
Abstract
Engineering jawbone tissues from pluripotent stem cells presents a challenge owing to the lack of protocols for selectively inducing the jawbone progenitor, the first pharyngeal arch (PA1) ectomesenchyme, and for recapitulating three-dimensional osteocyte networks. Here we present a method for generating jawbone-like organoids from human induced pluripotent stem cells through PA1 ectomesenchyme of the mandibular prominence (mdEM). A three-dimensional culture system enables sequential differentiation of induced pluripotent stem cells into neural crest cells and mdEM. The mdEM exhibits proximal–distal patterning from the centre outwards, mirroring mandibular development. The introduction of exogenous pharyngeal epithelial signals induces mandibular prominence-specific regional patterning in the mdEM. When cultured under osteogenic conditions, the mdEM forms jawbone-like organoids comprising osteoblasts and network-forming osteocytes embedded in self-produced mineralized bone matrices. Moreover, these organoids promote bone regeneration when transplanted into jaws containing bone defects and recapitulate phenotypes of osteogenesis imperfecta, a genetic disorder characterized by fragile bones, using patient-derived induced pluripotent stem cells. Our protocols establish a foundation for investigating human jaw embryology and pathophysiology.
Similar content being viewed by others

Main
The maxillary and mandibular bones, collectively referred to as jawbones, constitute the largest biomineralized organs in the oral region. These structures play a central role in mastication alongside teeth and also are crucial for maintaining respiration, pronunciation and facial aesthetics. Jawbones are highly vulnerable to irreversible damage caused by various pathological factors, including oral bacterial infections, excessive mastication, traumas, tumours and congenital malformations1. Such injuries significantly impact the quality of life. Despite this, no studies have yet described faithful human jawbone models that could serve as a foundation for studying jawbone pathophysiology and developing therapeutic interventions.
Recapitulating the sequence of developmental milestones using pluripotent stem cells (PSCs) presents a promising approach for generating embryonic cell populations of interest2,3. While the axial and appendicular skeletons originate from mesoderm, jawbones derive from cranial neural crest cell (NCC)-derived ectomesenchyme colonized in the first pharyngeal arch (PA1)4. During vertebrate embryogenesis, NCCs dynamically migrate throughout the cranial regions, differentiating into various cell types, including ectomesenchyme. Their regional specifications are constrained by the anterior–posterior (A–P) axial identity of NCCs, established by Hox genes5,6,7. Unlike other posterior arches expressing specific combinations of Hox genes, midbrain–anterior hindbrain (rhombomeres 1 and 2) NCCs giving rise to PA1 ectomesenchyme do not express Hox genes. In the intra-arch stage, PA1 divides into maxillary and mandibular prominences, with PA1 ectomesenchyme of the mandibular prominence (mdEM) exhibiting specific regional patterning along proximal–distal and oral–aboral axes8,9,10,11. NCC migration and mdEM patterning are guided by pharyngeal arch epithelial signals. Ultimately, mdEM gives rise to various mandibular structures, including the jawbone. While successful induction of NCCs and their derivatives from human and mouse PSCs has been reported12,13,14, directed differentiation of PSC-derived NCCs into mdEM with accurate developmental positional information remains elusive, posing a challenge in establishing jawbone models.
During mandibular bone formation, post-patterning mdEM first forms Meckel’s cartilage15, a guiding structure for developing the mandibular bone, and ossification starts sequentially. Unlike mesoderm-derived bones, which primarily form through endochondral ossification, mandibular bone undergoes different special ossification patterns. While the distal and proximal portions of the mandible are endochondrally ossified, the mandibular bone predominantly undergoes an intramembranous process15, wherein mdEM condenses, directly differentiates into osteoblasts16, secretes type-I collagen-rich extracellular matrices and becomes partially embedded within them. Eventually, these cells differentiate into osteocytes, forming three-dimensional (3D) cellular networks within mineralized bone matrices. Osteocytes, the most abundant cells in bone, play crucial roles in bone homeostasis, regulating the functions of other bone cells, calcium and phosphate metabolism, and responding to mechanical stress17,18,19. Detailed functional analyses of osteocytes remain challenging owing to their location within mineralized matrices. Few studies have achieved partial recapitulation of 3D osteocyte networks using human PSCs in vitro. While a protocol for generating bone-like nodules containing osteocytes from human induced pluripotent stem cells (iPSCs) has been reported20, its reliance on two-dimensional (2D) culture makes histological analysis of osteocyte biological functions difficult. Conversely, the 3D culture of human iPSCs has been shown to facilitate the generation of various functional organoids in vitro21,22. Thus, we focused on organoid systems to overcome the limitations of 2D culture and enable precise morphological assessment of jawbone tissues.
This study introduces a 3D culture system for directed differentiation of human iPSCs into mdEM and subsequent generation of jawbone-like organoids. Considering future clinical applications, we used xeno-free conditions for induction. Furthermore, the organoids exhibited bone regenerative capacity and recapitulated phenotypes of osteogenesis imperfecta (OI), a genetic disorder characterized by fragile bones, using patient-derived iPSCs, demonstrating the versatility of the organoids.
Results
3D induction of HOX-negative NCCs from iPSCs
To establish a stepwise induction protocol for generating jawbone organoids from iPSCs, our initial focus was on developing a 3D culture system conducive to the targeted induction of HOX-negative NCCs (Fig. 1a). Previous research from our team has demonstrated the efficient differentiation of human iPSCs into HOX-negative NCCs under xeno-free 2D culture conditions using a combination of TGF-β inhibitor, SB431542 (SB), and GSK3b inhibitor, CHIR99021 (CHIR)12. Thus, we sought to adapt these induction protocols to 3D-cultured iPSCs. Initially, dissociated human 1231A3 iPSCs (a feeder-free and xeno-free iPSC line) were aggregated on 96-well V-bottom ultra-low-cell-adhesion plates in medium containing the ROCK inhibitor, Y-27632 (Fig. 1b,c), and cultured with SB/CHIR under shaking conditions (to enhance nutrient absorption) for 4 days (Extended Data Fig. 1a). CD271 (also known as p75NTR, encoded by NGFR) is widely utilized to evaluate the induction efficiency of human PSC-derived NCC23,24. We observed the induction of CD271high+ NCCs within these aggregates, alongside undesired PAX6+ neuroectoderm (Extended Data Fig. 1b). To mitigate neuroectoderm derivation, we subjected the aggregates to BMP4 (ref. 25) treatment before SB/CHIR culture (Extended Data Fig. 1c,e). Although excessive BMP4 signalling induced E-cadherin (ECAD)+ cells on the aggregate surface (Extended Data Fig. 1d,f), treatment of 10 ng ml−1 BMP4 for 1 day effectively induced SOX10+CD271+TFAP2A+ NCCs (Fig. 1d and Extended Data Fig. 1d,f,g). Flow cytometry analysis confirmed the enrichment of CD271high+ NCCs (94.9 ± 1.9%) (Fig. 1e), with TFAP2A+ECAD+ non-neural ectoderm, PAX6+ neural ectoderm and Oct3/4+ (encoded by POU5F1) undifferentiated iPSCs observed at minimal levels (Fig. 1f). The high efficiency of NCC induction was consistent across different iPSC clones, including Ff-I 14s04, HLA-KO Ff-I 14s04, Ff-I 01s04, OI patient-derived iPSCs (OI-iPSCs; clone OI08-12)20 and mutation-rescued OI-iPSCs, achieved by adjusting initial cell density and BMP4 concentration for each cell line (Supplementary Fig. 1). qPCR analysis demonstrated that CD271high+ sorted cells from day 5 aggregates (d5 271H) expressed NCC markers (SOX10, NGFR, TFAP2A, RHOB and PAX3) at levels comparable to those from previously reported 2D protocols12 (2D 271H) (Fig. 1g), with downregulation of the pluripotent cell marker POU5F1 in both cell populations. Furthermore, the aggregates showed a gene expression pattern similar to that of d5 271H, reflecting the highly efficient induction of NCCs. RNA sequencing (RNA-seq) analysis revealed a gradual transition in cell population from iPSCs (day 0) to NCCs (day 5), passing through the neural plate border cell state (Supplementary Fig. 2a). Consistent with these findings, SOX10 and CD271 proteins were detected after 4-day induction (Supplementary Fig. 2b). Furthermore, the differentiation potential of the aggregates into NCC derivatives was confirmed by the induction of peripheral neurons (Fig. 1h), melanoblasts (Fig. 1i) and mesenchymal stem/stromal cells (MSCs) (Supplementary Fig. 3).

a, Schematic representation illustrating the regional specification of cranial NCCs along the A–P axis in an embryo. PA1-3, pharyngeal arch 1-3. b, Differentiation strategy for NCCs from iPSCs in a 3D culture system. Y, Y-27632; SB, SB431542; CHIR, CHIR99021. c, Bright-field images of 1231A3 aggregates on days 1, 2 and 5 in the optimized culture. d, Immunofluorescence staining for SOX10 and CD271, and TFAP2A in serial sections of day 5 aggregates. Nuclei stained with DAPI. Dashed boxes magnified on the right (n = 34 from 11 biologically independent experiments). e, Flow cytometry analysis of CD271 expression in day 5 aggregates. Results shown as mean ± s.d. (six biologically independent experiments). f, Immunofluorescence staining for TFAP2A and ECAD, PAX6, and Oct3/4 in day 5 aggregates (n = 3 from 3 biologically independent experiments). g, The expression of NCC markers (SOX10, NGFR, TFAP2A, and PAX3) and a PSC marker (POU5F1) in iPSCs (d0), day 5 aggregates (d5), CD271high+ NCCs sorted from day 5 aggregates (d5 271H) and CD271high+ NCCs sorted from previous 2D protocols (2D 271H) (n = 6 from 3 biologically independent experiments). h,i, Immunofluorescence staining for immature neuron markers (TUBB3 and PRPH) and mature neuron markers (NeuN, MAP2 and Neurofilament-H) in aggregates after peripheral neuron induction (h) and for melanoblast markers (TRP2 and MITF) and a melanocyte marker (MART-1) in aggregates after melanoblast induction (i). Dashed boxes magnified on the bottom. In h, magnified images represent z-stack projection of 15 confocal optical sections (step size, 0.5 μm) (except for NeuN/MAP2) (n = 6 from 3 biologically independent experiments). j,k, Gene expression analysis for NCC markers (SOX10 and NGFR) (j) and HOX genes (HOXA1, HOXA2, HOXB2 and HOXA3) (k) in iPSCs (d0), day 5 aggregates (d5) and RA-treated day 5 aggregates (RA+ d5) (n = 6 from 3 biologically independent experiments). In g, j and k, values are shown as mean ± s.d. P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. ND, not determined; NS, not significant. In c–f, h and i, representative data are shown. All scale bars, 100 μm.
To evaluate the A–P axial identity of the NCC aggregates, we prepared aggregates treated with retinoic acid (RA) to induce posterior fates13,26 as a positive control (Supplementary Fig. 4a–c) and conducted qPCR analysis. Non-RA-treated NCC aggregates exhibited minimal expression of HOX genes (HOXA1, HOXA2, HOXB2 and HOXA3)27 compared with that of RA-treated NCC aggregates (Fig. 1j,k), with RNA-seq analysis confirming these results (Supplementary Fig. 4d). In addition, non-RA-treated NCC aggregates expressed midbrain (OTX1 and OTX2) and midbrain–hindbrain border markers (PAX2, PAX7, EN1 and EN2) but lacked the expression of forebrain and spinal cord region markers (Supplementary Fig. 4d). These findings demonstrate the efficiency of our 3D culture system in inducing HOX-negative midbrain–anterior hindbrain NCCs from human iPSCs.
