
Abstract
The coronavirus membrane protein (M) is the main organizer of coronavirus assembly1,2,3. Here, we report on an M-targeting molecule, CIM-834, that blocks the assembly of SARS-CoV-2. CIM-834 was obtained through high-throughput phenotypic antiviral screening followed by medicinal-chemistry efforts and target elucidation. CIM-834 inhibits the replication of SARS-CoV-2 (including a broad panel of variants) and SARS-CoV. In SCID mice and Syrian hamsters intranasally infected with SARS-CoV-2, oral treatment reduced lung viral titres to nearly undetectable levels, even (as shown in mice) when treatment was delayed until 24 h before the end point. Treatment of infected hamsters prevented transmission to untreated sentinels. Transmission electron microscopy studies show that virion assembly is completely absent in cells treated with CIM-834. Single-particle cryo-electron microscopy reveals that CIM-834 binds and stabilizes the M protein in its short form, thereby preventing the conformational switch to the long form, which is required for successful particle assembly. In conclusion, we have discovered a new druggable target in the replication cycle of coronaviruses and a small molecule that potently inhibits it.
Similar content being viewed by others


Main
Three previously unknown betacoronaviruses that are highly pathogenic to man emerged during the past two decades. These were the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002, the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 and SARS-CoV-2 in 2019. The average case fatality rate was around 10% for SARS-CoV and around 36% for MERS-CoV4. The COVID-19 pandemic, caused by SARS-CoV-2, has resulted in about 7 million confirmed deaths and around 28 million estimated deaths5 so far. At least 10% of COVID-19 patients develop long COVID6, which is the continuation or development of new symptoms for several months after the initial infection. During and after the emergence of SARS-CoV in 2002, there have been efforts to develop inhibitors of the coronaviral main protease (Mpro or 3CLpro). This work laid the basis for the rapid development, 20 years later, of nirmatrelvir, a potent Mpro inhibitor with pan-coronavirus coverage7. When therapy with nirmatrelvir is initiated within five days of the onset of COVID-19 symptoms, the drug (which is combined with the pharmacokinetic (PK) booster ritonavir) reduces mortality and hospitalization by around 90%8. Remdesivir and molnupiravir are both prodrugs of nucleoside analogues that, as their 5′-triphosphate metabolite, target the viral RNA-dependent RNA polymerase (RdRp)9 and induce error catastrophe, respectively10. These molecules were originally developed for the treatment of other viral infections and have relatively broad-spectrum antiviral activity. They were therefore repurposed for the treatment of infections with SARS-CoV-2 (refs. 9,11). Remdesivir requires intravenous administration, but an oral form of the same nucleoside, obeldesivir12, is currently in clinical development. The use of nirmatrelvir/ritonavir, remdesivir and molnupiravir is relatively limited because of problems with drug–drug interactions, the need for intravenous administration or suboptimal efficacy, respectively.
In the context of COVID-19, and as a measure of preparedness for new highly pathogenic coronaviruses, it remains of utmost importance to identify new inhibitors and new druggable targets in the coronavirus replication cycle. Furthermore, antivirals with non-overlapping resistance profiles are ideally suited for the development of (fixed-dose) combinations to avoid the development of antiviral drug resistance. Complementary to target-based drug design, phenotypic antiviral screening enables the discovery of other targets in the viral replication cycle that are, so far, not known to be druggable. Screening large libraries of small molecules allows the identification of compounds that inhibit viral replication with no or minimal cytotoxicity. Once the potency and selectivity of such molecules has been improved through medicinal chemistry, the molecular mechanism can be unravelled, resulting in the potential identification of a new druggable target. This approach has resulted, for example, in the identification of highly potent hepatitis C virus inhibitors that target the viral NS5A protein13 and the discovery of a class of highly potent pan-serotype dengue virus inhibitors14,15. We have used the same strategy (high-throughput screening followed by hit optimization) to discover CIM-834, an orally efficacious, first-in-class small-molecule assembly inhibitor of SARS-CoV-2 and SARS-CoV that targets the viral M protein. The M protein is the most abundant structural protein in the viral envelope and is recognized as the main driver of coronavirus assembly1,2,3,16. As well as the M protein, a mature virion carries the spike protein (S), which mediates entry into the host cell17, the envelope protein (E), which functions as a viral ion channel18, and the nucleocapsid protein (N), which packages the viral RNA (vRNA) genome19. For genome replication, coronaviruses remodel host cellular membranes into double-membrane vesicles (DMVs)20,21,22. DMVs are protected microenvironments for the production of subgenomic mRNAs (encoding, among others, the E, M, N and S proteins) and progeny genomic RNA (gRNA). After export from the DMVs, gRNA associates with N, thereby forming viral ribonucleoprotein (vRNP) complexes23, whereas the envelope proteins M, E and S are directed towards the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), where virus assembly takes place16. The M protein appears as a mushroom-shaped dimer with two distinct conformations, a short form (Mshort, also termed Mcompact) and a long form (Mlong)1,2,3. A highly conserved hinge region (residues 106–116) is required for conformational changes1,24. A model has been proposed in which interactions between the M protein, the N protein and gRNA trigger the conformational switch from Mshort to Mlong and the oligomerization of M into higher-order oligomers. These conformational changes in M probably induce bending of the ERGIC membrane around the vRNP, resulting in virion formation1,2,25,26 (a graphical summary is shown in Fig. 4j). Furthermore, the M protein coordinates, together with the E protein, the retention and maturation of the S protein at the virion assembly sites27.
In this study, we show that our identified SARS-CoV-2 inhibitor, CIM-834, blocks the conformational switch of the coronavirus M protein from Mshort to Mlong, thereby preventing the assembly of infectious virions. Our work demonstrates that the M protein and the assembly process of coronaviruses are excellent druggable targets.
Discovery and antiviral profile of CIM-834
Using a phenotypic high-throughput, high-content, imaging-based SARS-CoV-2 antiviral assay in GFP-expressing VeroE6 (VeroE6–GFP) cells28, a diverse library of 350,000 small molecules was screened. One of the hits exerted low micromolar antiviral activity and was selected as the starting point for an extensive medicinal-chemistry program with more than 500 analogues. This led to the identification of CIM-834 (Fig. 1a), which was selected as a tool compound for mechanistic studies and to assess its efficacy in vivo. In VeroE6–GFP and A549ACE2+TMPRSS2 cells, CIM-834 inhibited the replication of a panel of SARS-CoV-2 isolates (variants of concern and contemporary isolates) with average half-maximal effective concentration (EC50) values of 112 nM and 84 nM, respectively (Fig. 1b,c), which is similar to the antiviral potency of nirmatrelvir (average EC50 values of 94 nM and 57 nM in VeroE6–GFP and A549ACE2+TMPRSS2 cells, respectively). GS-441524, the parent nucleoside of remdesivir, was included as a second reference compound and exhibited antiviral activity in the micromolar range, as expected29. CIM-834 also inhibited the replication of SARS-CoV (EC50 of 640 nM in VeroE6–GFP cells and 530 nM in VeroE6 cells) (Fig. 1b and Extended Data Table 1) and had low micromolar activity against HCoV-OC43, but it lacked activity against MERS-CoV and selected alphacoronaviruses (HCoV-229E, HCoV-NL63, feline infectious peritonitis virus (FIPV) and PEDV) (Extended Data Table 1). CIM-834 retained activity for SARS-CoV-2 viruses that are resistant to nirmatrelvir30 (Extended Data Table 1). The antiviral activity was further confirmed in primary human nasal epithelial cells cultured at the air–liquid interface (Fig. 1d–f). In these cultures, the virus yield of SARS-CoV-2 B.1.1.7 four days after infection was reduced by 4.1 and more than 5log10, at CIM-834 concentrations of 1 and 3 µM, respectively (Fig. 1f).

a, The structural formula of CIM-834. b, CIM-834 inhibits the replication of different SARS-CoV-2 variants and SARS-CoV in two different cell lines. Nirmatrelvir and GS-441524 were included for comparison. Box plots show the median and the 25th and 75th percentiles, and the whiskers show the maximum and minimum. The number of biological independent experiments is indicated above the x axis c, Dose–response curves of the antiviral activity (coloured lines) and cytotoxicity (dashed lines) on A549ACE2+TMPRSS2 cells (mean ± s.e.m.; n = 4 biologically independent experiments). d–f, CIM-834 inhibits SARS-CoV-2 B.1.1.7 replication in primary human nasal cells grown at the air–liquid interface (d). The reduction in vRNA levels after treatment is shown at day 2 (e) and day 4 (f); P-values are shown; Kruskal–Wallis test and Dunn’s comparison, mean ± s.e.m.; n = 6 biologically independent experiments. LLOQ, lower limit of quantification. Credit: d, created in BioRender, M. Laporte (2025). https://BioRender.com/q39i535.
Based on acceptable metabolic stability data in mouse and hamster liver microsomes (half-life of 45 min and 10 min, respectively), the PK properties of CIM-834 were next assessed in mice (in the absence of ritonavir) and in hamsters (in the presence of ritonavir) (Extended Data Fig. 1a). In both species, the compound showed low clearance, good oral bioavailability and favourable lung tissue distribution.
The efficacy of CIM-834, together with nirmatrelvir as a positive control, was first assessed in a SCID mouse SARS-CoV-2 B.1.351 infection model that we had validated previously for antiviral studies31 (Extended Data Fig. 1c). Nirmatrelvir (in the absence of ritonavir boosting) was devoid of antiviral activity at a dose of 100 mg per kg given twice a day but was effective at a three-fold higher dose. Oral treatment with CIM-834 (100 mg per kg) reduced infectious viral titres in the lung by more than 1.5log10 when given once a day, and more than 3log10 when given twice a day. Viral RNA levels were reduced by 2.3log10 and 2.5log10, respectively. There was no significant weight change in both drug-treated and vehicle-treated mice. The antiviral efficacy of CIM-834 in SCID mice was also assessed in a therapeutic set-up in which treatment was initiated at 24 h, 30 h or 48 h after infection (Fig. 2a). The earlier treatment was initiated after infection, the more pronounced the reduction of vRNA levels was in the lungs (Fig. 2c). Remarkably, treatment still reduced infectious titres by more than 3log10 when initiated 48 h after infection (Fig. 2b), although under these conditions, no significant reduction in vRNA was observed. This is in line (as further shown in this study) with the mechanism of action, which is inhibition of particle formation but not of vRNA production. The antiviral effect of CIM-834 (combined with the PK booster ritonavir) was next studied in hamsters intranasally infected with SARS-CoV-2 USA-WA1/2020. Nirmatrelvir was used as the positive control (Fig. 2e). Oral treatment with CIM-834/RTV resulted in a 3.4log10 and greater than 4log10, respectively, reduction in vRNA levels and infectious virus titres in the lungs at the end point (Fig. 2f). In vehicle-treated animals, severe peribronchial and perivascular inflammation with endothelialitis and transmural inflammation was observed, as well as limited bronchopneumonia. In the groups treated with CIM-834/RTV and nirmatrelvir, the lung parenchyma appeared nearly normal with some very focal perivascular inflammation without endothelialitis; the bronchi appeared normal (Fig. 2f,h and Supplementary Table 1). Hamsters that had been treated with CIM-834/RTV did not transmit the virus to untreated sentinel littermates, unlike our control animals (Fig. 2g).

