
Abstract
Neuropsychiatric disorders remain difficult to treat due to complex and poorly understood mechanisms. NeuroPainting is a high-content morphological profiling assay based on Cell Painting and optimized for human stem cell–derived neural cell types, including neurons, progenitors, and astrocytes. The assay quantifies over 4000 features of cell structure and organelle organization, generating a dataset suitable for phenotypic screening in neural models. Here, we show that, in studies of the 22q11.2 deletion—a strong genetic risk factor for schizophrenia—we observe cell-type-specific effects, particularly in astrocytes, including mitochondrial disruption, altered endoplasmic reticulum organization, and cytoskeletal changes. Transcriptomic analysis shows reduced expression of cell adhesion genes in deletion astrocytes, consistent with post-mortem brain data. Integration of RNA and morphology data suggests a link between adhesion gene dysregulation and mitochondrial abnormalities. These results illustrate how combining image-based profiling with gene expression analysis can reveal cellular mechanisms associated with genetic risk in neuropsychiatric disease.
Similar content being viewed by others


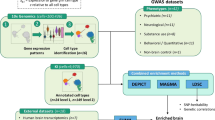
Introduction
Severe mental illnesses represent a significant global health crisis. Individuals suffering from mental illness face debilitating symptoms, including dramatic mood swings, persistent hallucinations, and increased risk of substance abuse and suicidality, contributing to severe personal and societal consequences such as unemployment, homelessness, and incarceration1,2. Current therapeutic approaches, largely unchanged for decades, rely on a combination of psychotherapy and pharmacotherapy. However, these treatments do not adequately address the needs of all patients, with more than half of people with schizophrenia and bipolar disorder continuing to experience debilitating functional impairments3,4,5,6. This inadequacy underscores a critical gap in our understanding of the biological mechanisms of these conditions, hindered further by the slow pace of innovation in psychiatric therapeutic development. The stalled advancement in therapeutic options is partly due to the complex nature of these disorders, which are influenced by a myriad of genetic and environmental factors7. There is a need for robust phenotyping approaches to increase our ability to extract meaningful biological insights associated with these conditions.
Chromosome 22q harbors the recurrent 22q11.2 microdeletion (22q11.2del), a well-studied copy number variant (CNV) that leads to the 22q11.2 deletion syndrome (also known as DiGeorge Syndrome), a multisystem disorder characterized by a range of neuropsychiatric, cardiac, and immune phenotypes8. Notably, 22q11.2del is the genetic factor most strongly associated with schizophrenia at the population level9. The cardiac effects of this deletion have been linked to the TBX1 gene within the canonical deletion interval10. However, the neuropsychiatric impacts remain largely unexplained by candidate genes within the deleted region, suggesting the possibility of non-canonical mechanisms underlying these phenotypes10,11,12,13. Additionally, like all other chromosomes, 22q contains thousands of common SNPs that contribute additively to human complex trait variation, including traits associated with the 22q11.2 deletion14. Given the immense clinical heterogeneity of 22q11.2del15, robust, multimodal phenotyping is crucial to uncover the molecular mechanisms driving this disorder. Such an approach will help identify affected pathways and assess the mutation’s impact across different cellular contexts, offering insights into its diverse clinical manifestations, with potential implications for schizophrenia more broadly.
Phenotypic assays such as Cell Painting16 that capture high-throughput, unbiased cellular morphological measurements are appealing because they do not require selecting a specific target or even a specific phenotype in advance, both of which can be flawed if based on incomplete mechanistic understanding. Cell Painting, and its adaptations, have been used in various applications to understand gene function, uncover cell-type-specific biology, and build large reference datasets for the scientific community17,18,19,20,21. Cell morphology profiling offers a significant advantage for functional genomics studies over methods like gene expression, as it is more cost-effective and easily scalable for both bulk and single-cell level analyses of mRNA. Cell morphology profiling also has the advantage of capturing downstream phenotypic outcomes, whereas mRNA levels reflect the potential for protein expression and may show only partial correlation with protein levels22. Cell morphology profiling, therefore, allows for a more direct assessment of functional cellular changes in response to genetic or environmental perturbations.
Recent studies indicate that disease-associated cellular phenotypes can be identified directly from patient samples in an unbiased manner, rather than relying on hypothesized phenotypes. Specifically, morphology phenotypes have been discovered for neurological and neuropsychiatric conditions by image-based profiling of primary skin fibroblasts. For example, mitochondrial morphology was disrupted in a study of 41 patients with mid-stage idiopathic Parkinson’s disease compared to controls23. Similarly, a deep learning-based analysis identified distinct morphological phenotypes in 46 Parkinson’s patients—32 sporadic and 14 with specific mutations—compared to healthy controls24. Machine learning also differentiated images of 12 spinal muscular atrophy patients from 12 healthy controls, though technical confounders could not be ruled out25. Wali et al. successfully used morphological profiling, including DNA, mitochondria, and acetylated α-tubulin stains, to distinguish 15 patients with hereditary spastic paraplegia and matched controls26. While associating a cell phenotype with a condition does not prove causation, using patient-derived cellular phenotypes provides a strong link to therapeutic hypotheses: reversing an unhealthy cellular phenotype through chemical modulation in vivo may be effective at reversing behavioral deficits.
A strength of studying primary cells, such as skin fibroblasts, is their ease of collection, allowing studies to reach sufficient power to elucidate cellular phenotypes. The use of primary somatic cells also presents a limitation. For some conditions, including psychiatric disorders, it is important to consider phenotypes in the appropriate tissues, as disease-associated genetic variation is often enriched in tissue-specific genes27,28. Wali et al. recognized this and extended their initial work on fibroblasts to patient-derived induced pluripotent stem cells (iPSCs) to validate phenotypes in neural cells29. While this shift to iPSCs allowed for the examination of more disease-relevant cell types, it also significantly reduced the study’s scale, limiting phenotyping to only six cell lines (three healthy/three cases) and focusing on a few cellular characteristics like mitochondria and neurite length. Although this approach was useful for identifying strong phenotypes from highly pathogenic mutations, it underscores the need for methods capable of detecting more subtle phenotypes associated with milder genetic mutations that may present with significant clinical heterogeneity30,31. To achieve this, it will be necessary to extend these studies to a larger number of cell lines and capture a broader range of cellular features. State-of-the-art microscopy approaches like Cell Painting, which can measure thousands of morphological features16,17, offer a promising solution.
Here, we established NeuroPainting, a high-dimensional phenotyping assay adapted from Cell Painting, for morphological discovery in neural cell types derived from human iPSCs. This robust and scalable system enabled us to examine cells from many donors simultaneously, increasing the likelihood of capturing individual variation known to exist at the genetic and clinical levels in psychiatric conditions. We discuss the optimization and development of the NeuroPainting assay and analytical pipelines and quantify cellular structure and organelles across different neuropsychiatric-relevant iPSC-based neural cell types, including neurons and astrocytes. We further use NeuroPainting to discover morphological phenotypes associated with the 22q11.2 deletion. We show that NeuroPainting can quantify differences among diverse cell types and demonstrate cell-type-specific morphological phenotypes linked to the 22q11.2 deletion, highlighting the utility of NeuroPainting for uncovering biology linked to genetic variants implicated in psychiatric disease. Lastly, we integrated RNA sequencing data with our NeuroPainting morphological data, pinpointing specific cell adhesion genes that may underlie morphological changes in astrocytes with 22q11.2 deletion and suggesting avenues for biological characterization and potential therapeutic strategies.
Results
NeuroPainting for high-dimensional morphological phenotyping of neural cell types at scale
We developed “NeuroPainting” by adapting the six original dyes of the Cell Painting assay to permit robust and scalable analyses of iPSC-derived neural cell types (Figs. 1a and S1a). We generated NeuroPainting profiles from stem cells, neuronal progenitor cells, neurons, and astrocytes from 44 cell lines, 22 of which harbor a 22q11.2 deletion and 22 neuro-typical controls with matched age and ancestry, which we previously characterized13 (Fig. 1b and Table S1). For each cell type, all lines were differentiated in concert using methods we previously described32,33,34 before being plated in 384-well microplates for staining and imaging. To reduce technical variation known to impact microscopy-based assays24, we randomized plate maps to ensure equal distribution of genotypes and cell lines across the plates. We plated cells at predetermined densities and matured them to a desired time point determined by preliminary experiments (Methods) before fixing and staining. Cells were then imaged at 20× using a Perkin Elmer Phenix high-content imaging system. We tested several densities for each of the cell types in our study to identify the optimal parameters. Our goal was to identify conditions where cells are dense enough to grow in a healthy, robust manner but not so crowded as to interfere with their typical morphology. By examining brightfield images from the 384-well plates, we settled on specific densities and fixation conditions for each individual cell type (iPSCs—10 k cells/well, fixation 24 h post plating; NPCs—15 k cells/well, fixation 24 h post plating; neurons—2.5 k cells/well, fixation 25 days post plating, astrocytes—3 k cells/well, fixation 48 h post plating, Fig. S1b).

