
Abstract
A multidimensional sleep health framework improves screening and treatment efficacy by simultaneously addressing multiple sleep domains. However, limited studies have used objective measures to evaluate the co-occurrence of diverse unhealthy sleep characteristics and their pleiotropic health effects. To represent real-world sleep patterns, we introduce the Unfavorable Sleep Profile (USP), an integrated multidimensional sleep health metric developed using accelerometer data in the UK Biobank (N = 85,233; aged 43–79 years). USP captures five domains: sleep timing, efficiency, duration, rhythmicity, and regularity. Phenome-wide association study found that USP was significantly associated with 76 out of 526 incident health outcomes over 7.9 years of follow-up. We identified several upstream environmental risk factors associated with USP, including low socioeconomic status. Whole-genome sequence analyses identified common variants in MEIS1 and rare coding variants in TTC1 associated with USP. We validated the USP framework in an independent cohort, the Multi-Ethnic Study of Atherosclerosis. Our findings underscore the importance of multidimensional sleep health assessment in predicting and potentially mitigating a wide array of health disorders and advance genetic insights into sleep health.
Similar content being viewed by others

Introduction
Sleep is a fundamental biological necessity, essential for human life and increasingly recognized as crucial to public health1. The American Heart Association has acknowledged sleep’s substantial and independent impact on overall health and cardiometabolic outcomes by incorporating sleep health as the eighth metric in the Life’s Essential 8 framework2. This inclusion aligns with endorsements from the European Academy of Neurology and the World Health Organization, emphasizing sleep’s vital role in brain health3. Globally, common sleep insufficiency and sleep disorders pose significant challenges4.
Sleep health is a complex, multidimensional construct that extends beyond individual sleep characteristics or specific sleep disorders4. The RU-SATED model proposed by Buysee describes sleep health through six domains: regularity, satisfaction, alertness, timing, efficiency, and duration4,5. Additionally, rhythmicity, which characterizes the strength of the overall sleep-wake rhythm in a 24 h cycle, has also been proposed as an important domain6,7. However, previous studies examining the associations between sleep and disease outcomes have predominantly focused on specific sleep characteristics such as sleep duration and insomnia8,9, overlooking the multidimensional nature of sleep health. Moreover, prior research has relied largely on subjective, self-reported measures of sleep, which often correlate poorly with objective measures obtained through the gold-standard polysomnography (PSG)10. The subjective sleep data also have limitations in capturing the regularity and rhythmicity of sleep patterns, as these domains require continuous, longitudinal 24-h measurements.
Actigraphy or accelerometer, which provides sleep-related data often comparable to PSG11, offers unique opportunities to investigate the associations between multidimensional sleep health and disease outcomes, particularly regarding regularity and rhythmicity12,13. Accelerometer data reduce reporting bias and provide objective, noninvasive, and continuous measurements of sleep-wake patterns in the home setting, enabling comprehensive analysis of sleep health across multiple domains. Recent studies using wearable devices have analyzed the relationships between objectively measured, separate sleep characteristics and chronic disease risks7,14,15. However, these studies, while informative, are limited by failing to consider sleep as a multidimensional construct, relatively small samples, and restricted range of disease outcomes. Thus, it is critical to adopt a multidimensional approach to sleep health and to investigate the association between sleep health and a broad range of future disease outcomes, using objectively measured data from a large sample.
Understanding environmental and genetic factors that influence sleep patterns can guide the development of tailored interventions and therapeutic strategies to improve sleep and, consequently, overall health. While genome-wide association studies have identified hundreds of common and low-frequency variants associated with sleep-related traits, these variants explain only a modest portion of the heritability16. Rare variants (RVs) may explain additional unaccounted heritability17 and provide insights into gene function in disease pathophysiology, potentially revealing new therapeutic targets18,19,20. Large-scale whole genome sequencing (WGS) studies and biobanks now allow for examination of associations between sleep health and both coding and noncoding RVs across the genome. However, the upstream factors influencing multidimensional sleep health remain underexplored.
In this study, we introduced a multidimensional sleep health marker termed the Unfavorable Sleep Profile (USP) to capture real-world sleep patterns. This marker consolidates multiple sleep dimensions into a single comprehensive metric by extracting and clustering a multitude of objective sleep characteristics measured by accelerometer, using data from the large UK Biobank cohort (587,152 nights of data from 85,233 participants). We employed a phenome-wide association study (PheWAS) to examine the associations between USP and future health outcomes. Additionally, we used multivariable logistic regression to investigate environmental factors associated with USP and conducted whole-genome sequence analyses to investigate genetic factors linked to USP (Fig. 1). We assessed the effects of common and low-frequency variants individually through single variant analysis and the aggregate effects of RVs using various coding and noncoding functional categories. Our study highlights the importance of adopting a multidimensional sleep health framework using objective sleep measures in understanding both the downstream health risks and the upstream environmental and genetic factors associated with sleep.

Left: Workflow for identifying sleep profiles and assessing the associated future health risks. A phenome-wide analysis was performed to identify associations between sleep profiles and incident disease phenotypes (phecodes). Right: Workflow for analyzing upstream factors associated with sleep profiles. Regression analysis examined associations between environmental factors and sleep profiles. Functionally-informed whole-genome association analysis investigated the genetic factors associated with sleep profiles.
Results
Description of the study participants
The study included 85,233 UK Biobank participants with valid accelerometer data. Participants had a median age of 63.48 years (interquartile range: 56.32, 68.55) at accelerometry assessment, with 56.5% being female and the majority being white (Table 1, Supplementary Data 1). Accelerometry monitoring last a median of 7 days, generating 587,152 person-nights of data. For genetic analyses, 80,007 individuals had whole-exome data available for coding variant association analysis, and 35,556 had whole-genome data for noncoding association analysis.
Construction of USP
We developed a multidimensional sleep health marker, the USP, by integrating multiple sleep dimensions into a single comprehensive metric. First, using exploratory factor analysis (EFA), we extracted factors representing sleep health domains through evaluating eigenvalues, visually inspecting scree plots, and ensuring interpretability (Supplementary Fig. 1, Methods). Adhering to the multidimensional sleep health framework4, we adopted a 5-factor model comprising 26 variables, with factors representing timing, efficiency, duration, rhythmicity, and regularity (Fig. 2a, Supplementary Data 2). This model explained 77% of the total variance, with factor independence confirmed through multiple orthogonal rotations (Supplementary Data 3–6). Notably, our analysis identified rhythmicity as a distinct dimension of sleep health, a finding not previously confirmed in prior studies, but one that supported recent calls to integrate circadian rhythm characteristics into the multidimensional sleep health framework6,7. As illustrated in Supplementary Fig. 2, a participant randomly selected from the lowest quintile of rhythmicity scores exhibited irregular, fragmented rest-activity cycles with low stability across days. The time series appeared highly random, with little distinction between day and night. In contrast, a participant randomly selected from the highest quintile of rhythmicity scores showed a robust and consistent rest-activity pattern, with clear separation between periods of rest and activity across the 24 h cycle.

a Factor analysis: Display of the 5-factor model results, labeled as timing, efficiency, duration, rhythmicity, and regularity. Numbers indicate factor loadings; purple dashed arrows denote negative correlations between the latent factor and its corresponding original variables. AD all days average, WD weekdays average, WE weekend days average, WASO wake after sleep onset, (sd) standard deviation, IV intra-daily variability, RA relative amplitude, IS inter-daily stability. b Latent profile analysis: Identification of the optimal model with two profiles, based on the best integrated completed likelihood values. The x-axis shows the five factors obtained from the factor analysis, and the y-axis shows the standardized factor scores. Source data are provided as a Source Data file.
Second, we performed latent profile analysis (LPA) to construct a multidimensional sleep health marker. Building on the 5-factor model identified in EFA, LPA determined the optimal model with two profiles: a favorable sleep profile (n = 69,225) and an unfavorable sleep profile (n = 16,008), based on the highest integrated completed likelihood values (Supplementary Data 7, Supplementary Fig. 3). Compared to favorable sleep profile, USP was characterized by later sleep timing, lower sleep efficiency, shorter sleep duration, weaker rhythmicity, and less regular sleep patterns with greater variability in both timing and duration (Fig. 2b, Supplementary Data 8). These findings revealed that sleep behaviors do not occur in isolation; instead, unhealthy sleep characteristics tend to cluster together and form distinct sleep profiles.
Association of USP with incident diseases and mortality
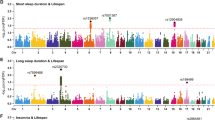
To systematically examine the future health risks associated with USP, we conducted a PheWAS using a Cox proportional hazards model analyzing 526 diseases across 16 groups defined by the Phecode Map21,22. Over an average follow-up period of 7.9 years, the PheWAS identified 76 significant associations with USP at the Bonferroni-corrected significance level of \(0.05/526=9.51\times {10}^{-5}\), after adjusting for multiple covariates (Fig. 3). These associations comprised 17 circulatory, 11 endocrine or metabolic, 9 respiratory, 7 mental, 7 genitourinary, 5 neurological, 4 dermatologic, 3 infectious, 3 hematopoietic, 3 digestive, 4 injuries and poisonings, and 1 each of symptom, sense organ, and musculoskeletal diseases (Fig. 4).

