
Abstract
Hybrid seed failure arising from wide crosses between plant species is a recurring obstacle in plant breeding, impeding the transfer of desirable traits. This postzygotic reproductive barrier primarily occurs in the endosperm, a tissue that nourishes the embryo and functions similarly to the placenta in mammals. We found that incompatible seeds show a loss of DNA methylation and chromatin condensation in the endosperm, similar to seeds lacking maternal RNA polymerase IV activity. This similarity is linked to a decrease in small interfering RNAs in the endosperm (sirenRNAs), maternal RNA polymerase IV-dependent short interfering RNAs that regulate DNA methylation. Several AGAMOUS-like MADS-box transcription factor genes (AGLs), key regulators of endosperm development, are targeted by sirenRNAs in cis and in trans. This finding aligns with the enrichment of AGL target genes among deregulated genes. We propose that hybrid seed failure results from reduced maternal sirenRNAs combined with increased AGL expression, leading to abnormal gene regulation in the endosperm.
Similar content being viewed by others

Main
Hybrid seed failure resulting from wide crosses between plant species poses a recurring challenge in plant breeding, obstructing the transfer of desirable traits between species. This postzygotic reproductive barrier predominantly manifests in the endosperm—an embryo-nourishing tissue serving a similar function as the placenta in mammals1. In most flowering plants, the endosperm is triploid, comprising one paternal and two maternal genomes. This specific genomic ratio is pivotal for endosperm viability, as evidenced by seed abortion upon hybridization of plants with differing ploidy2,3,4. Much like failures in interploidy hybridization, crosses between closely related species with equal ploidy often result in seed arrest. The term ‘effective ploidy’ has been used to characterize this phenomenon, wherein certain species exhibit behaviour reminiscent of polyploid species, despite their actual diploid nature5. Remarkably, seeds arrested from both interploidy and interspecies crosses exhibit similar phenotypic and molecular abnormalities6,7,8, implying the existence of a dosage-sensitive element contributing to this reproductive barrier within the endosperm. However, the precise nature of this component remains elusive.
In the Capsella genus, interspecies hybridizations between the inbreeder C. rubella (Cr) and the outbreeder C. grandiflora (Cg) lead to a loss of chromatin condensation and DNA methylation in the endosperm, particularly in the CHG and CHH contexts (H represents all bases except G). This chromatin change is associated with an accumulation of deregulated genes in pericentromeric regions, many of which are targeted by type I AGAMOUS-like MADS-box transcription factors (AGLs)9. Type I AGLs belonging to the Mγ and Mγ-interacting Mα* clades are key regulators of endosperm proliferation and cellularization10. These data suggest that hypomethylation in the hybrid endosperm facilitates the access of AGLs to hypomethylated regions, resulting in detrimental gene activation. Similarly, in Arabidopsis, interploidy hybridizations of diploid (2x) maternal plants with tetraploid (4x) paternal plants causes endosperm hypomethylation in the CHG and CHH contexts11, which has been linked to a disruption of the RNA-directed DNA methylation (RdDM) pathway in the hybrid endosperm12. This pathway initiates with the plant-specific DNA-dependent RNA polymerase IV (Pol IV) generating short non-coding transcripts that are transformed into double-stranded RNA by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) and further processed into 24-nucleotide short interfering RNAs (siRNAs) by DICER-LIKE3. These siRNAs, when incorporated into ARGONAUTE (AGO) proteins, guide DNA methyltransferases to target sequences, inducing DNA methylation13. Notably, both ovules and endosperm accumulate highly abundant siRNAs over a modest number of distinct loci, commonly referred to as sirenRNAs (small interfering RNAs in the endosperm) that are primarily of maternal origin14,15,16. In Arabidopsis, sirenRNAs target AGLs, and their abundance is reduced in triploid seeds derived from crosses between 2x and 4x plants17. Nonetheless, whether sirenRNAs constitute the dosage-sensitive component determining hybridization success remains an open question.
Previous work revealed that the loss of paternal Pol IV function can suppress the interploidy hybridization barrier in Arabidopsis11,14. Interestingly, affected genes in the endosperm differ depending on whether the nrpd1 mutation (disrupting Pol IV function) is maternally or paternally inherited, suggesting that maternal and paternal Pol IV-dependent siRNAs have distinct effects on endosperm development for reasons that remain to be fully elucidated11,18.
In this study, we demonstrate that the loss of maternal Pol IV function mirrors the loss of chromatin condensation and DNA methylation observed in Capsella hybrids. This resemblance in phenotype is correlated with a shared decrease in sirenRNAs, which regulate target genes in cis and trans by guiding DNA methylation. Among these sirenRNA targets are several AGLs, aligning with the enrichment of AGL target genes among deregulated genes. On the basis of these data, we propose that the response to interspecies and interploidy hybridizations is instigated by two factors: the depletion of maternally derived sirenRNAs and the heightened expression of AGLs targeting hypomethylated regions in hybrid endosperm.
Results
Capsella interploidy and interspecies hybrids share similar defects
To test whether interploidy and interspecies hybridization share a common molecular basis, we analysed the cross between 2x maternal and 4x paternal Cr plants (referred to as Cr × 4xCr; by convention the female parent is always mentioned first). As previously reported for paternal-excess interploidy crosses in Arabidopsis3, the resulting 3xCr seeds were dark and shrivelled and failed to germinate (Fig. 1a and Extended Data Fig. 1a; the term ‘paternal-excess’ is used to refer to crosses where the paternal parent has higher numerical or effective ploidy than the maternal parent). The defects of 3xCr seeds were related to failed endosperm cellularization (Extended Data Fig. 1b), resembling previously reported defects of interspecies paternal-excess hybrid seeds in Capsella, Arabidopsis and other species3,6,19,20. As in the case of interspecific hybrids21, 3x embryos could be rescued by removing them from the seeds and growing them in vitro (Extended Data Fig. 1c), supporting the view that the failure of 3x embryo survival is a consequence of a defect in endosperm development.

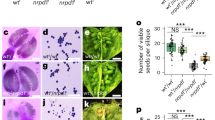
a, Cleared hybrid Capsella seeds at 4 to 6 DAP. The corresponding mature seeds are shown on the right side of each panel. Scale bars, 100 µm and 1 mm for cleared and mature seeds, respectively. The colour code reflects effective ploidy as indicated in the figure. b, Mature seed phenotypes of crosses Cr × Cbp and Cbp × Cr. Scale bars, 1 mm. c, Mature seed phenotypes of crosses Co × Cr and 4xCo × Cr. Scale bars, 1 mm. d, Germination data for the crosses shown in c. Shown is the fraction of germinating seeds from each cross, with each filled circle representing one biological replicate; there are three biological replicates per genotype. The numbers at the top represent total seed numbers. The asterisks represent statistical significance as calculated by one-way analysis of variance with post-hoc Tukey’s honestly significant difference test (***P < 0.001). e, Upset plot showing the overlap of upregulated genes between interploidy (Cr × 4xCr) and interspecies crosses of different Capsella species. f, Heat map and dendrogram show clustering of samples based on log2 fold changes (compared with the corresponding maternal parent) of genes upregulated in the analysed hybrids.
To determine whether phenotypic similarities in interploidy and interspecies hybrids are reflected by a similar molecular signature, we generated transcriptome data of hybrid seeds (six days after pollination (DAP)) resulting from incompatible crosses. These included Cr × 4xCr interploidy crosses and previously characterized paternal-excess interspecies crosses of Cr × Cg and C. orientalis (Co) × Cr21 with the corresponding intraspecific crosses. We furthermore included the cross of C. bursa-pastoris (Cbp) × Cg (Supplementary Data 1). Despite Cbp being an allotetraploid species, when used as the maternal parent in crosses with Cg, Cbp exhibited strikingly similar behaviour to Cr. The resultant seeds exhibited a paternal-excess phenotype, characterized by shrinkage and abortion, with embryos arrested at the torpedo stage (Fig. 1a). Correspondingly, reciprocal crosses of Cr × Cbp gave rise to normal, viable seeds (Fig. 1b), revealing that Cbp, despite being a tetraploid species, behaved like a diploid. On the basis of the cross-incompatibilities between the species, we assigned an effective ploidy order of Co < Cr = Cbp < Cg = 4xCr, consistent with previously published work21. Using the parent with higher effective ploidy as the maternal parent gave rise to maternal-excess phenotypes21 (Extended Data Fig. 1d), supporting the assigned order of effective ploidy. In line with theoretical predictions22, this order aligned with the breeding mode of the species: while Cg is an outbreeder, Cr, Co and Cbp are inbreeding species. However, whereas Co became an inbreeder as early as ~900,000 years ago, Cr and Cbp made the transition to inbreeding only ~200,000 years ago23,24. Previous work revealed that increased ploidy of the maternal Cr parent can restore the viability of hybrid Cr × Cg seeds21. Similarly, we found that increased ploidy of the maternal Co parent could suppress Co × Cr hybrid seed defects. While seeds derived from Co × Cr crosses aborted with similar morphological and transcriptional defects as Cr × Cg, Cbp × Cg and Cr × 4xCr hybrids21 (Fig. 1a,c,d), seeds obtained from crosses of 4xCo × Cr were phenotypically like non-hybrid seeds and able to germinate (Fig. 1c,d).
