
Abstract
Patients with Parkinson’s disease (PD) exhibit an increased risk of melanoma, implying shared yet incompletely understood molecular mechanisms. This study aimed to delineate these common and distinct pathways by analyzing gene expression profiles from the Gene Expression Omnibus. A total of 90 differentially expressed genes (DEGs) were commonly regulated, while 173 DEGs exhibited divergent regulation between PD and melanoma. Protein-protein interaction analysis identified SNCA as a central node within a 21-protein network. LASSO regression revealed 13 hub genes (e.g., CCNB1, CCNH, CORO1C, and GSN) with high diagnostic accuracy (AUC >0.93) across both conditions. Gene set enrichment analysis implicated copper-induced cell death (cuproptosis) in PD neurons and melanoma cells, linking this process to hub genes. RT-qPCR confirmed increased SNCA expression during cuproptosis. Additional analyses identified macrophage involvement and WNT-β-catenin signaling as relevant. These findings suggest cuproptosis as a potential therapeutic target in PD and melanoma.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder primarily characterized by the progressive loss of dopaminergic neurons in the substantia nigra, a brain region crucial for movement control1. These neurons are distinguished by their production of neuromelanin, a dark pigment that contributes to their characteristic appearance2.On the other hand, melanoma is a malignant tumor originating from melanocytes, the pigment-producing cells commonly found in the skin, which derive from neural crest cells3.
Accumulating epidemiological evidence has revealed a significant association between PD and melanoma, with PD patients exhibiting a higher incidence of melanoma compared to the general population4,5,6,7,8,9. Initially, it was hypothesized that this increased risk might be attributable to levodopa treatment, a standard treatment for PD. As levodopa is a precursor in melanin biosynthesis, it was speculated to promote both melanin production and melanoma proliferation, potentially accelerating the growth of preexisting melanoma lesions10,11,12,13. However, subsequent reviews and studies have largely dismissed a direct causal relationship between levodopa therapy and melanoma, suggesting instead that shared underlying mechanisms might contribute to the co-occurrence of these conditions7,8,9.
Recent research has unveiled several shared genetic variants between PD and melanoma, with particular attention given to the PARK2 gene, which encodes the Parkin protein. PARK2 mutations, frequently associated with early-onset PD, have also been identified in melanoma tissues14,15. Interestingly, Parkin protein is expressed in melanocytes but absent in most melanoma cell lines, and reintroducing Parkin in these cell lines significantly reduces their proliferation16. Additionally, the hallmark pathological feature of PD is the aggregation of the neuronal protein α-synuclein into Lewy bodies within the brain17,18. Recent investigations have detected α-synuclein aggregates in peripheral tissues, which are being explored as potential presymptomatic markers for PD19,20. Notably, phosphorylated α-synuclein has been observed in skin biopsies, raising the possibility of utilizing cutaneous detection for early diagnosis21,22. Furthermore, α-synuclein has been identified within melanocytes of PD patients and is highly expressed in both primary and metastatic melanoma, indicating a potential molecular connection between PD and melanoma23,24,25.
Despite shared molecular pathways, PD and melanoma exhibit fundamentally distinct cellular outcomes. In PD, the progressive degeneration and death of neurons are central to the disease’s devastating effects on the nervous system. In stark contrast, melanoma is driven by the unchecked proliferation and malignant transformation of melanocytes. This juxtaposition underscores the necessity of investigating the intricate regulatory mechanisms behind cell death and survival in these diseases. A focused exploration of programmed cell death (PCD), differentially expressed genes (DEGs), and key signaling pathways could provide invaluable insights into the divergent processes of neurodegeneration in PD and oncogenesis in melanoma, advancing our understanding of both diseases and opening potential therapeutic avenues.
In the present study, we utilized gene expression data from the Gene Expression Omnibus (GEO) database to perform an in-depth analysis of gene expression profiles in both PD and melanoma tissues. We focused on identifying key genes involved in PCD pathways and other potential hub genes or molecular networks implicated in both diseases. Furthermore, we aimed to validate the expression of these hub genes in melanoma cells. By identifying critical DEGs and elucidating their role in shared molecular pathways, our goal is to identify potential therapeutic targets that could inhibit neuronal death in PD, offering novel opportunities for clinical intervention and treatment development.
Results
Characteristics of datasets
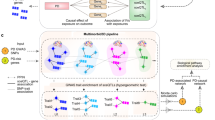
The study methodology is illustrated in Fig. 1, summarizing the analysis of six datasets from the GEO database. This includes three datasets for PD: GSE7621, GSE20141, and GSE49036, and three for melanoma: GSE100050, GSE114445, and GSE238207. Hierarchical clustering and principal component analysis (PCA) confirmed consistent dimensional distributions across the combined datasets (Fig. 2A, B for PD; 2D, E for melanoma). Additionally, two datasets were employed for external validation: GSE20292 for PD and GSE15605 for melanoma.

Study overall workflow.

A Hierarchical clustering heatmap of merged PD datasets. B Principal component analysis of merged PD datasets. C Volcano plot of DEGs in PD. D Hierarchical clustering heatmap of merged melanoma datasets. E Principal component analysis of merged melanoma datasets. F Volcano plot of DEGs in melanoma. G Venn diagram depicting the intersection of DEGs between PD and melanoma.
DEGs shared and distinct between PD and melanoma
Using predefined criteria, we identified 1097 DEGs in PD, including 419 upregulated and 678 downregulated genes (Fig. 2C), and 2897 DEGs in melanoma, with 1587 upregulated and 1310 downregulated genes (Fig. 2F) compared to control samples. Intersection analysis of DEGs between PD and melanoma datasets revealed several categories: 76 genes upregulated in PD but downregulated in melanoma, 97 genes downregulated in PD but upregulated in melanoma, and 44 genes consistently upregulated and 46 genes consistently downregulated across both conditions (Fig. 2G). The 90 consistently regulated genes were combined into a gene set termed com-DEGs, while the remaining 173 genes formed another gene set named dif-DEGs.
Enrichment analysis of PD and melanoma
Dynamic KEGG enrichment analysis of the merged PD dataset identified the top ten functional pathways, highlighting significant neurodegenerative and infectious disease pathways. Key pathways include those associated with amyotrophic lateral sclerosis, Huntington’s disease, and PD, alongside oxidative phosphorylation and proteasome pathways, which reflect the complex pathophysiology of PD. Notably, pathways related to neurodegeneration, proteasome function, and the synaptic vesicle cycle emphasize the importance of cellular and molecular mechanisms in PD (Fig. 3A). Conversely, the KEGG enrichment analysis of the merged melanoma dataset revealed pathways primarily involved in immune response and cell cycle regulation. These pathways include antigen processing and presentation, IL-17 signaling, rheumatoid arthritis, as well as the cell cycle and proteasome pathways, underscoring the critical role of immune interactions and cellular proliferation in melanoma progression (Fig. 3C).