Induction of mdEM with proximal–distal patterning
The intra-arch polarity of PA1 ectomesenchyme along the proximal–distal axis is characterized by Dlx and Hand genes4,5,6 (Fig. 2a). While Dlx2 is expressed in both maxillary ectomesenchyme (mxEM) and mdEM, Dlx5 expression is exclusive to mdEM, with further restriction of Hand2 towards the distal end. To selectively induce HOX-negative NCCs into mdEM rather than other NCC derivatives, we investigated the epithelial signals in developing PA1. Fgf8 is crucial for the survival and proliferation of PA1 ectomesenchyme and is detected throughout the nascent PA1 ectoderm28,29. Meanwhile, endothelin-1 (Edn1) is exclusively secreted from the mandibular prominence, driving PA1 ectomesenchyme towards an mdEM fate by activating Dlx5 and Hand2 (ref. 30). In addition, Bmp4 promotes distal mandibular development31,32,33. To determine the optimal conditions for inducing HOX-negative NCCs into mdEM, we tested the combinations of FGF8, EDN1 and BMP4 (Extended Data Fig. 2). We observed that FGF8 was essential for downregulating the NCC marker SOX10 and upregulating the early mesenchyme markers TWIST1 and PRRX1 (refs. 34,35) (Extended Data Fig. 2a). The combination of FGF8 with EDN1 induced common PA1 ectomesenchyme markers (DLX1 and DLX2) and mdEM markers (DLX5, DLX3 and GSC), with additional BMP4 further upregulating these genes and activating the distal marker HAND2 (Extended Data Fig. 2b). RNA-seq analysis confirmed these results and revealed minimal induction of melanocyte, sensory neuron, autonomic neuron and glia cell markers (Extended Data Fig. 3). Low expression of HOX genes indicated the absence of induction in other arches. Therefore, the concomitant activation of these pathways, referred to as FEDB, was used for further experiments (Fig. 2b).

a, Schematic model illustrating the regional specification of pharyngeal arch ectomesenchyme along the proximal–distal axis in an embryo. mdEM, mandibular prominence ectomesenchyme. b, Differentiation strategy of mdEM from NCC aggregates. c, Bright-field images and immunofluorescence staining for SOX10 and TWIST1 in d5 and d5 treated with FEDB (combination of FGF8, EDN1 and BMP4) for 4 days (d5 + FEDB). Nuclei stained with DAPI (n = 6 from 3 biologically independent experiments). d, Phalloidin staining of d5 and d5 + FEDB (n = 6 from 3 biologically independent experiments). e, Triple-colour immunofluorescence staining for DLX2, DLX5 and HAND2 in d5 + FEDB. Bottom panels show magnified views of the white dashed boxes in the top panels (n = 15 from 8 biologically independent experiments). White dotted circle, DLX5-weak and HAND2-negative area; yellow dotted line, HAND2-negative area. f, Positive rates of DLX2, DLX5 and HAND2 to DAPI calculated using ImageJ (n = 6 from 3 biologically independent experiments). g, Schematic model illustrating the regional specification of embryonic PA1 and induced mdEM. h, Schematic model illustrating the strategy to examine the effect of EDN1 signal on the bifurcation of maxillary prominence ectomesenchyme (mxEM) and mdEM. i, Immunofluorescence staining for SOX10 and TWIST1 in d5 treated with FGF8 and BQ-123 for 7 days (d5 + FQ) (n = 3 from 3 biologically independent experiments). j,k, Gene expression analysis for mdEM marker (DLX5) (j) and mxEM markers (POU3F3 and CYP26A1) (k) in aggregates (n = 6 from 3 biologically independent experiments). l, Schematic model illustrating the strategy to examine the importance of initial A–P identity in mdEM differentiation. m, Immunofluorescence staining for SOX10 and TWIST1 in RA+ d5 treated with FEDB for 4 days (RA+ d5 + FEDB) (n = 3 from 3 biologically independent experiments). n, Gene expression analysis for mdEM marker (DLX5) and HOX genes (HOXB2 and HOXB4) in aggregates (n = 6 from 3 biologically independent experiments). In j, k and n, values are shown as mean ± s.d. P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. In c–e, i and m, representative data are shown. All scale bars, 100 μm.
Immunostaining demonstrated that the FEDB condition effectively drove SOX10+ NCCs into TWIST1+ ectomesenchyme with a mesenchymal migratory morphology characterized by actin stress fibre formation36 (Fig. 2c,d). Notably, the aggregates contained DLX2 single-positive cells in the central zone, DLX2+DLX5+ cells in the intermediate zone and DLX2+DLX5+HAND2+ cells in the surface zone, suggesting that FEDB generated mdEM recapitulating the proximal–distal patterning of embryonic mdEM (Fig. 2e–g).
To further elucidate the effect of the EDN1 signal on the bifurcation of mxEM and mdEM, NCC aggregates were treated with a combination of FGF8 with the Endothelin Receptor Type A (EDNRA)4 antagonist, BQ-123 (referred to as FQ) (Fig. 2h). Immunostaining revealed differentiation into ectomesenchyme, albeit relatively less than with FEDB (Fig. 2i). EDN1 inhibition significantly increased the expression of mxEM markers (POU3F3 and CYP26A1)37, but not that of mdEM markers (Fig. 2j,k and Supplementary Fig. 5a,b). These findings suggest that EDN1 signalling is critical for maxillary–mandibular selection.
To examine the importance of initial A–P identity in mdEM differentiation, RA-treated posteriorized NCCs were cultured with FEDB (Fig. 2l). RA-treated NCCs differentiated into ectomesenchyme but exhibited minimal expression of mdEM marker and significantly increased HOX genes (HOXB2 and HOXB4) (Fig. 2m,n and Supplementary Fig. 5a,c), confirming that initial positional information in NCCs affects the patterning pathways.
Recapitulation of later-stage patterning using mdEM
To ascertain the induction of developmentally accurate mdEM, we investigated whether mdEM exhibited detailed regional patterning characteristic of later stages of development. In the later stage, epithelial domains undergo further subdivision along proximal, distal, aboral and oral axes, characterized by the expression of Fgf8, Bmp4, Edn-1 and Shh, respectively8,10,11 (Fig. 3a). In particular, Bmp4 signalling directs mdEM towards the distal cap domain11,38 (Fig. 3a,b). Conversely, Shh signalling specifies the oral domain, which is responsible for tooth development39. Moreover, the antagonistic interactions between Fgf8 and Bmp4 along the proximal–distal axis are involved in the specification of molars and incisors, respectively8. Therefore, we aimed to segregate mdEM into two distinct domains: the distal cap and the proximal-oral molar domain.

a,b, Schematic representation of major pharyngeal epithelial signals (a) and regional patterning of mdEM (b) in the mandibular prominence. The red dashed line corresponds to the coronal section. c, Strategies for inducing distal cap and proximal-oral domain from mdEM. LDN, LDN193189. d–f, Bright-field images and immunofluorescence staining for DLX5 (d), HAND1, GATA3 and HAND2 (e), and FOXF1 and PITX1 (f) in 1231A3 mdEM (d9, top) and aggregates after distal cap (d13, middle) or proximal-oral domain (d13, bottom) induction. Nuclei stained with DAPI (n = 6 from 3 biologically independent experiments). Scale bars, 100 μm. In d, dashed boxes magnified on the right. g, Gene expression analysis for distal cap marker (HAND1), distal region markers (HAND2, GATA3 and DKK1), proximal-oral domain-related genes (FOXF1, PITX1, GBX2 and BARX1) in d5, d9 and d13 (distal cap or proximal-oral domain) aggregates. Values shown as mean ± s.d. (n = 6 from 3 biologically independent experiments). P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. In d–f, representative data are shown.
The mdEM aggregates were transferred to a 96-well U-bottom ultra-low-cell-adhesion plates. For distal cap induction, mdEM aggregates were cultured with various induction media containing BMP4 (Extended Data Fig. 4a). Notably, we observed a pronounced upregulation of HAND1, the principal distal cap marker, alongside other distal markers (HAND2, GATA3 and DKK1) in response to elevated BMP4 concentrations (25 ng ml−1). These genes were further activated with decreased levels of the proximal signal, FGF8, albeit with an increase in apoptotic cells (Extended Data Fig. 4b). Replacing FGF8 with FGF2 enhanced cell viability, leading to the adoption of a condition termed BMP4high, EDN1 and FGF2low for distal cap induction (Fig. 3c). Immunostaining revealed decreased expression of DLX5 (Fig. 3d) and clear detection of HAND1 in the outer zone (Fig. 3e), akin to the mouse distal cap11. Expression of HAND2 and GATA3 was also distinctly observed.
For proximal-oral domain induction, mdEM aggregates were cultured with various induction media containing FGF8 and the Hedgehog signalling agonist, SAG (Extended Data Fig. 4c). This combination effectively induced proximal-oral-related genes (FOXF1, GLI1, PITX1, GBX2, BARX1, PAX9 and LHX6). Furthermore, these genes were upregulated when opposite regional signals, EDN1 and BMP4, were inhibited by BQ-123 and LDN193189 (LDN). Hence, the conditions FGF8, SAG, BQ-123 and LDN were selected for proximal-oral domain induction (Fig. 3c). Immunostaining revealed maintained expression of DLX5 and clear detection of FOXF1 and PITX1. Importantly, these marker expression patterns were mutually exclusive between distal cap and molar domain induction (Fig. 3d–f). qPCR and RNA-seq analyses confirmed these findings (Fig. 3g and Extended Data Fig. 5a).
Lastly, we assessed the odontogenic differentiation capacity of proximal-oral domain mdEM. Dental mesenchyme differentiation requires signalling from dental epithelium, including Shh, Fgf8, Wnt and Activin8,40,41,42. Treatment of proximal-oral domain mdEM with FGF8, SHH, CHIR and ACTIVIN-A (referred to as FSCA) resulted in the expression of dental mesenchyme markers represented by TFAP2B, TFAP2A, LHX6 and PAX9 (ref. 43) (Extended Data Figs. 5b and 6a,b). Gene Ontology (GO) analysis revealed a significant enrichment of genes related to ‘Odontogenesis’ and ‘Odontogenesis of dentin-containing tooth’ in the induction from NCCs (Extended Data Fig. 6c). In summary, mdEM successfully exhibited regional patterning similar to the embryonic mandible10,11,38,44, demonstrating developmentally faithful mdEM induction.
Generation of jawbone-like organoids
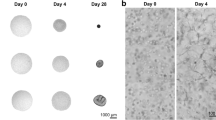
To mimic the intramembranous process, induced mdEM aggregates with condensed cell structures were cultured directly in xeno-free osteogenic induction medium, optimized for 3D culture (Fig. 4a). By day 38, we obtained white clusters with diameters of 1.0–1.5 mm (Fig. 4b). There was a significant increase in generally used osteogenic markers, ALP activity and calcium content (Fig. 4c). In addition, mineralized tissue formation was evidenced by Alizarin Red and von Kossa staining (Fig. 4d). qPCR analysis indicated peak expression of osteoblast markers at day 26, with a continued increase in bone matrix protein and osteocyte markers until day 38 (Extended Data Fig. 7a). Notably, the mature osteocyte marker, SOST, showed clear upregulation after day 19 (Fig. 4e), which was corroborated by RNA-seq analysis (Extended Data Fig. 7b). Conversely, some chondrogenic markers were also expressed (Extended Data Fig. 7c), and HOX gene expression remained marginal (Extended Data Fig. 7d), confirming maintenance of positional information in derivatives.

