
Abstract
Enzymatic reductions catalyzed by reductases generally depend on reduced nicotinamide cofactors as a hydride source. However, for industrial viability, it is more cost-effective to use water as the hydrogen source, bypassing the requirement for the cofactor. Here we report a hybrid photo-biocatalyst system based on infrared (IR) light and responsive reductive graphene quantum dots (rGQDs), for performing the direct transfer of hydrogen from water to prochiral substrates. The photo-biocatalyst, assembled from rGQDs and cross-linked aldo-keto reductase (AKR), mediates the synthesis of the pharmaceutical intermediate, (R)−1-[3,5-bis(trifluoromethyl)-phenyl] ethanol ((R)−3,5-BTPE), in 82% yield and >99.99% ee under IR illumination. Our photo-enzymatic systems can also be effectively used to drive the enzymatic reduction of imines and alkenes. Since the hybrid photo-biocatalysts are insoluble, they can be readily recovered and recycled. This work opens new avenues to create artificial photo-biocatalyst systems, enabling the facile coupling of renewable solar energy and sustainable chemical production.
Similar content being viewed by others
Introduction
Biocatalysis is widely applied in the pharmaceutical and fine chemical industries in the enantioselective production of valuable chiral chemicals under mild, aqueous conditions1,2,3. Prochiral ketone reductions catalyzed by ketoreductases, for example, are exquisitely enantioselective and cost-effective methods for the industrial production of the corresponding chiral alcohols2,4. Chiral amines have also served as a focus of attention because of the prevalence of nitrogen atoms in drugs, and imine reductases are used for their preparation5,6. Similarly, ene reductases, typically from the old yellow enzyme (OYE) family, reduce conjugated C = C double bonds7. All of these enzymes require efficient regeneration of NAD(P)H cofactors for their cost-effective use and this typically involves using a dehydrogenase in combination with a sacrificial cosubstrate, e.g. alcohol/alcohol dehydrogenase (ADH), glucose/glucose dehydrogenase (GDH) or formate/formate dehydrogenase (FDH), or a hydrogenase with atom-economic cosubstrate H2 (Fig. 1a)8,9,10,11,12. Indeed, enzyme-mediated cofactor regeneration is the cornerstone of bioreduction but often suffers from unfavorable kinetics13.

a Strategies for the bioreduction of unsaturated compounds and the catalytic mechanism of AKR-catalyzed 3,5-BTPE reduction. During the enzymatic catalysis, ternary AKR-NADPH-substrate complexes form and the NADPH binding induces protonation of Tyr to form the catalytically active Tyr-OH₂⁺ species. Then, the common coenzyme binding domain of AKR permits pro-R-hydride from NADPH transfer to the carbonyl through protonation of the carbonyl by the TyrOH2+. Finally, the oxidized coenzymes NADP+ and chiral product leave. b NIR-driven photo-enzyme-coupled catalysis for 3,5-BTAP reduction. c This work, strategies for the light-driven photo-enzymatic reduction of unsaturated compounds by rGQDs/reductase photo-biocatalyst system.
The ideal sacrificial cosubstrate is water, which can be used in conjunction with electricity (electrochemical)14,15, or light (photochemical)16, as the source of hydrogen atoms without the need for a second enzyme. The use of water as a hydrogen source not only provides an economical alternative but also represents a greener and more sustainable option. This approach aligns with the principles of green chemistry by minimizing environmental impact and enhancing enzyme compatibility. Much effort has been expended, therefore, regarding the mediation of electron transfer from water to NAD(P)+ by light-driven photocatalysts. This generally involves the use of semi-conductors such as Au/TiO217 or expensive precious metal complexes such as [Cp*Rh(bpy)(H2O)]2+ (M, Cp* = pentamethylcyclopentadienyl, bpy = 2,2’-bipyridyl) as electron mediators in order to avoid undesirable side reactions stemming from the radical nature of two non-catalyzed single electron transfer (SET) steps and convert these two SETs (and a protonation step) into a single step17,18,19,20,21,22,23,24,25,26,27,28,29,30,31. The electrochemical method suffers from the same issues, providing the enzymatic reductase system with complexity and reducing its economic viability14,32,33. In contrast, we envisaged a simplified biotransformation that would bypass the traditional cofactor-dependent catalytic pathway altogether.
Another problem associated with photocatalysis is the weak penetration of ultraviolet or visible light in various reaction media because biological tissues, including proteins and nucleic acids, absorb visible light much more strongly than chemical reaction media34. Moreover, although IR light is responsible for half of the energy of sunlight and has considerable penetration depth in biological tissues, its photon energy is relatively low and usually insufficient to directly stimulate photocatalysis35,36. This hurdle can be overcome by introducing an infrared light-responsive component35, such as rGQDs in which the p electrons are excited to a high-energy state (e.g. the lowest unoccupied molecular orbital, LUMO) and then transition back to the s orbital37,38.
rGQDs were recently developed as near-infrared (NIR) emissive nanomaterials39,40. We previously demonstrated41 that loading rGQDs onto TiO2 nanotubes photocatalyst enabled a greatly improved NADPH photo-regeneration efficiency in the presence of M without extra sacrificial reductant due to abundant Ti-O-Ti bonds formed between the interfaces, which promoted multi-electron transfer under NIR excitation. Indeed the new-to-nature reactivity of enzymes has been greatly extended via light promotion42,43. Hyster et al. demonstrated that cofactor or a charge-transfer complex, formed by cofactor and substrate in the active site, could harvest the incident photons to induce electron transfer from the cofactor to substrate44,45. Based on a NADPH recycling system, these photo-enzymatic modes work well on asymmetric dehalogenation46, C-C bond47,48 and C-N bond formations49. In contrast, upon visible light irradiation, ene-reductases (ER) could initiate single electron oxidation, which has been successfully applied in hydrosulfonylation of olefins50,51 and lactone synthesis, without the involving of cofactor recycling52.