a, SCID mice were intranasally infected with 105 times the tissue-culture infectious dose (TCID50) of SARS-CoV-2 B.1.351 and treated twice a day with CIM-834 or with nirmatrelvir. No ritonavir was used in either of the treated groups. Treatment was initiated just before infection (Day 0) or at the indicated times after infection. b,c, Lung infectious titres (b) and vRNA load (c) determined 3 days after infection. Individual data and median values are shown, along with P-values. d, Weight change for the different treatment groups at day 3 after infection as a percentage, normalized to the body weight at the time of infection (Kruskal–Wallis test and Dunn’s comparison, mean ± s.d.; n = 2 biologically independent experiments with 6 mice in each group per experiment). e, Syrian hamsters were intranasally infected with 104 times the TCID50 of SARS-CoV-2 USA-WA1/2020 treated for four consecutive days with vehicle, CIM-834/ritonavir (100 + 50 mg per kg, twice a day) or nirmatrelvir (300 mg per kg, twice a day). One day after infection, treated index hamsters were co-housed with untreated sentinel hamsters. f, Lung infectious titres, vRNA load and pathology scores of index hamsters at day 4 after infection. g, Lung infectious titres and vRNA load in untreated sentinel hamsters three days after the start of co-housing (Kruskal–Wallis test and Dunn’s comparison; n = 2 biologically independent experiments with, for each experiment, two hamsters for the mock-infected group, two hamsters in the group of sentinels co-housed with vehicle-treated hamsters and four hamsters in the other groups). h, Representative haematoxylin and eosin-stained images of hamster lungs at day 4 after infection (see supplementary Table 1 for all lung pathology scores). Blue and red arrows indicate inflamed bronchi and vascular regions, respectively; green arrows indicate limited bronchopneumonia (only observed in the vehicle group). Scale bars, 100 μm. Credit: a,e, created in BioRender, M. Laporte (2025). https://BioRender.com/q39i535.
CIM-834 targets the viral M protein
To identify the molecular target of CIM-834, in vitro resistance selection was performed by passaging SARS-CoV-2 B.1.1.7 in the presence of increasing concentrations of CIM-834 (Fig. 3a). This was done in three independent cultures and allowed, at passage 5 in each culture, the selection of virus that replicated in the presence of 1 µM CIM-834. The compound concentration could not be increased to 3 µM without losing the virus. Therefore, virus samples from passage 5 were used for whole-genome sequencing (using the ARTIC protocol Illumina). A good coverage was obtained for each sample throughout the genome (more than 100 reads), except for two small regions for which coverage was consistently less than 10 reads (around 200 nucleotides at the beginning of the Mpro gene and 400 nucleotides covering the ORF7a gene). Alignment of the sequences with those of the B.1.1.7 inoculum revealed a single non-synonymous mutation (C26916T) present in all three independent virus stocks in more than 97% of the reads and with good coverage in each sample (more than 900 reads). This mutation results in the amino acid change P132S in the intra-virion part of the M protein (Fig. 3b). To confirm that this change is responsible for the phenotypic resistance, the wild-type CCG codon at positions 26916–26918 was replaced by AGC in the background of a SARS-CoV-2 Wuhan strain (rWuhan; Fig. 3c). In primary human nasal cells cultured at the air–liquid interface, no significant differences in the growth kinetics of M(P132S) compared with the wild-type virus were observed (Fig. 3d). The rWuhan strain carrying the M(P132S) mutation remained fully susceptible to nirmatrelvir and GS-441524, whereas its susceptibility to CIM-834 was markedly reduced (a 58-fold increase in EC50) (Fig. 3e). As we were writing this manuscript, a preprint became available that reports on another SARS-CoV-2 inhibitor, JNJ-9676, which targets the M protein and also selects for M(P132S) (resulting in a 43-fold increase in EC50 compared with the wild type)32. Three other substitutions in M were reported that induce a 10-fold increase or more for JNJ-9676: M91K (a 10-fold increase), S99A (a 97-fold increase) and N117K (a 145-fold increase)32. We reverse-engineered these substitutions in the Omicron rBF.7 background (Fig. 3f,g) and found that M91K and N117K have only a small impact on the in vitro activity of CIM-834 (2.6-fold and 5.4-fold increases in EC50 values, respectively), whereas S99A (like P132S) results in complete resistance.

a, In vitro resistance selection was done by passaging SARS-CoV-2 B.1.1.7 in the presence of increasing concentrations of CIM-834 in A549ACE2+TMPRSS2 cells. b, The P132S substitution is located in the carboxy-terminal intravirion domain of the M protein. c, Introduction of M(P132S) in the background of SARS-CoV-2 (Wuhan and Omicron BF.7) through reverse genetics. d, Replication kinetics of rWuhan-WT and the M(P132S) mutant in human nasal epithelial airway cultures grown at the air–liquid interface. Infectious virus titres from apical washes collected at different time points were determined (mean ± s.e.m.; n = 6 biologically independent experiments). e, Susceptibility of rWuhan-WT and M(P132S) for CIM-834 and the reference inhibitors GS-441524 and nirmatrelvir, in A549ACE2+TMPRSS2 cells (individual and median values are shown; n = 6 biologically independent experiments). Two-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test was used to compare EC50 values. f, Level of compound resistance associated with other M substitutions reported in ref. 32. Mutations were reverse-engineered in the omicron BF.7 background (individual and median values are shown; n = 3 or n = 4 (P132S) biologically independent experiments with two technical repeats). Two-way ANOVA with Dunnett’s multiple comparisons test was used to compare EC50 values. g, Fold changes in EC50 values of M mutants versus wild-type virus (same data as in f; individual and mean values are shown; n = 3 or n = 4 (P132S) biologically independent experiments). Credit: b,c, created in BioRender, M. Laporte (2025). https://BioRender.com/t04g029.
CIM-834 inhibits SARS-CoV-2 assembly
Because the M protein has a pivotal role in virion assembly, we next explored whether CIM-834 acts at the late stages of the viral replication cycle. Time-of-drug-addition assays confirmed a late mechanism of action (Extended Data Fig. 2a–c). In single-cycle replication assays (read-out 10 h after infection), CIM-834 has, in contrast to nirmatrelvir, no effect on viral RNA synthesis but completely blocks the formation of infectious particles. Even when CIM-834 was first added 5 h after infection, intracellular loads of infectious viruses were decreased by 3log10. In a subsequent time-of-drug-addition experiment with a SARS-CoV-2-mNeonGreen reporter virus (Extended Data Fig. 2d–f), CIM-834 did not affect the expression of the reporter when analysed at the end of the first replication cycle (10 h after infection), but it fully prevented the next round of viral replication. Hydroxychloroquine and nirmatrelvir were included as reference molecules; they block SARS-CoV-2 endosomal entry (the dominant entry pathway in VeroE6 cells33) and genome replication, respectively.
To seek further evidence that CIM-834 interferes with viral assembly, the effect of CIM-834 on the formation of virus-like particles (VLPs) was studied in Huh7 cells transfected with M and E expression plasmids (Fig. 4a). Analysis of intracellular M protein levels revealed that CIM-834 inhibits intracellular oligomerization of the wild-type M, but not the M(P132S), protein (Fig. 4b–d). In the next step, we investigated whether the loss of oligomerization is caused by an effect of CIM-834 on the subcellular trafficking of the M protein. However, no effect of CIM-834 on the co-localization of M with TGN46, a marker for the trans-Golgi network, could be detected (Extended Data Fig. 2g).

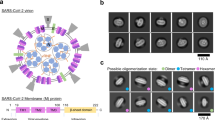
a, The set-up of the VLPs assay. b,c, Western blots were quantified and Moligomers/Mtotal was calculated for wild-type M (b) and M(P132S) (c) relative to the DMSO control (mean ± s.d.; n = 4 biologically independent experiments; ordinary one-way ANOVA and Dunnett’s test). d, Representative western blot (with tubulin as a loading control). e,f, VLP secretion quantified by extracellular M monomer detection by western blot, and calculation of Mmedium/Mlysate + medium relative to the DMSO control, for wild-type M (e) and M(P132S) (f) (mean ± s.d.; n = 4 biologically independent experiments; ordinary one-way ANOVA and Dunnett’s test). A representative western blot with extracellular M levels is shown below each graph. g, Overview of the perinuclear region in DMSO- or CIM-834 (1 µM)-treated cells 10 h after infection with SARS-CoV-2. Yellow arrowheads indicate virions and virion-assembly sites; orange arrowheads and dotted circles indicate extended membrane compartments. White boxes are magnified in Extended Data Fig. 2h. Asterisks indicate DMVs. N, nucleus. Section thickness, 70 nm. Scale bars: 500 nm in the main images, 200 nm in the zoomed images. h, Tomographic reconstructions. Yellow arrowheads indicate mature and assembling virions (top left); orange arrowheads indicate regions of high membrane curvature and invaginations in the DMV proximal membrane accumulations (bottom left). MT, microtubules; IF, intermediate filaments; zER, zippered ER. The tomographic volume of the DMSO- and CIM-834-treated samples is displayed in Supplementary Videos 1 and 2, respectively. Section thickness is an average of 5 tomographic slices of 0.78 nm each. Scale bars: for DMSO, both 50 nm; for CIM-834, left is 100 nm, right is 50 nm. i, Top, quantification of the number of DMVs per 1 µm2 of cytoplasmic area. Middle, percentage of cells showing (assembling) virions. Bottom, percentage of cells with DMV-proximal membrane stacks. Mean ± s.d. of n = 2 independent experiments with 8 infected cells per condition analysed. j, Proposed antiviral mechanism of CIM-834. Top, the viral M protein drives SARS-CoV-2 assembly; the vRNP associates with E, S and Mshort at the ERGIC membrane, which induces a conformation switch to Mlong, resulting in membrane curvature and subsequent virion assembly and release. Bottom, inhibition of coronavirus assembly by CIM-834; binding of CIM-834 to Mshort (see Fig. 5) prevents the conformational switch and, as a result, prevents virion assembly. TGN, trans-Golgi network. Credit: j, created in BioRender, M. Laporte (2025). https://BioRender.com/g67z787.
In line with M protein oligomerization being essential for viral assembly1, CIM-834 results in a concentration-dependent inhibition of VLP release (Fig. 4e) from cells transfected with wild-type M and E. When cells were transfected with the M(P132S) construct, CIM-834 was not able to significantly block VLP secretion (Fig. 4f), which corroborates the observation that M(P132S) is associated with the resistant phenotype (Fig. 3e–g). Together, these data indicate that CIM-834 prevents the assembly of infectious viral particles by inhibiting oligomerization of the M protein.