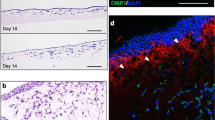
a, Stepwise induction strategy of jawbone-like organoids. b, Image of 1231A3-derived d38 cluster. c, Monitoring ALP activity and calcium content in clusters during osteogenic induction (n = 6 from 3 biologically independent experiments). d, Alizarin Red and von Kossa staining in serial sections of d38 clusters. Dashed boxes magnified on the right (Alizarin Red, n = 12; von Kossa, n = 6 from 3 biologically independent experiments). Scale bars, 100 μm. e, The expression of mature osteocyte marker (SOST) in clusters during osteogenic induction (n = 6 from 3 biologically independent experiments). f, HE and Azan staining in serial sections of d38 clusters (n = 52 from 12 biologically independent experiments). Scale bars, 100 μm. g, Triple-colour immunofluorescence staining for COLI, OCN and OPN in d38 tissues. Nuclei stained with DAPI (n = 9 from 3 biologically independent experiments). Scale bars, 100 μm. h–k, Immunofluorescence staining for SP7 and ALPL (h), PDPN (i), PHEX (j) and SOST (k) in d38 tissues. F-actin stained with phalloidin. Images represent the z-stack projection of 40 confocal optical sections (step size, 0.5 μm) (i–k) (n = 6 (h), n = 12 (i), n = 6 (j) and n = 6 (k) from 4 biologically independent experiments). Scale bars, 25 μm. l, Schematic model of generated jawbone-like organoids. m, Image of a kidney of NSG mice at 4 weeks after jawbone-like organoid transplantation. n, 3D reconstructed micro-CT image of the renal region at 4 weeks after transplantation. The dashed circle indicates an ectopic mineralized tissue. Scale bar, 1.0 mm (n = 6). o, HE and Azan staining in serial sections of transplanted tissues (n = 6). Scale bars, 100 μm. p, Immunofluorescence staining for human Vimentin in the transplanted tissues. F-actin stained with phalloidin. Images represent the z-stack projection of 40 confocal optical sections (step size, 0.5 μm) (n = 6). Scale bars, 25 μm. c,e, Box limits, upper/lower quartile; centre line, median; whiskers, max/min. P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. In b, d, f–k and m–p, representative data are shown. All specimens stained after decalcification (except for d).
Histological analysis of decalcified specimens revealed bone-like tissues with eosinophilic affinity and dense matrices stained with Azan Red (Fig. 4f). Moreover, major bone matrix protein, COLI, was abundantly expressed in the matrices, and non-collagenous bone proteins, OCN and OPN, were also detected (Fig. 4g and Extended Data Fig. 8a).
Subsequently, cellular characteristics in the bone-like tissues were examined. SP7+ osteogenic lineages appeared at day 12 and increased during differentiation (Extended Data Fig. 8b). The cell morphology gradually changed from fibroblastic to dendritic and PDPN+PHEX+ early osteocytes were detected around the surface area after 26 days of induction (Extended Data Fig. 8c). By day 38, ALP+SP7+ osteoblasts with cuboidal shapes lined the surface layer of the tissues (Fig. 4h), and a notable number of cells differentiated into PDPN+PHEX+ early osteocytes with dendritic processes extending from the surface to the inner area (Fig. 4i,j). Notably, within the bone matrices, SOST+ mature osteocytes formed 3D dendritic networks (Fig. 4k and Supplementary Video 1). Conversely, the central zone of the clusters exhibited immature chondrogenesis stained with Alcian Blue and not stained with Safranin O-stained, but these matrices showed minimal COLX expression, suggesting that the primary mechanism of osteogenesis is intramembranous ossification rather than endochondral ossification characterized by the appearance of hypertrophic cartilage (Extended Data Fig. 8d). In summary, our approach generated jawbone-like organoids from mdEM aggregates, recapitulating 3D network-forming osteocytes embedded in mineralized bone matrices (Fig. 4l). To examine the effect of initial patterning of mdEM on organoid induction, distal cap and proximal-oral mdEM were cultured in osteoinductive medium. Distal cap mdEM showed capacity to form bone tissues, unlike proximal mdEM (Supplementary Fig. 6). The generation of jawbone-like organoids via mdEM was reproduced using other iPSC clones (Supplementary Fig. 7).
We hypothesized that our jawbone-like organoids represent an early stage of jawbone development before vascularization. To assess the potential of these organoids to give rise to more mature bone tissues, we transplanted an organoid under the capillary-rich renal capsule of NOD/Scid/IL2Rγ (NSG) mice. Four weeks post-transplantation, white tissue was observed under the renal capsule (Fig. 4m). Micro-computed tomographic (micro-CT) analysis revealed highly mineralized tissue (Fig. 4n). Histological analysis confirmed the formation of vasculature and mature bone tissues (Fig. 4o and Extended Data Fig. 9a). In addition, the tissues consisted of human bone matrices and human Vimentin-positive donor cells with dense dendritic processes (Fig. 4p, Extended Data Fig. 9b and Supplementary Video 2). Hyaline cartilage similar to Meckel’s cartilage was sometimes observed, albeit in small amounts (Extended Data Fig. 9c). These findings demonstrate that jawbone-like organoids have the capacity to develop into mature human bone tissues in vivo.
Bone formation by transplantation of jawbone-like organoids
To evaluate the regenerative potential of jawbone-like organoids, three clusters of these organoids were directly transplanted into mandibular jawbone defects measuring 2.0 mm in diameter in NSG mice (Fig. 5a and Extended Data Fig. 10a,b). The bone graft group was prepared as a comparator of bone regenerative capacity. Four weeks post-transplantation, micro-CT analysis revealed that the amount of mineralized tissue was significantly increased in the organoid-grafted group compared with that of the non-grafted group and was similar to that of the bone graft group (Fig. 5b,c and Extended Data Fig. 10c–e). The mineralized tissue density in the organoid-grafted group was still inferior to that of the bone graft group (Fig. 5d). Histological examination using HE and Azan staining demonstrated that while some new bone formation was observed in the group without grafts, bone tissues in the transplanted group closely resembled the mature structure of the bone graft (Fig. 5e and Extended Data Fig. 10f). Immunostaining with antibodies specific to human COLI, OCN and OPN confirmed the presence of bone matrix proteins secreted by donor human cells derived from the jawbone-like organoids, further validating their contribution to bone regeneration (Fig. 5f). Moreover, within the bone matrices, both Vimentin-positive donor human osteocytes and -negative host mouse osteocytes formed dendritic cellular networks (Fig. 5g,h), and tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts were observed along the newly formed bone, indicating active bone remodelling (Fig. 5i). Whole-mount staining of the jawbone after transplantation revealed that the newly formed bones transitionally fused with host bones and were highly vascularized (Fig. 5j and Supplementary Video 3). These findings collectively suggest that transplanted jawbone-like organoids promote bone regeneration through successful osteogenesis with vascularization.

a, Images depicting a mandibular bone defect without graft or with three jawbone-like organoids or with bone graft. Yellow dotted lines denote the mandibular bone border. b, Coronal single-section and 3D reconstructed micro-CT images of mandibular bone defects at 4 weeks post-transplantation (n = 8). Scale bars, 1.0 mm. c,d, Quantification of newly formed mineralized tissue volume (c) and tissue mineral density (TMD) (d) in defects at 4 weeks post-transplantation. Data shown as mean ± s.d. (n = 8). P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test (c) and by two-tailed unpaired Student’s t-test (d). e–g, HE and Azan (e), triple-colour immunofluorescence staining for human COLI, OCN and OPN (f), and human Vimentin (g) in serial sections of transplanted tissues at 4 weeks post-transplantation. Nuclei stained with DAPI (f,g) (n = 8). Dashed boxes magnified on the right (e,g). Black arrowheads indicate the edge of the defects (e). White and yellow arrowheads indicate donor human cells and host mouse cells, respectively (g). Scale bars, 200 μm (e,f) and 100 μm (g). h, Immunofluorescence staining for human Vimentin in transplanted tissues. F-actin stained with phalloidin (n = 6). Images represent z-stack projection of 40 confocal optical sections (step size, 0.5 μm). Scale bar, 50 μm. i, TRAP staining in transplanted tissues (n = 6). Scale bars, 100 μm. j, Whole-mount immunofluorescence staining for human COLI and CD31 in transplanted tissues at 4 weeks post-transplantation. Mouse and human collagen fibres in bones visualized by SHG imaging. Images represent z-stack projection of 17 multiphoton optical sections (step size, 10 μm). The yellow arrowhead indicates the edge of the defect. Scale bars, 200 μm. In a, b and e–j, representative data are shown. All specimens stained after decalcification.
In vitro disease modelling using jawbone-like organoids
Currently, OI diagnosis primarily focuses on mesoderm-derived long bones45,46, with minimal attention given to the jawbone. Furthermore, although the severity of bone fragility likely involves osteocyte-to-bone matrix interactions, understanding the role of osteocytes in OI pathogenesis has been limited47. Therefore, we investigated the utility of jawbone-like organoids for histological analysis of OI.
First, mdEM was induced from OI-iPSCs obtained from a patient with autosomal dominant mutations of COLIA1, along with gene-corrected (rescued) OI-iPSCs (resOI-iPSCs)20 (Supplementary Fig. 8a). After 3 days of osteogenic induction, initial COLI production was observed in both OI- and resOI-iPSC-derived aggregates (Fig. 6a). Notably, collagen secreted from OI-iPSC-derived aggregates stained positive for collagen hybridizing peptide (CHP), indicating the production of misfolded triple-helical chains. By day 38, gene expression analysis indicated that the expression of COL1A1, later osteocyte markers (DMP1, PHEX, SOST and MMP13), and ER stress marker (BiP) were significantly lower in OI-organoids than resOI-organoids, suggesting that COLI mutation affects osteogenic differentiation in the organoids (Supplementary Fig. 8b). Azan blue-stained immature bone matrices were evident in OI-iPSC-derived jawbone-like organoids (OI-organoids) (Fig. 6b), with disrupted formation of osteocyte dendritic processes (Fig. 6c). These abnormal features were rectified in resOI-iPSC-derived organoids (Fig. 6a–c).

a, Immunofluorescence staining for COLI and CHP in OI- and gene-corrected (rescued) OI (resOI)-iPSC-derived aggregates after 3-day osteogenic induction. Nuclei stained with DAPI (n = 6 from 3 biologically independent experiments). Scale bars, 25 μm. b, HE and Azan staining in serial sections of d38 OI- and resOI-jawbone-like organoids (n = 6 from 3 biologically independent experiments). Scale bars, 100 μm. c, Phalloidin staining in OI- and resOI-organoids. Images represent z-stack projection of 40 confocal optical sections (step size, 0.5 μm). Dashed boxes magnified on the right (n = 6 from 3 biologically independent experiments). Scale bars, 25 μm. d, 3D reconstructed micro-CT images of ectopic mineralized tissues in the renal regions of NSG mice at 4 weeks after OI- or resOI-organoid transplantation. Scale bars, 1.0 mm. e, HE and Azan staining of the transplanted tissues (n = 6 from 3 biologically independent experiments). Scale bars, 100 μm. f, Quantification of mineralized tissue volume and TMD in OI- and resOI-organoid transplanted groups. g, Quantification of osteocytes and Azan Red-stained areas. h, TUNEL staining of the transplanted tissues in OI- and resOI-organoid transplanted groups (n = 6). Dashed boxes magnified below. Scale bars, 50 μm. i, Immunofluorescence staining for human Vimentin in the transplanted tissues. F-actin stained with phalloidin. Images represent z-stack projection of 40 confocal optical sections (step size, 0.5 μm) (n = 6). Scale bars, 25 μm. j, SHG imaging for collagen fibres in the bone tissues. F-actin stained with phalloidin (red). Images represent z-stack projection of 23 confocal optical sections (step size, 1.0 μm) (n = 6). Scale bars, 25 μm. k, Quantification of TUNEL-positive apoptotic cells. l, Quantification analysis of osteocyte dendritic process number. m, Quantification analysis of SHG intensity normalized to resOI. In f, g and k–m, data are shown as mean ± s.d. (n = 6; except for m, n = 5). P < 0.05 by two-tailed unpaired Student’s t-test. In a–e and h–j, representative data are shown. All specimens stained after decalcification.