We identified deregulated genes in the hybrids by comparing the hybrid transcriptome with that of the corresponding maternal parent (Fig. 1e), the paternal parent and both parents (Extended Data Fig. 1e,f). Our data revealed that all tested hybrids shared a large set of commonly upregulated genes, indicative of a common genetic basis. To test the molecular consequences of increased maternal ploidy, we profiled the transcriptomes of 4xCr × Cg and 4xCo × Cr seeds and found that most genes that were upregulated in Cr × Cg and Co × Cr hybrid seeds were either downregulated or unchanged in hybrid seeds derived from a 4x maternal plant (Fig. 1f). These data support the idea that a dosage-sensitive component generated by the maternal parent determines hybridization success. Together, these data strongly suggest that interploidy and interspecies hybridization barriers share a common molecular basis. Moreover, the fact that increased ploidy of the maternal parent can alleviate paternal-excess hybrid incompatibility implies the existence of a quantitative maternal factor determining hybridization success.
Incompatible hybrids lose chromatin condensation and CHG and CHH methylation
Previous work revealed that Cr × Cg hybrid endosperm has decondensed chromocenters, which relates to a preferential expression of genes localized in pericentromeric regions9. To test whether this phenomenon is a general pattern connected with hybrid incompatibility in the Capsella genus, we analysed the locations of upregulated genes in paternal-excess crosses in relation to their positions on the chromosomes. Strikingly, we found that in all interspecies and interploidy paternal-excess hybrids, there was a preferential location of upregulated genes close to pericentromeric regions (Fig. 2a). We tested whether this phenomenon corresponded to a loss of chromosome condensation and analysed chromocenter condensation in a range of paternal-excess incompatible hybrids. Consistent with the similar molecular phenotypes of interspecies and interploidy paternal-excess hybrid seeds, we found that all hybrid nuclei had significantly reduced chromocenter condensation compared with non-hybrid nuclei (Fig. 2b,c and Supplementary Figs. 1 and 2). Importantly, chromocenter condensation defects and gene deregulation were suppressed in the nuclei of 4xCr × Cg and 4xCo × Cr hybrids, suggesting that a dosage-sensitive maternal factor regulates chromatin condensation in the endosperm (Fig. 2a–c). While there were substantially fewer genes deregulated in the 4xCo × Cr than in the Co × Cr hybrids, deregulated genes remained enriched in pericentromeric regions (Fig. 2a). This suggests that despite visibly restored chromatin condensation upon increased maternal ploidy (Fig. 2c), these regions remained more accessible than in the wild type.

a, Percentage of genes in pericentromeric and non-pericentromeric regions that are upregulated in different incompatible Capsella hybrids. The numbers above the bars represent numbers of upregulated genes in pericentromeric and non-pericentromeric regions of a respective genotype. The asterisks represent statistical significance as calculated by two-sided chi-squared tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001; **P < 0.01; *P < 0.05). b, DAPI-stained chromocenters from endosperm nuclei at 4 DAP of different Capsella genotypes. Scale bars, 5 μm. c, Quantification of chromatin condensation given as mean circularity of chromocenters (Methods). The numbers below the boxes indicate the number of analysed nuclei. The asterisks represent statistical significance as calculated by two-sided Wilcoxon tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001; **P < 0.01; *P < 0.05). NS, not significant. d, Metagene plots showing DNA methylation levels of TEs in the endosperm of Cg × Cg, 4xCr × Cg and 4xCr × 4xCr 6 DAP seeds. e, Box plots showing methylation levels of TEs in the endosperm of 6 DAP seeds of the indicated genotypes. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001; **P < 0.01). f, Metagene plots showing DNA methylation levels of TEs in the endosperm of Cg × Cr 6 DAP compared with published data for Cr × Cr, Cg × Cg and Cr × Cg (ref. 9). g, Box plots showing methylation levels of TEs in the endosperm of 6 DAP seeds of the indicated genotypes. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001). In c,e,g, the horizontal lines show the median values, the box edges show the interquartile range and the whiskers show the full range excluding outliers.
Decondensation of pericentromeric regions in Cr × Cg and Co × Cr hybrid endosperm co-occurred with a loss of CHG and CHH methylation9, which was reversed by increasing the maternal ploidy (4xCr × Cg and 4xCo × Cr) and in the reciprocal maternal-excess Cg × Cr and Cr × Co hybrids (Fig. 2d–g and Extended Data Figs. 2a–d and 3). Similarly, the loss of chromatin condensation in the nuclei of interploidy hybrids was related to reduced CHG and CHH methylation on genes and transposable elements (TEs) (Extended Data Fig. 2e–h). The loss of DNA methylation on TEs was most prominent in pericentromeric regions (Extended Data Fig. 2i), consistent with preferential upregulation of genes in this region9. Together, these data reveal that interploidy and interspecies hybridization cause similar molecular defects consistent with the similar phenotypes of hybrid seeds.
Maternal nrpd1 causes loss of chromatin condensation and CHG and CHH methylation
The observed reduction of CHG and CHH methylation upon interploidy and interspecies hybridization9 (Fig. 2d–g and Extended Data Figs. 2 and 3) suggests a role of the RdDM pathway in establishing hybridization barriers in the endosperm. Pol IV is required to produce precursor RNAs, which are converted into 24-nucleotide siRNAs that guide de novo DNA methylation in all sequence contexts13. We thus tested whether a mutant in NRPD1, encoding the largest subunit of Pol IV, would cause a similar effect on chromatin condensation and the loss of DNA methylation. Indeed, we found that maternal loss of NRPD1 reduced chromatin condensation in the endosperm, while the loss of paternal NRPD1 had no effect, and the loss of maternal and paternal NRPD1 function did not enhance chromatin decondensation (Fig. 3a,b). Chromatin decondensation upon the loss of maternal NRPD1 function was associated with the loss of CHG and CHH methylation on genes and TEs in the endosperm, with similar regions being affected in interploidy and interspecies hybrids and in seeds depleted of maternal NRPD1 function (Fig. 3c,d and Extended Data Fig. 4a–d). This was also reflected by a large set of similarly deregulated genes in the endosperm of seeds lacking maternal NRPD1 function and that of interploidy and interspecies hybrids (Fig. 3e and Supplementary Data 1). Consistent with the similar molecular phenotype, endosperm cellularization was delayed in seeds derived from nrpd1 mutant maternal plants, leading to a 10–20% abortion rate (Extended Data Fig. 5a–c). Maternal nrpd1 also caused aggravated seed phenotypes in nrpd1 × Cg crosses and seed abortion in 4xnrpd1 × Cg crosses (Extended Data Fig. 5a,c,d). Previous studies found no evidence of NRPD1 being imprinted in Capsella21,25, suggesting that maternal Pol IV-dependent siRNAs are generated in maternal sporophytic tissues, consistent with previous work16,26. Together, we concluded that reduced dosage of maternal Pol IV-dependent siRNAs affects DNA methylation, chromatin condensation and gene expression in a similar manner as interploidy and interspecies hybridizations. These data raise the hypothesis that interploidy and interspecies hybrid endosperms are depleted of maternal Pol IV-dependent siRNAs, causing loss of DNA methylation, decondensation of pericentromeric regions and activation of genes. Similar to previous findings11,14, we found that paternal loss of NRPD1 function could suppress interploidy seed arrest (Extended Data Fig. 7e), supporting the idea that maternal and paternal Pol IV-dependent siRNAs have different functions.

a, DAPI-stained chromocenters from endosperm nuclei from Cr × Cr, Cr × nrpd1, nrpd1 × Cr and nrpd1 × nrpd1. Scale bars, 5 μm. b, Quantification of chromatin condensation given as mean circularity of chromocenters (Methods). The numbers below the boxes indicate the number of analysed nuclei. The asterisks represent statistical significance as calculated by two-sided Wilcoxon tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001). c, Metagene plots showing DNA methylation levels of TEs in the endosperm of Cr × Cr, nrpd1 × nrpd1, Cr × nrpd1 and nrpd1 × Cr 6 DAP seeds. d, Box plots showing methylation levels of TEs in the endosperm of 6 DAP seeds of the indicated genotypes. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests; P values were adjusted for multiple comparisons with Benjamini and Hochberg correction (***P < 0.001; **P < 0.01). In b and d, the horizontal lines show the median values, the box edges show the interquartile range and the whiskers show the full range excluding outliers. e, Upset plot showing the overlap of upregulated genes among Cr × 4xCr, nrpd1 × nrpd1, nrpd1 × Cr and Cr × Cg derived seeds. Genes were considered as upregulated on the basis of log2(fold change) > 1 and Padj < 0.05 compared with Cr × Cr.
