
Abstract
The high diversity and complexity of the eukaryotic transcriptome make it difficult to effectively detect specific transcripts of interest. Current targeted RNA sequencing methods often require complex pre-sequencing enrichment steps, which can compromise the comprehensive characterization of the entire transcriptome. Here we describe programmable full-length isoform transcriptome sequencing (PROFIT-seq), a method that enriches target transcripts while maintaining unbiased quantification of the whole transcriptome. PROFIT-seq employs combinatorial reverse transcription to capture polyadenylated, non-polyadenylated and circular RNAs, coupled with a programmable control system that selectively enriches target transcripts during sequencing. This approach achieves over 3-fold increase in effective data yield and reduces the time required for detecting specific pathogens or key mutations by 75%. We applied PROFIT-seq to study colorectal polyp development, revealing the intricate relationship between host immune responses and bacterial infection. PROFIT-seq offers a powerful tool for accurate and efficient sequencing of target transcripts while preserving overall transcriptome quantification, with broad applications in clinical diagnostics and targeted enrichment scenarios.
Similar content being viewed by others


Main
The diversity of the eukaryotic transcriptome is greatly expanded by alternative transcription and alternative splicing, and the enormous number of molecules of different types makes it difficult to characterize specific transcripts of interest. Although targeted RNA sequencing (RNA-seq)1,2,3,4 can improve the sensitivity of detecting target transcripts, these hybridization capture and amplicon-based methods rely on efficient probe or primer design and lose quantitative information during the enrichment process. Nanopore sequencing, offering long read lengths and native modification detection ability, has been widely applied in transcriptome studies. During the sequencing process, negatively charged DNA or RNA molecules are driven through the nanopore by a constant voltage5, where Oxford Nanopore Technologies (ONT) provides an adaptive sequencing feature that can reject sequencing fragments by reversing the drive voltage6. Recent studies have developed adaptive sequencing strategies to enrich target genomic regions and deplete unwanted genomes7,8,9,10,11, but no effort has been made in the field of transcriptome adaptive sequencing. The enrichment efficiency with the current RNA-seq protocol is also limited by the RNA molecule length, with rejected fragments inevitably including over half of the unwanted transcripts8. Moreover, canonical nanopore complementary DNA sequencing using oligo(dT) reverse transcription (RT) can only capture polyadenylated RNAs and is unable to profile non-polyadenylated transcriptomes.
To overcome these challenges, we developed the programmable full-length isoform transcriptome sequencing (PROFIT-seq) strategy. PROFIT-seq employs combinatorial RT and rolling circle amplification (RCA) to effectively capture the whole transcriptome, and implemented a user-friendly adaptive sequencing system for programmable enrichment of target transcripts while preserving the undisturbed abundance of all transcripts. Through extensive evaluations, we demonstrated that our method can effectively capture polyadenylated, non-polyadenylated and circular transcriptomes and enrich the target RNA panel while achieving reliable whole-transcriptome quantification. We next used PROFIT-seq for rapid detection of the microbiome composition and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection from sputum samples of patients with pneumonia or coronavirus disease 2019 (COVID-19). Finally, we used PROFIT-seq to explore the transcriptome landscape in colorectal polyp development, unveiling the complex association between the immune response and microbiome infection during intraepithelial neoplasia transformation. Overall, as a flexible targeted RNA-seq protocol with reliable whole-transcriptome quantification capabilities, PROFIT-seq can serve as a useful approach for rapid clinical diagnostics and target gene enrichment.
Results
Programmable total transcriptome sequencing with PROFIT-seq
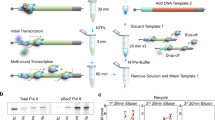
To effectively enrich various transcript types, we first employed a combinatorial RT strategy using double-stranded oligo(dT), double-stranded random primer and single-stranded random primer to capture full-length polyadenylated, non-polyadenylated and circular transcripts (Fig. 1a and Methods). Then, we employed splint-based circularization12 and the RCA assay following the Rolling Circle Amplification to Concatemeric Consensus (R2C2) protocol13 to increase the length of cDNAs and improve the power of discriminating finished and rejected reads. Here, the increased length of RCA products (>10 kb) compared with linear RT products (~1 kb) provided the basis for effective target enrichment. Finally, the library was constructed and sequenced on the MinION platform. Raw sequencing reads were divided into subreads using the circularization splint sequence, and consensus sequences were calculated to accurately represent the high-fidelity transcripts (Methods). To improve the applicability of current adaptive sampling tools, PROFIT-seq provides a user-friendly web interface for programmable real-time sequencing manipulation, allowing users to specify sequencing mode, run duration, channel number, barcodes and genes of interest, or upload a configuration file for multiple target submissions (Extended Data Fig. 1a–c).

a, A schematic overview of the PROFIT-seq method. First, the ribosomal RNA-depleted total RNA was reverse transcribed using a combinatorial RT strategy. Double-stranded oligo(dT) (dsdT), dsN and ssN were successively added to capture polyadenylated, non-polyadenylated and circular RNAs. The reverse-transcribed cDNA was then circularized and amplified using the RCA assay. The nanopore sequencing library was constructed, and PROFIT-seq was used for real-time control of the sequencing process. The acquired chunk data were basecalled and demultiplexed according to the sequencing time, channel number and detected barcodes. The basecalled sequences were subsequently aligned to the reference genome. Finally, PROFIT-seq determined whether the sequencing process should be continued or rejected according to the user-provided sequencing configuration. b, The length of rejected (purple) and finished (green) reads for canonical DNA, cDNA and PROFIT-seq runs. All bulk fast5 runs were simulated for sequencing all reads or rejecting all reads for 1 h. c, The elapsed time for raw signal basecalling, sequence alignment and pore manipulation for each acquired chunk of data. d, PROFIT-seq provides three manipulation modes, including enrichment or depletion of reads aligned to target regions and the balancing mode for dynamic determination of enriching targets based on the accomplished coverage. e, The performance of enriching chromosomes 1, 2, 5, 11 and 12 and depleting other chromosomes (middle) or balancing coverage of all chromosomes (right). Source numerical data are available in Source data.
To demonstrate the benefit of the PROFIT-seq protocol, we simulated different DNA, cDNA and PROFIT-seq sequencing runs for 1 h using the playback function of the MinKNOW software (Methods). As shown in Fig. 1b, the rejected reads in both runs had a median length of ~600 bp, with rejected reads in the cDNA run inevitably accounting for ~67.5% (554 of 820 bp) of full-length sequences, impeding the enrichment efficiency. In contrast, the finished reads in the PROFIT-seq run were 10-fold longer than rejected reads, indicating a more discriminative length difference. The real-time analysis efficiency of PROFIT-seq was verified by assessing the elapsed time for data processing steps. Based on the mapping rate of basecalled sequences, an optimized data acquisition interval of 0.4 s was determined (Extended Data Fig. 1d), and all data processing steps were able to be finished within every chunk acquisition period (Fig. 1c). Then, the enrichment effect was evaluated using three pore manipulation modes: (1) enrichment of target region, (2) depletion of target region and (3) balancing mode, where target regions are dynamically determined according to the sequenced coverage (Fig. 1d). PROFIT-seq successfully enriched target regions, with 72.10% of consensus reads aligning to target chromosomes in enrichment mode compared with 35.00% in control. In addition, the standard variance of reads aligned to different chromosomes decreased from 2.20 in control samples to 1.48 in balancing mode (Fig. 1e), demonstrating the successful pore manipulation using PROFIT-seq.
Combinatorial whole-transcriptome RT
As current nanopore cDNA sequencing protocols often employ oligo(dT)-based RT to capture polyadenylated RNAs, the diversity of non-polyadenylated transcripts is largely missing. In contrast, PROFIT-seq employs a combinatorial RT strategy with double-stranded oligo(dT), double-stranded random primer and single-stranded random primer to capture full-length polyadenylated, non-polyadenylated and circular RNAs14. To assess the performance of our combinatorial RT protocol, total RNA from HeLa cells was reverse transcribed using indexed combinatorial RT primers (Methods), and reads were demultiplexed on the basis of the aligned index primers in the first and last 150 bp of the sequences. In two replicates, 82.35% of the sequenced molecules originated from double-stranded oligo(dT) (dsdT) primers, while double-stranded random hexamers (dsN) and single-stranded random hexamers (ssN) accounted for 12.55% and 5.05%, respectively (Fig. 2a). The majority of combinatorial RT reads aligned to the exonic regions of GENCODE-annotated genes (Extended Data Fig. 2a). Compared with the oligo(dT) primer, random hexamers generated reads of similar length (Fig. 2b) but displayed a more even distribution across the 5′ end of transcripts (Fig. 2c) and captured more non-polyadenylated transcripts (for example, pseudogene, long non-coding RNA (lncRNA), miscellaneous RNA (miscRNA) and small nuclear RNA (snRNA)) (Fig. 2d). In particular, ssN primers captured more circular RNAs (circRNAs), aligning with our original intent of the combinatorial RT strategy. Compared with oligo(dT) libraries, the combinatorial RT sample also demonstrated an increase in the proportion of reads from circRNAs and other non-polyadenylated transcripts (Extended Data Fig. 2b,c), indicating that PROFIT-seq was able to efficiently capture non-polyadenylated RNAs.

