
Abstract
Enhancers are non-coding DNA regions that facilitate gene transcription, with a specialized subset, super-enhancers, known to exert exceptionally strong transcriptional activation effects. Super-enhancers have been implicated in oncogenesis, and their identification is achievable through histone mark chromatin immunoprecipitation followed by sequencing data using existing analytical tools. However, conventional super-enhancer detection methodologies often do not accurately reflect actual gene expression levels, and the large volume of identified super-enhancers complicates comprehensive analysis. To address these limitations, we developed the super-enhancer to gene links (SE-to-gene Links) analysis, a platform named “SEgene” which incorporates the peak-to-gene links approach—a statistical method designed to reveal correlations between genes and peak regions (https://github.com/hamamoto-lab/SEgene). This platform enables a targeted evaluation of super-enhancer regions in relation to gene expression, facilitating the identification of super-enhancers that are functionally linked to transcriptional activity. Here, we demonstrate the application of SE-to-gene Links analysis to public datasets, confirming its efficacy in accurately detecting super-enhancers and identifying functionally associated genes. Additionally, SE-to-gene Links analysis identified ERBB2 as a significant gene of interest in the lung adenocarcinoma dataset from the National Cancer Center Japan cohort, suggesting a potential impact across multiple patient samples. Thus, the SE-to-gene Links analysis provides an analytical tool for evaluating super-enhancers as potential therapeutic targets, supporting the identification of clinically significant super-enhancer regions and their functionally associated genes.
Similar content being viewed by others

Introduction
Enhancers are non-coding DNA elements that function as cis-regulatory sequences, facilitating gene transcription through interaction with transcriptional activators, thereby enhancing target gene expression1,2,3,4. While enhancers are generally categorized into active and poised/inactive states, active enhancers are frequently—but not exclusively—marked by H3K27ac histone modifications, which are associated with enhancer functionality5,6,7. A distinct subset of enhancers, termed super-enhancers (SEs), has been identified and is noted for exerting markedly stronger transcriptional activation than typical enhancers8,9,10. SEs are characterized by dense signal aggregation of transcription factors and H3K27ac in chromatin immunoprecipitation sequencing (ChIP-seq) data, often forming clusters within specific genomic loci. Studies have demonstrated that SEs are closely linked to cellular pluripotency and tissue specificity8,9,10,11,12, and are structurally supported by three-dimensional assemblies comprising DNA, mediators, transcription factors, and RNA polymerase II13.
Interest in SEs has grown in the oncology field, where aberrant SE formation or depletion has been implicated in tumorigenesis, metastasis, and therapeutic responses14,15. Thus, the accurate identification and analysis of SEs are considered promising strategies for drug discovery and the identification of therapeutic targets11. A commonly employed tool, rank ordering of super-enhancers (ROSE), identifies SEs based on ChIP-seq signal strength. This detection involves (1) defining enhancer regions based on significant ChIP-seq peak accumulation, (2) merging adjacent enhancers to form composite SEs, and (3) ranking SEs by signal intensity to distinguish those surpassing a threshold as SEs8,10. However, the ROSE-generated SE list merely represents enhancer clusters with enhanced marker signal intensity and does not guarantee that all identified SEs exhibit transcriptional superiority over typical enhancers12. Furthermore, the reliance on H3K27ac signal strength as an SE marker remains contentious, with studies both supporting and questioning its validity compared to other surrogate markers8,9,16. ROSE also frequently yields an extensive list of SE candidates, complicating detailed examination and prioritization based solely on signal intensity.
Critically, the SE identification approach by ROSE depends exclusively on enhancer-mark accumulation and does not incorporate RNA expression data for activation validation. A possible solution is to align SE regions with RNA expression data from the same sample to verify transcriptional activity. However, standardized methods for cross-validating ChIP-seq and RNA-seq data are currently lacking17,18, and the associations between enhancers and genes, as well as enhancers and promoters, remain subjects of ongoing debate19,20,21. Although ensemble approaches have been proposed to integrate multiple data sources22, such data may not always be available, requiring users to collect, analyze, and interpret information independently.
To overcome these challenges and provide an enhanced SE analysis framework, we introduce the “super-enhancer to gene” (SE-to-gene) Links, which integrates the peak-to-gene links methodology23, to examine the correlation of SEs with gene expression. SE-to-gene Links accepts RNA-seq and ChIP-seq data as inputs, and provides users with:
-
(1)
Comprehensive correlation analysis of enhancer groups within each SE.
-
(2)
Network-based evaluations among SEs to elucidate inter-SE relationships.
-
(3)
Gene-targeted SE analysis for specific user-defined genes of interest.
In contrast to existing SE databases that compile public SE data24,25,26,27,28,29, SE-to-gene Links is tailored to analyze SEs within user-specified sample groups. Additionally, SE-to-gene Links is not designed for SE prediction8,10,30,31,32,33,34; rather, it assesses ROSE-identified SEs by their correlation with gene expression within the user’s dataset. Significantly, SE-to-gene Links requires only ChIP-seq and RNA-seq inputs, eliminating the dependency on Hi-C or other spatial chromatin interaction data35,36. This feature ensures broad applicability across diverse research contexts, thereby enhancing its utility as a more precise tool for SE analysis.
