
Abstract
Lipoylation is a highly conserved post-translational modification (PTM) crucial for energy metabolism enzymes, with distinct pathways across organisms. Whereas bacteria like Escherichia coli inherit both salvage and de novo pathways, only the latter is found in eukaryotes. Here, we present a PTM-based strategy that achieves multiple metabolic benefits with a single intervention. By expressing E. coli-derived lipoate protein ligase A (LplA) from the salvage pathway, we enhance lipoylation and the activities of the pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase complexes and glycine cleavage system in mammalian, algal and fungal cells, leading to improved energy metabolism, cofactor supply, mitochondrial function, and overall cell physiology. Our approach specifically targets multiple metabolic hubs through PTM modulation. Beyond its fundamental significance, our finding presents a unified and efficient way to boost biosynthesis across organisms, demonstrated in antibody production in Chinese hamster ovary cells, fatty acids synthesis in cyanobacteria and diatoms, and organic acid production in fungi.
Similar content being viewed by others

Introduction
Protein lipoylation is a crucial and highly conserved post-translational modification (PTM) of key energy metabolism enzymes in bacteria, fungi, plants, animals and humans1,2. To date, four enzyme complexes are well known to have lipoylation modification, including pyruvate dehydrogenase (PDH), alpha-ketoglutarate dehydrogenase (OGDH), branched-chain alpha-ketoacid dehydrogenase (BCKDH), and H protein in the glycine cleavage system (GCSH)3,4. Additionally, several species-specific lipoylated proteins have been identified, which are closely related to the aforementioned proteins. For example, 2-oxoadipate dehydrogenase (OADH) in humans shares the same lipoylation domain in the E2 subunit of OGDH, which catalyzes the lysine degradation in mitochondria5. Acetoin dehydrogenase (AODH), found in bacteria of the Firmicutes and Proteobacteria phyla, is homologous to PDH, using acetoin as substrate instead of pyruvate6. Lipoate-binding proteins (LbpA2) in Hyphomicrobium denitrificans function as key components in the Hdr-like pathway of sulfur oxidation7. Among them, PDH and OGDH regulate the entry of different substrates, such as glucose, glycerol and glutamine, into the central metabolic pathway of tricarboxylic acid (TCA) cycle, followed by oxidative phosphorylation (OXPHOS) for energy generation3. Especially, PDH activity directly affects the distribution of carbon fluxes towards either the energetically efficient TCA cycle or the overflow metabolism, a phenomenon manifesting as acetate production in bacteria, the Crabtree effect in yeast and the Warburg effect in mammals8. It has been reported that the absence of lipoylation results in a notable decrease in enzyme activities, leading to impaired energy metabolism and diseases3,9. Besides, lipoylation has garnered attention due to its association with aberrant energy metabolism and cuproptosis10,11. Excessive copper leads to abnormal aggregation of lipoylated proteins, which disrupts energy production and ultimately results in cell death10. Therefore, lipoylation is crucial in maintaining energy metabolism and cellular activities in both prokaryotes and eukaryotes. However, there are few studies on enhancing energy metabolism and biosynthesis through modification of the lipoylation pathway.
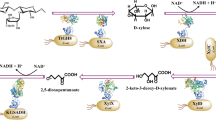
It is known that lipoylation can be catalyzed by a conserved de novo pathway via multi-enzymes from octanoyl-ACP in both bacteria and eukaryotes2 (Fig. 1a). In Escherichia coli and algae, octanoyl transferase, known as LipB or Lip2, and lipoate synthase, called LipA or Lip1, are responsible for transferring the octanoyl moiety to the protein lipoyl domain and inserting two sulfur atoms to form a complete lipoamide prosthetic group2. Yeast and mammalian cells require an extra lipoyl transferase, referred to as Lip3 or LIPT1, to transfer the lipoyl moiety from H protein to other proteins to be lipoylated1. Apart from the de novo pathway, bacteria like E. coli possess a so-called salvage pathway, which uses lipoate protein ligase A (LplA) to specifically catalyze the lipoylation with free lipoic acid12 (Fig. 1a). Paredes et al. suggested that the enzymes acetyl-coenzyme A synthetase medium-chain family member 1 (ACSM1) and LIPT1 can function jointly as LplA to catalyze a salvage pathway of lipoylation from lipoic acid in mammalian cells13. However, the addition of lipoic acid alone cannot recover lipoylation defects in yeast or mammalian cells caused by the absence of octanoyl transferase, lipoate synthase or lipoyl transferase14,15, suggesting the lack of a salvage pathway in these organisms in accordance with the literature. Engineering of energy metabolism is a major endeavor of metabolic engineering and synthetic biology. In general, engineering of multiple specific enzymes or metabolic pathways are required to improve energy metabolism, cofactor supply and biosynthesis16,17,18. The tuning and regulation of such complicated multiple pathways are challenging. Unlike other PTMs such as phosphorylation, methylation and acylation, lipoylation is highly conserved and targets several specific energy metabolic-related enzymes1. Therefore, modulation of lipoylation is a promising and potentially efficient PTM-based approach to engineer chassis cells for enhanced energy metabolism and biosynthesis.

a The de novo synthetic pathways and salvage pathway of lipoylation in E. coli, algae, yeast and mammals. b E2 subunits of PDH and OGDH are lipoylated to catalyze the decarboxylation of pyruvate and alpha-ketoglutarate (AKG). c Immunoblots assaying lipoylation of purified, recombinant DLAT and DLST proteins derived from CHO cells in vitro. Data were representative of at least three independent experiments. d Immunoblots assaying lipoylation of purified, recombinant E2 subunits of PDH, OGDH, and GCSH proteins derived from algae in vitro. Data were representative of at least three independent experiments. e Immunoblots assaying lipoylation of purified, recombinant E2 subunits of PDH protein derived from higher plants in vitro. Data were representative of at least three independent experiments. f Immunoblots assaying lipoylation of purified, recombinant Lat1 and Kgd2 proteins derived from yeast S. cerevisiae in vitro. Data were representative of at least three independent experiments. g Sequence logos generated via WebLogo for the conserved lipoyl domain in 17 PDH-E2 and 14 OGDH-E2 proteins from animal, yeast, higher plant, green algae, and cyanobacteria, bacteria (Supplementary Data 2). The conserved lipoylated lysine “K” is marked by a red arrow. h Sequence logos generated via WebLogo for the conserved lipoyl domain in 15 GCSH proteins from animal, yeast, higher plant, green algae, and cyanobacteria, bacteria (Supplementary Data 2). The conserved lipoylated lysine “K” is marked by a red arrow. Immunoblots assaying lipoylation in vitro were performed using lipoic acid and His-tag antibodies as primary antibodies (c–f). ACP acyl-carrier-protein, LDP lipoylation-dependent proteins, LipB/Lip2/LIPT2 octanoyl transferase, LipA/Lip1/LIAS lipoate synthase, Lip3/LIPT1 lipoyl transferase, LplA lipoate protein ligase A, PDH pyruvate dehydrogenase, AKG alpha-ketoglutarate, OGDH alpha-ketoglutarate dehydrogenase, DLAT/Lat1 dihydrolipoamide acetyltransferase, DLST/Kgd2 dihydrolipoamide succinyltransferase. Source data are provided as a Source Data file.
In this study, we show that the exogenous introduction of the bacterial small protein LplA can simultaneously modulate the activities of enzymes within well-defined central energetic pathways in mammalian, algae and fungal cells. This intervention enhances energy metabolism, mitochondrial function, cell growth and biosynthesis in the aforementioned organisms to inhabit only the de novo lipoylation pathway.
Results
E. coli-derived LplA enables the use of lipoic acid for salvage lipoylation in multiple species in vitro
To investigate whether LplA from E. coli has the ability to stimulate the lipoylation of energy metabolism proteins in eukaryotes and cyanobacteria, in vitro lipoylation assays of the E2 subunits of PDH, OGDH and GCSH (Fig. 1b) in multiple organisms were performed. Heterologous substrate proteins (Supplementary Data 1) are predominantly produced in an inactive state when expressed in E. coli BL21 (DE3) (Fig. 1c–f). This is due to the low endogenous concentration of LplA, which is insufficient to catalyze the lipoylation. In detail, the purified dihydrolipoamide acetyltransferase (DLAT) and dihydrolipoamide succinyltransferase (DLST) from CHO cells are much more effectively lipoylated using E. coli-derived LplA (Fig. 1c). Similarly, PDH-E2 protein from P. tricornutum and S. elongatus can also be significantly lipolyzed by the additional E. coli-derived LplA in the presence of lipoic acid (Fig. 1d and Supplementary Fig. 1a, c). Furthermore, E2 subunits of mitochondrial PDH from five common plant model species (Zea mays, Arabidopsis thaliana, Glycine max, Nicotiana tabacum, Oryza sativa) were separately assayed for lipoylation in vitro. Immunoblotting results exhibit that LplA can also significantly increase the lipoylation level of these five E2 subunits of PDH in the presence of lipoic acid (Fig. 1e and Supplementary Fig. 1e–i). Besides, LplA enables the lipoylation of Lat1 and Kgd2 proteins from S. cerevisiae (Fig. 1f). The above in vitro lipoylation assay across mammalian, algal and fungal proteins demonstrates the universal functionality of LplA at the protein level. Furthermore, multiple sequence alignment of PDH-E2 and OGDH-E2 proteins from bacteria, yeast, algae, higher plants and animals reveals a highly conserved lipoylation region, which mechanistically implies that LplA can catalyze the salvage lipoylation of PDH-E2 and OGDH-E2 proteins across species (Fig. 1g and Supplementary Data 2). Especially, in vitro assay shows that, in addition to PDH-E2 protein, E. coli-derived LplA can also catalyze the lipoylation of GCSH protein in algae (Fig. 1d and Supplementary Fig. 1b, d), which is essential for the oxidative cleavage of glycine into CO2, reducing power and one-carbon (1C) unit and photorespiration. Similar to PDH-E2 and OGDH-E2 proteins, GCSH proteins from bacteria, yeast, algae, higher plants and animals also possess a highly conserved lysine “K” in the lipoylation region (Fig. 1h).
Exogenous introduction of E. coli LplA enhances lipoylation level in multiple species in vivo
Though PDH is also present in plastids in algae, the eukaryotic lipoylated proteins, including PDH, OGDH, GCSH and BCKDH, are primarily located in mitochondria. Therefore, LplA is targeted for exogenous expression in the mitochondrial matrix across multiple species through a mitochondrial targeting sequence (MTS) (Supplementary Data 3). In mammalian cells, MTS of ornithine transcarbamylase (hsaOTC) and superoxide dismutase 2 (cgeSOD2) were employed in U2OS and CHO cells, respectively. Immunoblots were performed to confirm the successful expression of exogenous genes (Supplementary Fig. 2a, b). Immunofluorescence staining examined by confocal microscopy was performed to assess the colocalization of exogenously expressed LplA with mitochondria (Fig. 2a, c). The observation shows that exogenous MTS-GFP and MTS-GFP-LplA proteins are successfully expressed in the mitochondrial matrix both in U2OS and CHO cells. Without MTS, the GFP-LplA protein exhibits a diffuse distribution in the cytoplasm (Fig. 2c). The immunoblotting analysis demonstrates that the lipoylation levels of DLAT and DLST are significantly increased with mitochondrial-targeted LplA overexpression in both U2OS and CHO cells (Fig. 2b, d). Differently, the overexpression of endogenous LIPT1 cannot enhance lipoylation levels of DLAT and DLST in U2OS cells (Supplementary Fig. 2c). When LplA is expressed in cytoplasm without MTS, the lipoylation level dose not differ from that of the control groups (Fig. 2d). Additionally, the exogenous expression of LplA3M, which lacks lipoylation function due to mutations at three residues (N121A, D122A, and K133A), cannot catalyze the salvage pathway of lipoylation in vivo, resulting in no change in lipoylation levels of the targeted proteins (Fig. 2d). Due to the requirement of lipoic acid as a substrate for the salvage lipoylation pathway, the intracellular lipoic acid level in CHO cells was assessed. The result confirms that the lipoic acid pool is sufficient for the salvage pathway of lipoylation (Supplementary Fig. 2d, e). Furthermore, the addition of 10 μM lipoic acid to the medium does not affect the lipoylation levels of DLAT and DLST in WT cells (Supplementary Fig. 2f), indicating that mammalian cells lack the salvage pathway. Differently, supplementation with 10 μM lipoic acid enhances the lipoylation levels of DLAT and DLST by over two-fold in the MTS-GFP-LplA group in CHO cells (Supplementary Fig. 2g). These results indicate that targeted expression of LplA can catalyze the salvage lipoylation pathway in vivo.