a, The percentage of reads derived from different primers. Rep, biological replicates. The colours indicate dsdT, dsN and ssN, respectively. b, The length of different primed reads in combinatorial RT libraries. c, The coverage across the top 1,000 highly expressed transcripts in different primed reads. The x axis represents the relative position along the transcript, and the y axis is the per cent coverage of combined reads. d, The proportion of reads aligned to transcripts of different biotypes according to the GENCODE annotation. The colours represent different primers. The proportion of circRNA reads is calculated using CIRI-long. e, The identification of poly(A)+ and poly(A)− genes from Yang et al.16. Poly(A)+, poly(A)− and bimorphic genes were classified according to the relative abundance between the poly(A)+ and poly(A)− samples. f,g, The log-scaled gene expression levels of poly(A)+ and poly(A)− genes in combinatorial RT, oligo(dT) primed (f) and ONT direct RNA-seq data (g). Poly(A)+ and poly(A)− genes were classified according to the relative abundance between the poly(A)+ and poly(A)− samples from Yang et al. The colours represent the density of transcripts. h, Tracks of sequencing depth and reconstructed isoforms in the RPL34 and RPS2 loci. i, Tracks of sequencing depth and reconstructed isoforms in the MYC and SMARCE1 loci. The GENCODE v37 annotation and genomic coordinates are indicated above the tracks. Source numerical data are available in Source data.
To validate the quantification ability of the combinatorial RT method, two random and two oligo(dT) primed cDNA libraries were constructed and sequenced as described above. Compared with the HeLa Illumina total RNA-seq15,16, oligo(dT), combinatorial RT and random primed libraries exhibited high correlation of gene-level expression values (R = 0.65; Extended Data Fig. 2d), which demonstrated that combinatorial RT does not bias quantification. For the poly(A)+ transcriptome, the combinatorial RT library (R = 0.65) achieved similar transcript-level quantification accuracy to oligo(dT) libraries (R = 0.67; Extended Data Fig. 2e), while the random primed library was substantially biased (R = 0.49). Thus, these results demonstrated that combinatorial RT could combine the poly(A)+ and poly(A)− transcriptomes without introducing notable bias that skews the quantification analyses. To further investigate the performance of combinatorial RT, a total of 22,649 poly(A)+ and 302 poly(A)− genes were identified using the public poly(A)-enriched and poly(A)-depleted dataset16 (Fig. 2e). Among them, a total of 16,453 poly(A)+ and 111 poly(A)− genes were successfully recovered in the combinatorial RT dataset. The abundance of poly(A)+ genes correlated highly with oligo(dT) and ONT direct RNA-seq17, while poly(A)− genes were enriched by the combinatorial RT protocol (Fig. 2f,g), indicating that combinatorial RT can better characterize the non-polyadenylated transcriptome compared with canonical poly(A)+ RNA-seq approaches.
For instance, canonical oligo(dT) cDNA-seq only sequenced two protein-coding isoforms from the RPL34 gene, while combinatorial RT effectively captured the retained intron supported by the GENCODE v37 annotation18 (Fig. 2h). Similarly, oligo(dT) data only captured the major protein-coding isoform in the RPS2 gene, but combinatorial RT effectively reconstructed three alternative-spliced isoforms and one pseudogene from the same locus. Moreover, the combinatorial RT also effectively captured more transcript isoforms in lower-expressed transcription factors such as MYC and SMARCE1 (Fig. 2i), demonstrating enhanced isoform discovery sensitivity.
To examine the quantitative performance of the complete PROFIT-seq protocol, the HeLa transcriptome was sequenced using the combinatorial RT and R2C2 amplification. As shown in Extended Data Fig. 3a, the transcript expression levels from the PROFIT-seq library and combinatorial RT library without R2C2 amplification were significantly correlated (R = 0.87, P < 10−31, Pearson correlation test), indicating reliable quantitative capability. Consistent with previous studies13,19, the consensus reads with a high circular consensus sequence number (>5) exhibited a high accuracy rate (99.22%; Extended Data Fig. 3b). In addition, PROFIT-seq provides a lightweighted strategy for consensus calling and RCA chimeric filtration (Methods), showing comparable performance to the C3POa13 algorithm with similar read accuracy (Extended Data Fig. 3c–e), faster processing speeds (Wilcoxon rank sum test, P = 0.02; Extended Data Fig. 3f) and fewer RCA artefacts (Wilcoxon rank sum test, P = 0.02; Extended Data Fig. 3g).
Overall, these results demonstrated that PROFIT-seq can effectively sequence polyadenylated, non-polyadenylated and circular transcripts with high accuracy, which can better delineate transcriptome diversity compared with the canonical cDNA sequencing protocol.
Enrichment effect of PROFIT-seq on the target gene panel
Next, we evaluated the ability of PROFIT-seq to enrich the target gene panel in two colorectal polyp samples (JL01 and JL19). Here, a panel of 717 protein-coding genes, 1,055 lncRNAs and 606 circRNAs that are related to colorectal cancer were selected as enrichment targets on the basis of public databases20,21,22,23 (Fig. 3a). Each sequencing flow cell was divided into two sections, where half of the channels were sequenced with PROFIT-seq for target enrichment and the other half using the default program as a control. To validate the enrichment efficiency, the number of on-target and unwanted raw reads and consensus reads was calculated in a sliding window of 30 min. As shown in Fig. 3b,c, PROFIT-seq sequenced more molecules and produced more on-target raw and consensus reads in the first 12 h, and the number decreased gradually with the accelerated loss of pores during adaptive sequencing8. Finally, PROFIT-seq achieved an increased number of 1.9× target molecules and 1.68× target consensus reads compared to control runs, indicating the successful enrichment of cancer-related transcripts (Fig. 3d). Then, we investigated the number of cancer-related genes covered using the generated consensus reads. PROFIT-seq rapidly detected 363 target genes (26 mRNA, 286 lncRNA and 39 circRNA loci) within 6 h, while the control runs only sequenced 219 target genes (18 mRNA, 174 lncRNA and 21 circRNA loci) within that same amount of time (Fig. 3e,f). Finally, PROFIT-seq also generated a better number of recalled genes in the complete run. Here, 248 genes stably enriched with a >2-fold increase (Fig. 3g), and the overall enrichment efficiency was significantly correlated between the two samples (R = 0.27, P = 4.61 × 10−23, Spearman correlation test; Fig. 3h), suggesting the effective enrichment of the cancer-related gene panel.

a, The genomic location of the target gene panel (1,509 loci). The number of transcripts from different databases is indicated in the table. b,c, The absolute number of sequenced molecules (left) and consensus reads (right) aligned to target regions (b) or out-of-target regions (c). The colours represent PROFIT-seq and control runs, and the styles of lines indicate two biological replicates (JL01 and JL19). d, The cumulative number of sequenced molecules (left) and consensus reads (right) aligned to target regions. The colours represent PROFIT-seq and control runs, and the styles of lines indicate two biological replicates (JL01 and JL19). e, The cumulative number of genes covered by consensus reads during the sequencing run. The colours correspond to the PROFIT-seq (red) and control (grey) runs. The styles indicate two biological replicates. f, Bar plot indicating the number of recalled genes at 6 h in two biological replicates. g, The enrichment efficiency of each target gene at 6 h. The colours represent whether genes are 2-fold (red) or <2-fold (grey) enriched. h, The log-scale fold change (enrichment efficiency) of target genes between PROFIT-seq and control data in two polyp samples. The colours represent robustly enriched (red, ≥2-fold enrichment in both samples) and minorly enriched (grey) genes, respectively. R = 0.27, P = 4.61 × 10−23, Spearman correlation test. FC, fold change. i, The number of genes with more (orange) or fewer (blue) reconstructed isoforms in PROFIT-seq compared with control data. j,k, Tracks of sequencing reads and reconstructed isoforms in the GAS5 (j) and BID (k) loci. The numbers represent the discovery sensitivity of GENCODE-annotated transcripts in each sample. Source numerical data are available in Source data.
With the effective enrichment of cancer-related genes, we next examined the ability of PROFIT-seq to reveal RNA isoform diversity. Here, StringTie2 (ref. 24) was utilized for genome-wide transcript assembly using consensus PROFIT-seq reads. Compared with the control runs, PROFIT-seq dramatically improved the number of reconstructed transcripts in the target locus. As shown in Fig. 3i, 78.11% and 69.70% of cancer-related genes exhibited increased isoform diversity, while no similar effect was observed for non-target genes. For instance, only three isoforms from the GAS5 loci were identified in the control data, while seven and five isoforms were reconstructed in PROFIT-seq runs, respectively (Fig. 3j). Similarly, a notable increase in the aligned reads and reconstructed isoforms was also observed for the BID locus (Fig. 3k), indicating an increased discovery sensitivity of annotated genes. Taken together, these results suggested that PROFIT-seq was able to effectively enrich cancer-related genes and their splicing isoforms in clinical samples without prior experimental enrichment.
Unbiased quantification of targeted and non-targeted RNAs
As targeted reads are selectively enriched during the sequencing run, the quantification of targeted and non-targeted RNAs could be biased. To address this issue, we proposed an expectation–maximization (EM)-based algorithm that combines full-length consensus reads and rejected partial reads for effective transcript reconstruction and unbiased quantification (Fig. 4a). Briefly, basecalled reads were divided into full-length reads and partial fragments on the basis of the existence of RT primers and template-switching oligo sequences. Enriched full-length reads were aligned to the reference genome, and transcripts were assembled using StringTie2 (ref. 24). Next, partial fragments of rejected reads were extracted and realigned to the reconstructed transcripts. The expression value of each assembled transcript was measured by Salmon25 using full-length and partial reads, respectively, and the final expression values were determined on the basis of a modified EM algorithm (Methods).