Results
Design of the SE-to-gene Links analysis
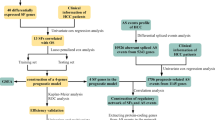
This platform is designed to facilitate the analysis of sample groups with ChIP-seq and RNA-seq data, such as patient cohorts for a particular disease. The workflow is structured as follows (Fig. 1):
-
1.
Input data integration: The platform accepts ChIP-seq and RNA-seq data as input for each sample in the dataset. Both data types undergo preprocessing to ensure quality and consistency, preparing them for cross-correlation analysis.
-
2.
Super-enhancer (SE) detection: Using the ROSE algorithm8,10, candidate SE regions are identified within each sample’s ChIP-seq data. This step provides a comprehensive list of SEs that are potential regulatory hotspots based on histone modifications or transcription factor binding sites.
-
3.
Correlation analysis: The platform applies a peak-to-gene linking method to assess correlations between SEs (identified from ChIP-seq peaks) and gene expression levels (from RNA-seq data) within a defined genomic range, typically ± 1 Mbp from transcription start sites23. Correlations are calculated and statistically evaluated to generate high-confidence peak-to-gene associations.
-
4.
Filtered SE prioritization: By applying statistical thresholds to the correlation results (e.g., false discovery rate and correlation coefficient), SE regions with significant gene associations are extracted from the broader SE list. This filtering process enables targeted selection of SEs that are more likely to contribute to gene regulatory networks.
-
5.
SE-to-gene Links network mapping: For advanced analyses, the platform can construct a network of SE-gene interactions, providing insights into SE clusters and their functional implications within the genomic landscape. This network visualization facilitates the identification of central SEs with potentially critical regulatory roles across multiple samples or patient cohorts (Fig. 2).

This pipeline integrates RNA-seq and ChIP-seq datasets to derive insights into super-enhancer (SE) networks. The process begins with the extraction of gene expression profiles from RNA-seq data and the generation of peak regions and BAM files from ChIP-seq data using the nf-core packages58. Subsequently, the peak-to-gene links (P2GL) analysis calculates correlation datasets between gene expression and ChIP-seq peak data. Simultaneously, the ChIP-seq peak and BAM data are processed using ROSE to identify super-enhancer regions, referred to as “ROSE SE.” The super-enhancer dataset is then refined by filtering it with the P2GL correlation data, resulting in a set of super-enhancers designated as “Filtered SE”. These filtered SEs, along with the P2GL correlation datasets, are utilized to construct an SE network graph, which provides deeper insights into SE-gene associations. Note that tools are denoted by circles. Aggregated individual datasets are depicted as overlapping squares, and cohesive data collections are represented by double-outlined squares.

a Identifying SE-associated peaks: Peaks within a predefined search window that show significant correlations with specific genes are identified using P2GL analysis. SEs containing these correlated peaks are then selected for further analysis. b Merging SE regions across samples: SE regions identified in multiple samples are consolidated to form a unified region, referred to as a “Merged SE”. This consensus SE region represents data integrated across all samples. c Building a multi-layer network: In the same chromosomal region, the genes identified through P2GL analysis, their associated peaks, and the corresponding merged SE are mapped as shown in the upper section. These components are represented as nodes in a three-layer network as illustrated in the bottom section.
Application of the SE-to-gene Links platform to colorectal cancer dataset
We utilized the public dataset GSE156614, comprising tumor tissue samples from colorectal cancer patients (n = 72)37. This analysis allowed us to examine correlations between candidate enhancer regions and genes located within ±1 M of transcription start sites. By applying a filter with a false discovery rate (FDR) threshold of 0.05 and r > 0.5, we identified significantly correlated peak-gene pairs, including an example of correlation between the peak region at chr4: 68,573,594–68,573,929 and the UGT2B17 gene (Supplementary Fig. 1, a full list is provided in Supplementary Data 1). Gene ontology (GO) analysis of the 1554 genes identified through this filtering, conducted with g:Profiler38, revealed multiple terms related to cellular development in GO:BP (Supplementary Table 1). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis further indicated significant enrichment in the Wnt and Hippo signaling pathways, both extensively linked to colorectal cancer (Supplementary Table 2)39,40,41,42,43,44,45,46.
Integrating peak-to-gene links data with ROSE output allows for a refined evaluation of SE regions. In tumor sample T01 from the GSE156614 dataset, 221 of the 1371 SEs identified by ROSE (16.1%) demonstrated significant overlap with peak-to-gene links data (Fig. 3a, b). Extending this SE-to-gene Links correlation analysis across all tumor samples facilitated the identification of overlapping SE regions. Merging these SE regions across samples produced a distribution graph, highlighting SE presence across genomic loci (Fig. 3c, a full list is provided in Supplementary Data 2), with chr7:748,439–998,341 standing out as a region with substantial SE concentration (Fig. 3d). Subsequent integrative genomics viewer (IGV) analysis of this locus identified several genes with ADAP1 and CYP2W1 showing confirmed peak-to-gene correlations (Fig. 3e). Notably, CYP2W1 has documented links to colorectal cancer47, while ADAP1 have reported associations with oncogenic processes48,49. To further validate the biological relevance of this locus, we performed an independent annotation using SEdb 2.0 and eRNAbase28,50, both of which provided comprehensive catalogs of SE regions annotated with tissue- and cell-type metadata. Notably, the SE region at chr7:748,439–998,341, along with several other merged SE regions, was recurrently annotated in the samples derived from colon or colorectal tissues, which is consistent with the origin of the GSE156614 dataset (Supplementary Table 3). These findings underscore the tissue-specific enrichment of the identified SE regions and further support the robustness and biological relevance of the merged SE identification strategy.