a Immunofluorescence of LplA localization in the mitochondria of U2OS cells. b Immunoblotting for lipoylated DLAT and DLST in U2OS cells. Relative lipoylation level is normalized to total DLAT and DLST, respectively. Data were represented as mean ± SD (n = 6, n biologically independent samples). Data were representative of at least three independent experiments. c Immunofluorescence of GFP or LplA localization in CHO cells. d Immunoblotting for lipoylated DLAT and DLST in CHO cells. Relative lipoylation level was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data were represented as mean ± SD (n = 9 in MTS-GFP, GFP-LplA and MTS-GFP-LplA groups, n = 8 in WT group, n = 4 in MTS-GFP-LplA3M group, n biologically independent samples). Data were representative of at least three independent experiments. e Immunoblotting for lipoylated PDH-E2, OGDH-E2 and GCSH of P. tricornutum and S. elongatus co-expressed with LplA in E. coli. The relative lipoylation levels are normalized to His-tag. Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. f Immunoblotting for lipoylated Lat1 and Kgd2 in S. cerevisiae. Relative lipoylation level was normalized to voltage-dependent anion channel protein (VDAC). Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. Lipoic acid (b, d–f), GAPDH (b, d, f), and His-tag (e) antibodies are used as primary antibodies for the lipoylation assay. Multiple unpaired two-sided t-test was used for the statistical analyses for (b, e), two-way ANOVA was used for (d, f). P adjustments were made for multiple comparisons. DLAT dihydrolipoamide acetyltransferase, DLST dihydrolipoamide succinyltransferase, PDH pyruvate dehydrogenase, OGDH alpha-ketoglutarate dehydrogenase, GCSH glycine cleavage system H. Source data are provided as a Source Data file.
In P. tricornutum, the signal sequence of N-terminal triosephosphate isomerase (TPI) and a C-terminal GAPDH (alias GapC3) fusion protein (TPI-GapC3) was used as MTS19 and appended to the N-terminal of LplA to direct its mitochondrial import (Supplementary Data 3). Differently, due to the absence of mitochondria in cyanobacteria, exogenous LplA is directly expressed in S. elongatus. Polymerase chain reaction (PCR) and immunoblotting results show that LplA is successfully expressed in both P. tricornutum and S. elongatus (Supplementary Fig. 3a–d). Given that anti-lipoic acid antibody is not applicable in algae, the substrate proteins are co-expressed with LplA in E. coli to verify their feasibility in vivo. The immunoblotting results show that endogenous and exogenous PDH_E2 can be distinguished in crude enzyme extracts of E. coli with a significant increase in lipoylation levels (Supplementary Fig. 3e, i). However, the GCSH protein cannot be distinguished in crude enzyme extracts (Supplementary Fig. 3f). Furthermore, the algae-derived substrate proteins, including PDH_E2, OGDH_E2, and GCSH, were purified and lipoylation assay was performed via immunoblotting. The results demonstrate that the substrate proteins can be significantly lipoylated by LplA from E. coli in the presence of lipoic acid (Fig. 2e and Supplementary Fig. 3g, h). Additionally, the exogenous expression of MTS-GFP, GFP-LplA and MTS-GFP-LplA proteins in yeast S. cerevisiae is examined using immunoblotting (Supplementary Fig. 3j). The lipoylation levels of Lat1 and Kgd2, which are the E2 subunits of PDH and OGDH in yeast, respectively, are significantly improved by 34% and 17% upon target expression of LplA in mitochondria (Fig. 2f).
Overexpression of LplA promotes overall cellular metabolic levels in U2OS cells
In mammals, the lipoyl swinging arms of DLAT and DLST in PDH and OGDH interact with the corresponding E1 and E3 units to facilitate the decarboxylation of pyruvate and alpha-ketoglutarate (α-KG), respectively20,21. Delipoylation of PDH and OGDH has been proven to inhibit their enzymatic activities, resulting in low efficiency of energy generation22,23. To examine the effects of improved lipoylation levels of PDH and OGDH upon exogenous expression of LplA (Fig. 1b), an Agilent Seahorse XFe96 extracellular flux analyzer is employed to measure OXPHOS via oxygen consumption rate (OCR) and glycolysis level via extracellular acidification rate (ECR) in U2OS cells. As shown in Fig. 3a, b, the basal respiration, ATP production, and maximal respiration are significantly enhanced by 48, 67, and 66% in the MTS-GFP-LplA group compared to the WT group, respectively. Besides, the glycolysis level and capacity are also enhanced by 49 and 19%, respectively (Fig. 3b). Additionally, the overexpression of endogenous LIPT1 in U2OS cells cannot enhance mitochondria respiration as well as lipoylation levels of DLAT and DLST in vivo (Supplementary Figs. 4a, 2c), indicating that enhancing endogenous lipoylation pathway through a single enzyme is not feasible.

a Oxygen consumption rate (OCR) represents for mitochondrial respiration in two engineered cells. Data were represented as mean ± SD (n = 6, n biologically independent samples). b Extracellular acidification rate (ECR) represents for glycolysis in two engineered cells. Data were represented as mean ± SD (n = 6, n biologically independent samples). Specific consumption rates of glucose (c), glutamine (d), pyruvate (e), and lactate (f) in U2OS cells after 10 and 18 h cultivation. Data were represented as mean ± SD (n = 6, n biologically independent samples). g Direction of 13C carbon atoms in glycolysis and TCA cycle. Fractional labeling ratio of pyruvate (h), lactate (i), AKG (j), succinate (k), malate (l), and glutamate (m) after 12- and 18-h cultivation of [U-13C] glucose addition. Data were represented as mean ± SD (n = 3, n biologically independent samples). n Heatmap of metabolites abundance in glycolysis, TCA cycle and amino acids biosynthesis after 24-h cultivation of [U-13C] glucose addition. Data were represented as mean ± SD (n = 8, n biologically independent samples). o Immunoblotting for mitochondrial fusion-related proteins. MFN1, MFN2, OPA1, and GAPDH antibodies are used as primary antibodies. Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. p Immunoblotting for mitobiogenesis-related proteins. NRF1, TFAM, and GAPDH antibodies are used as primary antibodies. Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. q Immunoblotting for mTORC1-related proteins. S6, pS6, S6K1, pS6k1, 4EBP1, p4EBP1, and GAPDH antibodies are used as primary antibodies. Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. Multiple unpaired two-sided t-test was used for the statistical analyses for (a–f, h–m, o–q). P adjustments were made for multiple comparisons. AKG alpha-ketoglutarate, Suc succinate, Mal malate, MFN1 mitofusion1, MFN2 mitofusion2, OPA1 optic atrophy 1, NRF1 nuclear respiratory factor 1, TFAM transcription factor A, S6 ribosomal protein S6, pS6 phosphorylated S6, S6K1 S6 kinase 1, pS6k1 phosphorylated S6K1, 4EBP1 4E-binding protein 1, p4EBP1 phosphorylated 4EBP1. Source data are provided as a Source Data file.
To monitor the cell growth and extracellular nutrients consumption during the cultivation, cells cultured for 10 and 18 h are harvested for phenotype detection. The results suggest that cell size is enlarged upon exogenous expression of LplA (Supplementary Fig. 4b). Due to the spatial limitations of adherent culture, a slight increase of cell numbers is observed in MTS-GFP-LplA cell line compared to MTS-GFP group (Supplementary Fig. 4c). Differently, the upregulation of lipoylation levels of DLAT and DLST in the MTS-GFP-LplA group leads to increased activation of PDH and OGDH, driving higher consumption of glucose and glutamine (Fig. 3c, d and Supplementary Fig. 4d, e), providing more substrates for the TCA cycle and increasing mitochondrial respiration (Fig. 3a). This reduces the demand for free pyruvate, resulting in excess pyruvate being re-secreted into the medium (Fig. 3e and Supplementary Fig. 4f). Since U2OS are cancer cells and predominantly utilize aerobic glycolysis for energy production, excess pyruvate is also converted to lactate. This results in the upregulation of lactate production upon exogenous expression of LplA (Fig. 3f and Supplementary Fig. 4g), which is consistent with the upregulation of glycolysis (Fig. 3b).
To further investigate the underlying molecular mechanisms, isotope-tracing experiments using [U-13C] glucose and targeted metabolomics are performed to detect the effect of LplA expression on carbon metabolism (Fig. 3g). The results confirm that exogenous expression of LplA accelerates the glucose metabolism by upregulating the lipoylation level of DLAT, leading to increased labeling ratio and pool size of pyruvate, lactate and citrate, which is consistent with the ECR data (Fig. 3h–k, n). Additionally, OGDH is activated by the upregulated lipoylation of DLST, which not only accelerates the TCA cycle from acetyl-CoA but also drives higher consumption of glutamine to directly convert to AKG (Fig. 3d). The larger pools of succinate, fumarate and malate, along with the smaller pools of glutamate and glutamine (Fig. 3n), demonstrate a more active TCA cycle, leading to higher OCR upon exogenous expression of LplA (Fig. 3a). With the depletion of glutamine in the medium and abundance of substrates in TCA cycle, the excess substrate flow into other pathways, such as glutamate synthesis, which explain the upregulation of labeling ratio of glutamate at 48 h.
Since the glycolysis pathway can be regulated by downstream metabolites and energy status, the expression levels of hexokinases (HK), phosphofructokinases (PFKP), and pyruvate kinases (PK) are examined using an immunoblotting assay. The results show that the expression levels of HK and PFKP have no significant difference (Supplementary Fig. 4i). Specifically, the upregulated expression level and downregulated phosphorylation level of PKM2 suggest increased enzyme activity (Supplementary Fig. 4i), providing more pyruvate for further aerobic glycolysis and the TCA cycle. Given that energy metabolism is closely related to mitochondrial function, immunoblotting analysis was employed to evaluate mitochondrial function and the regulation mechanism. Immunofluorescence staining results show that mitochondria exhibited an elongated morphology in LplA-expressed cells (Fig. 2a), suggesting the occurrence of mitochondrial fusion. This process is critical for maintaining mitochondrial function by diluting damaged mitochondrial DNA, redistributing metabolic intermediates, and improving mitochondrial respiration. Consequently, mitochondrial fusion-related proteins, including mitofusion1 (MFN1), mitofusion2 (MFN2), and optic atrophy 1 (OPA1), were examined by immunoblotting and the results performed significant improvement in the MTS-GFP-LplA group (Fig. 3o). Additionally, the expression level of nuclear respiratory factor 1 (NRF1) and transcription factor A (TFAM) exhibit an increasing trend upon exogenous expression of LplA, indicating that mitochondrial biogenesis is activated (Fig. 3p). These results suggest that overexpression of LplA promotes both mitochondrial fusion and biogenesis, thereby enhancing mitochondrial efficiency and resilience. In addition, the mechanistic target of rapamycin complex 1 (mTORC1) serves as a central regulator of cell growth and metabolism, with its activation intimately linked to cellular energy status and nutrient availability. The phosphorylation levels of 4E-binding protein 1 (4EBP1) and ribosomal protein S6 kinase 1 (S6K1) are significantly increased (Fig. 3q), suggesting the activation of the mTORC1 pathway and subsequent promotion of protein synthesis and cell enlargement.
Adherent and IgG-producing CHO cell line with boosted energy metabolism, cell growth and antibody production
The lipoylation levels of DLAT and DLST are significantly increased in LplA exogenously expressed CHO cells (Fig. 2d). Subsequently, PDH and OGDH activities are both found to increase by over 2.5-folds in MTS-GFP-LplA cells compared to the WT cells (Fig. 4a, b). Higher PDH and OGDH activities allow more substrates to enter the TCA cycle and to increase their fluxes, providing more dihydronicotinamide adenine dinucleotide (NADH) and dihydroflavine-adenine dinucleotide (FADH2) for OXPHOS-mediated ATP formation24. Besides, the expression level of hypoxia-inducible factor 1α (HIF-1α) protein, a transcription factor responding to anoxic environment, is significantly decreased by 29% compared to WT cells (Fig. 4c), suggesting that the activity of the Warburg effect is inhibited25. Seahorse assay of mitochondrial respiration reveals that the exogenous expression of LplA in mitochondria significantly improves OCR in comparison to the WT, MTS-GFP, and GFP-LplA cells. In more detail, the basal respiration, spare respiratory capacity and ATP production are upregulated by 51, 48, and 76%, respectively, in the MTS-GFP-LplA cell line compared to wildtype (Fig. 4d). Reactive oxygen species (ROS) is a byproduct of mitochondrial respiration due to electron leakage from the electron transport chain (ETC)26. Interestingly, the ROS level is significantly reduced in the MTS-GFP-LplA cells, implying an improved mitochondrial environment for energy generation. However, when lipoic acid was added to the medium, mitochondrial respiration decreases as the concentration of lipoic acid increases in MTS-GFP-LplA cells (Fig. 4f), accompanied by a significant increase in ROS level (Fig. 4g). This phenomenon suggests that intracellular lipoic acid is sufficient for the induced salvage lipoylation pathway, and additional lipoic acid bound with LplA imposes a metabolic burden on cells.

PDH activity (a), OGDH activity (b), immunoblotting for HIF-1α protein (c), mitochondrial respiration (d), and reactive oxygen species (ROS) level (e) in WT, MTS-GFP, GFP-LplA, and MTS-GFP-LplA cells. f The mitochondrial respiration of MTS-GFP-LplA cells upon 0, 10, and 20 μM lipoic acid addition in the medium. g The relative ROS level in WT, MTS-GFP, GFP-LplA, and MTS-GFP-LplA cells upon 0, 10, and 20 μM lipoic acid addition in the medium. HIF-1α and GAPDH antibodies are used as primary antibodies, and data were representative of at least three independent experiments in (c). Data were represented as mean ± SD (n = 3 in a, b, e, g, n = 5 in c, n = 8 in d, n = 6 in f, n biologically independent samples). One-way ANOVA was used for the statistical analyses for (a–c, e), two-way ANOVA was used for (d, f, g). P adjustments were made for multiple comparisons. PDH pyruvate dehydrogenase, OGDH alpha-ketoglutarate dehydrogenase, HIF-1α hypoxia-inducible factor 1α, ROS reactive oxygen species. Source data are provided as a Source Data file.