Subsequently, OI- and resOI-organoids were transplanted under the renal capsule of NSG mice. Micro-CT analysis after 4 weeks revealed that OI-organoids generated less, and lower mineralized tissues compared with those of resOI (Fig. 6d,f). Histological analysis demonstrated that OI-organoids formed Azan blue-stained immature bone tissue, with a large number of osteocytes exhibiting characteristics typical of OI bone features48 (Fig. 6e,g and Supplementary Fig. 8c). These osteocytes were partially apoptotic (Fig. 6h,k). Conversely, resOI-organoids exhibited recovery from these abnormal features. Furthermore, osteocyte 3D network formations were disoriented in OI, with a significantly lower dendrite number compared with that of resOI (Fig. 6i,l). The bone matrices were also disoriented in OI (Fig. 6j,m and Supplementary Videos 4 and 5), suggesting aberrant osteocyte-to-bone matrix interactions. These findings indicate that the combination of jawbone-like organoids and OI-iPSCs successfully recapitulated the phenotypes of OI.
Discussion
In this study, we developed a selective induction protocol from human iPSCs to mdEM via HOX-negative NCCs under manipulated extrinsic signalling conditions (Fig. 7). Following osteogenic induction, iPSC-derived mdEM yielded functional jawbone-like organoids with 3D network-forming osteocytes embedded in bone matrices. Several groups reported protocols to differentiate PSCs into NCCs using 3D culture systems24,49, but these do not describe later stages of differentiation, particularly mandibular NCC derivatives under xeno-free conditions. Most studies on iPSC-derived 3D bone organoids have focused on endochondral ossification, which is the ossification model of long bone50,51. These strategies have first produced cartilage organoids and subsequently induced ossification by in vitro osteogenic induction or in vivo transplantation. Although organoids express osteocyte markers, their main component is cartilage tissue without induction of bone tissue. Furthermore, the recapitulation of 3D network-forming osteocytes embedded in bone matrices has been lacking in previous PSC-derived bone models, hampering the investigation of human tissues. Approaches for generating bone organoids from primary human cells such as MSCs and osteoblasts51 rely on tedious protocols for isolating bone marrow cells, which are often difficult to obtain in sufficient quantities, especially from the jaw. Moreover, NCCs and mdEM that give rise to the jawbone are difficult to evaluate in vivo because they are transient cell populations during development. Therefore, jawbone-like organoids generated from iPS cell lines cultured in xeno-free condition offer advantages in studying human jawbone embryology, pathophysiology, therapeutic development and serving as a foundation for broader systemic bone research.

mdEM and jawbone-like organoids were generated from human iPSCs in a stepwise manner, guided by signalling environments during embryogenesis, as depicted in the diagram. Initial SB/BMP4 treatment for 1 day, followed by SB/CHIR treatment, effectively induced HOX-negative NCC aggregates from iPSC aggregates (days 0–5). Transient RA treatment induced HOX-positive posterior NCC aggregates. From days 5 to 9, the combination of FGF8, EDN1 and BMP4 directed HOX-negative NCC aggregates to mdEM, recapitulating the proximal–distal patterning of embryonic mdEM. The regional identity of mdEM or mxEM was modulated by adjusting EDN1 signal activity. HOX-positive NCC aggregates did not differentiate into mdEM, underscoring the importance of initial NCC positional identity. mdEM exhibited later-stage regional patterning in response to exogenous mandibular epithelial signals (FGF8, SAG, EDN1 and BMP4), bifurcating into distal cap and proximal-oral molar domain. From days 9 to 38, under osteogenic conditions, mdEM formed jawbone-like organoids consisting of osteoblasts and network-forming osteocytes embedded in self-produced mineralized bone matrices. PA1-3, pharyngeal arch 1-3; mx, maxillary prominence; md, mandibular prominence; mdEM, md ectomesenchyme; SB, SB431542 (TGF-β inhibitor); CHIR, CHIR99021 (GSK3b inhibitor); BQ-123 (Endothelin Receptor Type A (EDNRA) antagonist); LDN, LDN193189 (BMP inhibitor); SAG (Hedgehog signalling agonist).
Because the lack of clarity in the composition of the commercial osteoinductive medium makes it unsuitable for exploring the detailed molecular mechanisms of osteogenesis, this study focused on regenerative therapies and pathological analyses to demonstrate the versatility of jawbone-like organoids. For a deeper understanding of physiological phenomena of jaw, transcriptome analysis during the induction process of the organoid should be performed to discover essential molecular signals in jawbone formation and to update the induction protocol to a chemically defined one.
In NCC aggregate induction, BMP4 conditions were optimized for each cell line (Extended Data Fig. 1). Previous ectodermal organoids such as inner ear22 and skin organoids52,53 require the optimization of BMP4 concentration potentially owing to different endogenous BMP4 expression levels. Therefore, the differentiation protocol must be adapted to each cell line.
Although our 3D culture system successfully induced mdEM from PSCs, to our knowledge, no studies have achieved this in 2D culture. We hypothesized that scaffold stiffness influences the direction of NCC differentiation. Mechanical forces are crucial for NCC migration and differentiation54,55. In mouse embryos, shaping the mandibular prominence requires 3D mesenchymal intercalations guided by actomyosin polarity56. In our study, NCCs treated with FEDB in 2D culture expressed smooth muscle cell markers without upregulation of mdEM markers (Supplementary Fig. 9a–d). Moreover, inhibition of actin stress fibre formation by Y-27632 treatment significantly rescued the expression of EDNRA and mdEM markers in 2D cultured cells, albeit not to the extent observed in 3D culture (Supplementary Fig. 9e). Conversely, spheroid culture of NCCs under FEDB induction led to significant cell apoptosis (Supplementary Fig. 9f). These findings suggest that our aggregate culture may provide NCCs with stiffness for mdEM differentiation. Notably, EDNRA is among the mechanosensitive G-protein-coupled receptors54, yet their activation mechanisms in NCCs remain incompletely understood. Our protocol offers insights into the novel mechanisms of Endothelin signalling.
While jawbone-like organoids successfully recapitulated the initiation stage of mandibular bone osteogenesis before vascularization, they lacked immune cells, including monocytes, macrophages and osteoclasts, which are essential for later-stage bone tissue features such as bone remodelling. Prolonged culture periods increased bone matrix density and mineral deposition, leading to necrosis and chondrogenesis owing to limited nutrient and oxygen diffusion. To overcome these limitations, incorporating a vascularization system containing immune cells may prove beneficial. Recent advancements in organ-on-a-chip technology have enabled the replication of various vascularized tissues and vascular flow environments57,58,59. Bone marrow organoids generated from iPSCs have also enabled modelling the physiology of the human bone marrow microenvironment60. In addition, trabecular bone organoids generated by co-culturing primary mouse osteoblasts and osteoclasts have recapitulated bone remodelling in vitro61. Integration of jawbone-like organoids with these technologies might generate genuine jawbone tissues containing all bone components.
The PA1 ectomesenchyme is the main source of mesenchyme in various oral tissues, including teeth, salivary glands, tongues, temporomandibular joints and jawbones4,62,63. While several induction protocols for oral tissue organoids have been described, most of these organoids partially or completely relied on primary cells for generation and failed to incorporate human PSC-derived PA1 ectomesenchyme64,65. However, in this study, we successfully induced molar region mesenchyme and subsequent dental mesenchyme from human iPSC-derived mdEM by manipulating epithelial signalling (Fig. 3 and Extended Data Figs. 5b and 6). Advancements have also been made in dental epithelial induction from human iPSCs66 and tooth germ organoid formation through co-culturing of dissociated embryonic tooth germ-derived dental mesenchyme and dental epithelium in a collagen gel67. Thus, combining our dental mesenchyme and these protocols holds promise for generating teeth organoids from human iPSCs, contributing to further advancements in oral medicine.
Methods
Human iPSCs culture and maintenance
Human iPSCs, reprogrammed with episomal vectors, were cultured and maintained on iMatrix-511-coated (human laminin-511 E8 fragment; Nippi) cell culture plates in StemFit AK03N medium (Ajinomoto). Upon reaching approximately 80% confluency, typically every 4–5 days, the cells were detached and dissociated into single cells using Accutase (Innovative Cell Technologies). After centrifugation, the cells were resuspended in StemFit AK03N supplemented with 10 μM Y-27632 (ROCK inhibitor; FUJIFILM Wako) and seeded onto iMatrix-511-coated culture plates. The following day, the culture medium was replaced with fresh StemFit AK03N without Y-27632. The medium was subsequently changed every 2 days until the next passages. The study utilized the following human iPSC lines: 1231A3, HLA homozygous iPSCs (Ff-I 14s04, Ff-I 14s04 knockout of human leukocyte antigen (HLA)-A,B, and CIITA genes (HLA-KO Ff-I 14s04, clone Ff-I 14s04-AB II-KO-13), Ff-I 014s04) provided by CiRA Foundation, OI patient-derived iPSCs (OI-iPSCs, clone OI08-12, exhibiting a point mutation at the splicing donor site of intron 32 in COLIA1 (c.2235 + 1G > A), classified as Sillence Type III) and mutation-rescued OI-iPSCs (resOI-iPSCs)20.
NCC induction from iPSCs in 3D conditions
For HOX-negative NCC induction, dissociated iPSCs were promptly reaggregated in 100 μl of StemFit Basic03 (equivalent to AK03N without bFGF, Ajinomoto) supplemented with 10 μM Y-27632 in 96-well V-bottom ultra-low-cell-adhesion plates (Sumitomo Bakelite). The cell concentrations applied to aggregation culture were as follows: 1231A3 (1.0 × 104 cells per well), Ff-I 14s04, HLA-KO Ff-I 14s04, Ff-I 014s04, OI-iPSCs and resOI-iPSCs (3.0 × 103 cells per well). Aggregation was facilitated by centrifugation at 280 × g for 3 min (day 0). For NCC induction, these cell aggregates were maintained in a 37 °C incubator with 5% CO2. After 24 h (day 1), the culture medium was replaced with 150 μl of StemFit Basic03 containing 10 μM SB431542 (TGF-β inhibitor; FUJIFILM Wako) and BMP4 (R&D Systems) (1231A3, Ff-I 14s04, HLA-KO Ff-I 14s04 (10 ng ml−1), Ff-I 014s04, OI-iPSCs and resOI-iPSCs (2.5 ng ml−1)). After another 24 h (day 2), the aggregates were individually collected and transferred into 48-well non-coated plates (IWAKI) and cultured in 250 μl of StemFit Basic03 containing 10 μM SB431542 and 1 μM CHIR99021 (GSK-3 inhibitor, Axon Medchem) (SB/CHIR) on an orbital shaker (CS-LR; TAITEC) at 120 rpm from days 2 to 5. For HOX-positive NCC induction, 1 μM all-trans-RA was added (FUJIFILM Wako) from days 4 to 5.
Optimization of BMP4 treatment in NCC aggregate induction
To investigate the effect of BMP4 concentration on NCC induction, 1231A3 aggregates (day 1) were cultured in medium containing 10 μM SB and BMP4 (0 ng ml−1, 0.1 ng ml−1, 1 ng ml−1, 10 ng ml−1 and 100 ng ml−1) for 1 day before SB/CHIR treatment. On day 5, flow cytometry analysis using anti-CD271 antibody and immunostaining with anti-CD271, PAX6 and ECAD antibodies were performed, and 10 ng ml−1 was selected as the optimal concentration to efficiently produce NCC with the lowest neural and non-neural ectoderm formation.
To investigate the effect of BMP4 treatment duration on NCC induction, 1231A3 aggregates (day 1) were cultured in medium containing 10 μM SB and 10 ng ml−1 BMP4 for 6 h, 12 h, 1 day, 2 days, 3 days or 4 days before SB/CHIR treatment. On day 5, flow cytometry analysis and immunostaining were performed, and 1- or 2-day treatment was required for efficient NCC induction and 1 day was selected as the minimum duration.
