only one sample) in the 2012–2016 dataset (381) than in the 2011 dataset (299). Unique haplotypes were distributed among samples from many different participants and we found 7 or fewer unique haplotypes per participant across all samples in 2011 (13 or fewer unique haplotypes per participant in 2012–2016). The increased sequencing depth in the 2012–2016 dataset may partially explain the increase in singleton haplotypes detected—unique haplotypes made up a small portion of the reads in their respective samples than shared haplotypes did. (interquartile range (IQR) for the percentage of reads per unique haplotypes within a sample in 2011: 0.4–2.0%; IQR for the percentage of reads per shared haplotypes within a sample in 2011: 2.0–18.6%; IQR for percentage of reads per unique haplotype in 2012–2016: 0.3–1.3%, IQR for percentage of reads per shared haplotype in 2012–2016: 1.4–16.4%). We removed all unique (singleton) haplotypes from further analysis.
Defining molFOI
We defined individual clones (individual haplotypes at any locus) and infection events (all haplotypes within a sample). To allow for imperfect data, stochastic dropout of loci, and sequestration of parasites, we tested different conditions to allow for clones to disappear for short periods of time and still be considered part of an ongoing infection. We also required at least ten samples per participant per season to include in our sequencing, to minimize uneven numbers of samples between participants. In the 2011 dataset, we tested both the number of “skipped visits,” the number of samples without a given clone before it reappears (0−4), the number of days between appearances of a given clone (15, 30, 60—to correct for longer periods of time between visits, if a participant had fewer samples from a given period of time), and combinations of the two metrics. Although the number of new clones changed with this sensitivity testing, the overall conclusions of our analyses did not; thus, we defined clones as new if they are not detected for more than two visits in a row or more than 30 days. We also tested the sensitivity of our definitions of new infection events, requiring 1, 2, or 3 new clones present within a sample, or requiring new clones at 2 different loci present within a sample. We defined new infection events by the presence of 2 new clones within a sample. We also repeated a subset of the above combinations with a higher minimum read-threshold (50 read-pairs, instead of 10). Finally, we repeated all sensitivity tests with a higher threshold to retain haplotypes in the analyses: 50 read-pairs per haplotype per sample. We retained the 10 read-pair minimum threshold, and all analyses include data from all four loci, unless otherwise stated.
We repeated these sensitivity analyses on the 2012–2016 dataset, with only a few changes. Due to the change in sampling frequency in these years (biweekly to monthly), we increased the number of days between clone appearances in our tests (30, 60, or 90 days), and we defined clones as new if they are not detected for more than two visits in a row or more than 60 days. We also tested a higher read threshold here (500 read-pairs per haplotype per sample) and retained the 200 read-pair threshold.
We defined the molFOI as the total number of new clones present within a participant over a given period of time (generally one transmission season). To do this, we looked at all new infection events for a participant and filtered to only new clones present then, as opposed to clones carried over from the previous visit. We then counted the maximum number of new clones present at any one of our four sequenced loci. Finally, we summed these maximum numbers of new clones present from each new infection event over the time period of interest.
Spatial analysis
We used latitude and longitude coordinates of participant households, as well as the metrics of distance to clinic or water from these households, from earlier studies of this cohort17. We used the latitude and longitude coordinates for visualization purposes (Fig. 5a), along with coordinates of local water (Global Map of Mali © ISCGM/IGM). We performed the analyses in Fig. 5b, c using the distance data from the previous cohort study. We used the R packages “sf” and “tidyverse” for the visualization in Fig. 5a57,58,59. We used the “mat2listw” and “moran.mc” functions from the “spdep” R package for the Moran’s I analysis58.
Systems serology analysis
These analyses included the 201 participants that overlapped between the serological data and the genetic data. The distribution of participant ages in this subset of participants does not significantly differ from that of the full cohort (Kruskal–Wallis test, χ2(1) = 0.60, P = 0.44). We centered and scaled all data (Z-scored), using the “scale” function in R. For each feature, we removed any outlier points, defined as having a raw value outside of 1.5 times the interquartile range. We then created three different thresholds for selecting high and low molFOI participants (see Supplemental Table 2). We performed the same analyses on each subset of data. We identified features that were significantly different between the two groups, using Mann–Whitney tests with a Benjamini–Hochberg correction for multiple hypothesis testing18.