Manhattan plot-style visualization assessing the relationship between sleep profiles and future disease risk. A Cox proportional hazards model was applied to each phenotype using the two-sided Wald test, adjusted for age, sex, ethnicity, diet, smoking status, alcohol consumption, education, self-rated health, body mass index, season, moderate-to-vigorous physical activity, and Townsend deprivation index. Uncorrected P-values are plotted, with a blue dashed line indicating the Bonferroni-corrected significance threshold for α ≤ 0.05 and a red dashed line representing the Benjamini-Hochberg false discovery rate (FDR) cutoff at 0.05. USP: unhealthy sleep profile. Source data are provided as a Source Data file.

A Cox proportional hazards model was applied to each phenotype using the two-sided Wald test, adjusted for age, sex, ethnicity, diet, smoking status, alcohol consumption, education, self-rated health, body mass index, season, moderate-to-vigorous physical activity, and Townsend deprivation index. The significant threshold is defined by multiple comparisons using the Bonferroni correction (\(0.05/526=9.51\times {10}^{-5}\)). Uncorrected P-values are plotted. Left: Effect sizes from the Cox models are presented as hazard ratios (HRs) with 95% confidence intervals (CIs), using the favorable sleep profile as the reference. Right: Significance of the effect size. Source data are provided as a Source Data file.
USP was associated with a broad range of chronic diseases, such as heart failure (hazard ratio [HR] 1.35, 95% confidence interval [CI] 1.20–1.51), atrial fibrillation and flutter (HR 1.18, 95% CI 1.09–1.29), hypertension (HR 1.13, 95% CI 1.08–1.19), type 2 diabetes (HR 1.31, 95% CI 1.19–1.43), chronic bronchitis (HR 1.34, 95% CI 1.19–1.51), and chronic renal failure (HR 1.31, 95% CI 1.19–1.44) (Fig. 4, Supplementary Data 9). These findings align with previous research linking sleep disturbances to common chronic diseases23,24,25,26. In mental disorders, USP was associated with 7 conditions, including anxiety disorder and substance-related disorders. Among neurological diseases, USP was associated with an increased risk of migraine, abnormal movement, and abnormality of gait. Additionally, USP was associated with diagnosed sleep apnea (HR 1.70, 95% CI 1.45–1.99) and sleep disorders (HR 1.55, 95% CI 1.34–1.81), indicating the interconnectedness of poor sleep patterns.
We also expanded the evidence for associations between sleep profiles and several conditions that have not been extensively studied in previous sleep research. For instance, USP showed significant associations with electrolyte imbalance (HR 1.30, 95% CI 1.17–1.44), acidosis (HR 1.71, 95% CI 1.37–2.15), iron deficiency anemia (HR 1.38, 95% CI 1.24–1.53), blindness and low vision (HR 1.75, 95% CI 1.34–2.27), injury (HR 1.45, 95% CI 1.22–1.71), and fracture of the lower limb (HR 1.37, 95% CI 1.17–1.59).
In stratified analyses of the significant associations, most of the 76 disease phenotypes remained significant in participants aged 65 or older and in male participants (Supplementary Fig. 4, Supplementary Data 10). Among participants younger than 65 years and female participants, associations were directionally concordant with those observed in older adults and males. A subset of these associations did not reach statistical significance after Bonferroni correction, possibly due to the fewer disease events observed in younger and female participant groups. These findings were robust after excluding participants with incomplete covariate data or history of shift work (Supplementary Fig. 5–6, Supplementary Data 11–14).
To examine the individual associations between each of the five sleep domains constituting USP and health outcomes, we fitted restricted cubic spline models for the 526 disease phenotypes. In each model, the factor score of one domain was entered as the primary predictor, adjusting for the other four domains and relevant covariates. Overall, each individual dimension yielded fewer significant findings than USP. Specifically, independent associations were observed for 5 phenotypes with timing, 3 with efficiency, 64 with duration, 19 with rhythmicity, and 21 with regularity (Bonferroni-corrected \(P < 9.51\times {10}^{-5}\)). Later timing was associated with increased risks. Higher sleep efficiency consistently conferred lower risks. For sleep duration, short sleep was consistently associated with adverse health outcomes whereas long sleep was associated with some adverse outcomes. Lower rhythmicity and greater irregularity were broadly linked to adverse health outcomes (Supplementary Fig. 7).
As a sensitivity analysis, we constructed a continuous healthy sleep score by weighting the harmonized factor scores of the five domains according to their proportion of variance explained (Methods). In the PheWAS of 526 phenotypes, we identified 81 significant associations (Bonferroni-corrected \(P < 9.51\times {10}^{-5}\)), 47 of which overlapped with the 76 phenotypes identified using the binary USP (Supplementary Fig. 8). Given that aggregating the five dimensions into a single metric assumes linear relationships with health outcomes, which may not capture more complex, non-linear associations, we present the continuous score as a sensitivity analysis, while the binary USP derived from LPA clustering is used as the primary metric.
We further examined the relationship between USP and mortality risk using Cox regression analyses (Supplementary Data 15). USP was associated with increased risks of all-cause mortality (HR 1.32, 95% CI 1.22–1.42) and cardiovascular disease (CVD) mortality (HR 1.55, 95% CI 1.32–1.83). The associations remained significant, albeit attenuated, after excluding individuals who died within 1 year of accelerometer monitoring (Supplementary Data 16).
Population attributable fraction of USP
To assess the public health impact of USP, we examined the population attributable fraction (PAF) for all-cause mortality, CVD mortality, and the 76 USP-associated diseases. The PAF for USP was 5.66% for all-cause mortality and 9.25% for CVD mortality. The PAF estimates indicated that 2.3%–12.3% of incident cases across the 76 diseases could be associated with USP (Supplementary Data 17). Blindness and low vision, acidosis, sleep apnea, peripheral vascular disease, and type 2 diabetes with ophthalmic manifestations showed the highest PAFs. We also calculated the number of attributable cases per 100,000 person-years for USP across the 76 disease phenotypes, accounting for both PAF and incidence density. The greatest potential impact of eliminating USP was observed for common diseases including essential hypertension, chronic renal failure, anxiety disorder, pneumonia, osteoarthrosis, iron deficiency anemia, ischemic heart disease, and type 2 diabetes. The top 20 phenotypes with the highest numbers of attributable cases are presented in Table 2.
Upstream environmental factors associated with USP
We studied the associations between environmental factors and sleep profiles, identifying several risk factors significantly associated with USP at the nominal P < 0.05 level (Methods, Supplementary Data 18). Demographic risk factors included older age (adjusted odds ratio (OR) 1.06; 95% CI 1.01–1.11), male gender (OR 1.61; 95% CI 1.54–1.69), and non-white ethnicity (OR 1.99; 95% CI 1.78–2.22). Socioeconomic factors were associated with USP as well; higher education levels were protective (OR 0.94; 95% CI 0.89–0.99), while high deprivation increased the risk (OR 1.23; 95% CI 1.17–1.30). Meanwhile, being married or partnered was associated with lower risk (OR 0.70; 95% CI 0.65–0.76). Among lifestyle factors, both current smoking (OR 1.64; 95% CI 1.51–1.79) and heavy alcohol consumption (>28 standard drinks/week: OR 1.21; 95% CI 1.13–1.30) were significantly associated with increased risk of USP. Obesity exhibited a strong association with increased risk (OR 1.94; 95% CI 1.83–2.05), as did winter season (OR 1.15; 95% CI 1.08–1.23). We observed distinct seasonal variations in specific sleep characteristics. Sleep timing was phase-advanced in spring, accompanied by the highest sleep efficiency. Summer was characterized by the shortest sleep duration, while winter, despite being associated with longer sleep duration, showed evidence of circadian misalignment and poorer rhythmicity (Supplementary Data 19). Self-reported sleep measures not captured by accelerometer, including sleep apnea, daytime sleepiness, and ease of getting up, were also associated with USP.
USP polygenic score and USP-linked phenotypes
To investigate whether the associations between USP and various phenotypes could be explained by shared genetic components, we conducted a genome-wide association study (GWAS) for USP and derived a polygenic score (PGS) based on the identified genome-wide significant variants. We then assessed the associations between the USP PGS and 76 phenotypes previously found to be linked with USP at the phenotypic level. These analyses followed the same modeling approach and covariate adjustments as used in the initial USP PheWAS, ensuring consistency in the analytical framework. Among the tested phenotypes, 32 remained significantly associated with the USP PGS after Bonferroni correction (P < 6.6 × 10⁻⁴), covering domains such as metabolic disorders, cardiovascular conditions, respiratory diseases, and mental health traits (Supplementary Fig. 9, Supplementary Data 20).
Mendelian randomization analysis of USP and USP-linked phenotypes
To further assess the potential causal relationships between USP and its associated phenotypes, we performed two-sample Mendelian randomization (MR) analyses. Using whole-genome sequencing data from 83,347 individuals in the UK Biobank, variants associated with USP at a significance threshold of P < 5 × 10⁻⁶ were identified via the STAARpipeline27, resulting in 362 significant variants. After linkage disequilibrium clumping, 16 independent variants were retained as genetic instruments.
Using the inverse variance weighted (IVW) method to evaluate the potential causal effect of USP on 76 USP-linked phenotypes, we observed suggestive evidence for associations with five outcomes: other chronic nonalcoholic liver disease, iron deficiency anemias, anxiety disorder, iron deficiency anemias (unspecified or not due to blood loss), and migraine (Supplementary Fig. 10, Supplementary Data 21).