a SE ranking plot: Genomic regions classified as super-enhancers are ranked based on the H3K27ac ChIP-seq signal, highlighted with red markers indicating SEs with significant peak-to-gene correlations. b Pie chart showing the proportion of SEs significantly correlated with P2GL data. c Bar plot displaying ranked merged SEs identified in tumor samples. The merged SE region of interest (chr7:748,439–998,341) is highlighted with a red bar. d Stacked read plot for the chr7:748,439–998,341 region. e IGV visualization of the merged SE region (chr7:748,439–998,341). The regions of the ADAP1 and CYP2W1 genes are highlighted with red boxes.
In the most enriched region, chr1:1,109,435–1,174,178, our analysis revealed a super-enhancer region enriched with genes such as LOC148413, ATAD3A, NOC2L, and TMEM52. Among these, ATAD3A encodes a mitochondrial membrane protein implicated in maintaining mitochondrial function and structure51, while NOC2L is involved in transcriptional repression and nucleolar organization52. However, the functions of LOC148413 and TMEM52 are largely unknown. The identification of these genes indicates that SEgene may uncover previously unrecognized genomic regions associated with colorectal cancer. Finally, we assessed the robustness of the merged SE identification by performing ROSE analyses using a range of stitching distances (5, 10, 12.5 kb [default], 15, 20, 25, and 30 kb; Supplementary Table 4). Several high-ranking merged SE regions, such as chr1:1,109,435–1,174,178 and chr7:748,439–998,341, consistently appeared across the different parameters, though with slight boundary variations. These findings suggest that the SEgene platform provides stable and reproducible merged SE detection, with limited sensitivity to parameter variations.
Representation of network hubs in the SE-to-gene Links analysis
Understanding the broad influence of SEs on genomic regulation is essential, as SEs play a pivotal role in orchestrating the expression of genes that define cell identity and function. To investigate the extensive regulatory impact of SE regions on their target genes, we first identified SE regions for each cohort. When an SE is classified as “common” because it overlaps in at least two samples, it captures shared regulatory elements that may exert influence over large genomic domains across the entire cohort. This approach highlights the extensive reach of SEs, which can coordinate the expression of multiple genes across wide genomic regions, providing critical insights into their role as master regulators of gene expression and their potential contributions to genome-wide regulatory networks. Subsequently, we constructed a three-layer network hub connecting common SE regions, overlapping enhancer peaks, and target genes identified through SE-to-gene Links analysis in the GSE156614 dataset. The resulting subgraphs, categorized by node count, revealed a network structure characterized by several large, densely connected subgraphs alongside numerous smaller subgraphs comprising only a few nodes (full visualizations are available in Supplementary Data 3). Notably, the merged SE network for the chr7:748,439–998,341 genomic region confirmed the network architecture involving ADAP1 and CYP2W1 gene expression and their enhancer peaks associated with gene regulation (Fig. 4). This network hub visualization highlights the potential regulatory role of super-enhancer regions in modulating gene expression within the cohort.

The merged SE network was constructed using enhancer peaks identified within the chr7:748,439–998,341 genomic region, as highlighted in Fig. 3c. This visualization demonstrates the connections between the merged SE, enhancer peaks associated with gene regulation, and target genes, providing insights regarding the regulatory architecture underlying gene expression.
Application of the SE-to-gene Links platform to glioblastoma dataset
Building on the previous analysis, we further investigated the publicly available dataset GSE145646, which comprises tumor tissue samples from patients with glioblastoma (GBM), including H3K27ac ChIP-seq data (n = 34) and RNA-seq data (n = 32)53. Utilizing P2GL analysis, we explored the correlation between candidate enhancer regions and genes located within ±1 Mbp of transcription start sites (FDR > 0.1 and r > 0.5). GO analysis of the 345 genes identified through this filtering revealed multiple terms related to neural processes, including “axon guidance,” “neural crest cell migration,” “visceral motor neuron differentiation,” and “regulation of neuron apoptotic process” in the GO:BP (Supplementary Table 5). Subsequently, SE regions were evaluated by integrating the P2GL link data with the ROSE output. In the tumor sample GBM01 from the GSE145646 dataset, ROSE identified 1,267 SEs, of which eight (0.8%) significantly overlapped with the P2GL data (Supplementary Fig. 2a, b). By extending this SE and gene correlation analysis across all samples, we merged the data and generated a distribution graph (Supplementary Fig. 2c). Notably, chr12:57,818,379-57,907,246 emerged as a prominent SE-enriched domain (Supplementary Fig. 2d), included GLI1, METTL1, XRCC6BP1, DTX3, TSFM, DCTN2, and MARCH9. Notably, GLI1 is a member of the GLI family of zinc finger proteins and serves as a nuclear mediator of the Sonic Hedgehog signaling pathway, which regulates various aspects of early central nervous system development54. Aberrant activation of GLI1 has been implicated in tumorigenesis, cancer growth, and self-renewal of cancer stem cells. Similarly, METTL1 encodes a methyltransferase involved in RNA modification, and its high expression has been significantly associated with poor prognosis in patients with glioma, suggesting that it may serve as an independent risk factor55. These findings underscore the significance of the SE region identified on chromosome 12 and its potential regulatory influence on genes associated with GBM pathogenesis.