We envisaged that an accessible upconversion material such as rGQDs, comprising abundant conjugate structures with dangling carbon bonds, would form stable assemblies with enzymes through multiple forces (e.g. cation−π, anion−π, hydrophobic and π−π interactions). This would allow the short-range transfer of active hydrogen, generated by water splitting under IR illumination on GQDs, to the nearby enzyme-bound substrate without the intervention of a cofactor (Fig. 1b). In this work, in order to validate our envisaged cofactor-free, infrared light driven photoenzymatic catalysis, we choose the synthesis of (R)−3,5-BTPE, a chiral intermediate for the drug Aprepitant used in the clinical treatment of adverse reactions caused by chemotherapy, by reduction of the corresponding prochiral ketone39,53,54,55,56. Subsequently, other photoenzymatic reductions using an imine reductase, ene reductase, and carbonyl reductase is conducted and shown to be efficient for the reduction of an imine, cinnamaldehyde, and ketone, respectively.
Results
Preparation and characterization of rGQDs/AKR photo-biocatalyst
As shown in Fig. 2a, a microwave-assisted bio-orthogonal click reaction was utilized to crosslink prefunctionalized AKR protein (Gene ID: 897867), which typically binds cofactor NADPH in an extended anti-conformation to catalyze 4-pro-R hydride transfer57,58,59. The rGQDs/AKR hybrid catalyst was constructed by grafting rGQDs on crosslinked AKR (AKR-CLEs) through a simple self-assembly. The aggregation makes them convenient for further characterization. The hybrid material displayed stability comparable with that of rGQDs or AKR-CLEs which was confirmed by their Zeta potential (Fig. S9A). Corresponding XRD and FT-IR measurements (Fig. S9B and S10) indicated no recrystallization of rGQD and no new chemical bond formation. The morphology of rGQDs/AKR was studied by CLSM as presented in Fig. 2b, SEM (Fig. S11a) and TEM (Fig. S11b). The rGQDs/AKR photo-biocatalyst exhibited a regular coral-like structure, consistent with that of AKR-CLEs (Fig. S3a and S4). Subsequently, atomic force microscopy (AFM) examination revealed numerous tiny particles (rGQDs) scattered on the surface of AKR-CLEs (Fig. 2c). This characteristic was confirmed by the energy dispersive X-ray spectroscopy (EDS) diagram, in which a rough hybrid surface with nanosized carbon clusters was exhibited (Fig. 2d). Compared with the rGQDs precursor, there is no obvious change in carbon’s chemical states present in X-ray photoelectron spectroscopy (XPS) results (Fig. 2e). The resulting hybrid material retained the infrared light responsive ability. Its optical upconversion emissions at 525 nm, 545 nm and 661 nm are similar to rGQDs under 980 nm IR light excitation (Fig. 2f)60, are consistent with the rGQDs being anchored on the AKR.

a Schematic illustration for the synthesis of rGQDs/AKR, (b) CLSM image of rGQDs/AKR stained with FITC (λEX = 488 nm), (c) AFM image and (d) EDS element mappings of rGQDs/AKR, (e) XPS spectra: C1s spectra of rGQDs, rGQDs/AKR. (f) PL spectra of rGQDs, rGQDs/AKR with 980 nm excitation, (g) UV-Vis-NIR spectra of rGQDs, rGQDs/AKR, (h) Band structures of rGQDs, rGQDs/AKR, (i) EPR spectra of hydroxyl radicals.
The bandgaps were analyzed before and after the self-assembly of rGQDs and AKR-CLEs. As shown in Fig. 2g, the light absorption of the rGQDs and rGQDs/AKR ranged from the UV to the infrared region, consistent with potentially high sunlight utilization efficiencies. Assisted by the transformed Kubelka-Munk function versus the energy (Fig. S12) and the XPS VB spectra (Fig. S13), the bandgap structures of rGQDs and rGQDs/AKR were depicted, obviously both of them had suitable photoredox potentials for water splitting in theory (Fig. 2h). Therefore, we evaluated the photocatalytic water-splitting performance of rGQDs. As shown in Fig. S14, rGQDs exhibit photocatalytic activity for hydrogen production from water splitting under infrared light irradiation, with a H2 generation rate of 3.04 μmol·gcat−1·h−1. As shown in Fig. 2i, the presence of hydroxyl radicals from photocatalytic water splitting under illumination was successfully detected by ESR.
Molecular simulations and catalysis for rGQDs/reductase photo-biocatalyst
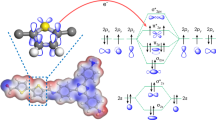
The molecular features of assembled rGQDs/AKR were characterized by molecular dynamics (MD) simulations with and without bound NADPH. Owing to the variety of conformations, rGQDs can bind to various sites on the enzyme. The two-layered rGQDs that was initially located at different positions relative to AKR-NADPH/AKR with a distance of 30 Å (Fig. S18 and S19) eventually formed stable binding complexes of rGQDs/AKR-NADPH and rGQDs/AKR within 400 ns MD simulations, as indicated by the converged RMSD (root mean squared deviation) and distance profiles (Fig. S20–23). The converged RMSD profiles confirmed the formation of stable binding complexes of rGQDs–AKR both with and without NADPH cofactor (Fig. S20 and S21). The equilibrated binding conformations showed extensive cation−π and anion−π interactions between the surface residues of AKR and rGQD, which have been reported to have an interaction strength of ~14–15 kcal·mol–1 (close to the magnitude of a hydrogen bond)61,62. Hydrophobic and π−π interactions also contributed to the stable binding. The difference in locations of rGQDs on the AKR surfaces can be ascribed to varied charge distribution due to the binding of NADPH. Notably, structure comparison of the equilibrated rGQDs/AKR-NADPH and rGQDs/AKR with the corresponding crystal structures of AKR-NADPH (PDB: 6KIY) and AKR (PDB: 6KIK) demonstrated that binding of rGQDs would not cause dramatic conformational changes to the overall structure and catalytic pocket domain of AKR, and the surface bound rGQDs would hardly hinder the binding and release of NADPH and substrate molecules (Fig. S24 and Fig. 3).