a, A schematic overview of the isoform assembly and quantification pipeline. First, full-length reads were aligned to the reference genome for isoform reconstruction. Then, the recovered partial fragments were aligned to the reconstructed isoforms, and Salmon was used for transcript quantification. The final expression levels were measured using an EM-based strategy by combining the full-length reads and recovered partial fragments. b, The percentage of full-length reads for the target (right) and non-target (left) transcripts in the JL01 sample. c, The expression levels of the target (red) and non-target (grey) genes in PROFIT-seq using recovered partial fragments. d, The expression levels of the target (red) and non-target (grey) genes before correction (left) or after correction using the EM algorithm (right) in the JL01 sample. e, Correlation of gene expression levels for Illumina total RNA-seq, control runs without pore manipulation and PROFIT-seq data using either the EM quantification strategy or only full-length reads. The colours represent the correlation coefficient calculated using the Spearman correlation test. f, The log-scale fold change of target gene expression values in PROFIT-seq and Illumina total RNA-seq data as controls. The colours indicate the expression level before correction (grey) or after correction using both full-length PROFIT-seq reads and recovered partial fragments (red). FC, fold change. Source numerical data are available in Source data.
In PROFIT-seq data, the percentage of full-length consensus reads in target transcripts was higher than that in non-target transcripts, indicating effective enrichment but also quantification bias (Fig. 4b and Extended Data Fig. 4a). Intriguingly, a high Spearman correlation (R = 0.687) between gene quantification results calculated from partial fragments and control runs was observed (Fig. 4c), which demonstrated the viability of expression level correction using these partial fragments. When using full-length reads only, the gene expression values of target genes were substantially overestimated (Fig. 4d and Extended Data Fig. 4b), and the correlation was notably improved after the adjustment by the EM quantification algorithm (R = 0.717), indicating the effective correction of quantification bias.
We further benchmarked performance of the EM-based quantification results against Illumina total RNA-seq data. As shown in Fig. 4e, the raw full-length PROFIT-seq data without quantification adjustment exhibited a weak correlation to the Illumina data, while adjusted gene quantification using both full-length and partial fragments was similar to that without sequencing manipulation. Notably, target gene expression levels in PROFIT-seq data exhibited a much lower dispersion level with Illumina and control samples (Fig. 4f and Extended Data Fig. 4c). Taken together, these results indicated that PROFIT-seq was able to effectively enrich target transcripts while maintaining unbiased transcript expression levels.
Rapid pathogen detection and variant identification
To demonstrate the performance of PROFIT-seq in detecting low-abundance pathogens, a total of 16 sputum samples were collected from 8 patients with pneumonia and 8 patients infected with SARS-CoV-2. All samples were then sequenced using a modified PROFIT-seq protocol that uses only dsN and ssN to capture pathogenic RNAs without poly(A) tails. Each flow cell was divided into two sections for PROFIT-seq and control sequencing for 24 h as described above. To explore the pathogenic composition of each sample, the genomic sequences of pneumonia-related pathogens26,27 (for example, Streptococcus pneumoniae, Staphylococcus aureus and Klebsiella pneumoniae) and SARS-CoV-2 were downloaded from the RefSeq28 database as enrichment targets (Fig. 5a). Then, both non-full-length reads and cleaned consensus reads were aligned to the National Center for Biotechnology Information (NCBI) non-redundant protein sequences (NR) database using the long-read mode of diamond2 (ref. 29).

a, A schema of sample collection and target enrichment. Each flow cell was divided into two sections, where half of the pores were used to enrich target pathogens and the other half were sequenced without manipulation as a control. Adapted from Servier Medical Art by Servier under a Creative Commons license CC BY 4.0. b, The yield of total and on-target sequenced molecules in control and adaptive run. The y axis represents the number of raw reads or bases of all reads (top row) or target transcripts (bottom row). The bar colours represent control and adaptive runs, respectively. c, The coverage of SARS-CoV-2 genome during the sequencing process. The colours represent PROFIT-seq (red) or control runs (blue). The dashed lines indicate the time taken for adaptive sampling runs to achieve equivalent coverage as 24 h of control runs. d, The performance of PROFIT-seq in reducing elapsed time for SARS-CoV-2 detection. The points represent individual samples. The error bars indicate 95% confidence intervals (P = 0.0007, two-sided Wilcoxon rank sum test, n = 8 biological replicates). e, The performance of PROFIT-seq in increasing data yield for SARS-CoV-2 detection. The points represent individual samples. The error bars indicate 95% confidence intervals (P = 0.0007, two-sided Wilcoxon rank sum test, n = 8 biological replicates). f, The sensitivity of S-protein variant calling using PROFIT-seq (red) or control (blue) data. g, The composition of pathogenic bacteria in the S01 and S06 samples. h, The composition of detected pathogens in the sputum samples of patients with pneumonia using PROFIT-seq (top) and control data (bottom). The colours in g and h correspond to different target pathogens. Pneumonia and COVID-19 samples were named as P and S, respectively. Source numerical data are available in Source data.
In agreement with previous studies, the total yield of raw reads in PROFIT-seq libraries was lower than unmanipulated control8, but a substantial enrichment in the number and yield of both raw reads and consensus reads from target pathogens was observed (Fig. 5b and Extended Data Fig. 5a). Notably, the amount of on-target data generated by the PROFIT-seq runs within 6 h was equivalent to that of control runs within 24 h (Extended Data Fig. 5b–d). Moreover, the total number of pathogen-derived consensus reads generated was also increased, with an average of 3.21- and 3.57-fold enrichment in the COVID-19 and pneumonia samples, respectively (Extended Data Fig. 5e). For the patients with COVID-19, we first evaluated the diagnostic efficiency measured by the coverage of the SARS-CoV-2 genome (minimum ten supporting consensus reads). Compared with the control runs, the coverage of the SARS-CoV-2 genome detected by PROFIT-seq exhibited a more rapid and efficient increase (Fig. 5c). Specifically, PROFIT-seq required much less time (24.40%) to achieve the same coverage (Fig. 5d) and was able to generate a higher yield (3.3-fold) of target pathogenic consensus reads within the same amount of time (Fig. 5e).
In particular, PROFIT-seq runs generated higher sequencing depth for the spike (S) protein, providing the basis for identifying high-confidence mutations (Extended Data Fig. 5f). Notably, all 22 representative Omicron mutations recorded in the 2019nCoVR database30 and the key mutations of Omicron BF.7 subvariants31 (for example, R346T, L452R and F486V) were successfully identified, which is consistent with the pandemic of BF.7 subvariants in the sampling region (Extended Data Fig. 5g). Of note, compared with the control runs, the PROFIT-seq method enabled the rapid and effective detection of S-protein mutations (Fig. 5f). The sensitivity of mutation identification was saturated within 6–12 h using PROFIT-seq, which surpassed the final sensitivity of 24 h sequencing in the control runs. Interestingly, although the S01 and S06 samples contained a low abundance of SARS-CoV-2, the majority of key mutations (~80%) could still be detected, indicating the significant advantage of PROFIT-seq in detecting low-abundance pathogen transcripts with highly accurate consensus sequences.
Next, we examined the composition of target bacterial pathogens in all samples. As shown in Extended Data Fig. 5f, although a high proportion (>60%) of coronavirus was observed in most COVID-19 samples, S01 and S06 showed a distinct pathogen composition, in which >70% of assigned reads were identified as Haemophilus influenzae (Fig. 5g). The concurrent infection of H. influenzae and SARS-CoV-2 in these two samples also explained the low efficiency in detecting SARS-CoV-2 variants. For pneumonia samples, PROFIT-seq also exhibited better performance in detecting pathogenic reads (Fig. 5h), revealing the diverse source of dominant pathogens in different patients with pneumonia (for example, K. pneumoniae in P13, S. aureus in P15, S. pneumoniae in P26 and H. influenzae in others). Overall, these results suggested the widespread application of PROFIT-seq in rapid characterization of target pathogens in clinical samples.
Revealing host–microbiome association in polyp development
To further demonstrate the applicability of PROFIT-seq in characterizing complex associations in disease, we performed PROFIT-seq on 18 colorectal polyp samples including 6 inflammatory polyps, 5 low-grade intraepithelial neoplasia (LIN) and 7 high-grade intraepithelial neoplasia (HIN) samples (Fig. 6a), targeting an immune- and tumour-related gene panel covering 106.69 Mb of genomic regions. In addition, reads that were unmapped or chimerically mapped to the reference genome were also enriched to detect unannotated transcripts, including pathogen RNAs or fusion transcripts. In addition, each sample was sequenced using Illumina total RNA-seq and PROFIT-seq without enrichment targets as the control for comparative analysis.