Maternal sirenRNAs are depleted in hybrid endosperm
To understand the role of maternal NRPD1 in hybrid incompatibility, we sequenced small RNAs (sRNAs) in manually dissected endosperm of 6 DAP seeds derived from crosses of Cr × Cr, Cg × Cg, Cr × Cg, Cg × Cr, 4xCr × Cg, nrpd1 × Cr and nrpd1 × nrpd1 (Supplementary Table 1). Profiles of sRNAs correlated well among biological replicates of the same genotype but were clearly distinct between genotypes (Extended Data Fig. 6a,b). Consistent with previous work14, we found that 24-nucleotide siRNAs were the predominant siRNA species in the endosperm (Extended Data Fig. 6c). Using ShortStack27, we identified 13,616 clusters accumulating 24-nucleotide siRNAs in the endosperm of Cr × Cr seeds. Of those clusters, only a few loci produced high levels of siRNAs in Cr × Cr endosperm, resembling the characteristics of siren loci15,16 (Fig. 4a and Supplementary Data 2). We identified 1,385 loci giving rise to 90% of the total siRNAs in Cr × Cr endosperm, which we will refer to as siren loci. As previously reported16, siren loci were longer than other loci expressing siRNAs (Extended Data Fig. 6d) and overlapped TEs, intergenic regions and genes (Extended Data Fig. 6e). SirenRNA loci were enriched for 24-nucleotide siRNAs (Extended Data Fig. 6f) with an adenine bias at the 5′ nucleotide (Extended Data Fig. 6g), suggesting interaction with AGO4-related AGO proteins28, consistent with previous observations in Brassica16. SirenRNA loci are distributed over the length of the chromosomes with an enrichment in pericentromeric regions (Extended Data Fig. 6h,i), but without a pronounced preference for any specific TE family (Extended Data Fig. 6j). We tested the percentage of maternally produced sirenRNAs by analysing the number of siren loci having reduced siRNAs in the nrpd1 × Cr cross. We found that out of 1,385 sirenRNA loci, 985 (71.12%) had at least twofold reduced siRNAs. Similarly, in the nrpd1 × nrpd1 endosperm, 1,220 siren loci (88.09%) were depleted by at least twofold (Fig. 4b–d). There are conflicting data on the origin of sirenRNAs; while they have been proposed to be predominantly maternally produced in Brassica16,29, biparental production was proposed in Arabidopsis18. Our data suggest that most likely both the maternal seed coat and the endosperm are the source of sirenRNAs in the endosperm, with a predominant fraction being maternally derived. Like in maternal nrpd1 endosperm, in Cr × Cg hybrid endosperm sirenRNA abundance was strongly depleted (Fig. 4b–d). Nearly all loci losing siRNAs in Cr × Cg endosperm corresponded to siren loci (Fig. 4c,d), strongly suggesting that maternally produced sirenRNAs are the dosage-sensitive component that becomes depleted in hybrid endosperm. Consistent with this idea, increased maternal genome dosage restored sirenRNAs in 4xCr × Cg hybrids (Fig. 4d). To further test the parental origin of sirenRNAs, we made a parental-specific analysis of our sRNA data and identified several loci with sufficient coverage of parental-specific reads. In the Cr × Cg endosperm, out of 158 sirenRNA loci (≥15 parental-specific reads), 103 clusters (65.19%) were maternally biased (maternal:paternal ratio, >4). Using the same criteria, in the Cg × Cr cross, out of 222 sirenRNA clusters, 184 clusters (82.88%) were maternally biased, and in the 4xCr × Cg cross, out of 227 sirenRNAs, 152 clusters (66.96%) were maternally biased (Fig. 4f,g). Mapping reads from Cr × Cr and Cg × Cg to their respective genomes revealed variations in the level and specificity of sirenRNAs. Specifically, when Cg × Cg reads were mapped to the Cr genome, there was an apparent decrease in sirenRNA levels, which was also observed when mapping Cr × Cr reads to the Cg genome. These data reveal that at least some sirenRNAs are species-specific and that Cg accumulates substantially higher levels of sirenRNAs than Cr (Fig. 4b,e). This was also reflected in substantially higher siRNA levels in Cg × Cr hybrids at loci losing siRNAs in the reciprocal Cr × Cg hybrid (Extended Data Fig. 7a).

a, Cumulative expression plot of 18–25-nucleotide siRNA loci in endosperm and leaves. Only loci with expression ≥2 reads per million (RPM) in combined replicates were analysed (n = 13,616 in endosperm and 13,495 in leaves). In endosperm and leaves, 0.99% and 27.6% of the most highly expressed 24-nucleotide-dominant loci account for 90% of siRNAs, respectively. b, Mean siRNA accumulation at siren loci in the endosperm of the indicated genotypes and Cr leaves. siRNA abundance at each locus is normalized by locus length (in kb) and library size. The mean of two replicates is presented. The black vertical lines represent medians. Individual measurements are shown as the rug below each density. The left plots show data obtained by mapping reads to the Cr genome; the right plots show data obtained by mapping reads to the Cg genome. RPKM, reads per kilobase million. c, Venn diagram showing the overlap among siren loci, loci with siRNA depletion in the Cr × Cg hybrid and loci with siRNA depletion in nrpd1 × Cr (log2(fold change) < 0, Padj < 0.1). The significance of overlap was calculated using the supertest function from the SuperExactTest package in R70 (***P < 0.001). d, Box plots showing the accumulation of siRNAs (18–25 nucleotides) on siren loci in different genotypes. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests (***P < 0.001). e, siRNA accumulation and CHH methylation in the endosperm of the indicated genotypes on selected genes overlapping siren loci (marked with violet lines). The blue boxes represent genes; the purple boxes represent TEs. f, Parental-specific accumulation of sRNAs on loci having at least 15 parental-specific reads in total. The numbers above the boxes indicate the number of such genes in each genotype. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests (***P <0.001). In d and f, the horizontal lines show the median values, the box edges show the interquartile range and the whiskers show the full range excluding outliers. g, Parental-specific sRNA accumulation in the endosperm of the indicated genotypes on selected genes. The blue boxes represent genes; the purple boxes represent TEs.
Siren loci were heavily methylated, corresponding to the production of high levels of sirenRNAs (Figs. 4e and 5a). The depletion of sirenRNAs in Cr × Cg and nrpd1 × Cr endosperm corresponded to increased mRNA levels of many genes overlapping those loci (Fig. 5b–d), indicating that sirenRNAs negatively regulate gene expression, consistent with previous work29,30. Transcriptional changes were related to a loss of CHG and CHH methylation; however, this loss was not significant in the hybrid endosperm (Figs. 4b and 5e).

a, Comparison of DNA methylation levels in genes (gene body), promoters and TEs overlapping siren loci with all genes and TEs in the Cr genome. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests (***P < 0.001). b, Expression of genes overlapping siren loci in Cr × Cr, Cr × Cg and nrpd1 × Cr seeds at 6 DAP. The asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests (***P < 0.001; **P < 0.01). c, Heat map and dendrogram show clustering of samples based on log2 fold changes (compared with the corresponding maternal parent) of genes overlapping siren loci. d, Scatter plots showing Pearson’s correlation (r) (two-sided) between the loss of siRNAs and changes in genes expression (left) and CHH methylation (right). e, Comparison of DNA methylation in Cr × Cr versus nrpd1 × Cr (left) and Cr × Cr versus Cr × Cg (right) endosperm on upregulated genes (in the respective crosses) overlapping siren loci. The asterisks indicate statistically significant differences as calculated by a two-sided Wilcoxon test (*P < 0.05). In a, b and e, the horizontal lines show the median values, the box edges show the interquartile range and the whiskers show the full range excluding outliers.
The depletion of maternal 24-nucleotide siRNAs in the endosperm of hybrid tomato seeds was related to decreased expression of Pol IV-subunit-encoding genes NRPD1 and NRPD2 (ref. 31). We tested the expression of RdDM components in hybrid seed transcriptomes and found significantly reduced NRPD1 transcript levels in Cr × Cg, Co × Cr and Cr × 4xCr endosperm (Extended Data Fig. 7c). However, we also found significantly reduced NRPD1 transcripts in 4xCr × Cr endosperm (Extended Data Fig. 7c), making it unlikely that reduced NRPD1 expression is causally responsible for the loss of sirenRNAs in the hybrids.
Depletion of sirenRNAs causes loss of methylation at trans targets
Previous work revealed that sirenRNAs can methylate other genomic sequences in trans29. We identified sirenRNA trans targets by mapping sirenRNAs to the genome masked for sirenRNA-producing loci by allowing two mismatches. We found that loci accumulating higher levels (≥4) of non-perfectly matching sirenRNAs had higher methylation levels in the CHG and CHH contexts (Fig. 6a). SirenRNAs targeted genes and TEs, with dosage-dependent effects on DNA methylation being most prominent on TEs (Fig. 6b). Together, these data strongly suggest that sirenRNAs guide DNA methylation in trans in the endosperm, like their proposed function in ovules29. Consistent with this idea, we found a decline of CHG and CHH methylation at trans targets in Cr × Cg hybrid endosperm that correlated with the dosage of depleted sirenRNAs (Fig. 6c,d). Conversely, the gain of CHG and CHH methylation upon increased maternal ploidy (4xCr × Cg and 4xCo × Cr) and in maternal-excess hybrids (Cg × Cr and Cr × Co) occurred on both maternal and paternal alleles (Extended Data Fig. 8), consistent with the idea that maternal siRNAs have trans-acting activity.