a–d The equilibrated overall binding complexes of rGQD-1/rGQD-2/rGQD-3/rGQD-4–AKR, respectively, with the intermolecular interactions shown in the enlarged illustrations. The cation−π and anion−π interactions between rGQDs and the surrounding residues are indicated by pink dotted lines. The NADPH binding domains are highlighted with brown and purple colors, with the purple regions indicating the locations of the nicotinamide group.
3,5-BTAP was selected as the substrate to explore the photo-enzymatic properties of rGQDs/AKR in molecular docking and MD simulations model. It is usually synthesized by enantioselective hydrogenation of 3,5-BTAP. As binding of rGQDs would not hinder the binding of substrate or change the conformation of AKR catalytic domain, simplified models were used without rGQDs included. Expectations for the rGQDs/AKR photo-biocatalyst system require that the enantioselective reduction of 3,5-BTAP can be initiated by the active hydrogen generated from water molecules by rGQDs through infrared light irradiation (Fig. 4). On the basis of stably anchored 3,5-BTAP in the catalytic pocket of AKR (Fig. 4a–c), we anticipated that the distribution of water molecules around the binding 3,5-BTAP, representing diverse access paths of active hydrogen to the carbon atom of 3,5-BTAP carbonyl group approximately, would play a critical role in producing the chiral 3,5-BTPE. More specifically, the position of the hydrogen atom of water that is located nearest to the carbon atom (C9) of 3,5-BTAP carbonyl group relative to the plane defined by the 3,5-BTAP atoms C6, C9, and C10 basically determines the chirality of 3,5-BTPE (Fig. 4). Hence, the improper dihedral of ∠C6-C10-C9-H was monitored throughout the 20 ns MD simulations, in which H means the hydrogen atom belonging to the water molecule nearest to 3,5-BTAP C7 atom. The positive and negative torsion angles correspond to the pro-(R) and pro-(S) binding forms of 3,5-BTAP, leading to the generation of (R)- and (S)-3,5-BTPE respectively. Distributions of the improper dihedral along MD simulations showed a clear preference for the pro-(R) binding forms in both AKR–NADPH–3,5-BTAP and AKR–3,5-BTAP systems (Fig. S25d and Fig. 4d), resulting in an excellent enantioselective hydrogenation result (e.e. (R) > 99%).

a Equilibrated binding conformation of AKR–3,5-BTAP, (b) RMSD profiles of the binding components, (c) Profile of the distance between the geometric centers of W21 sidechain and aromatic ring of 3,5-BTAP, (d) and (e) Value and distribution of the key improper dihedral ∠C6-C10-C9-H across the MD simulation. The access path of hydrogen atom (H) to the carbonyl carbon atom (C9) is indicated by a magenta dotted line. The π−π stacking and hyrogen bond interactions are indicated by cyan and pale green dotted lines.
Molecular docking calculations indeed found two potential binding conformations of 3,5-BTAP, i.e. the carbonyl-out and the carbonyl-in ones (Fig. S26). In both binding forms, 3,5-BTAP interacts with the catalytic pocket residues through hydrogen bonds. However, due to the electrostatic repulsive interactions between the carbonyl groups of 3,5-BTAP and AKR Q169 in the carbonyl-in binding form (Fig. S26b), it is energetically unfavorable relative to the carbonyl-out binding form. Interestingly, driven by the induced-fit effect, a fast conformational switch (within 500 ps) from the carbonyl-out to the carbonyl-in form of 3,5-BTAP was discovered in the MD simulation, corresponding well with the sudden decrease of the distance between the carbonyl oxygen atom of 3,5-BTAP and the hydroxyl oxygen atom of the AKR Y58 sidechain at the starting stage of MD simulation (Fig. S27a). A representative carbonyl-out form snapshot of the AKR–3,5-BTAP binding complex was retrieved from the MD trajectory (Fig. S27c). It shows that, though the aromatic ring of 3,5-BTAP tends to form π − π stacking interactions with the sidechains of W21 and Y198, their distances (4.56 Å) are obviously longer than a common π − π stacking interaction distance of 3.50 Å, indicating a transient unstable interaction. In addition, the inner trifluoromethyl group of 3,5-BTAP is located near a negatively charged carboxyl group of D53 and the hydrophobic sidechain of I242, which is energetically unfavorable (Fig. S27c). These factors are the driving force of the conformational change of the binding 3,5-BTAP.
Data in Fig. S27 demonstrates that the carbonyl-in binding complex equilibrated to a similar conformation to that in Fig. 4a within 20 ns. The hydrogen bond interaction with Y58 and the π−π stacking to W21 are stable, as indicated by their distance profiles (Fig. S27b and S28c). The value and distribution of the improper dihedral ∠C6-C10-C9-H across the MD simulation (Fig. S28d, e) also showed a clear preference for the pro-(R) binding form of 3,5-BTAP, agreeing well with our experimental result and validating the results shown in Fig. 4. Therefore, it is feasible and convenient to apply rGQDs/AKR as the photocatalyst for water splitting and enantioselective hydrogenation.