a Representative images of astrocytes stained with the Cell Painting dyes. Scale bar = 50 μm. b Schematic of strategy for capturing NeuroPainting high-dimensional image-based profiles from a large cohort of iPSCs. In batches of 44, iPSCs were differentiated into NPCs, neurons, and astrocytes and then plated in 384-well microplates in a randomized orientation for downstream image acquisition. CellProfiler was used to process images and extract morphological features. We imaged one batch for each cell type.
We created an image analysis and feature extraction pipeline specific to neural cell types in the freely available software, CellProfiler. We chose standard methods for feature extraction (as opposed to deep learning based methods) to improve the interpretation of morphological phenotypes - well-understood features of morphology include size, shape, intensity, and texture of various stains, which resulted in 4104 different cellular traits35. We grouped these traits based on each cellular compartment: Cells, Nuclei, Cytoplasm (“Cells” refers to the entire cell, which is segmented based on the outer membranes; “Nuclei” are segmented based on the Hoescht stain; “Cytoplasm” is the region of the “Cell” minus the ”Nuclei”) and seven measurement categories: AreaShape, Granularity, Intensity, Radial Distribution, Correlation, ObjectSkeleton, and Texture (Table S2). Measurements in an additional “Other” category were excluded from analysis, because it includes some non-biological features such as location within the well. All analytical pipelines are freely available via our GitHub repository (see Methods). We refer to the collection of single-cell features measured by these pipelines as NeuroPainting profiles. We then mean-averaged single-cell profiles to create a profile for each well. These well-level average profiles underwent a four-step preprocessing pipeline described in ref. 36, yielding 723 unique features (Table S3): (1) removal of low-variance features (coefficient of variation < 1e-3), (2) robust standardization using median absolute deviation, (3) rank-based inverse normal transformation (INT), and (4) correlation-based feature selection to eliminate redundant features with correlation > 0.9. All downstream analyses were performed on the resulting data matrix, and morphology traits were Gaussianized using the INT method to generate feature values.
Morphology-based classification of neural cell types
By eye, the NeuroPainted cell types displayed markedly different morphology (Fig. 2a). While, using our approach, each cell type is generated separately, the ability to classify cell types based on morphological profiles would be valuable for examining cultures or tissues with mixed cell types, and for studying the effect of mutations, or genetic or pharmacological perturbations on cell states and differentiation potential and trajectories. To explore whether we could use NeuroPainting to classify cell types, we compared profiles across stem cells, neuronal progenitors, neurons, and astrocytes from the neurotypical controls. Although the differentiation protocol for each cell type was distinct, each imaging batch contained all donors of a given cell type. This setup facilitated robust cross-donor comparisons—our primary goal—but we acknowledge that it could introduce batch effects across cell types.

a Representative image of cell types included in this study. Scale bar = 50 μm. b Accuracy of Random Forest classifiers in predicting cell type from image-based features. Each point represents the mean classification accuracy from 5-fold cross-validation using all features, grouped by cell type. Error bars represent 95% confidence intervals obtained from n = 1000 bootstrap replicates. c Barplot of feature categories for the 100 most important features based on our Random Forest classifier. The fractions of the features marked as important are indicated for each category. d Classification accuracy for each feature category, calculated as in (b). e Violin plot for Nuclei_RadialDistribution_MeanFrac_Mito_1of4 across cell types (p = 1.37e-35, ANOVA, n = 176 per cell type, two-sided). Feature values are INT scores for each morphology trait. f Violin plot for Cytoplasm_Correlation_RWC_AGP_ER across cell types (p = 1.64e-15, ANOVA, n = 176 per cell type, two-sided), presented like Fig. 2e. In both panels (e, f), boxplots within violins show the median (line), first and third quartiles (box limits), and whiskers extend to the most extreme data point within 1.5 times the interquartile range (IQR) from the box. g Schematic representation of two distinguishing cell morphology characteristics across the four cell types. (top) Nuclei_RadialDistribution_MeanFrac_Mito_1of4 is related to the intensity of the mitochondrial dye near the inner-most region of the cell. A greater value for this feature indicates more of a cell’s mitochondria are present near the nucleus. (bottom) Cytoplasm_Correlation_RWC_AGP_ER refers to the co-presence of the AGP and ER dyes for a given location in the cytoplasm of the cell. A greater feature value indicates that there is stronger overlap for the AGP and ER stains when compared to lower values.
We trained a Random Forest classifier to distinguish among the four cell types based on 723 features across 788 samples. The model achieved 97.41% overall accuracy (95% CI: 92.63–99.46%) with a Kappa of 0.962, indicating strong agreement between predicted and true cell types. Neurons were classified perfectly (sensitivity = 1.00), while astrocytes (0.92), progenitors (0.96), and stem cells (0.96) had slightly lower sensitivities. Positive predictive values (PPV) were similarly high, though neurons had a slightly lower PPV (0.95), while the other cell types achieved perfect values. Balanced accuracy ranged from 95.83% to 98.08%, confirming the model’s robust performance (Fig. 2b). Thus, the classifier confirmed that NeuroPainting accurately captures meaningful, biologically relevant differences in cell morphology.
To identify the most important features, we ranked their contributions to the model’s performance. Correlation and Radial Distribution features dominated the top 100, comprising 80% of the most informative features for classifying cell types (Fig. 2c). Correlation features capture the spatial relationship between pixel intensities across different channels, providing insights into colocalization of cellular components (CCs) such as organelles and cytoskeletal structures. This can indicate differences in organelle organization or intracellular signaling pathways between cell types. Radial Distribution features, on the other hand, describe how specific stains or structures are distributed relative to the cell center, offering information on nuclear positioning, cytoplasmic compartmentalization, and organelle organization. This result was unexpected, as we had anticipated a more prominent role for AreaShape features, given the observable differences in cell shape.
To further assess the contribution of each feature category, we retrained our classifier using only one of the six feature types at a time (AreaShape, Correlation, Granularity, Intensity, RadialDistribution, and Texture). The Correlation category achieved the highest performance (accuracy = 95.31%, Kappa = 0.93), followed by Texture (accuracy = 92.90%, Kappa = 0.89) and RadialDistribution (accuracy = 92.76%, Kappa = 0.89). AreaShape had the lowest accuracy (85.34%, Kappa = 0.78), while Granularity and Intensity both showed moderate performance, with accuracies around 91% and Kappa values of 0.87. These findings underscore the importance of Correlation and Texture features, while AreaShape features, although less powerful individually, still contribute meaningfully to distinguishing cell types (Fig. 2d).
From the top of our feature importance analysis, we highlighted two key features: Nuclei_RadialDistribution_MeanFrac_Mito_1of4, which relates to mitochondrial distribution around the nucleus, and Cytoplasm_correlation_RWC_AGP_ER, which measures the relative weighted correlation between actin/Golgi/plasma membrane and the endoplasmic reticulum (ER) in the cytoplasm. Nuclei_RadialDistribution_MeanFrac_Mito_1of4 specifically reflects the intensity of mitochondrial staining near the innermost region of the cell, where higher values indicate a greater concentration of mitochondria near the nucleus. In contrast, Cytoplasm_correlation_RWC_AGP_ER captures the co-localization of AGP and ER staining in the cytoplasm, with greater values indicating a stronger overlap between these two compartments. Both features exhibited statistically significant differences across cell types (Fig. 2e, f; ANOVA, p = 1.37e-35, p = 1.64e-15). To illustrate these morphological differences, we have included schematics that depict how mitochondrial distribution and AGP-ER correlation vary across different cellular contexts, highlighting the structural distinctions between cell types (Fig. 2g).