A Top ten functional pathways in KEGG enrichment analysis of the merged PD dataset. B Top ten functional pathways in GO enrichment analysis of the PD dataset. C Top ten functional pathways in KEGG enrichment analysis of the merged melanoma dataset. D Top ten functional pathways in GO enrichment analysis of the melanoma dataset.
The dynamic GO enrichment analysis of the PD dataset highlighted pathways centered on synaptic and vesicular functions, including axon terminus, synaptic vesicle, and vesicle-mediated transport in synapses. These results indicate the critical role of neuronal communication and mitochondrial functions in PD. Additionally, the analysis underscored the involvement of inner mitochondrial membrane protein complexes and mitochondrial protein-containing complexes, reflecting mitochondrial dysfunction in PD pathology (Fig. 3B). In parallel, the GO enrichment analysis of the melanoma dataset emphasized regulatory processes involved in chromosome segregation and cell cycle transitions. Key findings included negative regulation of chromosome segregation, mitotic phase transitions, and spindle checkpoint signaling, revealing the significance of precise cell division control in melanoma development and progression (Fig. 3D).
Enrichment analysis of shared DEGs and protein–protein interaction (PPI) network construction
A PPI network analysis of the differential expressed genes (dif-DEGs) was performed using the STRING database, resulting in a network of 58 nodes and 70 edges. KEGG enrichment analysis identified significant pathways, including adherens junction, breast cancer, endometrial cancer, and acute myeloid leukemia, indicating the involvement of these genes in cancer-related pathways (Fig. 4A). GO enrichment analysis revealed significant cellular component associations, particularly in chromosomal and spindle structures such as chromosomal region, kinetochore, and condensed chromosome, which are critical for maintaining genomic integrity during cell division. Additional highlights included complexes related to protein kinase activity and actin cytoskeleton dynamics, underscoring the roles of regulatory and structural proteins in cellular processes (Fig. 4B).

A KEGG enrichment analysis of dif-DEGs. B GO cellular components enrichment analysis of dif-DEGs. C GO biological processes enrichment analysis of dif-DEGs. D GO molecular function analysis of dif-DEGs. E Visualization of a key functional module within the PPI network, comprising 21 nodes and 42 edges, with highly connected genes indicated in red.
Biological processes enriched in the GO analysis focused on chromosome segregation, mitotic nuclear division, and cell cycle regulation, essential for accurate cell division and genome stability. Notably, the analysis also emphasized the regulation of neurotransmitter secretion and transport, linking these DEGs to synaptic function (Fig. 4C). Molecular function analysis underscored roles in protein kinase regulation, kinase activation, and metallochaperone activity, highlighting the regulatory functions of these DEGs within kinase signaling pathways (Fig. 4D).
Using Cytoscape software with the MCODE plug-in, a key functional module of the PPI network was constructed, yielding a network of 21 nodes and 42 edges, visualized and color-coded based on MCODE scores, with highly connected genes highlighted in red to indicate their potential central roles (Fig. 4E).
Additionally, PPI network analysis of common DEGs (com-DEGs) produced a network with 36 nodes and 35 edges. KEGG enrichment analysis of these genes identified systemic lupus erythematosus as a significant pathway (Supplementary Fig. 1A). GO enrichment analysis highlighted key cellular components related to cation channel complexes and transmembrane transport complexes (Supplementary Fig. 1B). Biological processes included muscle system processes, regulation of ion transmembrane transport, cardiac muscle contraction, and sensory perception of temperature and pain (Supplementary Fig. 1C). Molecular functions emphasized ion channel activities, voltage-gated channel activities, and structural constituents of chromatin (Supplementary Fig. 1D). A functional module constructed with Cytoscape and the MCODE plug-in yielded two networks, each with four nodes and six edges, with highly connected genes indicated in red, signifying their potential central roles in the network (Supplementary Fig. 1E).
Identification and validation of hub genes
To identify key hub genes, we employed LASSO regression on the merged PD dataset, which served as the training dataset. From the 21 genes in the critical functional module constructed with Cytoscape using the MCODE plug-in, we identified 13 hub genes: ACTN4, CCNH, CENPE, CENPK, CORO1C, FOXO3, RAD52, RMI2, SIRT2, ZWILCH, and ZWINT, which were significant for both the lambda.min and lambda.1se models; CCNB1 was selected exclusively for lambda.min model, and GSN solely for lambda.1se model (Fig. 5A, B). Notably, ACTN4, FOXO3, GSN, RAD52, and SIRT2 were upregulated in PD and downregulated in melanoma, while CCNB1, CCNH, CENPE, CENPK, CORO1C, RMI2, ZWILCH, and ZWINT exhibited the opposite trend (Fig. 2G).

A Mean squared error as a function of Log(λ) in Lasso regression. B Regression coefficients as a function of Log(λ) in Lasso regression. C Wilcoxon test results for the lambda.min and lambda.1se models in distinguishing PD patients from controls (P < 0.001). D ROC curve analysis showing AUC values of 0.934 for the lambda.min model and 0.93 for the lambda.1se model in PD. E Wilcoxon test results for melanoma samples indicate significant differences (P < 0.001). F ROC curve analysis demonstrating AUC values of 0.935 for both models in melanoma. G Individual gene analysis reveals AUC values greater than 0.638 for all hub genes in PD. H Individual gene analysis reveals AUC values greater than 0.666 for all hub genes in melanoma.
To assess the diagnostic performance of the LASSO regression models in differentiating PD patients from controls, we conducted Wilcoxon tests and generated ROC curves. In the training dataset, both models demonstrated significant differences between PD and control samples, with the lambda.min model (P = 2.8e-12) and the lambda.1se model (P = 5.3e-12) showing excellent diagnostic efficiency (Fig. 5C). The lambda.min model achieved an AUC of 0.934, and the lambda.1se model achieved an AUC of 0.93 (Fig. 5D). In the merged melanoma dataset, both models also revealed significant differences between melanoma and control samples, with P values of 6.2e-10 for both models (Fig. 5E), and both models achieved an AUC of 0.935 (Fig. 5F). Moreover, individual gene analysis indicated all hub genes had AUC values greater than 0.6, further validating their diagnostic potential (Fig. 5G, H).
We further validated these models using two external datasets. In the PD dataset GSE20292, both models demonstrated satisfactory diagnostic accuracy, with AUC values of 0.914 for the lambda.min model and 0.934 for the lambda.1se model (Supplementary Fig. 2A). Statistical analysis using the Wilcoxon test revealed significant differences, with P values of 6.7e-05 for the lambda.min model and 2.1e-05 for the lambda.1se model (Supplementary Fig. 2B). In the melanoma dataset GSE15605, both models also exhibited strong diagnostic performance, with AUC values of 0.829 for the lambda.min model and 0.815 for the lambda.1se model (Supplementary Fig. 2C). The differences between melanoma and control samples were statistically significant, with P values of 6.4e-05 for the lambda.min model and 0.00013 for the lambda.1se model (Supplementary Fig. 2D). Collectively, these results provide robust evidence that the identified hub genes and LASSO regression models possess strong discriminative ability for differentiating PD and melanoma patients from control groups across multiple independent datasets.