To confirm these results in cells infected with SARS-CoV-2, the effect of CIM-834 was investigated by both thin-section transmission electron microscopy and electron tomography (Fig. 4g–i and Extended Data Figs. 2h and 3). Infected vehicle-treated cells (at 10 h after infection) displayed clusters of DMVs in the perinuclear region, numerous virions in maturation in the ERGIC and fully assembled virions in the vicinity of small, fragmented Golgi stacks (Fig. 4g,h). These stacks were enriched at the centre of the DMV clusters, at the outer periphery and in the proximity of the nuclear envelope (Fig. 4g,h and Extended Data Fig. 3a–e). Infected and CIM-834 treated cells contained a similar number of DMVs of normal sizes and with similar distribution (Fig. 4g with quantification in Fig. 4i; Extended Data Figs. 2h and 3f–i), which is consistent with the lack of inhibition of viral RNA replication by CIM-834 (Extended Data Fig. 2b). However, neither assembly sites nor virions were detected in cells treated with CIM-834 (Fig. 4g with quantification in Fig. 4i; Extended Data Figs. 2h and 3f–i). Instead, extended membrane compartments were observed in the periphery of the DMV clusters that were not detected in surrounding uninfected cells or in control non-infected cells treated with CIM-834 or vehicle (data not shown). These structures are characterized by concentric membrane layers, containing rounded protrusions as well as circular invaginations about 40–60 nm in size (Fig. 4g,h). The organization of these layers, and the presence of coated budding sites at their margins, indicates that these membranes might be an extended Golgi derivative, possibly generated by the accumulation of viral proteins owing to stalled virion formation. In nirmatrelvir-treated samples, no cells producing DMV clusters, virions or concentric membrane stacks could be detected using the same screening method (Fig. 4i).
Taken together, these mechanistic studies demonstrate that CIM-834 has a late mechanism of action and blocks coronavirus particle assembly. This is in line with the observations that CIM-834 does not block S-mediated entry or have an inhibitory effect on the activity of essential enzymes, such as the viral Mpro (the target of nirmatrelvir) or PLpro proteases, or on the viral RdRp (Nsp12, the target of remdesivir and molnupiravir), or on either Nsp14 or Nsp10/Nsp16 (the viral RNA methyltransferases) (Extended Data Fig. 2i).
Induced fit of CIM-834 in Mshort
We next demonstrated the direct binding of CIM-834 to purified SARS-CoV-2 M protein by using offline affinity selection–mass spectrometry (AS–MS) (Extended Data Fig. 4). Reproducible recovery of CIM-834 was obtained with the M protein relative to a buffer-only control. Then, cryo-electron microscopy (cryo-EM) was used to understand the binding mode of CIM-834 to the M protein. We used two conformationally selective antigen-binding antibody fragments, Fab-B and Fab-E, to stabilize the M protein in the short or long form, respectively1. Both M-Fab complexes were incubated with CIM-834, and the resulting cryo-EM structures were resolved at resolutions of 3.2 Å and 3.3 Å, respectively (Extended Data Figs. 5 and 6 and Extended Data Table 2). Incubation of CIM-834 with M–Fab-E complexes allowed us to resolve a structure of M–Fab-E without further density when compared with the apo structure, indicating that CIM-834 does not bind to the long form of the M protein (Extended Data Fig. 7). However, when CIM-834 was incubated with M–Fab-B complexes, further density, resolved to around 3.5 Å, was found at the boundary of the intra-virion part and the transmembrane (TM) region (situated between TM helices 2 and 3 and the hinge region), close to the dimer interface of Mshort (Fig. 5a–d and Extended Data Figs. 7h and 8). Although the map did not enable unambiguous assignment of the inhibitor pose, subsequent atomic modelling revealed a plausible binding mode of CIM-834 at this position (the rationale for which is described in the Methods). The inhibitor pocket is formed by M residues 36, 39, 40, 55, 95, 96, 99, 112–117 and 134, which participate in van der Waals interactions. CIM-834 binding is stabilized by a pi-stacking interaction with Y95 and putative hydrogen bonds with the side chains of Q36, W92, S99, N117 and the backbone carbonyl of E115 (Fig. 5e–g). Compared with published Mshort structures1,34, the side chains of Y39, Y95, F96 and F100 are reorientated to prevent steric clashes with CIM-834 (Fig. 5h,i). Except for local conformational changes in the CIM-834 binding pocket, the compound-bound Mshort is similar to previously reported Mshort structures1,34 (Fig. 5j), deviating by root mean square deviation (r.m.s.d.) values of 1.2 Å or 1.4 Å across all Cα atom pairs. These data indicate that CIM-834 specifically binds to, and stabilizes, Mshort and thus prevents the transition of Mshort to Mlong (Supplementary Video 3), thereby preventing the multimerization required for the assembly of virions. Interestingly, position P132 does not directly interact with CIM-834 but is juxtaposed with the hinge region (Fig. 5e–g). P132S may increase the flexibility of the M protein and restore the ability for short-to-long transitions in the presence of the inhibitor. An analysis of the amino acids in proximity to the CIM-834 binding pocket, and their conservation in the M protein sequence of different human coronaviruses, largely explains the antiviral spectrum of CIM-834 (Extended Data Fig. 9). The reference genome M protein sequence (NC_045512) has greater than 98% identity with the canonical SARS-CoV-2 variants, and 90% identity compared with SARS-CoV. The similarity drops to 33–38% for other betacoronaviruses (HCoV-HKU-1, MERS-COV and HCoV-OC43) and decreases further to 25–26% for alphacoronaviruses (HCoV-NL63 and HCoV-229E) (Extended Data Fig. 9d). It is noteworthy that sequence conservation seems to be higher in the vicinity (within 5 Å) of the bound CIM-834 compound, with a 72% identity rate for betacoronaviruses and 52–56% for alphacoronaviruses. This higher-than-average conservation rate around CIM-834 holds when comparing other (non-human) coronaviral sequences (Extended Data Fig. 9e). Interestingly, P132 is 100% conserved in both betacoronavirus and alphacoronavirus sequences (Extended Data Fig. 9c).

a,b, Cryo-EM map (a) and atomic model (b) of SARS-CoV-2 M protein in complex with Fab-B and CIM-834, showing M protein protomers (blue and purple), Fab-B (pink) and bound inhibitor (yellow). c, Two orthogonal views of the sharpened cryo-EM map for CIM-834 shown at a sigma level of 4.6. d, Cartoon representation of a single M protomer. The bound CIM-834, situated between transmembrane helices 2 and 3 and the hinge region, is shown as a surface representation. e, Atomic model of the M dimer in complex with CIM-834 showing the positions of the resistance mutations M91, S99, N117 and P132 (orange). f,g, Zoomed-in view of the CIM-834 binding site, with residues within 5 Å of the compound shown as sticks. Putative hydrogen bonds are shown as dashed lines. h, Atomic model of the CIM-834 binding site, showing residues within 5 Å of CIM-834 as pale transparent shapes. i, The equivalent view to h showing a short-form M dimer (PDB: 8CTK) in the absence of the inhibitor. j, Superposition of the M-CIM-834 complex (this study) with previously determined short-form structures (PDB: 8CTK (ref. 34) and 7VGS (ref. 1).
Discussion
The coronavirus Mpro and RdRp are, so far, the only targets in the coronavirus replication cycle that have been extensively explored and validated for inhibition by small-molecule therapeutics9,11. This has resulted in the Mpro inhibitors nirmatrelvir and ensitrelvir, and the RdRp-targeting molecules remdesivir and molnupiravir9,11. However, there may be many more excellent druggable targets in the coronavirus replication cycle, perhaps enzymes (such as the helicase and the mRNA capping machinery) or non-enzymatic (currently unknown) targets. To identify small-molecule inhibitors with a different mechanism of action as well as new druggable targets, we screened a structurally diverse compound library in a cell-based anti-SARS-CoV-2 assay. After hit identification and a medicinal-chemistry effort, we prioritized one chemical series, from which we selected CIM-834 as a probe compound for target deconvolution and in vivo proof-of-concept studies. CIM-834 exerts equipotent in vitro antiviral activity against a broad panel of SARS-CoV-2 variants and is also active against SARS-CoV. No antiviral activity was observed against alphacoronaviruses or against MERS-CoV. Oral dosing efficiently reduces infectious virus titres in the lungs of SCID mice intranasally infected with SARS-CoV-2, even when the start of treatment was delayed until 24 h before the end point. Despite completely blocking infectious viral titres in the lung, CIM-834 did not significantly reduce viral RNA levels when treatment began 48 h after infection. This aligns with the mechanism of action of CIM-834, which blocks assembly but does not inhibit vRNA replication. By the time treatment was initiated (48 h after infection), several replication cycles had occurred, resulting in high vRNA levels, whereas infectious titres remained low because virion formation was prevented. Finally, CIM-834 also inhibits SARS-CoV-2 replication in the lungs of hamsters, and treated animals no longer transmit the virus to untreated sentinels.
Serial passaging of SARS-CoV-2 in vitro in the presence of increasing concentrations of CIM-834 led to the selection of a P132S substitution in the viral M protein. Introduction of M(P132S) into the wild-type virus confirmed the role of M(P132S) in phenotypic resistance. Because the M protein is known to orchestrate coronavirus assembly, we explored whether CIM-834 inhibits viral assembly. In single-cycle time-of-drug-addition studies, the addition of CIM-834 can be delayed until late in the replication cycle without losing its ability to block the formation of infectious viruses. The fact that CIM-834 does not affect vRNA replication (which occurs before virus assembly) in single-replication-cycle assays further supports a late mechanism of action.
CIM-834 was next shown to inhibit the assembly of VLPs in cells that co-expressed the M and E proteins. The SARS-2 M protein typically forms dimers that further assemble into higher-order oligomers1. All M forms (monomers, dimers and oligomers) were detectable in the lysates of transfected cells. Interestingly, inhibition of VLP assembly by CIM-834 coincided with the loss of M oligomers in the lysates. A similar reduction in M oligomers was observed when M was expressed alone in the presence of CIM-834 (data not shown). This, together with the observation that there is direct binding of CIM-834 to the M protein, indicates that CIM-834 exerts a direct effect on M protein homo-oligomerization. To provide proof of the inhibition of SARS-CoV-2 assembly by CIM-834 in infected cells, transmission electron microscopy and electron tomography studies were done. In treated cells, no virions or intermediate assembly structures were observed. Interestingly, in infected CIM-834-treated cells, DMVs, which are the sites of vRNA synthesis, are as abundant as they are in infected untreated cells. Furthermore, in CIM-834-treated cells, in close proximity to DMV-enriched regions, extended membrane compartments are visible, which might correspond to sites of stalled virion formation where structural proteins such as the M protein accumulate.
It has been proposed that homotypic M–M interactions are required for coronavirus assembly to create a lattice-like matrix and ensure the incorporation of the other structural proteins into the virus particle35,36. Moreover, it has been suggested that a conformational switch from Mshort to Mlong and multimerization may have a role in the correct bending of the particle membrane1,2. The conformational switch of Mshort to Mlong may be triggered by interactions with the N protein, newly formed gRNA and the accumulation of the M, S and E proteins in the ERGIC membrane. Interactions between these viral factors result in oligomerization of the M protein, enabling the curving of the ERGIC membrane and finally virion formation1,2,25,26. Using AS–MS, we demonstrate that CIM-834 interacts directly with the M protein. Single-particle cryo-EM and atomic modelling revealed the binding pocket of CIM-834 in Mshort near the hinge region. Our findings indicate that this induced fit of CIM-834 in Mshort prevents the switch to Mlong, thereby blocking M-oligomerization and subsequently membrane curvature, and thus virion assembly (the proposed mechanism is shown in Fig. 4j). P132S is not part of the CIM-834 binding pocket, instead being located around 10 Å away, juxtaposed with the hinge region of the protein. P132S may increase the flexibility of the M protein and restore its ability for short-to-long transitions in the presence of the inhibitor. Interestingly, a recent preprint37 reports that Mshort can bind Golgi-enriched anionic lipids, including ceramide-1 phosphate, in the same site as CIM-834. Molecular-dynamics simulations indicate that this shifts the conformational equilibrium towards Mshort. A model is proposed in which Mshort is stabilized (in the early endomembrane system) by these lipids, whereby their release in a later phase triggers short-to-long conformation and subsequent virus assembly37.