Sensitivity analyses using alternative MR methods yielded generally consistent effect directions with the IVW estimates. No significant heterogeneity (Cochran’s Q test P > 0.05) or horizontal pleiotropy (MR Egger intercept P > 0.05) was detected for these five outcomes, suggesting the validity of the instrumental variable assumptions. The strength of the instruments was supported by mean F-statistics exceeding 20 across analyses, indicating a low risk of weak instrument bias (Supplementary Data 22). Leave-one-out analyses further confirmed the robustness of these findings (Supplementary Fig. 11–15).
Common and rare genetic variants associated with USP
To investigate the genetic architecture of USP, we first performed a functionally informed analysis to detect coding variants associated with USP, analyzing whole-exome sequencing (WES) data from 80,007 individuals in the UK Biobank through STAARpipeline27 (Methods). This analysis included single variant association analysis of individual autosomal variant (minor allele count [MAC] ≥ 40) and variant set analysis of aggregated rare autosomal variants (minor allele frequency [MAF] < 1%) association. For rare variant analysis, STAARpipeline provides gene-centric analyses of seven coding functional categories of protein coding genes (Methods). In the single variant analysis, none achieved the genome-wide significant associations at a level of \(5\times {10}^{-9}\) (Fig. 5a, b). In the gene-centric coding analysis, a genome-wide significant association was detected between protein-truncating (PTV) and disruptive missense RVs in TTC1 and USP at the Bonferroni-corrected level \(0.05/{{\mathrm{20,000}}}=2.50\times {10}^{-6}\) (Methods, Fig. 5c, d, Supplementary Data 23). All 25 PTV and disruptive missense RVs in TTC1 were extremely rare variants with MAC < 5, indicating the associations were driven by the aggregated effect of multiple RVs and were independent of any previously reported sleep health-related common variants (Supplementary Data 24).

a Manhattan plots for WES single variant analysis. The horizontal line indicates the genome-wide significant level of \(5\times {10}^{-9}\). b Quantile-quantile plots for WES single variant analysis. c Manhattan plots for WES gene-centric coding analysis. The horizontal line indicates a genome-wide STAAR-O P-value threshold of \(2.5\times {10}^{-6}\). The significant threshold is defined by multiple comparisons using the Bonferroni correction (\(0.05/{{\mathrm{20,000}}}=2.50\times {10}^{-6}\)). d Quantile-quantile plots for WES gene-centric coding analysis. e Manhattan plots for WGS single variant analysis. The horizontal line indicates the genome-wide significant level of \(5\times {10}^{-9}\). f Quantile-quantile plots for WGS single variant analysis. g Manhattan plots for WGS gene-centric noncoding analysis. The horizontal line indicates a genome-wide STAAR-O P-value threshold of \(2.5\times {10}^{-6}\). The significant threshold is defined by multiple comparisons using the Bonferroni correction (\(0.05/{{\mathrm{20,000}}}=2.50\times {10}^{-6}\)). h Quantile-quantile plots for WGS gene-centric noncoding analysis. i Manhattan plots for WGS ncRNA analysis. The horizontal line indicates a genome-wide STAAR-O P-value threshold of \(2.5\times {10}^{-6}\). The significant threshold is defined by multiple comparisons using the Bonferroni correction (\(0.05/{{\mathrm{20,000}}}=2.50\times {10}^{-6}\)). j Quantile-quantile plots for WGS ncRNA analysis. In panels, (a−i) the chromosome number are indicated by the colors of dots. In all panels, STAAR-O is a two-sided test. USP: unfavorable sleep profile; WES: whole exome sequencing; WGS: whole genome sequencing; STAAR-O: omnibus test in the STAAR framework.
We then performed a functionally informed genetic analysis to detect noncoding variants associated with USP, using whole-genome sequencing (WGS) data from 35,556 individuals in the UK Biobank through STAARpipeline (Methods). Analogous to the coding genome analysis, we performed single variant analysis of variants with MAC ≥ 40, and gene-centric noncoding analysis of RVs with MAF < 1%, including seven noncoding functional categories of protein coding genes, as well as one noncoding functional category of ncRNA genes (Methods). For each variant set, we further incorporated 12 in-silico variant functional annotation scores that prioritize functional variants using multidimensional variant biological functions through the STAAR framework28 to enhance analytical power (Supplementary Data 25). The single variant analysis identified two intronic variants, rs113851554 and rs11679120 in the MEIS1 gene that reached genome-wide significance level of \(5\times {10}^{-9}\) (Fig. 5e, f, Supplementary Data 26). These two variants are previously reported to be associated with sleep health traits, such as, sleep duration29 and insomnia complaints30. In gene-centric noncoding analysis of USP, although no noncoding associations of protein-coding genes achieved genome-wide significance at the level of \(2.50\times {10}^{-6}\), the association between enhancer RVs overlaid with DNase Hypersensitivity (DHS) sites of CDK8 and USP approached significance with a P-value of \(3.19\times {10}^{-6}\), nearly reaching the defined threshold (Fig. 5g, h). The most significant variant of enhancer DHS RVs of CDK8 gene with USP, rs140365310, had an individual \(P=7.58\times {10}^{-3}\), indicating that the association was driven by the cumulative effects of multiple enhancer DHS RVs (Supplementary Data 27). The gene-centric noncoding analysis of ncRNA genes did not detect any significant associations at the genome-wide significance level \(2.50\times {10}^{-6}\) (Fig. 5i, j).
Sensitivity analyses of genome-wide significant loci from the primary analyses, excluding shift workers or individuals reporting sleep or psychiatric medication usage, and further adjusting for potential confounders (Methods), did not substantially alter the effect estimates of the identified signals (Supplementary Data 28, 29).
External validation of the USP framework
We analyzed actigraphy data from an independent cohort, the Multi-Ethnic Study of Atherosclerosis (MESA), to evaluate the external validity of the USP framework. Factor analysis captured the same five latent sleep dimensions (Supplementary Fig. 16). The factor structure and directionality of the loadings were consistent with those from the UK Biobank (Supplementary Data 30–33). The LPA also identified USP (Supplementary Data 34), consistent with the UK Biobank findings, with 470 (21.8%) participants in the MESA classified as USP, similar to the 18.9% observed in the UK Biobank. Individuals in USP exhibited later sleep timing, lower sleep efficiency, shorter sleep duration, weaker rhythmicity, and less regular sleep patterns, mirroring the patterns observed in the UK Biobank (Supplementary Fig. 17). We further examined USP associations with incident CVD events and mortality in MESA. While some results did not reach statistical significance, possibly due to MESA’s smaller sample size and shorter follow-up (mean: 4.9 years), all HR point estimates exceeded 1.0 and aligned with the UK Biobank findings, supporting the generalizability of the USP framework (Supplementary Data 35, 36).
Discussion
In this study, we analyzed objective accelerometer-derived sleep data from 85,233 UK Biobank participants and identified five sleep domains: timing, efficiency, duration, rhythmicity, and regularity. We developed the USP, a multidimensional sleep marker integrating these five domains to reflect real-world sleep patterns. Our analysis revealed that 18.9% of the participants exhibited USP, characterized by later sleep timing, lower sleep efficiency, shorter sleep duration, weaker rhythmicity, and greater sleep irregularity. Over an average follow-up period of 7.9 years, USP was associated with all-cause and CVD mortality, together with 76 out of 526 disease phenotypes (14%) across the human phenome. These associations were particularly pronounced in older adults and men. Furthermore, by transitioning to a Favorable Sleep Profile, an estimated 2.3% to 12.3% of cases across the 76 USP-associated diseases could potentially be prevented, as estimated by the PAF. We investigated the upstream environmental risk factors and examined the genetic architecture of USP. A few environmental factors were found to be related to USP, such as low socioeconomic status and obesity. Genetic analyses revealed that common variants in MEIS1 and rare coding variants in TTC1 were associated with USP. External validation in the MESA replicated the five-domain structure and identified a similar USP, supporting the applicability of the USP framework to diverse populations of adults in midlife and older age.
Recent studies have applied the RU-SATED model to investigate multidimensional sleep health31,32,33,34. However, the representative sleep characteristics used to define each dimension vary across studies, complicating the reproducibility of findings. The RU-SATED model outlines six dimensions of sleep health, including regularity, satisfaction, alertness/sleepiness, timing, efficiency, and duration. Among these, satisfaction can only be measured through self-report, while the other dimensions can be assessed using both subjective and objective measures. Napping plays an important role in overall sleep health and alertness. However, napping behavior is highly variable and can be influenced by a variety of factors, including individual sleep patterns, cultural norms, and the availability of time to nap4. As a result, without concurrent sleep diary entries during accelerometer wear, napping could not be reliably detected. Although baseline napping data are available in the cohort, they were self-reported and were collected ~5.7 years before accelerometer assessment for over 90% of participants, making them unsuitable for integration into the current analytic framework. Future studies may benefit from using concurrent sleep diary to collect information including nap-related behaviors alongside accelerometer monitoring, enabling a more accurate and comprehensive assessment of daytime sleepiness, total sleep duration, and rhythmicity.
We additionally identified rhythmicity as a distinct sleep health dimension, extending beyond prior studies and aligning with recent calls to incorporate circadian rhythm characteristics into the multidimensional sleep health framework6,7. The rhythmicity domain was characterized by established parametric and non-parametric measures that assess fragmentation and synchronization35. Accelerometer-based circadian rhythm measurement is based on the principle that movement increases during wake periods and decreases during sleep, a validated approach35. The recognition of rhythmicity advances the multidimensional framework of sleep health by incorporating the regulation of both homeostatic processes and the circadian system, a 24 h rhythm that regulates most physiological systems3. Although our study did not capture satisfaction and sleepiness dimensions, the composite USP, derived from five objectively measured dimensions, showed significant associations with diagnosed sleep disorders and future health risks. This suggests that USP provides a comprehensive measure of sleep health with potential for standardization and clinical application.