Application of the SE-to-gene Links analysis to lung adenocarcinoma without clinically actionable genetic alterations
As demonstrated in the analysis of the GSE156614 dataset, our platform successfully generated interpretable results. Building on this, we subsequently applied the platform to lung adenocarcinoma (LUAD) cases from the National Cancer Center Japan cohort56. This dataset focuses on LUAD cases lacking clinically actionable genomic alterations (non-CAGA) but exhibiting aberrant gene expression driven by structural abnormalities around super-enhancer regions56. We evaluated the overlap of SE regions extracted from the dataset and assessed the distribution of SE-associated genes (n = 118). Following the SE-to-gene Links analysis, SEs identified in individual samples were filtered and merged to evaluate the overall distribution of SE regions across the cohort (Fig. 5a, a full list is provided in Supplementary Data 4). Notably, the chr17:39,508,684–39,870,304 region exhibited the highest density of overlapping SEs, forming clusters indicative of super-enhancer activity (Fig. 5b). This region contains ERBB2, a gene previously implicated as a driver in non-CAGA LUAD cases (Table 1)56. Our analysis identified a strong correlation between H3K27ac CPM values within the merged SE region and the expression of ERBB2 and its neighboring genes (Fig. 6)56. In contrast, when SEs were ranked solely based on the ROSE algorithm across our LUAD cohort, the rankings of the SEs associated with ERBB2 were distributed across a broad range from low to high positions (Supplementary Fig. 3). This result corroborates prior findings that ERBB2-associated super-enhancer formation is prevalent in non-CAGA LUAD cases and highlights its potential role in driving aberrant gene expression across the cohort. The results underline the platform’s utility in identifying super-enhancer-gene associations at both individual56 and cohort-wide levels (this study). In particular, our platform identified ERBB2-associated SEs, supporting the hypothesis that super-enhancer-driven ERBB2 activation may be a shared mechanism across the cohort. While its viability as a drug target requires further validation, this finding demonstrates the platform’s capability to prioritize SEs with potential therapeutic relevance.

a Graphical representation of ranked merged SEs, highlighting the clustering of SEs across multiple samples. Several SE regions along the rank axis (X-axis) exhibit substantial overlap, reflected as elevated SE counts (Y-axis), indicating genomic loci of potential significance. b Stacked read plot providing a detailed visualization of the merged SE region at chr17:39,508,684–39,870,304, illustrating the dense aggregation of SEs within this genomic locus across multiple samples.

Genes identified by P2GL as having significant Pearson correlation coefficients with SE peaks are displayed. The X-axis represents log10-transformed TPM values, while the Y-axis represents log10-transformed CPM values. The figure shows gene-specific Pearson correlation coefficients for SEs within the merged SE region: (a) MIEN1, (b) STARD3, (c) GRB7, (d) MIR4728, (e) AC087491.1, and (f) PGAP3. This visualization underscores the strength of the associations between the merged SE region and the expression levels of each gene.
Discussion
The ROSE algorithm is widely used for SE identification. However, it has certain limitations. Primarily, ROSE relies on H3K27ac ChIP-seq data to define enhancers, which may not capture the full spectrum of enhancer activity, because some enhancers may not exhibit strong H3K27ac signals. Additionally, the process of stitching adjacent enhancers within a predefined distance can be arbitrary, potentially leading to inconsistencies in SE identification57. To address these limitations, we developed and validated the SE-to-gene Links analysis, a novel platform designed to integrate P2GL analysis with SE regions identified through ROSE. This approach facilitates the functional association of SEs with gene expression, enabling the prioritization of biologically relevant SEs from the large number of candidates identified by ROSE. By combining these methods, the platform provides a robust framework for understanding the regulatory role of SEs in gene expression. To demonstrate the utility of this approach, we first applied SE-to-gene Links analysis to the publicly available GSE156614 dataset, which includes colorectal cancer tissue samples37. This analysis successfully identified several SEs linked to genes with known roles in cancer, validating the platform’s capability to uncover biologically meaningful SE-gene associations (Fig. 3e). The identification of these cancer-related genes highlights the platform’s potential to uncover regulatory elements critical for tumor biology. To further evaluate its applicability, we tested the platform on LUAD samples from the National Cancer Center Japan cohort. The analysis captured enhancer-gene associations within the ERBB2 genomic region, consistent with previous studies demonstrating the role of the ERBB2 gene in cancer56. Notably, these associations were observed across multiple patients, reinforcing the platform’s ability to identify shared SE trends within patient cohorts (Fig. 5). This result emphasizes the platform’s sensitivity to clinically relevant enhancer-gene interactions, even in complex cancer contexts such as LUAD.