During the writing of this manuscript, another preprint became available that reports on JNJ-9676 as a new, orally efficacious SARS-CoV-2 inhibitor. JNJ-9676 also targets the M protein and acts, similar to CIM-834, late in the replication cycle32. The resistance profile of JNJ-9676 partly overlaps with that of CIM-834, with S99A and P132S being associated with a high level of resistance for both compounds. Whereas the N117K substitution is associated with a complete loss of susceptibly to JNJ-9676 (more than 100-fold), this is limited for CIM-834 (5.4-fold). JNJ-9676 binds partly in the same pocket as CIM-834. The binding involves hydrogen-bond interactions with the side chains of N117 and S99. Both N117 and S99 have van der Waals interactions with CIM-834, with N117 having an extra hydrogen bond with the inhibitor. In both cases, the Y95 side chain is reorientated to accommodate the bound compound. The conformation of the JNJ-9676-bound M protein seems to be markedly different from that of either the published short or long forms, however32. It is not uncommon in antiviral drug discovery that phenotypic screens lead to the identification of different chemotypes that bind in the same pocket of a viral protein. Examples of such promiscuous drug pockets are the HIV non-nucleoside reverse transcriptase inhibitor pocket38 and the hydrophobic groove of the RSV F protein, which is the target of RSV fusion inhibitors39.
Taken together, our data identify CIM-834 as a potent inhibitor of SARS-CoV-2 and SARS-CoV replication. The class of M-targeting molecules to which CIM-834 belongs is currently being further optimized. We demonstrate that CIM-834 blocks the assembly of infectious virions by locking the M protein in its short form. This detailed structural information may allow further rational improvements of its potency and may allow expansion of the antiviral spectrum to MERS-CoV and other coronaviruses. Because the conservation rates of the M protein and residues in the CIM-834 pocket are similar to those found in the active site of the viral Mpro, the identified binding site represents a viable target for drug development.
Drugs with a new mechanisms of antiviral action will be ideally suited for combination therapies with current protease and polymerase inhibitors. We demonstrate that in vitro combinations of CIM-834 with nirmatrelvir and GS-441524 are additive (Extended Data Fig. 10). Such combinations may also prevent the development of drug-resistant variants. Although a single mutation is sufficient for resistance to CIM-834, we aim in the optimization process to develop analogues with a higher barrier to resistance.
Our insight into the binding mode of CIM-834, together with phenotypic antiviral testing, allows us to further facilitate the optimization of this compound class towards a clinical candidate.
In conclusion, our work highlights the value of antiviral drug and target discovery that is based on high-throughput phenotypic antiviral screening. Such an approach also holds the promise of identifying new inhibitors and, equally importantly, also new druggable targets for other viral families with epidemic and pandemic potential.
Methods
Ethics
Animal housing conditions and experimental procedures were approved by the ethics committee of animal experimentation of KU Leuven (licence P001/2021).
Compound synthesis and chemical characterization of CIM-834
The synthesis and characterization is described in the Supplementary Methods.
Antiviral molecules other than CIM-834
GS-441524 was obtained from MedChem Express (HY-103586). Hydroxychloroquine was purchased from Cell Signaling Technology (85523S). Nirmatrelvir (PF-07321332) was from Wuxi.
Cell lines
VeroE6–GFP cells (African monkey kidney cell line expressing green fluorescent protein; provided by M. van Loock, Janssen Pharmaceutica40) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS) + 0.5 mg ml−1 geneticin. VeroE6–mCherry cells (generated as described in ref. 41) were maintained in DMEM, supplemented with 10% (v/v) heat-inactivated FBS and 10 μg ml−1 blasticidin. The A549ACE2+TMPRSS2 cells (a human lung carcinoma cell line overexpressing human ACE2 and human TMPRSS2 receptors), used for antiviral studies, were from InvivoGen (a549d-cov2r, A549-Dual hACE2-TMPRSS2 cells) and were cultured in DMEM supplemented with 10% v/v heat-inactivated FBS, 300 μg ml−1 hygromycin, 0.5 μg ml−1 puromycin and 10 μg ml−1 blasticidin. A549ACE2+TMPRSS2 cells, used for subcellular studies, were generated in-house from A549 obtained from ATCC (CCL-185), using lentiviral transduction with pWPI vectors encoding ACE2 (selected using 500 μg ml−1 geneticin) and TMPRSS2 (selected using 1 μg ml−1 puromycin) under EF1alpha promotor control. These cells were cultivated in DMEM supplemented with 10% (v/v) FBS, 100 U ml−1 penicillin, 10 µg ml−1 streptomycin and 1% non-essential amino acids. All assays using VeroE6–GFP, VeroE6–mCherry and A549ACE2+TMPRSS2 involving virus replication were performed in the respective cell growth medium containing 2% (instead of 10%) FBS. All cell cultures were done at 37 °C and 5% CO2. BHK-21 cells (Baby hamster kidney cell line obtained from ATCC, CCL10) were maintained in Glasgow MEM (Invitrogen) supplemented with 5% v/v FBS, 10% tryptose phosphate broth, 100 U ml−1 penicillin, 100 µg ml−1 streptomycin and 10 mM HEPES, pH 7.4. For transfection experiments, cells were maintained in Eagle’s minimal essential medium (EMEM), as described42. Huh7 cells were cultured in DMEM supplemented with 10% FBS and 1 mM Glutamax.
A549TMPRSS2+DPP4 cells (A549 obtained from ATCC (CCL-185), developed in-house to overexpress human TMPRSS2 and DPP-4) were cultured in DMEM supplemented with 5% FBS, penicillin (100 U ml−1), streptomycin (100 μg ml−1), blasticidin (10 µg ml−1) and geneticin (0.1 mg ml−1). DMEM with 5% FBS, penicillin (100 U ml−1) and streptomycin (100 μg ml−1) was used for MERS-CoV infection experiments. HCT-8 cells (human colorectal carcinoma cell line obtained from ATCC, CCL-244) were cultured in DMEM supplemented with 5% FBS. DMEM + 3% FBS was used for HCoV-OC43 infection experiments. CRFK cells (Crandell-Rees feline kidney cell line obtained from ATCC, CCL-94) were cultured in DMEM supplemented with 5% FBS, penicillin (100 U ml−1) and streptomycin (100 μg ml−1). The same medium was used for experiments involving FIPV infection. LLC-MK2 cells (a rhesus monkey kidney epithelial cell line, obtained from ATCC, CCL-7) were cultured in MEM with Hanks’ and Earle’s salts supplemented with 5% FBS. MEM with Hanks’ and Earle’s salts and 3% FBS was used for HCoV-NL63 infection experiments. Vero cells (ATCC, CCL-81) were cultured in DMEM supplemented with 5% FBS, penicillin (100 U ml−1) and streptomycin (100 μg ml−1). DMEM with penicillin (100 U ml−1), streptomycin (100 μg ml−1) and 1 µg ml−1 trypsin was used for PEDV infection experiments. All cell lines tested negative for mycoplasma contamination.
Viruses
The SARS-CoV-2 GHB strain (GHB-03021/2020, GISAID: EPI_ISL_407976 | 2020-02-03) was recovered from a nasopharyngeal swab taken from an asymptomatic patient returning from Wuhan, China. Virus stocks were inoculated on HuH-7 cells and then passaged seven times on VeroE6–eGFP cells. GHB has a ΔTQTNS deletion at residues 676–680 that is typical for SARS-CoV-2 strains passaged on VeroE6 cells43. SARS-CoV-2 strains belonging to the alpha, beta, delta and omicron variants of concern were recovered from nasopharyngeal swabs of human cases confirmed by quantitative PCR with reverse transcription (RT–qPCR). Virus stocks were generated by passaging the virus on Calu-3 cells followed by production of a screening virus stock on A549ACE2+TMPRSS2 cells. The sequences of the passage 0 of these strains are available through GISAID: Alpha/B.1.1.7 (hCoV19/Belgium/rega-12211513/2020; EPI_ISL_791333), beta/B.1.351 (hCoV19/Belgium/rega-1920/2021; EPI_ISL_896474); Delta/B.1.617.2 (EPI_ISL_2425097); Omicron BA.5 (EPI_ISL_14782497); and XBB1.5 (EPI_ISL_17273054). The sequences of BA.2.86 (SARS-CoV-2/hu/DK/SSI-H135) and EG.5.1 (SARS-CoV-2/hu/DK/SSI-H121) variants are available in the European Nucleotide Archive under the project number PRJEB67449 with accession numbers OY747653 and OY747654, respectively. The SARS-CoV-2 BavPat1/2020 strain was provided by C. Drosten (Charité Berlin) through the European Virology Archive (Ref-SKU: 026V-03883). This virus, which has a D614G mutation in the S protein and a mutation affecting the furin cleavage site, owing to passaging on VeroE6 cells, has been used previously for subcellular studies22. Recombinant SARS-CoV-2–mNeonGreen virus (Wuhan strain)44 was a gift from V. Thiel (University of Bern). SARS-CoV-2 USA-WA1/2020 (EPI_ISL_404895) was used for the hamster efficacy study. This virus was obtained through BEI Resources (ATCC, NR-52281, batch 70036318). This isolate is closely related to the prototypic Wuhan-Hu-1 2019-nCoV (GenBank accession 112 number: MN908947.3) strain, as confirmed by phylogenetic analysis.
rSARS-CoV-2 WT-USA-WA1, rSARS-CoV-2 WT-USA-WA1-3CLpro: L50F, rSARS-CoV-2 WT-USA-WA1- 3CLpro: T21I-D263G, rSARS-CoV-2 WT-USA-WA1-3CLpro: L50F- E166A L167V have been described previously30. SARS-CoV, strain 200300592, was obtained from the Centers for Disease Control and Prevention. Live SARS-CoV-2 and SARS-CoV work was done in the high-containment A3 and biosafety level 3 facilities of the KU Leuven Rega Institute (3CAPS) under licences AMV 30112018 SBB 219 2018 0892 and AMV 23102017 SBB 219 20170589, according to institutional guidelines. All work with live SARS-CoV (strain Frankfurt-1) and MERS-CoV (strain Jordan N3/2012) virus was done in a biosafety level 3 laboratory at the Leiden University Medical Center.
Other coronaviruses used in this study are: MERS-CoV isolate England 1, identifier: 1409231v, National Collection of Pathogenic Viruses, Public Health England, United Kingdom (studies performed at Jagiellonian University); HCoV-OC43 (ATCC VR-1558), FIPV strain 79-1146 (granted by G. Tekes, Justus Liebig University Giessen); HCoV-NL63 isolate Amsterdam 1 (GenBank: AY567487.2)45; PEDV CV777 (granted by C. M. Lia van der Hoek, University of Amsterdam46). A short description of the antiviral assays performed with these viruses is provided in Extended Data Table 1.