a, A summary of the sample collection sites and the sequencing strategy. A total of 18 colorectal polyp samples from three clinical stages were collected. All samples were sequenced using Illumina total RNA-seq and PROFIT-seq with and without enriching target panels, respectively. Adapted from Servier Medical Art by Servier under a Creative Commons license CC BY 4.0. b, The number of target genes that are significantly enriched (>2-fold) (orange), minorly enriched (1- to 2-fold) (yellow) or not enriched (grey). Only genes with more than five supporting reads in both control and adaptive sampling runs were included. c, The percentage of reads in filtered regions or unannotated transcripts, which includes reads that are discordantly mapped or unmapped to the reference genome. d, Principal component analysis representation of samples with different sequencing strategies. The colours represent clinical stages, and the shapes indicate sequencing strategies. The dashed circles correspond to the 3 s.d. ellipses (P = 0.001, ANOSIM test). The P value of ANOSIM test is overlaid. e, The fraction of BCR reads identified by TRUST4. The bar colours represent Illumina total RNA-seq (grey), PROFIT-seq without enrichment (yellow) and PROFIT-seq with enrichment of the target panel (orange), and the background colours indicate different clinical stages. Significant difference between PROFIT-seq and control (P = 1.53 × 10−5, Wilcoxon signed rank test). f, The diversity of BCR hypervariable CDR3 sequences measured by CPK individual CDR3 sequences in six inflammatory, five low-grade and seven high-grade polyp samples. In the control groups, significant differences among stages were observed (P = 0.036, Kruskal–Wallis H test) with Dunn’s post hoc test revealing significant changes between inflammatory and high-grade intraepithelial neoplasia (IN) (P = 0.027), low-grade IN and high-grade IN (P = 0.030). In the PROFIT-seq groups, significant differences among stages (P = 0.017, Kruskal–Wallis H test) were also observed, with Dunn’s post hoc test showing significant changes between inflammatory and high-grade CIN (P = 0.011) and low-grade CIN and high-grade CIN (P = 0.021). g, The diversity of BCR hypervariable CDR3 sequences measured by clonality in six inflammatory, five low-grade and seven high-grade polyp samples. In the control groups, significant differences among stages (P = 0.024, Kruskal–Wallis H test) with significant changes between inflammatory and low-grade CIN (P = 0.006, Dunn’s post hoc test) were observed. In the PROFIT-seq groups, significant differences among stages (P = 0.022, Kruskal–Wallis H test) with significant changes between inflammatory and low-grade CIN (P = 0.006, Dunn’s post hoc test) were also observed. h, The usage of immunoglobulin (IG) heavy-chain isotypes in the PROFIT-seq data of 18 polyp samples. The colours represent different isotypes of the IG heavy chain. i, A heat tree representation of detected microbiotas. The colours represent the normalized change level of bacterial taxa at different ranks in 18 samples measured using the test statistic of Kruskal‒Wallis H test. j, A dot plot representation of the abundance of pathogens related to colorectal cancer. The colours within each dot represent normalized abundance across all samples. For box plots, the middle lines represent the median, and the lower and upper bounds represent the first and third quartiles. The upper and lower whiskers represent the limits of 1.5 inner quantile ranges, and points outside this range are plotted as outliers. Source numerical data are available in Source data.
For highly supported genes with more than five aligned reads in both runs, 80.79% of targets were successfully enriched by PROFIT-seq, with 26.43% of the genes over 2-fold enriched (Fig. 6b). Overall, the target gene panels were significantly enriched (Wilcoxon rank sum test, P < 0.001) in all 18 PROFIT-seq runs (Extended Data Fig. 6a). Moreover, the percentage of consensus reads that mapped to unwanted ‘filtered regions’ was decreased (Fig. 6c), indicating the effective enrichment of the target gene panel. Moreover, PROFIT-seq generated more discordantly mapped or unmapped reads and fewer unwanted reads than Illumina total RNA-seq data, which validated the advantages of our combinatorial RT strategy in capturing and enriching the full spectrum of the transcriptome. Notably, the principal component analysis of gene expression levels demonstrated that samples from the same biological groups were similar regardless of the sequencing strategy (P = 0.001, ANalysis Of SIMilarity (ANOSIM) test; Fig. 6d), indicating that the PROFIT-seq strategy was able to preserve the real biological diversity between different samples.
For immune receptor repertoire analysis, the T cell receptor (TCR) and B cell receptor (BCR) sequences were de novo assembled using TRUST4 (ref. 32) and annotated with the international ImmunoGeneTics (IMGT) database33. As expected, the effective enrichment of BCR and TCR reads (P = 1.53 × 10−5 to 0.014, Wilcoxon signed rank test) was clearly observed in the PROFIT-seq data (Fig. 6e and Extended Data Fig. 6b), whereas the percentage of both BCR and TCR reads was also significantly higher than that of canonical Illumina total RNA-seq data (P = 7.63 × 10−6, Wilcoxon signed rank test). As a result, a gradual decrease in diversity (measured by clonotypes per kilo-reads, CPK) (Fig. 6f) and increase in clonality of hypervariable complementarity-determining region 3 (CDR3) of BCRs (Fig. 6g) were observed in both PROFIT-seq and control data (P = 7.63 × 10−6, Kruskal‒Wallis H test), which was consistent with the reported depletion of B cell diversity in colon cancer against adjacent non-cancerous samples34. To determine the specific BCR isotype that is related to the malignant transformation of polyps, the usage of different immunoglobulin heavy-chain (IGH) isotypes was assessed. As shown in Fig. 6h, PROFIT-seq revealed a significant disturbance in IGHA2 (P = 0.028, Kruskal‒Wallis H test) and IGHG2 (P = 0.045, Kruskal‒Wallis H test), suggesting the switch of pathogenic microbiota during the polyp malignant transformation process35,36. Notably, the IGH isotypes and IGHV and IGHJ family usage observed in PROFIT-seq were successfully confirmed by Illumina RNA-seq data (Extended Data Fig. 6c,d). Finally, the recombination pattern of the V and J segments was investigated. Although the combination varied in different samples, the relative usage of the top-utilized combination was more centralized in the high-grade intraepithelial neoplasia groups (Extended Data Fig. 6e), consistent with the decreasing diversity of BCR in low- and high-grade intraepithelial neoplasia samples.
Taking advantage of the ability to capture the full spectrum of the transcriptome, we further investigated the microbiota composition during the transition of polyps to neoplasia (Fig. 6i). Overall, the occurrence of common intestinal microbiota was detected, and a reduced alpha diversity of the polyp microbiome, although not significant (P = 0.17, Kruskal‒Wallis H test), was observed in the PROFIT-seq data (Extended Data Fig. 6f). In particular, the abundance of Fusobacterium, whose infection in colorectal cancer has been widely reported37, was significantly increased in high-grade intraepithelial neoplasia (P = 0.004, Dunn–Bonferroni post hoc test; Fig. 6k). Other colorectal cancer-related gut bacteria, including Bacteroides, Firmicutes, Enterococcus, Escherichia, Proteobacteria and Streptococcus38,39,40, were also effectively captured in the PROFIT-seq data. Together, the reduced microbial diversity and changes in gut microbiota composition indicated dysbiosis of the healthy microbiome during neoplasia progression. It should be noted that several fusion transcripts were successfully detected in the PROFIT-seq data. For instance, the fusion of two protein-coding genes, PCNP and RPS18 (Extended Data Fig. 6f), was frequently observed in two low-grade and two high-grade IN samples, and this PCNP–RPS18 fusion event was also reported in the ChiTaRS 5.0 database41. In summary, these results demonstrated the promising ability of PROFIT-seq to characterize the full spectrum of the transcriptome with polyadenylated and non-polyadenylated RNAs and to enrich target and unannotated transcripts in a programmable manner.
Discussion
In this study, we present the PROFIT-seq strategy for effective enriching of target transcripts while maintaining unbiased quantification of the whole transcriptome. PROFIT-seq utilizes combinatorial RT and RCA amplification (R2C2 protocol) for simultaneous profiling of polyadenylated, non-polyadenylated and circular transcripts, and provides a real-time and programmable approach for effectively enriching transcripts of interest. The in-depth performance evaluations demonstrated the potential of PROFIT-seq in rapidly and accurately identifying pathogens in pneumonia and COVID-19 clinical samples, as well as in effectively detecting complex transcriptome changes during the development of colorectal intraepithelial neoplasia.
The detection of specific transcripts is essential in clinical diagnostics, pathogen identification, variant detection and immune repertoire analysis. However, current targeted RNA-seq methods depend on complex experimental enrichment processes42,43,44, which limit their ability to simultaneously target different types of transcript. In addition, probe-based strategies1,2,4,45,46 and amplicon-based approaches3,47 rely on specific sequence panels to detect targets, which are ineffective for enriching pathogens without clearly defined targets. Furthermore, the rich information of the full transcriptome is permanently lost in these constructed libraries, introducing quantification bias and preventing the integration of targeted RNA-seq data across different targets.
To this end, PROFIT-seq overcomes these barriers in transcriptomics by enabling flexible and effective target enrichment while maintaining unbiased quantification. Utilizing a combinatorial RT strategy and nanopore adaptive sequencing, PROFIT-seq provides an easy-to-use assay for enriching target transcripts across various types without the need for complex experimental enrichment processes. Based on the ONT ReadUntil interface6, PROFIT-seq provides a user-friendly web interface that allows simplified control over time segments, pore channels, demultiplexed barcodes and target genes. This setup enables the simultaneous, programmable enrichment of multiple transcript targets.
One of the key limitations of current long-read sequencing techniques is their reliance on oligo(dT)-based RT, which preferentially captures polyadenylated mRNAs and neglects crucial non-polyadenylated transcripts, including lncRNAs, circRNAs and pathogen RNAs lacking poly(A) tails. Effective detection of these non-polyadenylated transcripts has emerged as a critical aspect of transcriptome analysis48,49,50,51. Based on our previous efforts in detecting circRNAs14, PROFIT-seq adapts a combinatorial RT strategy using double-stranded oligo(dT) and random primers as well as ssN, which enables the simultaneous detection of both non-polyadenylated and circular transcripts without biasing the expression levels of dominant mRNAs. Comprehensive evaluation using the HeLa transcriptome demonstrated that PROFIT-seq effectively captures the full spectrum of the transcriptome with unbiased quantification.