Our molecular simulation data provided pivotal information for the enantioselective process catalyzed by the rGQDs/AKR photo-biocatalyst system at the atomic level. These properties were evaluated by performing the photoenzymatic synthesis of (R)-3,5-BTPE in the absence of both NAD(P)H or NAD(P)+ and a noble metal complex (Fig. 5a). Figure 5b shows the yields of (R)-3,5-BTPE with different catalyst systems, namely (rGQDs, AKR, rGQDs/AKR, rGQDs/AKR and NADP+, rGQDs/AKR and NADP+ and M) under 18 h infrared light irradiation, using isopropanol as cosolvent (50 mW·cm−2). It clearly shows that no (R)-3,5-BTPE was observed with only AKR or rGQDs addition, but once the combination of rGQDs/AKR was provided, (R)-3,5-BTPE was produced in up to 82% yield with 99.99% ee, thus demonstrating the success of our method. We also observed that the yields (88%, 96%) were elevated when M and/or NADP+ were added to the rGQDs/AKR photo-biocatalyst system, but these were not significant compared to those in the absence of these two coenzymes. We conclude that, considering the high costs of both NADP+ and M, the photocatalytic synthesis of (R)-3,5-BTPE assisted by only rGQDs/AKR is an important step in the right direction. We also studied the effect of the wavelength of the light source. As shown in Fig. 5c, the yield of (R)-3,5-BTPE was 0 under dark conditions, showing that light illumination was the requirement for this photochemical reaction. The reaction system also worked well under the irradiation of visible light or simulated sunlight (AM1.5 G), with a yield of 63% and 86%, respectively. The lower yield under visible light compared with under IR light showed that our photo-biocatalyst had better responsiveness to IR light. To establish the optimum wavelength for illumination, the synthesis of (R)-3,5-BTPE was conducted using different monochromatic light sources (365 nm, 750 nm and 980 nm). As shown in Fig. 5d, it is apparent that the NIR 750 nm is the best, i.e. close to the optimum upconversion excitation wavelength of rGQDs (780 nm). The most suitable IR light intensity was 50 mW·cm−2 (Fig. 5e). When the light intensity is elevated to 100 or 200 mW·cm−2, the yields are all greatly decreased to 2.9% and 1.7%, respectively. The lower yields are a result of reductions in photo-biocatalyst activity caused by the excessive local heating through the photothermal effect of the rGQDs upon strong IR irradiation. This explanation is in agreement with the results of comparison experiments (Fig. 5f).

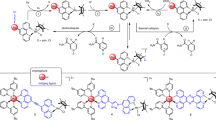
a HPLC spectrum of the product. (R)−3,5-BTPE yields of (b) using different catalyst systems including AKR, rGQDs, rGQDs/AKR, rGQDs/AKR + NADP+, and rGQDs/AKR + NADP+ + M (M denotes [Cp*Rh(bpy)(H2O)]2+) with IR light ( > 800 nm, 50 mW·cm-2) irradiation, (c) using different light sources, (d) using different wavelength LED lamps as light sources, (e) using infrared light (λ > 800 nm) as light sources with different light intensities, (f) using different rGQDs loading amounts at IR light irradiation. g Photo-enzymatic reduction of 1-methyl-3,4-dihydroisoquinolin and cinnamaldehyde with imine reductase AoIRED and ene reductase OYE1 as the corresponding biocatalyst, respectively. h Reduction of 3-chloro-1-phenylpropan-1-one using different catalyst systems, including rGQDs/carbonyl reductase (NaCBR), TiO2/NaCBR and TiO2-rGQDs/NaCBR, under corresponding photo-enzymatic reaction conditions. (i) Reuse of TiO2-rGQDs/NaCBR in cyclic catalysis. The error bars represent the standard deviations of three parallel measurements.
To assess the possible effect of rGQDs on enantioselectivity, an imine reductase AoIRED, displaying poor enantioselectivity in (1-methyl-3,4-dihydroisoquinolin) reduction, was examined63. First, distributions of the key improper dihedral ∠N1-C10-C7-H in the AoIRED–NADPH–DHIQ and AoIRED–DHIQ models were analyzed based on 20 ns MD simulations, which showed dramatically decreased enantioselectivity in both systems. The theoretical product 1-methyl-1,2,3,4-tetrahydroisoquinoline contained almost 60% S-configuration and 40% R-configuration, i.e. 20% ee (Fig. S29-30). The result was confirmed in the photo-enzymatic reduction using rGQDs/AoIRED as the photo-biocatalyst system and DMF as the cosolvent under IR irradiation. The corresponding product was obtained in 65% yield with 22% ee, predominantly in the S- configuration, consistent with the selected enzyme’s catalytic preference (Fig. 5g and S31). Additionally, in the presence of isopropanol cosolvent, cinnamaldehyde was reduced to form 3-phenylpropanol (65%) through the synergistic catalysis of ene reductase OYE1 with rGQDs under IR irradiation (Fig. 5g and S32-34)64. On the other hand, an electron mediator may negatively affect the conversion system due to possible ligand exchange with the active residue of the enzyme, leading to a significant drop in yield (Fig. 5g and S34b)65. These results strongly suggest that the enantioselectivity and hydrogenation activity of the photoenzymatic system are controlled separately by the enzyme and photocatalyst, respectively.