SARS-CoV-2 antiviral and toxicity assays
VeroE6–GFP cells were seeded at a density of 25,000 cells per well in 96-well plates (Greiner Bio One, 655090) and pretreated with three-fold serial dilutions of the compounds overnight in the presence of the MDR1 inhibitor CP-100356 (final concentration, 0.5 μM). The next day (day 0), cells were infected with SARS-CoV-2 inoculum at a multiplicity of infection (MOI) of 0.001 median TCID50 per cell. The number of fluorescent pixels of GFP signal determined by high-content imaging on day 4 after infection was used as a read-out. The percentage inhibition was calculated by subtracting the background (the number of fluorescent pixels in the untreated − infected control wells) and normalizing to the untreated − uninfected control wells (also background subtracted). The EC50 was determined by logarithmic interpolation. A similar protocol was used to determine the antiviral activity in A549ACE2+TMPRSS2 (InvivoGen) cells, but no MDR1 inhibitor CP-100356 was used and the cell viability was determined four days after infection using viability staining with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). The percentage of antiviral activity was calculated by subtracting the background and normalizing to the untreated − uninfected control wells (also background subtracted), and the EC50 was determined using logarithmic interpolation. In both cell lines, the potential toxicity of compounds was assessed in a similar set-up in treated − uninfected cultures in which metabolic activity was quantified at day 5 using the MTS assay. The 50% cytotoxic concentration (CC50, the concentration at which cell viability reduced to 50%) was calculated by logarithmic interpolation.
Antiviral assays with other coronaviruses
For the antiviral assays with HCoV-NL63, FIPV, PEDV, HCoV-OC43 and MERS-CoV, cells were seeded the day before infection. The next day, serial compound dilutions were mixed with virus (1:1 v/v), incubated for 30 min at room temperature and overlaid on confluent cells. The cells were then incubated for 2 h with the relevant virus at 37 °C (for FIPV, PEDV and MERS-CoV) or 32 °C (HCoV-OC43 and HCoV-NL63). Cells were washed twice with PBS, after which compound containing medium was added and the cells were further incubated at 37 °C (for FIPV, PEDV and MERS-CoV) or 32 °C (HCoV-OC43 and HCoV-NL63) for 3–5 days and cell-culture supernatant was collected for RT–qPCR. For the HCoV-229E assay, three-fold serial dilutions of the compounds in assay medium were made in 96-well plates. After that, Huh7 cell suspension was added to assay wells at a density of 10,000 cells per well and cells were infected with HCoV-229E at an MOI of 0.01. The cell viability was determined 5 days after infection using viability staining with MTS. For the SARS-CoV CPE reduction assays, VeroE6 cells (11,000 cells per well), and for MERS-CoV CPE reduction assays, HuH-7 (10,000 cells per well), were seeded in 96-well plates the day before infection. The next day, two-fold serial dilutions of compound in infection medium with 2% FCS (and 0.25 µM p-GP inhibitor CP-100356 for VeroE6 cells) were added to cells (eight dilution series in parallel), and half of the plate was infected with SARS-CoV Frankfurt-1 (300 plaque-forming units per well) or MERS-CoV Jordan-N3/2012 (200 plaque-forming units per well). The other half of the plate was mock infected to determine the possible cytotoxic effect of the compounds in parallel. Three (for SARS-CoV) or two (MERS-CoV) days after infection, the CellTiter 96 aqueous non-radioactive cell-proliferation kit (Promega) was used to measure the cell viability of infected (n = 4) and non-infected cells (cytotoxicity, n = 4) by absorbance measurement at 495 nm with an Envision plate reader (Perkin Elmer). The values of the wells were normalized to the average signal of uninfected untreated cells (100%). The EC50 and CC50 values were determined by nonlinear regression.
Activity in SARS-CoV-2-infected SCID mice
Nirmatrelvir (from Excenen, batch EXA5024) was formulated as a 100 mg ml−1 and 33.3 mg ml−1 stock (for 300 mg per kg and 100 mg per kg dosing, respectively) in a vehicle containing 43% absolute ethanol and 27% propylene glycol (Sigma) in sterile distilled water. CIM-834 was formulated as 20 mg ml−1 in 14% propylene glycol (Sigma), 1% Tween 80 (Sigma), 85% pH 5 citrate buffer. To evaluate in vivo efficacy, male SCID mice (CB-17/Icr-Prkdcscid/scid/Rj; Janvier Laboratories) 7–9 weeks old were treated by oral gavage with either the vehicle (n = 12, twice a day) or CIM-834 at 100 mg per kg (n = 12 twice a day and n = 12 once a day) or nirmatrelvir at 300 mg per kg (n = 12, twice a day) or 100 mg per kg (n = 6, twice a day), starting from day 0, just before infection with the beta variant B.1.351 (hCoV-19/Belgium/rega-1920/2021; EPI_ISL_896474, 2021-01-11). For virus infection, animals were anesthetized with isoflurane and inoculated intranasally with 40 µl containing 105 TCID50 SARS-CoV-2 beta variant (day 0). In the therapeutic set-up, animals were infected on day 0 and treatment with CIM-834 (100 mg per kg, twice a day) was initiated 24 h, 30 h or 48 h after infection. Mice were housed in individually ventilated cages with three mice per cage and monitored daily for weight changes and any clinical signs. At day 3 after infection, animals were euthanized by intraperitoneal injection of 100 µl Dolethal (200 mg ml−1 sodium pentobarbital, Vétoquinol SA), and the lungs were collected. Infectious viral lung loads were quantified by end-point virus titration. To prevent carry-over of the compound during determination of infectious virus titres, the cells were washed and given fresh medium, immediately after incubation for 2 h with lung homogenates. Cells were subsequently incubated at 37 °C for three days before TCID50 read-out.
Activity in SARS-CoV-2-infected Syrian hamsters
The hamster infection model of SARS-CoV-2 has been described previously47. Female Syrian hamsters (Mesocricetus auratus) 8–10 weeks old from Janvier Laboratories were kept in twos in individually ventilated isolator cages (IsoCage N Bio-containment System, Tecniplast) at 21 °C, 55% humidity and 12 h:12 h day:night cycles. Housing conditions and experimental procedures were approved by the ethics committee of animal experimentation of KU Leuven (licence P065−2020). For infection, animals were anesthetized with ketamine/xylazine/atropine and inoculated intranasally with 50 µl containing 1 × 104 TCID50 of SARS-CoV-2 USA-WA1/2020. Hamsters were treated by oral gavage with either the vehicle, CIM-834+Ritonavir (100 + 50 mg per kg) or nirmatrelvir (300 mg per kg), twice daily starting from day 0, directly preceding the infection with SARS-CoV-2. All the treatments continued until day 3 after infection. On day 4, hamsters were euthanized by intraperitoneal injection of 500 μl Dolethal (200 mg ml−1 sodium pentobarbital, Vétoquinol SA). Lungs were collected and viral RNA and infectious virus were quantified by RT–qPCR and end-point virus titration, and the left lung lobes were fixed in 4% formaldehyde for histopathological analysis. Two independent studies were performed, each with a total of n = 8 for the vehicle group, CIM compound group and nirmatrelvir group. In parallel, we assessed whether treatment of infected animals with CIM-834/Ritonavir could prevent transmission of infection to non-infected contact hamsters. For this, starting in the afternoon of day 1 after infection, each index infected hamster, treated with either vehicle (n = 4) or CIM-834 + Ritonavir (100 + 50 mg per kg, n = 8), was co-housed with untreated sentinel hamsters in one cage, and the co-housing continued until the day of euthanization, three days after the first exposure.
Histology
For histological examination, the lungs were fixed overnight in 4% formaldehyde and embedded in paraffin. Tissue sections (5 μm) were analysed after staining with haematoxylin and eosin and scored blindly for lung damage by a pathologist. The scored parameters, for which a score of 0–3 was attributed, were the following: congestion, intra-alveolar haemorrhagic, apoptotic bodies in the bronchus wall, necrotizing bronchiolitis, perivascular oedema, bronchopneumonia, perivascular inflammation, peribronchial inflammation and vasculitis. The sum of the different scores was then reported as a cumulative score.
In vitro resistance selection
SARS-CoV-2 B.1.1.7 was passaged in A549ACE2+TMPRSS2 (InvivoGen) cells in the presence of increasing concentrations of CIM-834. Selection was initiated at 0.03 μM (around 1/3 of its EC50) and, for each passage, the virus was cultured at the same concentration, and at 3×, 10× and 30× higher concentrations. The culture with the highest compound concentration that still showed virus breakthrough, as observed by a significant cytopathic effect, was then used for the next passage. At passage 5 (day 17), vRNA in the cell-culture medium was sequenced.
Genotypic analysis
Illumina sequencing
RNA was extracted from the cell-culture supernatant using a NucleoSpin RNA virus kit (Macherey-Nagel) according to the manufacturer’s instructions. Whole-genome sequencing was outsourced to Eurofins Genomics (ARTIC SARS-CoV-2 WGS, Konstanz), who performed reverse transcription, enrichment of the viral genome using a primer set similar to the ARTIC primers (more than 200 primer pairs, covering the full 29.9-kilobase viral genome), generation of libraries, Illumina sequencing (2 × 150 base-pair read mode) and sequence cleaning to remove adapters and poor-quality bases. Sequences were further analysed using Geneious Prime software (v.2022.2.1) by mapping to the SARS-CoV-2 reference sequence (NC_045512), and variant calling was performed as described by the software manufacturer (https://help.geneious.com/hc/ en-us/articles/360045070991-Assembly-of- SARS-CoV-2-genomes-from- tiled-amplicon-Illumina- sequencing-using-Geneious-Prime).
Generation and rescue of recombinant viruses
Reverse-engineered SARS‐CoV‐2 Wuhan with the M-protein P132S substitution was constructed by mutating nucleotide positions 26916–26918 from CCG to AGC in a bacterial artificial chromosome vector containing a full-length cDNA copy of the genome of SARS-CoV-2 strain SARS-CoV-2/human/NLD/Leiden-0008/2020 using the following primer pair for two-step en passant recombineering in Escherichia coli48:
5′-TCTTCTCAACGTGCCACTCCATGGCACTATTCTGACCAGAAGCTTGCTAGAAAGTGAACTCGTAATGGCGTATCACGAGGCCCTTTCGTC-3′ and
5′-CACGAAGGATCACAGCTCCGATTACGAGTTCACTTTCTAGCAAGCTTCTGGTCAGAATAGTGCCATTAGGGATAACAGGGTAATGGCCTG-3′.
With the design of the primers, a silent marker mutation was introduced at nucleotide positions 26919 and 26921 changing the CUU codon to UUG. Two independent mutant BACs were sequenced across the M-protein gene and 5 µg DNA was linearized with NotI. Full-length RNA was obtained by in vitro transcription with T7 RNA polymerase followed by lithium chloride precipitation according to the manufacturer’s protocol (mMessage-mMachine T7 kit, Ambion). Synthetic mRNA encoding the SARS-CoV-2 N protein was generated as described previously49. To launch SARS-CoV-2 wild type and mutant, 5 µg of full-length SARS-CoV-2 RNA and 10 µg of N-protein mRNA were electroporated into 2 × 106 BHK-21 cells using the AMAXA Nucleofector 2b (program A-031) and Nucleofection T solution kit (Lonza). Transfected cells were mixed with 4 × 106 VeroE6 cells, and cells were incubated at 37 °C until the full cytopathic effect was observed. From this passage 0 virus stock, a passage 1 stock was prepared on Calu-3 cells as described50. The presence of the AGC codon at nucleotide positions 26916–26918, encoding M(P132S), was confirmed by Illumina sequencing (see above). This sequencing also showed that no other mutations had been accidently introduced in the viral backbone.