Programmed cell death (PCD) in PD and melanoma
To investigate the role of PCD in PD and melanoma, we conducted single-sample gene set enrichment analysis (ssGSEA) using PCD-related gene sets from integrated datasets, employing the GSVA R package. Our analysis revealed a statistically significant but modest upregulation of necroptosis and pyroptosis in PD tissues (Fig. 6A). In contrast, melanoma tissues showed a pronounced increase in various PCD forms, including necroptosis and pyroptosis (Fig. 6B).

A ssGSEA results show upregulation of necroptosis and pyroptosis in PD tissues. B ssGSEA results indicate significant increases in various PCD forms, including necroptosis and pyroptosis, in melanoma tissues. C Correlation analysis of hub genes with PCD pathways in PD. D Correlation analysis of hub genes with PCD pathways in melanoma.
We examined the relationships between the identified hub genes and PCD pathways. In melanoma, significant correlations were observed between these hub genes and PCD pathways (Fig. 6D). Conversely, the associations in PD were less consistent, suggesting a more complex relationship between hub genes and PCD pathways (Fig. 6C).
Additionally, xCell analysis revealed a significant reduction in neuronal populations within PD tissues, indicative of neurodegeneration (Fig. 7A). Melanoma tissues exhibited reductions in both neuronal and melanocyte populations (Fig. 7B, C). Detailed xCell analysis results are shown in Supplementary Fig. 3. Among the hub genes, ACTN4, CCNB1, CCNH, CORO1C, FOXO3, GSN, and ZWINT were significantly associated with neurons in PD and melanocytes in melanoma, highlighting a parallel role of neurons in PD and melanocytes in melanoma, while indicating contrasting relationships between neurons in both conditions (Fig. 7D, E).

A xCell analysis showing a significant reduction in neuronal populations in PD tissues. B, C xCell analysis indicating decreased neuronal and melanocyte populations in melanoma tissues. D Association of hub genes with neuronal populations in PD. E Association of hub genes with melanocyte populations in melanoma. F Elevated cuproptosis levels in PD neurons. G Elevated cuproptosis levels in melanoma melanocytes. H Sankey diagram illustrating the relationships between hub genes and cuproptosis in PD neurons. I Sankey diagram illustrating the relationships between hub genes and cuproptosis in melanoma melanocytes.
Further investigation revealed significantly elevated cuproptosis in neurons affected by PD (Fig. 7F) and in melanocytes within melanoma (Fig. 7G). These findings suggest shared pathological mechanisms underlying neuronal degeneration in PD and melanocyte dysfunction in melanoma, indicating a potential avenue for therapeutic exploration. The Sankey diagram illustrates the relationships between various hub genes and cuproptosis in both conditions, with CCNB1, CCNH, CORO1C, and GSN associated with cuproptosis in PD neurons (Fig. 7H) and ACTN4, CCNB1, CCNH, CENPE, CENPK, CORO1C, GSN, RMI2, and ZWINT linked to cuproptosis in melanoma melanocytes (Fig. 7I). These results enhance our understanding of the complex interplay between PCD and cellular pathology in PD and melanoma, warranting further investigation into therapeutic interventions targeting these pathways.
Hub gene expression alterations during cuproptosis
To investigate the effects of cuproptosis on gene expression in A2058 melanoma cells, we treated the cells with elesclomol and CuCl2 at concentrations of 100 nM (low) and 200 nM (high), with PBS as a control. After a 24-h incubation, Western blot analysis demonstrated a significant reduction in FDX1 protein levels in elesclomol-treated cells, confirming cuproptosis induction (Fig. 8A, B).

A Western blot analysis showed reduced FDX1 protein levels in A2058 melanoma cells treated with 100 or 200 nM elesclomol and CuCl2 for 24 h, indicating successful induction of cuproptosis. B Quantification of the relative fluorescent intensity of FDX1 normalized to β-actin from Western blot, presented as mean ± SEM (n = 3). C RT-qPCR analysis revealed significant changes in hub gene expression post-cuproptosis induction, including upregulation of cell cycle-related genes (CCNB1, CCNH, CENPE, CENPK, ZWILCH, ZWINT), cytoskeletal organization genes (CORO1C, GSN), and the DNA repair gene (RMI2), as well as downregulation of FOXO3. Upregulation of the PD-associated gene SNCA and the sirtuin gene SIRT2 is also observed, with data presented as mean ± SEM (n = 3).
Subsequent RT-qPCR analysis revealed significant changes in the expression of various hub genes. Notably, genes associated with cell cycle regulation and mitosis, including CCNB1, CCNH, CENPE, CENPK, ZWILCH, and ZWINT, were upregulated. Additionally, genes involved in cytoskeletal organization (CORO1C, GSN) and DNA repair (RMI2) also exhibited increased expression. In contrast, FOXO3, associated with oxidative stress response, was downregulated. Furthermore, the PD-related gene SNCA and the sirtuin family gene SIRT2 were upregulated during cuproptosis induction (Fig. 8C). These results underscore the complex regulatory dynamics of gene expression during cuproptosis in melanoma cells and provide insights into the molecular pathways implicated in this copper-dependent cell death process.
Immune cell infiltration in PD and melanoma
xCell analysis revealed no significant changes in immune cell infiltration within PD tissues (Supplementary Fig. 3A). In contrast, melanoma exhibited marked alterations in immune cell types (Supplementary Fig. 3B). A further immune cell infiltration analysis using CIBERSORT indicated an increase in resting memory CD4 + T cells in PD (Fig. 9A). Conversely, melanoma showed significant increases in regulatory T cells, γδ T cells, activated memory CD4 + T cells, neutrophils, M1 macrophages, and M0 macrophages, while resting memory CD4 + T cells, activated natural killer (NK) cells, resting mast cells, eosinophils, resting dendritic cells, and activated dendritic cells decreased (Fig. 9B).

A CIBERSORT analysis in PD tissues. B CIBERSORT analysis in melanoma tissues. C Correlation of hub genes with immune cell populations in PD. D Correlation of hub genes with immune cell populations in melanoma. E Correlation of immune cell populations with PCD in PD. F Correlation of immune cell populations with PCD in melanoma.
The patterns of resting memory CD4 + T cells varied distinctly between PD and melanoma. Correlation analyses between the identified hub genes and infiltrating immune cell populations revealed that in PD, hub genes were predominantly associated with M2 macrophages (Fig. 9C), whereas in melanoma, they correlated with M1 macrophages (Fig. 9D).
Additionally, we explored the relationship between PCD and immune cell infiltration, reinforcing the notion of differing macrophage populations in PD and melanoma. In PD, hub genes were associated with M2 macrophages (Fig. 9E), while in melanoma, they were linked to M1 macrophages (Fig. 9F). Given that cuproptosis is associated with both neurons in PD and melanocytes in melanoma, the functional distinctions between M1 and M2 macrophages may significantly influence the divergent outcomes observed, specifically the neurodegeneration in PD versus melanocyte survival in melanoma. This differential immune response provides valuable insights into the pathophysiology of these conditions and potential therapeutic strategies.