To further illustrate the function of rGQDs in promoting solar light energy conversion, we integrated TiO2 with strong UV and low visible-light absorbing properties into the above photoenzymatic system. Under simulated sunlight, (S)-3-chloro-1-phenyl-1-propanol ((S)-CPPO), a key chiral intermediate for the synthesis of the chiral side chain of Fluoxetine and Atomoxetine, obtained in 72.3% yield, which is 2.7 times and 2.6 times of that without TiO2 and rGQDs, respectively (Fig. 5h and S35)66,67,68,69. What’s more, corresponding electrostatic assembled hybrid catalysts (TiO2-rGQDs/NaCBR) can be recycled with excellent chemical stability, maintaining 90% of its original catalytic efficiency and consistent enantioselectivity (99.9% ee) after 6-cycles (Fig. 5i).
An isotope-tracer experiment was carried out to confirm the hydrogen donor in the cofactor-independent photo-enzymatic reduction system mediated by rGQDs/AKR to synthesize (S)-1-(2-chlorophenyl) ethanol. The high-resolution mass spectrum (HRMS) analysis of the product obtained using D2O reveals that it was completely labelled with two deuteriums, confirming that water was the hydrogen resource (Fig. S36-37). In 3,5-BTAP and cinnamaldehyde reductions isopropanol, a more energetically favorable source of the hydrogen atoms than water, was used as a cosolvent. However, it is also feasible to use DMF as the sole co-solvent for 1-methyl-3,4-dihydroisoquinoline reduction or synthesize (S)-CPPO in the absence of a cosolvent. Hence, we conclude that with this hybrid photo-biocatalyst, water can be used as the sole source of hydrogen.
Discussion
In conclusion, we have successfully constructed a infrared light responsive hybrid rGQDs/AKR photo-reductase system that requires neither an expensive noble metal complex nor an expensive cofactor for its activity. Moreover, the two hydrogen atoms required for the reduction are provided by a molecule of water. The catalyst was essentially enantiospecific ( > 99.99%) in the synthesis of the chiral alcohol, (R)-3,5-BTPE. This custom-designed hybrid system provides active hydrogen atoms through photo-catalytic water decomposition and directly delivers them to the immobilized AKR for transfer to the ketone substrate. The rGQDs was also successfully combined with an imine reductase and an ene reductase to afford 1-methyl-1,2,3,4-tetrahydroisoquinoline and 3-phenylpropanol, respectively, via IR-driven photo-enzymatic reduction.
This methodology represents a paradigm shift in the development of enantioselective reductions of prochiral ketones by involving cofactor-independent photo-biocatalysts in combination with water as the sole source of hydrogen atoms and NIR light as the source of energy. We anticipate that this methodology can be used to design efficient hybrid-reductase systems that are sensitive to infrared light, and could also be used to study and control protein activity in cells and organisms. Since our hybrid system is a heterogeneous immobilized biocatalyst, it can also be readily recycled and/or used in continuous flow operation. In short, we believe that this unprecedented example of a hybrid photo-biocatalyst that requires only NIR radiation and water to perform sustainable, highly (enantio)selective reductions could represent the tip of an iceberg. It could form a basis for the development of various cofactor independent biotransformations using only water and light as the source of, respectively, reducing equivalents and energy.
Methods
Preparation of assembled photo-biocatalyst rGQDs/AKR and TiO2-rGQDs/NaCBR
The photo-biocomposite rGQDs/AKR was obtained by electrostatic self-assembly under mechanical oscillation. Typically, aldo-keto reductase aggregates were dispersed in a 5 mL rGQDs (5 mg·mL−1) suspension with a final concentration of 5 mg·mL−1. Then the mixture was shaken continuously at 10 °C for 4 h to obtain hybrid material rGQDs/AKR, which were collected by freeze-drying after centrifugation.
To prepare the bio-inorganic hybrid TiO2-rGQDs/NaCBR, the assembly of TiO2 and rGQDs was first performed by shaking the mixture of 200 mg of TiO2 and 40 mL of rGQDs suspension (5 mg·mL−1) at 60 °C overnight. After centrifugal separation and washing with 10 mL of deionized water three times, 50 mg of TiO2/rGQDs composite was obtained and then assembled with carbonyl reductase (5 mg·mL−1) in 5 mL PBS buffer, pH 7.0, at 10 °C for 4 h to give TiO2-rGQDs/NaCBR after collection by freeze-drying and centrifugation.
General procedure for the photo-enzymatic reduction reactions
All photo-enzymatic catalysis was carried out in a 45 mL custom-made quartz reactor (Fig. S15) at 20 °C. For (R)-3,5-BTPE synthesis, 10 mg of rGQDs/AKR were dispersed in 5 mL of phosphate-buffered saline (PBS buffer) (pH = 7.0, 100 mM), then 3,5-BTAP (32.3 mg, 0.125 mmol) and 3 μL of isopropanol cosolvent was added. After that, the reactor was irradiated at AM1.5 G with an optical filter (CEL-HXF300) for 18 h. When the photoenzymatic assays were performed in the presence of NADP+ and (or) electron mediator, 0.1 mM of NADP+, and (or) 0.25 mM of M were added to the reaction system in sequence. After the reactions, the target product was extracted from the supernatant in the reaction solution by 3 mL of anhydrous n-hexane. The yield and ee values were determined by high-performance liquid chromatography.