Although the SE-to-gene Links analysis offers significant advantages, it has several limitations. First, the reliance of the platform on enhancer-gene associations inferred through P2GL, which primarily considers proximal genomic regions, may not fully capture the complexity of three-dimensional chromatin interactions. SEs often regulate genes over long genomic distances through looping interactions. Without any direct chromatin conformation data, such as Hi-C or ChIA-PET, this analysis may miss critical long-range regulatory links. Integrating these data types could provide a more comprehensive understanding of SE-mediated gene regulation. For instance, Hi-C data can reveal topologically associating domains (TADs), which are regions of the genome that interact more frequently with themselves than with other regions, thereby influencing gene regulation. Incorporating Hi-C data into SEgene could help identify such domains and their interactions with SEs, leading to a more accurate understanding of gene regulation mechanisms. Second, the platform’s current dependency on ROSE for SE identification may introduce variability, as SE definitions can differ based on the specific algorithm or parameters used. Standardization of SE definitions across studies will be necessary to ensure the reproducibility and comparability of results11. Third, the analysis does not currently account for epigenetic or transcriptional dynamics over time. SE activity can be highly context-dependent and influenced by external stimuli or temporal changes, which are not captured in static datasets. Expanding the platform to include temporal or stimulus-dependent data could reveal dynamic regulatory relationships and enhance its utility in studying transient or context-specific SE functions.
Despite these limitations, SE-to-gene Links analysis provides a practical means for researchers to systematically prioritize SE regions for further functional studies. By leveraging enhancer-gene correlations identified within a cohort, researchers can narrow their focus to SEs that are not only active but also potentially critical in driving disease-specific gene expression. This ability is particularly valuable in clinical oncology, where understanding the regulatory differences between tumor and normal samples is key to identifying cancer-driving mechanisms56. A significant strength of this approach lies in its capacity to pinpoint SE regions associated with oncogenic transcription factor interactions. These interactions often represent key regulatory nodes in cancer biology, offering insights into the transcriptional programs driving tumorigenesis. By facilitating the identification of SEs with strong enhancer-gene links, the platform can prioritize SEs that are not only active in oncogenesis but may also serve as actionable therapeutic targets. In addition to its applicability in individual cohorts, this platform can also be used to analyze large public datasets systematically. For example, researchers could apply SE-to-gene Links analysis to select SEs associated with specific pathways or phenotypes across multiple datasets, enabling broader insights into SE function across diverse conditions. Such systematic analyses may lead to the discovery of conserved SEs that act as master regulators across cancers or other diseases, providing valuable targets for drug development.
Future studies could expand the utility of this platform by integrating additional data layers, such as chromatin accessibility, histone modification profiles, or proteomics. This integration would further enhance its ability to identify SEs with critical roles in gene regulation and disease. Moreover, extending this approach to non-coding RNA targets or long-range chromatin interactions could provide an even deeper understanding of the regulatory landscape shaped by SEs. SE-to-gene Links analysis is a versatile and powerful tool for unraveling the complex regulatory networks mediated by SEs. While limitations remain, its ability to prioritize functionally significant SEs makes it a valuable resource in both basic and translational research, particularly in the context of diseases such as cancer. As the field of regulatory genomics continues to evolve, this platform has the potential to play a central role in advancing our understanding of SE-mediated gene regulation and its implications for precision medicine.
Methods
Preprocessing of the ChIP-seq and RNA-seq datasets
For each ChIP-seq and RNA-seq sample, alignment and quantification were performed, resulting in peak data (CPM: counts per million) for ChIP-seq and expression data (TPM: transcripts per million) for RNA-seq. The nf-core pipelines (nf-core/chipseq and nf-core/rnaseq) were used for these processes under standard conditions58. Briefly, raw ChIP-seq fastq files were aligned using BWA59, with peaks identified through MACS260. For RNA-seq, TPM values were derived using Salmon61.
Acquisition of super-enhancer regions from individual ChIP-seq data
Super-enhancer (SE) lists are generated using the ROSE algorithm on processed ChIP-seq data, specifically utilizing bam and peak bed files8,10. The ROSE process designates input peak regions as enhancers, merges adjacent peaks within a specified distance, ranks the resultant enhancer clusters by signal intensity, and selects those surpassing a predefined threshold as SEs. Initially, ChIP-seq bam and peak bed files are converted to gff format using the awk command62. Following this conversion, SEs are identified with ROSE using the following command:
python ROSE_main.py -g HG38 -i PEAK_GFF_FILE -r $INPUT_BAM -c $IP_BAM -o $OUTPUT_DIR -s 12500 -t 2500.
In this command, “-g HG38”, “-I”, “-r”, “-c”, “-o”, “-s” and “-t” designate the genome, input peak file in gff format, input bam file, control bam file, output directory, stitching, and threshold parameters, respectively. This approach allows for the systematic identification of SE regions in the dataset.