Molecular pathways in PD and melanoma
We conducted a comprehensive analysis of molecular pathways using 50 Hallmark gene sets from MsigDB to identify distinct and overlapping mechanisms in PD and melanoma. In PD, key pathways such as coagulation, IL2-STAT5 signaling, K-Ras signaling, TNF-α signaling via NF-κB, and WNT-β-Catenin signaling were significantly upregulated, while the spermatogenesis pathway was notably downregulated (Fig. 10A). Similarly, melanoma exhibited upregulation of coagulation, IL2-STAT5 signaling, K-Ras signaling, and TNF-α signaling via NF-κB; however, the spermatogenesis and WNT-β-Catenin pathways showed opposite regulatory trends compared to PD (Fig. 10B). A complete analysis of all 50 Hallmark pathways is detailed in Supplementary Fig. 4.

A Upregulated and downregulated molecular pathways in PD. B Upregulated and downregulated molecular pathways in melanoma. C Correlation of hub genes with a molecular pathway in PD. D Correlation of hub genes with a molecular pathway in melanoma. E Correlation of molecular pathway with cell populations in PD. F Correlation of the molecular pathway with cell populations in MM.
Further investigation revealed strong associations between previously identified hub genes and spermatogenesis and WNT-β-Catenin signaling pathways, particularly in melanoma (Fig. 10C, D). Additionally, analysis of cell types identified by xCell indicated a strong correlation of neurons in PD and melanocytes in melanoma with the spermatogenesis and WNT-β-Catenin pathways (Fig. 10E, F). Notably, the WNT-β-Catenin and IL2-STAT5 signaling pathways were divergently associated with cuproptosis in PD and melanoma (Fig. 11A, B). These findings indicate that the divergent regulation of the WNT-β-Catenin pathway may represent critical molecular distinctions between PD and melanoma, potentially influencing contrasting cellular outcomes through interactions with cuproptosis.

A Correlation of molecular pathway with PCD in PD. B Correlation of molecular pathway with PCD in MM.
Discussion
In this study, we performed a comprehensive analysis of the molecular mechanisms shared and distinct between PD and melanoma. Our investigation identified 90 DEGs that are commonly regulated across both conditions, as well as 173 DEGs that display divergent regulation. Protein–protein interaction analysis revealed a prominent network centered around the SNCA gene, which encodes the key pathological protein alpha-synuclein, suggesting its significant role in both diseases. Notably, we uncovered cuproptosis as a critical mechanism of PCD in both PD neurons and melanoma melanocytes, with RT-qPCR confirming significant expression changes in the associated hub genes. Furthermore, our findings revealed distinct macrophage populations and the WNT-β-catenin pathway as important correlates of hub gene expression in both conditions, indicating their potential involvement in the cuproptosis mechanism.
Epidemiological studies have shown that while most cancer rates are lower in individuals with PD, melanoma is one of the few malignancies with an increased incidence in this population4,5,6,7,8,9,26,27. Although early theories proposed that this elevated melanoma risk was linked to levodopa therapy, recent comprehensive studies have largely dismissed a direct causal relationship, instead highlighting shared molecular mechanisms as likely drivers of this co-occurrence7,8,9,10,11,12,13. A hallmark of PD pathology is the aggregation of α-synuclein into Lewy bodies within the brain, and recent studies have detected α-synuclein aggregates in peripheral tissues, with phosphorylated forms found in skin biopsies, offering a promising approach for early, non-invasive PD diagnosis17,18,19,20,21,22. High α-synuclein expression in PD melanocytes and melanoma cells further suggests a molecular link between PD and melanoma23,24,25.
In this study, an intersection analysis of DEGs between PD and melanoma datasets uncovered both significant overlaps and important distinctions, indicating potential shared molecular signatures. These overlapping genes point to common biological pathways, with notable enrichment in pathways involving cation or monoatomic ion channels and the transmembrane transport of monoatomic or metal ions. Of particular interest, melanin, and neuromelanin are synthesized from tyrosine through the copper-dependent enzyme tyrosinase, highlighting the essential role of metal ion transport in maintaining appropriate copper levels in both dopaminergic neurons and melanocytes28,29. This copper homeostasis is crucial not only for supporting biosynthetic processes but also for preventing melanocyte death and preserving SOD1 activity, which has been shown to protect against neuronal loss in PD30,31. One of the most compelling findings of our study is the shared involvement of cuproptosis pathways in the neurons affected by PD and the melanocytes associated with melanoma. Disruptions in metal ion transport that lead to copper overload may contribute to the pathogenesis of PD through copper-dependent PCD in dopaminergic neurons. This finding aligns with evidence from Wilson disease—a genetic disorder marked by defective copper metabolism that causes copper accumulation in the brain and other organs. The neurological symptoms of Wilson's disease, such as tremors, rigidity, and bradykinesia, resemble those in PD, suggesting a significant role of copper dysregulation in movement-related brain damage32. Furthermore, elevated free copper in PD has been associated with oxidative stress, alpha-synuclein aggregation, and increased iron in affected brain regions, all of which are linked to intensified neuronal damage33,34. Together, these findings underscore the importance of copper homeostasis in both PD and melanoma, with potential implications for copper-focused therapeutic strategies in neurodegenerative and oncological contexts.
Protein–protein interaction analysis of differentially regulated genes in PD and melanoma revealed a network centered around the SNCA gene, which encodes α-synuclein. This network highlights SNCA as a pivotal gene potentially influencing both PD-related neurodegeneration and melanoma tumorigenesis. Interestingly, SNCA expression was downregulated in PD, likely due to feedback inhibition from accumulated α-synuclein, whereas it was upregulated in melanoma, where it may support melanocyte survival and proliferation. To further explore SNCA expression in the context of cuproptosis, we performed RT-qPCR in melanoma cells, confirming that SNCA is indeed upregulated during this process. This finding suggests that α-synuclein may contribute to maintaining optimal intracellular copper levels. Studies have shown that copper binds strongly to monomeric α-synuclein via the N-terminal amine and His50, and readily incorporates into amyloid fibers. This copper binding may mitigate copper dys-homeostasis in PD, protecting neurons from copper-induced oxidative stress35,36,37. These results indicate a potentially protective role for monomeric α-synuclein in melanin- or neuromelanin-producing cells. Therefore, therapeutic strategies that maintain α-synuclein in its soluble, monomeric state—rather than simply reducing its levels—may offer clinical benefits. Targeting pathways to prevent α-synuclein aggregation while preserving its normal, soluble form could present a promising direction for disease-modifying therapies in PD.