In conclusion, our results suggest that NeuroPainting provides a robust and biologically meaningful approach to classifying neural cell types. Beyond classification, it offers a nuanced understanding of how cell morphology diverges across lineages, complementing gene expression data. This capability opens the door to using NeuroPainting as a powerful readout in studies exploring functional consequences of genetic variation and treatment responses in neurobiology.
Cell-type-specific morphological signatures in neural cell types from individuals with 22q11.2 deletion syndrome
The 22q11.2 deletion is the most common chromosomal deletion, associated with various diseases including congenital heart defects, autoimmune disorders, and neuropsychiatric conditions such as schizophrenia8. While prior research has shown that this deletion alters gene expression and synaptic function in specific cell types13, the underlying mechanisms driving its broad phenotypic effects remain poorly understood. This highlights the need for high-throughput phenotyping to uncover insights. To address this, we employed NeuroPainting to investigate whether morphological signatures could differentiate control and deletion samples across neural cell types, potentially offering avenues for drug screening.
We generated NeuroPainting profiles from neuronal progenitor cells, neurons, and astrocytes derived from iPSCs of individuals with 22q11.2 deletion syndrome and matched controls. Correlation matrices revealed distinct clustering of control and deletion samples, indicating clear morphological differences among these groups across cell types (Fig. 3a). Pairwise Pearson correlations within and between conditions were calculated for each cell type. The mean correlation for control-control pairs was r = 0.17, 0.03, 0.007, and 0.14 for stem cells, progenitors, neurons, and astrocytes, respectively. These values dropped substantially in control-deletion comparisons, with r = −0.11, −0.02, −0.003, and −0.10. A two-sample T-test confirmed that these distributions were significantly different for stem cells, progenitors, and astrocytes, but not for neurons (t-statistic = 96.09, p = 4.68e-53; t-statistic = 14.70, p = 9.279608e-49; t-statistic = 7.28, p = 3.165349e-13; t-statistic = 33.86, p = 3.948375e-232) (Fig. S2a). This demonstrates that the 22q11.2 deletion induces distinct morphological changes in these cell types.

a Correlation matrix of NeuroPainting profiles for iPSCs, progenitors, neurons, and astrocytes from control and 22q11.2 deletion cell lines. The green and purple notation on the left of the plot indicates control versus deletion genotype. The rainbow notation corresponds to individual cell line aliases. b Barplot for significantly different features as measured by the Wilcoxon rank sum-test between control and 22q11.2 deletion carriers across each cell type. Features were significant if they passed an FDR threshold of 5%. c Venn diagram showing the number of significant features that overlap across the different cell types. d Dotplot of feature characteristics for 72 overlapping significant features. e Dotplot of feature values for Cytoplasm_Correlation_RWC_ER_Mito across each cell type (n = 22 per genotype, per cell type). Error bars represent the standard error of the mean. f Representative images for min/max values for Cytoplasm_Correlation_RWC_ER_Mito in control (top) and 22q11.2 (bottom) astrocytes. Minimum and maximum values are based on per-well INT transformed feature values. Images were selected based on the highest and lowest average feature value for both control and 22q11.2 deletion carriers. Scale bar is 50 μm.
Next, we performed a Wilcoxon Rank-Sum test to identify specific morphological features significantly impacted by the deletion. We identified 567, 387, 224, and 536 significant features in stem cells, progenitors, neurons, and astrocytes, respectively, that passed multiple hypothesis correction (Fig. 3b and Tables S4–S7, Wilcoxon, FDR < 0.05). Among these, 72 significant features overlapped across each cell type, suggesting some shared effects of the 22q11.2 deletion in different cellular contexts (Fig. 3c). Many feature categories, including texture, radial distribution, and correlation across several organelles were altered by the mutation in all cell types (Fig. 3d). The most prominent impact of the 22q11.2 deletion across cell types was associated with the AreaShape of the Nuclei (Fig. 3d).
The effect of the 22q11.2 deletion on Cytoplasm_Correlation_RWC_ER_Mito showed an interesting response: higher values for the deletion in stem cells and progenitors, roughly equivalent values for neurons, and lower values for astrocytes (Fig. 3e). This cell type-specific behavior parallels previous findings from gene expression analyses13. Cytoplasm_Correlation_RWC_ER_Mito describes the spatial relationship between the ER and mitochondria in the cytoplasm, with higher values indicating a stronger co-localization of these two compartments, whereas lower values suggest a more spatially distinct organization. To further illustrate these differences, representative images of astrocytes with the highest and lowest feature values per well were selected for both control and 22q11.2 deletion carriers (Fig. 3f). These images highlight how changes in ER-mitochondria interactions manifest in the most extreme cases, where maximum values correspond to cells with the greatest ER-mitochondria overlap, while minimum values reflect cells with the least spatial correlation between these structures.
Most significant features did not overlap, pointing to cell-type-specific effects of the 22q11.2 deletion on cell morphology. These mirrored earlier observations by our group when examining gene expression patterns in these same cell lines13. While many of the significant features were not shared across all cell types, we observed similar effects on feature compartments, categories, and channels from the 22q11.2 deletion (Fig. S2b). In stem cells, numerous texture-based features for AGP, RNA, and mitochondria across all cellular compartments were identified, indicating potential organelle dysfunction (Fig. S2c, p = 2.69e-08). Neuronal progenitor cells carrying a 22q11.2 deletion showed altered cell size (Fig. S2d, p = 0.000268), consistent with neuroimaging studies linking the mutation to changes in brain size37,38. Neurons with the 22q11.2 deletion exhibited asymmetry of their cell bodies when compared to control cells (Fig. S2e, p = 0.0239). In astrocytes, the deletion resulted in altered radial distributions of the ER, AGP, and mitochondria, potentially impacting cell shape, motility, and mitochondrial dynamics (Fig. S2f, p = 5.9e-6).
We next sought to determine whether the observed differences in NeuroPainting signatures could be leveraged to classify samples by genotype. For each donor and cell type, we averaged individual morphological features and applied a Random Forest classifier. The classifier achieved high accuracy across most cell types, with balanced accuracies of 0.95 for stem cells (p < 0.0001), 0.96 for astrocytes (p < 0.0001), and 0.84 for progenitors (p = 0.04). However, classification accuracy was notably lower in neurons (0.56, p = 0.30), consistent with fewer differentially significant features in this cell type. The area under the curve (AUC) scores further supported these findings, indicating strong discriminatory power in stem cells (AUC = 0.95) and astrocytes (AUC = 0.96), moderate performance in progenitors (AUC = 0.84), and poor performance in neurons (AUC = 0.54) (Fig. S2g). These results suggest that NeuroPainting signatures reliably capture genotype-specific morphological differences, with limited retrievability in neurons.
These findings highlighted widespread disruptions in cellular organization across cell types associated with the 22q11.2 deletion.
22q11.2 deletion regulates expression of cell-adhesion molecules in astrocytes
We further investigated the impact of the 22q11.2 deletion in astrocytes, which displayed nearly 408 significantly altered morphological features. Given increasing evidence of the critical role that glial cells, particularly astrocytes, play in psychiatric conditions through astrocyte-neuron interactions34,39,40,41, we aimed to understand how this deletion affects astrocyte function.
We generated genome-wide gene expression data from a subset of our cell lines (n = 12, control = 6/deletion = 6) using a pooled (cell village) approach33,42. This approach allowed us to culture and profile cells from multiple donors within the same experimental batch, reducing technical variability and enabling direct comparisons across genotypes (Fig. S3a). Uniform Manifold Approximation and Projection (UMAP) showed high homogeneity across individual cell lines, with no cell line clustering distinctly from others using Louvain unsupervised clustering (Fig. 4a). Nor did we observe dramatic clustering based on genotype, indicating that the presence of the deletion did not impair differentiation capacity broadly (Fig. 4b). The fact that overall expression profiles were not dramatically different suggested that a consistent change in expression of individual genes might be informative. As expected, we observed a consistent decrease in the expression of genes located in the Chr22q11.2 interval (Fig. S3b).