Statistical analysis
We used R 4.1.260 for all analyses, unless otherwise stated, with the packages “tidyverse,” “here,” and “RColorBrewer”59,61,62. All statistical tests were two-sided, when applicable. We tested for significance using “glm” or “kruskal.test,” as described in figure legends. We used the “dunn.test” R package to perform Dunn tests with Holm post hoc corrections for all analyses with multiple hypothesis testing, unless otherwise stated63. We used “cor_test” and “cor_to_ci” from the “correlation” package for the Spearman’s ranked correlation analysis in Fig. 4a, b64. We used the “lmer” function from the “lme4” R package65 to fit linear mixed-effects models; we then used the “anova” function and the pseudo-R2 procedure as previously described66 to compare models. We used “geom_quasirandom” from “ggbeeswarm” for all jittered beeswarm plots67. We used Adobe Illustrator 2024 to create Fig. 1 and Supplemental Fig. 1, to arrange panels in all figures, and add lines to denote significant comparisons between groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Supplementary Data 1 file contains a data dictionary and seven tables (three each for the 2011 cohort and 2012–2016 cohort). One table per cohort contains relevant metadata on the participants, including age (in years) and HBB genotype, among others. A second table lists all the samples that we sequenced per cohort, and each sample is identified by a unique ID, as well as the participant ID, visit date, and whether it generated sequencing data passing filters. A third table contains all the haplotypes called in our pipeline at each of the four loci per sample, with sample IDs that can be joined with the other tables to link to relevant metadata. The seventh table contains the raw systems serology dataset. Consent was obtained to publish these data, including multiple indirect identifiers. All raw sequencing data were submitted to the NCBI Sequence Read Archive under accession PRJNA1129562. Source data are provided with this paper.
Code availability
The code used to process sequencing data is available at https://github.com/broadinstitute/malaria-amplicon-pipeline.
References
Anacleto, O. et al. Genetic differences in host infectivity affect disease spread and survival in epidemics. Sci. Rep. 9, 4924 (2019).
King, K. C. & Lively, C. M. Does genetic diversity limit disease spread in natural host populations? Heredity 109, 199–203 (2012).
Springbett, A. J., MacKenzie, K., Woolliams, J. A. & Bishop, S. C. The contribution of genetic diversity to the spread of infectious diseases in livestock populations. Genetics 165, 1465–1474 (2003).
Slater, H. C. et al. The temporal dynamics and infectiousness of subpatent Plasmodium falciparum infections in relation to parasite density. Nat. Commun. 10, 1433 (2019).
Bousema, T., Kreuels, B. & Gosling, R. Adjusting for heterogeneity of malaria transmission in longitudinal studies. J. Infect. Dis. 204, 1–3 (2011).
Macià, D., Pons-Salort, M., Moncunill, G. & Dobaño, C. The effect of disease transmission on time-aggregated treatment efficacy estimates: a critical analysis of factors influencing the RTS,S and R21 malaria vaccine phase 3 trials. Lancet Infect. Dis. S1473-3099, 00090-00098 (2025).
Rodriguez-Barraquer, I. et al. Quantifying heterogeneous malaria exposure and clinical protection in a cohort of Ugandan children. J. Infect. Dis. 214, 1072–1080 (2016).
Nkhoma, S. C. et al. Population genetic correlates of declining transmission in a human pathogen. Mol. Ecol. 22, 273–285 (2013).
Tran, T. M. et al. An intensive longitudinal cohort study of Malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clin. Infect. Dis. 57, 40–47 (2013).
Crompton, P. D. et al. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. J. Infect. Dis. 198, 1265–1275 (2008).
Koepfli, C. & Mueller, I. Malaria epidemiology at the clone level. Trends Parasitol. 33, 974–985 (2017).
Mueller, I. et al. Force of infection is key to understanding the epidemiology of Plasmodium falciparum malaria in Papua New Guinean children. Proc. Natl. Acad. Sci. USA 109, 10030–10035 (2012).
Badoum, E. S. et al. Force of infection (FOI) and multiplicity of infection (MOI) in Plasmodium falciparum infected children aged 1.5-12 years living in the malaria endemic area of Banfora, Burkina Faso. Pathogens 13, 883 (2024).
Zhan, Q., Tiedje, K. E., Day, K. P. & Pascual, M. From multiplicity of infection to force of infection for sparsely sampled Plasmodium falciparum populations at high transmission. eLife 13, RP100076 (2024).
Briggs, J. et al. Sex-based differences in clearance of chronic Plasmodium falciparum infection. eLife 9, e59872 (2020).
Gong, L. et al. Evidence for both innate and acquired mechanisms of protection from Plasmodium falciparum in children with sickle cell trait. Blood 119, 3808–3814 (2012).
Doumbo, S. et al. Co-infection of long-term carriers of Plasmodium falciparum with Schistosoma haematobium enhances protection from febrile malaria: a prospective cohort study in Mali. PLoS Negl. Trop. Dis. 8, e3154 (2014).
Nziza, N. et al. Accumulation of neutrophil phagocytic antibody features tracks with naturally acquired immunity against malaria in children. J. Infect. Dis. 228, 759–768 (2023).