By integrating multiple dimensions of sleep, USP effectively captures the combined impacts of various sleep patterns. Our findings showed that USP was associated with multiple chronic diseases, advancing beyond previous studies that examined relationships between self-reported single sleep variables and specific chronic conditions36,37. Notably, our analyses identified associations between USP and both coronary atherosclerosis (HR 1.20, 95% CI 1.09–1.32, \(P=1.22\times {10}^{-4}\)) and type 2 diabetes (HR 1.31, 95% CI 1.19–1.43, \(P=1.40\times {10}^{-8}\)), associations that were not detected in a previous study of 6,785 adults utilizing commercial wearable devices to analyze six single-dimension sleep metrics15. The inclusion of rhythmicity characteristics in our analytic framework may have enhanced our ability to detect these associations, which aligned with established links between circadian disruption and type 2 diabetes38, as well as known circadian regulation of major cardiovascular functions25,39. Our sensitivity analyses provided further support, showing that poor rhythmicity alone was significantly associated with higher risks of heart failure and type 2 diabetes. In addition, insufficient sleep duration, another key dimension of USP, was independently linked to elevated risks of these cardiometabolic conditions. These results are consistent with prior evidence on the adverse health consequences of sleep deprivation. Sleep loss impairs insulin and glucose homeostasis, disrupts metabolic hormones such as leptin and ghrelin, alters adipose tissue function, and contributes to the development of type 2 diabetes mellitus37. Moreover, elevated cardiovascular risk has been linked to hyperactivation of the sympathetic nervous system and increased oxidative stress in response to insufficient sleep duration40.These results highlight the value of incorporating multiple sleep dimensions, including rhythmicity, in capturing complex sleep-health relationships.
The PheWAS found USP extensively associated with a broad spectrum of diseases, including mental, neurological, endocrine/metabolic, hematopoietic, infectious, respiratory, and cardiovascular diseases. USP’s associations with worsened mental health outcomes aligned with previous research on sleep’s role in cognitive function through neurophysiological changes and memory consolidation3,41,42,43,44,45. In addition to cognitive pathways, evidence also supports sleep’s essential role in emotion regulation. Sleep disruption not only worsens existing psychiatric conditions but also independently increases the risk of anxiety, depression, bipolar disorder, substance use disorders, and suicide3. These impairments in emotional regulation, recognition, and reasoning are thought to arise from neurophysiological alterations in cortical and subcortical structures affected by acute or chronic sleep deprivation41. Moreover, sleep disorders not only increase the risk of stroke and worsen patient outcomes but are also prevalent among stroke patients, highlighting the critical link between sleep and neurocognitive health1,46. Our results also supported the connections between poor sleep and metabolic diseases, as sleep disturbances affect glucose homeostasis and hormone regulation37. The significant associations between USP and various infectious and hematopoietic diseases reinforced previous evidence that sleep is crucial for immune function and that sleep deprivation induces a proinflammatory state47,48,49. Notably, both USP and the rhythmicity domain were associated with septicemia and urinary tract infection, whereas the other four domains showed no such associations. This may be explained by the role of circadian disruption in impairing immune function and increasing susceptibility to infection48. Our findings on USP’s relationship with cardiovascular diseases corroborated earlier research linking sleep patterns to conditions such as myocardial infarction and hypertension50. In respiratory health, USP was associated with conditions such as respiratory insufficiency, obstructive chronic bronchitis, and chronic airway obstruction, reflecting sleep’s influence on ventilation and respiratory control through impaired respiratory muscle function and increased upper airway resistance51,52,53.
Beyond well-documented sleep-health relationships, our analyses revealed novel associations between USP and less frequently studied conditions. For instance, USP was associated with an increased risk of electrolyte imbalances, possibly due to sleep-related alternations in chemoreceptor sensitivity to hypoxia and hypercapnia, leading to hypercapnia52,53. In addition, USP was associated with increased risk of lower limb fractures (HR 1.37, 95% CI 1.17–1.59,\(P=4.69\times {10}^{-5}\)) and injuries (HR 1.45, 95% CI 1.22–1.71, \(P=1.54\times {10}^{-5}\)). These risks may be partially explained by established mechanisms linking sleep deficiency to cognitive-motor impairments, including slowed reaction times and poor judgment, which have been associated with elevated risks of both workplace accidents and unintentional injuries3. These findings underscore the broader implications of sleep health, extending beyond chronic diseases to include critical considerations for public safety and injury prevention strategies.
Although prior literature has frequently reported associations between sleep disorders and both obesity and depression54,55,56,57, neither condition was reported among the USP-linked phenotypes in the present study. In our prospective analyses, USP was associated with an increased risk of incident obesity (HR = 1.10; 95% CI: 1.02–1.19; P = 0.017). However, this association did not meet the Bonferroni-corrected significance threshold and was therefore not highlighted among the primary findings. For depression, only 24 incident cases occurred at least 1 year after accelerometer wear, which did not meet the threshold for inclusion in the prospective analyses. Both conditions warrant further investigation in future research.
Given that sleep profiles affect health through multiple pathways, identifying upstream determinants holds significant potential for improving sleep and overall health. Historically marginalized populations face heightened risks from adverse environmental and social factors affecting health, including sleep health, across both developing and developed nations58. Our findings reflect these disparities, showing that individuals with higher Townsend deprivation index (TDI), lower levels of education, and those without partners were more likely to exhibit USP. While previous research has linked caffeine and alcohol to fragmented, poor-quality sleep59, this study found no evidence that caffeine or tea intake increases the risk of USP. These divergent findings may arise from differences in sleep profile definitions and changes in lifestyle habits over time. We also observed significant associations of smoking, overweight/obesity, and heavy drinking (>28 standard drinks/week) with USP. Current smoking was associated with a higher risk of USP, and nicotine, known to cause sleep fragmentation, may be a potential mechanism underlying this association60. These findings suggest potential sleep health benefits from weight management, reducing excessive alcohol intake, and smoking cessation. We also observed seasonal variations in sleep characteristics. Our findings align with previous studies, such as the observation of shorter sleep duration during summer months61. These seasonal differences may be driven by environmental factors, such as temperature, humidity, and photoperiod, which are known to influence sleep quality and circadian regulation4. Additionally, we found significant associations between self-reported sleep apnea, sleepiness, and ease of getting up with USP. This support the hypothesis that sleep patterns across different dimensions are interrelated and may influence each other synergistically62. Hence, investigating comprehensive sleep patterns is both valuable and essential for a thorough understanding of sleep health.
While our initial analyses identified phenotypic correlates of USP, prior evidence has suggested that these associations may be partially underpinned by shared genetic factors. Large-scale genomic studies of insomnia and related sleep traits have reported substantial polygenic overlap with psychiatric disorders56. Building on this evidence, we examined the polygenic architecture of USP and its pleiotropic relationships. Notably, the USP PGS showed significant associations with anxiety disorders, aligning with known genetic correlations between sleep and psychiatric conditions55,56,57. These findings indicate that shared genetic factors likely drive a substantial portion of the observed USP-phenotype associations. Mendelian randomization analyses further supported potential causal links, particularly suggesting USP as a contributor to anxiety risk.
These population-level genetic insights guided our subsequent variant discovery. We characterized the genetic architecture of USP through a functionally informed association analysis by integrating multiple variant functional annotations with WES and WGS data, utilizing single variant analysis and rare variant analysis of comprehensive coding and noncoding units provided in STAARpipeline. Two intronic variants in gene MEIS1, rs113851554 and rs11679120, have been detected to be associated with USP at genome-wide significance level. Previous research has recognized MEIS1 as a susceptibility gene for restless legs syndrome, a common sleep disorder63. We also detected associations that have not been previously reported for sleep health in genetic studies, including PTV and disruptive missense RVs in TTC1 (\(P=1.47\times {10}^{-6}\)), and enhancer DHS RVs of CDK8 (\(P=3.19\times {10}^{-6}\)). Previous studies have demonstrated that TTC1 is associated with depressive and anxiety disorders through its interaction with CRY264. Furthermore, the stability of CRY2, which is regulated by FBXL3, plays a crucial role in controlling human sleep-wake behavior65. This evidence supports our findings that TTC1 is linked to sleep health. CDK8 is a colorectal cancer oncogene66, but there are no studies that focus on its role in sleep health.