Since CHO cells are the most important mammalian host for manufacturing biopharmaceutical proteins27,28,29, LplA was specifically expressed in the mitochondria of an IgG-producing CHO cell line in suspension culture. Immunoblotting assay demonstrates the successful expression of LplA and LIPT1 in cells (Supplementary Fig. 5a). In batch culture, LplA expression significantly improves the cell growth in the exponential cell growth phase (after about 72 h) (Fig. 5a). The cell viability (VIA) remains consistently above 90% during the entire culture, and it is slightly higher in the MTS-LplA cells compared to the MTS-GFP group (Fig. 5a). The MTS-LplA cells enter the stationary phase at day 7. On day 6, the VCD of MTS-LplA cells reaches a cell density of 1.33 × 107 cells mL−1, which is 41% higher than that in the MTS-GFP cells (Fig. 5a). The specific growth rate of MTS-LplA and MTS-GFP cells is calculated to be 0.025 and 0.022 h−1, respectively, between day 3 and day 6, confirming the enhanced cell growth kinetics upon exogenous expression of LplA in batch culture. The production of antibody (Canakinumab) is monitored from day 4 to day 7. The results show that the overexpression of LplA can also upregulate the production of antibodies during the production phase. On the harvest day 7, the canakinumab titer achieves the highest level of 134.67 mg L−1 in MTS-LplA cells, which is 37% higher than that in the MTS-GFP cells (Fig. 5d). However, the overexpression of endogenous LIPT1 exhibits similar effects to the control group in terms of lipoylation levels, mitochondrial respiration, cell growth and antibody production (Supplementary Fig. 5b–e). These finding is consistent with the results in U2OS cells, indicating that enhancing endogenous lipoylation pathway through a single enzyme is not feasible.

Viable cell density (VCD) and viability (VIA) (a), canakinumab titer (b), immunoblotting for lip-DLAT, and lip-DLST from whole cells (c, d), in MTS-GFP and MTS-LplA cells during the cultivation. The solid and dashed lines in (a) represent VCD and VIA, respectively. The relative lipoylation levels in (c, d) are normalized to GAPDH. Data were represented as mean ± SD (n = 3, n biologically independent samples). e Immunoblotting for lip-DLAT and lip-DLST from isolated mitochondria on day 3. The relative lipoylation levels in (e) are normalized to VDAC. Data were represented as mean ± SD (n = 3, n biologically independent samples). Data were representative of at least three independent experiments. f OCR represents mitochondrial respiration on day 3. Data were represented as mean ± SD (n = 13 in MTS-GFP group, n = 12 in MTS-LplA group, n biologically independent samples). g ROS level in MTS-GFP and MTS-LplA cells during the cultivation. Data were represented as mean ± SD (n = 3, n biologically independent samples). h Mitochondrial membrane potential (MMP) in MTS-GFP and MTS-LplA cells during the cultivation. Data were represented as mean ± SD (n = 3, n biologically independent samples). The cell growth (i), canakinumab titer (j), ATP production (k), spare respiratory capacity (l), and immunoblotting for lipoylation of DLAT and DLST on day 3 (m) in MTS-GFP and MTS-LplA cells upon addition of 0, 1, 10 μM lipoic acid. Data were representative of at least three independent experiments. The relative lipoylation levels in (m) are normalized to GAPDH. Data were represented as mean ± SD (n = 4 in (j), n = 7 in (k, l), n = 4 in (m), n biologically independent samples). Lipoic acid and GAPDH antibodies are used as primary antibodies in (c, e, m). Multiple unpaired two-sided t-test was used for the statistical analyses for (b, f, g, h), two-way ANOVA was used for (e), one-way ANOVA was used for (j–l), unpaired two-sided t-test was used for (c, d, m). P adjustments were made for multiple comparisons. DLAT dihydrolipoamide acetyltransferase, DLST dihydrolipoamide succinyltransferase, VDAC voltage-dependent anion channel. Source data are provided as a Source Data file.
To clarify the relationship between lipoylation level, energy metabolism, and cell performance, cells during the batch culture are collected for immunoblotting and Seahorse Analyzer assays. DLAT is clearly lipoylated since day 1, whereas a clear lipoylation of DLST is observed starting from day 3 (Fig. 5b, c). The expression levels of Lip-DLAT and Lip-DLST proteins are upregulated from day 3 to day 5 in the MTS-LplA cells compared to MTS-GFP cells. Besides, mitochondria from the engineered cell lines are isolated on day 3, when the cell cycle is in an active state, and a lipoylation assay is performed. The lipoylation levels of DLAT and DLST are upregulated by 68 and 80%, respectively, in MTS-LplA cells (Fig. 5e), which is consistent with the lipoylation level of whole cells on day 3 (Fig. 5b, c). Consistent with this phenomenon, the OCR curve of MTS-LplA cells is significantly improved compared to that of MTS-GFP cells on day 3 (Fig. 5f). More specifically, the basal respiration, spare respiration capacity and ATP production increase by 48, 62, and 48% in MTS-LplA cells, respectively (Fig. 5f), compared to the MTS-GFP cells. When the cell cycle is poised to enter a stable phase, the lipoylation level in both groups begins to decline. Notably, the band of lip-DLST is almost disappeared in MTS-GFP cells, resulting in a 3.2-fold increase in Lip-DLST in MTS-LplA cells by day 5 (Fig. 5c). Corresponding to this phenomenon, the OCR curves as well as the OCR-related parameter, including the basal respiration, spare respiration capacity and ATP production, perform similarly in the two engineered cell lines on day 5 (Supplementary Fig. 5f). The reduction of mitochondrial respiration might be associated with the acidification of the medium mediated by lactate accumulation, which inhibited the OXPHOS pathway30. Different from the performance of lipoylation level and mitochondrial respiration, the ROS level exhibits a reduction in MTS-LplA cells throughout the cultivation process (Fig. 5g), which provides potential evidence for the ability of overexpressed LplA to scavenge free radicals in consistence with the intro study of LplA31. Additionally, the mitochondrial membrane potential (MMP) displays an increase trend in MTS-LplA cell line during the cultivation (Fig. 5h). The observed phenomenon confirms that inducing the salvage pathway for lipoylation can greatly augment the lipoylation level and energy metabolism along with improvement of mitochondrial environment. Additionally, different concentrations of lipoic acid at 1 and 10 μM are supplemented to the medium during the cultivation of IgG-producing cell lines. The results show that the addition of lipoic acid restricts cell growth, antibody production and mitochondrial respiration (Fig. 5i–l). Therefore, the PTM strategy of this study can be efficiently and economically implemented in a general medium free of lipoic acid.
13C NMFA reveals upregulation of energy metabolism upon targeted overexpression of LplA
To investigate the intracellular metabolism of CHO cells upon LplA overexpression, [U-13C] glucose is applied at the exponential growth phase (day 3) of a suspension culture (Supplementary Fig. 6a). Similar to the batch culture, the VCD and canakinumab titer significantly increase in MTS-LplA cell line during the cultivation (Supplementary Fig. 6a, b). After the addition of [U-13C] glucose, the fractional labeling rate of glucose is 40 and 35%, respectively, in the cultivation of MTS-LplA and MTS-GFP cells (Supplementary Fig. 6c).
Extracellular fluxes of metabolites in the medium are calculated after 48 h of adding [U-13C] glucose and normalized to the glucose flux in the MTS-GFP group. As shown in Fig. 6a, the consumption flux of glucose is increased by 6% with a 28% decrease in lactate production flux in the MTS-LplA group. The consumption flux of pyruvate has no significant difference between the two engineered cell lines. In contrast, the consumption of other substrates of the TCA cycle, including glutamine and asparagine, shows higher fluxes in the MTS-GFP cell line. Moreover, the other major amino acids in the medium, including glutamate, aspartate, serine, glycine and alanine, do not show significant differences in their consumption or production (Fig. 6b). After 24 h of adding [U-13C] glucose, the metabolites in the glycolysis pathway, such as glucose-6-phosphate (G6P) and 3-phosphoglycerate (3PG) reach the isotopic steady state in both engineered cells (Fig. 6c). While pyruvate and lactate request more than 72 h to reach the isotopic steady state. Differently, intermediates in the TCA cycle have not reached a steady state after 72 h. The labeling dynamics for TCA cycle intermediates perform approximately a 20% increase in MTS-LplA cells compared to MTS-GFP cells. Surprisingly, the normalized percentage of labeled succinate to initial labeled glucose in MTS-LplA cells reaches 29.28% after 48 h of addition of [U-13C] glucose, which is 2.96-fold higher than that in MTS-GFP cells, indicating an increased flux from α-ketoglutarate to succinate upon exogenous expression of LplA. Only six amino acids, including aspartate, alanine, glutamate, glutamine, serine, and glycine, are detected to be labeled after tracer addition. The labeling of these amino acids exhibits a linear increase over a 72-h period and did not reach isotopic steady state in both engineered cell lines. Above all, since metabolites of the TCA cycle and amino acid metabolism pathway do not reach isotopic steady state, non-stationary 13C MFA is applied to quantify the metabolic fluxes in a compartmented network of engineered CHO cells32.

Extracellular fluxes of glycolysis-related metabolites (a) and amino acids (b) into cells after 48 h of adding [U-13C] glucose. Asterisk symbol represents the metabolite is depleted after 24 h. Positive values represent the consumption fluxes, while negative values represent the production flux. Heatmap of isotopic labeling of intracellular metabolites (c) and MID data (d–r) after 24 and 48 h of adding [U-13C] glucose. s Metabolic flux distribution of engineered cell lines. Data were represented as mean ± SD (n = 3, n biologically independent samples). Multiple unpaired two-sided t-test was used for the statistical analyses for (a, b). P adjustments were made for multiple comparisons. G6P glucose-6-phosphate, 3PG 3-phosphoglycerate, Cit citrate, AKG alpha-ketoglutarate, Suc succinate, Fum fumarate, Mal malate, OAA oxaloacetate, Glu glutamate, Gln glutamine, Asp aspartate, Asn asparagine, Ser serine, Gly glycine, Lac lactate, Ala alanine. Source data are provided as a Source Data file.
The calculated extracellular fluxes (Fig. 6a, b) and intracellular mass isotopomers distributions (MID) data (Fig. 6d–r) collected after the 48-hour addition of [U-13C] glucose are combined with a CHO metabolic model to generate flux maps (Fig. 6s). For metabolites in glycolysis, such as G6P and 3PG, the MID data do not show obvious differences (Fig. 6d, e), which is consistent with metabolic flux in glycolysis pathway (Fig. 6s). Moreover, the flux of lactate synthesis is 22.3 nmol cell−1 h−1 in LplA overexpressed cells, indicating that 11% of glucose is transformed into lactate (calculated method was cited by literature33), which is reduced by 30.6% compared to the control group. The results suggest that the overexpression of LplA can alleviate lactate accumulation caused by the Warburg effect. Different from the metabolites’ performances in glycolysis, the intermediates in the TCA cycle exhibit significant differences between the two engineered cell lines, especially succinate (Fig. 6d–r). The M2 mass isotopomers of TCA-related metabolites along with glutamate, glutamate and aspartate were generated from [U-13C] glucose34. Pyruvate flux entering mitochondria is enhanced by 13.3% in the LplA overexpressed cell line. As expected, 33.2% of glucose entering cells is redirected through pyruvate into the TCA cycle in MTS-LplA cells during the exponential phase, which was improved by 48.88% compared to the MTS-GFP cells. As shown in Fig. 6j, succinate is predominantly M2 labeled in the MTS-LplA cell line after 48-h addition of tracer, indicating that more AKG is catalyzed through OGDH. Besides, the reaction rate of malate to oxaloacetate achieves 9.5 nmol cell−1 h−1 in the MTS-LplA cells, which is enhanced by 75.9% compared to the control group, suggesting that more NADH is generated through this pathway for ATP formation. Above all, the upregulation of glycolysis and TCA cycle flux, as well as downregulation of lactate production, provided more NADH for ATP synthesis in the electron transport chain (ETC).
