
Abstract
Multidrug-resistant bacterial pathogens like vancomycin-resistant Enterococcus faecium (VREfm) are a critical threat to human health1. Daptomycin is a last-resort antibiotic for VREfm infections with a novel mode of action2, but for which resistance has been widely reported but is unexplained. Here we show that rifaximin, an unrelated antibiotic used prophylactically to prevent hepatic encephalopathy in patients with liver disease3, causes cross-resistance to daptomycin in VREfm. Amino acid changes arising within the bacterial RNA polymerase in response to rifaximin exposure cause upregulation of a previously uncharacterized operon (prdRAB) that leads to cell membrane remodelling and cross-resistance to daptomycin through reduced binding of the antibiotic. VREfm with these mutations are spread globally, making this a major mechanism of resistance. Rifaximin has been considered ‘low risk’ for the development of antibiotic resistance. Our study shows that this assumption is flawed and that widespread rifaximin use, particularly in patients with liver cirrhosis, may be compromising the clinical use of daptomycin, a major last-resort intervention for multidrug-resistant pathogens. These findings demonstrate how unanticipated antibiotic cross-resistance can undermine global strategies designed to preserve the clinical use of critical antibiotics.
Similar content being viewed by others


Main
Antimicrobial resistance (AMR) is one of the greatest current public health threats, with 1.27 million deaths being attributed to bacterial AMR in 20191. Infections caused by multidrug-resistant bacteria are associated with frequent treatment failure and high rates of morbidity and mortality; the preservation of last-resort antibiotics for effective treatment is therefore of critical importance.
E. faecium is classified by the World Health Organization (WHO) as a high-priority pathogen as it is a leading cause of nosocomial infections4. The intrinsic antibiotic resistance of hospital-associated clones coupled with their ability to rapidly acquire additional antibiotic-resistance genes makes E. faecium infections increasingly difficult to treat5. In particular, strains resistant to vancomycin, the first-line antibiotic for invasive infections, have emerged and disseminated globally due to the acquisition of transferable van resistance genes6.
The lipopeptide daptomycin (DAP) is a WHO-designated last-resort antibiotic that is used ‘off label’ to treat invasive VREfm infections7. The increasing reports of DAP-resistant (DAP-R) VREfm are therefore of clinical concern. In VREfm, DAP resistance is thought to be largely driven by mutations in the regulatory systems LiaFSR/LiaXYZ and in the cardiolipin synthase (Cls)8,9,10. However, many VREfm are resistant to DAP through unknown mechanisms, indicating that other molecular pathways are involved9,11,12. Accordingly, we undertook a combined genomic and phenotypic analysis to investigate the DAP resistance mechanisms in VREfm. Here we show that DAP resistance is linked to the presence of specific rpoB mutations in VREfm, with resistance emerging de novo in E. faecium after exposure to rifaximin, a commonly prescribed antibiotic that is used prophylactically to prevent hepatic encephalopathy in patients with liver disease3. We identify a major mechanism of DAP resistance in VREfm and identified rifaximin, an antibiotic considered to be low-risk for the emergence of bacterial AMR13, as an important driver of last-resort antibiotic resistance.
Australian DAP-R VREfm are polyphyletic
Daptomycin susceptibility testing was performed on VREfm isolated during two unbiased state-wide ‘snapshot’ studies undertaken in 2015 (n = 294) and 2018 (n = 423) in Victoria, Australia. The proportion of DAP-R isolates was 16.6% (n = 49) in 2015 and 15.3% (n = 65) in 2018. Given this high rate of resistance, we included VREfm isolated in 2017 (n = 108) and 2018 (n = 173)14,15, and found that 28.4% (n = 80) of these isolates were also DAP-R. Overall, a surprisingly high 194 (19.4%) of all VREfm were DAP-R.
To investigate the relationship between DAP-R and DAP-susceptible (DAP-S) VREfm, a maximum-likelihood phylogeny was inferred from an alignment of 6,574 core genome single-nucleotide polymorphisms (SNPs) (n = 1,000; 998 study isolates and 2 controls) (Extended Data Fig. 1a). We identified 36 sequence types (STs) within the 1,000 isolates and 30 of these STs included at least one DAP-R VREfm; thus, DAP-R was polyphyletic. The largest clade of DAP-R strains (ST203), accounted for 41.2% of resistant isolates (n = 80 out of 194) suggestive of an expanding DAP-R lineage. The other predominant VREfm STs (ST80, ST796, ST1421 and ST1424) consisted of several groups of DAP-R isolates that showed no phylogenetic clustering, suggesting that the phenotype has arisen independently on multiple occasions.
We next sought to determine the genetic determinants leading to DAP resistance in VREfm. We systematically screened VREfm genomes for mutations in the regulatory genes liaFSR, liaXYZ, yycFG (walKR), cardiolipin synthase (cls) and the division site tropomyosin-like locus (divIVA), all previously linked to DAP resistance7,8,16. Notably, there were no significant associations between DAP resistance and mutations in these loci (Supplementary Table 1).
rpoB mutations lead to DAP resistance in VREfm
To identify genetic loci linked with DAP resistance, we performed a genome-wide association study (GWAS) with the 998 E. faecium study isolates and 2 controls (Fig. 1a). This analysis identified 142 mutations (in 73 genes) significantly (P < 1 × 10−10) associated with DAP resistance (as a binary variable with a breakpoint of 8 mg l−1). The top five significant amino acid substitutions were (1) I274S in an uncharacterized ABC efflux protein (P = 7.44 × 10−15); (2) G71S in an uncharacterized permease protein (P = 7.77 × 10−14); (3) V288L in a mannitol dehydrogenase protein (P = 6.08 × 10−12); (4) S491F in RpoB, the RNA polymerase β subunit (P = 1.57 × 10−13); and (5) T634K in RpoC, the RNA polymerase β′ subunit (P = 4.40 × 10−11).

a, Manhattan plot of 10,530 variants, displayed by their position on the reference genome and their association with DAP resistance, as determined using one-sided Fisher’s exact tests and a mixed-effects logistic regression model to correct for population structure; correction for multiple testing was performed using the Bonferroni method (dashed line). n = 998 study isolates and 2 control strains. b, The percentage of DAP-R strains with a RpoB substitution; the RRDR is shown in bold. The total number of VREfm containing each mutation is shown. c, Maximum-likelihood core-SNP-based phylogeny of clinical VREfm (n = 998 study isolates and 2 control strains) inferred from 6,574 SNPs. Overlaid are the results of in silico multi-locus sequence type (MLST), DAP phenotypic testing and RpoB substitutions associated with DAP resistance. In the first circle, ST is not shown for uncommon STs (n ≤ 5). The scale bar indicates number of nucleotide substitutions per site (top), with an approximation of SNP distance shown in parentheses. d, Rifampicin susceptibility testing results for the WT and isogenic rpoB mutants and complemented strains (designated by -C). n = 3. e, DAP susceptibility testing results for the WT and isogenic rpoB mutants and complemented strains. n = 3. The MIC for each strain is shown without error bars as there was no variation between the independent biological replicates.
To test the contribution of these amino acid substitutions to DAP resistance, we introduced each mutation into a clinical DAP-S ST796 VREfm isolate (Aus0233). None of the amino acid substitutions had any impact on DAP resistance except for the S491F substitution within RpoB, which resulted in a DAP-R phenotype (fourfold increase in DAP minimum inhibitory concentration (MIC), from 2 mg l−1 to 8 mg l−1). Notably, every clinical strain with the RpoB S491F substitution (n = 105, spanning different E. faecium genotypes) also contained the RpoC T634K substitution (n = 105), with no strains containing the RpoC T634K substitution in isolation. As the T634K substitution did not affect DAP susceptibility (MIC still 8 mg l−1), it might instead be compensatory for negative fitness effects associated with the RpoB S491F substitution. In support of this idea, in vitro competition assays (wild type (WT) versus RpoB(S491F) or WT versus RpoB(S491F)/RpoC(T634K)), showed that the RpoB S491F substitution posed a substantial fitness cost, with a significant (P < 0.0001) shift to the WT population (Extended Data Fig. 1b). However, in the RpoB(S491F)/RpoC(T634K) double mutant, the population dynamics remained stable, with no significant differences compared to the inoculum (Extended Data Fig. 1b). These data suggest the RpoC T634K substitution compensates for a negative fitness impact of RpoB S491F.
We next assessed whether other RpoB substitutions were associated with DAP resistance (Fig. 1b). We observed DAP resistance in 81.3% of VREfm strains (n = 13 out of 16) with a G482D substitution and 83.3% of VREfm strains (n = 10 out of 12) with a H486Y substitution. Isolates containing these mutations were spread across the phylogenetic tree, indicative of multiple independent acquisitions (Fig. 1c). No putative compensatory mutations in the RNA polymerase genes were identified. Clonal expansion was also observed for a dominant, DAP-R clone (ST203) containing the S491F substitution. Daptomycin-resistant isolates from this ST203 lineage were identified across ten geographically distinct hospital networks, indicating that they were not part of a singular hospital outbreak (Extended Data Fig. 1c).
The G482D, H486Y and S491F substitutions were located within the predicted rifampicin-resistance determining region (RRDR) of RpoB (spanning amino acids 467 to 493). Rifampicin susceptibility testing (rifampicin being a marker of rifamycin resistance17) confirmed that all of the isolates with a RRDR rpoB mutation had high-level rifampicin resistance (n = 169; median MIC, 256 mg l−1), while control isolates containing the WT RRDR displayed a median MIC of 8 mg l−1 (n = 169; Extended Data Fig. 1d).
To test whether the G482D and H486Y substitutions in RpoB also led to rifamycin and DAP resistance we introduced these substitutions into our rifamycin-susceptible, DAP-S, clinical strain of ST796 VREfm (Aus0233). Introduction of the G482D, H486Y or S491F RpoB substitutions resulted in high-level rifampicin resistance (>512 mg l−1) (Fig. 1d). The introduction of the G482D or H486Y RpoB substitutions also resulted in a DAP-R phenotype (fourfold increase in DAP MIC, from 2 mg l−1 to 8 mg l−1) (Fig. 1e). Reversing the mutations to recreate the WT rpoB allele resulted in susceptibility to rifampicin (MIC of 4 mg l−1) and DAP (MIC of 2 mg l−1), confirming that G482D, H486Y and S491F caused rifamycin resistance and DAP cross-resistance.
VREfm rpoB mutations are globally spread
To determine whether the RpoB substitutions associated with DAP resistance observed in Australian VREfm were representative of E. faecium globally, we analysed publicly available VREfm genome sequencing data (n = 3,476 international and n = 1,000 Australian) (Extended Data Fig. 2a). The majority (n = 3,378) of these VREfm isolates were healthcare associated, with 98 isolates from animal origin. In total, 630 (14.1%) VREfm isolates had an amino acid substitution in the RRDR of RpoB, with the S491F substitution being the most common, present in 77.9% (n = 461) of genomes with a RRDR RpoB substitution (Extended Data Fig. 2b). The S491F substitution was identified in VREfm genomes from 20 countries and across 21 different STs (Extended Data Fig. 2b,c). The G482D and H486Y substitutions were also common, found in 6.8% (n = 43) and 11.6% (n = 73) of strains with a RpoB substitution, respectively. The G482D and H486Y substitutions were also observed globally (7 and 10 countries, respectively) and across different STs (9 and 22, respectively) (Extended Data Fig. 2b,c). There was a significant association between RRDR RpoB substitutions and healthcare-associated VREfm18 (P < 0.001, Fisher’s exact test) (Extended Data Fig. 2d), suggesting that these RpoB substitutions conferring resistance to rifamycins and cross-resistance to DAP are enriched within the healthcare setting and are globally prevalent.
Rifaximin approval linked to S491F emergence
We used phylodynamic analyses to estimate the emergence date of VREfm with the S491F substitution globally. Within Australian VREfm, we observed the expansion of a dominant ST203 clone from 2015 to 2018 that carried the S491F substitution (Fig. 1c). As this clone comprised only vanA-VREfm, we genome sequenced all historical vanA-VREfm from our public health laboratory (n = 229), which consisted of every vanA-VREfm isolate collected from 2003 to 2014, to increase the potential to detect a molecular clock signal. We contextualized all Australian isolates (n = 1,229) with the international data (n = 3,476) and identified three distinct clusters containing the RpoB S491F substitution (Fig. 2a). The same ST203 clone formed the largest cluster (cluster 1; n = 219 taxa), consisting of isolates from Australia and the UK. Cluster 2 (n = 85 taxa) consisted of ST80 and ST78 isolates from Australia, Europe, South America, the UK and the USA, while cluster 3 (n = 68 taxa) consisted of ST80 isolates from Australia, Europe and the UK.