a, CHG and CHH DNA methylation at sirenRNA trans targets (50-bp genomic windows, with ≥1 RPM realigning sirenRNAs and ≤2 mismatches) in Cr × Cr endosperm. The numbers on the plots show the number of windows in each category; the breaking points correspond to 0.7, 0.9, 0.98 and 0.995 quantiles. Rand, random. b, CHG and CHH DNA methylation at sirenRNA trans targets overlapping genes (coding region +2 kb promoters; top) and TEs (bottom). The numbers on the plots show the number of windows in each category; the breaking points correspond to 0.5, 0.8 and 0.9 quantiles. c, Comparison of CHG and CHH DNA methylation levels in 50-bp windows differing in sirenRNA accumulation at trans targets. The x axis shows the difference in DNA methylation (Cr × Cr minus Cr × Cg), and the y axis shows the difference in sirenRNA accumulation in trans (Cr × Cr minus Cr × Cg). The numbers correspond to windows in each category; the breaking points correspond to 0.6, 0.85 and 0.95 quantiles. In a–c, the asterisks indicate statistically significant differences as calculated by two-sided Wilcoxon tests with Benjamini and Hochberg correction for multiple comparisons (***P < 0.001; *P < 0.05). d, Examples of genes and TEs with reduced sirenRNA accumulation at trans targets in Cr × Cg, nrpd1 × Cr and nrpd1 × nrpd1 compared with Cr × Cr endosperm. The tracks show levels of sirenRNAs and CHH methylation at the indicated genes and TEs. The blue boxes represent genes; the purple boxes represent TEs. e, Distance to the nearest TE upstream of the transcriptional start site of upregulated genes in Cr × Cg compared with non-upregulated genes. The asterisks indicate statistically significant differences as calculated by a two-sided Wilcoxon test (***P < 0.001). f, Methylation levels of TEs in the promoters of upregulated genes in the Cr × Cg hybrid. Statistical differences were calculated with two-sided Wilcoxon’s tests for paired samples. In e and f, the horizontal lines show the median values, the box edges show the interquartile range and the whiskers show the full range excluding outliers.
We next asked whether there is a connection between sirenRNAs, loss of DNA methylation at trans targets and changes in gene expression in the hybrid Cr × Cg endosperm. Consistent with sirenRNAs most prominently targeting TEs (Fig. 6b), we found that genes upregulated in Cr × Cg hybrids were closer to a TE upstream of their transcription start site than other genes (Fig. 6e). Moreover, the TEs in the promoters of those genes had reduced CHG and CHH methylation in Cr × Cg hybrids (Fig. 6f). Among the genes that were targeted by trans-acting sirenRNAs and lost DNA methylation we found many upregulated AGLs (Extended Data Fig. 4a,b and Supplementary Data 3), consistent with previous findings reporting targeting of AGLs by maternal Pol IV-dependent siRNAs30.
On the basis of these data, we propose that sirenRNAs in the Capsella endosperm guide non-CG DNA methylation in trans. Depletion of sirenRNAs in the Cr × Cg hybrid leads to loss of DNA methylation and increased expression of several trans targets, among them several genes with potential function in establishing hybridization barriers (Supplementary Data 3). These include SUVH7, which is involved in the triploid block in Arabidopsis32, YUC10, an auxin biosynthesis gene33 and ARF21—a centromeric ARF regulating endosperm cellularization in Arabidopsis34. Moreover, among those genes are nine AGLs including AGL38—the orthologue of PHE1 (Extended Data Fig. 9a,b), which activates key regulators of the triploid block in Arabidopsis35. Most of these genes had paternally biased expression in the hybrid endosperm, consistent with the paternal-excess phenotype (Extended Data Fig. 9c–f).
Putative AGL targets are upregulated in hybrid endosperm
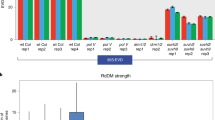
Previous work revealed that target sequence motifs of the AGL PHE1 are frequently located in helitron TEs35. Since helitrons in Arabidopsis and other Brassicaceae are concentrated in pericentromeric regions36,37, we tested whether pericentromeric regions in Capsella were enriched for PHE1 binding motifs. We found that the frequency of genes containing PHE1 binding motifs was significantly higher in pericentromeric regions than in chromosome arms (Fig. 6g). Importantly, in all analysed incompatible crosses, the promoter regions of upregulated genes located in pericentromeric regions were significantly enriched for PHE1 binding sites (Fig. 6g). On the basis of these data, we propose that the increased expression of genes in pericentromeric regions in hybrid endosperm is a consequence of the increased expression of AGLs and their ability to target pericentromeric regions due to the loss of DNA methylation9. DNA methylation was shown to antagonize the binding of PHE1 (ref. 35), supporting the proposed antagonism of DNA methylation and AGL binding. Our data (Extended Data Fig. 9a,b) and previously published data show that sirenRNAs negatively regulate AGLs in the endosperm30. Nevertheless, while the loss of maternal Pol IV function delays endosperm cellularization (Extended Data Fig. 5d), it does not cause complete endosperm cellularization failure38, suggesting an additional component causing this phenotype in paternal-excess hybrids. It has been shown that many AGLs have higher expression levels in Cg than in Cr6. Similarly, many PHE1-like (Mγ-type) AGLs are more highly expressed in 4xCr than in 2x plants, resembling the profile of Cg AGL genes (Fig. 6h). The same pattern applied for Mα*-like AGLs (Fig. 7c), which encode potential heterodimerization partners of Mγ-type AGLs10. On the basis of these data, we propose that the increased expression of type I AGL genes in Cg and 4xCr causes increased expression of AGL targets in hypomethylated regions (Fig. 7a). The allopolyploid Cbp behaved like Cr in crosses with Cg (Fig. 1a,c), which was reflected by reduced expression of several AGLs in Cbp compared with Cg. These data support the idea that increased dosage of AGLs in hybrid endosperm is a critical determinant of hybrid seed arrest. Similarly, the expression of Mγ- and Mα*-type AGLs in Co was lower than in Cg, resembling that of Cr. While several AGLs were more highly expressed in Co than in Cr, the CrPHE1 orthologue Carubv10020903m.g9 was nearly twice as strongly expressed in Cr (Fig. 7b), possibly connecting to the paternal-excess effect of Cr when crossed to Co (Fig. 1a). Furthermore, the majority of AGLs were upregulated to substantially higher levels in paternal-excess hybrid seeds than in nrpd1 seeds, supporting the idea that the increased expression of AGLs determines the phenotypic difference between paternal-excess hybrid seeds and seeds lacking maternal NRPD1 function (Fig. 7d). Importantly, increased maternal genome dosage suppressed AGL expression in 4xCr × Cg and 4xCo × Cr hybrids (Fig. 7d), which was connected with increased DNA methylation in the promoter regions of AGL genes (Extended Data Fig. 7b) and restored seed viability21 (Fig. 1d).

a, Distribution of PHE1 binding motifs in the Capsella genome in different groups of genes: all genes and genes upregulated in Cr × Cg, Cbp × Cg, Co × Cr, Cr × 4xCr, 4xCr × Cg and 4xCo × Cr. The numbers of pericentromeric (PC) and non-pericentromeric (NPC) genes upregulated in different genotypes are compared with the numbers of all pericentromeric/non-pericentromeric genes. The asterisks indicate statistically significant differences calculated by two-sided chi-square tests with Benjamini and Hochberg correction for multiple comparisons (***P < 0.001). The numbers show the number of genes in each category. b,c, Heat maps and dendrograms show clustering of genes based on the expression (RPKM) of type I γ-MADS-box (b) and α*-MADS-box (c) transcription factors in different Capsella genotypes. d, Heat map and dendrogram show clustering of genes based on log2(fold change) (in comparison to the maternal parent) of γ and α* type I MADS-box transcription factors in different Capsella hybrids. Genes marked in bold overlap siren loci; genes marked in purple are targeted by trans-acting sirenRNAs.
Together, on the basis of our data, we propose that seed abortion in response to paternal-excess interspecies and interploidy hybridizations is triggered by two factors: the depletion of maternally derived sirenRNAs and the increased expression of AGLs targeting hypomethylated regions in hybrid endosperm.
Discussion
In this study, we uncover a connection between the dosage of maternal Pol IV-dependent sirenRNAs and hybridization success in Capsella. Our findings demonstrate that depletion of sirenRNAs connects to chromatin decondensation and the loss of non-CG DNA methylation in the endosperm, resembling phenotypes of paternal-excess hybrid endosperm. Consistent with the similar molecular and chromatin phenotypes observed in hybrid paternal-excess and maternal nrpd1 mutant endosperm, there was a strong depletion of sirenRNAs in the hybrid, underscoring the impact of hybridization on sirenRNA production. This depletion of sirenRNAs in both hybrid and maternal nrpd1 mutant endosperm correlated with a substantial increase in mRNA levels of genes overlapping siren loci, highlighting the potential role of sirenRNAs as negative regulators of gene expression. Siren loci exhibit extensive methylation in wild-type endosperm (Figs. 4d,e and 5a). The transcriptional alterations observed in nrpd1 and hybrid endosperm align with a reduction of DNA methylation at siren loci, providing strong support for the role of sirenRNAs in directing DNA methylation. Our data also suggest, in line with previous findings in male meiocytes and ovules29,39, that sirenRNAs may act in trans by guiding DNA methylation to non-perfectly matching TEs and protein-coding genes. Genes targeted by sirenRNAs that undergo upregulation in hybrid endosperm encompass several genes with potential involvement in establishing hybridization barriers, including SUVH7, YUC10, ARF21 and PHE1 orthologues32,34,35. This strongly implies a direct connection between the depletion of sirenRNAs and the arrest of hybrid seed development.