Using LASSO regression, we identified a set of hub genes—ACTN4, CCNB1, CCNH, CENPE, CENPK, CORO1C, FOXO3, GSN, RAD52, RMI2, SIRT2, ZWILCH, and ZWINT—with significant diagnostic potential for both PD and melanoma. These genes showed a strong association with disease states and consistently high diagnostic performance, reflected by high AUC values across multiple datasets for both lambda.min and lambda.1se models, underscoring their robustness as biomarkers to distinguish PD and melanoma patients from healthy controls. Further investigation revealed that genes CCNB1, CCNH, CORO1C, and GSN are implicated in cuproptosis in PD-affected neurons, while ACTN4, CCNB1, CCNH, CENPE, CENPK, CORO1C, GSN, RMI2, and ZWINT are associated with cuproptosis in melanoma melanocytes. RT-qPCR analysis in melanoma cells confirmed upregulation of CCNB1, CCNH, CENPE, CENPK, CORO1C, GSN, RMI2, and ZWINT during cuproptosis. Interestingly, DEGs indicated downregulation of CCNB1, CCNH, CENPE, CENPK, CORO1C, RMI2, and ZWINT in PD, contrasting with the upregulation of GSN, highlighting disease-specific expression patterns that may further inform therapeutic targeting in these conditions.
The genes CCNB1 and CCNH, essential for cell cycle regulation, interact with CDKs to control mitotic entry and DNA repair, with their upregulation linked to accelerated growth rates38,39. This finding suggests that cyclins activating CDK1/cdc2, the primary G2/M phase kinase, may play a role in melanoma tumorigenesis38,39,40. Cyclin activation may also support melanoma cell survival during cuproptosis by regulating cell division and managing DNA repair, potentially buffering cuproptosis-induced cell stress. Conversely, in neurons affected by PD, cyclin expression appears downregulated, reducing their potential protective effect. In models of PD, such as neurons exposed to the toxin 1-methyl-4-phenylpyridinium ion (MPP + ), CCNB1 expression is decreased, indicating a possible link between cyclin downregulation and neurodegeneration41,42. Studies also show that CDK inhibitors can offer neuroprotection, suggesting a potential protective role of cyclins in mitigating neurodegeneration even in non-dividing neurons43,44. Together, these findings indicate that while neurons in PD may lack the proliferative benefit from cyclin activity, cyclins could still provide essential protective functions against degeneration.
Both CORO1C and GSN are key regulators of the actin cytoskeleton, playing critical roles in maintaining cell shape, motility, and migration. CORO1C is essential for the proper organization of cytoskeletal components, including actin filaments, microtubules, and vimentin intermediate filaments, and is involved in cell proliferation, migration, and lamellipodia formation45. Additionally, CORO1C is crucial for mitochondrial positioning within cells and facilitates endoplasmic reticulum-associated endosome fission45,46. GSN, a calcium-regulated actin-binding protein, modulates actin filaments by binding to their barbed ends, preventing monomer exchange while promoting filament nucleation and severing47. In response to copper stress, which can disrupt cytoskeletal integrity through oxidative modifications, CORO1C and GSN may stabilize actin filaments, potentially buffering cells against cuproptosis-induced damage. Emerging evidence links CORO1C to PD pathogenesis through interactions with LRRK2, a gene associated with PD. Variants within or near CORO1C may influence the penetrance of LRRK2 mutations, and decreased CORO1C expression observed in PD could impair LRRK2 function48. Meanwhile, GSN, encoding gelsolin, is implicated in the formation of Lewy bodies in PD, with studies indicating that gelsolin can accelerate α-synuclein aggregation49,50,51. Interestingly, reduced levels of gelsolin have been observed in plasma extracellular vesicles from individuals with dementia with Lewy bodies compared to controls and Alzheimer’s samples, suggesting a role for gelsolin in Lewy body formation52. Gelsolin also modulates neuroinflammation, a critical component of neurodegenerative diseases, by inhibiting LPS-induced inflammatory processes53. While GSN mRNA levels appear upregulated in PD, previous findings show that soluble gelsolin levels are reduced, likely due to its aggregation with α-synuclein, leading to decreased availability of functional gelsolin54.
The immune cell infiltration profiles in PD and melanoma revealed striking differences. While PD tissues showed minimal changes in immune cell populations, melanoma tissues were marked by significant alterations in immune cell infiltration, particularly an increase in pro-inflammatory cells such as regulatory T cells, γδ T cells, and M1 macrophages. This highlights the divergent roles of the immune system in neurodegenerative versus oncogenic contexts. The pronounced immune response in melanoma aligns with its immunogenic nature, where cancer progression is closely linked to the evasion of immune surveillance and the recruitment of immunosuppressive cells to the tumor microenvironment55. In contrast, the subtle immune changes in PD suggest that neuroinflammation, although present, may not be the primary driver of neuronal death. Interestingly, cuproptosis and related hub genes in melanoma correlated with M1 macrophages, indicating that macrophage polarization plays distinct roles in the pathophysiology of the disease. M1 macrophages, generally recognized for their anti-tumor properties, may increase melanoma in response to tumor growth56. Meanwhile, PD shows limited immune cell infiltration compared to melanoma, M2 macrophages associated with cuproptosis, and related hub genes indicate potential therapeutic targets. Studies have suggested that M1 macrophage activation can drive neurodegeneration in PD, while M2 macrophages may inhibit M1 states and promote repair and healing57. Additionally, research has identified central nervous system (CNS)-resident macrophages, specifically border-associated macrophages, as critical mediators of α-synuclein-related neuroinflammation, further highlighting immune cell specialization in PD pathology58.
Our pathway analysis reveals that the WNT-β-Catenin signaling pathway exhibits inverse regulatory trends and divergent associations with cuproptosis in PD and melanoma, while maintaining consistent patterns in relation to neurons in PD and melanocytes in melanoma. These findings suggest that the divergent regulation of WNT-β-Catenin signaling may underlie the contrasting cellular outcomes in PD-affected neurons and melanoma cells. In melanoma, increased nuclear β-catenin activity through the WNT-β-Catenin pathway correlates with improved prognosis and decreased cellular proliferation, highlighting its therapeutic potential in oncology59,60. Conversely, in PD, disruptions in WNT-β-Catenin signaling, such as gene hypermethylation and reduced protein expression, compromise dopaminergic neurons, while pathway activation has been shown to support neuronal growth and mitochondrial function61,62.