a, Maximum-likelihood, core-SNP-based phylogeny for 4,476 VREfm inferred from 9,277 core-genome SNPs, demonstrating the presence of the S491F RpoB substitution in international VREfm. Overlaid is the region of isolation for each strain and the presence of the S491F substitution. The coloured branches indicate the three VREfm clusters identified with core genome MLST (cgMLST) used as the input for Bayesian evolutionary analyses. The scale bar indicates the number of nucleotide substitutions per site (top); an approximation of SNP distance is shown in parentheses. b, Bayesian phylodynamic analyses showing the MCC trees of the three VREfm clusters with the timing (years on x axis) of emergence for each lineage. The presence of the S491F RpoB trait for each isolate is shown in purple. Overlaid onto the MCC trees is the first instance of FDA approval for rifaximin (2004) and for hepatic encephalopathy (HE) (2010). c, Violin plots of the most recent common ancestor (MCRA) for each cluster, representing when the RpoB S491F substitution first emerged, with 95% highest-posterior density (HPD) intervals—2006 (HPD 1993–2012) for cluster 1, 2000 (HPD 1989–2008) for cluster 2, and 2004 (HPD 2001–2010) for cluster 3. Overlaid onto violin plots is the FDA approval date for rifaximin (2004).
To model the evolutionary trajectories of these clusters, we used core-genome SNP diversity and year of isolation (Extended Data Fig. 3a). The substitution rate (the number of expected nucleotide substitutions per site per year) was consistent with other estimates for healthcare-associated VREfm18,19,20,21 (Fig. 2b). The year of emergence for the most recent common ancestor (MRCA) of each cluster with S491F was around 2006 (Fig. 2b), a time period that coincides with the first clinical use of rifaximin. As rifampicin was approved for clinical use in the USA in 197122, several decades before the estimated emergence of the S491F-containing VREfm strains, we considered this rifamycin unlikely to have had a major role in the spread of resistance. Analysis of the three maximum-clade credibility (MCC) trees (Fig. 2b) indicated that each E. faecium lineage has continued to expand since its emergence, consistent with the growing use of rifaximin globally, in particular since 2010 when it was approved for the prevention of hepatic encephalopathy3 (Fig. 2b,c). Notably, in all three VREfm clusters, the S491F substitution has been stably maintained after its acquisition, suggesting sustained selective pressure on the bacterial population, as rpoB mutations usually carry a fitness cost23 (Extended Data Fig. 3b–d). These data show the S491F substitution emerged in VREfm at least three times since the early 2000s, with the predicted dates of emergence closely correlated with the clinical introduction of rifaximin.
Rifaximin linked with DAP-R VREfm carriage
Rifaximin is a non-absorbable oral agent with direct antimicrobial activity in the gastrointestinal tract, predominantly prescribed to prevent recurrent hepatic encephalopathy in patients with liver cirrhosis—a cohort with frequent VREfm gastrointestinal colonization3,24,25. As the Bayesian phylodynamic analyses showed a correlation between the S491F RpoB substitution in VREfm and the use of rifaximin, we posited that rifaximin use in patients with VREfm colonization may enrich for VREfm RpoB substitutions and, therefore, DAP resistance. To test this hypothesis, we assessed the association between rifaximin exposure and DAP-R VREfm in a retrospective patient cohort at a quaternary referral healthcare centre in Melbourne, Australia (Supplementary Table 2). The E. faecium isolates from patients with current or previous rifaximin exposure (rifaximin group) and without previous exposure (control group) were assessed (1) genomically for RpoB substitutions (G482D, H486Y and S491F) that result in DAP resistance; and (2) phenotypically for DAP resistance, by study investigators blinded to patient details and the exposure groups. Genomically clustered isolates likely to represent patient-to-patient transmission were excluded from the analysis, with the included isolates being phylogenetically distributed and representative of E. faecium strains identified in state-wide surveys26 (Methods and Extended Data Fig. 4a).
Among the patient cohort colonized with VREfm, compared with unexposed patients (n = 116), patients with recent exposure to rifaximin (n = 96) were more likely to be colonized with VREfm with RpoB substitutions (P < 0.001), including substitutions associated with DAP resistance (G482D, H486Y or S491F; P < 0.001) and DAP-R VREfm (P < 0.001) (Fig. 3 and Supplementary Table 3). After adjusting for potential confounding effects, including age, sex, underlying comorbidities and exposure to other antimicrobials, recent rifaximin exposure remained an independent predictor of DAP-R RpoB substitutions (odds ratio (OR) = 8.69; 95% confidence interval (CI) = 2.95–30.84; P < 0.001) and DAP-R VREfm (OR = 6.47; 95% CI = 2.34–20.80; P < 0.001) (Supplementary Table 4a,b). Given that almost all patients who received rifaximin had underlying chronic liver disease (with hepatic encephalopathy being the predominant indication for rifaximin prescribing in Australia), we assessed the association between rifaximin and DAP resistance first within the subgroup of patients with liver disease (n = 127) (Supplementary Table 5a–c) and, second, in an independent cohort of patients without liver disease who had received rifaximin (n = 57) as antimicrobial prophylaxis after haematopoietic stem cell transplantation (HSCT) in Germany (Supplementary Table 6a–c). The association between rifaximin exposure and DAP-R RpoB substitutions was again identified in both patient groups—(1) patients with chronic liver disease (P = 0.001); and (2) HSCT patients without liver disease (P < 0.001) (Extended Data Fig. 4b and Supplementary Tables 5c and 6c). Among patients with liver disease colonized with VREfm, recent rifaximin was an independent predictor of DAP-R VREfm (OR = 4.37; 95% CI = 1.70–12.84; P = 0.004). We conducted other sensitivity analyses to verify the robustness of these associations and assess for other confounders, including a subgroup analysis that excluded patients who received rifaximin from the analysis (Supplementary Table 7), but these did not identify any other associations between DAP resistance and underlying disease or antibiotic exposure.

Summary of the percentage of VREfm with any rpoB SNP, DAP-associated rpoB SNP (S491F, G482D and H486Y) or DAP-R in patients in the control (n = 116) or rifaximin (n = 96) groups. Data were analysed using one-sided Fisher’s exact tests. Exact P values were as follows: P < 2.2 × 10−16 (any rpoB SNP); P = 3.044 × 10−14 (DAP-associated rpoB SNP) and P = 1.463 × 10−10 (DAP-R).
Six patients from the control group carried DAP-R VREfm—three (50%) of these had isolates with no mutations in rpoB, suggesting an alternative mechanism for resistance, while the remaining three patients had previously been admitted to the liver ward for inpatient care, suggesting that they may have acquired DAP-R VREfm through healthcare-associated transmission.
While only one representative VREfm isolate from each patient was included in the above analysis, the de novo emergence of DAP-R VREfm carrying the G482D RpoB substitution was observed in one patient from whom multiple isolates had been collected during rifaximin therapy, consistent with rifaximin exposure driving the de novo emergence of DAP-R VREfm.
These data showed a strong clinical association between recent rifaximin exposure and patient gastrointestinal carriage of VREfm with DAP-resistance-associated RpoB substitutions, suggesting that exposure to rifaximin drives DAP-R VREfm.
Rifaximin use leads to de novo DAP-R VREfm
We used a mouse VREfm gastrointestinal colonization model to test whether rifaximin exposure caused de novo emergence of rpoB mutations that confer cross-resistance to DAP. Mice were colonized with a DAP-S (MIC 2 mg l−1) clinical VREfm ST796 isolate (Aus0233) containing a WT rpoB allele before being administered a human-equivalent dose of either rifaximin, rifampicin, DAP or vehicle (Extended Data Fig. 5a,b). Rifampicin was chosen as a comparison as it is a rifamycin that is used in clinical practice. After 7 days of rifamycin treatment, we observed rifamycin-resistant VREfm in significantly more mice receiving rifampicin (80% of mice) or rifaximin (90% of mice) than in mice that received DAP (0% of mice) (P < 0.0001 and P < 0.0001; unpaired t-test) or vehicle (0% of mice) (P < 0.0001 and P < 0.0001; unpaired t-tests) (Extended Data Fig. 5c).
For each mouse, we then determined the percentage of individual VREfm isolates that were rifamycin resistant or DAP-R. There were significantly more rifamycin-resistant VREfm isolated from mice receiving rifaximin or rifampicin than mice receiving the vehicle control (P < 0.0001 and P < 0.0001; unpaired t-test) or DAP (P < 0.001 and P < 0.001; unpaired t-tests) (Extended Data Fig. 5d). Similarly, there were significantly more DAP-R VREfm in mice receiving rifaximin or rifampicin than in mice receiving the vehicle control (P < 0.05 and P < 0.05; unpaired t-tests) or DAP (P < 0.05 and P < 0.05; unpaired t-tests) (Fig. 4a,b). DAP-R VREfm accounted for between 0% and 41% of the gastrointestinal VREfm population in mice given rifaximin and between 0% and 36% in mice given rifampicin, demonstrating conclusively that rifamycin administration drives the emergence of VREfm with resistance to rifamycins and cross-resistance to DAP.

a, The percentage of total mice (n = 5 mice for vehicle, n = 10 mice for rifampicin, n = 10 mice for rifaximin, n = 10 mice for DAP) with DAP-R VREfm strains. b, The percentage of VREfm from each mouse (n = 50 colonies from each individual mouse) that were resistant to DAP after 7 days of antibiotic treatment. The points represent the percentage of DAP-R VREfm from each individual mouse. The percentage was calculated from DAP MIC values (either resistant or susceptible) from the 50 VREfm colonies isolated from each mouse. For all box plots, the centre line shows the median, the box limits show the 25th and 75th percentiles, the upper and lower whiskers extend from the hinge to the largest and smallest values at most 1.5× interquartile range from the hinge. c, Overview of the RpoB substitutions identified in the rifampicin-resistant colonies. Each point represents a single VREfm isolate. Isolates are separated by each RpoB substitution and grouped into either DAP-S or DAP-R. The RpoB substitutions coloured in red had an association with DAP resistance. n values represent the number of isolates containing each mutation for rifaximin (left) and rifampicin (right). Data in a and b were analysed using two-sided unpaired t-tests (vehicle versus rifampicin or vehicle versus rifaximin and rifampicin verses DAP or rifaximin verses DAP). Exact P values are provided when the P value is above P < 0.0001.
We next performed whole-genome sequencing (WGS) on 150 randomly selected VREfm isolates from each group (rifaximin or rifampicin; n = 300 total) to identify all of the mutations present in the rifamycin-resistant isolates. This collection consisted of 100 rifaximin- or rifampicin-resistant isolates taken after the last day of treatment and 50 isolates from each group collected before rifamycin administration. No substitutions in RpoB were identified in any E. faecium isolate taken before rifaximin or rifampicin exposure. However, after exposure, VREfm carrying mutations within rpoB were commonly identified. The S491F substitution was most prevalent (n = 53 (rifaximin) and n = 63 (rifampicin)), with all isolates carrying this substitution being DAP-R (Fig. 4c). H486Y was the second most common RpoB substitution identified (n = 12 (rifaximin) and n = 28 (rifampicin)), with all isolates again being DAP-R. The G482D substitution was the third most common RpoB substitution (n = 15 (rifaximin) and n = 6 (rifampicin)), with 19 isolates DAP-R. Other RpoB substitutions, including V135F, L471V, Q473L and H486R, were identified; however, all of the VREfm isolates carrying these substitutions were DAP-S. Importantly, the proportions of each RpoB substitution observed in VREfm collected from the gastrointestinal tract of mice administered rifamycin closely matched the proportions of each mutation observed in our collection of human clinical VREfm isolates, with the S491F substitution most identified, followed by H486Y and then G482D. These mouse model data strongly suggest that exposure to rifaximin is driving the de novo emergence of DAP resistance in colonizing strains of E. faecium in humans.
rpoB mutations upregulate the prd locus
We next used a multiomics approach to understand how amino acid substitutions in the β subunit of the bacterial RNA polymerase (RpoB) cause resistance to DAP, a cell membrane active antibiotic. We first compared the lipidomes of the three isogenic DAP-R RpoB mutants (G482D, H486Y or S491F) to that of WT VREfm. We also included an isogenic RpoB mutant that did not confer resistance to DAP (Q473L; DAP MIC 2 mg l−1). Principal component analysis (PCA) of lipid species detected clearly separated the DAP-R RpoB mutants from the WT and DAP-S Q473L strain, indicating distinct lipid profiles (Fig. 5a). While the same classes of lipid species were observed in the WT and DAP-R RpoB mutants, there were significant reductions in anionic cardiolipins (CLs) and phosphatidylglycerols (PGs), as well as an increase in digalactosyldiacylglycerols (DGDGs) and cationic lysyl-PGs (Lys-PGs) in the RpoB mutants (Extended Data Fig. 6a–d). CL, PG, DGDG and Lys-PG profiles returned to WT when each of the three RpoB mutations was reverted to WT, demonstrating that the differences observed in the DAP-R strains were due to the RpoB substitutions (Supplementary Table 8).