Peak-to-gene links analysis for the ChIP-seq and RNA-seq dataset
The peak-to-gene links (P2GL) approach, initially described by Corces et al.23, evaluates Pearson correlation coefficients between ATAC-seq peaks and gene expression. Here, it is adapted to assess correlations between ChIP-seq peaks and gene expression. The platform calculates Pearson correlation coefficients between expression values (log2(TPM + 1)) and peak values (log2(CPM)) for peaks within a user-defined search window around the transcriptional regulatory region. Given that enhancer-gene interactions typically fall within ±1 Mbp63,64,65, this distance is set as the default, though other values, such as 0.5 Mbp, are also implemented. Correlation significance is determined using randomly selected peaks from other chromosomes, followed by FDR adjustments, to extract significant gene-peak correlations. To obtain quantitative peak counts across samples, we utilize the SCREEN-cCRE dataset66 as a reference for enhancer locations. The specific steps are as follows:
-
1.
Preparation of ChIP-seq CPM table: Using human cCRE-peak data from GRCh38 as enhancer reference regions, we count ChIP-seq bam files using featureCounts (v2.0.0)67 to create a table of CPM values for each peak.
-
2.
Preparation of RNA-seq TPM table: RNA-seq TPM data per gene is compiled into a table. Using BioMart68, we append gene symbols with genomic positions and transcription orientations.
-
3.
Exclusion of lower 25% variance data: The bottom 25% by peak variance in both ChIP-seq and RNA-seq tables is excluded from the analysis.
-
4.
Identification of candidate gene-peak sets: Gene-peak pairs within the specified distance (default: ± 1 Mbp) are designated as candidates.
-
5.
Calculation of correlation coefficients: For all candidate pairs, Pearson correlation coefficients (r) are calculated, with ChIP-seq CPM on the x-axis and RNA-seq TPM on the y-axis.
-
6.
Generation of random null distribution: For each gene, 10,000 peaks from other chromosomes are randomly sampled to compute correlation coefficients, establishing a null distribution and allowing for the calculation of mean and standard deviation.
-
7.
P-value and FDR correction: Using these statistics, p-values are derived for each gene-peak pair, and FDR correction is applied via the Benjamini-Hochberg method69 to identify significant gene-peak correlations.
Evaluation of super-enhancer regions using P2GL data
The ROSE algorithm merges adjacent enhancer peaks within a specified distance (default: 12.5 kb), ranks these merged regions by signal intensity, and designates SEs based on an inflection point threshold8,10. We evaluated SE regions from ROSE by intersecting them with P2GL-identified enhancer regions (FDR < 0.05, r > 0.5) to identify SEs that show positive correlations with gene expression. Specifically, SE regions from ROSE and P2GL-identified peaks with gene expression correlations are cross-referenced using bedtools70 through pybedtools71 in Python, thereby extracting SEs functionally associated with transcription.
Visualization of network hubs in SE-to-gene Links analysis
To explore the interactions between multiple SE regions and their associated target genes, we classified SE regions as “common SEs” if they were shared across at least two samples within a cohort. Leveraging the SE-gene analysis framework, we devised a network-based approach to represent these associations, integrating the concept of constructing networks informed by gene correlation data72,73. For network analysis and visualization, we employed the Python library NetworkX, ensuring a systematic and scalable approach to graph generation74. A search range of ± 1 Mbp, FDR threshold of <0.05, and a Pearson correlation coefficient (r) cutoff of >0.4 were applied to define robust SE-gene correlations. Furthermore, only SEs duplicated in more than 10 samples within the region defined as common SE were adopted as common SE nodes in this analysis. These parameters accounted for the complexity of multi-SE-to-gene relationships inherent to this model. In the constructed network, each common SE served as a root node connected to correlated enhancer peaks as intermediate nodes, which were further linked to their associated target genes. The network structure inherently reflects the multi-enhancer-to-gene relationships modeled by the P2GL analysis, resulting in some genes being associated with multiple SE nodes. This design facilitates a comprehensive and detailed representation of SE-gene interactions, offering valuable insights into the regulatory architecture within the cohort.
Data availability
All data analyzed in this study are included in the manuscript and its supplementary materials. This study did not generate any data types requiring mandatory deposition in public databases. The processed dataset used for SEgene analysis is publicly available via Figshare at the following https://doi.org/10.6084/m9.figshare.28171127. This dataset includes: (1) GSE156614_tumor_RNA.csv and GSE156614_tumor_ChIP.tsv, containing transcriptomic and epigenomic data used for P2GL analysis; (2) GSE156614_tumor_2000000.tsv, presenting integrated P2GL results; and (3) GSE156614_SE_table.txt, summarizing ROSE analysis of super-enhancer regions.
Code availability
The code for reproducing the SE-to-gene Links analysis is available in the GitHub repository (https://github.com/hamamoto-lab/SEgene). A comprehensive Jupyter notebook is included in the repository in order to provide users with step-by-step instructions for data preprocessing, analysis, and visualization.
References
Banerji, J., Rusconi, S. & Schaffner, W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308 (1981).
Pennacchio, L. A., Bickmore, W., Dean, A., Nobrega, M. A. & Bejerano, G. Enhancers: five essential questions. Nat. Rev. Genet. 14, 288–295 (2013).