SirenRNAs, originating predominantly from the maternal source (ref. 16 and our own data), probably guide methylation in the endosperm post-fertilization. Since heightened maternal genome dosage can ameliorate hybrid seed defects while concurrently inducing DNA hypermethylation (Fig. 2d,e and Extended Data Fig. 3), we posit that dosage of maternally provided sirenRNAs is insufficient to repress sirenRNA targets in paternal-excess hybrids. Consistently, interploidy Cr × 4xCr seeds exhibit similar molecular aberrations to Cr × Cg hybrids, strongly corroborating the notion that the relative dosage of maternal sirenRNAs to their targets is a key determinant of hybridization success. The loss of RDR2, which together with Pol IV is required for siRNA production13,40, causes strong seed defects in Cg, differing from the mild defects of rdr2 mutants in Cr41. These data align with our finding that Cg produces higher levels of sirenRNAs than Cr, suggesting that sirenRNA levels adapt to the expression strength of their targets. Among those targets are AGL genes that we found to scale in expression with the effective ploidy of Capsella species (Co < Cr = Cbp < Cg = 4xCr). The differential expression levels of AGLs in Capsella are probably driven by mating system divergence. According to theoretical predictions, genes promoting endosperm growth are more strongly expressed in outbreeding than in inbreeding species22. Consistent with these predictions, Cg is an outbreeder and has the strongest AGL expression levels, followed by the recent inbreeders Cr and Cbp and finally by the ancient inbreeder Co (Fig. 7b).
The maternal nrpd1 mutant exhibited a phenotype mirroring that of paternal-excess hybrids in terms of alterations in DNA methylation and chromatin condensation. The seeds of nrpd1 mutants cellularize later than wild-type seeds and also abort, albeit at a lower frequency than interspecies hybrids (Extended Data Fig. 5). One probably decisive difference between hybrid and nrpd1 endosperm is the extent of AGL deregulation, which may account for the difference in seed abortion frequency. While AGLs targeted by sirenRNAs were also upregulated in nrpd1 endosperm, most experienced higher upregulation in the hybrid endosperm (Fig. 7d). This discrepancy is probably attributed to the amplified AGL expression in the paternal parents of hybrids, inducing a more pronounced response in hybrid endosperm than in the nrpd1 mutant.
Given that AGL target genes are notably concentrated in pericentromeric regions, the combined effects of diminished DNA methylation and increased AGL activity probably account for the heightened expression of AGL targets in paternal-excess hybrids. The same scenario applies to interploidy Cr × 4xCr seeds, which exhibit a substantial overlap in hyperactivated AGL target genes with paternal-excess hybrids. Mutations in several AGLs were shown to weaken barriers associated with interploidy and interspecies hybridization35,42,43, providing further support for this scenario. Since Mγ AGLs have strongly expanded in Capsella and most of them are highly upregulated in the hybrids (Fig. 7d), directly testing the functional relevance of AGLs in establishing interspecies hybridization barriers in Capsella is challenging, but this remains an important task of future studies. Taken together, on the basis of our findings, we propose that maternal sirenRNAs serve as a dosage-sensitive factor that decisively influences the success of hybridization between plant species.
Methods
Plant material and growth conditions
The following accessions of different Capsella species have been used in this study: Cr 48.21, 4xCr 48.21, Cbp 27.4, Cg 23.5 and Co 1719. Tetraploid Co seeds were provided by M. Lascoux44. The seeds were surface sterilized with 30% bleach and 70% ethanol, rinsed with distilled water and sown on agar plates containing ½ Murashige and Skoog medium and 1% sucrose. The seeds were then stratified for two days in the darkness at 4 °C and moved into a growth chamber with a long-day photoperiod (16 h and 22 °C light, 8 h and 19 °C darkness) with a light intensity of 110 µE. Seven-day-old seedlings were transferred to pots filled with sterile soil, and the plants were grown in a growth chamber with 60% humidity and daily cycles of 16 h light at 21 °C and 8 h darkness at 18 °C with a light intensity of 150 µE. For germination tests, dry seeds were stored for 30 days to break dormancy. The seeds were then surface sterilized and sown on agar plates as described above. The seeds were stratified for two days at 4 °C and then moved to a growth chamber and scored after seven days for seedling establishment.
Histological and fluorescence analyses
Manually pollinated siliques were harvested after 4–7 DAP and processed for clearing and Feulgen staining as previously described33. The siliques were opened at the side with needles and incubated overnight in fixing solution (ethanol:acetic acid (3:1)) at 4 °C. The samples were washed with distilled water three times for 15 minutes and then incubated for 1 h in freshly prepared 5 N HCl. After incubation, the samples were washed again with distilled water three times for 15 minutes and incubated with Schiff reagent (Sigma-Aldrich) for 3–4 h. The samples were washed with cold (4 °C) distilled water three times for 10 minutes and then washed in different concentrations of ethanol (10%, 30%, 50%, 70% and 95%) for 10 minutes in each solution. The samples were then washed with 99.5% ethanol until they remained colourless (at this point samples can be stored in this solution overnight at 4 °C). The samples were incubated in 99.5% ethanol:LR White (London Resin White + catalyst, Sigma Aldrich) 3:1 and 2:1, 15 minutes in each solution. The samples were incubated in 1:1 ethanol:LR White for 1 h and then in LR White overnight. Seeds were taken out of the siliques, mounted on a microscope slide in a drop of LR White + catalyst and baked at 60 °C for 16 h for polymerization. The slides were watched under a two-photon microscope with an excitation wavelength of 800 nm and emission from 518 nm and onwards.
Capsella grandiflora sequencing and genome assembly
DNA was extracted from young leaf material of Cg using the CTAB method45. DNA was sized on the BluePippin system (Sage Science) to remove small fragments and then sequenced on one Oxford Nanopore PromethION flow cell. The pore version used was R9.4.1, and the PromethION release version was 19.05.1. A total of 49.2 Gb was sequenced, corresponding to about 200× coverage of the estimated genome size of approximately 250 Mb (roughly calculated by flow cytometry analysis). Additionally, Cg DNA was used to prepare an Illumina overlap library. DNA was sheared using a Covaris M220 (Covaris) to 450 bp. The sheared DNA was sized using a BluePippin prep system and used to prepare a PCR-free Illumina sequencing library. The library was sequenced on the Illumina NovaSeq SP platform using an SP flow cell and 2 ×250 bp protocol.
A selection of the longest Oxford Nanopore PromethION sequence reads, together representing 60× haploid genome coverage, were assembled using Minimap2 (v.2.16-r922, with settings m, 1,600; K2G; I8G)46 and Miniasm (v.0.2-r137-dirty, with settings R; c, 2; m, 500; s, 4,000)47. A consensus sequence was generated through three iterations of Racon (v.1.4.10, with the default settings)48, using all sequence reads. The Illumina read pairs were then used to polish the consensus through three rounds of BWA-mem (v.0.7.17, with the default parameters)49 and Pilon (v.1.22, with the default parameters)50. Finally, purge_dups51 was used (v.15082019, with the default parameters) to generate a haploid representation of the heterozygous Cg assembly.
RNA-seq
Seeds derived after manual pollination were dissected out of siliques at 6 DAP and collected in 20 µl of RNAlater solution (Sigma-Aldrich). Each sample consisted of 10–15 siliques. RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions. 500 ng of RNA was used for RNA-seq library preparation using the NEBNext Poly(A)mRNA Magnetic Isolation Module and NEBNext Ultra RNA LibraryPrep Kit for Illumina. Three biological replicates were generated for each genotype. The libraries were sequenced on an Illumina HiSeq X machine in 150-bp paired-end mode.
RNA-seq analysis
Adapter trimming was performed using Trim galore with the following parameters: three_prime_clip_R1, 15; three_prime_clip_R2, 15; clip_R1, 10; clip_R2, 10. Sequencing reads were aligned to the Cr genome v.1.0 (Phytozome) using HISAT2 (ref. 52). Reads were assigned to genes with featureCounts from the Bioconductor Rsubread package53. Differentially regulated genes were detected using DESEQ2 (ref. 54). Genes were considered as upregulated on the basis of log2(fold change) > 1 and Padj < 0.05 in comparison to the maternal parent (for incompatible hybrids) or to Cr × Cr (for crosses with the nrpd1 mutant). Pericentromeric regions were defined as in ref. 55.
Bisulfite sequencing
For bisulfite sequencing (BS-seq), seeds derived after manual pollination were dissected out of siliques at 6 DAP, and the endosperm was manually dissected as previously described25. Manually pollinated seeds 6 DAP were removed from siliques and put on a microscopic slide covered with a piece of tissue paper soaked with a drop of RNAlater solution (Sigma-Aldrich). The seeds were gently squashed with another microscopic slide to release the endosperm, and the embryos and seed coats were removed with tweezers. Tissue paper with the endosperm was put in an Eppendorf tube and frozen in liquid nitrogen. Approximately 600 seeds were used per replicate. The samples were stored at −70 °C and then used for genomic DNA isolation using the DNeasy Plant Mini Kit (Qiagen). Biological duplicates were generated for each genotype. Libraries were prepared with the Accel-NGS Methyl-Seq DNA Library Kit from Illumina, and the sequencing was performed on an Illumina NovaSeq 6000 machine in 150-bp paired-end mode.