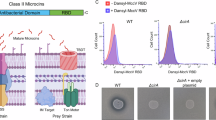
a, PCA denoting segregation of the total lipid classes obtained for the WT and RpoB mutants. b, The 25 significantly differentially expressed loci identified in both RNA-seq and proteomics. The fold change value for RNA-seq is shown in the colour scale (log2-transformed fold change). S+T, the double RpoB(S491F)/RpoC(T634K) mutant. c, Daptomycin susceptibility testing results for the WT and rpoB backgrounds (S491F, G482D or H486Y) with prdR, prdA or prdB deleted (n = 3). The MIC for each strain is shown without error bars as no variation between independent biological replicates was observed. d, PCA denoting the segregation of the total lipid classes obtained for the WT, S491F mutant, WTEV and WTprdR. e, The zeta potential (measured in mV). The points represent each biological replicate (n = 3), and the lines represent the median and interquartile range. Data were analysed using one-way analysis of variance (ANOVA) with correction for multiple testing using the Dunnett method, comparing WT versus RpoB mutant and WTEV versus WTprdR. f, Binding of BoDIPY–DAP, represented as relative fluorescence units (RFU). The points represent each biological replicate (n = 5) and the lines represent the median and interquartile range. Data were analysed using one-way ANOVA with correction for multiple testing using the Dunnett method, comparing WT versus RpoB mutant and WTEV versus WTprdR. Exact P values are provided when the P value is above P < 0.0001.
Given the central role of RpoB in transcription, we posited that the lipidome differences of the DAP-R RpoB mutants were caused by alterations in gene expression. We modelled the RNA polymerase complex structure in E. faecium to assess the structural impact of changes imparted by Q473L, G482D, H486Y and S491F substitutions on transcription. All four substitutions were predicted to be present at the rifamycin active site and in direct interaction with nucleic acids at the transcription replication fork, with probable changes in stability or altered interactions with nucleic acid templates (Extended Data Fig. 6e). The S491F substitution defined changes to a bulkier hydrophobic side chain, with predicted mild reductions in protomer stability and affinities to rifampicin, other RNA polymerase subunits and nucleic acids within the replication fork. Thus, S491F is likely to directly impact the rate of transcription and rate of gene expression. Conversely, the G482D substitution resulted in the introduction of a bulkier negatively charged side chain, which led to steric clashes and increased electrostatic potential of the RNA-binding cleft. The G482D substitution was predicted to have the largest detrimental effect on protein stability, which may reduce the amount of active RNA polymerase and gene expression. It was also predicted to increase nucleic acid binding affinity of the mutant complex, further reducing RNA polymerase activity and processing. Finally, the DAP-R substitution H486Y and DAP-S substitution Q473L were predicted to confer similar effects on protein stability, and rifampicin- and nucleic-acid-binding affinity. However, the H486Y substitution, but not the Q473 substitution, was predicted to increase RNA polymerase complex stability, reducing the dynamic flexibility required for enzyme activity, potentially leading to changes in gene expression. Overall, the DAP-R substitutions S491F, H486Y and G482D were all characterized by distinct interactions from the WT RNA polymerase while the DAP-S substitution Q473L retained the original WT interactions.
Given the predicted changes in RNA polymerase transcriptional activity, we used a combination of RNA sequencing (RNA-seq) and data-independent acquisition (DIA) based proteomics and identified 26 loci that were significantly differentially expressed in both RNA-seq and proteomics in all three DAP-R strains (G482D, H486Y and S491F), but not in the DAP-S strain (Q473L) or the WT VREfm (Extended Data Fig. 7a and Supplementary Table 9). No differences in expression (by transcriptomics or proteomics) of the liaFSR, liaXYZ or yycFG (walKR) regulons nor cls or divIVA genes were observed, indicating that the mechanism of RpoB-mediated DAP resistance was independent of previously described systems7,8,16. As compensatory rpoC mutations can alter the kinetic parameters of the RNA polymerase enzyme, we hypothesized that the number of dysregulated genes would decrease in the RpoBC double mutant (RpoB(S491F) and RpoC(T634K)) compared with the single RpoB(S491F) mutant, leaving genes that are possibly associated with DAP resistance. Indeed, only six loci were differentially expressed (on the basis of RNA-seq and proteomics) in the RpoBC mutant, compared with 44 loci identified in the single RpoB(S491F) mutant (Fig. 5b and Extended Data Fig. 7a). These included genes encoding a cold-shock protein (CspA, AGS75480 or EFAU233_01583), a hypothetical protein of unknown function (AGS75874 or EFAU233_01826), a potassium uptake transporter (K+ transporter, AGS74117 or EFAU233_00176) and a DNA-binding transcriptional regulator of the PadR family in a putative operon with two hypothetical membrane proteins (AGS74325, AGS74326 and AGS74327, or EFAU233_00444, EFAU233_00445 and EFAU233_00446). All six of these loci were significantly upregulated (on the basis of RNA-seq and proteomics) in the three DAP-R RpoB mutants (G482D, H486Y and S491F), as well as in the RpoBC double mutant.
To understand their potential role in DAP resistance, each of these six genes were deleted from the VREfm Aus0233 WT and RpoB(S491F) mutant. The dltC gene, linked to DAP resistance in Staphylococcus aureus, was also deleted as it was differentially expressed in the G482D, H486Y and S491F strains, but not in the RpoBC double mutant. Of these 7 genes, only deletions of the PadR-family regulator (named here prdR, phenotypic resistance to DAP regulator) or the hypothetical membrane proteins (named here prdA and prdB) increased DAP susceptibility (by fourfold) in the WT or S491F backgrounds (Fig. 5c and Extended Data Fig. 7b). Similar fourfold increases in DAP susceptibility were observed when the prdRAB genes were deleted from the RpoB(G482D) or RpoB(H486Y) background (Fig. 5c).
Clinical paired VREfm isolates representative of the G482D, H486Y and S491F mutations were analysed using DIA proteomics to examine the abundance of PrdR, PrdA and PrdB in the cell. In DAP-R clinical strains carrying the S491F (ST1421 and ST203), G482D (ST80) or H486Y (ST203) mutations, the production of PrdRAB was significantly (P < 0.05) increased compared with DAP-S (WT rpoB allele) strains of the same ST (Extended Data Fig. 7c,d and Supplementary Table 9). The increased expression of PrdRAB in both genetically distinct clinical VREfm harbouring the G482D, H486Y or S491F mutations and in isogenic strains suggested that the prdRAB operon was responsible for RpoB-mediated DAP resistance.
PrdR overexpression leads to DAP-R VREfm
We hypothesized that the overexpression of the prdRAB operon in the RpoB mutants was leading to the observed changes in the cell membrane and resistance to DAP. Thus, an overexpression vector with the prdR gene, along with an empty vector (EV) control, were introduced separately into the WT VREfm. Plasmid-mediated overexpression of prdR increased the WT DAP MIC fourfold to 8 mg l−1. Proteomic analysis (WTEV versus WTprdR) confirmed that PrdR was expressed at a similar level in the WTprdR strain as in the RpoB mutants (2.5-fold and 2.2-fold in S491F, respectively) and PrdR specifically controlled the expression of the PrdAB membrane proteins (Extended Data Fig. 8a and Supplementary Table 9). PCA of the WT, WTEV, WTprdR and RpoB(S491F) lipidomes supported the overexpression of the prdR gene driving the changes in the lipid species, with the WTprdR and RpoB(S491F) lipidomes clustering separately than the WT and WTEV lipidomes (Fig. 5d and Supplementary Table 8). Reductions in the same anionic phospholipids (CL and PG) and increases in cationic Lys-PG species were observed in the WTprdR strain at levels similar to those seen in the RpoB(S491F) mutant (Extended Data Fig. 8b–d). Lipidomic analyses of the previously described paired clinical isolates demonstrated the DAP-R clinical strains carrying RpoB mutations had similar significant differences in charged phospholipids (decreases in CL and PG, increases in Lys-PG), compared with DAP-S strains containing the WT rpoB allele (Extended Data Fig. 8e,f and Supplementary Table 8). These data indicated that overexpression of the prdRAB operon in DAP-R VREfm carrying RpoB mutations leads to changes in the abundance of charged lipid species in the cell membrane.
Given the decreases in anionic phospholipids (CL and PG) and increases in cationic phospholipids (Lys-PG), we tested whether strains with RpoB mutations have differences in overall cell membrane charge and DAP binding. Both the DAP-R isogenic and clinical RpoB mutants (G482D, H486Y and S491F) had a significantly reduced negative charge associated with the cell membrane compared with their paired isolate (Fig. 5e and Extended Data Fig. 9a), respectively, and consequently bound to less fluorescent DAP (Fig. 5f and Extended Data Fig. 9c). Complementation of the RpoB mutations reversed cell membrane charge and DAP binding to WT levels (Extended Data Fig. 9b,d). No significant difference was observed in membrane charge or DAP binding for the DAP-S Q473L strain (Fig. 5e,f). Furthermore, the overexpression of the prdR gene resulted in a significantly reduced negative charge across the cell membrane compared with the WTEV strain, with a similar charge to the DAP-R RpoB mutants (Fig. 5f). Moreover, binding of DAP was significantly decreased compared with the WTEV strain (Fig. 5f). These experiments show that the increased expression of the prdRAB operon in VREfm carrying DAP-resistance-associated RpoB mutations leads to changes in the abundance of charged phospholipids and a decrease in the cell surface negative charge, which in turn reduces DAP binding to the cell membrane.
Discussion
Here we show that specific amino acid substitutions within the RRDR of RpoB (G482D, H486Y and S491F) lead to clinical DAP resistance in E. faecium. These mutations are spread in E. faecium populations worldwide, with their prevalence equal to well-characterized amino acid substitutions such as LiaFSR/LiaXYZ and Cls, that have been considered the dominant mechanisms of DAP resistance in VREfm16,27,28. Our multiomics analyses show that G482D, H486Y and S491F RpoB substitutions mediate DAP resistance through a conserved mechanism that is independent of previously described systems (Lia operons or Cls). We showed that each RpoB substitution leads to transcriptional dysregulation of a previously uncharacterized genetic locus, which we have named the phenotypic resistance to DAP, or prd, operon. Upregulation of the prd operon, which consists of a transcriptional regulator (PrdR) and two putative membrane proteins (PrdAB), leads to VREfm cell membrane remodelling and a decreased negative cell surface charge, that reduces DAP binding and, ultimately, renders VREfm resistant to DAP. The G482D, H486Y and S491F RpoB substitutions should therefore be considered to be clinically relevant and major mediators of DAP resistance in VREfm.
Exposure to rifamycin antimicrobials is well known to enrich for bacteria with RpoB substitutions23,29,30. Our data suggest that the clinical use of rifaximin is likely to be responsible for driving VREfm isolates with rpoB mutations that are resistant to not only rifamycins, but also DAP. Three lines of evidence support this hypothesis: Bayesian phylodynamic analyses show that the emergence of phylogenetically distinct VREfm lineages carrying the S491F substitution is temporally linked to the clinical approval of rifaximin in the early 2000s. Independent retrospective patient cohort studies demonstrated a robust association between recent rifaximin exposure and the carriage of DAP-R E. faecium with RpoB substitutions. Lastly, animal experiments demonstrated that administrating rifaximin to VREfm-colonized mice leads to the de novo emergence of DAP-R VREfm strains within the gastrointestinal tract, carrying the same substitutions within RRDR of RpoB as seen in patients.