a UMAP projection of single-cell RNA-sequencing (scRNAseq) data from astrocytes colored by donor. b UMAP projection of astrocytes scRNAseq data colored by genotype. c Volcano plot for DEGS (Wald test with Benjamini–Hochberg correction, Log2FC cutoff = 0.05, padj > 0.05). d Gene set enrichment for downregulated genes (Enrichment significance was assessed using a hypergeometric test with Benjamini–Hochberg correction.). e. Metagene score analysis for genes in downregulated pathways (Two-sample T-test, p = 1.065e-6, n = 48, 24 control/24 22q11.2). f Heatmap of logCPM expression for cell-adhesion genes. g Metagene score analysis for top 25 SNAP-a genes (NRXN1, SLC1A2, RNF219-AS1, NTM, ZNF98, GPC5, GRM3, HPSE2, NKAIN3, SLC4A4, CTNND2, NCKAP5, SGCD, LSAMP, GPM6A, LRRC4C, LRRTM4, EPHB1, PREX2, RORA, TMEM108, ARHGAP24, SYNE1, TENM2, AC091826.2) (Two-sample T- test-test, p = 0.0111, n = 48, 24 control/24 22q11.2). In both panels (e, g), boxplots within violins show the median (line), first and third quartiles (box limits), and whiskers extend to the most extreme data point within 1.5 times the IQR from the box.
We measured differentially expressed genes (DEGS) using DESeq2 by generating pseudo-bulked expression profiles for each donor across sequencing replicates (Methods). We identified 1358 DEGS based on our threshold (Fig. 4c, Table S8, Log2FC > 0.5, padj < 0.05). Of these, 450 were upregulated and 908 were downregulated. We next performed a gene-set enrichment analysis (GSEA) on both upregulated and downregulated genes in 22q11.2 deletion astrocytes. GSEA of upregulated genes revealed significant enrichment in just two pathways related to epithelial and branching morphogenesis (padj. < 0.01). However, we observed downregulation of 10 pathways in our data (padj. < 0.01). Among these, several pathways related to neuronal development as well as axon generation and guidance were identified (Fig. 4d). We observed a strong decrease in the expression of genes in these pathways by applying a metagene approach (Fig. 4e, Two-sample T-test, t = 6.5022, df = 23.712, p = 1.065e-06). Notably, many of the genes that comprise the leading edge for our significant pathways (ALCAM, CDH2, FN1, NCAM1, PTPRM, RELN, SEMA3E) behave as cell-adhesion molecules and have been linked to schizophrenia and other psychiatric conditions (Fig. 4f)43,44,45,46,47,48,49. Recent studies have shown that disruption of cell-adhesion gene expression in astrocytes is associated with schizophrenia, and these genes harbor a high degree of genetic risk for psychiatric outcomes39,50. To assess whether the impact of the 22q11.2 deletion on astrocyte gene expression was consistent with emerging evidence from findings in brain tissue from persons with idiopathic schizophrenia, we generated a metagene score based on the top gene loadings in astrocytes from the synaptic-neuron astrocyte program (SNAP) identified in Ling et al.39. We observed a significant decrease in the expression of these genes in 22q11.2 astrocytes compared to control cells (Fig. 4g, two-sample T-test, t = 2.6843, df = 33.663, p = 0.01119).
These results suggested that the 22q11.2 deletion significantly impacts astrocyte gene expression patterns relevant to psychiatric conditions.
Gene-morphology correlations in 22q11.2 deletion highlight changes in mitochondrial features
We next sought to explore the relationship between these changes in gene expression and changes in cell morphology to nominate genes that may play a role in our 22q11.2 NeuroPainting signatures. To do this, we generated donor-level (n = 12, control = 6/deletion = 6) Gaussianized profiles for both RNA-seq and our morphological profiles in astrocytes. For our analyses, we focused on all DEGs (1358) and all significant morphological features (536) from our earlier results.
A Canonical correlation analysis (CCA) identified strong associations between gene expression patterns and morphological features. The first canonical variable (CV1) represents a weighted linear combination of morphological features that maximally correlates with a corresponding gene expression variable. CV1 effectively separated samples by condition, highlighting systematic differences in how gene expression relates to morphology in deletion versus control samples (Fig. 5a). Importantly, this axis does not measure a single morphological feature but rather captures an aggregate pattern of morphological variation that is most strongly linked to gene expression differences. To further understand these relationships, we examined the top 100 genes and top 100 morphological features based on their loadings in the first canonical variable (CV1). These features and genes contribute most strongly to the latent structure captured by CCA, meaning they are key drivers of the observed variation in gene expression-morphology associations rather than direct measures of morphological change.

a Scatter plot of first CCA variables (CV1) for RNA-seq and NeuroPainting profiles, colored by condition (control = green, 22q11.2 = purple). b Heatmap of correlation coefficients between gene expression and morphology feature values for the top 100 genes and the top 100 features from CCA loadings. Correlation difference is measured as the absolute value between the control and deletion coefficients. c Scatter plot of absolute differences in correlation coefficients against fold change coefficients between control and 22q11.2 deletion astrocytes for gene-feature pairs. d Barplot for morphology feature categories for cell adhesion gene-feature pairs (chi-square test, Two-sided, χ² = 28.655, df = 6, p-value = 7.069e-05). e The Dotplot highlights the absolute difference in correlation coefficients between control and 22q11.2 astrocytes for cell adhesion gene-mitochondrial feature pairs.
Then we more thoroughly examined the specific relationships between these 100 genes and 100 features to identify relationships between these pairs that were altered by the presence of the 22q11.2 deletion. We computed pairwise correlations for each gene-feature pair, separately for control and deletion conditions, and identified 530 gene-feature pairs with significantly different correlations (R2) between control and deletion samples (Fig. 5b, p-value < 0.05).
We were particularly interested in gene-feature pairs whose correlations displayed dramatic changes. For example, whether there were pairs with strong positive correlations in control samples, but in the presence of the deletion, showed strong negative correlations. Changes in correlation were quantified as the absolute difference in correlation coefficients between conditions. We observed many gene-feature correlations that were dramatically different between the control and 22q11.2 deletion astrocytes. Notably, features related to RadialDistribution and Granularity were disproportionately affected (chi-square test, χ² = 23.669, df = 5, p = 0.00025). This suggested that many of our gene expression changes were associated with these feature categories.
To investigate whether these changes in gene-morphology correlations were driven by underlying alterations in gene expression, we examined the relationship between log2 fold change (log2FC) in gene expression and the observed differences in correlation coefficients using Spearman’s rank correlation test. We found a significant negative correlation (rho = −0.364, p-value < 1e-05), suggesting that genes with larger changes in expression (either upregulated or downregulated) tended to exhibit greater changes in their correlations with morphological features in the deletion samples (Fig. 5c).
Next, we wanted to explore the biological context of these relationships. To do so, we conducted a pre-ranked GSEA including the genes from our significant gene-feature pairings. We identified two pathways that were significantly enriched in our list (FDR-adjusted p < 0.05). The pathway glial cell apoptotic process (GO:0034349) was enriched, driven by three key genes: PRKCH, AKAP12, and TNFRSF21. Enrichment of this pathway suggests the 22q11.2 deletion may be rendering astrocytes more vulnerable to apoptosis. Additionally, the collagen-containing extracellular matrix pathway (GO:0062023) included nine genes (LTBP2, LAMA4, SBSPON, NAV2, COLEC12, NTN4, MMP23B, MEGF9, and SFRP2), many of which are implicated in cell adhesion processes51,52,53,54. This finding corroborates our earlier results that 22q11.2 deletion may alter genes associated with cell adhesion and membrane structure.
Then we sought to identify specific morphological changes that might be linked to the genes in our GSEA. When we examined the features associated with the extracellular matrix pathway, we observed a strong enrichment of mitochondrial-associated features. (Fig. 5d, chi-square test, χ² = 28.655, df = 6, p-value = 7.069e-05), suggesting that changes in the expression of these nine genes elicited a specific disruption of mitochondrial function and organization relative to all other feature types. When examining individual gene-feature pairs, we found pronounced differences in correlation coefficients between control and 22q11.2 deletion samples for cell adhesion genes and mitochondrial features (Fig. 5e).
Discussion
Abnormal cell morphology is widely recognized as a hallmark of various pathological conditions, often reflecting underlying disruptions in fundamental cellular processes such as cytoskeletal organization, adhesion, intracellular trafficking, and signaling pathways55. In the context of neurological disorders, changes in neuronal and glial morphology can directly impact cellular function, connectivity, and network activity, contributing to deficits in neurodevelopment and synaptic plasticity56. For example, alterations in astrocyte morphology can disrupt their roles in neurotransmitter regulation and metabolic support, leading to imbalances that exacerbate neuronal dysfunction57. Similarly, changes in neuronal shape and dendritic complexity have been linked to cognitive dysfunction in disorders such as schizophrenia and autism spectrum disorder, where glial and neuronal deficits play a crucial role in disease pathology55.