For 1-methyl-3,4-dihydroisoquinolin reduction, 10 mg of AoIRED and 5 mg of rGQDs were dispersed in 5 mL of PBS buffer (pH = 7.0, 100 mM), then 1-methyl-3,4-dihydroisoquinolin (18.2 mg, 0.125 mmol) and 100 μL of DMF were added. For cinnamaldehyde reduction, 10 mg of OYE1 and 5 mg of rGQDs were dispersed in 5 mL of PBS buffer (pH = 7.0, 100 mM), then cinnamaldehyde (16.5 mg, 0.125 mmol) and 3 μL of isopropanol were added. For 3-chloro-1-phenylpropan-1-one reduction, 10 mg of NaCBR and 5 mg of rGQDs were dispersed in 5 mL of PBS buffer (pH = 7.0, 100 mM), then 3-chloro-1-phenylpropan-1-one (21.0 mg, 0.125 mmol) was added. The above reaction mixtures were irradiated at IR irradiation (CEL-HXF300, >800 nm, 50 mW·cm−2) for 18 h. After the reactions, the target products were extracted from the supernatant in the reaction solutions by 3 mL of anhydrous n-hexane. The yield and ee values were determined by high-performance liquid chromatography.
To detect the recycling efficiency of TiO2-rGQDs/NaCBR, 15 mg of TiO2-rGQDs/NaCBR was dispersed in 5 mL of PBS buffer (pH = 7.0, 100 mM) at a quartz reactor. Then, 3-chloro-1-phenylpropan-1-one (21.0 mg, 0.125 mmol) was added. The reaction mixture was stirred and irradiated at AM1.5 G (CEL-HXF300, 50 mW·cm−2) for 18 h at 20 °C. After that, the TiO2-rGQDs/NaCBR was separated from the reaction mixture by centrifugation (10000 x g, 15 min) and reused for the next cycle of reactions under standard reaction conditions. The liquid supernatant was extracted by anhydrous n-hexane and the yield was determined by HPLC.
Data availability
All data supporting the present study are available in the manuscript, source data file, and supplementary information. Source data are provided with this paper.
References
Ghosh, S. et al. Exploring emergent properties in enzymatic reaction networks: design and control of dynamic functional systems. Chem. Rev. 124, 2553–2582 (2024).
Bell, E. L. et al. Biocatalysis. Nat. Rev. Methods Prim. 1, 46 (2021).
Kissman, E. N. et al. Expanding chemistry through in vitro and in vivo biocatalysis. Nature 631, 37–48 (2024).
Reetz, M. T., Qu, G. & Sun, Z. Engineered enzymes for the synthesis of pharmaceuticals and other high-value products. Nat. Synth. 3, 19–32 (2024).
Arnodo, D. et al. Asymmetric Reduction of Cyclic Imines by Imine Reductase Enzymes in Non-Conventional Solvents. Chemsuschem 17, e202301243 (2024).
Lin, X. et al. Engineered imine reductase for asymmetric synthesis of dextromethorphan key intermediate. Org. Lett. 26, 4463–4468 (2024).
Livada, J., Vargas, A. M., Martinez, C. A. & Lewis, R. D. Ancestral sequence reconstruction enhances gene mining efforts for industrial Ene Reductases by expanding enzyme panels with thermostable catalysts. ACS Catal. 13, 2576–2585 (2023).
Hartley, C. J. et al. Engineered Enzymes that Retain and Regenerate their Cofactors Enable Continuous-Flow Biocatalysis. Nat. Catal. 2, 1006–1015 (2019).
Bruffy S. K. et al. Biocatalytic asymmetric aldol addition into unactivated Ketones. Nat. Chem. https://doi.org/10.1038/s41557-024-01647-1 (2024).
Choi, S. Y. et al. Sustainable Production and Degradation of Plastics Using Microbes. Nat. Microbiol. 8, 2253–2276 (2023).
Lauterbach, L., Lenz, O. & Vincent, K. A. H2-driven cofactor regeneration with NAD(P)+-reducing hydrogenases. FEBS J. 280, 3058–3068 (2013).
Peng, Y. et al. State-of-the-Art light-driven hydrogen generation from formic acid and utilization in enzymatic hydrogenations. ChemSusChem 18, e202401811 (2025).
Bachosz, K., Zdarta, J., Bilal, M., Meyer, A. S. & Jesionowski, T. Enzymatic cofactor regeneration systems: a new perspective on efficiency assessment. Sci. Total Environ. 868, 161630 (2023).
Lee, Y. S., Gerulskis, R. & Minteer, S. D. Advances in electrochemical cofactor regeneration: enzymatic and non-enzymatic approaches. Curr. Opin. Biotechnol. 73, 14–21 (2022).
Choi, D. S., Kim, J., Hollmann, F. & Park, C. B. Solar-assisted eBiorefinery: photoelectrochemical pairing of oxyfunctionalization and hydrogenation reactions. Angew. Chem. Int. Ed. 59, 15886–15890 (2020).
Xu, Z., Zhou, F., Chen, H. & Wang, J. Regeneration of NAD(P)H and its analogues by photocatalysis with ionized carbon nitride. ACS Catal. 14, 5868–5878 (2024).
Mifsud, M. et al. Photobiocatalytic chemistry of oxidoreductases using water as the electron donor. Nat. Commun. 5, 3145 (2014).
Ma, Y. et al. Photobiocatalysis: More than just an interesting lab curiosity?. Chem. Catal. 4, 101077 (2024).
Bachar, O., Meirovich, M. M., Zeibaq, Y. & Yehezkeli, O. Protein-mediated biosynthesis of semiconductor nanocrystals for photocatalytic NAD(P)H regeneration and chiral amine production. Angew. Chem. Int. Ed. 61, e202202457 (2022).
Rudzka, A., Antos, N., Reiter, T., Kroutil, W. & Borowiecki, P. One-pot sequential two-step photo-biocatalytic deracemization of sec-alcohols combining photocatalytic oxidation and bioreduction. ACS Catal. 14, 1808–1823 (2024).