Our study has potential limitations that warrant consideration when interpreting the results. Although our findings are substantiated by extensive orthogonal experimental evidence, the retrospective nature of our patient cohorts may introduce biases typical of such studies. These include selection and information biases, potentially arising from incomplete or imprecise patient medical records, such as antibiotic exposure history and underlying comorbidities. We implemented stringent genomic screening criteria to exclude clustered VREfm isolates from our cohort studies. However, possible undetected transmission events within the healthcare setting probably impact the assumption of independence in our logistic regression analyses, resulting in overly narrow confidence intervals. While our retrospective cohorts demonstrate a highly robust association between rifaximin use and DAP-R VREfm, these data alone cannot establish causation. Our conclusions are therefore based on the convergence of evidence from diverse experimental sources within our study, including phylodynamic modelling, controlled animal experimentation, extensive genomic epidemiology and independent retrospective patient cohorts. Future prospective multicentre studies would further validate our findings and address potential uncertainties in the reported effect size and confidence intervals.
Notwithstanding these limitations, we propose that patients with chronic liver disease receiving rifaximin are a primary source for the emergence and spread of DAP-R VREfm with rpoB mutations. Furthermore, our HSCT cohort analysis suggests the phenomenon extends beyond patients with liver disease, such that any patient colonized with VREfm and receiving rifaximin is at risk of DAP-R E. faecium emerging within the gut. These findings have important clinical implications. First, our data suggest that DAP should be avoided for empiric therapy of invasive VREfm infections in patients receiving or recently treated with rifaximin due to the higher risk of DAP resistance. Second, to preserve DAP efficacy, hospitals should consider maintaining isolation precautions for rifaximin-treated, VREfm-colonized patients, avoiding cohorting with other VREfm-colonized patients where possible. Lastly, while effective for hepatic encephalopathy prophylaxis, consideration should be given to keeping rifaximin as a second-line option behind other therapies for this indication, and its use for prophylaxis after HSCT should be reconsidered, given the propensity to induce rpoB mutations and subsequent DAP resistance.
Coordinated global efforts are underway to preserve last-resort antimicrobials through stringent stewardship protocols, limiting the use of these critically important medicines31. This strategy assumes that restricted antibiotic use correlates with reduced opportunities for pathogens to develop resistance. Our findings challenge this assumption, demonstrating that exposure to prophylactic rifaximin can lead to DAP-R VREfm emergence without direct DAP exposure.
Our data underscore the potential for unanticipated antimicrobial cross-resistance after the implementation of new treatment regimens, even those perceived to be low-risk and highlight the complex interplay between antimicrobial use and bacterial resistance mechanisms. The potential far-reaching impacts of seemingly benign antimicrobial regimens on AMR necessitates careful consideration, especially regarding the use of prophylactic antibiotics. Our research reinforces the need for judicious use of all antibiotics, and emphasizes the delicate balance required in managing AMR while meeting clinical needs.
Methods
Media and reagents
E. faecium was routinely cultured at 37 °C in brain–heart infusion (BHI) broth (Becton Dickson) or BHI agar (BHIA), BHI solidified with 1.5% agar (Becton Dickson). For electroporation, E. faecium was cultured in BHI supplemented with 3% glycine and 200 mM sucrose (pH 7.0). Escherichia coli was cultured in Luria broth. Broth microdilution (BMD) MICs were performed in cation-adjusted Mueller Hinton with TES broth (CAMHBT) (Thermo Fisher Scientific). A concentration of 10 mg l−1 chloramphenicol (Sigma-Aldrich) was used for plasmid selection in E. faecium and E. coli. The following antibiotics were used at variable concentrations for susceptibility testing: rifampicin (Sigma-Aldrich), rifaximin (Sigma-Aldrich) and DAP (Cubicin).
Oligonucleotides were purchased from Integrated DNA Technologies and are listed in Supplementary Table 11. Plasmids were purified using the Monarch Plasmid Miniprep Kit (NEB). PCR products and gel extractions were purified using the Monarch DNA Gel Extraction Kit (NEB). Genomic DNA was purified using the Monarch Genomic DNA Purification Kit (NEB). Phusion and Phire DNA polymerase was purchased from NEB.
Bacterial isolates
A list of the bacterial strains used in this study is provided in Supplementary Table 10. Australian bacterial strains were collected across three data projects in the Microbiological Diagnostic Unit Public Health Laboratory (MDU PHL). Two unbiased cross-sectional surveys of VREfm were conducted between 10 November 2015 and 9 December 2015 (n = 331)26 and between 1 November 2018 and 30 November 2018 (n = 323) in the state of Victoria (referred to as the 2015 and 2018 snapshot, respectively). During this period, all VREfm-positive isolates (including screening and clinical samples) collected by laboratories across the state were sent to the MDU PHL. Moreover, this project included vanA-VREfm collected from the Controlling Superbugs study14,15, a 15-month (April 2017 to June 2017 and October 2017 to October 2018) prospective study including 8 hospital sites across 4 hospital networks, resulting in 346 VREfm isolates (308 patients) sent for WGS at MDU PHL. The VREfm were isolated from patient samples (including screening and clinical samples) routinely collected from hospital inpatients. For the ‘historical vanA-VREfm,’ every vanA isolate collected within MDU PHL was included. This resulted in an additional 229 isolates, sampled between 2003 and 2014. Collection of bacterial isolates for this study was approved by the Melbourne Health Human Research Ethics Committee (HREC), endorsed by the corresponding HREC at each participating site (HREC/13/MH/326) and the University of Melbourne Human Research Ethics Committee (22536). Approvals included a waiver of consent in accordance with the National Statement on Ethical Conduct in Human Research 2007 (Australia).
For publicly available isolates, our aim was to capture the diversity of E. faecium circulating globally by including isolates that formed part of several key studies involving hospital-associated VREfm (as of January 2021). To be included, isolates needed to have short-read data available, with geographical location (by country), year of collection and source (human or animal). Reads were included only if they had a sequencing depth of >50×. To capture the diversity of VREfm circulating in the USA, isolates from human sources were downloaded from the PathoSystems Resource Integration Center32. All isolates were confirmed to be E. faecium with the Kraken2 database (v.2.1.2)33. The final number of international isolates comprised those from Africa (n = 8), Asia (n = 25), Europe (n = 2,941), North America (n = 424) and South America (n = 78) (Supplementary Table 10).
Antibiotic susceptibility testing
Daptomycin susceptibility testing was performed using the BMD MIC method according to CLSI guidelines. In a 96-well plate, a twofold dilution series (from 32 to 0.5 mg l−1) of DAP was made in 100 μl volumes of CAMHBT, additionally supplemented with 50 mg l−1 Ca2+. An inoculum of 100 μl E. faecium broth culture adjusted to 1 × 106 colony-forming units (CFU) per ml in CAMHBT was then added to each well. After incubation for 24 h, the MIC was defined as the lowest antimicrobial concentration that inhibited visible growth. All assays were performed in biological triplicate, with the median MIC reported. In accordance with recent guidelines34, isolates with a DAP MIC ≥ 8 mg l−1 were considered to be DAP-R. A DAP-sensitive strain (AUS0085)35 and a DAP-R strain (DMG1700661)36 were used as controls.
Rifampicin susceptibility testing was performed using the BMD method in CAMHBT. High-level rifampicin resistance was defined with a MIC > 32 mg l−1. All susceptibility testing was performed in triplicate.
WGS analysis
Genomic DNA was extracted from a single colony using the JANUS automated workstation (PerkinElmer) and Chemagic magnetic bead technology (PerkinElmer). Genomic DNA libraries were prepared using the Nextera XT kit according to the manufacturer’s instructions (Illumina). WGS was performed using the Illumina NextSeq platform, generating 150 bp paired-end reads. The short reads of isolates sequenced at MDU-PHL are available at the NCBI Sequence Read Archive (BioProject: PRJNA565795 (controlling superbugs), PRJNA433676 (2015 snapshot) and PRJNA856406 (2018 snapshot) and PRJNA856406 (historical vanA isolates)).
Phylogenetic analysis
De novo assemblies of the genomes were constructed using Spades37 (v.3.13). In silico MLST was determined using the program mlst with the efaecium database38 (https://github.com/tseemann/mlst). The 1,000 Australian genomes as well as the 4,705 Australian and international VREfm were mapped to the reference E. faecium genome AUS0085 isolated from a human bacteraemia infection in Victoria, Australia (NCBI: CP006620)35 using snippy (https://github.com/tseemann/snippy) (v.4.4.5), applying a minfrac value of 10 and mincov value of 0.9. This reference was selected as it was a publicly available complete genome collected locally and DAP-S. A maximum-likelihood phylogenetic tree was inferred using IQ-TREE (v.2.1.2)39 with a general time-reversible (GTR + G4) substitution model, including invariable sites as a constant pattern and 1,000 bootstrap replicates. Recombination masking was not performed for species maximum-likelihood trees due to the small size of the resulting core alignment. All trees were mid-point rooted and visualized in R (v.4.0.3; https://www.r-project.org/) using phangorn40 (v.2.5.5), ape41 (v.5.4), ggtree42 (v.2.3.4) and ggplot (v.3.3.2).
The genome assemblies of all isolates were screened for acquired AMR determinants using abriTAMR (https://github.com/MDU-PHL/abritamr)43.
GWAS analysis of DAP resistance
A GWAS approach was applied to identify genetic variants of DAP resistance in E. faecium. A genotype matrix of SNPs was constructed and used as input to homoplasyFinder44 (v.0.0.0.9) to determine the consistency index at each locus and kept mutations that had an index of ≤0.5 (indicating at least two independent acquisitions across the phylogeny). We then ran GWAS using DAP resistance as a binary trait, where isolates were categorized as resistant if their DAP MIC was ≥8 mg l−1. To correct for population structure, we used the factored spectrally transformed linear mixed models (FaST-LMM) implemented in pyseer45 (v.1.3.6), which computes a kinship matrix based on the core genome SNPs as a random effect. P values were corrected for multiple-hypothesis testing using the Bonferroni correction method.
Competition assay
For competition assays, overnight cultures of WT and corresponding RpoB(S491F) or RpoB(S491F)/RpoC(T634K) mutants were diluted to an optical density at 600 nm (OD600) of 0.5 in BHI and equal volumes added to an overnight culture. Serial dilutions of each co-culture were performed at times 0 and 24 h on BHIA. Colonies were then replica plated onto BHIA and BHIA rifampicin 20 µg ml−1 to determine the proportion of WT to mutant.
cgMLST and clustering
cgMLST alleles for each isolate were defined using the public E. faecium cgMLST scheme46 and chewBBACA (v2.0.16)47, implemented locally in the COREugate pipeline (v.2.0.4) (https://github.com/kristyhoran/Coreugate). The pipeline determines the alleles of each core gene for every isolate as defined by the specific pathogen scheme. The E. faecium cgMLST scheme contains 1,423 genes. The number of allelic differences between each isolate within this core set of genes is then determined. The cgMLST clusters were determined using single linkage clustering and a pairwise allelic difference threshold of ≤250. This threshold was chosen as it maximized diversity within clusters, to improve temporal sampling depth, while still clustering based on maximum-likelihood tree structure.