Enhanced cell growth, nutrition consumption, and antibody production of cells with LplA overexpression in fed-batch culture
In view of the fact that the fed-batch strategy is commonly employed in industry for recombinant protein production, engineered CHO cells were grown in fed-batch culture over a period time of 10 days to examine the effects of LplA exogenous expression. As shown in Fig. 7a, MTS-LplA cells grow more efficiently and reach the stationary growth phase one day earlier than the MTS-GFP cells. Similar to the batch culture (Fig. 5a, d), VCD and canakinumab titers are also significantly improved with the exogenous expression of LplA during the fed-batch culture. More specifically, a peak VCD of 2.38 × 107 cells mL−1 and an antibody titer of 595.67 mg L−1 are obtained in MTS-LplA cells on day 8 and harvest day 10 (Fig. 7a, b), which are significantly improved by 14 and 37% compared to the MTS-GFP cells, respectively. Besides, qIgG is upregulated to the highest level of 289.91 ng cell−1 h−1 in the LplA overexpressed cell line during the day 8 and day 9 (Fig. 7c). During the fed-batch culture, glucose is supplemented to a concentration of 30 mmol L−1 every two days when the glucose concentration falls below 10 mmol L−1 (Fig. 7d). With the synergetic consumption of glucose and glutamine, lactate, and pyruvate concentrations perform rapid increase in the medium within the first two days (Fig. 7d, e, g, h). This phenomenon can be explained by the attributed to the saturation of the TCA cycle flux, the extra pyruvate from glycolysis is transformed to lactate or secreted to the medium directly. Then the MTS-LplA cells consume more pyruvate with a slight increase in lactate in Batch I. During this period, the qGluc of two engineered cell lines remains at a similar level (~50 nmol cell−1 h−1) with no significant difference. However, −qLac is decreased by 20% in MTS-LplA cells in Batch I (Fig. 7k). It was reported the theoretical ratio of glucose to lactate (qLac/qGluc) was 1.5–1.7 when other nutritious were sufficient in the medium35. In Batch I of this research, the ratio of qLac/qGluc in MTS-LplA cells is 0.84, which is 39% lower than that in MTS-GFP cells, indicating that the overexpression of LplA can induce more pyruvate to the TCA cycle instead of lactate biosynthesis. According to Fig. 7g, h, pyruvate and glutamine are deficient after Batch II in the medium. This implies that cells consume more lactate and glutamate to supply energy and intermediates in Batch III and Batch IV (Fig. 7e, g, i). Though qGluc is downregulated in the MTS-LplA cells compared to MTS-GFP cells in Batch II and Batch III, cells upon LplA overexpression have higher capability of qLac and qGlu (Fig. 7k, l). In particular, qLac and qGlu values of the MTS-LplA cells exhibit a significant increase in Batch III, with a notable elevation to 31.59 and 4.68 nmol cell−1 h−1, respectively. To verify the feasibility of this strategy in antibody manufacturing, we collaborated with WUXI BIOLOGICS to cultivate the engineered cell lines under industrial conditions. As shown in Supplementary Fig. 7, cells in the MTS-LplA group show higher VCD, VIA, and canakinumab titer compared to the control group, as expected. The antibody titer reached the highest level of 2.32 g L−1 on the harvested day, which is 20% higher than that in MTS-GFP cells.

a VCD during the fed-batch culture. b Canakinumab titer measured from day 5 to day 10. c qIgG from day 6 to day 10. d Glucose consumption and addition in the medium. e Lactate production and consumption in the medium. f The ratio of qLac/qGluc before glucose addition. g Pyruvate, h glutamate, i Glutamine concentration in the medium. Specific consumption rate of glucose (j), lactate (k), and glutamate (l) during the fed-batch culture. Data were represented as mean ± SD (n = 3, n biologically independent samples). Multiple unpaired two-sided t-test was used for the statistical analyses for (b, c, j–l). P adjustments were made for multiple comparisons. Source data are provided as a Source Data file.
Therefore, the cell phonotype observed in fed-batch culture of engineered cell lines confirms again that the overexpression of LplA can accelerate nutrients consumption rate, including glucose, pyruvate, lactate, and glutamate, which in turn provide sufficient precursors for the TCA cycle and the energy metabolism, leading to upregulation of cell growth and antibody productivity.
E coli-derived LplA improves growth, respiration and fatty acid synthesis of algae cell lines
Cyanobacteria and diatom are two of the most promising chasses for sustainable production of biofuels and biochemicals. To characterize the effects of LplA on physiological traits, P. tricornutum and S. elongatus are grown under photomixotrophic conditions with the addition of glycerol (100 mM) and glucose (5.5 mM) as sole organic carbon source, respectively. As shown in Fig. 8a, b, both LplA and MTS-LplA groups exhibited better growth rate than the WT strains. When lipoic acid is added to the medium, there is no significant difference in cell growth and lipoylation levels in Se_WT group (Supplementary Fig. 8a–c). Though several lipoylation bands are markedly increased upon lipoic acid addition in the Se_LplA group, cell growth remains similar in the presence or absence of lipoic acid (Supplementary Fig. 8a–c). After inducible expression of LplA, algal cells at the exponential phase (typically at 48 h) are harvested to further characterize the respiration and photosynthesis, which are measured as OCR and oxygen exchange rates (OER) using a Clark-type oxygen electrode at 20 °C. Surprisingly, exogenous expression of LplA promotes both the respiratory rate and photosynthetic rate, with better performance in the MTS-LplA group in P. tricornutum (Fig. 8c, e). In S. elongatus, the respiratory rate is significantly improved in the LplA strain, whereas the photosynthetic rate is slightly decreased compared to the Se_WT strain (Fig. 8d, h). Furthermore, the intracellular ratio of NADH/NAD+, an important cofactor and product of respiration, is also higher in both LplA and MTS-LplA groups (Fig. 8f, i). To verify the effect of LplA on modulating photorespiratory processes, algal cells are cultivated under high-light and high-temperature autotrophic conditions. Under low-light and high-CO2, the autotrophic growth of LplA group is markedly improved compared to the WT group in both P. tricornutum and S. elongatus (Supplementary Fig. 8d, h), which is consistent with mixotrophic growth condition (Fig. 8a, b). However, under high-light and high-temperature stress, the growth of LplA-expressing cells is inhibited compared to WT cells (Supplementary Fig. 8e, i). Specifically, the Se_LplA group displays a pronounced yellow color (Supplementary Fig. 8g), indicating reduced chlorophyll content, a critical determinant of photosynthetic efficiency. Non-photochemical quenching (Y(NPQ)) is carried out using IMAGING-PAM Max version in P. tricornutum. The results show that Y(NPQ) is significantly elevated in LplA-expressing strain compared to WT under identical stress conditions (Supplementary Fig. 8f). Further quantification revealed a significant decline in oxygen evolution rates in the Se_LplA group under these stress conditions compared to WT (Supplementary Fig. 8j). This paradoxical increase in NPQ capacity, coupled with reduced photosynthetic oxygen evolution, implies that the exogenous LplA expression in both cyanobacteria and diatoms amplifies photorespiratory flux under high-light and high-temperature conditions, ultimately destabilizing photosynthetic efficiency and growth. The above results demonstrate the excellent potential of exogenously expressed LplA in enhancing photorespiration.

Cell growth curve (a, b), respiratory activities (c, d), photosynthesis activities (e, h), ratio of NADH/NAD+ (f, i), and targeted free fatty acid content (g, j) in cultured P. tricornutum (a, c, e–g) and S. elongatus (b, d, h–j). Data were represented as mean ± SD (n = 3 in f, g, i, j, n = 4 in c–e, h, n biologically independent samples). k Exogenous expression of E. coli LplA in cyanobacteria and diatoms increases the lipoylation level of key metabolism enzymes (E2 subunit of PDH and OGDH, GCSH) and then enhances the energy metabolism, cofactor supply, and fatty acid biosynthesis. The black pentagrams indicate proteins that are modified using lipoic acid. Blue lines represent the glucose degradation pathway. Orange lines represent the glycerol degradation pathway. Green lines represent the Calvin–Benson–Bassham cycle. One-way ANOVA was used for the statistical analyses for (f), an unpaired two-sided t-test was used for (c–e, h–j), two-way ANOVA was used for (g). P adjustments were made for multiple comparisons. Chl chlorophyll, F6P fructose-6-phosphate, Ru5P ribulose-5-phosphate, SBP sedoheptulose-1,7-bisphosphate, DHAP dihydroxyacetone phosphate, PGA 3-phospho-D-glycerate, AcCoA acetyl-CoA, Pyr pyruvate, PDH pyruvate dehydrogenase, OGDH oxoglutarate dehydrogenase, TCA tricarboxylic acid cycle, FAS fatty acid synthesis. Source data are provided as a Source Data file.
More acetyl-CoA and NADH generated from enhanced energy metabolism can support fatty acid synthesis by providing more substrate and cofactors (Fig. 8k), which is verified by the fatty acid analysis. The polyunsaturated fatty acids (PUFA) docosahexaenoic acid (C22:6n3, DHA) and dihomo-γ-linolenic acid (20:3n-6, DGLA) of P. tricornutum cells are of high nutritional value to human health. Analysis of the free fatty acid (FFA) contents of P. tricornutum shows that overexpression of LplA significantly increases the production of DHA and DGLA (Fig. 8g). Similarly, overexpression of LplA promotes the production of trans-linoleic acid (C18:2n6t, TLA) in S. elongatus (Fig. 8j). These results demonstrate that the heterogeneous expression of LplA can improve cellular growth, energy metabolism, cofactor supply, and lipid production in diatom and cyanobacteria by increasing the lipoylation level of key metabolic enzymes, including PDH, OGDH, and GCSH (Fig. 8k).
Discussion
Lipoylated proteins are essential for the energy metabolism of both bacteria and eukaryotes. The general consensus is that only the de novo pathway of lipoylation exists in mitochondria of eukaryotes2. Recent research has reported that LIPT1 and Lip3 from human and yeast can function as a “half-ligase” to transfer the lipoyl moiety from lipoy-AMP to proteins, but cannot utilize the lipoic acid to form lipoy-AMP15,36. However, the overexpression of LIPT1 in mammalian cells cannot enhance the lipoylation levels of DLAT and DLST, thereby having no effect on mitochondrial respiration and cell phenotype (Supplementary Figs. 2c, 4a, 5b–e). Differently, plants possess the biosynthetic pathway for lipoic acid in both mitochondria and plastids37. However, previous studies have suggested that a fully functional salvage pathway is missing and only short putative “ligases” lacking an important C-terminal domain are present in higher plants, leading to the hypothesis that these are transferases rather than ligases2,38. Furthermore, the expression of Arabidopsis-derived-LplA (AtLplA) failed to restore the growth of TM137, a lipoylation-deficient mutant lacking lipoate protein ligase and octanoyltransferase, indicating that this short ligase cannot replace the bifunctional LplA in the presence of lipoic acid in E.coli38. In this study, we report that the exogenous introduction of E. coli-derived-LplA can function in multiple organisms, triggering a series of metabolic changes by activating a hereto unreported salvage pathway of lipoylation, ultimately promoting energy generation in vivo.
When LplA was targeted to be expressed in mitochondria of mammalian cells, the lipoylation of its interacting proteins, DLAT and DLST, were initially enhanced (Fig. 2b, d). Subsequently, the activities of PDH and OGDH increased significantly (Fig. 4a, b), accelerating the flux of the TCA cycle and strengthening the following OXPHOS process. Upon exogenous expression of LplA, more glucose and glutamine were transported into cells in both U2OS and CHO cell lines (Figs. 3c, 7d, h, i). Since U2OS cells are cancer model cells, which predominantly utilize aerobic glycolysis for energy production even in the presence of sufficient oxygen. Upregulated lipoylation levels not only enhance OXPHOS but also enhance the activity of PKM2, leading to an increase in glycolysis levels and lactate production (Fig. 3a, b, d). Differently, qLac/qGluc ratio is lower in MTS-LplA cells (Fig. 7f), indicating that the Warburg effect is inhibited and more pyruvate enters the TCA cycle rather than being used for lactate biosynthesis. Furthermore, the lactate switch is activated to facilitate more substrates entering the TCA cycle during the suspension culture of CHO cells (Fig. 7e), contributing to the energy generation and environmental improvement. The increased 13C labeling ratio of intermediates and metabolic fluxes in the TCA cycle also provides the evidence that more substrates and cofactor like NADH is directed to OXPHOS for energy generation (Figs. 3d, e, 6), reflecting an over 50% upregulation of ATP production in both cell lines (Figs. 3a, 4d, k). The enhanced energy availability promotes the phosphorylation and activation of mTORC1 (Fig. 3q), driving protein synthesis, lipid biosynthesis, and other anabolic pathways, thereby supporting cellular growth and proliferation. The above results reveal that the exogenous introduction of the salvage pathway of lipoylation can reprogram intracellular metabolic pathways by enhancing OXPHOS flux and alleviating the Warburg effect. Unexpectedly, the ROS level is significantly reduced in the LplA overexpression group (Figs. 4e, 5g), which might be attributed to the ability of LplA to directly hydrolyze superoxide anion or hydrogen peroxide31. Additionally, the improvement of mitochondrial metabolism also promotes the mitobiogenesis and mito-fusion (Figs. 3o, p), providing a more favorable mitochondrial environment for energy metabolism and forming a virtuous cycle39.
Similarly, the heterologous expression of E. coli LplA in the eukaryotic alga P. tricornutum and the cyanobacterium S. elongatus increases the cell growth and respiration rate (Fig. 8c, d) by specifically upregulating the lipoylation of PDH-E2 and OGDH-E2, which promotes higher enzyme activities of PDH and OGDH, thereby supplying more NADH from NAD+, as determined by the NADH/NAD+ ratio results (Fig. 8g, h). Furthermore, in vitro and in vivo assays also show that the lipoylation levels of the GCSH proteins from P. tricornutum and S. elongatus are upregulated in the presence of E. coli LplA (Figs. 1d, 2e and Supplementary Fig. 1a–d). GCSH is one of the four subunits that constitute the glycine cleavage complex (GCS), which is involved in the oxidative cleavage of glycine into CO2, supplying reducing power such as NADH and a one-carbon (1 C) unit, 5,10-CH2-THF, that is needed for photorespiration and biosynthetic processes of methionine, thymidylate, and purines40,41. Stress cultivation under high-light and high-temperature has demonstrated the excellent potential of exogenous expression of LplA in enhancing photorespiration (Supplementary Fig. 8d–j). In particular, photorespiratory metabolism allows plants and algae to possess resistance to numerous abiotic stresses41.