Bioinformatic analysis of BS-seq data
For DNA methylation analysis, the 150-bp-long paired-end reads were first quality trimmed by removing the first 5 bases from the 5′ end and the last 15 bases from the 3′ end. Reads were mapped to the Cr reference genome in paired-end mode (score_min L, 0–0.6) using Bismark v.0.16.3 (ref. 56). The mapped reads were deduplicated, and cytosine methylation values calculated using the Bismark Methylation Extractor.
Differentially methylated regions in the CG, CHG and CHH contexts were calculated using 50-bp windows across the genome as units. Only hypomethylated regions (Cr × Cr > Cr × Cg, Cr × Cr > Cr × 4xCr and Cr × Cr > nrpd1 × Cr) were considered. Windows with differences in fractional methylation below the first decile (Fisher’s exact test P < 0.01) were selected, and these were merged if they occurred within 300 bp.
Parental-specific analysis of RNA-seq and BS-seq data
Parental gene expression and parental methylation analyses in plant crosses were performed using the allele-specific alignment sorter SNPsplit57. A masked Cr genome with SNP positions masked by the ambiguity base ‘N’ were constructed to run SNPsplit in the crosses with Cg and Co. To define SNPs, we performed an ordinary Illumina resequencing of Cg and Co plants. Reads from both species were quality trimmed with trimgalore (clip_R1, 15; clip_R2, 15; three_prime_clip_R1, 5; three_prime_clip_R2, 5) and mapped to the Cr reference genome using BWA49 in PE mode. SNP calling was performed using freebayes58(iXu; G, 20; F, 0.9), and masked genomes were built using bedtools maskfasta59. The RNA-seq analysis used hisat2 (ref. 52), and the BS-seq analysis used bismark (v.0.16.3)56 as aligners, before the SNPsplit parental read sorting. Gene expression was quantified with htseq-count60.
Small RNA sequencing
Seeds derived after manual pollination were dissected out of siliques at 6 DAP and squeezed on facial tissue paper to release the endosperm. Seed coats and embryos were removed with forceps. A drop of RNAlater solution (Sigma-Aldrich) was added to the tissue containing the absorbed endosperm, transferred to an Eppendorf tube and frozen in liquid nitrogen. Each sample consisted of endosperm from about 600 seeds. Small RNAs were extracted using the mirVana miRNA Isolation Kit (ThermoFisher Scientific) following the manufacturer’s instructions for sRNA. Libraries were prepared using the NEBNext Multiplex Small RNA Library Prep Set for Illumina with 50–100 ng of input for each sample. Size selection was performed on a 6% polyacrylamide gel, and bands corresponding to about 150 bp were cut and purified from the gel for further analysis. Sequencing was performed on a NovaSeq 6000 machine in 150-bp paired-end mode.
Bioinformatic analyses of sRNA-seq data
Adapters were removed from the first read of the 150-bp long read pair of each library using cutadapt, and the resulting 18–25-bp long reads were selected. Reads belonging to chloroplasts, mitochondria and structural non-coding RNAs (tRNAs, snRNAs, rRNAs or snoRNAs) were removed using bowtie (v.1).
The remaining reads were mapped to the Cr or Cg genome (see above), and sRNA loci were annotated with ShortStack27,61 using filtered reads from all generated libraries and sRNA from leaves38. The options used for ShortStack were mismatches, 0; mmap, u; mincov, 0.5 rpm; and pad, 75. Replicates were checked for consistency by principal component analysis using the vegan v.2.6-4 package in R62. Clusters with differential accumulation of sRNAs were identified with DESEQ2 (ref. 54). Genes and TEs were considered as depleted of sRNAs on the basis of log2(fold change) < 0 and Padj < 0.1 in comparison to the maternal parent (for incompatible hybrids) or to Cr × Cr (for crosses with the nrpd1 mutant).
Parental-specific analysis of sRNA-seq data
Parental-specific analysis of sRNA was performed by mapping the 18–25-nucleotide sRNA population to the genomes of both parents with no mismatches (bowtie v.0)63 and selecting the reads that mapped exclusively to one parent and not to the other and vice versa. As parental genomes for Co and Cg, we used own-built ‘pseudogenomes’ in which the above-described SNPs between these species and Cr were substituted into the Cr reference genome.
Endosperm nuclei spreading
Endosperm nuclei spreading was performed as previously described9. Manually pollinated seeds were harvested 4 DAP and incubated overnight in a solution containing 2.5 mM 8-hydroxyquinoline, 100 μM oryzalin and 100 μM colchicine. The seeds were subsequently fixed in ethanol:acetic acid (3:1) for at least five hours at 4 °C. Following fixation, the seeds were washed with 10 mM citrate buffer and incubated for five hours in an enzyme solution comprising 0.3% cytohelicase, 0.3% pectolyase and 0.3% cellulase in 10 mM citrate buffer. After enzymatic digestion, five to ten seeds were placed on a slide, squashed with a needle, spread using 60% acetic acid and fixed on the slide with ethanol:acetic acid (3:1). The slides were mounted using Vectashield mounting medium containing DAPI (BioNordika AB). The experiment was conducted with three independent biological replicates.
Quantification of chromocenter condensation
Each endosperm nucleus was saved as a single .tif file and then analysed in Fiji64. Particle Analysis was run after threshold adjustment with MaxEntropy. Only particles larger than 0.05 µm2 were considered. The circularity of the particles was used as a proxy for chromocenter condensation as previously described65,66,67 with values ranging between 0 and 1 (1 corresponds to a perfect circle). Each nucleus was represented by the mean circularity of all chromocenters. For each genotype, around 50 nuclei were analysed.
Motif search
PHE1 binding motifs were previously identified in ref. 35. The motif file was used for scanning promoter sequences (1 kb upstream of the ATG) of Capsella genes with the findMotifsGenome.pl function from HOMER68.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The original data files for RNA-seq and DNA BS-seq can be obtained from the NCBI Gene Expression Omnibus (GSE246468). The assembled Cg genome sequence has been deposited at DDBJ/ENA/GenBank under the accession number JAVXYZ000000000. Source data are provided with this paper. The source data for Fig. 6a,c are also available via figshare at https://doi.org/10.6084/m9.figshare.27143508 (ref. 69).
References
Coughlan, J. M. The role of hybrid seed inviability in angiosperm speciation. Am. J. Bot. 110, 1–14 (2023).
Sekine, D. et al. Dissection of two major components of the post-zygotic hybridization barrier in rice endosperm. Plant J. 76, 792–799 (2013).
Scott, R. J., Spielman, M., Bailey, J. & Dickinson, H. G. Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 125, 3329–3341 (1998).
Pennington, P. D., Costa, L. M., Gutierrez-Marcos, J. F., Greenland, A. J. & Dickinson, H. G. When genomes collide: aberrant seed development following maize interploidy crosses. Ann. Bot. 101, 833–843 (2008).
Johnston, S. A. & Hanneman, R. E. Jr. Manipulations of endosperm balance number overcome crossing barriers between diploid Solanum species. Science 217, 446–448 (1982).
Rebernig, C. A., Lafon-Placette, C., Hatorangan, M. R., Slotte, T. & Kohler, C. Non-reciprocal interspecies hybridization barriers in the Capsella genus are established in the endosperm. PLoS Genet. 11, e1005295 (2015).
Roth, M., Florez-Rueda, A. M. & Stadler, T. Differences in effective ploidy drive genome-wide endosperm expression polarization and seed failure in wild tomato hybrids. Genetics 212, 141–152 (2019).
Tonosaki, K. et al. Overcoming the species hybridization barrier by ploidy manipulation in the genus Oryza. Plant J. 93, 534–544 (2018).
Dziasek, K. et al. Hybrid seed incompatibility in Capsella is connected to chromatin condensation defects in the endosperm. PLoS Genet. 17, e1009370 (2021).
Qiu, Y. & Kohler, C. Endosperm evolution by duplicated and neofunctionalized type I MADS-box transcription factors. Mol. Biol. Evol. 39, msab355 (2022).
Martinez, G. et al. Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat. Genet. 50, 193–198 (2018).
Satyaki, P. R. V. & Gehring, M. Paternally acting canonical RNA-directed DNA methylation pathway genes sensitize Arabidopsis endosperm to paternal genome dosage. Plant Cell 31, 1563–1578 (2019).
Cuerda-Gil, D. & Slotkin, R. K. Non-canonical RNA-directed DNA methylation. Nat. Plants 2, 16163 (2016).
Erdmann, R. M., Satyaki, P. R. V., Klosinska, M. & Gehring, M. A small RNA pathway mediates allelic dosage in endosperm. Cell Rep. 21, 3364–3372 (2017).
Rodrigues, J. A. et al. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl Acad. Sci. USA 110, 7934–7939 (2013).