This study has several limitations. First, the observational design precludes establishing causal relationships between sleep profiles and health outcomes. However, we strengthened the analyses by investigating longitudinally the associations between USP and incident diseases to establish temporality, and applied the phecode scheme exclusion criteria to the control cohort to minimize contamination. We also limited enrollment to individuals whose first diagnostic event occurred at least 1 year after accelerometer assessment to address potential reverse causation. In addition, we conducted MR analyses to explore potential causal relationships, providing complementary evidence. Second, potential volunteer bias may affect generalizability, as participants in the accelerometer study may differ from non-participants. Nevertheless, similar sleep dimensions have been identified in other studies using accelerometer, with results replicated across diverse groups without significant gender differences6, suggesting that our multidimensional sleep measures are likely representative. Third, although we validated the USP framework in an independent multi-ethnic cohort (MESA), both UK Biobank and MESA participants were predominantly older adults. Given that sleep patterns can vary across the lifespan, future research is needed to characterize multidimensional sleep profiles in younger populations. Fourth, the up to 7 day accelerometer data may not capture long-term sleep patterns. While previous research has suggested that some sleep characteristics tend to remain relatively stable in adults over periods of several years15, those characteristics did not encompass all the sleep variables included in USP. Additionally, though we accounted for many covariates including seasonality, other unobserved factors (e.g., subclinical disease) may also have influenced sleep patterns. Fifth, while actigraphy provides practical sleep-wake estimates based on movement patterns, its indirect nature and reliance on algorithmic processing, including parameters choices for classifying transitions between wakefulness and sleep and procedures for non-wear detection, might introduce measurement variability. We followed GGIR protocols used in previous UK Biobank studies67,68. These analytical choices, though empirically validated, may influence parameter estimates and should be corroborated with complementary neurophysiological measures in future research. Finally, the use of Electronic Health Record (EHR) diagnosis codes may result in incomplete case ascertainment.
This study offers several key strengths. Our proposed multidimensional sleep health metric USP can serve as a foundation for future sleep health research by capturing the combined effects of real-world sleep patterns. As one of the largest studies analyzing objectively measured sleep patterns, our findings provide robust evidence with clinical relevance. We performed comprehensive evaluations of the associations between integrated sleep behaviors and a broad range of future health outcomes using longitudinal data, advancing beyond previous cross-sectional studies. Furthermore, our whole-genome sequence analysis, incorporating both common and rare variants (coding and noncoding), expands understanding of sleep health’s genetic architecture.
In summary, this study emphasizes the importance of considering the multidimensional nature of sleep health and its extensive impact on well-being. Considering that sleep profiles affect health via multiple mechanisms, implementing large-scale screening and sleep promotion programs may lead to considerable public health gains. The genetic architecture underlying our defined sleep profile provides insights into the biological foundations of sleep health and potential therapeutic strategies.
Methods
Study participants
The UK Biobank is a large prospective cohort study of over 500,000 middle-aged adults in Great Britain69. The initial assessment was conducted during 2006–2010. The UK Biobank has received approval from the North West Multi-centre Research Ethics Committee (MREC) as a Research Tissue Bank (RTB). Written informed consent was obtained from all participants. Between May 2013 and December 2015, about 240,000 participants were invited to wear the Axivity AX3 wrist-worn triaxial accelerometer on their dominant wrists for seven consecutive days. A total of 103,720 participants returned their activity monitors with data covering at least three complete 24-h periods. After excluding those who withdrew from the study, we obtained raw activity monitor data for 103,626 participants (data-field 90001) in the form of binary Continuous Wave Accelerometer (cwa) files. After applying data quality control procedures recommended by the UK Biobank accelerometer working group and following data processing approaches used in previous studies based on UK Biobank accelerometry data, a total of 85,233 participants were retained for the final analysis67,68 (Supplementary Fig. 18).
For the whole-exome and whole-genome sequence analyses (UK Biobank Field #23158 and #24304), a series of quality control procedures were implemented to filter out low-quality variants and samples (see the corresponding section below for details). We did not use any study design that required randomization or blinding.
Accelerometer-based sleep variables
The GGIR R package was utilized to extract raw accelerometer data, enabling thorough quantitative assessments of sleep patterns70. This tool uses an algorithm validated against polysomnograms in an external cohort to accurately detect sleep periods without the need for a sleep diary, thus minimizing potential biases11. Median z-angle changes over 5-min rolling windows across a 24-h period were calculated to ensure activity monitor orientation insensitivity. Inactivity bouts lasting ≥30 min were documented, with consecutive bouts <60 min apart combined into blocks. The sleep period time-window (SPT-window) was defined by the onset and conclusion of the longest continuous inactivity block.
Building on the SPT, variables such as sleep onset time, wake-up time, sleep midpoint, sleep duration, and sleep efficiency were derived. Utilizing the time series of raw accelerometer data and an extended cosine model, circadian rhythm analyses were conducted to obtain metrics such as intra-daily variability (IV), inter-daily stability (IS), relative amplitude (RA), amplitude, acrophase, midline estimating statistic of rhythm (MESOR), and pseudo-F71,72. Detailed definitions and significance of the sleep variables extracted in this study are provided in Supplementary Data 37. Sleep variables derived from the SPT-window underwent thorough screening to exclude outliers, ensuring the inclusion of valid data for statistical analysis. Based on the data quality metrics provided by the UK Biobank accelerometer working group, the exclusion criteria were as follows: (1) data not well calibrated; (2) data with poor wear time; (3) unreliable data size; (4) data affected by daylight savings crossover; (5) data not calibrated on own data; (6) data with interrupted recording periods >0; and (7) data with excessively numerous recording errors (>Q3 + 1.5 × IQR). Individuals were additionally excluded if their mean sleep duration was shorter than 3 h or longer than 12 h, if they had fewer than 5 or >30 mean sleep episodes per night, or if they had fewer than 3 valid days of data67,68. Detailed configuration parameters for GGIR used in this study can be found in Supplementary Data 38.
Although accelerometry algorithms provide daily sleep-wake characteristics, we focused on summary measures across the observation period to examine habitual sleep patterns. These summary measures are commonly used in sleep health research due to their interpretability and their effectiveness as predictors of key health outcomes. As accelerometers only capture lack of movement, GGIR refers to classified sleep periods as sustained inactivity bouts (SIB), making it unable to accurately identify naps. Therefore, in the absence of sleep diary entries during accelerometer wear, nap-related variables cannot be extracted. We selected accelerometer summary variables for EFA based on their relevance to current research on sleep health and their clinical or scientific significance6,72.
Phenotypes and mortality
This work used data provided by patients and collected by the NHS as part of their care and support. We leveraged the full available historical EHR data spanning multiple decades to identify the earliest diagnosis dates for each participant and phenotype. Diagnosis events were assessed in subjects starting 1 year after accelerometer monitoring through linkage to EHRs. Events extracted from hospital inpatient data, death register, and cancer register were categorized into phenotypes (phecodes) using their corresponding International Classification of Diseases (ICD) codes with the Phecode Map 1.221,22. The phecode map provides exclusion criteria for each phenotype, identifying similar conditions that may suggest the likelihood of undiagnosed patients with the phenotype under consideration. An example of applying phecode exclusion criteria is demonstrated in a type 2 diabetes study using EHRs21. To define cases of type 2 diabetes, patients with ICD codes mapping to phecode 250.2 “Type 2 diabetes” were included. For the control cohort, only participants without phenotypes in the “Diabetes” group (phecodes 249-250.99) were included, preventing contamination by diseases such as “Type 1 diabetes” (phecode 250.1) and “Secondary diabetes mellitus” (phecode 249). Additionally, participants with signs and symptoms commonly associated with type 2 diabetes, like “Abnormal glucose” (phecode 250.4), were excluded to avoid including those who may be undiagnosed. In this study, all subjects meeting any exclusion criteria for a phenotype were excluded from the analysis of that phenotype to avoid including likely or potential prior cases. Detailed exclusion criteria for each selected phenotype are provided in Supplementary Data 39. Subjects excluded from one phecode analysis were not excluded from others unless they also met the exclusion criteria for those phecodes.
Among the 502,250 participants in the UK Biobank, we identified 1695 specific phecodes. Of the 85,233 subjects meeting the inclusion criteria for this study, we retained samples where genetic sex matched the recorded sex and excluded individuals with mismatched sex-specific diagnosis codes or those who withdrew. Consequently, 1633 disease phenotypes were extracted from EHRs at the time of data download on January 8, 2024, and grouped according to the phecode map, with an average follow-up of 7.92 years. To exclude participants with subclinical disease at the time of accelerometer monitoring, we limited enrollment to those whose first diagnostic event occurred at least 1 year after accelerometer wear, resulting in 526 phecodes with at least 200 cases.
Date of death and primary cause of death were obtained through linkage to national death registries. Cardiovascular disease (CVD) mortality was defined by ICD-10 codes: I00-I99.
Covariates
Most participants completed a touchscreen questionnaire and underwent anthropometric assessments at initial recruitment. Subsequently, a subset of included participants in the present study also engaged in first repeat assessments, imaging visit, and first repeat imaging visit (n = 7464, n = 22,822, and n = 2611, respectively). The time differences and number of participants between accelerometry and the four recruitment assessments for the included study participants are detailed in Supplementary Data 40 and Supplementary Fig. 19. Covariates included age, sex, ethnicity (white or non-white), TDI, education (no qualification, any other qualification, degree or above), diet (poor, ideal), physical activity, smoking status (never, previous, current), alcohol consumption (never, previous, current), self-rated health (excellent/good, fair/poor), body mass index (BMI), and season (spring for March to May, summer for June to August, autumn for September to November, and winter for December to February; UK Meteorological Office definitions) (Supplementary Data 41). A healthy diet was defined as consuming at least 5 out of 10 recommended food groups9,73. Intake goals for each food group are detailed in Supplementary Data 42. BMI categories were classified as normal/underweight (<25 kg/m²), overweight (25–30 kg/m²), and obese (≥30 kg/m²). Physical activity was quantified using accelerometer-derived moderate-to-vigorous physical activity (MVPA) duration in minutes, with a threshold of 100 mg for MVPA classification.