These results indicate that the heterologous expression of E. coli LplA can effectively enhance energy and cofactor supply in algae. This strategy could be further extended to higher plants and cash crops due to the highly conserved lipoyl domain present in all E2 subunits of α-ketoacid dehydrogenase complex and GCSH proteins (Fig. 1g, h), as further proved by in vitro assay of lipoylation in five common plant model species, including Z. mays, A. thaliana, G. max, N. tabacum, and O. sativa (Fig. 1e and Supplementary Fig. 1e–i). Interestingly, unlike in cyanobacteria, the heterologous expression of E. coli LplA in P. tricornutum not only increases the respiratory rate but also enhances the photosynthetic rate when glycerol is used as an organic carbon source (Fig. 8e). This phenomenon is reasonable because, while glycolytic routes in cyanobacteria are a reverse to the Calvin–Benson–Bassham (CBB) cycle, glycerol metabolism routes in P. tricornutum are parallel to an operating CBB cycle42,43 (Fig. 8k).
In synthetic biology, the provision of energy is a persistent limitation in the biosynthesis of energy-dependent products. Here, the application of LplA in multiple species not only boosts the energy metabolism, but also enhances cell growth and biosynthesis. For example, exogenous expression of LplA can significantly improve cell growth and canakinumab titer in both batch and fed-batch culture of CHO cells (Figs. 5a, d, 7a, b). To note, the specific consumption rate of nutrients in the medium and specific production rate of antibody are significantly increased (Fig. 7c, k, i), revealing that overexpression of LplA indeed improves the cellular metabolism at the individual cell level and provides sufficient energy and precursors for the synthesis, packaging and secretion of antibodies. Especially in an industrial setting, the antibody titer is increased by 20% at the level of over 2 g L−1 (Supplementary Fig. 7). Besides, TLA, DHA and DGLA, which are PUFAs of high nutritional value to human health, are significantly improved by over 20% in P. tricornutum and S. elongatus (Fig. 8g, j). As for fungi, S. cerevisiae and A. niger are the most common host cells in industrial applications for producing various extracellular enzymes, organic acids, and alcohols. In this study, the exogenous expression of LplA in yeast S. cerevisiae significantly improves the cell growth after 12-h cultivation (Supplementary Fig. 9a). The basal respiration in whole cells and ATP production in isolated mitochondria are increased by 78 and 123%, respectively (Supplementary Fig. 9b, c). In A. niger, the oxygen transfer rate detected in a RAMOS bioreactor system shows that the respiration in LplA overexpressed cell lines is significantly boosted (Supplementary Fig. 10a). During the fermentation of A. niger, the mycelia dry weight and citric acid are significantly increased upon LplA overexpression in both flask and DASGIP systems (Supplementary Fig. 10b, c, f) with higher consumption of O2 and production of CO2 (Supplementary Fig. 10d, e). We notice that Pietikäinen et al. and Nolan Bick et al. reported that introduction of E. coli-derived LplA can restore lipoylation levels, cellular respiration and cell growth in lipoylation-deficient yeast and human cells15,44. However, their focus is on clarifying protein functions and restoring the normal function of defective mitochondria, the aforementioned cases in this study suggest that overexpression of LplA can not only improve the mitochondrial function and energy metabolism, but also holds substantial potential for biosynthesis in multi-species chassis cells, especially the processes that require additional energy supply.
Targeting PTMs is an appealing approach for metabolic engineering and synthetic biology due to their ability to rapidly respond to external environmental stimuli and reduce the cost of protein synthesis within biological cells. However, modulation of PTMs in practical applications has been challenging due to the redundancy and non-specificity of the modification sites of the most studied forms of PTMs, including phosphorylation, methylation, and acylation45,46. In this work, we successfully engineered chassis cells by precisely modulating PTM of multiple well-defined47 energy metabolism, cofactor supply and biosynthesis through the induction of salvage lipoylation with a small protein across species.
Methods
Lipoylation activity assay in vitro
All proteins used for lipoylation assay in vitro, including EcLplA, cgeDLAT, cgeDLST, PtPDH_E2, SePDH_E2, PtGCSH, SeGCSH, ZmPDH_E2, AtPDH_E2, GmPDH_E2, NtPDH_E2, OsPDH_E2, SceLat1, and SceKgd2, were expressed by using the pET-28a (+) system in E. coli BL21 (DE3). The related sequences are listed in Supplementary Data 1. In detail, the bacteria were cultured at 37 °C in LB medium containing 50 μg/mL kanamycin and induced by 0.2 mM isopropyl β-D-thiogalactoside (IPTG) when OD600 reached 0.6. After collecting bacterial pellet by centrifugation (10,000 × g, 5 min, 4 °C), the pellets were resuspended in lysis buffer (500 mM NaCl, 50 mM Tris-HCl, 20 mM imidazole, pH 7.5) and lysed by high-pressure homogenization. Then the lysates were centrifuged at 12,000×g for 5 min at 4 °C, and the target proteins were purified from the supernatant by nickel column. Finally, the purified proteins were concentrated using a spin filter (Millipore) and stored in storage buffer for further experiment47. The lipoylation reaction system mixture (100 μL), including 0.5 mM lipoic acid, 1 mM ATP, 1 mM MgCl2, 0.1 mM dithiothreitol, 50 μM targeted proteins, and 20 μM LplA, was incubated at 37 °C for 20 h. The reaction was stopped by adding 10 μL 36% trichloroacetic acid to completely denature and precipitate all proteins. The lipoylation level was detected by immunoblotting. The mixture was boiled with SDS-PAGE loading buffer (Takara Bio, Japan) at 98 °C for 10 min. Proteins were loaded on SDS-polyacrylamide gel electrophoresis (PAGE) gels and transferred onto nitrocellulose (NC). The NC membranes were blocked in 5% skimmed milk in tris-buffered saline with tween 20 (TBST) at room temperature for 1 h and incubated overnight at 4 °C with the following primary antibodies: anti-lipoic acid antibody (Abcam, ab58724, UK, 1:2000), anti-His-Tag antibody (Proteintech, 66005-1, USA, 1:1000). Anti-rabbit immunoglobulin G (IgG) (LI-COR, 926-32211, USA, 1:10,000) and anti-mouse IgG (LI-COR, 926-68070, USA, 1:10,000) were applied as secondary antibodies. ChemiDoc MP Imaging system (Bio-Rad, 12003154, USA) was used for imaging.
Generation of mammalian cell lines
The U2OS cell line (ATCC, HTB96, USA), adherent CHO-K1 cell line (ATCC, CCL61, USA) and engineered industrial IgG-producing CHO cell line, which was constructed in cooperation with WUXI BIOLOGICS (Wuxi, China), were applied for the targeted overexpression of LplA or LplA3M in mitochondria. For U2OS transfection, the mitochondria targeting sequence (MTS) of ornithine transcarbamylase (hsaOTC) was added to the N-terminal of GFP, LplA, or LIPT1. For CHO transfections, MTS of superoxide dismutase 2 (cgeSOD2) was added to the N-terminal of GFP, LplA, or LIPT1. The above-mentioned MTS and engineered gene sequences are listed in Supplementary Data 3 and 4, respectively.
For adherent U2OS and CHO cell transfection, the pLenti-CMV-mEGFP plasmid was kindly provided by Prof. Peiguo Yang (Westlake University, China). Before transfection, HEK293T cells were incubated at 5 × 105 cells/well in six-well plates for 24 h. Then 1 μg of target plasmid, 500 ng of psPAX2 and 500 ng of pMD2.G were applied for lentivirus package via lipo 3000 reagent (Thermo, USA). The supernatant was collected after 48 h and filtered through 0.22 μm filters. The virus was concentrated via a universal virus concentration kit (Beyotime, C2901S, China). Then, 200 μL virus mixed with 2 μL polybrene (Solarbio, China) were added to 2 mL medium for U2OS and CHO cells cultivation. The positive cells were sorted according to GFP signal by using CytoFLEX SRT-2 (Beckman Coulter, USA).
To prepare the suspension host cells for transfection, CHO cells were seeded in the serum-free CD CHO medium (Gibco, USA) containing 4 mM glutamine for 1 to 3 days. For each transfection, 1.0 × 107 cells were centrifuged at 290 × g for 5 min and resuspended with 300 μL of DPBS (Corning Hyclone, USA). Then 2 μg of target plasmid and 20 μg of canakinumab expression plasmids (6.7 μg of heavy chain and 13.3 μg of light chain) were mixed with 250 μL of DPBS. The plasmid reagent mixture and host cells were mixed carefully and transferred to a 4 mm electroporation cuvette for electroporation. Electroporation was performed at 280 V, once pulse and pulse length of 20 ms. The transfected CHO cells were selected using 6 μg mL−1 of blasticidin, 400 μg mL−1 of zeocin and 500 μg mL−1 of geneticin (Invitrogen, USA) in serum-free CD CHO medium and cultured in a shaking incubator at 36.5 °C, 75% humidity, 6% CO2 and 225 rpm.
Cultivation of mammalian cells
For the cultivation of U2OS and adherent CHO cells, the Dulbecco’s modified eagle medium (DMEM) and Kaighn’s modification of Ham’s F-12 (F12K) medium (ATCC, 30-2004) were applied, respectively with supplantation of 10% fetal bovine serum (FBS) (Cell Max, USA) and 1% Penicillin-Streptomycin solution (PS) (Thermo, USA). Cells for phenotype measurement were seeded in 100 mm plates at 5 × 106 cells (U2OS cells) or 2 × 106 cells (CHO cells) and cultured for 24 h in a CO2 incubator at 37 °C and 5% CO2.
For batch and fed-batch cultivations of suspension CHO cells, the engineered stable cell lines were seeded at a cell density of 2 × 105 cells mL−1 in spin tubes containing 10 mL selected medium. After passaging two to three generations, the cells were transferred into HyClone™ ActiPro™ cell culture medium (Cytiva, China) (HA medium) with 0.1 mM aurintricarboxylic acid and passaged for 3 days. Then, the HA medium with 2 mM uridine was applied as production medium. The cells at an initial cell density of 3–5 × 105 cells mL−1 were cultured in 250 mL baffled flasks containing 50 mL production medium in a shaking incubator. In the fed-batch culture, glucose and amino acids were fed according to the growth and nutrient profiles in the medium.
Transformation of Phaeodacylum tricornutum and Synechococcus elongatus strains
The P. tricornutum strain CCAP1055/1 (CCMP 2561) was obtained from the Provasoli-Guillard National Center for Marine Algae and Microbiota (NCMA, USA). P. tricornutum transformation was accomplished through bacterial conjugation48. Briefly, the LplA coding gene from E. coli was amplified and inserted into an induced conjugation plasmid pEF1a-conj-XVE-LexA35S (a gift from Dr. Tianjun Cao from Westlake University, China), which includes estrogen receptor-based transactivator XVE sequence, LexA operator sequence and CaMV35S promoter, followed by 3xHA-GFP reporter genes, wherein XVE comprises the DNA-binding domain of LexA (X), VP16 transcriptional activation domain (V) and the regulatory region of human estrogen receptor (E). LplA was added to the N-terminal of GFP reporter. If needed, an MTS of TPI-GapC349 was added to the N-terminal of the LplA sequence. Therefore, plasmids with or without MTS were consecutively transformed into the conjugation-competent E. coli strain EPI30050. Then 1 × 108 cells of P. tricornutum were mixed with 200 μL of E. coli EPI300 and plated on BSR (blasticidin S HCl, 5 µg mL−1)-containing plates for the selection of positive colonies50,51. The sequences of TPI-GapC3, GFP and LplA are listed in Supplementary Data 3 and 4.
S. elongatus sp. PCC 7942 was obtained from the Institute of Hydrobiology (IHB) of the Chinese Academy of Sciences (CAS). The LplA encoding gene from E. coli was inserted into neutral site 3 (NS3) of the pAM4941 (pCV0053) plasmid52. The generated constructs were then transformed into Syn7942 cells by natural transformation53. Briefly, neutral site genes were replaced with antibiotic resistance cassettes via homologous recombination. Transformation efficiency is especially high in the exponential growth phase. Therefore, 200 ml Syn7942 cultures were inoculated in a 1 L Erlenmeyer flasks from a preculture with an OD750 of 0.15 on the day prior to transformation. On the day of transformation, the cells were harvested and resuspended in 600 µL fresh BG11. A volume of 300 µL of the cell suspension was mixed with 3 µg plasmid DNA and incubated for 6 h at 30 °C. Cells were plated on agar plates without antibiotics and kept in a climate chamber at 30 °C and 60 µE m−2 s−1. On the third day, antibiotics were added for selection pressure. After 2 weeks, single colonies were streaked on new BG11 agar plates with antibiotics for segregation. For a solid medium, 1% agar was supplemented.