Grover, J. W. et al. Abundant expression of maternal siRNAs is a conserved feature of seed development. Proc. Natl Acad. Sci. USA 117, 15305–15315 (2020).
Lu, J., Zhang, C., Baulcombe, D. C. & Chen, Z. J. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl Acad. Sci. USA 109, 5529–5534 (2012).
Satyaki, P. R. V. & Gehring, M. RNA Pol IV induces antagonistic parent-of-origin effects on Arabidopsis endosperm. PLoS Biol. 20, e3001602 (2022).
Ishikawa, R. et al. Rice interspecies hybrids show precocious or delayed developmental transitions in the endosperm without change to the rate of syncytial nuclear division. Plant J. 65, 798–806 (2010).
Lafon-Placette, C. & Kohler, C. Endosperm-based postzygotic hybridization barriers: developmental mechanisms and evolutionary drivers. Mol. Ecol. 25, 2620–2629 (2016).
Lafon-Placette, C. et al. Paternally expressed imprinted genes associate with hybridization barriers in Capsella. Nat. Plants 4, 352–357 (2018).
Brandvain, Y. & Haig, D. Divergent mating systems and parental conflict as a barrier to hybridization in flowering plants. Am. Nat. 166, 330–338 (2005).
Slotte, T. et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45, 831–835 (2013).
Douglas, G. M. et al. Hybrid origins and the earliest stages of diploidization in the highly successful recent polyploid Capsella bursa-pastoris. Proc. Natl Acad. Sci. USA 112, 2806–2811 (2015).
Hatorangan, M. R., Laenen, B., Steige, K. A., Slotte, T. & Kohler, C. Rapid evolution of genomic imprinting in two species of the Brassicaceae. Plant Cell 28, 1815–1827 (2016).
Grover, J. W. et al. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 94, 575–582 (2018).
Axtell, M. J. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19, 740–751 (2013).
Mi, S. et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133, 116–127 (2008).
Burgess, D., Chow, H. T., Grover, J. W., Freeling, M. & Mosher, R. A. Ovule siRNAs methylate protein-coding genes in trans. Plant Cell 34, 3647–3664 (2022).
Kirkbride, R. C. et al. Maternal small RNAs mediate spatial–temporal regulation of gene expression, imprinting, and seed development in Arabidopsis. Proc. Natl Acad. Sci. USA 116, 2761–2766 (2019).
Florez-Rueda, A. M., Fiscalini, F., Roth, M., Grossniklaus, U. & Stadler, T. Endosperm and seed transcriptomes reveal possible roles for small RNA pathways in wild tomato hybrid seed failure. Genome Biol. Evol. 13, evab107 (2021).
Wolff, P., Jiang, H., Wang, G., Santos-Gonzalez, J. & Kohler, C. Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. eLife 4, e10074 (2015).
Batista, R. A., Figueiredo, D. D., Santos-Gonzalez, J. & Kohler, C. Auxin regulates endosperm cellularization in Arabidopsis. Genes Dev. 33, 466–476 (2019).
Butel, N., Qiu, Y., Xu, W., Santos-Gonzalez, J. & Kohler, C. Parental conflict driven regulation of endosperm cellularization by a family of Auxin Response Factors. Nat. Plants 10, 1018–1026 (2024).
Batista, R. A. et al. The MADS-box transcription factor PHERES1 controls imprinting in the endosperm by binding to domesticated transposons. eLife 8, e50541 (2019).
Yang, L. & Bennetzen, J. L. Structure-based discovery and description of plant and animal Helitrons. Proc. Natl Acad. Sci. USA 106, 12832–12837 (2009).
Hu, K. et al. Helitron distribution in Brassicaceae and whole genome Helitron density as a character for distinguishing plant species. BMC Bioinform. 20, 354 (2019).
Wang, Z. et al. Polymerase IV plays a crucial role in pollen development in Capsella. Plant Cell 32, 950–966 (2020).
Long, J. et al. Nurse cell-derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 373, eabh0556 (2021).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Dew-Budd, K. J. et al. Mating system is associated with seed phenotypes upon loss of RNA-directed DNA methylation in Brassicaceae. Plant Physiol. 194, 2136–2148 (2023).
Hehenberger, E., Kradolfer, D. & Köhler, C. Endosperm cellularization defines an important developmental transition for embryo development. Development 139, 2031–2039 (2012).
Josefsson, C., Dilkes, B. & Comai, L. Parent-dependent loss of gene silencing during interspecies hybridization. Curr. Biol. 16, 1322–1328 (2006).
Duan, T., Sicard, A., Glemin, S. & Lascoux, M. Separating phases of allopolyploid evolution with resynthesized and natural Capsella bursa-pastoris. eLife 12, RP88398 (2023).
Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus 12, 13–15 (1990).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Koenig, D. et al. Long-term balancing selection drives evolution of immunity genes in Capsella. eLife 8, e43606 (2019).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27, 1571–1572 (2011).
Krueger, F. & Andrews, S. R. SNPsplit: allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Res. 5, 1479 (2016).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://arxiv.org/abs/1207.3907 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Anders, S., P Pyl, T. P. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Johnson, N. R., Yeoh, J. M., Coruh, C. & Axtell, M. J. Improved placement of multi-mapping small RNAs. G3 6, 2103–2111 (2016).
Oksanen, J. et al. vegan: community ecology package. R package version 2.6-4 (2022).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Snoek, B. L. et al. Genetic dissection of morphometric traits reveals that phytochrome B affects nucleus size and heterochromatin organization in Arabidopsis thaliana. G3 7, 2519–2531 (2017).
Recoules, L., Tanguy Le Gac, N., Moutahir, F., Bystricky, K. & Lavigne, A. C. Recruitment of the histone variant macroH2A1 to the pericentric region occurs upon chromatin relaxation and is responsible for major satellite transcriptional regulation. Cells 12, 2175 (2023).
Natale, F. et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 8, 15760 (2017).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Dziasek, K. et al. Dosage sensitive maternal siRNAs determine hybridization success in Capsella. figshare https://doi.org/10.6084/m9.figshare.27143508 (2024).
Wang, M. H., Zhao, Y. Z. & Zhang, B. Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923 (2015).
Acknowledgements
We thank T. Duan and M. Lascoux from Uppsala University for kindly sharing tetraploid Co seeds. We thank H. Bente for valuable suggestions about sRNA library preparation and M. Incarbone and members of the Köhler group for critical reading and helpful comments on the paper. This work was funded by the Knut and Alice Wallenberg Foundation (grant numbers 2018-0206 (C.K.) and 2019-0062 (C.K.)) and the Max Planck Society.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
K.D. and C.K. conceptualized the project, developed the methodology and provided supervision. K.D. and K.W. conducted the experiments. Y.Q. performed the phylogenetic analysis. D.R. and K.N. generated the genome assembly of Cg. J.Z. generated the genetic material used in this study. J.S.-G. performed the bioinformatic analyses. C.K. acquired the funding and administered the project. K.D. and C.K. wrote the original paper draft. All authors reviewed and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Effects of interploidy and interspecies crosses in Capsella.
a, Germination of triploid Cr seeds shown as the fraction of germinating seeds from each cross. Each filled circle represents one biological replicate; there are three biological replicates per genotype. Numbers on top represent total seed number. Asterisks represent statistical significance as calculated by one way ANOVA with post hoc Tukey’s HSD test (*** p < 0.001). Red dots show values for three biological replicates. Whiskers show the full range excluding outliers. b, Feulgen staining of diploid and triploid Cr seeds at 6 DAP (scale bars represent 200 µm). c, Cr × 4xCr embryos rescued on Murashige-Skoog medium. d, Seed phenotypes of Cr × Cr, Cg × Cr and Cr × Co seeds. Scale bars represent 1 mm. e, f. Upset plot showing the overlap of upregulated genes in comparison to the paternal parent (e) and both parents (f) between interploidy (Cr × 4xCr) and interspecies crosses of different Capsella species.
Extended Data Fig. 2 DNA methylation in the endosperm of different Capsella Cr and Cg hybrid crosses.
a. Metagene plots show DNA methylation levels of genes in the endosperm of Cg × Cg, 4xCr × Cg and 4xCr × Cr 6 DAP seeds. b, Boxplots show methylation level of genes in the endosperm of Cg × Cg, 4xCr × Cg and 4xCr × Cr 6 DAP seeds. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (** p-value <0.01,*** p-value < 0.001, n.s. – not significant). c, Metagene plots show DNA methylation levels of genes in the endosperm of Cg × Cr at 6 DAP in comparison to published data for Cr × Cr, Cg × Cg, Cr × Cg1. d, Boxplots show methylation level of genes in the endosperm Cg × Cr at 6 DAP in comparison to published data for Cr × Cr, Cg × Cg, Cr × Cg9. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (*** p-value < 0.001). e, f, Metagene (e) and box plots (f) show DNA m2ethylation levels of genes in the endosperm of diploid and triploid 6 DAP Cr seeds. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (*** p-value < 0.001). g, h, Metagene (g) and box plots (h) show DNA methylation levels of TEs in the endosperm of diploid and triploid 6 DAP Cr seeds. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (*** p-value < 0.001). i. Boxplots show CHG (left panel) and CHH (right panel) methylation levels on TEs in chromosome arms and pericentromeric regions in the endosperm of Cr × Cr, Cr × Cg and Cg × Cg and Cr × 4xCr at 6 DAP. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test (*** p-value < 0.001). b, d, f, h, i, Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers.