Phylodynamic analysis of the emergence of the S491F(RpoB) mutation in VREfm lineages
To investigate the emergence of the S491F mutation in RpoB in three different lineages, as defined with cgMLST, we undertook further analysis on these clusters/lineages. From the species-level maximum-likelihood tree (Fig. 2c), three lineages/clusters were identifiable by cgMLST due to their size (n > 50) and presence of the S491F mutation. The three clusters were analysed independently, such that individual core-genome SNP alignments were generated, as this increased the length of the core alignment and number of sites considered. Snippy (https://github.com/tseemann/snippy) (v.4.4.5) was used to generate the alignments for each cluster to the corresponding reference genome (AUSMDU00004024 (CP027517.1) for cluster 1; AUSMDU00004055 (CP027506.1) for cluster 2; and AUSMDU00004142 (CP027501.1) for cluster 3). Each core alignment used a within ‘cluster reference’ (complete genome of the same cluster) to maximize core-SNP alignment length. The reference for each cluster was chosen as they were a locally collected closed genome. Recombination was removed from the final alignment using Gubbins48 (v.2.4.1) to ensure that modelling was informed only by SNPs with tree-like evolution within the core genome. Maximum-likelihood trees for each of the three clusters were inferred from the core-SNP alignments (cluster 1, n = 219 taxa, 329 SNPs; cluster 2, n = 85 taxa, 541 SNPs; cluster 3, n = 68 taxa, 764 SNPs) with IQ-tree (v.2.1.2)49 with a general time-reversible (GTR + G4) substitution model, including invariable sites as a constant pattern. Phylogenetic uncertainty was determined through 1,000 nonparametric bootstrap replicates.
To investigate temporal signal in the three clusters of VREfm genomes, we first used TempEst50 (v.1.5). A root-to-tip regression analysis was performed on the root-to-tip branch distances within the three, cluster maximum-likelihood phylogenies as a function of year of collection, with the position of the root optimized according to the heuristic residual mean squared method.
The frequency of the emergence of the rpoB mutation in VREfm was inferred using a discrete trait model implemented in BEAST51 (v.1.10.4). Under this model the SNP alignments are used to infer the evolutionary process (that is, phylogenetic tree, time and nucleotide substitution model parameters) for the three clusters. The alignments all shared the HKY substitution model and a constant-size coalescent population prior19. To avoid ascertainment bias due to using a SNP alignment, the number of constant sites was considered for the likelihood calculations. The molecular clock was a relaxed clock with an underlying lognormal distribution. The molecular clock was calibrated using isolation dates for each genome by year of collection and the mean clock rate is shared between all three alignments, but the model allows for the individual alignments to have different standard deviations of the log-normal distribution and also different branch rates. The mean molecular clock rate requires an explicit prior distribution, for which we used a Gamma distribution and a 0.95 quantile range of 4.9 × 10−6 and 1.1 × 10−4 substitutions per site per year. This informative prior means that it acts as an additional source of molecular clock calibration. The median substitution rate was similar for cluster 1 and cluster 2, at 9.7 × 10−7 (95% highest posterior density (HPD) 6.88 × 10−7–1.24 × 10−6) and 1.25 × 10−6 (95% HPD 7.68 × 10−7–1.74 × 10−6), respectively, but slightly faster for cluster 3 at 3.86 × 10−6 (95% HPD 2.23 × 10−6–5.69 × 10−6). Cluster 1 (n = 219) was inferred from a core alignment of 1,869,554 bp containing 329 SNP sites; cluster 2 (n = 85) was inferred from an alignment of 1,524,024 bp containing 541 SNP sites; and cluster 3 (n = 68) was inferred from an alignment of 1,860,780 bp containing 764 SNP sites.
The presence or absence of the S491F mutation in rpoB was used as a binary trait52,53. The trait model was shared between the three alignments, with the different Markov jumps and rewards (that is, changes of trait state and time spent in each state, respectively) recorded for each of the three alignments. The posterior distribution of model parameters was sampled using a Markov chain Monte Carlo of 100,000,000 iterations, sampling every 100,000 iterations. Two independent runs were run for the models. We assessed sufficient sampling from the stationary distributions by verifying the effective sample size of key parameters was around or above 200. The final MCC trees were visualized in R (v.4.0.3, https://www.r-project.org/) using ggtree42 (v.2.3.4). The Markov jumps for the rpoB trait for each alignment were visualized in R (v4.0.3, https://www.r-project.org/).
Construction of isogenic mutants using allelic exchange and of pRAB11prdR
The rpoCT634K, rpoBG482D, rpoBH486Y, rpoBS491F, rpoBQ473L, ABC transporter (I274S), permease protein (G71S) or mannitol dehydrogenase (V288L) mutations were recombined into the chromosomal copy of each gene in ST796 VREfm (Ef_aus0233) by allelic exchange. Deletions of the CpsA, K+ transporter, hypothetical protein, DltC or PrdRAB were also completed using allelic exchange. The region encompassing each gene was amplified by splice overlap extension (SOE)-PCR and recombined into pIMAY-Z54 using the seamless ligation cloning extract (SLiCE)55 method and transformed into E. coli IM08B54. The construct was transformed into electrocompetent VREfm55, with allelic exchange performed as described previously56. Reversion of rpoBG482D, rpoBH486Y or rpoBS491F mutations were completed using allelic exchange with a construct containing the respective wild-type allele. To construct a vector containing prdR, the vector pRAB11 was used. The prdR gene was amplified using Aus0233 genomic DNA. The prdR product was gel extracted, SLiCE cloned into amplified pRAB11, and transformed into IM08B, yielding pRAB11:prdR. The plasmid and EV were then electroporated into Aus0233.
Genome sequencing and analysis of all mutants was conducted as described, with resulting reads mapped to the Ef_aus0233 reference genome and mutations identified using Snippy (https://github.com/tseemann/snippy) (v.4.4.5).
VREfm in vivo gastrointestinal colonization experiments
Female C57BL/6 mice at 6–8 weeks of age were purchased from WEHI and maintained in a specific-pathogen-free facility at the Peter Doherty Institute for Infection and Immunity. The facility operates a 12 h–12 h light–dark cycle and maintains ambient temperature (18–23 °C) and humidity (40–60%). Animals were administered a standard mouse chow diet (Barastoc irradiated mouse cubes) and provided with water ad libitum. All animal handling and procedures were performed in a biosafety class 2 cabinet. Animal procedures were performed in compliance with the University of Melbourne guidelines and approved by The University of Melbourne Animal Ethics Committee (application IDs: 20094 and 28528). Animals were randomly assigned into cages on reception. After acclimatization, the cages were randomly assigned to treatment groups.
Experimental group sizes (treatment versus controls) were calculated using a power of 80%, an attrition rate of 15% and a type I error of 5%. The dose for each antibiotic was calculated using the US Food and Drug Administration (FDA) human conversion formula to ensure that each mouse was given a human-equivalent dose57. To establish gastrointestinal colonization of VREfm, mice were administered ceftriaxone (410 mg kg−1 day−1; AFT Pharmaceuticals) through subcutaneous injection once daily for 4 days, followed by an antibiotic wean period of 24 h. Mice were then inoculated with 106 VREfm in 100 μl PBS by oral gavage. Then, 3 days after VREfm inoculation, single-housed mice were administered either rifaximin (113 mg kg−1 administered twice daily; Sigma-Aldrich), rifampicin (123 mg kg−1 administered once day; Sigma-Aldrich) or vehicle (Corn oil with 10% DMSO) through oral gavage; or DAP (50 mg kg−1 administered once daily; Cubicin) through subcutaneous injection (this results in similar exposure (AUC0–24) to that observed in humans receiving 8 mg kg−1 of intravenous DAP58). The above antibiotic dosing protocol was followed for 7 days. Faecal samples were collected at specific timepoints throughout the experiment to determine VREfm gut colonization and for downstream rifamycin and DAP resistance analysis. Investigators were blinded to treatment groups with faecal samples de-identified on collection from individual mice before being resuspended in PBS to a normalized concentration (100 mg ml−1). Serial dilutions of each de-identified faecal sample were performed, and the samples were plated onto Brilliance VRE agar (Thermo Fisher Scientific) for VREfm CFU enumeration.
For rifamycin and DAP analysis, VREfm colonies (n = 50 per de-identified faecal sample per mouse) from the Brilliance VRE agar plates were replica plated onto BHIA with and without rifampicin 20 µg ml−1 to determine the proportion of rifampicin-resistant VREfm in each mouse. Fifty colonies per mouse were then screened for DAP resistance, with a single colony being resuspended in PBS, then diluted 1/100 into CAMHBT containing 50 mg l−1 Ca2+ and 1/100 in CAMHBT containing 50 mg l−1 Ca2+ and 8 mg l−1 DAP. All suspected DAP-R colonies were confirmed using a DAP BMD MIC as before.
To determine which mutations were present in the rifamycin-resistant isolates, a random selection of 300 colonies, 150 from rifaximin-treated mice and 150 from rifampicin-treated mice (50 pre and 100 post for each treatment), were sampled for WGS as described above.
Analysis of patients receiving rifaximin for hepatic encephalopathy prophylaxis
To examine the potential association between rifaximin exposure and DAP-R VREfm, we analysed VREfm collected between 2014 and 2022 from a single quaternary hospital institution in Melbourne. In total, 225 patients were assessed for previous exposure to rifaximin, which was defined as at least a single dose administered before the collection date for the VREfm isolate and grouped into a rifaximin-exposed group and an unexposed control group. Only a single isolate was selected at random for testing and analysis from patients who had multiple samples with VREfm isolates. The VREfm isolates underwent WGS and DAP and rifampicin susceptibility testing as before. Patients with VREfm isolates that were assessed as genetically clustered with other VREfm isolates in the cohort and likely represented direct transmission were excluded. Genetic clustering was defined using an international standard SNP cut-off (7 SNPs)59,60 using a split k-mer (k = 15) analysis (https://github.com/simonrharris/SKA) (v.1.0), a reference-free pairwise method that compares the entire genome (unlike traditional core-genome based comparisons). Medical records from patients were reviewed for comorbidity and antibiotic prescribing data. Potential associations were assessed through univariate analysis using Pearson’s χ2 tests or Fisher’s exact tests for categorical data, and Student’s t-tests (parametric) or Wilcoxon rank-sum tests (nonparametric) for continuous data. To determine predictors of DAP-R VREfm, multivariable logistic regression with backward stepwise elimination procedure was used, excluding variables with P > 0.10 and reincluding variables with P < 0.05. Exposure to rifampicin and DAP were forced into the models as variables a priori. A P value of <0.05 was considered to be statistically significant. Cases with missing data (for example, incomplete medical record data due to interhospital transfer) were excluded. Several sensitivity analyses were also performed to assess for independent associations after excluding potential confounders, including analysis with exclusion of variables with <10 outcomes, assessing associations with antimicrobial exposure separately from demographic and comorbidity data, and modifying the rifaximin exposure variable to include (1) any previous exposure to rifaximin (including both recent and distant exposure), and (2) any previous exposure to rifamycin antibiotics (including rifampicin, rifabutin and rifaximin). The genomic relationships of VREfm isolates were visualized in a maximum-likelihood phylogenetic tree as before, using a core-SNP alignment of 12,886 sites. The mutations in RpoB were determined using snippy (https://github.com/tseemann/snippy) (v.4.4.5) as described.
Data were obtained from medical records with approval from the Austin Health Human Research Ethics Committee (HREC/92971/Austin-2023), which included a waiver of consent in accordance with the National Statement on Ethical Conduct in Human Research 2023 (Australia).
Analysis of patients receiving rifaximin for HSCT prophylaxis
To examine the potential association between rifaximin exposure and the presence of DAP-associated rpoB substitutions in VREfm independent of underlying chronic liver disease, we analysed isolates collected from patients undergoing HSCT from a hospital institution in Regensburg, Germany. In this cohort, rifaximin is used for gut decontamination to reduce the risk of gastrointestinal graft-versus-host disease. Liver cirrhosis is a contraindication to HSCT. Notably, no patient received prophylactic DAP treatment, which is a major risk factor for DAP-R VREfm. There were 68 patients initially assessed for recent exposure to rifaximin, which was defined as at least a single dose administered within 90 days before the isolate collection date. Only a single isolate was retained for testing and analysis from patients who had multiple samples. In this instance, the isolate included was randomized. The isolates underwent WGS using ion-torrent next-generation sequencing technology (Thermo Fisher Scientific) and Nanopore sequencing (Oxford Nanopore). Patients with isolates that were assessed as genetically clustered with other isolates in the cohort and likely represented direct transmission were excluded (as above). Statistical and phylogenetic analyses were undertaken as described above.
Data were obtained from medical records with approval from the local ethics committee (ethical committee of the University of Regensburg, 21-2521−101). Stool samples were collected from patients after obtaining written informed consent, and the study was performed in accordance with the Declaration of Helsinki.