Recent advances in nanopore adaptive genomic sequencing have shown great potential for enriching target genomic regions. However, the efficiency of adaptive transcriptome sequencing using the current RNA-seq protocol is often limited by the relatively short length of RNA molecules. PROFIT-seq addresses this by incorporating the RCA amplification assay to generate concatemeric reads containing multiple passes of a single cDNA template. The extended length of RCA-amplified cDNAs provides the basis for generating a discriminative difference in the length of fully sequenced transcripts and rejected fragments, thereby enhancing enrichment efficiency. Recognizing the potential for RCA to produce chimeric artefacts52, we implemented a computational filter based on subread length and similarity to minimize these artefacts in the consensus sequences (Extended Data Fig. 3g). Extensive evaluations using both simulated data and real clinical samples confirmed that PROFIT-seq effectively enriches target mRNA and pathogen gene panels. For example, in the diagnosis of SARS-CoV-2 infection, PROFIT-seq reduced sequencing time by ~75% or achieved a 3.3-fold increase in yield, while maintaining high accuracy and sensitivity in detecting key S-protein mutations. In addition, PROFIT-seq successfully profiled immune repertoire sequences in colorectal polyp samples, further demonstrating its accuracy and applicability in the field of rapid pathogen surveillance and transcriptome-based clinical applications.
An important advantage of PROFIT-seq is its ability to achieve unbiased quantification of both targeted and untargeted transcripts. By leveraging adaptive sequencing methods, which enrich target transcripts while recording all sequenced molecules, we employed the EM algorithm to integrate both full-length and partially recovered fragments, thereby obtaining corrected expression values. Comparative analyses with unselected control runs and Illumina total RNA-seq showed a high correlation in quantification results, preserving the true biological diversity between sample groups and underscoring the utility of PROFIT-seq.
In summary, PROFIT-seq effectively captures polyadenylated transcriptomes while enabling targeted enrichment or depletion of specific RNAs. Its capabilities make it a valuable tool for rapid diagnosis, targeted RNA-seq and transcriptome-based clinical applications. PROFIT-seq can be scaled with high-throughput sequencing platforms such as PromethION to increase yield, further enhancing its versatility across a range of clinical diagnostic scenarios. This method will provide critical insights into the complex interactions between host and pathogen transcriptomes during disease development.
Methods
Ethics statement
The 16 sputum and 18 colorectal polyp samples were collected from the China–Japan Union Hospital of Jilin University. The clinical information of the patients is presented in Supplementary Table 1. The clinicopathological features of colorectal polyp samples were confirmed by pathologists. The study was approved by the Ethics Committee of the China–Japan Union Hospital of Jilin University (no. 20220628006), Informed consent was obtained from all donors before enrolling in the study. All sample collection processes were part of routine diagnostic or surgical procedures, and no patients received financial compensation.
Cell culture
HeLa cells (#CCL-2, ATCC) were cultured in Dulbecco’s Modified Eagle Medium (Gibco) containing 10% foetal bovine serum (Gibco) at 37 °C in an incubator containing 5% CO2.
RNA extraction and ribosomal RNA depletion
Total RNA was extracted using TRIzol-LS (Invitrogen, 10296010) for sputum samples and TRIzol (Invitrogen, 15596018) for HeLa cells and polyp samples. The integrity of RNAs was confirmed using an Agilent 5200 Bioanalyzer. Ribosomal RNAs were removed using the KAPA RiboErase Kit Human/Mouse/Rat (KAPA Biosystems, KK8481) according to the manufacturer’s instructions.
Combinatorial RT
The extracted RNA was reverse transcribed using the SMARTer PCR cDNA Synthesis Kit (Clontech, 634925 and 634926) with minor modifications. To capture both polyadenylated and non-polyadenylated RNAs, three types of RT oligo, that is, (1) double-stranded oligo(dT) (RT.dT.ds, oligos: 5′-CTACACGACGCTCTTCCGATCTTTTTTTTTTTTTTTTTTTTTTVN-3′ and 5′-AGATCGGAAGAGCGTCGTGTAG-3′), (2) dsN (RT.N6.ds, oligos: 5′-CTACACGACGCTCTTCCGATCTNNNNNN-3′ and 5′-AGATCGGAAGAGCGTCGTGTAG-3′) and (3) ssN (RT.N6.ss, oligo: 5′-CTACACGACGCTCTTCCGATCTNNNNNN-3′), were used.
Here, denatured RNA was first incubated to RT.dT.ds oligos with NEBNext Quick Ligation Module (NEB, E5056) for 10 min at 25 °C in a 15 μl mixture containing 10 μl of RNA, 3 μl of 10× NEBNext Quick Ligation Reaction Buffer, 0.5 μl of RT.dT.ds oligos (1.4 µM) and 1.5 μl of T4 DNA Ligase. Then, 0.5 μl of RT.N6.ds oligos (1.4 µM) was added to the mixture and incubated for another 10 min at 25 °C. Subsequently, a mixture containing 10 μl of nuclease-free water, 8 μl of 5× first-strand buffer, 2 μl of dithiothreitol (100 mM), 2 μl of dNTP (10 mM), 1 μl of SMARTer II A oligo (12 μM), 1 μl of RNase inhibitor and 1 μl of SMARTer reverse transcriptase (100 U) was added to the samples followed by incubation at 42 °C for 60 min. Next, another 0.5 μl of RT.N6.ss (50 µM) was added to the reaction and incubated at 25 °C for 10 min, 42 °C for 60 min and then 70 °C for 10 min. The first strand of cDNA was purified with Agencourt AMPure XP magnetic beads (Beckman, A63880). To obtain sufficient cDNA product, PCR amplification was performed using 5 μl of cDNA with LongAmp Taq 2× Master Mix (NEB, M0287) and both the forward (5′-AAGCAGTGGTATCAACGCAGAGTAC-3′) and reverse primers (5′-CTACACGACGCTCTTCCGATCT-3′) (95 °C for 3 min; followed by 14–15 cycles of 95 °C for 10 s, 60 °C for 20 s and 65 °C for 60 s; with a final extension at 65 °C for 1 min). The PCR product was purified with AMPure XP beads with a 0.8× bead-to-sample ratio. For sputum samples, only RT.N6.ds and RT.N6.ss were added to capture bacterial RNAs that lack poly(A) tails, and 18–20 cycles of PCR amplification were performed to obtain sufficient cDNA product.
Indexed combinatorial RT
To evaluate the effect of individual primers, different index sequences were added to three types of RT oligo: (1) double-stranded oligo(dT) (RT.dT.ds, oligos: 5′-CTACACGACGCTCTTCCGATCTAACGTGATCGCTGATCTTTTTT TTTTTTTTTTTTTTTVN-3′ and 5′-GATCAGCGATCACGTTAGATCGGAAGAG CGTCGTGTAG-3′), (2) dsN (RT.N6.ds, oligos: 5′-CTACACGACGCTCTTCCGATCTCAGATCTGAGTACAAGNNNNNN-3′ and 5′-CTTGTACTCAGTCTGAGATCGGAAGAGCGTCGTGTAG-3′) and (3) ssN (RT.N6.ss, oligo: 5′-CTACACGACGCTCTTCCGATCTGATAGACACAATGGAANNNNNN-3′). Then, combinatorial RT was performed as previously described. The indexed combinatorial libraries were constructed using the SQK-LSK114 ligation sequencing kit and sequenced using R10.4.1 (FLO-PRO114M) flow cell on an ONT PromethION 24 sequencer from Beijing Huigene Biotechnology. The R10.4.1 data were basecalled using dorado 0.7.2 with dna_r10.4.1_e8.2_400bps_sup@v4.1.0 model. The products of each primer were identified by aligning the index sequences to the front and end 150 bp sequences.
Splint-based circularization
The cDNA libraries were constructed using the R2C2 protocol13. Briefly, the amplified cDNA was circularized with the DNA splint using the 2× NEBuilder HIFI DNA Assembly Master Mix (NEB, E2621S). First, ~100 ng of cDNA was mixed with 100 ng of DNA splint (5′-AGATCGGAAGAGCGTCGTGTAGTGAGGCTGATGAGTTCCATANNNNNTATATNNNNNATCACTACTTAGTTTTTTGATAGCTTCAAGCCAGAGTTGTCTTTTTCTCTTTGCTGGCAGTAAAAGTATTGTGTACCTTTTGCTGGGTCAGGTTGTTCTTTAGGAGGAGTAAAAGGATCAAATGCACTAANNNNNTATATNNNNNGCGATCGAAAATATCCCTTTAAGCAGTGGTATCAACGCAGAG-3′). The total volume of cDNA and DNA splint was adjusted to 10 μl, and the same volume of NEBuilder HiFi DNA Assembly Master Mix was added. The circularization reaction was incubated at 50 °C for 60 min. Non-circularized DNA was digested using 0.3 μl of Exonuclease III (NEB, M0206L) and 1 μl of Exonuclease I (NEB, M0293L) at 37 °C for 60 min. Circularized DNA was purified using AMPure XP beads and eluted in 20 μl of elution buffer (10 mM Tris, pH 8.0).