Beyond neurological disorders, morphological changes are also widely used as diagnostic markers in other pathological contexts. For instance, alterations in red blood cell morphology are routinely assessed in hematological disorders, where shape and size variations serve as indicators of underlying disease states58. The generalizability of morphology-based diagnostics across multiple disease types highlights the broader significance of structural changes as both biomarkers and potential contributors to disease progression59.
This study presents the development and application of NeuroPainting, a high-dimensional phenotyping assay adapted from Cell Painting, to identify morphological phenotypes in iPSC-derived neural cell types, including stem cells, astrocytes, neural progenitors, and neurons. Our results demonstrate the assay’s robustness and utility to uncover insights into neuropsychiatric disorders, particularly those linked to the 22q11.2 deletion syndrome, a major genetic risk factor for schizophrenia. Through this work, we sought to address several key challenges in cellular and molecular psychiatric research, including the need for disease-relevant cellular models and the ability to capture complex, cell-type-specific phenotypes.
NeuroPainting facilitates the implementation of scalable genetic and chemical screening assays, helping to catalyze target and therapeutic discovery in neuropsychiatric research. The classification of over 4000 cellular traits into well-defined categories such as size, shape, and texture across different cellular compartments allows for high-resolution analysis of neural cell morphology. Moreover, the extensive public dataset we created serves as a gold standard for the research community, enabling others to compare and validate their results, thus setting a benchmark for phenotypic screening in neuropsychiatric research.
We showed that NeuroPainting can successfully distinguish cell types based on morphological profiles. It can thus be leveraged to explore possible alterations in cell state (for instance, following genetic or pharmacological perturbations), and whether genetic mutations or drug treatments impact differentiation potential. In approaches where multiple cell types are present in the population that is being screened, this tool could also reveal shifts in cell type composition based on cellular morphology. It will be important to expand our platform to other neural cell types, which have been implicated in brain disorders, such as microglia, oligodendrocytes, medium spiny neurons, interneurons, and many others. Future studies should explore whether our parameters are sufficient to capture these cell types effectively, as we focused our analyses on more common, easy-to-generate cellular models.
The application of NeuroPainting to iPSC-derived neural cells from individuals with the 22q11.2 deletion syndrome revealed diverse, cell-type-specific morphological signatures associated with this genetic mutation. Notably, the deletion’s effects were most pronounced in stem cells and astrocytes, with many morphological features significantly altered in these cell types. The morphological signatures identified in astrocytes, particularly those related to the radial distribution and localization of mitochondria, ER, and AGP, point to biological mechanisms that may underlie the neuropsychiatric symptoms associated with the 22q11.2 deletion. The altered relationship between the ER and mitochondria, as indicated by the significant differences in Cytoplasm_Correlation_RWC_ER_Mito, could represent altered mitochondria-associated ER membranes (MAMs), which play a crucial role in diverse cellular functions, including metabolic regulation, signal transduction, autophagy, and apoptosis60.
We provided an investigation of transcriptional dysregulation in iPSC-derived astrocytes with a 22q11.2 deletion, building on our past findings of 22q11.2 deletion-associated transcriptional changes in iPSC-derived neural progenitor cells and excitatory neurons13. Our findings suggest that the deletion reduces the expression of many neurodevelopmental gene sets, including many genes that play a role in cell adhesion and migration. This result was interesting as recent studies have implicated altered expression of astrocytic cell adhesion genes in schizophrenia40,61. This indicates that the Synaptic Neuron Astrocyte Program (SNAP) is reduced in (living) human astrocytes with 22q11.2 deletion. Previously, decreased SNAP expression had only been observed in astrocytes from post-mortem brains of individuals with idiopathic schizophrenia39. Additionally, the integration of NeuroPainting and transcriptomic data identifies cell morphological phenotypes downstream of these changes in gene expression that are associated with mitochondrial morphology. These results validate many previous reports of mitochondrial dysfunction in 22q11.2 syndrome and neuropsychiatric disorders more broadly62,63. Our hope is that the broader context of linking gene expression to these mitochondrial phenotypes may provide an inroad into understanding these pathological mechanisms. In this instance, we defined a relationship between what is emerging as a robust cellular program altered in persons with schizophrenia, with a morphological trait that is screenable in vitro.
Our study has limitations that should be addressed in future research. While we reliably identified morphological signatures in stem cells, progenitors, and astrocytes, we encountered greater difficulty with neurons. This could be due to the complex morphology of neurons, resulting in perhaps noisier data with higher variability between samples. Larger sample sizes may help mitigate this challenge. It is also possible that the effects of the 22q11.2 deletion may produce more subtle phenotypes in neurons compared to the other cell types studied. Our results suggest that the 22q11.2 deletion alters the behavior of cell adhesion molecules in astrocytes. Given that adhesion signaling often involves interactions between neighboring cells, the use of the cell village approach may introduce cell non-autonomous effects, such as differential adhesion dynamics between control and deletion astrocytes. Future studies using isolated cultures or co-culture systems may help clarify whether these differences arise intrinsically within deletion astrocytes or as a consequence of interactions with control cells. Additionally, although our approach captures thousands of morphological features, it does not include specific phenotypes commonly analyzed in neuroscience, such as synaptic phenotypes. Integrating NeuroPainting with synapse-specific markers could enable more targeted analysis, combining broad-scale morphological profiling with traditional, well-established measurements familiar to the neuroscience field.
This study demonstrates the power of NeuroPainting to uncover morphological phenotypes associated with neuropsychiatric conditions. The cell-type-specificity of these phenotypes, coupled with the integration of gene expression data, provides a deeper understanding of the underlying mechanisms of 22q11.2 deletion syndrome. This study showed that the 22q11.2 deletion reduces the expression of cell adhesion genes, which mirrors recent findings from post-mortem brain studies of schizophrenia. Excitingly, we link these changes to specific morphological traits, including altered mitochondrial phenotypes. These relationships may provide insights into ways that transcriptional changes in the human brain may impact cell function. Future studies could expand the application of NeuroPainting to additional neural cell types and could explore phenotypes associated with other genetic mutations or chemical perturbations. Ultimately, this approach holds promise for the development of therapeutic strategies targeting the cellular and molecular bases of neuropsychiatric disorders.
Methods
Human pluripotent stem cell (hPSC) lines cohort and derivation
We used a cohort of cell lines we previously described in ref. 39. Briefly, we assembled a scaled discovery sample set through highly collaborative, multi-institutional efforts with the Stanley Center Stem Cell Resource (Broad Institute), the Swedish Schizophrenia Cohort (Karolinska Institute), the Northern Finnish Intellectual Disability Cohort (NFID), Umea University, Massachusetts General Hospital (MGH), McLean Hospital, and GTEx. These cell lines represented a mix of male (n = 23) and female (n = 21) as indicated from self-reported surveys at the time of sample collection.
iPSC culture
Human iPSCs were maintained on plates coated with Geltrex (Life Technologies, A1413301) in StemFlex media (Gibco, A3349401) and passaged with Accutase (Gibco, A11105). All cell cultures were maintained at 37 °C, 5% CO2.
Neuronal progenitor and excitatory neuron differentiation
On D0 iPSCs were passaged and replated at a density of 1 M cells per well in a 6-well plate. On D1 iPSCs were differentiated in Neural Induction Medium (NIM) [500 mL DMEM/F12 (1:1) (Gibco, Cat # 11320-033), 5 mL Glutamax (Gibco, Cat # 35050-061), 7.5 mL Dextrose (20%, SIGMA, Cat # 1181302), 5 mL N2 supplement (Invitrogen, Cat # 17502048)] supplemented with SB431542 (10 μM, Stemcell Technologies, Cat # 72234), XAV939 (2 μM, Stemcell Technologies, Cat # 72672) and LDN-193189 (100 nM, Stemcell Technologies, Cat # 72147) along with doxycycline hyclate (2 μg.mL-1, Sigma, Cat # D9891). Media was replaced on D2 and D3 with the same media used for D1, with the addition of Zeocin (1 μg.mL). On D4, cells are co-cultured with mouse glia to promote neuronal maturation and synaptic connectivity (TransnetYX, Cat # C57EASTWB). From D4 onward, cells are maintained in Neurobasal media (500 mL Neurobasal [Gibco, 21103-049], 5 mL Glutamax [Gibco, 35050-061], 7.5 mL Dextrose [20%, SIGMA, Cat # 1181302], 2.5 mL MEM-NEAA [Invitrogen, Cat # 11140050]) supplemented with B27 (50X, Gibco, 17504-044), BDNF, CTNF, GDNF (10 ng.mL–1, R&D Systems 248-BD/CF, 257-NT/CF, and 212-GD/CF) and doxycycline hyclate (2 µg.mL–1, Sigma, D9891). From day 4–5, Neurobasal media was complemented with the antiproliferative agent floxuridine (10 µg.mL–1, Sigma-Aldrich, Cat # F0503-100MG). To capture neuronal progenitor cells, we harvest D4 samples prior to adding them to the mouse glia.