Despite the comprehensive nature of this study, several limitations should be considered. First, our analysis primarily relied on publicly available gene expression datasets, which may introduce variability stemming from differences in sample collection, processing, and experimental conditions. This heterogeneity could affect the reproducibility and generalizability of our findings. Second, while we identified cuproptosis as a novel shared pathway of PCD between PD and melanoma, the functional validation of cuproptosis in these diseases remains limited. Further in vitro and in vivo studies are necessary to establish causal relationships and fully elucidate the biological significance of cuproptosis in PD and melanoma pathogenesis. Third, although we investigated macrophage populations and WNT-β-catenin signaling in relation to hub genes associated with cuproptosis, further research is needed to fully characterize their roles in cuproptosis regulation and their therapeutic potential. Another limitation of our study lies in the inconsistency of the brain regions sampled across the PD datasets. While most PD datasets were derived from the frontal cortex, the validation dataset was sourced from the Globus Pallidus interna (GPi), a region implicated in PD but not specifically the substantia nigra, which is central to PD pathology. Although datasets from the same experimental platform (GPL570) were used to ensure methodological consistency, the lack of a substantia nigra-specific validation dataset may limit the generalizability of our findings. Future studies incorporating more uniform, region-specific datasets, particularly from the substantia nigra, would strengthen the validation of the molecular mechanisms identified in this study. Additionally, the merged analysis approach for both PD and melanoma datasets, while enhancing statistical power, may have overlooked disease-specific nuances that could be better captured through an intersection-based approach. However, due to the relatively small sample sizes in the PD datasets derived from human tissues, the intersection-based approach did not yield robust differentially expressed genes (DEGs). Future studies with larger sample sizes may address this limitation. Despite the application of batch effect correction, the potential for residual batch effects from combining datasets with different technical characteristics remains a consideration.
This integrative study elucidates the intricate molecular connections between PD and melanoma, highlighting both shared and distinct disease mechanisms. Central to our findings is the role of cuproptosis, a copper-driven pathway of programmed cell death, in both conditions. The hub genes identified in this study, closely associated with cuproptosis, exhibit significant diagnostic potential for both diseases. Moreover, the divergent macrophage populations and WNT-β-catenin signaling patterns observed in PD and melanoma are correlated with these hub genes, underscoring their relationship with cuproptosis. These interactions are critical in determining the distinct fates of PD neurons and melanoma melanocytes, suggesting that modulating cuproptosis may be pivotal in protecting PD neurons from degeneration while simultaneously inhibiting the malignant proliferation of melanoma cells. These findings pave the way for future research aimed at elucidating the precise roles of cuproptosis, along with the associated immune profiles and WNT-β-catenin signaling, in disease progression. Ultimately, this work contributes to the development of targeted therapies that address both the unique and overlapping aspects of PD and melanoma, positioning cuproptosis and its related pathways at the forefront of innovative treatment strategies.
Methods
Acquisition and preparation of datasets
Gene expression datasets were retrieved from the Gene Expression Omnibus (GEO), a comprehensive functional genomics database maintained by the National Center for Biotechnology Information (NCBI). We conducted specific queries using the terms “Parkinson’s disease” and “melanoma” to identify relevant gene expression profiles derived from human tissue studies. For PD, we selected datasets derived from human brain tissue, ensuring methodological consistency and compatibility across experimental platforms. This approach guarantees that the datasets are comparable and that the findings are both robust and reproducible across various PD-related contexts. For melanoma, we chose datasets derived from primary melanoma tissues or control normal tissues, with consistent methodologies and experimental platforms. This ensures that the datasets capture the most biologically relevant and accurate gene expression profiles for melanoma while maintaining methodological consistency. In total, datasets GSE7621, GSE20141, and GSE49036 were selected for PD, and GSE238207, GSE100050, and GSE114445 were chosen for melanoma, based on their comprehensive coverage of disease-specific gene expression data. Detailed experimental metadata, including sample sizes and experimental conditions, are provided in Table 1. To address potential batch effects resulting from merging these datasets, we applied the sva (version 3.50.0). Specifically, we employed the ComBat function implemented in the sva package to adjust for batch effects and minimize unwanted variation attributable to batch differences.
Analysis of DEGs and identification of shared DEGs
We conducted differential expression analysis in the R programming environment (version 4.3.3), using stringent statistical thresholds (adjusted P value <0.05 and absolute log2 fold change >0.5) to identify significantly altered genes (DEGs). This analysis was applied across both PD and melanoma datasets, generating hierarchical clustering heatmaps, volcano plots, and principal component analysis (PCA) plots using the tinyarray (version 2.3.2) and ggplot2 (version 3.5.0) R packages. To identify shared and distinct DEGs between PD and melanoma, we utilized the Venn Diagram R package (version 1.7.3), classifying DEGs based on their up- or downregulation in each disease. This comparative analysis provided insights into convergent and divergent molecular mechanisms underlying PD and melanoma pathogenesis.
Dynamic enrichment analysis of DEGs
To explore the biological functions of the DEGs, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses using the enrichplot R package (version 1.22.0). GO analysis classified DEGs according to their involvement in biological processes, molecular functions, and cellular components, while KEGG pathway analysis identified key signaling pathways overrepresented by DEGs. This dynamic enrichment approach allowed us to uncover shared biological processes and pathways between PD and melanoma, providing a deeper understanding of the molecular underpinnings of these diseases. Further enrichment analysis of shared DEGs was performed using the clusterProfiler R package (version 4.10.1).
Construction of the protein–protein interaction (PPI) network of shared DEGs
To elucidate the interactions between shared DEGs, we constructed a protein–protein interaction (PPI) network using the STRING database (https://string-db.org/), setting a minimum interaction score of 0.4 to ensure high-confidence interactions. The resulting PPI network was visualized using Cytoscape software (version 3.10.2), and significant molecular complexes were identified using the MCODE plug-in (version 2.0.3), with customized parameters to detect key functional modules within the network. These analyses provided insights into the functional architecture of shared DEGs, revealing potential regulatory mechanisms.
Identification of hub genes using least absolute shrinkage and selection operator (LASSO) regression
Hub genes were identified from shared DEGs using LASSO regression analysis with the glmnet R package (version 4.1.8), applied to the PD datasets as the training set. Hub genes were then validated across melanoma datasets, with differential expression assessed using the Wilcoxon test. The diagnostic utility was evaluated through Receiver Operating Characteristic (ROC) curve analysis using the ROCR R package (version 1.0.11), with Area Under the Curve (AUC) values calculated to determine the diagnostic efficacy of the identified hub genes. Validation was conducted using additional external GEO datasets to confirm the robustness of these genes across diverse experimental conditions.
Correlation analysis of PCD
We examined the involvement of PCD pathways in PD and melanoma using gene sets associated with 14 types of PCD63. Single-sample gene set enrichment analysis (ssGSEA) was performed using the GSVA R package (version 1.40.1) to assess PCD pathway activation across datasets. This analysis allowed us to quantify the contribution of each PCD mechanism to the pathophysiology of PD and melanoma, highlighting potential therapeutic targets.
Analysis of hub gene expression during cuproptosis
To explore the role of hub genes in cuproptosis, a copper-dependent cell death pathway, we treated A2058 melanoma cells with elesclomol and CuCl2 to induce cuproptosis. Protein expression of key cuproptosis markers was confirmed by Western blot analysis, while hub gene expression was assessed via RT-qPCR. This dual approach provided critical insights into the regulation of cuproptosis by identified hub genes in melanoma cells.