Cultivation of algae
The WT and mutants of P. tricornutum were grown in f/2 medium (30‰ salinity, w/v) at 18 °C under constant light of 60 µmol photons m−2 s−1 unless otherwise indicated, on a shaker at a speed of 130 rpm. Light was supplied by cool white fluorescent bulbs (Phillips, F17T8/TL84) or LED strip light. For a solid medium, 1% agar was supplemented. Besides, the WT and mutants of S. elongatus were grown in BG11 medium at 28 °C. All further setups were conducted as described above for diatom cells.
Construction and cultivation of Saccharomyces cerevisiae strains
The yeast S. cerevisiae PK2-1C strain and the sgRNA-cas9-ATG14 plasmid were kindly provided by Dr. Yajie Wang from Westlake University. For S. cerevisiae mutants’ construction, MTS of sceSOD2 was added to the N-terminal of GFP or LplA. All the sequences had been optimized with codons according to the yeast system. The involved sequences are listed in the Supplementary Data 3 and 4. The target genes were transformed into the genome of WT by chemical transformation as the Quick & Easy Yeast Transformation Mix kit (Takara Bio, 631851, Japan). Both wildtype and modified strains grow on yeast extract peptone dextrose (YPD) or extract peptone glycerol (YPG) medium. The liquid culture of the strain was carried out in a shaker at 220 rpm and 30 °C. Solid plate culture is carried out under the condition of adding agar to the YPD medium.
Construction and cultivation of Aspergillus niger strains
The A. niger strain used in this study is kindly provided by Prof. Ping Zheng’s from Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences54. The plasmid pPTRI for gene overexpression was bought from the Shengwufeng website. The protoplasts were washed with ice-cold SC solution (182.2 g/L sorbitol, 7.35 g/L CaCl2·2H2O) and collected by centrifugation at 1500 × g at 4 °C for 10 min. For transformation, 4 × 106 protoplasts, 20 µL of 0.4 M aurintricarboxylic acid ammonium salt (ATA), 100 µL of 20% PEG-4000, and 5 µg plasmid were mixed and incubated at room temperature for 10 min. Then 1.5 mL 60% PEG-4000 was added and incubated for 20 min. Next, 5 mL of 1.2 M sorbitol was added to the mixture and centrifuged at 3000 × g for 10 min. The pellet was resuspended in 1 mL of 1.2 M sorbitol prior to plate spreading. The plates were incubated in a moist chamber at 30 °C for 3–4 days55. For plate cultivations, 1.5% (w/v) agar was added, and 1.22 g L−1 uridine was supplemented for auxotrophic strains. 2 mg L−1 pyrithiamine (hydrobromide) was added for isolating transformants when necessary. For batch cultivation, A. niger strains were grown at 30 °C on Complete Medium (CM) or Minimal Medium (MM) supplemented with the required carbon source56.
Confocal imaging
For the immunofluorescence analysis of the engineered U2OS and CHO cells expressing LplA, cells were seeded in a confocal dish overnight. For U2OS cells, the cells were incubated with Mito-Tracker Orange at a final concentration of 100 nM for 30 min at 37 °C. After washing with PBS, the cells were incubated with Hoechst 33342 for 5 min, washed again with PBS, and then observed under a Zeiss confocal microscope. For CHO cells, cells were washed with PBS three times and fixed with 4% paraformaldehyde (PFA) for 30 min. After rinsing three times with PBS, cells were incubated with blocking buffer (3% bovine serum albumin and 0.2% Triton X-100) for 60 min. Subsequently, three rinses were performed using wash buffer (0.1% TX-100 in PBS), the primary antibody of Tom20 (Santa Cruz, sc-17764, USA, 1:500) were performed for incubation at 4 °C overnight. Then the cells were washed with wash buffer three times and incubated with anti-mouse Alexa Flour 647 (Thermo, A21235, USA, 1:1000) and Alexa Flour 488 (Thermo, A28175, USA, 1:1000) as secondary antibodies for 60 min at room temperature. After washing with the wash buffer three times, immunofluorescence staining analysis was performed with a Laser scanning confocal microscope (Zeiss, LSM980, Germany).
Whole cell extracts preparation and immunoblotting
For immunoblot assay, U2OS and CHO cells were lysed by radio immunoprecipitation assay lysis buffer (RIPA) containing 1 mM phenylmethanesulfonylfluoride (PMSF) and incubated on ice for 30 min. The supernatant was collected by centrifugation at 12,000 × g at 4 °C for 30 min. For cyanobacteria, the cells were harvested and resuspended in potassium-phosphate buffer (pH 7.2) with 0.5 mL of glass beads (diameter 0.2 mm). After homogenization, the supernatant was collected by centrifugation at 10,000 × g at 4 °C for 5 min57. For diatom, cells were harvested and resuspended in the protein extraction buffer (RIPA, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 10 mM dithiothreitol [DTT]). After sonication by Bioruptor® PLUS sonicator (Diagenode, Belgium) combined with the Bioruptor® Water cooler at 4 °C (High model, 60 cycles of 10 s ON/20 sec OFF), supernatant were collected by centrifugation at 4000 × g at 4 °C for 10 min48. For S. cerevisiae, proteins were extracted by a commercial kit (Solarbio, BC3780, China). After centrifuging at 12,000 × g at 4 °C for 30 min, the supernatant was collected. Protein concentration was measured via Pierce BCA protein assay (Thermo Fisher). Residual supernatant was boiled with SDS-PAGE loading buffer (Takara Bio, Japan) at 98 °C for 10 min for further immunoblotting assay. The following antibodies are used as primary antibodies: lipoic acid (Abcam, ab58724, USA, 1:2000), GAPDH (Proteintech, 60004, USA, 1:5000), DLAT (CST, 12362, USA, 1:1000), DLST (CST, 11954, USA, 1:1000), LplA (GeneBio Systems, CSB-PA510777HA01ENU, Canada, 1:1000), GFP (Proteintech, HRP-66002, USA, 1:1000), LIPT1 (Invitrogen, PA5-57064, USA, 1:1000), α-Flag (Sigma-Aldrich, F1804, Germany, 1:1000), HIF-1α (Thermo Fisher, A300-286A, USA, 1:1000), OPA1 (Abcam, ab157457, USA, 1:1000), MFN1 (CST, D6E2S, USA, 1:1000), MFN2 (CST, 9482, USA, 1:1000), NRF1 (Proteintech, 66832, USA, 1:1000), TFAM (Proteintech, 22586, USA, 1:1000), Phos-S6 (Ser235/236) (CST, 4858, USA, 1:1000), S6 Ribosomal Protein (CST, 2217, USA, 1:1000), Phos-4E-BP (CST, 2855, USA, 1:1000), 4EBP1 (CST, 9644, USA, 1:1000), Phos-p70S6K (Thr389) (CST, 9205, USA, 1:1000), p70S6K (CST, 3447, USA, 1:1000), HK1 (CST, 2024, USA, 1:1000), PFKP (CST, 8164, USA, 1:1000), PKM2 (CST, 4053, USA, 1:1000), and pPKM2 (Tyr105) (Affinity Biosciences, DF2975, AUS, 1:1000) For CHO and yeast cells, anti-rabbit immunoglobulin G (IgG) (LI-COR, 926-32211, USA, 1:10,000) and anti-mouse IgG (LI-COR, 926-68070, USA, 1:10,000) were applied as secondary antibodies. For U2OS and algal cells, horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG were applied as secondary antibodies. ChemiDoc MP Imaging system (12003154, Bio-Rad) was used for imaging and ImageJ (v1.8.0, National Institutes of Health, USA) was used for relative quantification analysis.
Detection of lipoylation activity in algae via in vivo assay
Firstly, targeting proteins (GCSH, OGDH-E2 and PDH_E2) in pET-duet-1 were expressed with or without 0.1 mM of lipoic acid. All further steps about protein purification were similar as with the other proteins described herein. About 10 μg of targeting protein were loaded on SDS-PAGE, followed by immunoblotting with antibodies against lipoic acid (Abcam, ab58724, USA, 1:2000) and His-Tag (Proteintech, 66005-1, USA, 1:1000), respectively. Protein quantifications were determined using the immunoblotting signal strength of the His-Tag antibody.
Mitochondrial extraction from S. cerevisiae and CHO cells
The mitochondrial extraction from S. cerevisiae cells were carried out using Yeast Mitochondria Isolation Kit (Abcam, ab178779, USA) according to the manufacturer’s instructions. Mitochondria were extracted from CHO cells according to the previously reported protocols58. In brief, 2 × 107 cells were collected and resuspended in 11 mL ice-cold RSB buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl (pH 7.5)). After homogenization, 8 mL 2.5 × MS buffer (525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl (pH 7.5), 2.5 mM EDTA (pH 7.5)) was added to the homogenate and mixed by inverting. Then 10 mL 1 × MS buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl (pH 7.5), 1 mM EDTA (pH 7.5)) was added and centrifuged at 1300 × g for 5 min at 4 °C. Transfer the supernatant and repeat centrifugation (1300 × g, 5 min, 4 °C) two more times. Then the supernatant was centrifuged at 12,000 × g for 15 min at 4 °C to pellet the mitochondria. Washing the pellet via centrifugation (12,000 × g, 5 min, 4 °C) and resuspending the mitochondria in 1 × MS buffer. The purified mitochondria were collected and lysed for further immunoblotting assay. VDAC (Abcam, ab154856, USA, 1:2000) was used as a housekeeping protein.
PDH and OGDH activity assay
Totally 1 × 106 cells were collected for PDH and OGDH activity detection using commercial Kits (Abcam, ab 287387 and ab185440, USA), respectively. Briefly, cells were incubated with 100 μL ice-cold PDH/OGDH assay buffer on ice for 10 min. After centrifugation at 10,000 × g at 4 °C for 5 min, the protein concentration in supernatant was quantification using Pierce BCA protein assay. A total of 50 μg protein of each sample was added to a 96-well plate, and the volume were adjusted to 50 μL with PDH/OGDH assay buffer. Then the reaction mixture containing 46 μL PDH/OGDH assay buffer, 2 μL PDH/OGDH developer and 2 μL PDH/OGDH substrate was added to each well. The enzymatic activity was measured at 450 nm in kinetic mode for 60 min at 37 °C. The activities of PDH and OGDH are calculated as follows:
B represents NADH amount from standard curve (nmol), ΔT represents reaction time (min), C represents total protein concentration (mg).
Reactive oxygen species (ROS) and mitochondrial membrane potential (MMP) detection
ROS and MMP were measured using CellROX orange reagent (Thermo Fisher, C10443, USA) and IMAGE-IT TMRM reagent (Thermo Fisher, I34361, USA), respectively. Adherent cells were seeded in six-well plates at 5 × 105 cells and cultured overnight under CO2 incubation at 37 °C and 5% CO2. Then cells were washed with PBS, digested with trypsin and washed with PBS again three times for further incubation. For suspension cells, cells were collected every day at a density of 1 × 106 cells and washed with PBS three times. Then cells were resuspended in culture medium with 5 μM CellROX reagent or 0.1 μM TMRM reagent and incubated at 37 °C for 30 min. Finally, cells were washed and resuspended in PBS and measured with flow cytometry (Beckman, USA) at an excitation wavelength of 561 nm and emission wavelength of 585 nm.
Mitochondrial respiration test in whole cells
An XFe96 extracellular flux analyzer (Agilent, USA) with a 96-well plate was used to measure oxygen consumption rate (OCR)59. U2OS, adherent and suspension CHO, S. cerevisiae cells were seeded at a density of 2 × 104, 5 × 104, 1 × 104, and 2 × 105 cells per well, respectively. Before assay, U2OS and CHO cells were equilibrated for 1 h in a non-CO2 incubator at 37 °C with XF Base medium supplemented with 10 mM glucose, 2 mM pyruvate, and 4 mM L-glutamine. S. cerevisiae cells were incubated in YPG medium at 30 °C. For the mitochondrial stress test and glycolysis analyses, reagents were injected according to the protocols of the Seahorse XF Cell Mito Stress Test kit and the Seahorse Glycolysis Stress Test kit, respectively. The results were normalized using CyQUANT Cell Proliferation Kit (Invitrogen, C7026, USA).
Respiratory rate and photosynthetic rate measurements for P. tricornutum and S. elongatus
Respiration and photosynthesis were measured as O2 exchange rates using a Clark-type oxygen electrode at 20 °C (Chlorolab2+, Hansatech Instruments Ltd, Norfolk, UK). Actinic light was provided by light-emitting diodes with an emission maximum around 650 nm. For each measurement, cells were concentrated by centrifugation and resuspended to 107 cells mL−1 with fresh f/2 medium plus 0.1 M glycerin. O2 evolution rate (OER, photosynthetic rate) was measured at 1000 μmol photons m−2 s−1, whereas O2 consumption rate (OCR, respiratory rate) was measured in the dark. O2 changing rate was presented as nmol O2 evolved per minute per 106 cells. The O2 concentration in fresh f/2 medium (plus 0.1 M glycerin) without diatom cells was used as a control. Two biological replicates and two technical replicates were conducted for every strain. For S. elongatus, cyanobacteria cells were concentrated by centrifugation and resuspended to an OD750 of 2.0 with fresh BG11 medium plus 5.5 mM glucose. The O2 concentration in fresh BG11 medium (plus 5.5 mM glucose) without diatom cells was as control. Two biological replicates and two technical replicates were conducted for every strain. All further steps were conducted as described above for diatom cells.