Astrocyte differentiation
On D0 iPSCs were passaged and replated at a density of 1 M cells per well in a 6-well plate. On day 1, iPSCs were differentiated in N2 medium [500 mL DMEM/F12 (1:1) (Gibco, Cat # 11320-033), 5 mL Glutamax (Gibco, Cat # 35050-061), 7.5 mL Dextrose (20%, SIGMA, Cat # 1181302), 5 mL N2 supplement B (StemCell Technologies, Cat # 07156)] supplemented with SB431542 (10 μM, Stemcell Technologies, Cat # 72234), XAV939 (2 μM, Stemcell Technologies, Cat # 72672) and LDN-193189 (100 nM, Stemcell Technologies, Cat # 72147) along with doxycycline hyclate (2 μg.mL-1, Sigma, Cat # D9891). Media was replaced on D2 with the same media used for D1, with the addition of Zeocin (1 ug/mL). Starting on day 3, human induced neural progenitor-like cells were harvested with Accutase (Innovative Cell Technology, Inc., Cat # AT104-500) and re-plated at 15,000 cells cm−2 in Astrocyte Medium (ScienCell, Cat # 1801) with Y27632 (5 mM) on geltrex-coated plates. Cells were maintained for 30 days in Astrocyte Medium (ScienCell, Cat # 1801).
Cell-type-specific assay conditions: cell seeding, fixation, and staining
For each batch of imaging, cells were detached from 6-well NUNC plates using Accutase (StemcellTech; cat#07920) for generating single-cell suspensions. Following detachment, cells were centrifuged at 300xg for 5:00 and re-suspended in the appropriate medium for each individual cell type. After each cell line was counted to determine cell solution concentration and viability, the desired cell solution volume was aliquoted into a 96-deep well low attachment plate following a specific plate map to ensure that wells from any given cell line were not predominantly on the edge wells or too close together. To disperse a high number of cell lines across a 384-well plate in a semi-random fashion, we optimized the use of an Agilent Bravo liquid handling device. Here, using an 8-channel head, cell solutions were transferred from the 96-well low attachment plate and distributed into a geltrex-coated Perkin Elmer Cell Carrier 384-well plate at the determined density per well. Each cell line was plated into 8 distinct wells on the final screening plate in four-well quadrants. The ideal seeding densities were determined through pilot experiments and evaluated based on visual assessment of the cells. The final densities and staining conditions used in this study are as follows iPSCs—10 k cells/well, fixation 24 h post plating; NPCs—15 k cells/well, fixation 24 h post plating; astrocytes—3 k cells/well, fixation 48 h post plating; neurons—2.5 k cells/well, fixation 25 days post plating.
NeuroPainting and imaging
Once cells were seeded in 384-well plates, cells were treated for 30 min with 0.5 uM MitoTracker Deep Red FM—Special Packaging (Thermo Fisher cat#: M22426) dye at 37 °C. Following the MitoTracker treatment, cells were fixed with 16% paraformaldehyde diluted to a final concentration of 4% (Thermo Fisher cat#: 043368.9 M) for 20 min in the dark at RT. After three washes with 1× HBSS cells were permeabilized and stained using a solution of 1× HBSS (Thermo Fisher cat#: 14175095), 0.1% Triton-X-100 (Sigma Aldrich cat#: X100-5ML), 1% Bovine Serum Albumin, 8.25 nM Alexa Fluor 568 Phalloidin (Thermo Fisher cat#: A12380), 0.005 mg/mL Concanavalin A, Alexa Fluor 488 Conjugate (Thermo Fisher cat#: C11252), 1ug/mL Hoechst 33342, Trihydrochloride, Trihydrate (Thermo Fisher cat#: H3570), 6uM SYTO 14 Green Fluorescent Nucleic Acid Stain (Thermo Fisher cat#: S7576), and 1.5 ug/mL Wheat Germ Agglutinin, Alexa Fluor 555 Conjugate (Thermo Fisher cat#: W32464) for 1 h at RT in the dark. Following the staining, plates were washed 3× with 1× HBSS and sealed until imaging. Cell Painted plates were imaged on a Perkin Elmer Phenix Automated Microscope under a standardized protocol.
Quantification of cellular morphology traits and their quality control
The segmentation of individual cells in the image into their cellular compartments (whole cell, cytoplasm, and nuclei) and subsequent quantification of morphology traits for each cellular compartment was done using CellProfiler 4.2.4 (Stirling et al. 2021; pipelines are available at https://github.com/broadinstitute/NeuroPainting). Subsequently, cells missing measurements for more than 5% of traits were removed. Morphology traits a priori known to be problematic, not measured across all cells or non-variable across cells, were removed using the Caret v6.0-86 package. QC-ed cells were then segregated into two groups based on the number of neighbors: isolated cells having no neighbors and colony cells having one or more neighbors. Individual morphology traits were then summarized to well-level measurement by averaging them across all cells per well, resulting in a well-by-trait matrix. Following this, each morphology trait was Gaussianized across all four plates using the INT method.
Cell village creation, scRNA-sequencing, and donor assignment
IPSCs from each donor were mixed in equal proportions (1 M cells per donor) and differentiated as a pool following the Ngn2 astrocyte differentiation method described earlier. At D30 of the differentiation, astrocyte village cells were harvested, and 60,000 cells were prepared using 10× Chromium Single Cell 3’ Reagents v3 and sequenced on an Illumina NovaSeq 6000 with an S2 flow cell, generating paired-end reads of 2 × 100 bp. Raw sequencing data were aligned and processed following the Drop-seq workflow. Human reads were aligned to the GRCh38 reference genome and filtered for high-quality mapped reads (mapping quality ≥ 10). To determine the donor identity of each droplet, variants were filtered through multiple quality controls, ensuring only high-confidence A/T or G/C sites were included in the VCF files. Once the single-cell libraries and VCF reference files were filtered and quality-checked, the Dropulation algorithm was applied. This algorithm analyzes each droplet (or cell) independently, assigning a probability score to each variant site based on the observed versus expected allele. Donor identity is determined by computing the diploid likelihood at each UMI, summed across all sites, to identify the most likely donor for each droplet. To ensure consistency in differential expression testing downstream, we remove any cells that are clustered separately by individual donors.
Random forest classification for cell types
A random forest classifier was used to classify cell types based on morphological profiles from control samples. The data was split into 70% training and 30% testing sets, and a 5-fold cross-validation was employed to optimize hyperparameters. The number of features considered at each split (mtry) was tuned using a grid search with values ranging from 2 to 721. The final model was built using the best-performing mtry, with 500 trees, a maximum of 60 terminal nodes, and a minimum node size of 5. Model performance was evaluated on the test set using a confusion matrix, accuracy, sensitivity, specificity, and Kappa statistics. Receiver operating characteristic-area under the curve (ROC-AUC) scores were computed for each cell type, and predicted probabilities were visualized using boxplots. All analyses were performed in R (version 4.0.2) with the randomForest, caret, and pROC packages, and a random seed of 42 to ensure reproducibility.
Differential feature analysis
To identify differential features between control and deletion conditions across various cell types, we conducted a Wilcoxon rank-sum test using the well-level NeuroPainting profiles. The dataset was grouped by cell type to ensure comparisons were made within each cell type. For each group, a pairwise Wilcoxon rank-sum test was performed to compare control and deletion conditions for each numeric feature, with p-values recorded. To address multiple comparisons, we applied a false discovery rate (FDR) correction to the p-values within each cell type using the Benjamini–Hochberg (BH) method.