For Ff-I 14s04 and HLA-KO Ff-I 14s04 cell lines, treatment with 10 ng ml−1 BMP4 for 1 day BMP4 was suitable for NCC induction similar to 1231A3. For Ff-I 014s04, OI-iPSCs and resOI-iPSCs lines, 2.5 ng ml−1 BMP4 for 1 day BMP4 was suitable for NCC induction, while 10 ng ml−1 induced non-neural ectoderm frequently.
NCC induction from iPSCs in 2D conditions
In 2D conditions, NCCs were induced from 1231A3 iPSCs following previously established protocols12. In brief, iPSCs were seeded onto iMatrix-511-coated culture plates at a density of 3.6 × 103 cells per cm2 in StemFit AK03N medium and maintained in culture for 4 days. For NCC induction, the cells were cultured in SB/CHIR medium for 10 days, with medium changes every 2 days.
Differentiation of NCC aggregates
NCC aggregates at day 5 were transferred into 96-well U-bottom ultra-low-cell-adhesion plates (Sumitomo Bakelite).
For peripheral neuron differentiation, NCC aggregates were cultured in Neurobasal medium (Thermo Fisher Scientific) supplemented with B27 supplement (Thermo Fisher Scientific), N-2 supplement (Thermo Fisher Scientific), 2 mM l-glutamine (FUJIFILM Wako) and 10 ng ml−1 each of BDNF, GDNF, NT-3 and NGF (FUJIFILM Wako) for 14 days, with medium changes every 3 days.
For melanoblast differentiation, NCC aggregates were cultured in Stemfit Basic03 supplemented with 1 μM CHIR99021, 25 ng ml−1 BMP4 and 100 nM Endothelin-3 (TOCRIS) for 6 days, with medium changes every 3 days.
For MSC differentiation, NCC aggregates were dissociated and seeded onto fibronectin-coated (Millipore) plates at a density of 1.0 × 104 cells per cm2. The cells were cultured with StemFit Basic03 supplemented with 10 μM SB431542, 20 ng ml−1 each of EGF and FGF2 (FUJIFILM Wako) (SB/EF) and passaged before reaching subconfluency. Expanded cells (passage number 2 (PN2)) were seeded onto fibronectin-coated plates at a density of 1.0 × 104 cells per cm2 in SB/EF medium. After 24 h, the medium was replaced with PRIME-XV MSC Expansion XSFM medium (FUJIFILM Irvine Scientific). Passages were performed every 3 or 4 days. At PN2, MSC differentiation was confirmed by FACS analysis using human MSC markers. To confirm the differentiation potencies of the induced MSC-like cells, osteogenic, chondrogenic and adipogenic induction were performed, and the cells were stained with Alizarin Red, Alcian Blue and Oil Red O.
Induction of mdEM from HOX-negative NCC aggregates
HOX-negative NCC aggregates (day 5) were cultured in 350 μl of StemFit Basic03 supplemented with 100 ng ml−1 FGF8b (FUJIFILM Wako), 50 nM Endothelin-1 (PEPTIDE INSTITUTE) and 2.5 ng ml−1 BMP4 (referred to as FEDB medium) on an orbital shaker at 120 rpm from days 5 to 9. To quantify the DLX2-, DLX5- and HAND2-positive cells, dissociated mdEM were seeded onto culture plates, fixed immediately after 20 min and immunostained for these markers. Positive rates to DAPI were calculated using ImageJ and quantified from 5 fields per specimen (40× objectives).
To assess the impact of EDN1 signalling on the bifurcation of mxEM and mdEM, NCC aggregates were cultured in 350 μl of StemFit Basic03 supplemented with 100 ng ml−1 FGF8b and 10 μM BQ-123 (Endothelin A receptor antagonist; Selleck) on an orbital shaker at 120 rpm from days 5 to 12, with a medium change on day 9.
To investigate the significance of the initial A–P identity of NCCs in mdEM differentiation, RA-treated NCCs were cultured with 350 μl FEDB medium on an orbital shaker at 120 rpm from days 5 to 9.
To evaluate the effects of FGF8, EDN1 and BMP4 on 2D-induced NCCs under 2D conditions, CD271high+ sorted NCCs were seeded onto fibronectin-coated plates at a density of 1.0 × 104 cells per cm2 and cultured with FEDB medium for 4 days.
To examine the effects of FGF8, EDN1 and BMP4 on 2D NCCs in a 3D setting, CD271high+ sorted NCCs were rapidly aggregated in 100 μl FEDB medium at a density of 1.0 × 104 cells per well in 96-well V-bottom ultra-low-cell-adhesion plates and cultured for 4 days.
Induction of mdEM derivatives
mdEM aggregates (day 9) were transferred into 96-well U-bottom ultra-low-cell-adhesion plates (Sumitomo Bakelite).
For the induction of the distal cap domain, mdEM aggregates were cultured in 200 μl of StemFit Basic03 supplemented with 50 nM Endothelin-1, 20 ng ml−1 FGF2 and 25 ng ml−1 BMP4 from days 9 to 13, with a medium change on day 11.
For the induction of the proximal-oral domain, mdEM were cultured in 200 μl of StemFit Basic03 supplemented with 100 ng ml−1 FGF8b, 400 nM SAG (Smoothened Agonist, Selleck), 10 μM BQ-123 and 500 nM LDN193189 (ALK2/3 inhibitor, TOCRIS) from days 9 to 13, with a medium change on day 11. Subsequently, for dental mesenchyme induction, the proximal-oral domain induction medium was replaced with 200 μl of StemFit Basic03 supplemented with 100 ng ml−1 FGF8b, 400 nM SAG, 3 μM CHIR99021 and 30 ng ml−1 Activin A (R&D). Dental mesenchyme induction was performed from days 13 to 17, with a medium change on day 15.
Generation of jawbone-like organoids from mdEM
mdEM aggregates (day 9) were transferred into 96-well U-bottom ultra-low-cell-adhesion plates. The mdEM were cultured in 200 μl of StemFit Basic03 supplemented with 20 ng ml−1 EGF and FGF2, 50 ng ml−1 BMP2 (R&D), 3 μM CHIR99021 and 400 nM SAG for 3 days. On day 12, the differentiation medium was replaced with 200 μl MSCgo Osteogenic Differentiation Medium (SARTORIUS) and the clusters were cultured for 7 days. On day 19, the differentiation medium was replaced with 200 μl of StemFit Basic03 supplemented with 10 mM β-glycerophosphate disodium salt hydrate (Sigma), 250 μM l-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma), 100 nM dexamethasone (Sigma), 50 ng ml−1 BMP2 (R&D), 3 μM CHIR99021 and 400 nM SAG, and the clusters were cultured for 7 days. From days 26 to 38, the clusters were cultured in MSCgo Osteogenic Differentiation Medium, with medium changes every 2 days.
Chondrogenic induction of mdEM
To induce chondrogenic differentiation in 3D conditions, mdEM aggregates (day 9) were transferred into 96-well U-bottom ultra-low-cell-adhesion plates and cultured in MSCgo Chondrogenic XF (SARTORIUS) for 21 days.
Flow cytometry analysis and cell sorting
Cell aggregates or 2D cultured cells were dissociated using Accutase and resuspended in FACS buffer (1% human serum albumin in PBS) with appropriate antibodies, followed by a 60 min incubation at 4 °C. An isotype control was included in all experiments to eliminate non-specific background signals. Flow cytometry and sorting were performed on a BD FACSAria II_v1.87 (BD Biosciences). Resultant data were collected using FACSDiva_v9.0.1 and analysed using FlowJo_v10.8.1 software (BD Biosciences). Gating strategies are indicated in Supplementary Fig. 10. Details of the antibodies used are provided in Supplementary Table 3.
RT-qPCR analysis
RNA was extracted from cell aggregates or 2D cultured cells by lysing them with RLT Plus Buffer. Total RNA was purified using the RNeasy Micro Kit (QIAGEN) and treated with the RNase-Free DNase Set (QIAGEN) to eliminate genomic DNA. Subsequently, 0.5 μg of total RNA was reverse transcribed into single-stranded cDNA using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. RT-qPCR was performed using the THUNDERBIRD Next SYBR qPCR Mix (TOYOBO) and gene-specific primers on the QuantStudio 12K Flex Real-Time PCR System _v1.2.4 and StepOnePlus Real-Time PCR System_v2.3 (Applied Biosystems). The cycling parameters were as follows: initial denaturation (95 °C, 1 min), followed by 45 cycles of amplification and SYBR signal detection (95 °C denaturation, 3 s; 60 °C annealing, 30 s; 72 °C extension, 30 s). Data were analysed using the 2−ΔΔCt method and visualized using GraphPad Prism 9_v9.5.1 software. Primer sequences are provided in Supplementary Table 1.
RNA-seq analysis
Total RNA was extracted using the RNeasy Micro Kit and treated with the RNase-Free DNase Set to eliminate genomic DNA contamination. Subsequently, 10 ng of total RNA underwent reverse transcription to generate single-stranded cDNA, using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). For NGS library construction targeting the Ion AmpliSeq Transcriptome, the Ion AmpliSeq Transcriptome Human Gene Expression Panel, Chef-Ready Kit (Thermo Fisher Scientific) and the Ion Chef (Thermo Fisher Scientific) instruments were utilized, following the manufacturer’s protocol. In brief, a reverse transcription master mix was prepared by combining 5X VILO Reaction Mix and 10X SuperScript III Enzyme Mix. The mix was then aliquoted into reaction plates, with 10 ng of total RNA added to each. The plates were loaded into a thermal cycler and subjected to incubation at 42 °C for 30 min, followed by 85 °C for 5 min, and then held at 10 °C.
During library preparation on the Ion Chef instruments, the following consumables and cartridges were loaded: Ion AmpliSeq Chef Solutions DL8 cartridge, Ion AmpliSeq Chef Reagents DL8 cartridge, Ion AmpliSeq Tip Cartridge L8, PCR Frame Seal, IonCode 96 Well PCR Plate, PCR Plate Frame, Empty Tip Cartridge L8 and Enrichment Cartridge. Notably, IonCode 96 Well PCR Plates used for reverse transcription reactions contained an IonCode barcode adapter. In addition, the tube containing the Ion AmpliSeq Transcriptome Human Gene Expression Panel was inserted into an Ion AmpliSeq Chef Reagents DL8 cartridge. Multiplex PCR was performed in the thermal cycler within the Ion Chef, with 13 cycles of 16 min annealing/extension reactions. Subsequent steps involved primer digestion with FUPA reagents, ligation of IonCode barcode adapters, library purification using magnetic beads, equalization of library concentrations and mixing in a single tube.
Following library preparation, the Ion Chef facilitated template preparation and ISP beads loading onto the Ion 540 Chip Kit. NGS sequencing was conducted using the Ion GeneStudio S5 Prime sequencer (Thermo Fisher Scientific). Read counts were computed utilizing the AmpliSeq RNA Plugin, and data analysis was performed using R studio_v2023.06.1+524 software with the ‘DESeq2’ package for normalization to detect significantly (P ≤ 0.05) differentially expressed genes68. Differentially expressed genes were identified at a false discovery rate <0.05 with pairwise comparison. The raw and processed RNA-seq data were deposited in the NCBI GEO database under the accession numbers GSE261638, GSE261639, GSE261851, GSE261849, GSE261738 and GSE261850.
Pathway analysis
Pathway analysis was performed using the integrated Differential Expression and Pathway analysis (iDEP) software69,70 (version 9.6). Raw gene counts were input into the software, and the ‘DESeq2’ package within iDEP generated fold-change values, which were then used for gene set enrichment analysis (GSEA), independent of selected differentially expressed genes. GSEA was executed in pre-ranked mode, using an algorithm based on the fast gene set enrichment analysis (FGSEA) package_v3.21 (ref. 71). The minimum and maximum gene set size was set to 60 and 2,000, respectively. Applying a false discovery rate of 0.2, GO Biological Process gene sets were used to identify the top 15 pathways between FSCA-treated cells (dental mesenchyme) and NCCs (Supplementary Table 4).