Patrick, S. M. et al. Household living conditions and individual behaviours associated with malaria risk: a community-based survey in the Limpopo River Valley, 2020, South Africa. Malar. J. 22, 156 (2023).
Markwalter, C. F. et al. Plasmodium falciparum infection in humans and mosquitoes influence natural Anopheline biting behavior and transmission. Nat. Commun. 15, 4626 (2024).
Taylor, S. M., Parobek, C. M. & Fairhurst, R. M. Impact of haemoglobinopathies on the clinical epidemiology of malaria: a systematic review and meta-analysis. Lancet Infect. Dis. 12, 457–468 (2012).
Allison, A. C. Protection afforded by sickle-cell trait against subtertian malarial infection. Br. Med. J. 1, 290–294 (1954).
Aidoo, M. et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 359, 1311–1312 (2002).
Stanisic, D. I. et al. Acquisition of antibodies against Plasmodium falciparum merozoites and malaria immunity in young children and the influence of age, force of infection, and magnitude of response. Infect. Immun. 83, 646–660 (2015).
Kusi, K. A. et al. Variations in the quality of malaria-specific antibodies with transmission intensity in a seasonal malaria transmission area of Northern Ghana. PLoS ONE 12, e0185303 (2017).
Wu, L. et al. Antibody responses to a suite of novel serological markers for malaria surveillance demonstrate strong correlation with clinical and parasitological infection across seasons and transmission settings in The Gambia. BMC Med. 18, 304 (2020).
Clark, T. D. et al. Factors determining the heterogeneity of malaria incidence in children in Kampala, Uganda. J. Infect. Dis. 198, 393–400 (2008).
Kreuels, B. et al. Spatial variation of malaria incidence in young children from a geographically homogeneous area with high endemicity. J. Infect. Dis. 197, 85–93 (2008).
Band, G. et al. Malaria protection due to sickle haemoglobin depends on parasite genotype. Nature 602, 106–111 (2022).
Hamilton, W. L. et al. A fourth locus in the Plasmodium falciparum genome associated with sickle haemoglobin. Preprint at https://doi.org/10.1101/2023.09.14.557461 (2023).
MalariaGEN et al. An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Res. 6, 42 (2021).
Cohen, J. M. et al. Mapping multiple components of malaria risk for improved targeting of elimination interventions. Malar. J. 16, 459 (2017).
Gul, D. et al. Investigating differences in village-level heterogeneity of malaria infection and household risk factors in Papua New Guinea. Sci. Rep. 11, 16540 (2021).
Bell, G. J. et al. Impacts of ecology, parasite antigenic variation, and human genetics on RTS,S/AS01e malaria vaccine efficacy. Curr. Epidemiol. Rep. 8, 79–88 (2021).
Helb, D. A. et al. Novel serologic biomarkers provide accurate estimates of recent Plasmodium falciparum exposure for individuals and communities. Proc. Natl. Acad. Sci. USA 112, E4438–E4447 (2015).
Tran, T. M. et al. Naturally acquired antibodies specific for Plasmodium falciparum reticulocyte-binding protein homologue 5 inhibit parasite growth and predict protection from malaria. J. Infect. Dis. 209, 789–798 (2014).
Douglas, A. D. et al. The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nat. Commun. 2, 601 (2011).
Healer, J. et al. Neutralising antibodies block the function of Rh5/Ripr/CyRPA complex during invasion of Plasmodium falciparum into human erythrocytes. Cell. Microbiol. 21, e13030 (2019).
Wang, L. T. et al. Natural malaria infection elicits rare but potent neutralizing antibodies to the blood-stage antigen RH5. Cell 187, 4981–4995.e14 (2024).
Wright, G. J. & Rayner, J. C. Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLOS Pathog. 10, e1003943 (2014).
Feng, G. et al. Induction, decay, and determinants of functional antibodies following vaccination with the RTS,S malaria vaccine in young children. BMC Med. 20, 289 (2022).
Suscovich, T. J. et al. Mapping functional humoral correlates of protection against malaria challenge following RTS,S/AS01 vaccination. Sci. Transl. Med. 12, eabb4757 (2020).
Das, J. et al. Delayed fractional dosing with RTS,S/AS01 improves humoral immunity to malaria via a balance of polyfunctional NANP6- and Pf16-specific antibodies. Medicine 2, 1269–1286.e9 (2021).
Senkpeil, L. et al. Innate immune activation restricts priming and protective efficacy of the radiation-attenuated PfSPZ malaria vaccine. JCI Insight https://doi.org/10.1172/jci.insight.167408 (2024).