Differential gene expression
Differential gene expression analysis was conducted using DESeq2 (v1.34.0), a statistical package designed for analyzing count data from RNA sequencing experiments. Pseudo-bulk RNA expression profiles for each cell line were generated by summing the individual cell counts across each sequencing reaction, and these counts were used as input for DESeq2. The data were pre-processed to ensure quality control, including filtering out lowly expressed genes that had fewer than 10 reads in at least 50% of the samples. Normalization of the count data was performed using the DESeq2 default method, which estimates size factors for each sample to account for differences in sequencing depth. These size factors were used to normalize the read counts across samples, ensuring comparability. The DESeq2 model was fitted to the data using the negative binomial distribution to estimate the dispersion for each gene. Differential expression was determined by comparing the expression levels between the conditions of interest. For this study, the primary comparison was between 22q11.2 deletion and control, with the design formula in DESeq2 specified as ~condition, where condition represents the categorical variable of interest. DESeq2 calculates the log2FC for each gene, representing the ratio of gene expression between the two conditions. Statistical significance was assessed using the Wald test, and p-values were adjusted for multiple testing using the BH procedure to control the FDR. Genes with an adjusted p-value (padj) of less than 0.05 were considered significantly differentially expressed. A total of 835 genes were identified as differentially expressed between the conditions, with thresholds of log2FC > |1| and padj < 0.05.
Metagene score calculation
To derive the metagene score, we first selected a set of genes based on their ranking from a predefined gene list and extracted their corresponding log-transformed counts per million (logCPM) values from the gene expression matrix. The metagene score for each sample was then calculated as the sum of the logCPM values for these selected genes, normalized by the total logCPM sum across all expressed genes in that sample. This approach ensures that the score reflects the relative expression of the selected genes while accounting for sample-wide differences in expression levels. Finally, the computed metagene scores were combined with sample metadata, including condition labels (control or deletion), for downstream analysis and visualization.
CCA
We performed CCA to investigate the relationship between RNA sequencing (RNA-seq) gene expression data and morphological features (NeuroPainting). We used inverse normalized transformed data, including the DEGs (1358) and significant features (536). To reduce noise and multicollinearity, we first applied principal component analysis (PCA) to both datasets, retaining the top 5 principal components (PCs) from each. For RNA-seq data, PCA was performed on z-score expression values, while for morphological features, PCA was applied to z-scored feature values. The CCA was then conducted using the cancor() function in R, with the top five PCs from each dataset serving as input. We extracted the loadings from the first canonical variable (CV1) to identify the top contributing genes and morphological features for subsequent analysis, providing insight into the shared variance between the two datasets.
Morphology and gene expression correlation analysis
We performed a fixed-effects linear regression analysis to investigate the relationship between gene expression and morphological features in astrocytes with and without the 22q11.2 deletion. The model included gene expression as the predictor and various morphological traits as the response variable, with genotype as an interaction term to test for differential effects between control and deletion groups. Specifically, we fit the following model for each gene-feature pair:
The interaction term was used to assess whether the correlation between gene expression and morphology varied between the control and deletion groups. p-values for the interaction term were calculated, and significant gene-feature pairs were identified. Coefficients for expression and the interaction term were extracted to assess the strength and direction of the gene-morphology relationships across groups.
Gene set enrichment analysis (GSEA)
We performed a pre-ranked GSEA using the clusterProfiler package in R (v4.6.0). We employed the gseGO() function with the Gene Ontology (GO) database, querying both the biological process (BP) and CC ontologies. Gene symbols were mapped to Entrez IDs using the bitr() function and the org.Hs.eg.db annotation package (v3.16.0). To ensure accurate mapping, we filtered out genes without valid Entrez identifiers, resulting in a final gene list for enrichment analysis. The GSEA was performed using default parameters, with the following settings: minimum gene set size of 10, maximum gene set size of 500, and permutation type set to gene labels. Significance thresholds were determined using the BH procedure to control the FDR. Pathways with an adjusted p-value (FDR) < 0.05 were considered statistically significant. Enrichment scores (ES) and normalized enrichment scores (NES) were used to rank the significance of enriched pathways, and results were visualized using the enrichplot package.
Identification of significant gene-feature pairs
We calculated Pearson correlation coefficients for gene expression and morphology for both control and deletion groups, deriving the absolute difference between these two correlation coefficients for each pair. Significant gene-feature pairs were defined as those with an interaction p-value < 0.01 and a correlation difference > 1.0. Fisher’s Z-test was applied to evaluate whether the correlation differences were statistically significant.
Enrichment analysis for feature categories and cellular compartments
To assess whether certain morphological features or cellular compartments were overrepresented among the significant gene-feature pairs, we conducted a Chi-squared independence test. We grouped features by their respective categories (e.g., granularity, texture, and correlation) and compartments (e.g., nuclei, cytoplasm, and cells) and compared their distribution in our set of gene-pairs to their overall distribution amongst the full feature set.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw image data has been deposited in the Cell Painting Gallery on the Registry of Open Data on AWS as dataset cpg0038-tegtmeyer-neuropainting at no cost and no need for registration. Sequencing data, including gene expression matrices, have been deposited in the Broad Institute Single-cell Portal under accession SCP2865. Source data are provided with this paper.
Code availability
Source code to reproduce and build upon the presented results is available at https://github.com/broadinstitute/NeuroPainting.
References
Bessonova, L., Ogden, K., Doane, M. J., O’Sullivan, A. K. & Tohen, M. The economic burden of bipolar disorder in the United States: a systematic literature review. Clinicoecon. Outcomes Res. 12, 481–497 (2020).
Kadakia, A. et al. The economic burden of schizophrenia in the United States. J. Clin. Psychiatry 83, 14458 (2022).
Cohen, B. M. & Öngür, D. The urgent need for more research on bipolar depression. Lancet Psychiatry 5, e29–e30 (2018).
Kuperberg, M. et al. Psychotic symptoms during bipolar depressive episodes and suicidal ideation. J. Affect. Disord. 282, 1241–1246 (2021).
McIntyre, R. S. et al. Bipolar disorders. Lancet 396, 1841–1856 (2020).
Potkin, S. G. et al. The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 6, 1 (2020).
Paul, S. M. & Potter, W. Z. Finding new and better treatments for psychiatric disorders. Neuropsychopharmacology 49, 3–9 (2024).
McDonald-McGinn, D. M. et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Prim. 1, 15071 (2015).
Marshall, C. R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2016).
Yagi, H. et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 362, 1366–1373 (2003).
Motahari, Z., Moody, S. A., Maynard, T. M. & LaMantia, A.-S. In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects?. J. Neurodev. Disord. 11, 7 (2019).
Khan, T. A. et al. Neuronal defects in a human cellular model of 22q11.2 deletion syndrome. Nat. Med. 26, 1888–1898 (2020).
Nehme, R. et al. The 22q11.2 region regulates presynaptic gene-products linked to schizophrenia. Nat. Commun. 13, 3690 (2022).
Loh, P.-R. et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 47, 1385–1392 (2015).
Olsen, L. et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry 5, 573–580 (2018).
Bray, M.-A. et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 11, 1757–1774 (2016).
Cimini, B. A. et al. Optimizing the Cell Painting assay for image-based profiling. Nat. Protoc. 18, 1981–2013 (2023).
Rohban, M. H. et al. Systematic morphological profiling of human gene and allele function via Cell Painting. Elife 6, e24060 (2017).
Tegtmeyer, M. et al. High-dimensional phenotyping to define the genetic basis of cellular morphology. Nat. Commun. 15, 347 (2024).
Vigilante, A. et al. Identifying extrinsic versus intrinsic drivers of variation in cell behavior in human iPSC lines from healthy donors. Cell Rep. 26, 2078–2087.e3 (2019).
Seal, S. et al. Cell Painting: a decade of discovery and innovation in cellular imaging. Nat. Meths. 22, 254–268 (2025).
Buccitelli, C. & Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644 (2020).
Antony, P. M. A. et al. Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization. Sci. Rep. 10, 1569 (2020).
Schiff, L. et al. Integrating deep learning and unbiased automated high-content screening to identify complex disease signatures in human fibroblasts. Nat. Commun. 13, 1–13 (2022).
Yang, S. J. et al. Applying deep neural network analysis to high-content image-based assays. SLAS Discov. 24, 829–841 (2019).