Immunocytochemistry
Cells cultured on dishes were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.3% Triton X-100 (Sigma) in PBS at room temperature (RT) for 15 min. Subsequently, the cells were treated with Blocking One solution (Nacalai Tesque) at RT for 1 h. The cells were incubated with primary antibodies at 4 °C overnight and then with secondary antibodies at RT for 2 h. The antibodies were diluted in 10% (v/v) Blocking One in PBS-T. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:5,000; DOJINDO). Fluorescence signals were observed using a fluorescence microscope (BZ-X810 with BZ-X800 Viewer_v01.02.03.02; Keyence). The primary and secondary antibodies used are listed in Supplementary Table 2.
Alizarin Red and von Kossa staining
Fixed samples were embedded in paraffin and sectioned into 8-μm-thick slices. Alizarin Red staining was performed by incubating sections with 1% Alizarin Red at RT for 3 min. Von Kossa staining was conducted using the Calcium Stain Kit (Modified Von Kossa; ScyTek Laboratory) following the provided manual. The osteogenic potential was assessed using light microscopy.
Histological analysis of in vitro samples
For haematoxylin–eosin (HE), Azan and Safranin O/Fast green staining, samples were fixed with 4% PFA, decalcified with Decalcifying Solution B (FUJIFILM Wako) at RT overnight, paraffin-embedded and cut into 8-μm-thick sections. Sections were stained using standard protocols, followed by observation under light microscopy.
For immunofluorescence analysis, cell aggregates and decalcified samples were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek) and cryosectioned into 20-μm-thick sections. These sections were washed with PBS, permeabilized with 0.3% Triton X-100 in PBS at RT for 20 min, blocked with Blocking One Histo solution (Nacalai Tesque) at RT for 1 h, and incubated with primary antibodies at 4 °C overnight and then with secondary antibodies or Alexa Fluor 594 phalloidin for F-actin staining at RT for 4 h. The antibodies were diluted in antibody working solution (Can Get Signal Immunostain solution B, TOYOBO). Following DAPI counterstaining, the sections were mounted with VECTASHIELD Mounting Medium (Vector) and subjected to confocal microscopy (FV3000; Olympus). Confocal imaging data were analysed using FV31S-SW software_v2.6.1.243 (Olympus). The primary and secondary antibodies used are listed in Supplementary Table 2.
To visualize denatured COLI, cryosections were blocked and incubated with 15 µM 5-FAM-conjugated CHP (F-CHP) (3Helix) in PBS solution overnight at 4 °C and then incubated with secondary antibodies and DAPI as described above. Fluorescence signals were detected using a confocal microscope.
Cell viability
To assess cell viability, 2 µl of Cyto3D Live-Dead Assay (TheWell BIOSCIENCE) reagent was added to each culture well, and the aggregates were incubated at 37 °C for 15 min. Fluorescence signals were observed using a fluorescence microscope.
To identify apoptotic cells, sectioned samples were subjected to terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining (DeadEnd Fluorometric TUNEL System; Promega). Following DAPI counterstaining, fluorescence signals were visualized using a confocal microscope. TUNEL-positive cells were quantified using ImageJ_v1.54f software (NIH).
Transplantation
NOD/Scid/IL2Rγ (NSG) mice were housed in a 12 h/12 h light/dark cycle at 21–25 °C and 45–55% humidity. For renal subcapsular transplantation, 8-week-old male NSG mice were anaesthetized, and the left kidney was exposed. A jawbone-like organoid was transplanted under the renal capsule of each mouse without artificial materials (n = 6 per group). Mice were euthanized with CO2 4 weeks post-transplantation, and transplanted tissues were collected after micro-CT scanning.
For transplantation into jawbone defects, 12-week-old male NSG mice were anaesthetized, and a 10 mm skin incision was made to expose the right hemimandible, followed by the creation of a 2-mm-diameter circular defect at the mandible angle using a micro engine with round steel burs (VOLVER i7, NAKANISHI). Three jawbone-like organoids were transplanted into each defect without artificial materials (n = 8 per group), as illustrated in Fig. 5a. Jawbones for bone graft were collected from the mandible angle of other NSG mice. Mice were fed a soft diet for 2 weeks post-surgery before transitioning to a regular diet. Mandibular bones were collected and scanned by micro-CT 4 weeks post-transplantation.
Micro-CT analysis
Before euthanasia, renal regions and jawbones were scanned using a micro-CT system (inspeXio SMX-100CT; Shimadzu) with the following parameters: voxel size of 30 μm, tube voltage of 60 kV, tube current of 0.2 mA and exposure time of 480 ms. The CT slice resolution was 1,024 × 1,024 pixels. Bone morphometrics were analysed using 3D imaging software (TRI/3D-BON-FCS; RATOC System Engineering) according to the manufacturer’s instructions. Using a calibration curve based on known density phantoms scanned under the same conditions, the CT value of each tissue was converted to the tissue mineral density (TMD).
Histological analysis of in vivo samples
Collected kidneys and jawbones were fixed in 4% PFA, followed by a decalcification period of 7 days. The specimens were paraffin-embedded and cut into 8-μm-thick serial slices, with jawbone samples cut in the frontal plane.
HE, Azan and TRAP staining (TRAP/ALP Stain Kit, FUJIFILM Wako) were performed according to standard protocols and observed via light microscopy. The number of osteocytes in lacuna was counted manually aided by ImageJ software on HE-stained sections (20× objectives) and quantified from 4 fields per specimen. Quantitative assessment of the Azan Red-stained area was conducted utilizing ImageJ software.
For immunofluorescence analysis, deparaffinized sections were permeabilized and treated with LAB solution (Polyscience) for antigen activation at RT for 15 min. The sections were blocked with Blocking One Histo solution at RT for 1 h and incubated with primary antibodies diluted in antibody working solution at 4 °C overnight. For mouse antibodies against human OCN, the sections were blocked using the Mouse on Mouse kit (ReadyProbes Mouse-on-Mouse IgG Blocking Solution (30X), Invitrogen). Afterwards, the sections were incubated with secondary antibodies diluted in antibody working solution at RT for 4 h. To eliminate tissue autofluorescence, sections were treated with the Vector TrueVIEW Autofluorescence Quenching kit (Vector) at RT for 5 min. Following counterstaining with DAPI, the sections were mounted, and fluorescence signals were detected using a confocal microscope. Details of the primary and secondary antibodies used are provided in Supplementary Table 2.
To visualize F-actin filaments, phalloidin staining was performed on 20-μm-thick cryosections.
3D analysis of osteocytes
To visualize the 3D structure of phalloidin-stained osteocyte dendritic processes, 40 optical serial z-axis sections (0.5 µm step size) were acquired using a confocal microscope and reconstructed using IMARIS_v9.7.2 software (Oxford Instruments). The number of dendritic processes per osteocyte was counted in rotating the 3D image using the Filament Tracer function of IMARIS. The diameter for spots was set to 1 μm. Measurements were performed on randomly selected ten osteocytes per specimen.
Second harmonic generation imaging
Second harmonic generation (SHG) images were obtained using an upright multiphoton excited microscopy system (AX/AXR_v6.01, Nikon). Cryosections of 20 μm thick stained with phalloidin were used. The excitation wavelength was 880 nm. The detection wavelength for the acquisition of collagen-derived SHG signals was 446 nm. All SHG images were quantified using NIS-Elements AR_v6.02.03 software (Nikon) and reported as mean fluorescence intensity. The mean pixel intensities were calculated from five distinct regions of interest. These measurements served as technical replicates for individual samples.
Whole-mount immunostaining
Methods of whole-mount staining were conducted with reference to previous reports72. In brief, O.C.T. compound-embedded specimens of mouse jawbones were iteratively sectioned using a cryostat until the transplanted bone defect was fully exposed along coronal axis. Jawbone was then reversed, and the procedure was repeated on the opposite face of the bone until a thick slice of bone was obtained. O.C.T. compound covering the sample was removed, and slices were washed with PBS, blocked in Blocking One solution overnight at 4 °C and incubated with primary antibodies diluted in 10% (v/v) Blocking One in PBS-T at 4 °C for 3 days, followed by overnight washing in PBS. The slices were incubated with secondary antibodies diluted in 10% Blocking One in PBS-T at RT for 3 days and successively washed in PBS overnight and cleared in RapiClear 1.52 (SunJin Lab). The cleared slices were mounted on glass slides with RapiClear 1.52 and iSpacer (SunJin Lab) and imaged under the multiphoton excited microscopy system.
Statistics
Statistical analyses were performed using GraphPad Prism 9 software. One-way analysis of variance (ANOVA) with Tukey’s test or two-way ANOVA with Bonferroni’s test was used to compare multiple groups, while the two-tailed Student’s t-test was used for comparisons between two groups. The number of biological replicates is indicated in the figure legends. P values < 0.05 were considered statistically significant.
Ethics statement
The experimental protocols involving human subjects received approval from the Ethics Committee of the Department of Medicine and Graduate School of Medicine, Kyoto University, and the Ethics Committee of Shiga Medical Center for Children. Written informed consent was obtained from all donors. All procedures involving mice were conducted in strict accordance with the Regulations on Animal Experimentation at Kyoto University (protocol number 16-73, 22-155, 24-225).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are included within the paper and its Supplementary Information. The raw and processed RNA-seq data were deposited in the NCBI GEO database. The RNA-seq dataset focusing on NCC differentiation (Supplementary Fig. 2) is available under the accession number GSE261638 (ref. 73). The RNA-seq dataset focusing on the effect of RA on NCC (Supplementary Fig. 4) is available under the accession number GSE261639 (ref. 74). The RNA-seq dataset focusing on mdEM differentiation (Extended Data Fig. 3) is available under the accession number GSE261851 (ref. 75). The RNA-seq dataset focusing on regional patterning using mdEM (Extended Data Fig. 5) is available under the accession number GSE261849 (ref. 76). The RNA-seq dataset focusing on osteogenic differentiation (Extended Data Fig. 7) is available under the accession number GSE261738 (ref. 77). The RNA-seq dataset focusing on the effect of FEDB on 2D cultured NCC (Supplementary Fig. 9) is available under the accession number GSE261850 (ref. 78). Source data are provided with this paper.
References
Soares, A. P. et al. Uncovering the unique characteristics of the mandible to improve clinical approaches to mandibular regeneration. Front. Physiol. 14, 1152301 (2023).
Nakajima, T. et al. Modeling human somite development and fibrodysplasia ossificans progressiva with induced pluripotent stem cells. Development 145, dev165431 (2018).
Loh, K. M. et al. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467 (2016).
Minoux, M. & Rijli, F. M. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development 137, 2605–2621 (2010).
Selleri, L. & Rijli, F. M. Shaping faces: genetic and epigenetic control of craniofacial morphogenesis. Nat. Rev. Genet. 24, 610–626 (2023).
Depew, M. J., Lufkin, T. & Rubenstein, J. L. Specification of jaw subdivisions by Dlx genes. Science 298, 381–385 (2002).
Gavalas, A. et al. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development 125, 1123–1136 (1998).
Neubuser, A., Peters, H., Balling, R. & Martin, G. R. Antagonistic interactions between FGF and BMP signaling pathways: a mechanism for positioning the sites of tooth formation. Cell 90, 247–255 (1997).
Yuan, Y. et al. Spatiotemporal cellular movement and fate decisions during first pharyngeal arch morphogenesis. Sci. Adv. 6, eabb0119 (2020).
Xu, J. et al. Hedgehog signaling patterns the oral-aboral axis of the mandibular arch. eLife 8, e40315 (2019).