Reverse-engineered SARS‐CoV‐2 BF.7 (Omicron) was constructed, starting from the seven-plasmid reverse-genetics system51 for strain SARS-CoV-2/USA_WA1/2020 (a gift from P. Y. Shi through the World Reference Center for Emerging Viruses and Arboviruses). An RNA extract was made from a clinical isolate of SARS‐CoV‐2 Omicron subvariant BF.7 (donated by P. Maes, Rega Institute), using the QIAamp viral RNA kit (Qiagen). Next, cDNA was made with random hexamer primers and the Superscript IV RT First-Strand synthesis system (Invitrogen). The cDNA was used as a template to make seven PCR fragments covering the viral genome and containing an overlap with the target plasmids, using Platinum SuperFi PCR (Invitrogen). Using the NEBuilder HiFi DNA assembly cloning kit (New Biolabs), the WA1 sequences in the seven plasmids51 were replaced by the Omicron-encoding PCR fragments. The M-protein substitution P132S was introduced by Q5 site-directed mutagenesis (New England Biolabs). M-protein substitutions M91K, S99A and N117K were introduced by site-directed mutagenesis using Platinum SuperFi II DNA polymerase (Invitrogen).
All plasmids were validated by Sanger sequencing (Macrogen). Preparation of viral DNA, in vitro transcription and electroporation of BHK-21 cells was carried out as previously described51, except for the use of an ECM 830 Square Wave electroporation system (850 V, 3 pulses of 0.30 ms, a 3 s interval, BTX). The electroporated cells were added to A549ACE2+TMPRSS2 cells (InvivoGen) in medium containing 10% FCS. After incubation for 6 h at 37 °C, the medium was replaced by medium with 0.2% FCS. Four days later, the virus stocks were collected, passaged once on A549ACE2+TMPRSS2 (InvivoGen) and subjected to whole-genome sequencing (Oxford Nanopore Technologies, by B. Vanmechelen, Rega Institute) to verify the desired sequence.
SARS-CoV-2 studies in human nasal airway epithelial cultures
Previously described procedures52 were followed. In brief, human airway nasal epithelial cells (MucilAir pool of donors, product code EP02) were obtained from Epithelix in an air–liquid set-up. After arrival, the inserts were washed with pre-warmed 1× PBS and maintained in MucilAir medium (Epithelix, EP04MM) at 37 °C and 5% CO2 for at least four days before use. On the day of the experiment, the cultures were pretreated with basal medium containing compounds at different concentrations for 1 h before infection with 100 μl SARS-CoV-2 inoculum (1,000 TCID50 per insert) at the apical side for 1.5 h, after which the viral inoculum was removed. Viral release from the cultures was measured by washing the apical sides with 250 µl Mucilair medium and determination of the viral load by RT–qPCR or titration. Compound-containing medium in the basolateral side of the cultures was refreshed on day 2 after infection. All incubations from start of the infection were done at 35 °C and 5% CO2.
Quantification of vRNA using RT–qPCR
Viral RNA from apical washes (air–liquid culture inserts) or from cells was isolated using the Cells-to-cDNA II buffer kit (Thermo Fisher Scientific, AM8723). In brief, 5 μl wash fluid was added to 50 μl lysis buffer or cells were lysed in Cell lysis buffer (2,500 cells per μl Cell Lysis II Buffer); samples were incubated at room temperature for 10 min and then at 75 °C for 15 min. Next, 150 μl nuclease-free water was added to the mixture before RT–qPCR. In parallel, a ten-fold serial dilution of corresponding virus stock was extracted using the same protocol, and it could then later function as a standard curve. The amount of viral RNA was quantified by RT–qPCR using an iTaq universal probes one-step kit (Bio-Rad, 1725141) and a commercial mix of primers for the N-protein gene (forward primer, 5′-GACCCCAAAATCAGCGAAAT-3′; reverse primer, 5′-TCTGGTTACTGCCAGTTGAATCTG-3′) and probes (5′-FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1-3′) from IDT Technologies (10006606). The reaction (a final volume of 20 μl) consisted of 10 μl one-step reaction mix 2×, 0.5 µl RT enzyme, 1.5 μl of a primers and probes mix, 4 μl nuclease-free water and 4 μl of sample. The RT–qPCR was executed on a Lightcycler 96 thermocycler (Roche), starting at 50 °C for 15 min and 95 °C for 2 min, followed by 45 cycles of 3 s at 95 °C and 30 s at 55 °C. By correlating the Ct values of the ten-fold serial dilution of the virus stock with known titre (standard curve), the amount of vRNA from the samples could be expressed as a TCID50 equivalent per cell-culture insert (TCID50 equivalent per insert).
Time-of-drug-addition assay
VeroE6 cells were seeded in 12-well plates the day before infection. Cells were infected with an MOI of 1 TCID50 per cell (SARS-CoV-2 GHB) and compounds were added 0, 3, 5 and 7 h after infection. At 10 h after infection, virus-containing supernatant was removed and cells were either lysed for quantification of vRNA levels by RT–qPCR (see above) or cells were collected for determination of intracellular infectious virus titres by plaque assay (see below). For the time-of-drug-addition assay with reporter cells and reporter virus, VeroE6–mCherry cells were seeded in 96-well plates the day before infection. Cells were infected with SARS-CoV-2–mNeonGreen virus (at a final MOI of 0.1 TCID50 per cell) and compounds were added −0.5, 0, 2, 4, 6 and 8 h after infection. At 10 h after infection, cells were imaged using high-content imaging and the number of infected cells was calculated, after which the percentage inhibition relative to the DMSO-treated control was calculated.
Determination of infectious virus titres
Cells were trypsinized and resuspended in PBS, after which they were freeze-thawed 3× on liquid nitrogen to release intracellular viral particles. Cellular debris was removed by centrifugation (5 min at 13,400 rpm and 4 °C) and 50 µl of the supernatant was used to determine the infectious titre by plaque assay.
For this, 5-fold dilutions of the cell lysate supernatant were added to VeroE6 cell monolayers in 12-well plates and incubated at 37 °C for 1 h. Subsequently, the inoculum mixture was replaced with 0.8% (w/v) methylcellulose in DMEM supplemented with 2% FBS. After three days of incubation at 37 °C, the overlays were removed, the cells were fixed with 3.7% PFA and stained with 0.5% crystal violet, and plaques were counted visually.
Plasmid construction for SARS-CoV-2 expression of M and E protein
The coding sequences of the SARS-CoV-2 M and E proteins were cloned between the BamHI and EcoRI restriction sites of a pCDNA3.1(+) vector, containing a coding sequence for an amino-terminal V5 tag or a carboxy-terminal HSV tag for the M and E proteins, respectively. To this end, cDNA obtained after reverse transcription of RNA extracted from SARS-CoV-2 (strain LillehCoV-19/France/HDF-IPL/2020, GenBank MW575140) infected cells was amplified by PCR using Q5 High-Fidelity 2X Master Mix (New England Biolabs) with forward primer 5′-CGGGATCCGCAGATTCCAACGGTACTATTA-3′ and reverse primer 5′-CAGAATTCTTACTGTACAAGCAAAGCAATA-3′ for M, and forward primer 5′-TCGGATCCGCCACCATGTACTCATTCGT-3′ and reverse primer 5′-TGGAATTCGACCAGAAGATCAGGAACTC-3′ for E. To allow better visualization of the M protein on western blot, an M-N5Q glycosylation site mutant was used in all VLP studies. This M-N5Q glycosylation site mutant was generated by site-directed mutagenesis PCR with forward primer 5′-CGGGATCCGCAGATTCCCAAGGTACTATTACCGTT-3′ and reverse primer 5′-CAGAATTCTTACTGTACAAGCAAAGCAATA-3′. The M(P132S) mutation was introduced by fusion PCR using the primers 5′-CTTTCTAGAAGCGATCTGGTCAGAATAGTGCCAT-3′ and 5′-TTCTGACCAGATCGCTTCTAGAAAGTGAACTCGT-3′. For E, a silent mutation removing the intrinsic EcoRI restriction site was inserted at nucleotide position 195 (G > A) by fusion PCR using the primers 5′-GAATTTAGATTTTTAACACGAGAGTA-3′ and 5′-AATCTAAATTCTTCTAGAGTTCCTGA-3′.
SARS-CoV-2 VLP assay and western blot analysis
Huh-7 cells53 were seeded in 100-mm dishes at a concentration of 2 × 106 cells per dish. The next day, cell-culture medium was changed to medium containing various concentrations of CIM-834 (0.2, 1.0 or 5.0 µM) or DMSO. Cells were co-transfected with 2 µg of plasmid encoding for wild-type or mutant (M(P132S)) protein and 1 µg of E-HSV-encoding plasmid using TransIT-LT1 Transfection Reagent (Mirus Bio). Then, 48 h after transfection, VLPs and cell lysates were collected and stored as described previously54 for MERS-CoV VLPs. VLP and lysate samples were resuspended in reducing Laemmli loading buffer and separated on a 12% polyacrylamide gel by SDS–PAGE. M proteins were visualized by immunoblotting using a primary anti-V5 antibody (Thermo Fisher Scientific, R96025, clone SV5-Pk1, 1:1,000) and secondary HRP-labelled goat anti-mouse IgG antibodies (Jackson ImmunoResearch, 115-035-146, 1:10,000), as described previously54. Tubulin was used as a loading control and stained using a mouse anti-β-tubulin IgG1 antibody (Sigma-Aldrich, T5201, clone TUB 2.1, 1:2,000). Western blot bands were quantified using Image J and its band quantification function. For unprocessed blots, see Supplementary Fig. 1.
Sample preparation for thin section and tomography TEM
A549ACE2+TMPRSS2 cells (generated in-house) were cultured in 24-well plates on either glass coverslips for microwave-assisted chemical processing or on 3-mm flat sapphire discs 50 μm thick (Engineering Office M. Wohlwend, Switzerland) for high-pressure freezing. One hour after infection with SARS-CoV-2 (strain BavPat1/2020), the cells were treated with 1 µM CIM-834 or nirmatrelvir or a DMSO control. Mock-inoculated cells treated with DMSO or nirmatrelvir or CIM-834 were processed in parallel. Cells were collected at 10 h after infection by fixation in two steps. First, by adding a 2× concentrated EM fixative to the cell-culture medium in a 1:1 ratio for 15 min at room temperature, and subsequently, a 1× concentrated fixative for 120 min at room temperature. The composition of the 1× fixative was 2.5% glutaraldehyde and 1% formaldehyde in 50 mM Na-cacodylate buffer (pH 7.2) containing 50 mM KCl, 2.6 mM MgCl2, 2.6 mM CaCl2 and 2% sucrose. Plates were plunged in a bath of 6% EM-grade formaldehyde in PHEM buffer (pH 7.4) for virus inactivation for 30 min at room temperature and kept at 4 °C until further processing on the next day. Samples on coverslips were further processed at room temperature using a PELCO BioWave Pro+ microwave equipped with vacuum chamber (set to 20 hg) and water-cooling platform (set at 23 °C). Samples were rinsed six times with 100 mM Na-cacodylate for 10 min each, then post-fixed in a microwave in 65 mM Na-cacodylate buffer, 1% OsO4 at 150 mW, cycling power on and off every 2 min for seven cycles, under vacuum. Samples were then rinsed four times in double-distilled water (twice for 10 min on the bench and twice for 40 s each time in the microwave at 250 mW, vacuum off) and subsequently treated in 1% uranyl acetate in double-distilled water in a BioWave for seven cycles at 2 min each, cycling power on (100 mW) and off at each cycle, under vacuum. Dehydration with an ethanol series (50%, 70%, 90% and 2 × 100%) was then performed for 40 s each step, at 100 mW, vacuum off. Coverslips were finally transferred to BEEM capsules on fresh 100% Epon 812 resin and polymerized at 60 °C for 48–76 h.