Wali, G., Berkovsky, S., Whiten, D. R., Mackay-Sim, A. & Sue, C. M. Single cell morphology distinguishes genotype and drug effect in hereditary spastic paraplegia. Sci. Rep. 11, 16635 (2021).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Nehme, R., Pietiläinen, O. & Barrett, L. E. Genomic, molecular, and cellular divergence of the human brain. Trends Neurosci. 47, 491–505 (2024).
Wali, G. et al. Pharmacological rescue of mitochondrial and neuronal defects in SPG7 hereditary spastic paraplegia patient neurons using high throughput assays. Front. Neurosci. 17, 1231584 (2023).
Nehme, R. & Barrett, L. E. Using human pluripotent stem cell models to study autism in the era of big data. Mol. Autism 11, 21 (2020).
Tegtmeyer, M. & Nehme, R. Leveraging the genetic diversity of human stem cells in therapeutic approaches. J. Mol. Biol. 434, 167221 (2022).
Nehme, R. et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 23, 2509–2523 (2018).
Wells, M. F. et al. Natural variation in gene expression and viral susceptibility revealed by neural progenitor cell villages. Cell Stem Cell 30, 312–332.e13 (2023).
Berryer, M. H. et al. Robust induction of functional astrocytes using NGN2 expression in human pluripotent stem cells. iScience 26, 106995 (2023).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 22, 433 (2021).
Arevalo, J. et al. Evaluating batch correction methods for image-based cell profiling. Nat. Commun. 15, 6516 (2024).
Rogdaki, M. et al. Magnitude and heterogeneity of brain structural abnormalities in 22q11.2 deletion syndrome: a meta-analysis. Mol. Psychiatry 25, 1704–1717 (2020).
Ching, C. R. K. et al. Mapping subcortical brain alterations in 22q11.2 deletion syndrome: Effects of deletion size and convergence with idiopathic neuropsychiatric illness. Am. J. Psychiatry 177, 589–600 (2020).
Ling, E. et al. A concerted neuron-astrocyte program declines in ageing and schizophrenia. Nature 627, 604–611 (2024).
Pietiläinen, O. et al. Astrocytic cell adhesion genes linked to schizophrenia correlate with synaptic programs in neurons. Cell Rep. 42, 111988 (2023).
Rapino, F. et al. Small-molecule screen reveals pathways that regulate C4 secretion in stem cell-derived astrocytes. Stem Cell Rep. 18, 237–253 (2023).
Mitchell, J. M. et al. Mapping genetic effects on cellular phenotypes with ‘cell villages’. Preprint at bioRxiv https://doi.org/10.1101/2020.06.29.174383 (2020).
Zhong, X., Drgonova, J., Li, C.-Y. & Uhl, G. R. Human cell adhesion molecules: annotated functional subtypes and overrepresentation of addiction-associated genes. Ann. N. Y. Acad. Sci. 1349, 83–95 (2015).
Mørch, R. H. et al. Inflammatory markers are altered in severe mental disorders independent of comorbid cardiometabolic disease risk factors. Psychol. Med. 49, 1749–1757 (2019).
Halperin, D. et al. CDH2 mutation affecting N-cadherin function causes attention-deficit hyperactivity disorder in humans and mice. Nat. Commun. 12, 6187 (2021).
Shiwaku, H. et al. Autoantibodies against NCAM1 from patients with schizophrenia cause schizophrenia-related behavior and changes in synapses in mice. Cell Rep. Med. 3, 100597 (2022).
Kho, S.-H. et al. DNA methylation levels of RELN promoter region in ultra-high risk, first episode and chronic schizophrenia cohorts of schizophrenia. Schizophrenia 8, 81 (2022).
Habela, C. W., Song, H. & Ming, G.-L. Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol. Cell. Neurosci. 73, 52–62 (2016).
Yang, Y., Guan, W., Sheng, X.-M. & Gu, H.-J. Role of Semaphorin 3A in common psychiatric illnesses such as schizophrenia, depression, and anxiety. Biochem. Pharmacol. 226, 116358 (2024).
Pietiläinen, Olli et al. Astrocytic cell adhesion genes linked to schizophrenia correlate with synaptic programs in neurons. Cell reports 42, 111988 (2023).
Gallegos, L. L. et al. A protein interaction map for cell-cell adhesion regulators identifies DUSP23 as a novel phosphatase for β-catenin. Sci. Rep. 6, 27114 (2016).
Kimura, S., Lok, J., Gelman, I. H., Lo, E. H. & Arai, K. Role of A-kinase anchoring protein 12 in the central nervous system. J. Clin. Neurol. 19, 329–337 (2023).
Galea, C. A., Nguyen, H. M., George Chandy, K., Smith, B. J. & Norton, R. S. Domain structure and function of matrix metalloprotease 23 (MMP23): role in potassium channel trafficking. Cell Mol. Life Sci. 71, 1191–1210 (2014).
Brinkmann, E.-M. et al. Secreted frizzled-related protein 2 (sFRP2) redirects non-canonical Wnt signaling from Fz7 to Ror2 during vertebrate gastrulation. J. Biol. Chem. 291, 13730–13742 (2016).
Bernstein, H.-G. et al. Glial cell deficits are a key feature of schizophrenia: implications for neuronal circuit maintenance and histological differentiation from classical neurodegeneration. Mol. Psychiatry 30, 1102–1116 (2025).
He, C. & Duan, S. Novel insight into glial biology and diseases. Neurosci. Bull. 39, 365–367 (2023).
Rahman, M. M. et al. Emerging role of neuron-Glia in neurological disorders: at a glance. Oxid. Med. Cell. Longev. 2022, 3201644 (2022).
Constantino, B. T. Reporting and grading of abnormal red blood cell morphology. Int. J. Lab. Hematol. 37, 1–7 (2015).
Marchi, G. et al. Red blood cell morphologic abnormalities in patients hospitalized for COVID-19. Front. Physiol. 13, 932013 (2022).
Zhao, W.-B. & Sheng, R. The correlation between mitochondria-associated endoplasmic reticulum membranes (MAMs) and Ca transport in the pathogenesis of diseases. Acta Pharmacol. Sin. https://doi.org/10.1038/s41401-024-01359-9 (2024).
Sheikh, M. A. et al. Systemic cell adhesion molecules in severe mental illness: potential role of intercellular CAM-1 in linking peripheral and neuroinflammation. Biol. Psychiatry 93, 187–196 (2023).
Li, J. et al. Association of mitochondrial biogenesis with variable penetrance of schizophrenia. JAMA Psychiatry 78, 911–921 (2021).
Purcell, R. H. et al. Cross-species analysis identifies mitochondrial dysregulation as a functional consequence of the schizophrenia-associated 3q29 deletion. Sci. Adv. 9, eadh0558 (2023).
Acknowledgements
With thanks to Kris Dickson for reviewing and editing the manuscript, and members of the Nehme lab and the Carpenter-Singh lab for helpful comments and discussion. This work was supported by the Stanford Maternal and Child Health Research Institute through the Uytengsu-Hamilton 22q11 Neuropsychiatry Research Award Program, NIH R01MH128366 to RN, NIH R35 GM122547 (to A.E.C.), SFARI (890477) to R.N., the Broad Institute Next Generation Award and the Stanley Center for Psychiatric Research Gift to RN, and the Broad Institute SPARC award to R.N. and S.S.
Author information
Authors and Affiliations
Contributions
R.N. and S.S. conceived the work. M.T. and R.N. wrote the manuscript with input from all authors. M.T. performed the experiments with help from D.L., K.B., and D.H., M.T., P.V.R., C.T.-C., B.A.C., S.S., G.W., R.P., and Y.H. processed and analyzed Cell Painting data. R.N., A.E.C., and S.S. supervised the work and analyses. All authors edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
B.C., S.S., and A.E.C. serve as scientific advisors for companies that use image-based profiling and Cell Painting (A.E.C.: Recursion, SyzOnc, Quiver Bioscience, B.C.: Marble Therapeutics, and S.S.: Waypoint Bio, Dewpoint Therapeutics, Deepcell) and receive honoraria for occasional scientific visits to pharmaceutical and biotechnology companies. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhiping Pang and Toru Takumi for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tegtmeyer, M., Liyanage, D., Han, Y. et al. Combining phenomics with transcriptomics reveals cell-type-specific morphological and molecular signatures of the 22q11.2 deletion. Nat Commun 16, 6332 (2025). https://doi.org/10.1038/s41467-025-61547-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61547-x