Vincentz, J. W. et al. Exclusion of Dlx5/6 expression from the distal-most mandibular arches enables BMP-mediated specification of the distal cap. Proc. Natl Acad. Sci. USA 113, 7563–7568 (2016).
Kamiya, D. et al. Induction of functional xeno-free MSCs from human iPSCs via a neural crest cell lineage. npj Regen. Med. 7, 47 (2022).
Mica, Y., Lee, G., Chambers, S. M., Tomishima, M. J. & Studer, L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 3, 1140–1152 (2013).
Srinivasan, A. & Toh, Y. C. Human pluripotent stem cell-derived neural crest cells for tissue regeneration and disease modeling. Front. Mol. Neurosci. 12, 39 (2019).
Parada, C. & Chai, Y. Mandible and tongue development. Curr. Top. Dev. Biol. 115, 31–58 (2015).
Salhotra, A., Shah, H. N., Levi, B. & Longaker, M. T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 21, 696–711 (2020).
Qin, L., Liu, W., Cao, H. & Xiao, G. Molecular mechanosensors in osteocytes. Bone Res. 8, 23 (2020).
Robling, A. G. & Bonewald, L. F. The osteocyte: new insights. Annu. Rev. Physiol. 82, 485–506 (2020).
Bolamperti, S., Villa, I. & Rubinacci, A. Bone remodeling: an operational process ensuring survival and bone mechanical competence. Bone Res. 10, 48 (2022).
Kawai, S. et al. In vitro bone-like nodules generated from patient-derived iPSCs recapitulate pathological bone phenotypes. Nat. Biomed. Eng. 3, 558–570 (2019).
Hayashi, R. et al. Generation of 3D lacrimal gland organoids from human pluripotent stem cells. Nature 605, 126–131 (2022).
Koehler, K. R. et al. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 35, 583–589 (2017).
Lee, G., Chambers, S. M., Tomishima, M. J. & Studer, L. Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc. 5, 688–701 (2010).
Lee, G. et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 25, 1468–1475 (2007).
Marchant, L., Linker, C., Ruiz, P., Guerrero, N. & Mayor, R. The inductive properties of mesoderm suggest that the neural crest cells are specified by a BMP gradient. Dev. Biol. 198, 319–329 (1998).
Fattahi, F. et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 531, 105–109 (2016).
Rothstein, M., Bhattacharya, D. & Simoes-Costa, M. The molecular basis of neural crest axial identity. Dev. Biol. 444, S170–S180 (2018).
Creuzet, S., Schuler, B., Couly, G. & Le Douarin, N. M. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc. Natl Acad. Sci. USA 101, 4843–4847 (2004).
Trumpp, A., Depew, M. J., Rubenstein, J. L., Bishop, J. M. & Martin, G. R. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 13, 3136–3148 (1999).
Sato, T. et al. An endothelin-1 switch specifies maxillomandibular identity. Proc. Natl Acad. Sci. USA 105, 18806–18811 (2008).
Zuniga, E., Rippen, M., Alexander, C., Schilling, T. F. & Crump, J. G. Gremlin 2 regulates distinct roles of BMP and Endothelin 1 signaling in dorsoventral patterning of the facial skeleton. Development 138, 5147–5156 (2011).
Alexander, C. et al. Combinatorial roles for BMPs and Endothelin 1 in patterning the dorsal-ventral axis of the craniofacial skeleton. Development 138, 5135–5146 (2011).
Tucker, A. S., Khamis, A. A. & Sharpe, P. T. Interactions between Bmp-4 and Msx-1 act to restrict gene expression to odontogenic mesenchyme. Dev. Dyn. 212, 533–539 (1998).
Soo, K. et al. Twist function is required for the morphogenesis of the cephalic neural tube and the differentiation of the cranial neural crest cells in the mouse embryo. Dev. Biol. 247, 251–270 (2002).
Doufexi, A. E. & Mina, M. Signaling pathways regulating the expression of Prx1 and Prx2 in the chick mandibular mesenchyme. Dev. Dyn. 237, 3115–3127 (2008).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
Jeong, J. et al. Dlx genes pattern mammalian jaw primordium by regulating both lower jaw-specific and upper jaw-specific genetic programs. Development 135, 2905–2916 (2008).
Barron, F. et al. Downregulation of Dlx5 and Dlx6 expression by Hand2 is essential for initiation of tongue morphogenesis. Development 138, 2249–2259 (2011).
Cobourne, M. T. & Sharpe, P. T. Tooth and jaw: molecular mechanisms of patterning in the first branchial arch. Arch. Oral Biol. 48, 1–14 (2003).
Prochazka, J. et al. Migration of founder epithelial cells drives proper molar tooth positioning and morphogenesis. Dev. Cell 35, 713–724 (2015).
Yu, T. & Klein, O. D. Molecular and cellular mechanisms of tooth development, homeostasis and repair. Development 147, dev184754 (2020).
Zhang, Y. D., Chen, Z., Song, Y. Q., Liu, C. & Chen, Y. P. Making a tooth: growth factors, transcription factors, and stem cells. Cell Res. 15, 301–316 (2005).
Jing, J. et al. Spatiotemporal single-cell regulatory atlas reveals neural crest lineage diversification and cellular function during tooth morphogenesis. Nat. Commun. 13, 4803 (2022).
Rao, P., Jing, J., Fan, Y. & Zhou, C. Spatiotemporal cellular dynamics and molecular regulation of tooth root ontogeny. Int. J. Oral Sci. 15, 50 (2023).
Marini, J. C. et al. Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 3, 17052 (2017).
Forlino, A. & Marini, J. C. Osteogenesis imperfecta. Lancet 387, 1657–1671 (2016).
Pathak, J. L., Bravenboer, N. & Klein-Nulend, J. The osteocyte as the new discovery of therapeutic options in rare bone diseases. Front. Endocrinol. 11, 405 (2020).
Mahr, M. et al. Increased osteocyte lacunae density in the hypermineralized bone matrix of children with osteogenesis imperfecta type I. Int. J. Mol. Sci. 22, 4508 (2021).
Seto, Y., Ogihara, R., Takizawa, K. & Eiraku, M. In vitro induction of patterned branchial arch-like aggregate from human pluripotent stem cells. Nat. Commun. 15, 1351 (2024).
Lamande, S. R. et al. Modeling human skeletal development using human pluripotent stem cells. Proc. Natl Acad. Sci. USA 120, e2211510120 (2023).
Zhao, D., Saiding, Q., Li, Y., Tang, Y. & Cui, W. Bone organoids: recent advances and future challenges. Adv. Health. Mater. 13, e2302088 (2024).
Lee, J. et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature 582, 399–404 (2020).
Lee, J. et al. Generation and characterization of hair-bearing skin organoids from human pluripotent stem cells. Nat. Protoc. 17, 1266–1305 (2022).
Canales Coutino, B. & Mayor, R. Neural crest mechanosensors: seeing old proteins in a new light. Dev. Cell 57, 1792–1801 (2022).
Zhu, Y. et al. Matrix stiffness modulates the differentiation of neural crest stem cells in vivo. J. Cell. Physiol. 234, 7569–7578 (2019).
Tao, H. et al. Oscillatory cortical forces promote three dimensional cell intercalations that shape the murine mandibular arch. Nat. Commun. 10, 1703 (2019).
Ingber, D. E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 23, 467–491 (2022).
Zhang, B., Korolj, A., Lai, B. F. L. & Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 3, 257–278 (2018).
Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371 (2022).
Frenz-Wiessner, S. et al. Generation of complex bone marrow organoids from human induced pluripotent stem cells. Nat. Methods 21, 868–881 (2024).
Park, Y. et al. Trabecular bone organoid model for studying the regulation of localized bone remodeling. Sci. Adv. 7, eabd6495 (2021).
Han, D. et al. A TGFβ-Smad4-Fgf6 signaling cascade controls myogenic differentiation and myoblast fusion during tongue development. Development 139, 1640–1650 (2012).
Yuan, Y. & Chai, Y. Regulatory mechanisms of jaw bone and tooth development. Curr. Top. Dev. Biol. 133, 91–118 (2019).
Tanaka, J. et al. Human induced pluripotent stem cell-derived salivary gland organoids model SARS-CoV-2 infection and replication. Nat. Cell Biol. 24, 1595–1605 (2022).
Wang, Y. & Sun, Y. Engineered organoids in oral and maxillofacial regeneration. iScience 26, 105757 (2023).
Alghadeer, A. et al. Single-cell census of human tooth development enables generation of human enamel. Dev. Cell 58, 2163–2180 e2169 (2023).
Nakao, K. et al. The development of a bioengineered organ germ method. Nat. Methods 4, 227–230 (2007).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ge, S. X., Son, E. W. & Yao, R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 19, 534 (2018).
Ge, X. iDEP web application for RNA-seq data analysis. Methods Mol. Biol. 2284, 417–443 (2021).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2021).
Gomariz, A. et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun. 9, 2532 (2018).
Motoike, S. et al. RNA-seq data of “Time course gene expression pattern during neural crest cell (NCC) differentiation from induced pluripotent stem cells (iPSCs) in three-dimensional (3D) conditions”. GSE261638. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261638 (2025).
Motoike, S. & Ikeya, M. RNA-seq data of “The expression of regional markers in neural crest cell (NCC) aggregates treated with or without retinoic acid (RA)”. GSE261639. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261639 (2025).
Motoike, S. & Ikeya, M. RNA-seq data of “Generation of mandibular prominence ectomesenchyme (mdEM) from human induced pluripotent stem cell (iPSC)-derived neural crest cell (NCC) aggregates”. GSE261851. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261851 (2025).
Motoike, S. & Ikeya, M. RNA-seq data of “Recapitulation of regional patterning using human induced pluripotent stem cell (iPSC)-derived mandibular prominence ectomesenchyme (mdEM)”. GSE261849. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261849 (2025).
Motoike, S. & Ikeya, M. RNA-seq data of “Induction of osteogenic lineage cells from human induced pluripotent stem cell (iPSC)-derived neural crest cell (NCC) aggregates through mandibular prominence ectomesenchyme (mdEM)”. GSE261738. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261738 (2025).
Motoike, S. & Ikeya, M. RNA-seq data of “Evaluation of the effects of FGF8, Endothelin-1, and BMP4 (FEDB) on induced pluripotent stem cell (iPSC)-derived neural crest cells (NCCs) induced in two-dimensional (2D) conditions”. GSE261850. NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE261850 (2025).
Acknowledgements
We extend our gratitude to the current and former members of the M.I. laboratory, especially D. Kamiya and Z. Al-Akashi for their support with data analyses, to S. Kawai for provision of materials and to the Common Equipment Management Office of CiRA for their support throughout this study. We would like to thank Editage (www.editage.jp) for English language editing. This study was supported by Japan Agency for Medical Research and Development (AMED) under grant numbers 22bm0104001h0010 and JP23bm1323001, Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (JSPS) under grant numbers JP22H03275 and JP23H03028, and KOSÉ Cosmetology Research Foundation for M.I. This study was also supported by KAKENHI grant numbers 21K16993 and 23K15998, Grant-in-Aid for JSPS Fellows under grant number JP21J01564, Seeds Development Grant for Young Scientists of Japanese Society of Periodontology (JSP), the Future Development Funding Program of Kyoto University Research Coordination Alliance, and a grant from the iPS Cell Research Fund for S.M.