Correlation analysis of cell components
Cellular composition in PD and melanoma tissues was characterized using the CIBERSORT (version 0.1.0) and xCell (version 1.1.0) R packages. These analyses enabled the identification of immune and other cell types infiltrating target tissues, offering insights into the immunological landscape of PD and melanoma. The cellular localization of hub genes and molecular pathways was then correlated with specific cell types, particularly neurons in PD and melanocytes in melanoma.
Correlation analysis of molecular pathways
We performed pathway enrichment analysis using hallmark gene sets from the Molecular Signatures Database (MsigDB), covering 51 key molecular pathways. This analysis provided a comprehensive view of the molecular pathways implicated in PD and melanoma pathogenesis, with particular emphasis on pathways involved in cell death and cell-type-specific processes, such as neuronal death in PD and melanocyte proliferation in melanoma.
Data availability
The datasets used in this study are available in online repositories. The specific repository and accession numbers are provided in the article’s supplementary material. In particular, the datasets GSE7621, GSE20141, GSE20292, GSE49036, GSE100050, GSE114445, GSE15605, and GSE238207 can be accessed at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/). Experimental data supporting the findings of this study are included in the manuscript. The original experimental data are available from Q.W. and Y.Y. upon reasonable request.
Code availability
The code used in this study is available from the corresponding author upon reasonable request.
Abbreviations
- AUC:
-
Area Under the Curve
- DEGs:
-
Differentially Expressed Genes
- com-DEGs:
-
Differentially Expressed Genes with the Same Expression in PD and Melanoma
- dif-DEGs:
-
Differentially Expressed Genes with Opposing Expression in PD and Melanoma
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- GPi:
-
Globus Pallidus Interna
- GSVA:
-
Gene Set Variation Analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- LRRK2:
-
Leucine-Rich Repeat Kinase 2
- MCODE:
-
Molecular Complex Detection (Cytoscape Plug-in)
- MPP+:
-
1-Methyl-4-Phenylpyridinium Ion
- MsigDB:
-
Molecular Signatures Database
- PCA:
-
Principal Component Analysis
- PCD:
-
Programmed Cell Death
- PD:
-
Parkinson’s Disease
- PPI:
-
Protein-Protein Interaction
- ROC:
-
Receiver Operating Characteristic
- RT-qPCR:
-
Reverse Transcription Quantitative Polymerase Chain Reaction
- ssGSEA:
-
Single-Sample Gene Set Enrichment Analysis
References
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).
Vila, M. Neuromelanin, aging, and neuronal vulnerability in Parkinson’s disease. Mov. Disord. 34, 1440–1451 (2019).
Long, G. V., Swetter, S. M., Menzies, A. M., Gershenwald, J. E. & Scolyer, R. A. Cutaneous melanoma. Lancet 402, 485–502 (2023).
Bertoni, J. M. et al. Increased melanoma risk in Parkinson disease: a prospective clinicopathological study. Arch. Neurol. 67, 347–352 (2010).
Dalvin, L. A. et al. Parkinson disease and melanoma: confirming and reexamining an association. Mayo Clin. Proc. 92, 1070–1079 (2017).
Gao, X., Simon, K. C., Han, J., Schwarzschild, M. A. & Ascherio, A. Family history of melanoma and Parkinson disease risk. Neurology 73, 1286–1291 (2009).
Freedman, D. M. et al. Associations between cancer and Parkinson’s disease in U.S. elderly adults. Int J. Epidemiol. 45, 741–751 (2016).
Liu, R., Gao, X., Lu, Y. & Chen, H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 76, 2002–2009 (2011).
Olsen, J. H., Friis, S. & Frederiksen, K. Malignant melanoma and other types of cancer preceding Parkinson disease. Epidemiology 17, 582–587 (2006).
Fiala, K. H., Whetteckey, J. & Manyam, B. V. Malignant melanoma and levodopa in Parkinson’s disease: causality or coincidence?. Parkinsonism Relat. Disord. 9, 321–327 (2003).
Skibba, J. L., Pinckley, J., Gilbert, E. F. & Johnson, R. O. Multiple primary melanoma following administration of levodopa. Arch. Pathol. 93, 556–561 (1972).
Pfutzner, W. & Przybilla, B. Malignant melanoma and levodopa: is there a relationship? Two new cases and a review of the literature. J. Am. Acad. Dermatol 37, 332–336 (1997).
Dizdar, N. et al. L-dopa pharmacokinetics studied with microdialysis in patients with Parkinson’s disease and a history of malignant melanoma. Acta Neurol. Scand. 100, 231–237 (1999).
Levin, L. et al. Parkin somatic mutations link melanoma and Parkinson’s disease. J. Genet. Genomics 43, 369–379 (2016).
Inzelberg, R., et al. Parkinson disease (PARK) genes are somatically mutated in cutaneous melanoma. Neurol. Genet. 2, e70 (2016).
Hu, H. H. et al. PARKIN inactivation links Parkinson’s disease to melanoma. J. Natl Cancer Inst. 108 (2016).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Morris, H. R., Spillantini, M. G., Sue, C. M. & Williams-Gray, C. H. The pathogenesis of Parkinson’s disease. Lancet 403, 293–304 (2024).
Beach, T. G. et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702 (2010).
Gelpi, E. et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 29, 1010–1018 (2014).
Bellomo, G. et al. alpha-Synuclein seed amplification assays for diagnosing synucleinopathies: the way forward. Neurology 99, 195–205 (2022).
Gibbons, C. H. et al. Skin biopsy detection of phosphorylated alpha-synuclein in patients with synucleinopathies. JAMA 331, 1298–1306 (2024).
Dean, D. N. & Lee, J. C. Linking Parkinson’s disease and melanoma: interplay between alpha-synuclein and Pmel17 amyloid formation. Mov. Disord. 36, 1489–1498 (2021).
Matsuo, Y. & Kamitani, T. Parkinson’s disease-related protein, alpha-synuclein, in malignant melanoma. PLoS ONE 5, e10481 (2010).
Welinder, C., et al. Analysis of alpha-synuclein in malignant melanoma - development of a SRM quantification assay. PLoS ONE 9, e110804 (2014).
Chen, C., Zheng, H. & Hu, Z. Association between Parkinson’s disease and risk of prostate cancer in different populations: an updated meta-analysis. Sci. Rep. 7, 13449 (2017).
Becker, C., Brobert, G. P., Johansson, S., Jick, S. S. & Meier, C. R. Cancer risk in association with Parkinson disease: a population-based study. Parkinsonism Relat. Disord. 16, 186–190 (2010).
D’Mello, S. A., Finlay, G. J., Baguley, B. C. & Askarian-Amiri, M. E. Signaling pathways in melanogenesis. Int. J. Mol. Sci. 17, 1144 (2016).
Nagatsu, T., Nakashima, A., Watanabe, H., Ito, S. & Wakamatsu, K. Neuromelanin in Parkinson’s disease: tyrosine hydroxylase and tyrosinase. Int. J. Mol. Sci. 23, 4176 (2022).