Panigrahi, A. & O’Malley, B. W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 22, 108 (2021).
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020).
Creyghton, M. P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936 (2010).
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011).
Spitz, F. & Furlong, E. E. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626 (2012).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Hamamoto, R. et al. Analysis of super-enhancer using machine learning and its application to medical biology. Brief. Bioinform. 24, bbad107 (2023).
Pott, S. & Lieb, J. D. What are super-enhancers?. Nat. Genet. 47, 8–12 (2015).
Zhang, J. et al. Super enhancers-Functional cores under the 3D genome. Cell Prolif. 54, e12970 (2021).
Sengupta, S. & George, R. E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 3, 269–281 (2017).
Thandapani, P. Super-enhancers in cancer. Pharm. Ther. 199, 129–138 (2019).
Zhang, T. T., Zhang, Z. Q., Dong, Q., Xiong, J. & Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 21, 45 (2020).
Höllbacher, B., Balázs, K., Heinig, M. & Uhlenhaut, N. H. Seq-ing answers: Current data integration approaches to uncover mechanisms of transcriptional regulation. Comput. Struct. Biotechnol. J. 18, 1330–1341 (2020).
Moore, J. E., Pratt, H. E., Purcaro, M. J. & Weng, Z. A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome Biol. 21, 17 (2020).
Hariprakash, J. M. & Ferrari, F. Computational biology solutions to identify enhancers-target gene pairs. Comput. Struct. Biotechnol. J. 17, 821–831 (2019).
Tao, H. et al. Computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles. Brief. Bioinform. 22, bbaa405 (2021).
Xu, H., Zhang, S., Yi, X., Plewczynski, D. & Li, M. J. Exploring 3D chromatin contacts in gene regulation: The evolution of approaches for the identification of functional enhancer-promoter interaction. Comput. Struct. Biotechnol. J. 18, 558–570 (2020).
Gao, T. & Qian, J. EAGLE: An algorithm that utilizes a small number of genomic features to predict tissue/cell type-specific enhancer-gene interactions. PLoS Comput. Biol. 15, e1007436 (2019).
Corces, M. R. et al. The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018).
Chen, C. et al. SEA version 3.0: a comprehensive extension and update of the Super-Enhancer archive. Nucleic Acids Res. 48, D198–D203 (2020).
Khan, A. & Zhang, X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res 44, D164–D171 (2016).
Qian, F. C. et al. SEanalysis: a web tool for super-enhancer associated regulatory analysis. Nucleic Acids Res. 47, W248–W255 (2019).
Qian, F. C. et al. SEanalysis 2.0: a comprehensive super-enhancer regulatory network analysis tool for human and mouse. Nucleic Acids Res. 51, W520–W527 (2023).
Wang, Y. et al. SEdb 2.0: a comprehensive super-enhancer database of human and mouse. Nucleic Acids Res. 51, D280–D290 (2023).
Wei, Y. et al. SEA: a super-enhancer archive. Nucleic Acids Res. 44, D172–D179 (2016).
Bu, H., Hao, J., Gan, Y., Zhou, S. & Guan, J. DEEPSEN: a convolutional neural network based method for super-enhancer prediction. BMC Bioinform. 20, 598 (2019).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ji, Q. Y., Gong, X. J., Li, H. M. & Du, P. F. DeepSE: detecting super-enhancers among typical enhancers using only sequence feature embeddings. Genomics 113, 4052–4060 (2021).
Khan, A. & Zhang, X. Integrative modeling reveals key chromatin and sequence signatures predicting super-enhancers. Sci. Rep. 9, 2877 (2019).
Tian, S. Z. et al. MCIBox: a toolkit for single-molecule multi-way chromatin interaction visualization and micro-domains identification. Brief. Bioinform. 23, bbac380 (2022).
Gong, Y. et al. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat. Commun. 9, 542 (2018).
Perez-Rathke, A. et al. CHROMATIX: computing the functional landscape of many-body chromatin interactions in transcriptionally active loci from deconvolved single cells. Genome Biol. 21, 13 (2020).
Li, Q. L. et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat. Commun. 12, 6407 (2021).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 (2006).
Fearon, E. R. Molecular genetics of colorectal cancer. Annu Rev. Pathol. 6, 479–507 (2011).
Harada, N. et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 (1999).
Harvey, K. F., Zhang, X. & Thomas, D. M. The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257 (2013).
Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 (2004).
Morin, P. J. beta-catenin signaling and cancer. Bioessays 21, 1021–1030 (1999).
Pan, D. J. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 19, 491–505 (2010).
Yu, F. X., Zhao, B. & Guan, K. L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828 (2015).
Chung, F. F., Mai, C. W., Ng, P. Y. & Leong, C. O. Cytochrome P450 2W1 (CYP2W1) in colorectal cancers. Curr. Cancer Drug Targets 16, 71–78 (2016).
Oshimori, N. Cancer stem cells and their niche in the progression of squamous cell carcinoma. Cancer Sci. 111, 3985–3992 (2020).