Lipidomic analyses
Cultures of VREfm (n = 5) were grown to mid-exponential phase (OD600 = 0.6) and washed in PBS. The protein content for each sample was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and was normalized to 100 μg. Cells were lysed using the Bertin Precellys 24 homogenizer set at 6,000 rpm for 40 s and lipids were subjected to monophasic extraction as described previously61. Lipidomic samples were analysed using ultra-high-performance liquid chromatography (UHPLC) coupled to tandem mass spectrometry (MS/MS) using the Vanquish UHPLC system linked to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific), with separate runs in positive- and negative-ion polarities. Solvent A comprised 60:40 (v:v) acetonitrile/water with 5 mM medronic acid and 10 mM ammonium acetate and solvent B comprised 90:10 (v:v) isopropanol:acetonitrile with 10 mM ammonium acetate. 10 µl of each sample was injected into the Acquity UPLC HSS T3 C18 column (1 × 150 mm, 1.8 µm; Waters) at 50 °C at a flow rate of 60 μl min−1 for 3 min using 3% solvent B. During separation, the percentage of solvent B was increased from 3% to 70% in 5 min and from 70% to 99% in 16 min. Subsequently, the percentage of solvent B was maintained at 99% for 3 min. Finally, the percentage of solvent B was decreased to 3% in 0.1 min and maintained for 3.9 min.
All MS experiments were performed using an electrospray ionization source. The spray voltages were 3.5 kV in positive-ionization mode and 3.0 kV in negative-ionization mode. In both polarities, the flow rates of sheath, auxiliary and sweep gases were 25 and 5 and 0 arbitrary unit(s), respectively. The ion-transfer tube and vaporizer temperatures were maintained at 300 °C and 150 °C, respectively, and the ion funnel RF level was set at 50%. In the positive-ionization mode from 3 to 24 min, a top-speed data-dependent scan with a cycle time of 1 s was used. Within each cycle, full-scan MS spectra were acquired first in the Orbitrap at a mass resolving power of 120,000 (at m/z 200) across an m/z range of 300–2,000 using quadrupole isolation, an automatic gain control (AGC) target of 4 × 105 and a maximum injection time of 50 ms, followed by higher-energy collisional dissociation-MS/MS at a mass resolving power of 15,000 (at m/z 200), a normalized collision energy (NCE) of 27% in positive mode and 30% in negative mode, an m/z isolation window of 1, a maximum injection time of 35 ms and an AGC target of 5 × 104.
Identification and quantification of lipids and statistical analysis
LC–MS/MS data were searched using MS Dial v.4.90. The mass accuracy settings were 0.005 Da and 0.025 Da for MS1 and MS2. The minimum peak height was 50,000 and the mass slice width was 0.05 Da. The identification score cut-off was 80%. In positive-ionization mode, [M + H]+, [M + NH4]+ and [M + H-H2O]+ were selected as ion forms. In negative-ionization mode, [M-H]− and [M + CH3COO]− were selected as ion forms. All lipid classes available were selected for the search. PC, Lys-PC, DG, TG, CE and SM were identified and quantified from positive-ionization mode while PE, LPE, PS, LPS, PG, LPG, PI, LPI, PA, LPA, Cer and CL were identified and quantified in negative-ionization mode. The retention-time tolerance for alignment was 0.1 min. Lipids with a maximum intensity of less than fivefold of average intensity in blank were removed. All other settings were set as the default. All lipid LC–MS features were manually inspected and reintegrated when needed. These four types of lipids, (1) with only sum composition except SM, (2) lipids identified due to peak tailing, (3) retention time outliner within each lipid class, (4) LPA and PA artefacts generated by in-source fragmentation of LPS and PS, were also removed. The shorthand notation used for lipid classification and structural representation follows the nomenclature proposed previously62. Relative quantification of the lipid species was achieved using the MS intensity of each lipid ion at apex of the LC peak and normalized to the protein quantity in each sample.
RNA-seq transcriptomics analysis
Cultures were grown to mid-exponential phase (OD600 = 0.6) and total RNA was extracted using the Direct-zol RNA Miniprep kit (Zymo Research) according to the manufacturer’s instructions. Cells were lysed using the Bertin Precellys 24 homogenizer set at 6,000 rpm for 40 s. The samples were treated with TURBO DNase (Thermo Fisher Scientific) followed by clean-up using the RNA clean and concentrator kit (Zymo Research) according to the manufacturer’s instructions. The absence of DNA contamination was checked by PCR and RNA integrity and purity was checked using the Bioanalyser RNA kit (Agilent). Five sequencing libraries from independent RNA extractions were made for each of the VREfm strains using the Stranded Total RNA with Ribo-Zero Plus (Illumina) kit and sequenced on a single lane of the Illumina NovaSeq 6000 platform. Raw paired-end reads were quality trimmed using TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) (v.0.6.2). Bases with a quality score <20 and reads shorter than 50 bp after trimming were discarded. rRNA was removed by the BBDuk script in BBtools (https://sourceforge.net/projects/bbmap/) (v.39.01). The resulting reads were aligned to the Aus0233 reference genome by Bowtie263 (v.2.5.1) using the --no-mixed flag and read counts were generated using htseq-count64 (v.0.12.4) using the options -r pos -t CDS -m union --nonunique none. Differentially expressed genes were detected using Degust (v.4.1.1). Genes with log2[fold change] > 1.5 and adjusted P < 0.05 were considered differentially expressed.
Proteomic analysis
Pelleted snap-frozen bacterial cells (OD600 = 0.6) were solubilized in 4% SDS, 100 mM Tris pH 8.5 by heating them for 10 min at 95 °C. The protein concentrations were assessed by a bicinchoninic acid protein assay (Thermo Fisher Scientific) and 100 µg of each biological replicate prepared for digestion using S-trap Mini Columns (Protifi) according to the manufacturer’s instructions. In brief, the samples were reduced with 10 mM DTT for 10 min at 95 °C and then alkylated with 40 mM IAA in the dark for 1 h. The samples were acidified to 1.2% phosphoric acid and diluted with seven volumes of S-trap wash buffer (90% methanol, 100 mM tetraethylammonium bromide pH 7.1) before being loaded onto S-traps and washed three times with S-trap wash buffer. The samples were then digested with trypsin before being collected by centrifugation after the addition of 100 mM tetraethylammonium bromide, followed by 0.2% formic acid and then 0.2% formic acid/50% acetonitrile. The samples were dried and further cleaned up using C18 Stage65,66 tips to ensure the removal of any particulate matter.
C18 enriched proteome samples were resuspended in 2% acetonitrile (aq) containing 0.01% trifluoroacetic acid (buffer A*) and separated using the Vanquish Neo UHPLC (Thermo Fisher Scientific) system with a single-column chromatography set up composed of a ACQUITY UPLC Peptide BEH C18 Column (300 Å, 1.7 µm, 1 mm × 100 mm, Waters) at a flow rate of 50 μl min−1. Proteome samples were loaded directly on to the ACQUITY column with buffer A (0.1% formic acid, 2% DMSO) coupled directly to an Orbitrap 480 mass spectrometer (Thermo Fisher Scientific) and the buffer composition altered from 2% buffer B (0.1% formic acid, 77.9% acetonitrile, 2% DMSO) to 26% B over 70 min, then from 26% B to 99% B over 2 min and then was held at 99% B for 1.5 min. The mass spectrometer was operated in a data-independent mode automatically switching between the acquisition of a single Orbitrap MS scan (370−1,050 m/z, maximal injection time of 50 ms, an AGC set to a maximum of 300% and mass resolving power of 120,000 (at m/z 200) and the collection of 16.5 m/z DIA windows between 375 and 1,015 m/z (200–2,000 m/z, NCE 32%, maximal injection time of 54 ms, an AGC set to 1,000% and a mass resolving power of 30,000 (at m/z 200). Identification and label free quantification (LFQ) analysis were accomplished using Spectronaut (Biognosys) v.16 (16.0.220606.53000) using directDIA based analysis with minor modifications: protein LFQ method set to MaxLFQ, single hit proteins excluded and imputation disabled. Data were searched against the E. faecium Aus0004 proteome35 (UniProt: UP000007591) with carbamidomethyl (C) allowed as a fixed modification and acetyl (protein N-term) as well as oxidation (M) allowed as variable modifications. Data outputs from Spectronaut were processed using Perseus (v.1.6.0.7)67 with missing values imputed based on the total observed protein intensities with a range of 0.3 σ and a downshift of 1.8 σ. Statistical analysis was undertaken in Perseus using two-tailed unpaired t-tests and ANOVA. Proteins with log2[fold change] > 1 and adjusted P < 0.05 were considered to be differentially expressed.
Computational modelling
In predicting the potential effects of substitutions Q473L, G482D, H486Y and S491F, the full E. faecium DNA-dependent RNA polymerase was initially modelled using advanced homology modelling in Maestro (Schrodinger suite). BLAST-pdb was used to identify the M. tuberculosis homologue (PDB: 5UHC)68 as the template, as it had the best sequence identities across all RNA polymerase subunits. Modelling was performed based on the consensus between sequence alignments from MAFFT-DASH69, T-COFFEE70 and Clustal-W71 (within Maestro), which were manually optimized to minimize sequence gaps. The final RNA polymerase model, bound to rifampicin and the DNA replication fork was next subjected to loop refinement and minimization, and iteratively assessed for model quality within Maestro.
The modelled structure was used as input within in silico biophysical predictors Dynamut272, mmCSM-lig (https://biosig.lab.uq.edu.au/mmcsm_lig/), mmCSM-NA73 and mCSM-PPI273, which predicted the effects of mutations Q473L, G482D, H486Y and S491F on β-subunit stability, and affinities to rifampicin, nucleic acids within the replication fork, and other RNA polymerase subunits, respectively. During interpretation, all values were collectively considered to assess potential protein-level implications to wild-type function. In doing so, the affinity values for mutations located beyond 12 Å of the binding partner were presumed negligible.
Estimation of zeta potential
The zeta potential was measured on cells grown to exponential phase (OD600 = 0.6) and washed in PBS. The zeta potential measurements were performed in PBS to minimize the influence of pH. Each experiment was performed under identical experiment conditions (n = 5), 25 °C with 2 min of equilibration. The zeta potential was measured with a Zetasizer (Malvern).
Determination of cell-associated DAP with BoDipy labelling
BoDipy fluorescent dye (4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-S-indacene) (Invitrogen) was used to label DAP with minor modifications74. In brief, 50 μl DAP (50 mg ml−1) was mixed with 100 μl BoDipy (10 mg ml−1) and was made up to a final volume of 1 ml in 200 mM sodium bicarbonate (pH 8.5). The reaction was incubated for 1 h at 37 °C and unbound BoDipy was removed by dialysis at 4 °C using a Slide-A-Lyzer cassette (Thermo Fisher Scientific), with a 2.0 kDa cut-off according to the manufacturer’s instructions. The antibiotic activity of BoDipy–DAP was confirmed by BMD (described above). To measure cell-associated DAP, cultures were grown to exponential phase (OD600 = 0.6) 50 mg l−1 CaCl2. Each culture was incubated with BoDipy–DAP in darkness (10 min) and washed to remove unbound BoDipy–DAP. The amount of bound BoDipy–DAP was measured with excitation at 490 nm and emission at 528 nm using an Ensight microplate reader (PerkinElmer). Biological replicates (n = 5) were completed on separate days.
Data visualization and statistics
All figures were generated in R (v.4.0.3, https://www.r-project.org/) using tidyverse (v.1.3.1), patchwork (v.1.1.1), ggnewscale (v.0.4.5) and maps (v.3.4.2). Statistical analyses were performed using R (v.4.0.3, https://www.r-project.org/) and GraphPad Prism (v.9.3.1). Specific tests are provided together with each corresponding result in the text.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the Article and its Supplementary Information. The genomics data presented in the study are deposited under BioProject accessions PRJNA565795, PRJNA433676 and PRJNA856406. The MS proteomics data have been deposited in the Proteome Xchange Consortium via the PRIDE partner repository75 under dataset identifiers PXD039832 and PXD039831. Correspondence and requests for materials should be addressed to G.P.C. or B.P.H. Source data are provided with this paper.
References
Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Cairns, K. A. et al. Therapeutics for vancomycin-resistant enterococcal bloodstream infections. Clin. Microbiol. Rev. 36, e0005922 (2023).