Determination of ATP concentration in isolated mitochondria from S. cerevisiae
The mitochondria isolated from different strains were resuspended in MgCl2-free MiR05 medium and transferred into the chambers of the Oxygraph-2k (Oroboros Instruments, Austria) for oxygen consumption rate detection60. After 10 min of incubation, 0.1 mM EGTA, 15 μM EDTA, 5 mM pyruvate, 0.5 mM malate, 10 mM glutamate, and 50 mM succinate were injected into the chamber. When oxygen consumption approached a steady rate, 2 mM ADP was added to trigger ATP synthesis. After 15 min incubation, the mitochondrial respiration was inhibited with 0.5 μM rotenone and 2.5 μM antimycin A. The data were recorded and analyzed using the software DatLab (v8.1.1, Oroboros Instruments, Austria).
Isotope labeling experiments in U2OS and CHO cells
For U2OS, cells were plated 24 h before the isotope labeling experiment in 100 mm dishes. On the day of experiments, the medium was replaced with phenol red-free DMEM with 1 mM glutamine, 10% FBS and 25 mM 100% [U-13C] glucose. After 12-h and 18-h incubation, cells were washed once with ice-cold PBS and harvested for further analysis. For CHO cells, 100% [U-13C] glucose was added to three replicates of each cell line on day 3 (exponential phase) to a final concentration of 5 g L−1. Samples were collected after 24, 48 h, and 72 h. All collected samples were immediately cold-quenched and stored at −80 °C for further analysis.
Determination of growth and extracellular exchange rates in U2OS and CHO cells
The viable cell density (VCD) and viability (VIA) were monitored by Countess 3 (Invitrogen, USA) during the cultivation of U2OS and suspension CHO cells. The concentrations of extracellular glucose, pyruvate, lactate, ammonium, glutamate, glutamine, and canakinumab titer were measured via Cedex (Roche, Bio Hit, Switzerland).
A high-performance liquid chromatography (HPLC) system (Infinity 1260 II, Agilent, USA) equipped with a DAD detector and Agilent Poroshell HPH-C18 column (4.6 × 100 mm, 2.7 μm) was employed for the absolute quantification of extracellular amino acids. To this end, 20 μL supernatant were added into 200 μL methanol (precooled to −80 °C) and vortexed for 10 s. After incubation at −80 °C for 20 min, the solution was dried under N2 flow. Then the sample was dissolved in 200 μL 0.1 M HCl and centrifuged at 15,000 × g for 5 min at 4 °C. The upper clear liquid was transferred into a vial for HPLC analysis. The detailed method, including online derivatization and gradient program, was performed according to the protocol described by Agilent (https://www.agilent.com.cn/cs/library/applications/compendium-%20aminoacid-advancebio-5994-0033EN-us-agilent.pdf).
The specific growth rate (μ), specific consumption rates (qi) of metabolites were defined as follows:
Glutamine equation included degradation in the culture medium. The specific consumption rate of glutamine (qGln) was defined as follows:
The degradation rate constant (k) of glutamine was determined as 0.0031 h−1 (Supplementary Fig. 11).
Extraction and determination of intracellular metabolites in U2OS and CHO cells
On the harvested day, U2OS cells were quickly washed twice with ice-cold PBS, and the collected suspension CHO cells were quenched using 0.9% (w/v) NaCl solution (precooled to 0 °C)61. Intracellular metabolites of the quenched cell were extracted using 80% methanol (precooled to −80 °C) and incubated overnight at −80 °C. Then the samples were centrifuged at 15,000 × g for 20 min at 4 °C. The upper clear liquid was dried into a pellet by N2 flow. The dried metabolites were dissolved in 200 μL acetonitrile/methanol/water (2:2:1) solution for HPLC-QTOF analysis.
Intracellular metabolites were quantitated using the Agilent 1290 Infinity II UHPLC system tandem with 6545 Q/TOF-MS system. Chromatographic separation was achieved on an ACQUITY UPLC BEH Amide column (100 mm × 2.1 mm, 1.7 μm). The mobile phase consisted of 15 mM ammonium acetate, 0.3% NH3·H2O in water (A) and 15 mM ammonium acetate, 0.3% NH3·H2O in 9:1 acetonitrile/water (B) at a flow rate of 0.3 mL/min. In detail, the column was eluted with 95% mobile phase B for 1 min, followed by a linear gradient to 50% mobile-phase B over 8 min, held at 50% for 3 min, a linear gradient to 95% mobile phase B over 0.5 min, then 1.5 min at 95% mobile-phase B. The sample volume injected was 5 μL. MS data were acquired using electrospray ionization in both positive and negative ion mode over 50–1250 m/z. The parameters used were as follows: sheath gas temperature, 350 °C; sheath gas flow, 11 L min−1; capillary voltage, 4000 V; nozzle voltage, 1000 V; gas temperature, 350 °C; nebulizer gas, 30 psi; drying gas flow rate, 8 L/min; fragmentor, 110 V; skimmer, 65 V. Raw data were processed using Profinder 10.0 (Agilent, USA) for peak detection, alignment, and integration.
Network model of CHO cells metabolism and 13C metabolic flux analysis
A compartmentalized metabolic network model of CHO cell metabolism, provided in Supplementary Data 5, was employed for metabolic flux analysis62. Totally 84 reactions and 51 metabolites were included in the model. Isotopic non-stationary 13C-MFA was performed using INCA (v2.3, Vanderbilt University, USA)32.
Growth characterization of P. tricornutum and S. elongatus
Growth curves were monitored every 12 or 24 h by measuring the optical density (OD)53. In short, the optical density of the culture was determined by photometrical analysis (P4 UV-visible spectrophotometer, MAPADA, China) at 750 nm. Samples were diluted with ultrapure water by 1:10 when samples showed an OD750 value above 0.5. At least three biological replicates were conducted for every strain. For P. tricornutum strains, cells were continually grown in f/2 medium with 0.1 M glycerin at 18 °C. Normally, the cell density of P. tricornutum was indicated as an absorbance at 750 nm (OD750) with fresh f/2 medium, which was used to inoculate the cultures, as a blank reference. All further steps were conducted as described above for cyanobacterial cells.
Determination of the intracellular ratio of NAD+/NADH in algae
All the algae cultures used for the NAD+/NADH determination experiment were grown photo-mixotrophically in BG11 medium or f/2 medium. For diatom, cells were cultured in f/2 medium and ~2 × 106 algal cells were collected by centrifugation at 3500 × g at −9 °C for 10 min. Then the pellets were washed with 1 mL 20 mM cold phosphate-buffered saline (20 mM KH2PO4, 20 mM K2HPO4, and 150 mM NaCl) and disrupted by Bioruptor® PLUS sonicator (Diagenode, Belgium) combined with the Bioruptor® Water cooler (High model, 60 cycles of 10 s ON/20 s OFF). The supernatant was collected via centrifugation at 12,000 × g for 1 min at −9 °C. For the cyanobacteria cell, the pellet was resuspended in 50 μL extraction buffer with precooled glass beads (Ø = 0.18 mm). The mixture was vortexed four times for 1 min in the cold room (4 °C) and intermittently chilled on the ice for 1 min. Then 150 μL extraction buffer was added again and the mixture was centrifuged at 3500 × g for 10 min at −9 °C. The liquid phase was transferred as much as possible into a new reaction cup and centrifuged at maximum speed for 30 min at −9 °C. Then the supernatant was applied for NAD+/NADH ratio detection via the NAD+/NADH Assay kit (Beyotime, S0175, China).
Determination of free fatty acids in algae by GC-MS
For fatty acids profiling analysis, total lipids were extracted and analyzed by gas chromatography–mass spectrometry (GC-MS). Cells with a total OD750 of 3 in the logarithmic growth phase were harvested by centrifugation at 3000 × g for 5 min at 4 °C. Then 1 mL of Buffer I (2 M NaOH dissolved in MeOH: H2O (1:1, v/v) solution) was added to each tube. Cells were resuspended and transferred to 10 mL glass tubes. 20 μL of C19:0 (1 mg mL−1, 72332-1G-F, Sigma-Aldrich, Germany) was added as internal reference and vortexed to mix. Each tube was incubated at 100 °C for 3 min. After cooling to room temperature, 3 mL of 2 M HCl solution (Pure methanol as solvent) was added to each tube. The mixture was incubated at 85 °C for 30 min. The samples were then cooled to room temperature again, followed by the mixture with 2 mL of pure hexane. 1.8 mL of supernatant was subsequently collected in a 15 mL conical centrifuge tube after centrifugation at 4000 × g for 10 min at 4 °C. After drying under a nitrogen stream in a Nitrogen Evaporator (Yooning, DC150-2, China), 100 μL of hexane was added to resuspend the dried remains. The mixture was transferred to a 1.5 mL centrifuge tube, followed by centrifugation at 12,000 × g for 10 min. About 80 μL of the supernatant was transferred to a brown sampling bottle as samples prepared for the following detection by GC-MS (Agilent, 8890-7077, USA) equipped with HP-88 column (60 m × 0.25 mm × 0.2 μM) (Agilent, USA). Helium was used as the carrier gas, and the stigma pressure was 16.884 psi. The total flow rate at the outlet of the shunt was 44 mL min−1, and the septum purge flow rate was 3 mL min−1. The injector was set at 240 °C using the no-split injection mode, and 1 μL was injected. The temperature gradient was linearly increased from an initial temperature of 50 to 240 °C at a rate of 20 °C min−1 and finally held for 13 min. The total running time was 22.5 min. Standard products were purchased as fatty acid methyl esters from SUPELCO (CRMSupelco 37 Component FAME Mix, USA) and stored at −20 °C prior to use. The fatty acids were quantitatively analyzed by determining integrated peak areas, and relative (percentage) contents of detected fatty acids were calculated.
Growth characterization of S. cerevisiae
The optical density (OD) of culture was determined by photometrical analysis (P4 UV-Visible Spectrophotometer, MAPADA, China). Normally, the cell density of S. cerevisiae was indicated as the absorbance at 600 nm (OD600) with fresh YPG medium, which was used to inoculate the cultures, as a blank reference. Samples were diluted with YPG medium above 0.2 when inoculating the strain to a 48-well deep well plate of Microscreen high-throughput microbial growth analysis system (JIELING, China).
Determination of respiratory rate, citric acid titer and dry weight in A. niger
For the cultivation of A. niger, a liquefied corn medium was applied for citric acid production. Briefly, 200 g of corn flour were mixed with 1 g of CaCl2 in 500 mL of distilled water, and the pH was adjusted to 6.2 with Ca (OH)2 solution. After adding 300 μL of α-amylase, the mixture was heated at 70 °C for 1 h, followed by heating at 90 °C for 1 h. Then the mixture was boiled to inactive α-amylase and filtered through gauze. Finally, water was added to reach a total volume of 1 L (pH 5.5–6.0), and the medium was sterilized at 121 °C for 15 min. For real-time respiratory rate detection, the WT and engineered A. niger strains were seeded at 105 spores mL−1 in 20 mL liquefied corn media, and cultivated at 34 °C and 180 rpm for 96 h in a bioreactor equipped with a respiration activity monitoring system (RAMOS) (Zang Tech GmbH, Germany). For the citric acid fermentation, the inoculum was cultured in 50 mL liquefied corn medium at a concentration of 105 spores mL−1 in shaking flasks. The cultures were maintained at 34 °C and 220 rpm for 96 h. For bioreactor cultivation, strains were seeded at 105 spores mL−1 in a DASGIP system (Eppendorf, Germany) with 500 mL liquefied corn medium and cultured at 34 °C for 96 h. The aeration rate was coupled to dissolved oxygen concentration above 60%. The samples were collected every 24 h for dry weight and citric acid analysis.
Supernatants and mycelia were separated by centrifugation at 3500 × g for 20 min. Fungal biomass was determined by weighing the fungal dry mycelia. Samples were collected in pre-weighted falcon tubes and incubated at 50 °C until dry, and then the dry mycelia were weighted. Supernatants were boiled at 95 °C for 10 min and filtered through a 0.22-μm sterile filter membrane. Extracellular citric acid was detected after a 20-fold dilution with sterile distilled water using HPLC equipped with a UV detector (Agilent, USA) and an Aminex HPX-87H column (300 × 7.8 mm, Agilent, USA)54. The mobile phase is 5 mM H2SO4 with 0.4 mL/min of flow rate. The sample volume injected is 10 μL. The separation is carried out at 55 °C for 30 min. The absorbance wavelength is 210 nm.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this work are available within the paper and its Supplementary Information files. A Reporting Summary for this article is available as a Supplementary Information file. Source data are provided with this paper.
References
Rowland, E. A., Snowden, C. K. & Cristea, I. M. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 42, 76–85 (2018).
Cronan, J. E. Lipoic acid attachment to proteins: stimulating new developments. Microbiol. Mol. Biol. R. 88, 23 (2024).
Lin, C. H. et al. Protein lipoylation: mitochondria, cuproptosis, and beyond. Trends Biochem. Sci. 49, 729–744 (2024).