Cells prepared on sapphires for high-pressure freezing were prefixed and exported from BSL3 in 6% PFA, similarly to the samples on coverslips. The samples were then rinsed six times with PHEM buffer (100 mM), immersed in 100 mM PHEM with 15% BSA as a cryo-protectant and high-pressure frozen using a BalTec HPM-010 with carriers forming a 40 µm-deep cavity (3-mm aluminium carriers, type B 0/0.3 mm and type 748 0.04/0.020 mm, Engineering Office M. Wohlwend). High-pressure frozen samples were then subjected to freeze substitution in a Leica AFS II using cryo-tubes with screw caps and rubber sealing rings, containing 1 ml of fixative cocktail composed of 0.2% OsO4, 1% uranyl acetate and 5% water in acetone. The AFS chamber temperature was increased over the course of 24 h as follows: 1 h at −90 °C; 8 h at −90 °C to −80 °C; 8 h at −80 °C to −50 °C; 2 h at −50 °C to −20 °C; 2 h at −20 °C to 0 °C. Samples were then rinsed for 5 min with dry acetone on ice and further processed using a microwave, rinsed twice with ethanol for 40 s at 250 mW and infiltrated with increasing Epon 812 concentrations in ethanol (10%, 30%, 50%, 70%, 90% and 2 × 100% for 3 min each in a BioWave vacuum cycling system). At the last 100% step in Epon 812, sapphires were transferred to AFS plastic moulds and polymerized at 60 °C for 48–72 h. Ultrathin and semithin sections of 70 nm or 300 nm, respectively, were generated for both chemically fixed and high-pressure frozen samples using a UC7 Leica ultramicrotome and a 30° diamond knife (Diatome) and collected on Pioloform-coated slot grids. Grids were post-stained for 5 min with 3% uranyl acetate in 70% methanol and 2 min with lead citrate. To locate the infected cells, the Serial-EM Navigator functionality and a procedure adapted from ref. 55 were used to map the central section of a ribbon of 5 on a JEOL 1400 equipped with a TemCam-F416 camera. Cells displaying a DMV cluster were identified and stored in the map, and navigator maps and grids were then transferred to a JEOL 2100+ equipped with a Matataki sCMOS camera to acquire montages, covering the whole perinuclear region of the selected cells at a magnification of ×6,000 or ×15,500.
For tomography, semithin sections of 200 nm or 300 nm were screened and imaged using a Tecnai F30 microscope (Thermo Fisher Scientific) equipped with a Gatan OneView camera. Target positions were manually selected and acquired at ×15,500 magnification (−60° to +60° per axis; increment, 1°) by single-axis tomography (0.78 nm per pixel). The tilt series were reconstructed using IMOD56,57,58. Segmentation of selected tomograms was done manually using the brush segmentation tool in Amira-Avizo software v.2020.1 (ThermoFisher), Volume renderings and animations were also computed with the same software.
Offline affinity selection mass spectrometry
We incubated 5 µM M protein for 30 min at 25 °C in buffer only (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.001% LMNG, 0.0001% CHS, 0.00033% GDN, supplemented with 2% DMSO) or in buffer with CIM-834 (in DMSO, 2% final concentration). Unbound compound was removed from the bound compound to protein by Zeba Spin Desalting columns, 7 K MWCO, according to the manufacturer’s protocol. All experiments were done in triplicate. Mass-spectrometry measurements were done on an Agilent 6546 Quadrupole Time-of-Flight for liquid chromatography with tandem mass spectrometry. For data analysis, the Agilent Mass Hunter Qualitive Analysis software (target screening workflow) and the Find by Formula method for compound identification by mass/isotope pattern matching was used. An EIC peak area of [M + H]+, [M+Na]+ and [M + K]+ masses was extracted using a mass error tolerance window of 10 ppm.
Sample preparation for cryo-EM to study CIM-834–M interactions
For both the Fab–E and Fab–B complexes, M protein was combined with CIM-834 and either Fab–E or Fab–B, resulting in a final concentration of 44.44 µM M protein, 100 µM CIM-834 and Fab–E/B in a 2.2 molar excess of M protein. All components were diluted in a buffer solution comprising 20 mM HEPES-NaOH at pH 7.7, 150 mM NaCl, 0.0025% LMNG and 0.00025% CHS. Samples were mixed and incubated for 15 min on ice before vitrification. Approximately 3.5 µl of the sample was pipetted onto glow-discharged R1.2/1.3 200 mesh holey copper carbon grids (Quantifoil) and then plunge-frozen in liquid ethane using a Vitrobot mark IV (Thermo Fisher Scientific). Both datasets were collected at the Netherlands Center for Electron Nanoscopy. Grids were loaded into a Titan Krios electron microscope (Thermo Fisher Scientific) operating at 300 kV, equipped with a K3 direct electron detector and Bioquantum energy filter (Gatan). The slit width of the energy filter was set to 20 eV. Imaging was done at a nominal magnification of ×81,000 and ×105,000 for Fab–E and Fab–B, respectively, in super-resolution mode using EPU software (Thermo Fisher Scientific). A total of 5,058 and 5,037 movies were recorded for the Fab–E and Fab–B complexes, respectively. Detailed data-acquisition parameters are summarized in Extended Data Table 2.
Single-particle image processing to study CIM-834–M interactions
For the Fab–E and Fab–B complexes, patch motion correction, using an output F-crop factor of 0.5, and patch CTF estimation were performed in cryoSPARC59. Micrographs with a CTF estimated resolution of worse than 10 Å were discarded, leaving 5,058 and 5,036 images for further processing, respectively. The blob picker tool was then used to select 4,327,548 and 4,497,785 particles, respectively, which were then extracted in an 80-pixel box (Fourier binned 4.5 × 4.5). For the Fab–E complex, two rounds of 2D classification were done, resulting in 270,970 particles being selected for further processing. Ab initio reconstruction generated one well-defined reconstruction of the Fab–E complex. Particles belonging to this class were then re-extracted in a 300-pixel box. During extraction, particles were Fourier binned by a non-integer value, resulting in a final pixel size of 1.00 Å. Subsequently, non-uniform refinement was done on the extracted particles with C2 symmetry imposed60, yielding a final reconstruction with global resolution of 3.3 Å. Similarly, for the Fab–B complex, two rounds of 2D classification were done, resulting in 187,608 particles being selected for further processing. Ab initio reconstruction generated one well-defined reconstruction of the Fab–B complex. To identify all Fab–B complex particles from the dataset, all particles picked by the blob picking tool (4,497,785 particles) were used for five rounds of heterogeneous refinement, using the reconstruction of the Fab–B complex from the initial ab initio job as a reference volume for high-resolution particles to be further isolated. This was followed by two final rounds of ab initio to further isolate the best-quality particles for final reconstruction. The remaining 73,185 particles were then re-extracted in a 360-pixel box. During extraction, particles were Fourier binned by a non-integer value, resulting in a final pixel size of 0.836 Å. Subsequently, non-uniform refinement was done on the extracted particles with C2 symmetry imposed60, yielding a reconstruction with a global resolution of 3.3 Å. Reference-based motion correction was successfully run on 72,998 of the re-extracted particles. These particles were then subjected to a final non-uniform refinement, with C2 symmetry imposed60, yielding a final reconstruction with a global resolution of 3.2 Å. The gold standard Fourier shell correlation (FSC) criterion (FSC = 0.143) was used for calculating all resolution estimates, and 3D-FSC plots were generated in cryoSPARC61. To enable model building, globally refined maps were filtered by local resolution in cryoSPARC.
Modelling to study CIM-834–M interactions
Initially, a previously determined SARS-CoV-2 M structure (PDB: 8CTK; ref. 34) and an Alphafold2 model62 of Fab–B were rigid-body fitted into the M–Fab–B-CIM map using the UCSF Chimera Fit in map tool63. An initial round of molecular-dynamics flexible fitting was then done on the combined model using Namdinator64 and then manually adjusted in Coot65. During the latter step, the CIM-834 compound was added using the ligand builder tool. When selecting the pose of CIM-834, the following were taken into account. First, the morphology of the density. The flat plate-like morphology of the density orientated away from the M two-fold symmetry axis was consistent with the piperidine and pyrimidine rings of CIM-834. The density for the opposing end of the density was thinner and less well resolved (Extended Data Fig. 6), consistent with the increased flexibility of the molecule beyond the amide position. Second, the positioning of hydrogen-bonding partners. The selected orientation of CIM-834 positions the pyridazine ring in proximity to S99 and N117, allowing for potential direct or water-mediated hydrogen bonds. The model was then refined by performing iterative cycles of manual model building using Coot65 and real-space refinement using Phenix66; eLBOW was used to generate ligand restraints for CIM-834 (ref. 67). Model validation was done using Molprobity68,69,70.
Analysis and visualization to study CIM-834–M interactions
Interactions between M and CIM-834 were calculated using LigPlot71 and UCSF ChimeraX72. The UCSF Chimera MatchMaker tool was used to obtain root mean square deviation values using default settings. Figures were generated using UCSF ChimeraX72, and structural-biology applications used in this project were compiled and configured by SBGrid73.
Spike pseudotyped vesicular stomatitis virus entry assay
A human codon-optimized gene encoding the spike protein of SARS-CoV-2 S ancestral Wuhan-Hu-1 (GenBank: NC_045512.2) was synthesized by GenScript. SARS-CoV-2 S pseudo-typed vesicular stomatitis virus (VSV) and neutralization assays were reframed as described previously74. In brief, to produce SARS-CoV-2 S VSV, confluent HEK-293T cells were transfected with pCAGGS expression vectors encoding SARS-CoV-2 S with a C-terminal 18-residue cytoplasmic tail truncation, to increase cell surface expression. Then, 48 h after transfection, cells were infected with VSV G pseudotyped VSVΔG bearing the firefly luciferase reporter gene. Then, 24 h later, supernatant containing SARS-CoV-2 S pseudotyped VSV was collected and clarified, and the virus was titrated on Vero-E6 cells. For the virus-neutralization assay, CIM-834 and control monoclonal antibody 87G7 were serially diluted three-fold and five-fold, respectively, before pre-incubating with equal volumes of SARS-CoV-2 S pseudotyped VSV at room temperature for 1 h. After incubation, Vero-E6 cells were inoculated with the mixture and cells were incubated at 37 °C for 20 h. Subsequently, cells were washed once with PBS and lysed with passive lysis buffer (Promega). Firefly luciferase expression was measured on a Berthold Centro LB 960 plate luminometer with d-luciferin as a substrate (Promega). The percentage of virus neutralization was calculated as the ratio of the reduction in luciferase read-out in the presence of compound normalized to luciferase read-out in its absence. The half-maximal inhibitory concentrations (IC50) were determined using four-parameter logistic regression (GraphPad Prism v.8.3.0).