Bass, N. M. et al. Rifaximin treatment in hepatic encephalopathy. N. Engl. J. Med. 362, 1071–1081 (2010).
Arias, C. A. & Murray, B. E. The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278 (2012).
Top, J., Willems, R. & Bonten, M. Emergence of CC17 Enterococcus faecium: from commensal to hospital-adapted pathogen. FEMS Immunol. Med. Microbiol. 52, 297–308 (2008).
Arthur, M. & Courvalin, P. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 37, 1563–1571 (1993).
Montero, C. I., Stock, F. & Murray, P. R. Mechanisms of resistance to daptomycin in Enterococcus faecium. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.00774-07 (2008).
Diaz, L. et al. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob. Agents Chemother. 58, 4527–4534 (2014).
Lellek, H. et al. Emergence of daptomycin non-susceptibility in colonizing vancomycin-resistant Enterococcus faecium isolates during daptomycin therapy. Int. J. Med. Microbiol. 305, 902–909 (2015).
Kelesidis, T., Tewhey, R. & Humphries, R. M. Evolution of high-level daptomycin resistance in Enterococcus faecium during daptomycin therapy is associated with limited mutations in the bacterial genome. J. Antimicrob. Chemother. 68, 1926–1928 (2013).
Werth, B. J. et al. Defining daptomycin resistance prevention exposures in vancomycin-resistant Enterococcus faecium and E. faecalis. Antimicrob. Agents Chemother. 58, 5253–5261 (2014).
Turner, A. M., Lee, J. Y. H., Gorrie, C. L., Howden, B. P. & Carter, G. P. Genomic insights into last-line antimicrobial resistance in multidrug-resistant Staphylococcus and vancomycin-resistant Enterococcus. Front. Microbiol. 12, 637656 (2021).
Shayto, R. H., Abou Mrad, R. & Sharara, A. I. Use of rifaximin in gastrointestinal and liver diseases. World J. Gastroenterol. 22, 6638–6651 (2016).
Sherry, N. L. et al. Pilot study of a combined genomic and epidemiologic surveillance program for hospital-acquired multidrug-resistant pathogens across multiple hospital networks in Australia. Infect. Control Hosp. Epidemiol. 42, 573–581 (2021).
Gorrie, C. L. et al. Key parameters for genomics-based real-time detection and tracking of multidrug-resistant bacteria: a systematic analysis. Lancet Microbe 2, e575–e583 (2021).
Miller, W. R., Bayer, A. S. & Arias, C. A. Mechanism of action and resistance to daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb. Perspect. Med. 6, a026997 (2016).
Goldstein, B. P. Resistance to rifampicin: a review. J. Antibiot. 67, 625–630 (2014).
Rios, R. et al. Genomic epidemiology of vancomycin-resistant Enterococcus faecium (VREfm) in Latin America: revisiting the global VRE population structure. Sci. Rep. 10, 5636 (2020).
Duchêne, S. et al. Genome-scale rates of evolutionary change in bacteria. Microb. Genom. 2, e000094 (2016).
Lebreton, F. et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 4, e00534-13 (2013).
Raven, K. E. et al. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res. 26, 1388–1396 (2016).
Sensi, P. History of the development of rifampin. Rev. Infect. Dis. 5, S402–S406 (1983).
Merker, M. et al. Transcontinental spread and evolution of Mycobacterium tuberculosis W148 European/Russian clade toward extensively drug resistant Tuberculosis. Nat. Commun. 13, 5105 (2022).
Goel, A., Rahim, U., Nguyen, L. H., Stave, C. & Nguyen, M. H. Systematic review with meta-analysis: rifaximin for the prophylaxis of spontaneous bacterial peritonitis. Aliment. Pharmacol. Ther. 46, 1029–1036 (2017).
Lee, R. A. et al. Daptomycin-resistant Enterococcus Bacteremia is associated with prior daptomycin use and increased mortality after liver transplantation. Open Forum Infect. Dis. 9, ofab659 (2022).
Lee, R. S. et al. The changing landscape of vancomycin-resistant Enterococcus faecium in Australia: a population-level genomic study. J. Antimicrob. Chemother. 73, 3268–3278 (2018).
Bender, J. K. et al. Update on prevalence and mechanisms of resistance to linezolid, tigecycline and daptomycin in enterococci in Europe: towards a common nomenclature. Drug Resist. Updat. 40, 25–39 (2018).
Tran, T. T. et al. Whole-genome analysis of a daptomycin-susceptible Enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob. Agents Chemother. 57, 261–268 (2013).
Lee, J. Y. H. et al. Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat. Microbiol. 3, 1175–1185 (2018).
Guérillot, R. et al. Convergent evolution driven by rifampin exacerbates the global burden of drug-resistant Staphylococcus aureus. mSphere 3, e00550-17 (2018).
Majumder, M. A. A. et al. Antimicrobial stewardship: fighting antimicrobial resistance and protecting global public health. Infect. Drug Resist. 13, 4713–4738 (2020).
Snyder, E. E. et al. PATRIC: the VBI PathoSystems Resource Integration Center. Nucleic Acids Res. 35, D401–D406 (2007).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Humphries, R. M. The new, new daptomycin breakpoint for Enterococcus spp. J. Clin. Microbiol. 57, e00600-19 (2019).
Lam, M. M. et al. Comparative analysis of the complete genome of an epidemic hospital sequence type 203 clone of vancomycin-resistant Enterococcus faecium. BMC Genom. 14, 595 (2013).
Li, L. et al. Daptomycin resistance occurs predominantly in vana-type vancomycin-resistant Enterococcus faecium in Australasia and is associated with heterogeneous and novel mutations. Front. Microbiol. 12, 749935 (2021).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Homan, W. L. et al. Multilocus sequence typing scheme for Enterococcus faecium. J. Clin. Microbiol. 40, 1963–1971 (2002).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Sherry, N. L. et al. An ISO-certified genomics workflow for identification and surveillance of antimicrobial resistance. Nat. Commun. 14, 60 (2023).
Crispell, J., Balaz, D. & Gordon, S. V. HomoplasyFinder: a simple tool to identify homoplasies on a phylogeny. Microb. Genom. 5, e000245 (2019).
Lees, J. A., Galardini, M., Bentley, S. D., Weiser, J. N. & Corander, J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics 34, 4310–4312 (2018).
de Been, M. et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J. Clin. Microbiol. 53, 3788–3797 (2015).
Silva, M. et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 4, e000166 (2018).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Minin, V. N. & Suchard, M. A. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 56, 391–412 (2008).
Minin, V. N. & Suchard, M. A. Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. B 363, 3985–3995 (2008).
Monk, I. R., Tree, J. J., Howden, B. P., Stinear, T. P. & Foster, T. J. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6, e00308-15 (2015).
Zhang, Y., Werling, U. & Edelmann, W. in DNA Cloning and Assembly Methods (eds Valla, S. & Lale, R.) 235–244 (Humana Press, 2014).
Pidot, S. J. et al. Increasing tolerance of hospital Enterococcus faecium to handwash alcohols. Sci. Transl. Med. 10, eaar6115 (2018).
Nair, A. B. & Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 7, 27–31 (2016).
Heine, H. S., Bassett, J., Miller, L., Purcell, B. K. & Byrne, W. R. Efficacy of daptomycin against Bacillus anthracis in a murine model of anthrax spore inhalation. Antimicrob. Agents Chemother. 54, 4471–4473 (2010).
Higgs, C. et al. Optimising genomic approaches for identifying vancomycin-resistant Enterococcus faecium transmission in healthcare settings. Nat. Commun. 13, 509 (2022).
Maechler, F. et al. Split k-mer analysis compared to cgMLST and SNP-based core genome analysis for detecting transmission of vancomycin-resistant enterococci: results from routine outbreak analyses across different hospitals and hospitals networks in Berlin, Germany. Microb. Genom. 9, 000937 (2023).
Kralj, T. et al. Multi-omic analysis to characterize metabolic adaptation of the E. coli lipidome in response to environmental stress. Metabolites 12, 171 (2022).
Liebisch, G. et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 61, 1539–1555 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Rappsilber, J., Ishihama, Y. & Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and lc/ms sample pretreatment in proteomics. Anal. Chem. 75, 663–670 (2003).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Lin, W. et al. Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol. Cell 66, 169–179 (2017).
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M. & Katoh, K. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10 (2019).
Magis, C. et al. T-Coffee: tree-based consistency objective function for alignment evaluation. Methods Mol. Biol. 1079, 117–129 (2014).
Higgins, D. G. & Sharp, P. M. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 73, 237–244 (1988).
Rodrigues, C. H. M., Pires, D. E. V. & Ascher, D. B. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 30, 60–69 (2021).
Nguyen, T. B., Myung, Y., de Sá, A. G. C., Pires, D. E. V. & Ascher, D. B. mmCSM-NA: accurately predicting effects of single and multiple mutations on protein-nucleic acid binding affinity. NAR Genom. Bioinform. 3, lqab109 (2021).
Pader, V. et al. Staphylococcus aureus inactivates daptomycin by releasing membrane phospholipids. Nat. Microbiol. 2, 16194 (2016).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
This work was supported by a National Health and Medical Research Council (NHMRC) of Australia Ideas grant (GNT1185213) to G.P.C., C.L.G. and I.R.M., and NHMRC project grant (GNT1160745) to B.P.H., T.P.S. and G.P.C. T.P.S. was supported by an NHMRC Investigator grant (GNT1194325). G.P.C. was supported by a University of Melbourne MDHS (Medicine, Dentistry and Health Sciences) mid-career seeding ideas grant and B.P.H. by an NHMRC Investigator Grant (GNT1196103). J.C.K. is supported by an NHMRC Early Career Fellowship (GNT1142613). A.M.T. and N.L.S. are supported by an Australian Government Research Training Program scholarship. D.J.I. is supported by an NHMRC Investigator Grant (GNT1195210). S.D. is supported by the Inception program (Investissement d’Avenir grant ANR-16-CONV-0005), an Investigator grant from the NHMRC (GNT2017284), and the Australian Research Council (FT220100629). The Controlling Superbugs study was supported by the Melbourne Genomics Health Alliance (funded by the State Government of Victoria, Department of Health and Human Services and the ten member organizations). N.E.S. is supported by an Australian Research Council Future Fellowship (FT200100270) and an ARC Discovery Project Grant (DP210100362). This study was also supported by Deutsche Forschungsgemeinschaft Projektnummer (324392634 – TRR 221 B13) to A.G., E.H. and D.W. We thank the staff at the Melbourne Mass Spectrometry and Proteomics Facility of The Bio21 Molecular Science and Biotechnology Institute for access to MS instrumentation and staff within the Microbiological Diagnostic Unit Public Health Laboratory for technical assistance with antimicrobial susceptibility testing and WGS.