RCA and nanopore sequencing library construction
Circularized DNA was amplified using RCA in a 4 × 50 μl reaction containing 5 μl of 10× Phi29 buffer, 2.5 μl of 10 mM dNTP, 1 μl of exonuclease-resistant random hexamers NNNN*N*N (100 μM) with two 3′-terminal phosphorothioate modifications, 0.5 μl of bovine serum albumin, 2 μl of DNA, 29 μl of ultrapure water and 1 μl of Phi29 DNA polymerase (NEB, M0269L). Reactions were incubated at 30 °C overnight. Every two reactions were pooled, and DNA was extracted using AMPure XP beads with a 0.5 bead-to-sample ratio. For debranching of the RCA products, the beads were washed two times with 75% ethanol, and T7 Endonuclease I (NEB: M0302L) reaction mix containing 52 μl of ultrapure water, 6 μl of NEB buffer 2 and 2 μl of T7 Endonuclease I (10 units μl−1) was added to the beads. The beads were resuspended and incubated on a thermal shaker at 37 °C for 40 min under constant agitation at 1,800 rpm to debranch the RCA product. The beads were then placed on a magnet, and the DNA in the supernatant was extracted again using AMPure XP beads with a 0.5 bead-to-sample ratio. DNA fragments with more than 7 kb were size-selected on a BluePippin system (0.75% (w/v) agarose gel cassette, dye-free, S1 marker, low range, 1–10 kb).
The nanopore sequencing library was constructed following the ligation sequencing gDNA—native barcoding (LSK-109 with EXP-NBD104/114) protocol. All constructed libraries were sequenced with FLO-MIN106 (R9.4.1) flow cells using the ONT MinION device.
Programmable sequencing using PROFIT-seq
The MinKNOW (v21.11.9) interface and PROFIT-seq program were used to control the sequencing process. PROFIT-seq requires the path to reference sequences as input and will start a flask web server for input enrichment jobs and monitor the sequencing process. Users are allowed to specify parameters including manipulation mode (enrichment, depletion or balancing coverage), job name, start time, job duration, channel range, barcode names and target regions or gene names directly. In addition, users can upload a configuration file for batch job submission. Manipulation of reads that are multi-mapped or unmapped to the reference sequences can also be specified in the configuration file. After submission of each enrichment job, the enrichment targets and sequencing progress will be presented on the webpage. The start and termination of the enriching program can also be directly controlled from the webpage.
During the sequencing process, the PROFIT-seq program uses the read-until6 (v3.0.0) package to collect chunk data with an interval of 0.4 s. Then, collected raw data were basecalled in real time using the R9.4 fast basecalling model with guppy server (v5.1.15) and ont-pyguppy-client-lib (v5.1.15). The basecalled sequences were aligned to the reference genome using mappy (v2.17), a Python interface to minimap2 (ref. 53). Then, PROFIT-seq determines whether each sequence should be continued to be sequenced or rejected according to the time segment, channel number, demultiplexed barcode and aligned region provided in the job configuration, and the status of each pore is manipulated using the ReadUntilClient function. The information and decision of each basecalled chunk data point are recorded in the MinKNOW output directory. In this study, all sequencing runs were performed on a Dell Precision T5820 desktop workstation with an Intel Xeon W-2133 (six-core) processor, 64 G of random-access memory and one GeForce RTX 2080 graphic card.
Simulation data
To simulate the sequencing process, nanopore bulk fast5 of genomic DNA (http://s3.amazonaws.com/nanopore-human-wgs/rel6/MultiFast5Tars/FAF14035-3976726082_Multi_Fast5.tar) and transcriptome (https://s3.amazonaws.com/nanopore-human-wgs/rna/bulkFiles/PLSP61583_20171106_FAH20412_MN18458_sequencing_run_Notts_cDNA_Run1_96243.fast5) data were downloaded from the Oxford Nanopore Human Reference Datasets54,55 (https://github.com/nanopore-wgs-consortium/NA12878). For simulation of the PROFIT-seq library, we first constructed a PROFIT-seq library using the combinatorial RT and RCA with HeLa RNAs, and then the library was sequenced without adaptive sequencing (FAR58149) as control. During the process, all current signals were recorded in a bulk fast5 file, which could be used for simulation using the playback function of the MinKNOW software.
In the playback process, the continuous raw signal of a complete read is split into two small reads when an ‘unblock’ signal is received, so that the next read still contains similar sequence to the rejected reads. In real nanopore adaptive sequencing runs, the next read should be randomly selected. Thus, only completed reads (end reason ‘signal positive’ in older versions of MinKNOW or ‘read completed’ in the latest version) were kept for further analysis.
PROFIT-seq data analysis
The raw nanopore sequencing data were basecalled and demultiplexed using ont-guppy (v5.1.15) using the R9.4 high accuracy model. The sequencing adapters were trimmed from basecalled reads using a modified version of Porechop (https://github.com/artic-network/Porechop). Next, the Parasail56 library was used to align splint adapters and determine the boundaries of RNA sequences, whereas sequences with intact 5′ and 3′ flanking adapters were determined as full-length RNAs. The consensus sequence of captured RNA was computed using the adaptive banded partial order alignment57. The consensus sequence of splint adapters was also calculated to identify unique molecular identifier sequences for each consensus sequences. To mitigate chimeric RCA artefacts, all subreads’ lengths were required to fall within 80–120% of the consensus sequence, with subread similarity exceeding 50%. Finally, the consensus sequence was oriented according to the strandness of the detected adapter sequence, and the poly(A) tails were trimmed to reduce the false-positive alignment rate of genomic A-rich regions. Finally, the overlap between consensus sequences was identified using minimap2 with the ‘-x ava-ont’ options. Then, consensus reads with >80% consensus sequence overlaps and >80% unique molecular identifier similarity were considered as RCA duplicates and removed from downstream analysis. The deduplicated consensus sequences were referred to as ‘consensus reads’, and nanopore reads that do not contain more than one complete pass of cDNA are removed from most downstream analyses except for whole-transcriptome quantification.
For nanopore sequencing data analysis, the GRCh38.p13 human reference genome and GENCODE18 Release 37 annotation were downloaded from the GENCODE project. The cleaned reads were aligned to the reference genome using minimap2 (ref. 53) (v2.17) with the ‘-x splice’ option, and StringTie2 (ref. 24) (v2.1.5) was applied for full-length transcript reconstruction. Considering that partially sequenced reads can better represent the relative abundance of non-target transcripts, PROFIT-seq uses a modified EM algorithm by taking into account partially sequenced reads to estimate the real abundance of all transcripts. For a panel of \(n\) target transcripts, the relative expression level \({p}_{i}\) of transcript \(i\) is estimated as
where \({s}_{i,{{\mathrm{fl}}}}\) represents the number of full-length reads assigned to isoform \(i\) using Salmon25 (v0.14.0). Then, the maximization step is performed by combining the assigned full-length reads and partially sequenced reads. The number of final assigned reads \({s}_{i}\) for target transcript \(i\) is estimated by
where \({s}_{i,{{\mathrm{fl}}}+{{\mathrm{nonfl}}}}\) represents the total number of assigned full-length and partial reads. Finally, the expression level of target transcripts is measured by the counts per million as
where \({s}_{j}\) represents the number of partial reads assigned to a non-target transcript \(j\). Similarly, the expression values of non-target transcripts are calculated using
In addition, FusionSeeker58 (v1.0.1) was used to identify fusion transcripts from the alignment bam. For circRNA analysis, CIRI-long14 (v1.1.0) was employed to identify circRNAs from the consensus reads. For estimation of read accuracy, consensus reads were aligned to the reference genome using minimap2 with the option ‘-x splice--cs’, and the per read accuracy was calculated from the reported cs string. For RCA chimeric analyses, subreads for called consensus sequences were extracted and aligned to the reference genome using minimap2 with option ‘-x splice’. The RCA chimeric reads were determined if subreads from the same RCA concatemers were aligned to different genomic regions with a distance larger than 1 kb.
To estimate the abundance of microbiota in sputum and polyp samples, reads that could not be mapped to the reference genome were extracted using the SAMtools59 fastq command with the ‘-f 4’ option. The unmapped reads were aligned against the NCBI NR database using the frame-shift alignment mode of DIAMOND29 (v2.0.11) with the 1 × 10−6 e-value cutoff. In addition, bacterial reads were also identified using Kraken2 (ref. 60) (v2.0.9) with a confidence level of 0.05. For SARS-CoV-2, the NC_045512.2 reference genome61 was downloaded from the NCBI database, and cleaned reads were aligned to the SARS-CoV-2 genome as described above. Finally, bcftools59 was used for variant calling, and mutations with >80% allele frequency were kept and annotated using the ANNOVAR62 program.
For analysis of the immune receptor repertoire, TRUST4 (ref. 32) (v1.0.7) was used to identify TCR and BCR sequences. The reference immunoglobulin (IG) and TCR sequences were downloaded from the international IMGT information system33.
Illumina RNA-seq and data analysis
To construct the Illumina RNA-seq libraries, total RNA was extracted as described above. The total RNAs were shipped to Beijing Annoroad Gene Technology and sequenced using an Illumina NovaSeq 6000 sequencer according to the manufacturer’s instructions. In summary, a total of 854,012,543 paired-end 150 bp reads were generated with an average size of 14.23 Gb of data for each library.