Nagatani, K., Abe, Y., Homma, T., Fujii, J. & Suzuki, T. Copper chelation by d-penicillamine alleviates melanocyte death induced by rhododendrol without inhibiting tyrosinase. Biochem. Biophys. Res. Commun. 663, 71–77 (2023).
Trist, B. G. et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 134, 113–127 (2017).
Bandmann, O., Weiss, K. H. & Kaler, S. G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 14, 103–113 (2015).
Bisaglia, M. & Bubacco, L. Copper ions and Parkinson’s disease: why is homeostasis so relevant?. Biomolecules 10, 195 (2020).
Montes, S., Rivera-Mancia, S., Diaz-Ruiz, A., Tristan-Lopez, L. & Rios, C. Copper and copper proteins in Parkinson’s disease. Oxid. Med. Cell Longev. 2014, 147251 (2014).
Walke, G., Kumar, R. & Wittung-Stafshede, P. Copper ion incorporation in alpha-synuclein amyloids. Protein Sci. 33, e4956 (2024).
Bacchella, C. et al. Copper binding and redox activity of alpha-synuclein in membrane-like environment. Biomolecules 13, 287 (2023).
Dudzik, C. G., Walter, E. D. & Millhauser, G. L. Coordination features and affinity of the Cu(2)+ site in the alpha-synuclein protein of Parkinson’s disease. Biochemistry 50, 1771–1777 (2011).
Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Lim, S. & Kaldis, P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093 (2013).
Weber, R. G., Rieger, J., Naumann, U., Lichter, P. & Weller, M. Chromosomal imbalances associated with response to chemotherapy and cytotoxic cytokines in human malignant glioma cell lines. Int. J. Cancer 91, 213–218 (2001).
Wang, F. et al. Damage of neuroblastoma cell SH-SY5Y mediated by MPP+ inhibits proliferation of T-cell leukemia Jurkat by co-culture system. Int. J. Mol. Sci. 15, 10738–10750 (2014).
Pizarro, J. G. et al. Effects of MPP+ on the molecular pathways involved in cell cycle control in B65 neuroblastoma cells. Pharm. Res. 61, 391–399 (2010).
Wang, L. et al. Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J. Neurosci. Res. 88, 1970–1984 (2010).
Fei, X. F. et al. Olomoucine inhibits cathepsin L nuclear translocation, activates autophagy and attenuates toxicity of 6-hydroxydopamine. Brain Res. 1264, 85–97 (2009).
Behrens, J. et al. Coronin 1C-free primary mouse fibroblasts exhibit robust rearrangements in the orientation of actin filaments, microtubules and intermediate filaments. Eur. J. Cell Biol. 95, 239–251 (2016).
Hoyer, M. J., et al. A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175, 254–265.e214 (2018).
Nag, S. et al. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc. Natl Acad. Sci. USA 106, 13713–13718 (2009).
Lai, D. et al. Genomewide association studies of LRRK2 modifiers of Parkinson’s disease. Ann. Neurol. 90, 76–88 (2021).
Haltia, M. et al. Gelsolin variant and beta-amyloid co-occur in a case of Alzheimer’s with Lewy bodies. Neurobiol. Aging 12, 313–316 (1991).
Wisniewski, T., Haltia, M., Ghiso, J. & Frangione, B. Lewy bodies are immunoreactive with antibodies raised to gelsolin related amyloid-Finnish type. Am. J. Pathol. 138, 1077–1083 (1991).
Welander, H. et al. Gelsolin co-occurs with Lewy bodies in vivo and accelerates alpha-synuclein aggregation in vitro. Biochem. Biophys. Res. Commun. 412, 32–38 (2011).
Gamez-Valero, A., Campdelacreu, J., Rene, R., Beyer, K. & Borras, F. E. Comprehensive proteomic profiling of plasma-derived extracellular vesicles from dementia with Lewy bodies patients. Sci. Rep. 9, 13282 (2019).
Barbalace, M. C. et al. Unraveling the protective role of oleocanthal and its ixidation product, oleocanthalic acid, against neuroinflammation. Antioxidants 13, 1074 (2024).
Alberio, T. et al. Verification of a Parkinson’s disease protein signature in T-lymphocytes by multiple reaction monitoring. J. Proteome Res. 13, 3554–3561 (2014).
Marzagalli, M., Ebelt, N. D. & Manuel, E. R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 59, 236–250 (2019).
Boutilier, A. J. & Elsawa, S. F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 22, 6995 (2021).
Moehle, M. S. & West, A. B. M1 and M2 immune activation in Parkinson’s disease: foe and ally?. Neuroscience 302, 59–73 (2015).
Schonhoff, A. M., et al. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat. Commun. 14, 3754 (2023).
Yu, F. et al. Wnt/beta-catenin signaling in cancers and targeted therapies. Signal Transduct. Target Ther. 6, 307 (2021).
Atkinson, J. M., et al. Activating the Wnt/beta-catenin pathway for the treatment of melanoma-application of LY2090314, a novel selective inhibitor of glycogen synthase kinase-3. PLoS One 10, e0125028 (2015).
Liu, J. et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 7, 3 (2022).
Singh, S. et al. Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/beta-catenin signaling in rat model of Parkinson’s disease. Free Radic. Biol. Med. 129, 73–87 (2018).
Xiao, L. et al. Exploring a specialized programmed-cell death patterns to predict the prognosis and sensitivity of immunotherapy in cutaneous melanoma via machine learning. Apoptosis 29, 1070–1089 (2024).
Acknowledgements
This study involved the reanalysis of publicly available datasets and cell-based experiments using established cell lines, neither of which required specific ethical approval. This work was supported by the National Natural Science Foundation of China (Grant No. 82201446 to Y.Y.), the Natural Science Foundation of Jiangsu Province of China (Grant No. BK20220249 to Y.Y.), the Boxi Youth Natural Science Foundation (Grant No. BXQN2023007 to Q.W.), and the Suzhou Basic Research Pilot Program (Grant No. SSD2024043 to Y.Y.). We sincerely acknowledge the authors who contributed the datasets GSE7621, GSE20141, GSE20146, GSE49036, GSE100050, GSE114445, GSE15605, and GSE238207 to the public domain, facilitating this research.
Author information
Authors and Affiliations
Contributions
Q.W. played a central role in experiment design and execution, data analysis, and manuscript preparation, including drafting and revisions. H.L. and Y.Y. participated in the experiment design and manuscript revisions. Y.H.D. and Y.X. contributed to manuscript revisions. All authors reviewed and approved the final manuscript. Each author made a substantial contribution to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Duan, Y., Xu, Y. et al. Linking Parkinson’s disease and melanoma: the impact of copper-driven cuproptosis and related mechanisms. npj Parkinsons Dis. 11, 74 (2025). https://doi.org/10.1038/s41531-025-00928-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-00928-x
This article is cited by
-
Revisiting the alpha-synuclein paradox in melanoma-Parkinson’s disease connection: more than a tale of two cell fates
Cellular and Molecular Life Sciences (2025)