Van Duzer, A., Taniguchi, S., Elhance, A., Tsujikawa, T. & Oshimori, N. ADAP1 promotes invasive squamous cell carcinoma progression and predicts patient survival. Life Sci. Alliance 2, e201900582 (2019).
Song, C. et al. eRNAbase: a comprehensive database for decoding the regulatory eRNAs in human and mouse. Nucleic Acids Res. 52, D81–D91 (2024).
Chen, L., Li, Y., Zambidis, A. & Papadopoulos, V. ATAD3A: a key regulator of mitochondria-associated diseases. Int. J. Mol. Sci. 24, 12511 (2023).
Lu, S., Chen, Z., Liu, Z. & Liu, Z. Unmasking the biological function and regulatory mechanism of NOC2L: a novel inhibitor of histone acetyltransferase. J. Transl. Med. 21, 31 (2023).
Xu, L. et al. Topography of transcriptionally active chromatin in glioblastoma. Sci. Adv. 7, eabd4676 (2021).
Zhu, H. & Lo, H. W. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) family of transcription factors in gene regulation and diseases. Curr. Genom.11, 238–245 (2010).
Li, L. et al. Prognostic role of METTL1 in glioma. Cancer Cell Int. 21, 633 (2021).
Kaneko, S. et al. Mechanism of ERBB2 gene overexpression by the formation of super-enhancer with genomic structural abnormalities in lung adenocarcinoma without clinically actionable genetic alterations. Mol. Cancer 23, 126 (2024).
Zhou, R. W. & Parsons, R. E. Etiology of super-enhancer reprogramming and activation in cancer. Epigenet. Chromatin 16, 29 (2023).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
ROSE Bitbucket issue page, <https://bitbucket.org/young_computation/rose/issues/26/unexpected-running-result-using-rose#comment-27835299> (2016).
Cao, Q. et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 49, 1428–1436 (2017).
He, B., Chen, C., Teng, L. & Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl, Acad. Sci. USA 111, E2191–E2199 (2014).
Maston, G. A., Evans, S. K. & Green, M. R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet 7, 29–59 (2006).
Consortium, E. P. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Smedley, D. et al. BioMart–biological queries made easy. BMC Genom.10, 22 (2009).
Benjamini, Y. & Hochberg, Y. controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B-Stat. Methodol. 57, 289–300 (1995).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Dale, R. K., Pedersen, B. S. & Quinlan, A. R. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27, 3423–3424 (2011).
Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003).
Lee, H. K., Hsu, A. K., Sajdak, J., Qin, J. & Pavlidis, P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 14, 1085–1094 (2004).
Hagberg, A. A., Schult, D. A. & Swart, P. J. In Proc. 7th Python in Science Conference (SciPy 2008) (ed T. Vaught G. Varoquaux, J. Millman (Eds.)) 11-15 (Pasadena, 2008).
Acknowledgements
We thank all members of the Hamamoto laboratory for their kind assistance. We also thank Dr. Tazro Ohta (Institute for Advanced Academic Research, Chiba University) for valuable technical advice, and appreciate the DBCLS BioHackathon BH23.9 (2023) and BH24.6 (2024) for providing a platform for development and technical exchange. This work was supported by the Cabinet Office BRIDGE (programs for bridging the gap between R&D and the ideal society (Society 5.0) and generating economic and social value), the AMED Innovative Cancer Medical Practice Research Project (Grant Number JP22ck0106643), JST CREST (Grant Number JPMJCR1689), JSPS Grant-in-Aid for Scientific Research on Innovative Areas (Grant Number JP18H04908), JST AIP-PRISM (Grant Number JPMJCR18Y4), and MEXT subsidy for Advanced Integrated Intelligence Platform to R.H. and the JSPS Grant-in-Aid for Scientific Research (Grant Numbers JP21H03550, JP24K03039), the Takeda Science Foundation and the Research Grant of the Princess Takamatsu Cancer Research Fund (19-25108) to S.K.
Author information
Authors and Affiliations
Contributions
N.S.: Conceptualization, data curation, methodology, writing—original draft preparation, review, and editing. H.M.: Formal analysis, investigation. K.A.: Visualization, data interpretation. K.T.: Visualization, data interpretation. S.T.: Data interpretation, project administration. N.K.: Writing—review and editing. M.K.: Writing—review and editing. R.H.: Conceptualization, supervision, project administration. S.K.: Conceptualization, supervision, project administration, writing—original draft preparation, review, and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All methods were performed following the Ethical Guidelines for Medical and Health Research Involving Human Subjects. The study was approved by the institutional review board of the National Cancer Center Japan (2005-109, 2016-496, 2019-018). In addition, this study was conducted under the Declaration of Helsinki. All patients provided written informed consent. During the implementation, refinement, and visualization stages of the analysis pipeline, large language models such as ChatGPT (https://chat.openai.com/chat) and Claude (https://claude.ai) were utilized to support Python coding and figure generation. All authors have read and approved the final version of the manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shinkai, N., Asada, K., Machino, H. et al. SEgene identifies links between super enhancers and gene expression across cell types. npj Syst Biol Appl 11, 49 (2025). https://doi.org/10.1038/s41540-025-00533-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-025-00533-x

{kind=link}