For analysis of Illumina RNA-seq data, raw sequencing reads were assessed using FastQC (v0.11.9). Quality control was performed using TrimGalore (v0.6.6) with the ‘--stringency 6’ option. The cleaned reads were mapped to the GRCh38 reference genome using HISAT63 (v2.1.0) with default parameters. Then, StringTie2 (ref. 24) and Salmon25 were also employed for transcript assembly and quantification. All comparable analysis of Illumina libraries was performed at the gene level. The bacterial reads were identified using kraken2 (ref. 60) (v2.0.9) with a confidence level of 0.05. The TRUST4 (ref. 32) (v1.0.7) pipeline was used to identify TCR and BCR sequences as described for the nanopore sequencing data analysis.
Statistics and reproducibility
For evaluation of the combinatorial RT strategy, two experimental replicates are performed to ensure the reproducibility of results. For the assessment of PROFIT-seq protocol, two biological replicates were conducted to ensure the reproducibility of evaluation results. A total of 16 sputum samples (8 patients with pneumonia and 8 patients with COVID-19) and 18 colorectal polyp samples (6 inflammatory polyps, 5 low-grade intraepithelial neoplasia and 7 high-grade intraepithelial neoplasia) were collected to ensure the reproducibility of results and demonstrate the application of PROFIT-seq under different scenarios. No statistical method was used to pre-determine sample size, and no data were excluded from the analyses.
Protocol
A step-by-step protocol for the experimental and computational procedure to perform PROFIT-seq can be found in the protocols.io repository64.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Illumina RNA-seq and PROFIT-seq data generated in this study have been deposited in the Genome Sequence Archive in National Genomics Data Center65, China National Center for Bioinformation (GSA-Human: HRA003930) and are publicly accessible at https://bigd.big.ac.cn/gsa-human. The generated HeLa RNA-seq data are available in the SRA database under accession number PRJNA1133093. Details of these datasets are included in Methods and Supplementary Tables 1 and 2. Source data have been deposited in Zenodo at https://doi.org/10.5281/zenodo.12697811 (ref. 66). The human reference genome GRCh38.p13 (GENCODE Release 37) was downloaded from the GENCODE18 project (https://gencodegenes.org). The NC_045512.2 SARS-CoV-2 reference genome61 was downloaded from the NCBI database. The reference immunoglobulin and TCR sequences were downloaded from the international IMGT information system33. The tumour-related genes, lncRNAs and circRNAs were downloaded from COSMIC20, lnCAR21, CSCD2 (ref. 23) and MiOncoCirc22, respectively. The previously published datasets including poly(A) and total HeLa transcriptome (SRR3476958, SRR3479116, SRR1637089 and SRR1637090)15, HeLa direct RNA-seq (SRR24298524, SRR24298525 and SRR24298526)17 and poly(A) plus/minus RNA-seq (SRR067391, SRR067392, SRR067393, SRR067394, SRR067395 and SRR067396)16 were downloaded from the SRA database and re-analysed in this study. Bulk fast5 of genomic DNA (FAF14035) and cDNA (MN18458) sequencing runs were downloaded from the Oxford Nanopore Human Reference Datasets (https://github.com/nanopore-wgs-consortium/NA12878)54,55. Source data are provided with this paper.
Code availability
PROFIT-seq is implemented in Python and can be freely accessed on GitHub at https://github.com/bioinfo-biols/PROFIT-seq. The software is packaged with sample datasets and has been extensively tested with the latest version of MinKNOW (v23.04.6) on Linux. The detailed software installation guide has been included in our GitHub repository at https://github.com/bioinfo-biols/PROFIT-seq/wiki.
References
Mercer, T. R. et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat. Protoc. 9, 989–1009 (2014).
Sheynkman, G. M. et al. ORF Capture-Seq as a versatile method for targeted identification of full-length isoforms. Nat. Commun. 11, 2326 (2020).
Scolnick, J. A., Dimon, M., Wang, I. C., Huelga, S. C. & Amorese, D. A. An efficient method for identifying gene fusions by targeted RNA sequencing from fresh frozen and FFPE samples. PLoS ONE 10, e0128916 (2015).
Wang, F. et al. TEQUILA-seq: a versatile and low-cost method for targeted long-read RNA sequencing. Nat. Commun. 14, 4760 (2023).
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365 (2021).
Loose, M., Malla, S. & Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 13, 751–754 (2016).
Kovaka, S., Fan, Y., Ni, B., Timp, W. & Schatz, M. C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. 39, 431–441 (2021).
Payne, A. et al. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 39, 442–450 (2021).
Weilguny, L. et al. Dynamic, adaptive sampling during nanopore sequencing using Bayesian experimental design. Nat. Biotechnol. 41, 1018–1025 (2023).
Edwards, H. S. et al. Real-time selective sequencing with RUBRIC: Read Until with Basecall and Reference-Informed Criteria. Sci. Rep. 9, 11475 (2019).
Ulrich, J. U., Lutfi, A., Rutzen, K. & Renard, B. Y. ReadBouncer: precise and scalable adaptive sampling for nanopore sequencing. Bioinformatics 38, i153–i160 (2022).
Volden, R. & Vollmers, C. Single-cell isoform analysis in human immune cells. Genome Biol. 23, 47 (2022).
Volden, R. et al. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl Acad. Sci. USA 115, 9726–9731 (2018).
Zhang, J. et al. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat. Biotechnol. 39, 836–845 (2021).
Gao, Y. et al. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat. Commun. 7, 12060 (2016).
Yang, L., Duff, M. O., Graveley, B. R., Carmichael, G. G. & Chen, L. L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 12, R16 (2011).
Tavakoli, S. et al. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 14, 334 (2023).
Frankish, A. et al. Gencode 2021. Nucleic Acids Res. 49, D916–D923 (2021).
Cole, C., Byrne, A., Adams, M., Volden, R. & Vollmers, C. Complete characterization of the human immune cell transcriptome using accurate full-length cDNA sequencing. Genome Res. 30, 589–601 (2020).
Tate, J. G. et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Zheng, Y. et al. lnCAR: a comprehensive resource for lncRNAs from cancer arrays. Cancer Res. 79, 2076–2083 (2019).
Vo, J. N. et al. The landscape of circular RNA in cancer. Cell 176, 869–881.e813 (2019).
Feng, J. et al. CSCD2: an integrated interactional database of cancer-specific circular RNAs. Nucleic Acids Res. 50, D1179–D1183 (2022).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Wu, B. G. & Segal, L. N. The lung microbiome and its role in pneumonia. Clin. Chest Med. 39, 677–689 (2018).
Thibeault, C., Suttorp, N. & Opitz, B. The microbiota in pneumonia: from protection to predisposition. Sci. Transl. Med. 13, eaba0501 (2021).
O'Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Song, S. et al. The global landscape of SARS-CoV-2 genomes, variants and haplotypes in 2019nCoVR. Genomics Proteom. Bioinf. 18, 749–759 (2020).
Qu, P. et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7 and BA.2.75.2. Cell Host Microbe 31, 9–17.e3 (2023).
Song, L. et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat. Methods 18, 627–630 (2021).
Lefranc, M. P. IMGT, the International ImMunoGeneTics Information System. Cold Spring Harb. Protoc. 2011, 595–603 (2011).
Yu, K., Ravoor, A., Malats, N., Pineda, S. & Sirota, M. A pan-cancer analysis of tumor-infiltrating B cell repertoires. Front. Immunol. 12, 790119 (2021).
Spencer, J. & Sollid, L. M. The human intestinal B-cell response. Mucos. Immunol. 9, 1113–1124 (2016).
Ferrante, A., Beard, L. J. & Feldman, R. G. IgG subclass distribution of antibodies to bacterial and viral antigens. Pediatr. Infect. Dis. J. 9, S16–S24 (1990).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017).
Peters, B. A. et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome 4, 69 (2016).
Song, M., Chan, A. T. & Sun, J. Influence of the gut microbiome, diet and environment on risk of colorectal cancer. Gastroenterology 158, 322–340 (2020).
Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 21, 1325 (2021).
Balamurali, D. et al. ChiTaRS 5.0: the comprehensive database of chimeric transcripts matched with druggable fusions and 3D chromatin maps. Nucleic Acids Res. 48, D825–D834 (2020).
Jeck, W. R. & Sharpless, N. E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 32, 453–461 (2014).
Panda, A. C. et al. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 45, e116 (2017).
Xiao, M. S. & Wilusz, J. E. An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3′ ends. Nucleic Acids Res. 47, 8755–8769 (2019).
Karamitros, T. & Magiorkinis, G. A novel method for the multiplexed target enrichment of MinION next generation sequencing libraries using PCR-generated baits. Nucleic Acids Res. 43, e152 (2015).
Pinkard, O., McFarland, S., Sweet, T. & Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 11, 4104 (2020).
Yang, M. et al. Direct metatranscriptome RNA-seq and multiplex RT-PCR amplicon sequencing on Nanopore MinION—promising strategies for multiplex identification of viable pathogens in food. Front. Microbiol. 11, 514 (2020).
McKellar, D. W. et al. Spatial mapping of the total transcriptome by in situ polyadenylation. Nat. Biotechnol. 41, 513–520 (2023).
Li, X. et al. Novel method of full-length RNA-seq that expands the identification of non-polyadenylated RNAs using nanopore sequencing. Anal. Chem. 94, 12342–12351 (2022).
Salmen, F. et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat. Biotechnol. 40, 1780–1793 (2022).
Begik, O. et al. Nano3P-seq: transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing. Nat. Methods 20, 75–85 (2023).
Zhang, K. et al. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 24, 680–686 (2006).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
Workman, R. E. et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 16, 1297–1305 (2019).