Covariate data from the interview conducted closest in time prior to the accelerometry assessment were used as the baseline for this analysis. Exceptions included self-reported sex and TDI, which were obtained only at baseline, as well as self-reported ethnicity (assumed unchanged). Smoking status, alcohol consumption, and education remained stable between baseline and additional visits, while diet, self-rated health, and BMI exhibited some variability over time (Supplementary Figs. 20–22). In the final analysis, time-varying covariates comprised 90.53% from the initial assessment, 7.94% from the first repeat assessment, and 1.53% from the imaging visit (Supplementary Data 43). The number and combinations of missing covariates are shown in Supplementary Data 44 and Supplementary Fig. 23. The highest proportion of missing covariates does not exceed 1% (0–0.74%). We performed multiple imputations to assign missing covariate values using the mice package in R.
Statistical analyses
EFA was employed to extract representative sleep domains using selected sleep variables from accelerometer. We used the principal function in the psych package with a “quartimax” rotation (an orthogonal rotation), without imposing specific domains on the data. The number of factors was determined by examining eigenvalues, visually inspecting scree plots, and considering our hypotheses. Variables were assigned to a factor if they had a loading >0.50. If a variable loaded on multiple factors, we assigned it to the factor with the highest loading. If the highest and second highest loadings were close, we considered the factor’s interpretability for the assignment. Subsequently, we used the standardized factor scores (mean of 0 and standard deviation of 1) derived from the factor analysis as new variables for LPA using the mclust package in R to identify distinct sleep patterns in the participants. The number of latent profiles was selected based on the integrated completed likelihood criterion.
In the PheWAS analysis, a Cox proportional hazards model was conducted for each phecode with at least 200 cases. This threshold was determined through a power analysis via simulation to detect a 0.2 log (hazard ratio) effect size with 80% power74. The endpoint was the diagnosis of the phenotype, with censoring by death or the end of data collection. The timescale used was years since the accelerometer measurement. The independent variable was the sleep profile identified from LPA. To test the proportionality of hazards assumption, Schoenfeld residuals were examined using the cox.zph function. For variables displaying significant (P < 0.05) non-constant violations, a second accelerated failure time model was rerun and compared with the original model to determine the optimal fit. Bonferroni and FDR adjusted significance thresholds were utilized. To check for sex- and age-specific effects, we further conducted subgroup analyses based on sex and age (<65 years, ≥65 years).
To examine the associations between upstream environmental factors and sleep profiles, we used multivariable logistic regression analysis with USP as the dependent variable.
We also conducted several sensitivity analyses. First, we analyzed the relationship between USP and outcomes in a population without missing covariates. Second, we further excluded individuals with a history of shift work from the complete data set. Third, to address potential reverse causality, we excluded individuals who died within 1 year after wearing the accelerometer and analyzed the relationship between sleep profile and risks of all-cause and CVD mortality. Fourth, we applied restricted cubic spline models to examine the shape and strength of associations between each of the five sleep domains constituting USP and 526 disease phenotypes, allowing for potential non-linear relationships. Fifth, we derived a continuous healthy sleep score by linearly combining the harmonized factor scores of the five USP domains, weighted by their proportion of variance explained in the factor analysis. We then conducted PheWAS using Cox models to estimate hazard ratios per standard deviation increase in the healthy sleep score, with covariate adjustment consistent with the binary USP analyses. Finally, follow-up analyses of genome-wide significant loci from the primary analyses were conducted, excluding shift workers and users of sleep or psychiatric medications. These analyses incorporated additional covariate adjustments for smoking status, alcohol consumption, marital status, education, season of accelerometer wear, TDI, BMI, sleep apnea, sleepiness, and ease of getting up, in addition to the initial adjustments for age, sex, and 10 principal components75,76,77. Definitions of additional covariates and the list of medications can be found in the Supplementary Note. All P-values were two-sided and analyses were conducted using R v4.3.3.
Calculating PGS
PGS for USP were derived using individual-level genotype data from the UK Biobank dataset ukb22418. Variant-level quality control (QC) was performed using PLINK, excluding SNPs with minor allele frequency (MAF) < 1%, minor allele count (MAC) < 100, genotype missingness >10%, or Hardy-Weinberg equilibrium \(P < 1\times {10}^{-15}\). Individuals with >10% missing genotypes were also excluded.
Following QC, the dataset was randomly partitioned into a training set (80%) for GWAS and a tuning set (20%) for PGS parameter optimization. GWAS was conducted in the training set using Regenie78 under a logistic regression framework with Firth correction to account for case-control imbalance. GWAS summary statistics from this analysis were used to construct the USP PGS using a clumping and thresholding (C + T) approach implemented in RICE79. Linkage disequilibrium (LD) clumping was performed in PLINK80 with an r² threshold of 0.1 within a 500 kb window. PRSs were generated at nine GWAS P-value thresholds: \(P < 5\times {10}^{-8}\), \(5\times {10}^{-7}\), \(5\times {10}^{-6}\), \(5\times {10}^{-5}\), \(5\times {10}^{-4}\), \(5\times {10}^{-3}\), 0.05, 0.5, and 1.0. The optimal P-value threshold (\(P < 0.05\)) for inclusion of SNPs in the final score was selected based on performance in the tuning set. The final PGS was then computed in the study cohort and used to assess the association between polygenic risk of USP and phenotypes identified in the PheWAS analysis.
MR
We conducted two-sample MR analyses using the TwoSampleMR81 R package to investigate the potential causal effects of USP-associated genetic variants on the USP-linked phenotypes. Genetic instruments for the exposure (USP) were derived from our single variant analysis based on whole genome sequencing (WGS) data from the UK Biobank (Field #24304), as described below. For the outcomes, GWAS summary statistics were downloaded from the GWAS Catalog. To ensure consistency with the disease phenotype definitions used in our PheWAS, we selected outcome GWASs based on phecode-matched traits and included only those conducted in European ancestry populations82. Genetic variants reaching a P < 5 × 10⁻6 threshold in the USP WGS single variant analysis were first clumped to ensure independence (r² < 0.01 within 10 Mb, using 1000 Genomes EUR as the reference panel). The alleles of variants were harmonized between the exposure and outcome datasets using the default allele alignment method (action = 2) to account for palindromic variants based on allele frequencies.
For each of the phenotype, we applied five MR methods: IVW, MR Egger, weighted median, weighted mode, and simple mode. IVW served as the primary analysis method, while the others were used as sensitivity analyses to assess the robustness of the results under different assumptions about instrument validity. We further conducted heterogeneity testing using Cochran’s Q test, evaluated horizontal pleiotropy via the MR Egger intercept, and calculated the mean F-statistic to assess instrument strength. Additionally, leave-one-out analyses were performed to identify influential variants that might disproportionately affect the results.
Whole exome sequence analysis of coding variants associated with USP
We used the PLINK format files for WES data of UK Biobank participants (UK Biobank Field #23158). Quality control measures were performed in the following steps. We first removed the variants with Hardy-Weinberg Equilibrium \(P < 1\times {10}^{-15}\). Second, we removed variants for which > 10% of all genotypes for that variant had a read depth < 10 (ukb23158_500k_OQFE.90pct10dp_qc_variants.txt). We finally excluded the variants with > 10% missing genotypes and the samples with 10% missing genotypes.
We used STAARpipeline to perform genetic association analysis of USP. STAARpipeline is a regression-based framework that allows for adjustment of covariates, population structure, and relatedness by fitting linear and logistic mixed models for quantitative and dichotomous traits27,28. Specifically, we fitted a logistic mixed model adjusting for age, sex, the first 10 ancestral principal components to account for population structure, and a variance component for a sparse genetic relatedness matrix to account for sample relatedness83.
For single variant analysis, we calculated individual P-values of variants with MAC ≥ 40. We first used the normal approximation to calculate the P-value, and when it is <0.05, we applied the saddlepoint approximation to recalculate it84,85. The gene-centric coding analysis of variants, including both single-nucleotide variants (SNVs) and indels, provided seven coding functional categories of protein coding genes, including putative loss of function (stop gain, stop loss and splice) variants, missense variants, disruptive missense variants, putative loss of function and disruptive missense variants, synonymous variants, protein-truncating RVs (stop gain, stop loss, splice, frameshift deletion and frameshift insertion), and protein-truncating RVs and disruptive missense RVs. The putative loss of function, missense, synonymous, and protein-truncating RVs were defined by GENCODE VEP categories86,87. The disruptive variants were further defined by MetaSVM88, which measures the deleteriousness of missense mutations. For each variant set, we calculated the STAAR-Burden P-value. Same as the single variant analysis, we first used the normal approximation to calculate the P-value, and when it is <0.05, we applied the saddlepoint approximation to recalculate it.
Whole genome sequence analysis of noncoding variants associated with USP
We used the pVCF format files for WGS data of UK Biobank participants (UK Biobank Field #24304)89. We followed the same quality control procedure in previous study of UK Biobank WGS data90. We kept all variants with pass indicated by QC label and AAScore >0.5, where AAScore was generated by GraphTyper, the software used by the UK Biobank to perform genotype calling.