Extended Data Fig. 3 DNA methylation in the endosperm of different Capsella Co and Cr hybrid crosses.
a, b, Metagene (a) and box plots (b) show DNA methylation levels of TEs in the endosperm of Cr × Co, Co × Cr, 4xCo × Cr, 4xCo × 4xCo, Co × Co and Cr × Cr at 6 DAP. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test (*** p-value < 0.001). c, d, Metagene (c) and box plots (d) show DNA methylation levels of TEs in the endosperm of Cr × Co, Co × Cr, 4xCo × Cr, 4xCo × 4xCo, Co × Co and Cr × Cr at 6 DAP. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test (*** p-value < 0.001). b, d, Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers.
Extended Data Fig. 4 Maternal loss of NRPD1 mimics DNA methylation changes seen in paternal-excess hybrids.
a, Metagene plots show DNA methylation levels of genes in the endosperm of Cr × Cr, nrpd1 × nrpd1, Cr × nrpd1, and nrpd1 × Cr 6 DAP seeds. b, Boxplots show methylation level of genes in the endosperm at 6 DAP of the indicated genotypes. Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (*** p-value < 0.001, ** pvalue < 0.01, * p-value < 0.05). c, d, Venn diagrams show the overlap of genes (c) and TEs (d) losing DNA methylation in the endosperm derived from Cr × Cg, Cr × 4xCr and nrpd1 × Cr crosses. Asterisks indicate statistically significant differences calculated using the supertest function from R package SuperExactTest (*** p-value < 0.001)70.
Extended Data Fig. 5 Seed and endosperm phenotypes in reciprocal crosses with nrpd1.
a, Seed phenotypes of Cr × Cg, nrpd1 × Cg and nrpd1 × Cr seeds. Scale bar represents 1 mm. Barplot shows number of aborted and partially aborted seeds of three biological replicates of each genotype. Numbers above the bars indicate number of analysed seeds. b, Pictures of cleared seeds of Cr × Cr and nrpd1 × Cr at 6 DAP. Inset shows enlarged regions of cellularized (Cr × Cr) and uncellularized (nrpd1 × Cr) endosperm. Scale bars represents 100 µm (upper row) and 10 µm (lower row). c, Quantification of endosperm cellularization of indicated genotypes at 6 DAP. Numbers correspond to numbers of analysed seeds. d, Seed phenotypes of 4xCr × Cg and 4xnrpd1 × Cg seeds. Scale bar represents 1 mm. Barplot shows number of aborted and partially aborted and viable seeds of three biological replicates of each genotype. Numbers above the bars indicate number of analysed seeds. e, Seed phenotypes of Cr × 4xCr and Cr × 4xnrpd1 seeds. Scale bar represents 1 mm. Barplot shows number of aborted and partially aborted and viable seeds of three biological replicates of each genotype. Numbers above the bars indicate number of analysed seeds.
Extended Data Fig. 6 Quality of small RNA-Seq data and characterization of sirenRNAs.
a, Non-metric MultiDimensional Scaling (NMDS) of small RNA (sRNA) sequencing samples. b, Correlogram showing the correlation score matrix across all sequenced sRNA libraries. c, Barplot shows size distribution of sRNAs in all sequenced libraries. d, Length distribution of siRNA loci. Siren loci are larger than other siRNA expressing loci, including non-siren 24 nt-dominant, 21 nt-dominant and 22 nt-dominant loci. e, Genomic features overlapping with 24 nt-dominant clusters (n=28 075) and siren loci (n=1 385) in Cr. The genome was subdivided into 50-bp windows (n= 2 497 626) that were assigned to either exons, introns, intergenic regions, transposable elements and edges (50bp regions at the boundaries of gene bodies and repeats). f, Frequency distribution of sirenRNA clusters after dominating size class. 24 nt siRNAs dominate in the majority of sirenRNA clusters defined by ShortStack. g, 5′ nucleotide of sirenRNAs (18–24 nt) in Cr × Cr endosperm. h, Number of siren loci normalized per size of respective genomic regions (in bp) – non-pericentromeric and pericentromeric for each of the eight Cr chromosomes. i, Distribution of siren loci along the eight chromosomes of the Cr genome. Blue bars indicate siren loci, black boxes indicate positions of pericentromeric regions. j, TE families overlapping with24 nt-dominant and siren loci compared to the genome-wide frequencies in Cr.
Extended Data Fig. 7 sirenRNA accumulation and DNA methylation of selected AGLs and expression of RdDM pathway genes.
a, siRNA accumulation and CHH methylation in the endosperm of indicated genotypes on selected genes overlapping siren loci: Carubv10025009m.g (AGL33), Carubv10011966m.g (YUC10), Carubv10028580m.g (AGL62), Carubv10010156m.g (AGL28), and Carubv10002555m.g (γ-type AGL). Blue boxes represent genes, purple boxes represent TEs. b, Boxplots show DNA methylation on γ and α* type I MADS box TFs in 6 DAP endosperm of different Capsella genotypes. Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test for paired samples, p-values were adjusted with Benjamini and Hochberg correction (*** p-value < 0.001, ** p-value < 0.01, * p-value < 0.05). c, Barplot shows log2 fold change expression level of indicated genes in comparison to respective maternal plants. Asterisks indicates statistical significance as calculated with Wald test in DESEQ2 (*** padj < 0.001, ** p- padj < 0.01, padj < 0.05).
Extended Data Fig. 8 Parental-specific DNA methylation in the hybrid endosperm of Capsella crosses.
a, b, Boxplot show parental-specific DNA methylation levels of genes (a) and TEs (b) in the endosperm of Cr × Cg, 4xCr × Cg and Cg × Cr at 6 DAP. Maternal and paternal methylation values in each genotype are compared to the respective parent in the Cr × Cg cross. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (p-value: ***< 0.001, **< 0.01, *< 0.05, n.s. – not significant). c, d. Boxplot show parental-specific DNA methylation levels of genes (c) and TEs (d) in the endosperm of Co × Cr, 4xCo × Cr and Cr × Co at 6 DAP. Maternal and paternal methylation values in each genotype are compared to the respective parent in Co × Cr cross. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test, p-values were adjusted for multiple comparisons with Benjamini and Hochberg correction (p-value: ***< 0.001, **< 0.01, *< 0.05, n.s. – not significant). a-d, Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers.
Extended Data Fig. 9 Characterization of sirenRNAs trans-targets and parental-specific analysis of gene expression in Cr × Cg.
a, Heatmaps show expression of AGL genes (RPKM) targeted by trans-acting sirenRNAs, accumulation of sirenRNAs at trans AGL targets and corresponding CHG and CHH methylation. b, Tracks show sirenRNA accumulation at trans AGL targets and corresponding CHH and CHG methylation levels (same set of genes as on panel a). The purple line above the tracks marks cis acting siren RNAs. Light green and navy-blue bars on sirenRNA tracks show reads mapping in cis at siren loci. c-e, Barplots show the proportion of maternal and paternal reads in genes upregulated in Cr × Cg (c), γ-type AGLs upregulated in Cr × Cg (d) and selected orthologs of genes that have been previously reported as imprinted and involved in endosperm development or the triploid block in Arabidopsis (e). Expression data are obtained from whole seeds at 6 DAP. Black horizontal line on each plot represents the expected 2:1 ratio (maternal:paternal) for biallelic genes in the endosperm. f, Boxplot show the ratio of paternal:maternal read counts for all genes and genes upregulated in Cr × Cg. Boxes show median values and the interquartile range. Whiskers show the full range excluding outliers. Asterisks indicate statistically significant differences as calculated by the two-sided Wilcoxon test (*** p-value < 0.001).
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2 and Table 1.
Supplementary Data 1
Gene expression in all analysed crosses. The table shows log2 fold changes of gene expression in the endosperm of the indicated crosses.
Supplementary Data 2
List of siren loci and overlapping genomic features. The table shows the positions and normalized read counts over siren loci in the endosperm of the indicated genotypes.
Supplementary Data 3
List of potential trans targets affected in the Cr × Cg hybrid. The table shows differences in sirenRNA accumulation (Diff_RPM > 0), differences in CHH and CHG methylation (diff_CHH > 0.01 OR diff_CHG > 0.05), and differences in RNA accumulation (log2 fold change > 1, p < 0.05) in Cr × Cg compared with Cr × Cr.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dziasek, K., Santos-González, J., Wang, K. et al. Dosage-sensitive maternal siRNAs determine hybridization success in Capsella. Nat. Plants 10, 1969–1983 (2024). https://doi.org/10.1038/s41477-024-01844-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41477-024-01844-3
This article is cited by
-
Long-distance transport of siRNAs with functional roles in pollen development
Nature Plants (2026)
-
Epigenetic Remodeling During Seed Development: The Integrated Roles of Small RNAs, DNA Methylation, and Imprinted Genes
Plant Molecular Biology Reporter (2026)
-
REM transcription factors and GDE1 shape the DNA methylation landscape through the recruitment of RNA polymerase IV transcription complexes
Nature Cell Biology (2025)