Author information
Authors and Affiliations
Contributions
S.M., M.K. and M.I. contributed to the conception of the study. S.M. and M.I. designed the research, generated figures and drafted the paper. S.M., J.T. and M.I. reviewed the paper. S.M. carried out all experiments, with assistance from Y.I. in cell cultures and animal experiments. J.T. provided patient-derived iPSCs and clinical information. All authors provided critical feedback on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Mark Grimes, Karl Koehler and Antoine Zalc for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Optimization of BMP4 treatment in NCC aggregate induction.
a,c,e, Overview of the approach for testing the effect of BMP4 concentration (a,c) and treatment duration (e) on NCC induction efficiency in 1231A3 cell line. Aggregates were treated with the combination of SB and CHIR (a) or BMP4 (0, 0.1, 1, 10, 100 ng ml-1) (c) for 1 day. SB, SB431542; CHIR, CHIR99021. b,d,f, Immunofluorescence staining for CD271 / PAX6 or ECAD, and flow cytometry analysis of CD271 in day 5 aggregates. Nuclei stained with DAPI. b, Only SB/CHIR condition induced not only CD271high+ NCCs but also PAX6+ neuroectoderm. d, The absence of BMP4 (0 ng ml-1) induced not only NCCs but also neuroectoderm. Conversely, excessive BMP4 (100 ng ml-1) induced ECAD+ non-neural ectoderm on the surface of aggregates. In total, 10 ng ml-1 was employed. f, Short-term BMP4 treatment (6 and 12 h) induced NCCs and neuroectoderm. Prolonged treatment (3 and 4 days) induced non-neural ectoderm. 1-day treatment was employed. g, Optimization of BMP4 activity is required for highly efficient NCC induction. In b, d and f, n = 3 from three biologically independent experiments. Flow cytometry data shown as mean ± s.d. Scale bars, 100 μm. Representative data are shown.
Extended Data Fig. 2 Concomitant activation of FGF8, EDN1, and BMP4 upregulates mdEM markers effectively.
a,b, Gene expression analysis for NCC marker (SOX10) and early pharyngeal arch mesenchyme markers (TWIST1 and PRRX1) (a), and common PA1 ectomesenchyme markers (DLX1 and DLX2), mdEM markers (DLX5, DLX3 and GSC) and distal region marker (HAND2) (b) in 1231A3 iPSCs (d0), day 5 NCC aggregates (d5) and d5 treated with combinations of FGF8, Endothelin-1 (EDN1) and BMP4 for 4 days (d9). Values presented as mean ± s.d. (n = 6 from three biologically independent experiments). P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. NS, not significant. FGF8 treatment decreased SOX10 and upregulated TWIST1, PRRX1, DLX1 and DLX2, suggesting that it is required for ectomesenchyme differentiation of NCCs. The concomitant activation of FGF8, EDN1 and BMP4 upregulates mdEM markers effectively. BMP4 treatment was crucial for HAND2 expression.
Extended Data Fig. 3 Selective induction of mdEM from NCC aggregates.
a-c, RNA-seq analysis of 1231A3 iPSCs (d0), NCC aggregates (d5) and mdEM (d9). Heatmap illustrating gene expression levels of pluripotent stem cell (PSC), NCC, pan-ectomesenchyme (EM), mdEM, and maxillary ectomesenchyme (mxEM) markers (a); melanocyte, glia, sensory neuron and autonomic neuron markers (b); HOX genes (c). log2(Transcripts Per Million (TPM) + 1) values used for gene expression evaluation. (n = 3 from three biologically independent experiments). The cell population transited from iPSCs to mdEM via NCCs.
Extended Data Fig. 4 Investigation of optimized protocols for distal cap and proximal-oral (molar) domain induction.
a, Gene expression analysis for distal cap marker (HAND1) and other distal region markers (HAND2, GATA3 and DKK1) in 1231A3 NCC aggregates (d5), mdEM (d9) and mdEM cultured with various combinations of FGF8 (20 and 100 ng ml-1) or FGF2 (20 ng ml-1), EDN1 (50 nM), and BMP4 (2.5 and 25 ng ml-1) for 4 days (d13). Values shown as mean ± s.d. (n = 3 from three biologically independent experiments). b, Cell viability analysis of mdEM cultured with FGF8 or FGF2 (20 ng ml-1), EDN1 (50 nM), and BMP4 (25 ng ml-1) from day 5 to 9. Live cells (green) and dead cells (red) distinguished using a LIVE/DEAD Viability/Cytotoxicity kit. Apoptotic cells detected by TUNEL staining. Nuclei stained with DAPI. (n = 3 from three biologically independent experiments). Representative data are shown. Scale bars, 100 μm. c, Gene expression analysis for proximal-oral (molar) domain-related markers (FOXF1, GLI1, PITX1, GBX2, BARX1, PAX9 and LHX6) in NCC aggregates (d5), mdEM (d9), and mdEM cultured with various combinations of FGF8 (100 ng ml-1), SAG (400 nM), BQ-123 (BQ, 10 μM), LDN193189 (LDN, 500 nM), EDN1 (50 nM) and BMP4 (2.5 ng ml-1) for 4 days (d13). Values shown as mean ± s.d. (n = 3 from three biologically independent experiments). In a and c, P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. NS, not significant.
Extended Data Fig. 5 mdEM recapitulates the regional patterning of mandibular prominence.
a,b, RNA-seq analysis of 1231A3 iPSC-derived NCC aggregates (d5), mdEM (d9), distal cap (d13), and proximal-oral domain (d13) (a) and dental mesenchyme induction (d17) (b). Heatmap visualizing the gene expression levels of ectomesenchyme (EM), mdEM, aboral domain, distal domain, proximal-oral domain and dental mesenchyme markers. (n = 3 from three biologically independent experiments). Z-scores used to evaluate gene expression levels.
Extended Data Fig. 6 Dental mesenchyme induction from mdEM of the proximal-oral domain.
a, Bright-field image and immunofluorescence staining for TFAP2B and LHX6, TFAP2A and PAX9 in day 17 aggregates after dental mesenchyme induction. Nuclei stained with DAPI. Dashed boxes magnified to the right. (n = 3 from three biologically independent experiments). Representative data are shown. Scale bars, 100 µm. b,c, RNA-seq analysis of NCC aggregates (d5) and dental mesenchyme (d17). b, Volcano plot for differential expression genes. Some dental mesenchyme markers were selected in the plot. (n = 3 from three biologically independent experiments). c, Gene-Set Enrichment Analysis (GSEA). Top 15 enriched (activated and suppressed) pathways according to GO terms for biological processes shown. False discovery rate (FDR)–adjusted P-values (adj. P-value) (one-tailed) are calculated using the Benjamini-Hochberg method. adj. P-value < 0.05. (n = 3 from three biologically independent experiments). NES, normalized enrichment score.
Extended Data Fig. 7 Directed differentiation of osteogenic lineage from mdEM.
a, Gene expression analysis of bone matrix proteins (COLIA1, SPP1 and IBSP), osteoblast markers (SP7, RUNX2, ALP and DLX5), early osteocyte markers (CAPG, PDPN, DMP1 and PHEX) and mature osteocyte markers (MEPE, TNFSF11 and MMP13) in 1231A3 iPSC-derived mdEM (d9) and each time point of osteogenic induction. Box limits, upper/lower quartile; centre line, median; whiskers, Max./Min. (n = 6 from three biologically independent experiments). P < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test. NS, not significant. b–d, RNA-seq analysis of 1231A3 NCC aggregates (d5), mdEM (d9) and day 38 clusters (d38). Heatmap visualizing gene expression levels of bone matrix proteins, osteoblasts, early osteocytes and mature osteocytes (b); chondrogenic markers (c); and HOX genes (d). log2(TPM + 1) values were used to evaluate gene expression levels. (n = 2 from three biologically independent experiments).
Extended Data Fig. 8 mdEM gradually produce bone matrices and differentiate into osteogenic cells.
a–c, Immunofluorescence staining for COLI (a), SP7 (b), PDPN and PHEX (c) in 1231A3 iPSC-derived mdEM (d9) and at each time point of osteogenic induction. Nuclei stained with DAPI. F-actin stained with Phalloidin (c). (n = 3 from three biologically independent experiments). d, HE, Alcian blue / Fast red, and Safranin O / Fast green staining, and immunofluorescence staining for COLII and COLX in day 38 clusters after osteogenic induction and aggregates after chondrogenic induction. In a–d, representative data are shown. All specimens stained after decalcification. Scale bars; 100 µm (a,b,d), 25 µm (c).
Extended Data Fig. 9 Jawbone-like organoids form bone tissues composed of human bone matrix proteins.
a, Von Kossa, HE, Azan and Safranin O / Fast green staining and immunofluorescence staining for CD31 in bone-like tissues formed under NSG mouse renal capsule (n = 6). Nuclei stained with DAPI. Scale bars; 50 μm (CD31), 200 µm (the others). b, Triple-colour immunofluorescence staining for human bone matrix proteins (human COLI, OCN and OPN) in bone-like tissues. (n = 6). c, HE, Alcian blue / Fast red and Safranin O / Fast green staining in tissues formed under NSG mouse renal capsule. In a–c, representative data are shown. All specimens stained after decalcification (except for Von Kossa staining). Scale bars, 200 µm.
Extended Data Fig. 10 Jawbone regeneration by transplanted jawbone-like organoids and bone graft.
a, Schematic representation of jawbone-like organoid (d38) transplantation into mandibular bone defects in NSG mice. b, A micro-CT scanning image of bone defect before transplantation. c–e, Individual micro-CT scanning images of jawbones with no graft (c), with jawbone-like organoid transplantation (d) and with bone graft (e). (n = 8). f, HE and Azan staining in serial sections of bone-grafted tissues at 4 weeks post-transplantation (decalcified specimens). (n = 6). Scale bars, 200 μm. Representative data are shown.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and Tables 1–3.
Supplementary Table 4
Gene set enrichment analysis (GSEA). Top 15 enriched (activated and suppressed) pathways according to GO terms for biological processes.
Supplementary Video 1
Reconstructed 3D-view video of osteocyte networks in a jawbone-like organoid. 3D reconstitution of 40 confocal optical sectioned image. Nuclei and F-actin stained with DAPI (blue) and phalloidin (red), respectively. Scale bar, 20 µm.
Supplementary Video 2
Reconstructed 3D-view video of osteocyte networks in a jawbone-like organoid-derived tissue. 3D reconstitution of 40 confocal optical sectioned image (step size, 0.5 μm). Nuclei and F-actin stained with DAPI (blue) and phalloidin (red), respectively. Scale bar, 20 μm.
Supplementary Video 3
Reconstructed 3D-view video of transplanted jawbone-like organoid and vascularization. 3D reconstitution of 17 multiphoton optical sectioned image (step size, 10 μm). Whole-mount immunofluorescence staining for human COLI (green) and CD31 (red) in transplanted tissues at 4 weeks post-transplantation. Mouse and human collagen fibres in bones visualized by SHG (blue) imaging.
Supplementary Video 4
Reconstructed 3D-view video of bone matrices and osteocyte networks in an OI-organoid-derived tissue. 3D reconstitution of 23 multiphoton optical sectioned image (step size, 1.0 μm). Collagen fibres in bones visualized by SHG (blue) imaging. F-actin stained with phalloidin (red).
Supplementary Video 5
Reconstructed 3D-view video of bone matrices and osteocyte networks in a resOI-organoid-derived tissue. 3D reconstitution of 23 multiphoton optical sectioned image (step size, 1.0 μm). Collagen fibres in bones visualized by SHG imaging. F-actin stained with phalloidin (red). 3D reconstitution of 23 multiphoton optical sectioned image (step size, 1.0 μm). Collagen fibres in bones visualized by SHG (blue) imaging. F-actin stained with phalloidin (red).
Supplementary Data 1
Source data for Supplementary figures.
Source data
Source Data
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Motoike, S., Inada, Y., Toguchida, J. et al. Jawbone-like organoids generated from human pluripotent stem cells. Nat. Biomed. Eng 9, 1816–1834 (2025). https://doi.org/10.1038/s41551-025-01419-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41551-025-01419-3
This article is cited by
-
Bridging skeletal stem cell diversity and human skeletal modeling
Journal of Bone and Mineral Metabolism (2026)