We used STAARpipeline27 to perform genetic association analysis of USP. We fitted the null model in the same way as the WES analysis. For single variant analysis, we calculated individual P-values of variants with MAC ≥ 40. The gene-centric noncoding analysis provided eight genetic categories of SNVs, including promoter or enhancer overlaid with CAGE or DHS sites, UTR, upstream, downstream of protein coding genes, and noncoding RNA genes. The promoter RVs were defined as RVs in the +/- 3-kilobase (kb) window of transcription start sites with the overlap of CAGE sites or DHS sites. The enhancer RVs were defined as RVs in GeneHancer predicted regions with the overlap of CAGE sites or DHS sites91,92,93,94. We defined the UTR, upstream, downstream, and ncRNA RVs by GENCODE Variant Effect Predictor (VEP) categories86,87. For the UTR mask, we included RVs in both 5’ and 3’ UTR regions. For the ncRNA mask, we included the exonic and splicing ncRNA rare SNVs. We considered the protein-coding gene for the first seven categories provided by Ensembl95 and the ncRNA genes provided by GENCODE86,87. We incorporated nine annotation Principal Components (aPCs)28 and three integrative scores (CADD96, LINSIGHT97, and FATHMM-XF98) as weights in constructing STAAR-Burden statistics28.
Genome build
All genome coordinates are given in NCBI GRCh38/UCSC hg38.
Replication of the USP framework
To further examine the robustness and generalizability of the USP framework, we validated our findings in an independent multi-ethnic cohort, the MESA (BioLINCC Application ID: 15959). We selected the MESA cohort for validation due to its diverse demographic composition and independent data collection setting, which provided a meaningful test of the framework’s robustness across different populations and measurement environments. This cohort included 2237 participants who wore wrist-worn actigraphy devices (Actiwatch Spectrum, Philips Respironics) for up to seven consecutive days between 2010–2012. All recordings were scored by trained technicians at the Boston Sleep Reading Center. Institutional Review Boards of all participating MESA sites approved the study, and all participants signed informed consent. The validation analysis included 2152 participants aged 54–93 years with complete data. The sample comprised 37.4% white, 11.3% Chinese, 27.8% African American, and 23.4% Hispanic individuals.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings from this study are available within the manuscript and its supplementary information. The UK Biobank data were obtained under application 91486 (release date: 8 Jan 2024). The MESA data were obtained under application 15959 (BioLINCC Application ID). The functional annotation data are publicly available at the Functional Annotation of Variant-Online Resource (FAVOR) site99 (https://favor.genohub.org) and the FAVOR database100 (https://doi.org/10.7910/DVN/1VGTJI). Source data are provided with this paper.
Code availability
GGIR is implemented as an open-source R package available at https://github.com/wadpac/GGIR. GGIR v3.0.2 was used to extract raw accelerometer data. createUKBphenome is implemented as an open-source R package available at https://github.com/umich-cphds/createUKBphenome. createUKBphenome was used to extract and map ICD-coded hospital records in the UK Biobank to phecodes, enabling harmonized case/control phenotypes for phenome-wide association analyses. vcf2agds is implemented as a collection of applets in the UK Biobank Research Analysis Platform (RAP) available at https://github.com/drarwood/vcf2agds_overview. vcf2agds was used to preprocess the UK Biobank WGS data. PLINK2 is implemented as part of the swiss-army-knife applet in the UK Biobank RAP available at https://ukbiobank.dnanexus.com/panx/tool/app/swiss-army-knife. PLINK2 was used preprocess the UK Biobank WES data. STAAR is implemented as an open-source R package available at https://github.com/xihaoli/STAAR. STAARpipeline is implemented as an open-source R package available at https://github.com/xihaoli/STAARpipeline, and as an applet in the UK Biobank RAP available at https://github.com/xihaoli/staarpipeline-rap. STAARpipelineSummary is implemented as an open-source R package available at https://github.com/xihaoli/STAARpipelineSummary, and as an applet in the UK Biobank RAP available at https://github.com/li-lab-genetics/staarpipelinesummary_varset-rap and https://github.com/li-lab-genetics/staarpipelinesummary_indvar-rap. STAAR v0.9.7, STAARpipeline v0.9.7, and STAARpipelineSummary v0.9.7 were used for performing WES and WGS data analysis in the UK Biobank RAP. The following tools were used to construct the USP PGS: Regenie v4.1 https://rgcgithub.github.io/regenie; PLINK 1.9 https://www.cog-genomics.org/plink; and RICE https://github.com/jwilliams10/RareVariantPRS/blob/main/DNANexus/CT_Binary.R. R package TwoSampleMR v0.6.14 (https://mrcieu.github.io/TwoSampleMR) was used to conduct the two-sample MR analyses.
References
Grandner, M. A. & Fernandez, F. X. The translational neuroscience of sleep: a contextual framework. Science 374, 568–573 (2021).
Lloyd-Jones, D. M. et al. Life’s essential 8: updating and enhancing the American Heart Association’s construct of cardiovascular health: a presidential advisory from the American Heart Association. Circulation 146, e18–e43 (2022).
Lim, D. C. et al. The need to promote sleep health in public health agendas across the globe. Lancet Public health 8, e820–e826 (2023).
Nieto, F. J. & Petersen, D. Foundations of Sleep Health. https://www.thensf.org/foundations-of-sleep-health/ (2021).
Buysse, D. J. Sleep health: can we define it? does it matter?. Sleep 37, 9–17 (2014).
Wallace, M. L. et al. Multidimensional sleep health domains in older men and women: an actigraphy factor analysis. Sleep 44, zsaa181 (2021).
Wallace, M. L. et al. which sleep health characteristics predict all-cause mortality in older men? an application of flexible multivariable approaches. Sleep 41, zsx189 (2018).
Svensson, T. et al. Association of sleep duration with all- and major-cause mortality among adults in Japan, China, Singapore, and Korea. JAMA Netw. Open 4, e2122837 (2021).
Han, H. et al. Sleep duration and risks of incident cardiovascular disease and mortality among people with type 2 diabetes. Diabetes Care 46, 101–110 (2023).
Matthews, K. A. et al. Similarities and differences in estimates of sleep duration by polysomnography, actigraphy, diary, and self-reported habitual sleep in a community sample. Sleep. health 4, 96–103 (2018).
van Hees, V. T. et al. Estimating sleep parameters using an accelerometer without sleep diary. Sci. Rep. 8, 12975 (2018).
Fischer, D., Klerman, E. B. & Phillips, A. J. K. Measuring sleep regularity: theoretical properties and practical usage of existing metrics. Sleep 44, zsab103 (2021).
Phillips, A. J. K. et al. Irregular sleep/wake patterns are associated with poorer academic performance and delayed circadian and sleep/wake timing. Sci. Rep. 7, 3216 (2017).
Qin, S., Leong, R. L. F., Ong, J. L. & Chee, M. W. L. Associations between objectively measured sleep parameters and cognition in healthy older adults: a meta-analysis. Sleep. Med. Rev. 67, 101734 (2023).
Zheng, N. S. et al. Sleep patterns and risk of chronic disease as measured by long-term monitoring with commercial wearable devices in the all of us research program. Nat. Med. 30, 2648−2656 (2024).
Lane, J. M. et al. Genetics of circadian rhythms and sleep in human health and disease. Nat. Rev. Genet 24, 4–20 (2023).
Wainschtein, P. et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet 54, 263–273 (2022).
Plenge, R. M., Scolnick, E. M. & Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 12, 581–594 (2013).
Plenge, R. M. Disciplined approach to drug discovery and early development. Sci. Transl. Med. 8, 349ps315 (2016).
Szustakowski, J. D. et al. Advancing human genetics research and drug discovery through exome sequencing of the UK biobank. Nat. Genet. 53, 942–948 (2021).
Wu, P. et al. Mapping ICD-10 and ICD-10-CM codes to phecodes: workflow development and initial evaluation. JMIR Med. Inform. 7, e14325 (2019).
Wei, W. Q. et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PloS ONE 12, e0175508 (2017).
Cappuccio, F. P., D’Elia, L., Strazzullo, P. & Miller, M. A. Quantity and quality of sleep and incidence of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 33, 414–420 (2010).
Gangwisch, J. E. et al. Short sleep duration as a risk factor for hypertension: analyses of the first national health and nutrition examination survey. Hypertension 47, 833–839 (2006).
Fan, M. et al. Sleep patterns, genetic susceptibility, and incident cardiovascular disease: a prospective study of 385 292 UK biobank participants. Eur. heart J. 41, 1182–1189 (2020).
Joglar, J. A. et al. 2023 ACC/AHA/ACCP/HRS Guideline for the diagnosis and management of atrial fibrillation: a report of the American College of Cardiology/American Heart association joint committee on clinical practice guidelines. Circulation 149, e1–e156 (2024).
Li, Z. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat. Methods 19, 1599–1611 (2022).
Li, X. et al. Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet 52, 969–983 (2020).
Doherty, A. et al. GWAS identifies 14 loci for device-measured physical activity and sleep duration. Nat. Commun. 9, 5257 (2018).
Hammerschlag, A. R. et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat. Genet 49, 1584–1592 (2017).
Wallace, M. L. et al. Heightened sleep propensity: a novel and high-risk sleep health phenotype in older adults. Sleep. health 5, 630–638 (2019).
Dong, L., Martinez, A. J., Buysse, D. J. & Harvey, A. G. A composite measure of sleep health predicts concurrent mental and physical health outcomes in adolescents prone to eveningness. Sleep. Health 5, 166–174 (2019).
DeSantis, A. S. et al. A preliminary study of a composite sleep health score: associations with psychological distress, body mass index, and physical functioning in a low-income African American community. Sleep. health 5, 514–520 (2019).