Author information
Authors and Affiliations
Contributions
G.P.C., B.P.H. and C.L.G. conceived and planned the overall study design and supervised the project. A.M.T., L.L., J.Y.H.L. and G.P.C. performed all AST analyses. A.M.T. and I.R.M. designed plasmid constructs and generated all isogenic mutants. A.M.T., C.L.G. and A.H. analysed all WGS data and performed comparative genomic analyses. A.M.T., C.L.G., D.J.I. and S.D. performed phylodynamic (BEAST) analyses. G.P.C., L.L. and A.M.T. designed and performed all animal experimentation. J.C.K. led the collection and analysis of patient data from the Melbourne cohort, which was performed by J.C.K., S.V., N.I., B.C. and N.E.H. E.H. led the collection and analysis of patient data from the Regensburg cohort, which was performed by J.C.K., S.V., E.H., D.W. and A.G. G.P.C., L.L., N.E.S. and T.P.S. designed RNA-seq and DIA proteomics experiments, which were performed by A.M.T., with data then analysed by A.M.T., N.E.S., T.P.S. and C.J.W. Lipidomics experiments were designed by G.P.C., A.M.T., B.A.E. and G.E.R. and performed by A.M.T. and S.N., with data then analysed by A.M.T., G.E.R. and B.A.E. In vitro PrdRAB assays were performed by A.M.T. and L.K.S. In silico modelling of RNA polymerase was performed by S.P. and D.B.A. J.Y.H.L., T.S. and N.L.S. provided critical clinical and/or bioinformatics insights for the study. A.M.T., G.P.C. and C.L.G. co-wrote the manuscript with critical feedback from J.C.K., B.P.H. and T.P.S. and input from all of the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Cesar Arias, Judith Lok and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Specific RpoB substitutions are associated with DAP resistance in VREfm.
a. Maximum-likelihood core-SNP-based phylogeny of clinical VREfm (n = 1000) inferred from 6574 SNPs, demonstrating the interspersing of daptomycin resistance. Overlaid are the results of in silico MLST and daptomycin phenotypic testing. In the first heat map, ST is not shown for uncommon STs (n ≤ 5). The scale bar indicates number of nucleotide substitutions per site (top), with an approximation of SNP distance (in parentheses). ST=sequence type. SNP=single nucleotide polymorphism; MLST=multi-locus sequence type. b. Competition assays for the WT and RpoB S491F (dark purple) or WT and RpoB S491F/RpoC T634K (light purple) mutant pairs, with the percentage of rifampicin resistant (RIFR) to rifampicin susceptible (RIFS) isolates determined by plate count, shown on the y-axis. The x-axis denotes time in hours (either 0 or 24). Differences were assessed using two-way analysis of variance (ANOVA). All data points for independent biological replicates (n = 6) are displayed. Horizontal lines depict mean and error bars show the standard deviation. Exact P values are provided when the P value is above P < 0.0001. c. Maximum-likelihood core-SNP-based phylogeny of ST203, RpoB S491F VREfm (n = 80) inferred from 1960 SNPs, demonstrating the spread of isolates across different hospital networks. Overlaid are the year of isolation and hospital network for each isolate, represented as hospital 1 through 10 (H1-H10). The scale bar indicates number of nucleotide substitutions per site. d. Rifampicin susceptibility data for the clinical strains containing a mutation in the RpoB RRDR region (n = 169 isolates) versus a random selection of clinical strains containing a wild-type RRDR (n = 169 isolates). Bars represent the count of isolates containing each MIC. Three independent replicates were performed for each isolate. RRDR=rifampicin resistance determining region; MIC=minimum inhibitory concentration.
Extended Data Fig. 2 DAP resistance RpoB substitutions are present globally.
a. Map of 4,476 VREfm genomes included. Circle size corresponds to total number of genomes and colour corresponds to region of isolation. Country coordinates are the country centroid position. This map is derived from the public domain project Natural Earth and available from www.naturalearthdata.com (“world”). b. The frequency of RpoB substitutions within the rifampicin resistance determining region (RRDR) in 4,476 VREfm genomes, sampled from 43 MLSTs. Bars are coloured by the number of isolates from each region of isolation containing the mutation. The identified daptomycin resistance associated mutations are coloured in red. c. The frequency of various MLSTs identified in the 630 VREfm carrying RpoB mutations in the rifampicin resistance determining region (RRDR). The identified daptomycin resistance mutations are coloured in red. d. Maximum-likelihood core-SNP-based phylogeny of clinical (n = 4,378) and animal VREfm (n = 98) inferred from an alignment of 8,435 SNPs, demonstrating the presence of RRDR RpoB mutations in clinical VREfm isolates. Overlaid is a heatmap showing the presence of at least one substitution in the RRDR of RpoB. VREfm that were animal-associated are highlighted in pink.
Extended Data Fig. 3 The RpoB S491F substitution is maintained in the VREfm population.
a. Linear regression of root-to-tip distance as a function of sampling time. The slope is a crude estimate of the substitution rate (substitution/site) for the recombination-free SNP alignment, with the x-intercept indicative of the age of the root node and the r2 is a measure of clocklike behaviour. The x-axis is time (in years) and the y-axis displays the root-tip-divergence. The points represent an individual isolate in each cluster. b-d. Markov jump counts for the number of transitions between the binary trait of wild-type (WT) rpoB allele and RpoB S491F substitution. The number of Markov jumps for each cluster is shown on the x-axis, with the height of each bar representing the posterior probability for each individual jump.
Extended Data Fig. 4 Rifaximin prophylaxis is associated with DAP resistance in VREfm colonized patients.
a. Maximum-likelihood, core-SNP-based phylogeny for VREfm inferred from 14,420 core-genome SNPs, demonstrating which isolates were from the “control” (n = 116) or “rifaximin” (n = 96) groups from the Melbourne cohort. The scale bar indicates number of nucleotide substitutions per site. b. Summary of the percentage of HSCT patients (Germany cohort; n = 22 control patients and n = 35 rifaximin patients) with a VREfm isolate with any rpoB SNP or daptomycin (DAP) associated rpoB SNP (G482D, H486Y, or S491F). Data was analysed using a Fisher’s exact test (one-sided). The y-axis represents the number (shown as percent) of patients in the control or rifaximin group containing a rpoB mutation.
Extended Data Fig. 5 Rifaximin drives daptomycin resistance in VREfm colonized mice.
a. Timeline of the mouse experiment. VREfm-colonized mice (n = 5 independent mice for vehicle, n = 10 independent mice for rifampicin, n = 10 independent mice for rifaximin, n = 10 independent mice for daptomycin) received a human-equivalent dose of vehicle, rifampicin, or rifaximin (twice per day for rifaximin) for 7 days by oral gavage or subcutaneous injection with daptomycin for 7 days. CRO=ceftriaxone; DAP=daptomycin; RIFAX=rifaximin; RIF=rifampicin. Figure to scale. b. The colony forming units (CFU) for the duration of the mouse experiment. Each point represents the average VREfm CFU/g of faeces and error bars represent standard error of the mean. n = 5 independent mice for vehicle and n = 10 independent mice for DAP, RIF, and RIFAX. The x-axis is day of collection and y-axis is the CFU/g faeces of VREfm. c. Percentage of mice with rifampicin-resistant VREfm strains for each treatment group. d. Percentage of VREfm from each mouse (n = 50 colonies from each individual mouse) that were resistant to rifampicin after 7 days of antibiotic treatment. Points represent the percentage of rifampicin-resistant VREfm from each individual mouse. Percentage was calculated from rifampicin MIC values (either resistant or susceptible) from the 50 VREfm colonies isolated from each mouse. For all box plots, the lower and upper hinges depict the 25th and 75th percentiles. The upper and lower whiskers extend from the hinge to the largest and smallest values at most 1.5 × IQR from the hinge. The centre line in the box shows the median. Data in c and d were analysed using an unpaired t-test (two-sided) (vehicle versus rifampicin or vehicle versus rifaximin and rifampicin versus daptomycin or rifaximin versus daptomycin). Exact P values are provided when the P value is above P < 0.0001. The y-axes represent the percentage of rifampicin-resistant VREfm isolates.
Extended Data Fig. 6 Cell membrane lipid profile and molecular modelling of RpoB mutants.
a-d. The main lipid species differentially produced by the WT and RpoB mutants shown as normalized abundance (intensity/total protein). Bars represent the median value and error bars represent the SEM. Each point represents an independent biological sample (n = 5). Data was analysed with a Two-way ANOVA (WT versus RpoB mutant) and P values were corrected for multiple testing using the Dunnett method. Exact P values are provided when the P value is above P < 0.0001; ns=not significant. The y-axis is the normalized intensity (Normalized Int.) for each lipid identified. e. Structural localization of missense rpoB mutations. All studied mutations were located within interaction proximity of the ligand rifampicin binding site (grey), and the nucleic acids of the replication fork (dark red).
Extended Data Fig. 7 PrdR is over-expressed in VREfm with RpoB substitutions.
a. Intersection between RNAseq and proteomics analyses displayed as an UpSet plot (n = 5 independent biological replicates for RNAseq and n = 5 independent biological replicates for proteomics). The bar represents the count of each locus that was identified in each sample. RNA seq significance (FDR < 0.05, log2FC > 1 or log2FC < −1) and proteomics significance (adjusted p-value < 0.05, log2FC > 1 or log2FC < −1). b. Locus map of the prdR locus in the VREfm AUS0233 genome ([proteins] AGS74325, AGS74326, AGS74327 or EFAU233_00444, EFAU233_00445, EFAU233_00446). c. Proteomics comparing the abundance of the PrdRAB locus across five different clinical strain pairs (n = 5 independent biological replicates for each strain). Bars are the fold-change (log2) from five independent biological replicates showing proteins with statistically significant (<0.05) P-values. The y-axis is the log2 fold-change. Data was analysed using an unpaired t-test of daptomycin-susceptible clinical strain versus daptomycin-resistant clinical strain (with a RpoB mutation). d. Protein abundance changes of the clinical VREfm strain pairs (n = 5 independent biological replicates each isolate) containing mutations in RpoB from different genetic backgrounds, demonstrating the conserved upregulation of the PrdRAB operon. The x-axis is the log2 fold-change and the y-axis is the -log10 P value.
Extended Data Fig. 8 PrdR over-expression changes the cell membrane lipid profile.
a. Protein abundance changes for the prdR deletant mutant, compared to the WT strain, demonstrating the specificity of the regulator for the prdAB membrane proteins (n = 5 independent biological replicates for proteomics). The x-axis is the log2 fold-change and the y-axis is the -log10 P value. b-d. The lipid species differentially produced by the WT, S491F mutant, WT EV, and WTprdR shown as normalized abundance (intensity/total protein). Bars represent the median value and error bars represent the SEM. Each point represents independent biological replicates (n = 5). Data was analysed with a Two-way ANOVA ((WT versus RpoB mutant and WT EV versus WTprdR) and P values were corrected for multiple testing using the Dunnett method. Exact P values are provided when the P value is above P < 0.0001; ns=not significant. The y-axis is the normalized intensity (Normalized Int.) for each lipid identified. e-f. Lipid species differentially produced by the ST203 and ST80 RpoB S491F clinical strain pairs, shown as normalized abundance (intensity/total protein). Bars represent the median value and error bars represent the SEM. Each point represents independent biological replicates (n = 5). Data was analysed with a Two-way ANOVA (DAP-S versus DAP-R) and P values were corrected for multiple testing using the Dunnett method. Lipid species with a significant difference are denoted. The y-axis is the normalized intensity (Normalized Int.) for each lipid identified.
Extended Data Fig. 9 Cell membrane charge and daptomycin binding.
a-b. Zeta potential (measured in mV) is shown on the y-axis. Points represent each independent biological replicates (n = 3) and lines represent the median and interquartile range. Data was analysed with a one-way ANOVA [daptomycin-susceptible clinical strain versus daptomycin-resistant clinical strain (with a RpoB mutation) (ST203 [H486Y], ST80 [G482D], ST1421 [S491F]) or WT versus RpoB complement (-C)] and P values were corrected for multiple testing using the Dunnett method. c-d. Binding of BoDIPY-DAP, represented as relative fluorescence units (RFU) is shown on the y-axis. Points represent each independent biological replicates (n = 4 for the clinical strains and n = 5 for the RpoB complement strains) and lines represent the median and interquartile range. Data was analysed with a one-way ANOVA [daptomycin-susceptible clinical strain versus daptomycin-resistant clinical strain (with a RpoB mutation) (ST203 [H486Y], ST80 [G482D], ST1421 [S491F]) or WT versus RpoB complement (-C)] and P values were corrected for multiple testing using the Dunnett method. For a-d exact P values are provided when the P value is above P < 0.0001; ns=not significant.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Turner, A.M., Li, L., Monk, I.R. et al. Rifaximin prophylaxis causes resistance to the last-resort antibiotic daptomycin. Nature 635, 969–977 (2024). https://doi.org/10.1038/s41586-024-08095-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-08095-4
This article is cited by
-
Die Darm-Leber-Achse bei Leberzirrhose
Die Gastroenterologie (2026)
-
Genomic insights into the spread of vancomycin- and tigecycline-resistant Enterococcus faecium ST117
Annals of Clinical Microbiology and Antimicrobials (2025)
-
Increased risk of antimicrobial resistance in patients with cirrhosis and hepatic encephalopathy using rifaximin
Nature Communications (2025)
-
Genomic surveillance of vancomycin-resistant Enterococcus faecium: a study on Resistome, Plasmidome, and mobilome profiling
Current Genetics (2025)
-
Four-channel carbon dot fluorescent sensor array with a pattern recognition approach for the discrimination and quantification of rifamycin antibiotics
Microchimica Acta (2025)