Guelbéogo, W. M. et al. Variation in natural exposure to Anopheles mosquitoes and its effects on malaria transmission. eLife 7, e32625 (2018).
Guglielmo, F. et al. Quantifying individual variability in exposure risk to mosquito bites in the Cascades region, Burkina Faso. Malar. J. 20, 44 (2021).
Mbewe, R. B. et al. Genotyping of Anopheles mosquito blood meals reveals nonrandom human host selection: implications for human-to-mosquito Plasmodium falciparum transmission. Malar. J. 22, 115 (2023).
O’Meara, W. P. et al. Mosquito exposure and malaria morbidity: a microlevel analysis of household mosquito populations and malaria in a population-based longitudinal cohort in Western Kenya. J. Infect. Dis. 221, 1176–1184 (2020).
Hill, A. V. S. et al. Common West African HLA antigens are associated with protection from severe malaria. Nature 352, 595–600 (1991).
Kearney, E. A. et al. Anopheles salivary antigens as serological biomarkers of vector exposure and malaria transmission: a systematic review with multilevel modelling. eLife 10, e73080 (2021).
Oneko, M. et al. Safety, immunogenicity and efficacy of PfSPZ Vaccine against malaria in infants in western Kenya: a double-blind, randomized, placebo-controlled phase 2 trial. Nat. Med. 27, 1636–1645 (2021).
Murphy, S. C. et al. PfSPZ-CVac efficacy against malaria increases from 0% to 75% when administered in the absence of erythrocyte stage parasitemia: a randomized, placebo-controlled trial with controlled human malaria infection. PLoS Pathog. 17, e1009594 (2021).
Juraska, M. et al. Genotypic analysis of RTS,S/AS01E malaria vaccine efficacy against parasite infection as a function of dosage regimen and baseline malaria infection status in children aged 5-17 months in Ghana and Kenya: a longitudinal phase 2b randomised controlled trial. Lancet Infect. Dis. S1473-3099, 00179–8 (2024).
Osoro, C. B. et al. Policy uptake and implementation of the RTS,S/AS01 malaria vaccine in sub-Saharan African countries: status 2 years following the WHO recommendation. BMJ Glob. Health 9, e014719 (2024).
LaVerriere, E. et al. Design and implementation of multiplexed amplicon sequencing panels to serve genomic epidemiology of infectious disease: a malaria case study. Mol. Ecol. Resour. 22, 2285–2303 (2022).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Pebesma, E. Simple features for R: standardized support for spatial vector data. R. J. 10, 439–446 (2018).
Pebesma, E. & Bivand, R. Spatial Data Science: With Applications in R (Chapman and Hall/CRC, 2023).
Wickham, H. et al. Welcome to the {tidyverse}. J. Open Source Softw. 4, 1686 (2019).
R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2021).
Müller, K. Here: a simpler way to find your files. https://CRAN.R-project.org/package=here (2020).
Neuwirth, E. RColorBrewer: ColorBrewer palettes. https://CRAN.R-project.org/package=RColorBrewer (2022).
Dinno, A. Dunn.Test: Dunn’s test of multiple comparisons using rank sums. https://CRAN.R-project.org/package=dunn.test (2017).
Makowski, D., Ben-Shachar, M. S., Patil, I. & Lüdecke, D. Methods and algorithms for correlation analysis in R. J. Open Source Softw. 5, 2306 (2020).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Nakagawa, S. & Schielzeth, H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 4, 133–142 (2013).
Clarke, E., Sherrill-Mix, S. & Dawson, C. Ggbeeswarm: categorical scatter (violin point) plots. https://CRAN.R-project.org/package=ggbeeswarm (2023).
Acknowledgements
We thank the participants in the study and their caregivers for making this study possible. We thank the Kalifabougou clinic and study team. We thank Charlotte Switzer for helpful discussions. We thank Shanping Li for help with the original cohort samples. This project was supported by an NIH R01 award (R01AI141544, to DEN). The original Mali cohort study was funded by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.L., C.O.B., P.D.C., T.M.T., D.E.N. Formal analysis: E.L. Funding acquisition: C.O.B., D.E.N. Investigation: Z.M.J., M.S., N.N. Methodology: E.L., Z.M.J., M.S. Project administration: D.E.N. Resources: N.N., G.A., P.D.C., B.T., T.M.T. Supervision: C.O.B., G.A., D.E.N. Validation: E.L., Z.M.J., M.S. Visualization: E.L. Writing—original draft preparation: E.L., D.E.N. Writing—review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
LaVerriere, E., Johnson, Z.M., Shieh, M. et al. Marked heterogeneity in malaria infection rate in a Malian longitudinal cohort. Nat Commun 16, 6512 (2025). https://doi.org/10.1038/s41467-025-61462-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61462-1