Wallace, M. L. et al. Multidimensional sleep and mortality in older adults: a machine-learning comparison with other risk factors. J. Gerontol. Ser. A. Biol. Sci. Med. Sci. 74, 1903–1909 (2019).
Di, J. et al. Joint and individual representation of domains of physical activity, sleep, and circadian rhythmicity. Stat. Biosci. 11, 371–402 (2019).
Ayas, N. T. et al. A prospective study of sleep duration and coronary heart disease in women. Arch. Intern Med. 163, 205–209 (2003).
Koren, D. & Taveras, E. M. Association of sleep disturbances with obesity, insulin resistance and the metabolic syndrome. Metab. Clin. Exp. 84, 67–75 (2018).
Stenvers, D. J., Scheer, F., Schrauwen, P., la Fleur, S. E. & Kalsbeek, A. Circadian clocks and insulin resistance. Nat. Rev. Endocrinol. 15, 75–89 (2019).
Van Laake, L. W., Lüscher, T. F. & Young, M. E. The circadian clock in cardiovascular regulation and disease: Lessons from the Nobel Prize in Physiology or Medicine 2017. Eur. heart J. 39, 2326–2329 (2018).
Tobaldini, E. et al. Short sleep duration and cardiometabolic risk: from pathophysiology to clinical evidence. Nat. Rev. Cardiol. 16, 213–224 (2019).
Goldstein, A. N. & Walker, M. P. The role of sleep in emotional brain function. Annu Rev. Clin. Psychol. 10, 679–708 (2014).
Klinzing, J. G., Niethard, N. & Born, J. Mechanisms of systems memory consolidation during sleep. Nat. Neurosci. 22, 1598–1610 (2019).
Irwin, M. R. & Vitiello, M. V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 18, 296–306 (2019).
Insel, P. S., Mohlenhoff, B. S., Neylan, T. C., Krystal, A. D. & Mackin, R. S. Association of sleep and β-amyloid pathology among older cognitively unimpaired adults. JAMA Netw. Open 4, e2117573 (2021).
Helakari, H. et al. Human NREM sleep promotes brain-wide vasomotor and respiratory pulsations. J. Neurosci. 42, 2503–2515 (2022).
Bassetti, C. L. A. et al. EAN/ERS/ESO/ESRS statement on the impact of sleep disorders on risk and outcome of stroke. Eur. Respir. J. 55, 1901104 (2020).
Prather, A. A., Pressman, S. D., Miller, G. E. & Cohen, S. Temporal links between self-reported sleep and antibody responses to the influenza vaccine. Int J. Behav. Med. 28, 151–158 (2021).
Besedovsky, L., Lange, T. & Haack, M. The sleep-immune crosstalk in health and disease. Physiol. Rev. 99, 1325–1380 (2019).
Besedovsky, L., Lange, T. & Born, J. Sleep and immune function. Pflug. Arch. 463, 121–137 (2012).
James, S. M., Honn, K. A., Gaddameedhi, S. & Hpa, V. D. Shift work: disrupted circadian rhythms and sleep-implications for health and well-being. Curr. Sleep. Med. Rep. 3, 104 (2017).
McNicholas, W. T., Hansson, D., Schiza, S. & Grote, L. Sleep in chronic respiratory disease: COPD and hypoventilation disorders. Eur. Respir. Rev. 28, 190064 (2019).
Dempsey, J. A., Veasey, S. C., Morgan, B. J. & O’Donnell, C. P. Pathophysiology of sleep apnea. Physiol. Rev. 90, 47–112 (2010).
Phillipson, E. A. Control of breathing during sleep. Am. Rev. Respir. Dis. 118, 909–939 (1978).
Chaput, J. P. et al. The role of insufficient sleep and circadian misalignment in obesity. Nat. Rev. Endocrinol. 19, 82–97 (2023).
Watanabe, K. et al. Genome-wide meta-analysis of insomnia prioritizes genes associated with metabolic and psychiatric pathways. Nat. Genet 54, 1125–1132 (2022).
Madrid-Valero, J. J. & Gregory, A. M. Behaviour genetics and sleep: a narrative review of the last decade of quantitative and molecular genetic research in humans. Sleep. Med. Rev. 69, 101769 (2023).
Jansen, P. R. et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet 51, 394–403 (2019).
Slopen, N., Lewis, T. T. & Williams, D. R. Discrimination and sleep: a systematic review. Sleep. Med. 18, 88–95 (2016).
Baranwal, N., Yu, P. K. & Siegel, N. S. Sleep physiology, pathophysiology, and sleep hygiene. Prog. Cardiovasc Dis. 77, 59–69 (2023).
Wetter, D. W. & Young, T. B. The relation between cigarette smoking and sleep disturbance. Prev. Med. 23, 328–334 (1994).
Obradovich, N., Migliorini, R., Mednick, S. C. & Fowler, J. H. Nighttime temperature and human sleep loss in a changing climate. Sci. Adv. 3, e1601555 (2017).
Vyazovskiy, V. V., Walton, M. E., Peirson, S. N. & Bannerman, D. M. Sleep homeostasis, habits and habituation. Curr. Opin. Neurobiol. 44, 202–211 (2017).
Winkelmann, J. et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat. Genet 39, 1000–1006 (2007).
Kovanen, L., Kaunisto, M., Donner, K., Saarikoski, S. T. & Partonen, T. CRY2 genetic variants associate with dysthymia. PloS one 8, e71450 (2013).
Hirano, A. et al. A Cryptochrome 2 mutation yields advanced sleep phase in humans. Elife 5, e16695 (2016).
Firestein, R. et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455, 547–551 (2008).
Jones, S. E. et al. Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat. Commun. 10, 1585 (2019).
Jones, S. E. et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 10, 343 (2019).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Migueles, J. H., Rowlands, A. V., Huber, F., Sabia, S. & van Hees, V. T. GGIR: A Research community–driven open source R package for generating physical activity and sleep outcomes from multi-day raw accelerometer data. J. Meas. Phys. Behav. 2, 188–196 (2019).
Van Someren, E. J. et al. Bright light therapy: improved sensitivity to its effects on rest-activity rhythms in Alzheimer patients by application of nonparametric methods. Chronobiol. Int. 16, 505–518 (1999).
Yang, L. et al. Association of accelerometer-derived circadian abnormalities and genetic risk with incidence of atrial fibrillation. NPJ Digit. Med. 6, 31 (2023).
Said, M. A., Verweij, N. & van der Harst, P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK biobank study. JAMA Cardiol. 3, 693–702 (2018).
Brooks, T. G. et al. Diurnal rhythms of wrist temperature are associated with future disease risk in the UK biobank. Nat. Commun. 14, 5172 (2023).
Lane, J. M. et al. Biological and clinical insights from genetics of insomnia symptoms. Nat. Genet 51, 387–393 (2019).
Wang, H. et al. Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat. Commun. 10, 3503 (2019).
Lane, J. M. et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat. Genet 49, 274–281 (2017).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet 53, 1097–1103 (2021).
Williams, J. et al. Integrating common and rare variants improves polygenic risk prediction across diverse populations. medRxiv https://doi.org/10.1101/2024.11.05.24316779 (2024).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–d1012 (2019).
Lin, X., Dey, R., Li, X. & Li, Z. Scalable analysis of large multi-ancestry biobanks by leveraging sparse ancestry-adjusted sample-relatedness. Res. Sq. 12, rs.3.rs-5343361 (2024).
Jiang, L., Zheng, Z., Fang, H. & Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet 53, 1616–1621 (2021).
Dey, R., Schmidt, E. M., Abecasis, G. R. & Lee, S. A fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am. J. Hum. Genet 101, 37–49 (2017).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–d773 (2019).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet 24, 2125–2137 (2015).
Li, X. et al. Streamlining large-scale genomic data management: insights from the uk biobank whole-genome sequencing data. medRxiv 28, 2025.01.27.25321225 (2025).
Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740 (2022).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Forrest, A. R. et al. A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Kinsella, R. J. et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxf.) 2011, bar030 (2011).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014).
Huang, Y. F., Gulko, B. & Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet 49, 618–624 (2017).
Rogers, M. F. et al. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics 34, 511–513 (2018).
Zhou, H. et al. FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res. 51, D1300–d1311 (2023).
Zhou, H., Arapoglou, T., Li, X., Li, Z. & Lin, X. FAVOR Essential Database (Harvard Dataverse, 2022).
Acknowledgements
This study was conducted using the UK Biobank Resource (Application ID: 91486) and data from the Multi-Ethnic Study of Atherosclerosis (MESA) Sleep (BioLINCC Application ID: 15959). We thank all participants and investigators involved in UK Biobank and MESA. This work was supported by grant 24DTR055 from the Beijing Philosophy and Social Science Foundation (X.Y.L.).
Author information
Authors and Affiliations
Contributions
X.Y.L., Z.L. and X.H.L. designed the study. X.Y.L. acquired data. S.Z. and Z.L. conducted the main analysis, X.Y.L., Z.L., X.H.L., and S.Z. interpreted data. S.Z., Z.L., X.H.L. and X.Y.L. drafted the manuscript. M.Z. and Y.Y. contributed to the data collection and analysis. All authors critically reviewed the manuscript, suggested revisions as needed, and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tiina Paunio, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, S., Zhang, M., Yuan, Y. et al. Health risks and genetic architecture of objectively measured multidimensional sleep health. Nat Commun 16, 7026 (2025). https://doi.org/10.1038/s41467-025-62338-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62338-0