Lai, S. C. et al. Quantitative site-specific chemoproteomic profiling of protein lipoylation. J. Am. Chem. Soc. 144, 10320–10329 (2022).
Tort, F., Ferrer-Cortes, X. & Ribes, A. Differential diagnosis of lipoic acid synthesis defects. J. Inherit. Metab. Dis. 39, 781–793 (2016).
Spalding, M. D. & Prigge, S. T. Lipoic acid metabolism in microbial pathogens. Microbiol. Mol. Biol. R. 74, 200 (2010).
Cao, X. Y. et al. Lipoate-binding proteins and specific lipoate-protein ligases in microbial sulfur oxidation reveal an atpyical role for an old cofactor. eLife 7, 22 (2018).
Hartley, F., Walker, T., Chung, V. & Morten, K. Mechanisms driving the lactate switch in Chinese hamster ovary cells. Biotechnol. Bioeng. 115, 1890–1903 (2018).
Lambrechts, R. A. et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 11, 21 (2019).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254 (2022).
Tsvetkov, P. et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 15, 681–689 (2019).
Solmonson, A. & DeBerardinis, R. J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 293, 7522–7530 (2018).
Paredes, F. et al. Poldip2 is an oxygen-sensitive protein that controls PDH and αKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc. Natl Acad. Sci. USA 115, 1789–1794 (2018).
Habarou, F. et al. Biallelic mutations in LIPT2 cause a mitochondrial lipoylation defect associated with severe neonatal encephalopathy. Am. J. Hum. Genet. 101, 283–290 (2017).
Pietikäinen, L. P., Rahman, M. T., Hiltunen, J. K., Dieckmann, C. L. & Kastaniotis, A. J. Genetic dissection of the mitochondrial lipoylation pathway in yeast. BMC Biol. 19, 14 (2021).
Choi, K. R. et al. Systems metabolic engineering strategies: integrating systems and synthetic biology with metabolic engineering. Trends Biotechnol. 37, 817–837 (2019).
Cheong, S., Clomburg, J. M. & Gonzalez, R. Energy- and carbon-efficient synthesis of functionalized small molecules in bacteria using non-decarboxylative Claisen condensation reactions. Nat. Biotechnol. 34, 556–561 (2016).
Sacco, S. A. et al. Overexpression of peroxisome proliferator-activated receptor γ co-activator-1a (PGC-1a) in Chinese hamster ovary cells increases oxidative metabolism and IgG productivity. Metab. Eng. 79, 108–117 (2023).
Liaud, M. F., Lichtlé, C., Apt, K., Martin, W. & Cerff, R. Compartment-specific isoforms of TPI and GAPDH are imported into diatom mitochondria as a fusion protein: evidence in favor of a mitochondrial origin of the eukaryotic glycolytic pathway. Mol. Biol. Evol. 17, 213–223 (2000).
Perham, R. N. Swinging arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annu. Rev. Biochem. 69, 961–1004 (2000).
Tsai, C. S., Burgett, M. W. & Reed, L. J. Alpha-keto acid dehydrogenase complexes .10. kinetic study of pyruvate-dehydrogenase complex from bovine kidney. J. Biol. Chem. 248, 8348–8352 (1973).
Mathias, R. A. et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 (2014).
Rowland, E. A. et al. Sirtuin lipoamidase activity is conserved in bacteria as a regulator of metabolic enzyme complexes. mBio 8, 15 (2017).
Moller, J. et al. Regulation of pyruvate dehydrogenase complex related to lactate switch in CHO cells. Eng. Life Sci. 21, 100–114 (2021).
Kim, J. W., Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Chen, R. et al. Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Front. Cell Dev. Biol. https://doi.org/10.3389/fcell.2018.00132 (2018).
Majewska, N. I., Tejada, M. L., Betenbaugh, M. J. & Agarwal, N. N-glycosylation of IgG and IgG-like recombinant therapeutic proteins: why is it important and how can we control it?. Annu. Rev. Chem. Biomol. 11, 311–338 (2020).
Donaldson, J., Kleinjan, D. J. & Rosser, S. Synthetic biology approaches for dynamic CHO cell engineering. Curr. Opin. Biotechnol. 78, 9 (2022).
Saldanha, M., Padhye, K., Warke, V. G., Dandekar, P. & Jain, R. A feed enrichment strategy targeting the tricarboxylic acid cycle for increasing monoclonal antibody production and alleviating ammonia accumulation in Chinese hamster ovary cell culture. Biochem. Eng. J. 192, 11 (2023).
Buchsteiner, M., Quek, L. E., Gray, P. & Nielsen, L. K. Improving culture performance and antibody production in CHO cell culture processes by reducing the Warburg effect. Biotechnol. Bioeng. 115, 2315–2327 (2018).
Zhang, X., Hu, Z., Pörtner, R. & Zeng, A.-P. Hybrid biological hydrogel provides favorable bioenergetic, adhesive, and antioxidative effects on wound healing. ACS Biomater. Sci. Eng. https://doi.org/10.1021/acsbiomaterials.5c00072 (2025).
Young, J. D. INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335 (2014).
Kirsch, B. J. et al. Metabolic analysis of the asparagine and glutamine dynamics in an industrial Chinese hamster ovary fed-batch process. Biotechnol. Bioeng. 119, 807–819 (2022).
Wan, Q. F., Wang, Y. L. & Tang, H. R. Quantitative <SUP>13</SUP>C traces of glucose fate in hepatitis B virus -infected hepatocytes. Anal. Chem. 89, 3293–3299 (2017).
Zeng, A. P., Hu, W. S. & Deckwer, W. D. Variation of stoichiometric ratios and their correlation for monitoring and control of animal cell cultures. Biotechnol. Progr. 14, 434–441 (1998).
Cronan, J. E. Progress in the enzymology of the mitochondrial diseases of lipoic acid requiring enzymes. Front. Genet. 11, 6 (2020).
Yasuno, R. & Wada, H. The biosynthetic pathway for lipoic acid is present in plastids and mitochondria in Arabidopsis thaliana. FEBS Lett. 517, 110–114 (2002).
Ewald, R. et al. Lipoate-protein ligase and octanoyltransferase are essential for protein lipoylation in mitochondria of Arabidopsis. Plant Physiol. 165, 978–990 (2014).
Kawano, I., Bazila, B., Ježek, P. & Dlasková, A. Mitochondrial dynamics and cristae shape changes during metabolic reprogramming. Antioxid. Redox Signal. 39, 684–707 (2023).
Zhang, H., Li, Y. C., Nie, J. L., Ren, J. & Zeng, A. P. Structure-based dynamic analysis of the glycine cleavage system suggests key residues for control of a key reaction step. Commun. Biol. 3, 12 (2020).
Bauwe, H., Hagemann, M. & Fernie, A. R. Photorespiration: players, partners and origin. Trends Plant Sci. 15, 330–336 (2010).
Makowka, A. et al. Glycolytic shunts replenish the Calvin-Benson-Bassham cycle as anaplerotic reactions in Cyanobacteria. Mol. Plant 13, 471–482 (2020).
Villanova, V. et al. Investigating mixotrophic metabolism in the model diatom Phaeodactylum tricornutum. Philos. Trans. R Soc. Lond. B Biol. Sci. https://doi.org/10.1098/rstb.2016.0404 (2017).
Bick, N. et al. Engineered bacterial lipoate protein ligase A (lplA) restores lipoylation in cell models of lipoylation deficiency. J. Biol. Chem. https://doi.org/10.1016/j.jbc.2024.107995 (2024).
Ramazi, S. & Zahiri, J. Post-translational modifications in proteins: resources, tools and prediction methods. Database https://doi.org/10.1093/database/baab012 (2021).
Peng, Z. Y. et al. Protein acylation links metabolism and the control of signal transduction, transcription regulation, growth, and pathogenicity in Actinobacteria. Mol. Microbiol. 119, 151–160 (2023).
Nie, J. L. et al. A globular protein exhibits rare phase behavior and forms chemically regulated orthogonal condensates in cells. Nat. Commun. https://doi.org/10.1038/s41467-025-57886-4 (2025).
Cao, T. et al. An unexpected hydratase synthesizes the green light-absorbing pigment fucoxanthin. Plant Cell 35, 3053–3072 (2023).
Liaud, M. F., Lichtle, C., Apt, K., Martin, W. & Cerff, R. Compartment-specific isoforms of TPI and GAPDH are imported into diatom mitochondria as a fusion protein: evidence in favor of a mitochondrial origin of the eukaryotic glycolytic pathway. Mol. Biol. Evol. 17, 213–223 (2000).
Bai, Y. et al. Green diatom mutants reveal an intricate biosynthetic pathway of fucoxanthin. Proc. Natl Acad. Sci. USA 119, e2203708119 (2022).
Karas, B. J. et al. Designer diatom episomes delivered by bacterial conjugation. Nat. Commun. 6, 6925 (2015).
Taton, A. et al. Broad-host-range vector system for synthetic biology and biotechnology in cyanobacteria. Nucleic Acids Res. 42, e136 (2014).
Wang, Y. et al. Pyruvate:ferredoxin oxidoreductase and low abundant ferredoxins support aerobic photomixotrophic growth in Cyanobacteria. Elife https://doi.org/10.7554/eLife.71339 (2022).
Zhang, L. H. et al. Disruption or reduced expression of the orotidine-5′-decarboxylase gene pyrG increases citric acid production: a new discovery during recyclable genome editing in Aspergillus niger. Microb. Cell. Fact. 19, 12 (2020).
Kowalczyk, J. E. et al. Combinatorial control of gene expression in Aspergillus niger grown on sugar beet pectin. Sci. Rep. 7, 12 (2017).
de Vries, R. P. et al. A new black Aspergillus species, A. vadensis, is a promising host for homologous and heterologous protein production. Appl. Environ. Micro. 70, 3954–3959 (2004).
Burgstaller, H. et al. Synechocystis sp. PCC 6803 requires the bidirectional hydrogenase to metabolize glucose and arginine under oxic conditions. Front. Microbiol. 13, 16 (2022).
Clayton, D. A. & Shadel, G. S. Isolation of mitochondria from cells and tissues. Cold Spring Harb. Protoc. 2014, pdb.top074542 (2014).
Desousa, B. R. et al. Calculation of ATP production rates using the Seahorse XF Analyzer. EMBO Rep. https://doi.org/10.15252/embr.202256380 (2023).
Arias-Reyes, C., Raduan, F. A., Pinto-Aparicio, R., Joseph, V. & Soliz, J. Mitochondrial plasticity in the retrosplenial cortex enhances ATP synthesis during acclimatization to hypoxia in mice but not in rats. MitoFit Preprints https://doi.org/10.26124/mitofit:2023-0006 (2023).
Dietmair, S., Timmins, N. E., Gray, P. P., Nielsen, L. K. & Kromer, J. O. Towards quantitative metabolomics of mammalian cells: development of a metabolite extraction protocol. Anal. Biochem. 404, 155–164 (2010).
Ahn, W. S. & Antoniewicz, M. R. Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab. Eng. 13, 598–609 (2011).
Acknowledgements
We are grateful to Prof. Peiguo Yang from Westlake University for providing the plasmids for U2OS and adherent CHO cells transfection, Ruihui Li and Yiran Jia for their assistance in cultivating CHO cells, Zhengyang Xiao for his help in metabolic flux analysis, WuXi Biologics for construction of IgG-producing CHO cell lines and industrial cultivation of suspension CHO cell lines, Dr. Tianjun Cao and Dr. Xiaobo Li for supporting wild-type algae strains and technical assistance in diatom mutant construction, Xin Cui for technical assistance in using Clark-type oxygen electrode, Dr. Yajie Wang from Westlake University for providing the yeast strain, Dr. Ping Zheng from CAS Tianjin Institute of Industrial Biotechnology for providing the A. niger strain for citric acid production, the instruments support from Advanced Biomedical Technology Core Facilities in Westlake University including the Flow Cytometry Platform, Optical Microscope Platform and Mass Spectrometry & Metabolomics Core Facility. This research was supported by a grant from National Key Research and Development Program of China (No. 2022YFA0912000) to A.Z., a grant from the National Natural Science Foundation of China (32200310) to Y.W., a project (WU2022A002) from the Center of Synthetic Biology and Integrated Bioengineering of Westlake University.
Author information
Authors and Affiliations
Contributions
A.Z. and Z.H. conceived the study. R.Y., Y.W., M.K., Z.Z., J.M., Z.H. and A.Z. designed experiments. R.Y., Y.W., M.K., Z.Z., J.M., and K.S. performed experiments and data analysis. R.Y., Y.W., M.K., and A.Z. wrote the manuscript with input from the other authors. All authors read, corrected, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors A.Z., Z.H., R.Y., and Y.W. are inventors on PCT patent application PCT/CN2023/103401, “Regulation of cellular energy metabolism by lipoate protein ligase and its applications”, submitted by Westlake University. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jiazhang Lian and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, R., Wang, Y., Kong, M. et al. Boosting energy metabolism and biosynthesis in diverse organisms by a common bacterial salvage lipoylation protein. Nat Commun 16, 7540 (2025). https://doi.org/10.1038/s41467-025-62638-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62638-5