Özgen, F. F., Runda, M. E. & Schmidt, S. Photo-biocatalytic Cascades: combining chemical and enzymatic transformations fueled by light. Chembiochem 22, 790–806 (2021).
Schmermund, L. et al. Photo-biocatalysis: biotransformations in the presence of Light. ACS Catal. 9, 4115–4144 (2019).
Lee, S. H., Choi, D. S., Kuk, S. K. & Park, C. B. Photobiocatalysis: ACtivating Redox Enzymes by Direct Or Indirect Transfer of Photoinduced Electrons. Angew. Chem.-Int. Ed. 57, 7958–7985 (2018).
Broumidis, E. & Paradisi, F. Engineering a dual-functionalized polyhipe resin for photobiocatalytic flow chemistry. Angew. Chem. Int. Ed. 63, e202401912 (2024).
Tahir, M., Khan, A. A., Tasleem, S., Mansoor, R. & Fan, W. K. Titanium carbide (Ti3C2) MXene as a promising co-catalyst for photocatalytic CO2 conversion to energy-efficient fuels: a Review. Energy Fuels 35, 10374–10404 (2021).
Cheng, B. C., Wan, L. & Armstrong, F. A. Progress in scaling up and streamlining a nanoconfined, enzyme-catalyzed electrochemical nicotinamide recycling system for biocatalytic synthesis. Chemelectrochem 7, 4672–4678 (2020).
Reeve, H. A. et al. A Hydrogen-driven biocatalytic approach to recycling synthetic analogues of NAD(P)H. Chem. Commun. 58, 10540–10543 (2022).
Seel, C. J. & Gulder, T. Biocatalysis fueled by light: on the versatile combination of photocatalysis and enzymes. Chembiochem 20, 1871–1897 (2019).
Brown, K. A. et al. Photocatalytic Regeneration of Nicotinamide Cofactors by Quantum Dot-Enzyme Biohybrid Complexes. ACS Catal. 6, 2201–2204 (2016).
Cheng, W. H., de la Calle, A., Atwater, H. A., Stechel, E. B. & Xiang, C. X. Hydrogen from Sunlight and Water: A Side-by-Side Comparison between Photoelectrochemical and Solar Thermochemical Water-Splitting. ACS Energy Lett. 6, 3096–3113 (2021).
Reischauer, S., Pieber, B. Emerging Concepts in Photocatalytic Organic Synthesis. Iscience, 24.https://doi.org/10.1016/j.isci.2021.102209 (2021).
Wang, C. et al. Formate-Mediated Electroenzymatic Synthesis via Biological Cofactor NADH. Angew. Chem. Int. Ed. 63, e202408756 (2024).
Jia, C. C. et al. Facile Assembly of a Graphitic Carbon Nitride Film at an Air/Water Interface for Photoelectrochemical NADH Regeneration. Inorg. Chem. Front. 7, 2434–2442 (2020).
Lyu, S. et al. Subcutaneous Power Supply by NIR-II Light. Nat. Commun. 13, 6596 (2022).
Ravetz, B. D. et al. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 565, 343–346 (2019).
Sun, R. J., Zang, J. Y., Lai, R. C., Yang, W. X. & Ji, B. T. Near-Infrared-to-Visible Photon Upconversion with Efficiency Exceeding 21% Sensitized by InAs Quantum Dots. J. Am. Chem. Soc. 146, 17618–17623 (2024).
Shen, J., Zhu, Y., Chen, C., Yang, X. & Li, C. Facile Preparation and Upconversion Luminescence of Graphene Quantum Dots. Chem. Commun. 47, 2580–2582 (2011).
Jia, D. et al. Direct Electron Transfer from Upconversion Graphene Quantum Dots to TiO2 Enabling Infrared Light-Driven Overall Water Splitting. Res. (Wash. D. C.) 2022, 9781453 (2022).
Wang, Z., Hu, Y., Zhang, S. & Sun, Y. Artificial Photosynthesis Systems for Solar Energy Conversion and Storage: Platforms and Their Realities. Chem. Soc. Rev. 51, 6704–6737 (2022).
Hasan, M. T. et al. Near-Infrared Emitting Graphene Quantum Dots Synthesized from Reduced Graphene Oxide for in vitro/in vivo/ex vivo Bioimaging Applications. 2D Mater. 8, 035013 (2021).
Qiao L. et al. Near-infrared Light-driven Asymmetric Photolytic Reduction of Ketone Using Inorganic-enzyme Hybrid Biocatalyst. Int. J. Biol. Macromolecules 264. https://doi.org/10.1016/j.ijbiomac.2024.130612 (2024).
Emmanuel, M. A. et al. Photobiocatalytic Strategies for Organic Synthesis. Chem. Rev. 123, 5459–5520 (2023).
Xu, Y., Liu, F., Zhao, B. & Huang, X. Repurposing Naturally Occurring Enzymes Using Visible Light. Chin. J. Chem. 42, 3553–3558 (2024).
Sandoval, B. A. et al. Photoenzymatic Reductions Enabled by Direct Excitation of Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc. 143, 1735–1739 (2021).
Emmanuel, M. A., Greenberg, N. R., Oblinsky, D. G. & Hyster, T. K. Accessing Non-natural Reactivity by Irradiating Nicotinamide-dependent Enzymes with Light. Nature 540, 414–417 (2016).
Peng, Y. et al. Photoinduced Promiscuity of Cyclohexanone Monooxygenase for the Enantioselective Synthesis of α-Fluoroketones. Angew. Chem. Int. Ed. 61, e202211199 (2022).