Daily, J. Parasail: SIMD C library for global, semi-global and local pairwise sequence alignments. BMC Bioinf. 17, 81 (2016).
Gao, Y. et al. abPOA: an SIMD-based C library for fast partial order alignment using adaptive band. Bioinformatics 37, 2209–2211 (2021).
Chen, Y. et al. Gene fusion detection and characterization in long-read cancer transcriptome sequencing data with FusionSeeker. Cancer Res. 83, 28–33 (2023).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Hou, L. & Zhang, J. Real-time and programmable transcriptome sequencing with PROFIT-seq. Preprint at https://doi.org/10.17504/protocols.io.5jyl8p19rg2w/v1 (2024).
Chen, T. et al. The Genome Sequence Archive Family: toward explosive data growth and diverse data types. Genomics Proteom. Bioinf. 19, 578–583 (2021).
Zhang, J. Real-time and programmable transcriptome sequencing with PROFIT-seq. Zenodo https://doi.org/10.5281/zenodo.12697811 (2024).
Acknowledgements
This work was supported by grants from the National Key R&D Project (2021YFC2301300) to Z.Z., the National Natural Science Foundation of China (32025009, 32130020 and 32200530), the National Key R&D Project (2021YFA1300500, 2021YFA1302000, 2022YFC2703200 and 2022YFC2303801), the China National Postdoctoral Program for Innovative Talents (BX20220296), the China Postdoctoral Science Foundation (CPSF) (2022M723134) and the Special Research Assistant of the Chinese Academy of Sciences. The illustrations in Fig. 5a and Fig. 6a were partly generated using templates from Servier Medical Art (https://smart.servier.com/).
Author information
Authors and Affiliations
Contributions
F.Z. conceived the project. J.Z. implemented the algorithm and performed data analysis. L.H., Z.C., P.J. and Z.Z. performed the experiments and generated sequencing data. L.M., S.Y. and Y.L. collected clinical samples. J.Z. and F.Z. wrote the manuscript with the contribution of all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Functions of PROFIT-seq.
a, Example of a TOML-formatted configuration file for batch job submission. b, Schematic representation of chunk data processing. c, PROFIT-seq provides flexible functions to customize the sequencing duration, channel number, demultiplexed barcode, and aligned region for each individual task. d, Length of basecalled sequences (left), mapping rate (middle), and number of reads with different end reasons (right). All molecules were set to be rejected, and runs were simulated with different chunk acquisition intervals ranging from 0.1 to 1.0 s. Colors in the right panel represent different end reasons, including adaptive sampling voltage reversal (purple), read completed (green), device changed mux (blue), read became blocked (red) and unblock voltage reversal (orange). The middle lines represent the median, and the lower and upper bounds represent the first and third quartiles, respectively. The upper and lower whiskers represent the limits of 1.5 inner quantile ranges, and points outside this range are plotted as outliers. Source numerical data are available in source data.
Extended Data Fig. 2 Combinatorial RT enables effective capture of the full transcriptome spectrum.
a, Percentage of reads aligned to different genomic regions. Colors represent different genomic regions according to the GENCODE v37 annotation. b, Percentage of ribosomal RNA reads in PROFIT-seq and oligo(dT) data. c, Bar plots indicate the fraction of reads from non-poly(A) containing transcript in oligo(dT) and random primed cDNAs in HeLa cells. Colors indicate oligo(dT) (orange) and random primed (blue) reads, respectively. d, Proportion of reads aligned to transcripts of different biotypes according to the GENCODE annotation as a supplementary to Fig. 2d. Colors represent PROFIT-seq (red) and oligo(dT) (grey) libraries respectively. e, Log scaled gene expression levels in oligo(dT), combinatorial RT, and random primed cDNA libraries. The x-axis represents the log scaled gene expression levels from Illumina total RNA-seq dataset. Source numerical data are available in source data.
Extended Data Fig. 3 Accurate reconstruction of target transcripts using PROFIT-seq.
a, Expression level of target genes in adaptive sampling runs and unmanipulated controls. R = 0.87, P < 10-31, Pearson correlation test. b. Accuracy of consensus reads grouped by the copy number of full-length repetitive cDNA segments. All consensus reads were aligned to the reference genome using minimap2 with option ‘-x splice’, and the accuracy and error rate were calculated from the reported CIGAR values of 500 randomly subsampled reads. The middle lines of the boxes indicate the median and the lower and upper bounds represent the first and third quartiles. The upper and lower whiskers represent the limits of 1.5 inner quantile ranges, and points outside this range are plotted as outliers. c, Percentage of consensus reads generated by the PROFIT-seq script and C3POa (v3.1) using different parameter sets. d, Overlap between the results of PROFIT-seq script and C3POa. e, Per read accuracy of the PROFIT-seq script and C3POa results. Consensus reads were aligned to the reference genome using minimap2 with the option ‘-x splice–cs’, and the per read accuracy was calculated from the reported cs string. f, Run time for the PROFIT-seq script and C3POa. Colors indicates different tools. P = 0.02, Wilcoxon rank sum test. g, The percentage of RCA chimeric reads in PROFIT-seq and C3POa results. All subreads were extracted and aligned to the reference genome using minimap2 with option ‘-x splice’. The RCA chimeric reads were determined if subreads from the same RCA concatemers were aligned to different genomic region with a distance larger than 1 kb. Compared to C3POa, a significant lower chimeric rate of PROFIT-seq was observed in default mode (P = 0.02, Wilcoxon rank sum test). Source numerical data are available in source data.
Extended Data Fig. 4 An EM-based quantification strategy to reduce bias in quantification.
a, Percentage of full-length reads for the target (right) and non-target (left) transcripts in the JL19 sample. b, Expression levels of the target (red) and non-target (grey) genes before EM-based correction (left) or after correction (right) in the JL19 sample. c, Correlation of target gene expression levels for Illumina total RNA-seq, control runs without pore manipulation, and PROFIT-seq data with/without EM-based quantification correction. Colors represent the correlation coefficient calculated using the spearman correlation test. Source numerical data are available in source data.
Extended Data Fig. 5 Effective enrichment of target pathogens using PROFIT-seq.
a, The yield of total and on-target consensus reads in control and adaptive run. The y-axis represents number of raw reads or bases of all reads (top row) or target transcripts (bottom row). Bar colors represents control and adaptive runs respectively. b, The absolute number of assigned consensus reads for each pathogen in PROFIT-seq and control runs of the 16 sputum samples. Colors represent different stages of a 6-hour window during the 24-hour run. c-d, Number of reads assigned to target pathogens during sequencing of pneumonia (c) and COVID-19 (d) samples. Colors represent PROFIT-seq (red) or control runs (blue) respectively. e, Enrichment efficiency of target pathogens in COVID-19 and pneumonia samples. f, Tracks of sequencing depth of the spike protein in the SARS-CoV-2 genome. The genome structure and spike protein domains are indicated above the tracks. Colors represent PROFIT-seq (red) or control runs (blue) respectively. g, Heatmap presentation of the S-protein variants detected in 8 COVID-19 samples. Colors indicate the number of supporting reads for each mutation. h, Composition of pathogenic microbiota in 8 sputum samples of COVID-19 patients using PROFIT-seq (upper) and control data (bottom). Source numerical data are available in source data.
Extended Data Fig. 6 Changes of the immune repertoire in inflammatory polyps and low- or high-grade IN samples.
a, Expression level of target genes in adaptive sampling runs and unmanipulated controls. The x and y axis represent the expression level of target genes, the dashed lines represent the threshold of five supporting reads in control and adaptive sampling runs, respectively. P < 0.001, Wilcoxon rank sum test. b, Fraction of T cell receptor (TCR) reads identified by TRUST4. Bar colors represent Illumina total RNA-seq (gray), PROFIT-seq without enriching (yellow), and PROFIT-seq enriching target panel (orange), and background colors indicate different clinical stages. Significant difference between PROFIT-seq and control, P = 0.014, Wilcoxon signed rank test. c, Usage of IG heavy chain isotypes in Illumina total RNA-seq of 18 polyp samples. Colors represent different isotypes of IG heavy chains. d, Utilization of 7 IGHV and 6 IGHJ families in PROFIT-seq and Illumina total RNA-seq data. The middle lines represent the median and the lower and upper bounds represent the first and third quartiles. The upper and lower whiskers represent the limits of 1.5 inner quantile ranges, and points outside this range are plotted as outliers. e, Heatmap presentation of VJ combinations in all samples. Colors indicate the relative utilization values normalized across 18 samples. f, Alpha diversity of colorectal polyp microbiome in different stages. g, Schematic view of the PCNP-RPS18 fusion event. The supporting reads are indicated below the gene structure. Source numerical data are available in source data.
Supplementary information
Supplementary Tables
Supplementary Table 1. Sample metadata of 18 colorectal polyp samples. Supplementary Table 2. Sample metadata of 16 sputum samples. Supplementary Table 3. Flowcell metrics of PROFIT-seq libraries.
Source data
Source Data Figs. 1–6 and Extended Data Figs. 1–6
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Hou, L., Ma, L. et al. Real-time and programmable transcriptome sequencing with PROFIT-seq. Nat Cell Biol 26, 2183–2194 (2024). https://doi.org/10.1038/s41556-024-01537-1
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41556-024-01537-1
This article is cited by
-
Circular RNA discovery with emerging sequencing and deep learning technologies
Nature Genetics (2025)