Huang, X. et al. Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 584, 69–74 (2020).
Gao, X., Turek-Herman, J. R., Choi, Y. J., Cohen, R. D. & Hyster, T. K. Photoenzymatic Synthesis of α-Tertiary Amines by Engineered Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc. 143, 19643–19647 (2021).
Zhang, Z. et al. Photoenzymatic Enantioselective Intermolecular Radical Hydroamination. Nat. Catal. 6, 687–694 (2023).
Shi, Q. et al. Single-Electron Oxidation-Initiated Enantioselective Hydrosulfonylation of Olefins Enabled by Photoenzymatic Catalysis. J. Am. Chem. Soc. 146, 2748–2756 (2024).
Jiang, L. et al. Photoenzymatic Redox-Neutral Radical Hydrosulfonylation Initiated by FMN. ACS Catal. 14, 6710–6716 (2024).
Yu, J. et al. Repurposing Visible-Light-Excited Ene-Reductases for Diastereo- and Enantioselective Lactones Synthesis. Angew. Chem. Int. Ed. 63, e202402673 (2024).
Wang, N., Li, J., Sun, J., Huang, J. & Wang, P. Bioreduction of 3,5-Bis(trifluoromethyl)Acetophenone Using Ionic Liquid as a Co-solvent Catalyzed by Recombinant Escherichia coli Cells. Biochem.Eng. J. 101, 119–125 (2015).
Chen, K. et al. Carbonyl Reductase Identification and Development of Whole-cell Biotransformation for Highly Efficient Synthesis of (R)-[3,5-bis(trifluoromethyl)phenyl] Ethanol. Microb. Cell Factories 15, 191 (2016).
Tang, J. et al. Development of an Efficient and Cost-Effective Enzymatic Process for Production of (R)-[3,5-bis(trifluoromethyl)phenyl] Ethanol Using Carbonyl Reductase Derived from Leifsonia sp. S749. Appl. Biochem. Biotechnol. 188, 87–100 (2019).
Wang, X. et al. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem 2, 621–654 (2017).
Luo, Z. Y. et al. Precision Engineering of the Co-immobilization of Enzymes for Cascade Biocatalysis. Angew. Chem. Int. Ed. 63, e202403539 (2024).
Li, H. et al. Rapidly and Precisely Cross-Linked Enzymes Using Bio-Orthogonal Chemistry from Cell Lysate for the Synthesis of (S)-1-(2,6-Dichloro-3-fluorophenyl) Ethanol. ACS Sustain. Chem. Eng. 8, 6466–6478 (2020).
Zhang, J. et al. Controlled Chemical Assembly of Enzymes in Cell Lysate Enabled by Genetic-encoded Nonstandard Amino Acids. Mater. Chem. Front. 6, 182–193 (2022).
Cao, L. et al. Carbon Dots for Multiphoton Bioimaging. J. Am. Chem. Soc. 129, 11318–11319 (2007).
Xiaozhen, F., Xing, L., Zhenglin, H., Kaiyuan, Z. & Guosheng, S. DFT Study of Common Anions Adsorption at Graphene Surface due to Anion-π Interaction. J. Mol. Modeling 28, 225 (2022).
Dougherty, D. A. The Cation−π Interaction. Acc. Chem. Res. 46, 885–893 (2013).
Aleku, G. A. et al. Stereoselectivity and Structural Characterization of an Imine Reductase (IRED) from Amycolatopsis orientalis. ACS Catal. 6, 3880–3889 (2016).
Kuai, L. et al. Titania Supported Synergistic Palladium Single Atoms and Nanoparticles for Room Temperature Ketone and Aldehydes Hydrogenation. Nat. Commun. 11, 48 (2020).
Ranaware, V. et al. Highly-efficient and Magnetically-separable ZnO/Co@N-CNTs Catalyst for Hydrodeoxygenation of Lignin and its Derived Species under Mild Conditions. Green. Chem. 21, 1021–1042 (2019).
van der Worp, H. B. Fluoxetine and Recovery after Stroke. Lancet 393, 206–207 (2019).
Capuozzo, A. et al. Fluoxetine Ameliorates Mucopolysaccharidosis Type IIIA. Mol. Ther. 30, 1432–1450 (2022).
Tang, Y. P. et al. Efficient Synthesis of a (S)-Fluoxetine Intermediate Using Carbonyl Reductase Coupled with Glucose Dehydrogenase. Bioresour. Technol. 250, 457–463 (2018).
Jung, E. H. et al. Effects of paroxetine on the pharmacokinetics of atomoxetine and its metabolites in different CYP2D6 genotypes. Arch. Pharmacal Res. 43, 1356–1363 (2020).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (22005268, 22309039, 22206042, 22078079, 22378091, 22378092, 22178078), the University Leading Talents Program of Zhejiang Province (4095C502222140203, 4095C502222140201), the Zhejiang Provincial Natural Science Foundation of China (LQ22B060007), and the Medical Health Science and Technology Project of the Zhejiang Provincial Health Commission (2023KY1009).
Author information
Authors and Affiliations
Contributions
Z.G.W. contributed to the theoretical calculation. W.Y., P.G., A.M.W. and R.A.S. conceived and designed the overall study and provided comments and feedback on the discussion. L.Q. and X.Y.L. performed the analysis and drafted the manuscript with input from all co-authors. Y.J.J. and X.T.P. contributed to the data generation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiang Sheng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, A., Li, X., Qiao, L. et al. Cofactor-independent photo-enzymatic reductions with water mediated by reductive graphene quantum dots. Nat Commun 16, 8310 (2025). https://doi.org/10.1038/s41467-025-61908-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61908-6