Immunofluorescence
Huh-7 cells were seeded on coverslips in 24-well plates at a concentration of 8 × 104 cells per well. The next day, 1 µM CIM-834 or DMSO was added to the medium and cells were transfected with a total of 250 ng plasmid encoding for V5-tagged WT-M using the TransIT-LT1 transfection reagent (Mirus Bio). Then, 16 h after transfection, cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, followed by permeabilization with 0.1% Triton-X100 in PBS for 3 min at room temperature. After a blocking step with 10% normal horse serum for 15 min at room temperature, V5-tagged M proteins were visualized by incubation with monoclonal anti-V5 antibodies (Thermo Fisher Scientific, 37-7500, clone 2F11F7, 1:500) in 10% normal horse serum, followed by incubation with Alexa Fluor 488-conjugated donkey anti-mouse IgG secondary antibodies (Jackson ImmunoResearch, 715-545-151, 1:500). The trans-Golgi network was visualized using polyclonal sheep anti-human TGN46 antibodies (AHP500, BioRad,1:500), followed by incubation with Cy3-conjugated donkey anti-sheep IgG antibodies (Jackson ImmunoResearch, 713-165-147, 1:500). Nuclei were visualized using 1 µg ml−1 of 4′,6-diamidino-2-phenylindole (DAPI), and coverslips were mounted in Mowiol mounting medium. Images were acquired with an LSM 880 confocal laser scanning microscope (Zeiss) using a ×63 oil-immersion objective lens. The extent of colocalization between M and TGN46 was quantified by calculating the Pearson’s correlation coefficient using the JACoP plug-in of ImageJ. For both treated (1 µM CIM-834) and untreated (DMSO) conditions, 25 cells were analysed.
Expression and purification of recombinant proteins
The coding sequences for SARS-CoV-2 Wuhan-Hu-1 (GenBank: NC_045512.2) were used for recombinant protein expression. Nsp3 (PLpro domain), nsp15 (Mpro domain), nsp14 (N7-MTase), nsp10 and nsp16 (2′O-MTase) were cloned in fusion with an N-terminus hexa-histidine tag, as previously described75. The proteins were expressed in E. coli and purified by affinity using IMAC cobalt bead. In brief, after cell sonication in a buffer containing 50 mM Tris, pH 6.8, 300 mM NaCl, 10 mM imidazole, 5 mM MgCl2 and 1 mM BME, supplemented with 0.25 mg ml−1 lysozyme, 10 μg ml−1 DNase and 1 mM PMSF, the proteins were purified through affinity chromatography with HisPur Cobalt resin 480 (Thermo Scientific), washed with an increased concentration of salt (1 M NaCl) and imidazole (20 mM), before elution in a buffer supplemented with 250 mM imidazole. The MTases were subsequently purified by size-exclusion chromatography (GE Superdex S200) in a final buffer of 50 mM Tris, pH 6.8, 300 mM NaCl, 5 mM MgCl2 and 1 mM BME. The proteins were concentrated using Vivaspin 20 centrifugal concentrators with a 10 kDa MWCO (GE Healthcare, VS2001), dialysed against the elution buffer in the absence of imidazole, and stored at −20 °C in a buffer containing 50% glycerol. The replication/transcription complex endowed with RdRp activity was purified and assembled, according to ref. 76, with variations described in ref. 77.
Determination of Mpro inhibition by CIM-834
A fluorescent substrate comprising the cleavage site of SARS-CoV-2 Mpro (Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2) and buffer composed of 20 mM HEPES, 120 mM NaCl, 0.4 mM EDTA, 4 mM DTT, 20% glycerol, pH 7.0, was used for the inhibition assay. In the fluorescence resonance energy transfer (FRET)-based cleavage assay78, the fluorescence signal of the Edans generated as a result of the cleavage of the substrate by Mpro was observed at an emission wavelength of 460 nm with excitation at 360 nm using a Tecan Spark multimode microplate reader. The compound was diluted with 100% DMSO to prepare a stock solution. The IC50 of CIM-834 was determined after incubation of 0.5 µM of SARS-CoV-2 Mpro and CIM-834 at various concentrations from 0 to 500 µM in reaction buffer at 37 °C for 10 min. At the end, the FRET substrate at a final concentration of 50 µM was added to each well at a final total volume of 100 µl to initiate the reaction. The IC50 value was calculated with the help of the GraphPad Prism 9.2.0 software. Inhibitory activity of the compound was measured in triplicates and data are presented as mean ± s.d. Two positive control assays (nirmatrelvir and ensitrelvir) against Mpro were also performed simultaneously.
Determination of inhibition of PLpro by CIM-834
Activity and inhibition were determined by FRET, performed in black 384-well HiBase non-binding plates (Greiner Bio One, 784900), as previously described75. In brief, increasing concentrations of inhibitor were incubated with purified PLpro protein (55 nM) in the presence of 5 μM of a fluorescent synthetic peptide (Dabcyl-FTLKGG↓APTK-Edans, Genscript) in HEPES buffer (20 mM, pH 6.5) containing 120 mM NaCl, 0.4 mM EDTA, 4 mM DTT and 10% glycerol. The final concentration of DMSO was adjusted to 0.5%. Cleavage of the fluorogenic peptide separates the Edans/Dabcyl fluorophore–quencher pair. The time courses of the enzymatic reaction were followed for 40 min by monitoring the increase in fluorescence emission at 493 nm (excitation at 335 nm) using a Tecan Safire2 fluorimeter. Enzymatic activities were estimated by calculating the slope of the linear part of the reaction curve and were normalized with respect to the activity measured in the absence of inhibitor.
RNA synthesis inhibition and IC50 determination
The compound concentrations leading to 50% inhibition of polymerase-mediated RNA synthesis was determined in IC50 buffer (50 mM HEPES, pH 8.0, 10 mM KCl, 2 mM MnCl2, 2 mM MgCl2 and 10 mM DTT) containing seven increasing concentrations of compound (from 1 µM to 100 µM) and 150 nM of nsp12 (ref. 76) in complex with 450 nM nsp8 and 450 nM nsp8L7 (ref. 77).
Reactions were done in a volume of 40 µl on a 96-well Nunc plate. All experiments were robotized by using a BioMek I5 automate (Beckman). Then, 2 µl of each diluted compound in 100% DMSO was added in wells to the chosen concentration (5% DMSO final concentration). For each assay, the (nsp8L7 + nsp8) mix was distributed in wells after an 8-min incubation at room temperature to pre-form the active complex. Nsp12 was then added to wells and incubated for 8 min. Reactions were started by adding the UTP + poly(A) template mix and were incubated at 30 °C for 20 min, using 350 nM of poly(A) template and 750 µM of UTP final concentration.
Reaction assays were stopped by adding 20 µl EDTA (100 mM). Positive and negative controls consisted of a reaction mix with 5% DMSO (final concentration) or EDTA (100 mM) instead of compounds, respectively. Reaction mixes were then transferred to a Greiner plate using a Biomek I5 automate (Beckman). PicoGreen fluorescent reagent was diluted to 1/800 in TE buffer, according to the manufacturer’s instructions, and 60 μl of reagent was distributed into each well of the Greiner plate. The plate was incubated for 5 min in the dark at room temperature and the fluorescence signal was then read at 480 nm (excitation) and 530 nm (emission) using a Tecan Safire2 and/or a ClarioStar.
The IC50 was determined using this equation: percentage of active enzyme = 100/(1 + I2/IC50), where I is the concentration of inhibitor and 100% of activity is the fluorescence intensity without the inhibitor. The IC50 was determined from curve fitting using Prism software. For each measurement, results were obtained in triplicate.
N7 and 2′O-MTase assay
Purified SARS-CoV-2 nsp14 (N7-MTase) and nsp10/nsp16 (2′O-MTase) were incubated in a reaction mixture containing 40 mM Tris-HCl (pH 8.0), 1 mM DTT, 1 mM MgCl2, 2 μM SAM and 0.1 μM 3H-SAM (Perkin Elmer), in the presence of either 0.7 μM GpppAC4 or 7m GpppAC4 synthetic RNA79. The compounds were suspended in 50% DMSO (2.5% final DMSO concentration). Reactions were carried out at 30 °C and stopped after 30 min by ten-fold dilution in ice-cold water. Samples were transferred to a diethylaminoethyl filter mat (Perkin Elmer) using a Filtermat Harvester (Packard Instruments). The RNA-retaining mats were washed twice with 10 mM ammonium formate (pH 8.0), twice with water and once with ethanol. They were soaked with scintillation fluid (Perkin Elmer), and 3H-methyl transfer to the RNA substrates was determined using a Wallac MicroBeta TriLux liquid scintillation counter (Perkin Elmer).
Metabolic stability in mouse, hamster and human liver microsomes
Compound microsomal stability was determined using mouse microsomal fractions (Gibco; final protein concentration, 0.5 mg ml−1), with hamster microsomal fractions (Xenotech; final protein concentration, 0.5 mg ml−1) and with human microsomal fractions (Xenotech; final protein concentration, 0.5 mg ml−1) at a substrate concentration of 1 μM with or without NADPH (final concentration, 1 mM). Incubations were done at 37 °C for 120 min in duplicates. At various time points (5, 15, 30, 60 and 120 min), 25 μl of the reaction mixture was sampled and quenched in 300 μl of acetonitrile containing internal standard. The resulting suspension was vortexed and centrifuged, and the supernatant was diluted with water (two-fold). The resulting solution was analysed by liquid chromatography with tandem mass spectrometry to determine the half-life of the compound.
Statistics and reproducibility
All statistical comparisons in the study were done in GraphPad Prism (v.6.07, v.8.3.0, v.9.2.0, v.10.1.2 and v.10.2.3) and the statistical tests used are indicated in the figure legends. The 2D EMs for quantification of DMVs were acquired from three independent preparations. Tomography was acquired on a selection of technical duplicates of the samples previously characterized in 2D, with the exception of the HPF samples that were prepared in duplicates in one preparation.
Preparation of figures
All graphs were prepared using GraphPad Prism (v.6.07, v.8.3.0, v.9.2.0, v.10.1.2 and v.10.2.3). Figures and schemes were created using BioRender.com and Adobe Illustrator 28.1 (Windows).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the article or the Supplementary Information. The synthesis and chemical characterization of the compound described in this paper is provided as in the Supplementary Methods. Uncropped images of the western blots shown in Fig. 4d–f are in Supplementary Fig. 1. The atomic model of the SARS-CoV-2 M protein dimer (short form) in complex with Fab-B and CIM-834 has been deposited to the Protein Data Bank under the accession code 9EXA. The globally refined cryo-EM maps of SARS-CoV-2 M protein dimer (long form) in complex with Fab-E and incubated with CIM-834, and the SARS-CoV-2 M protein dimer (short form) in complex with Fab-B and CIM-834, have been deposited in the Electron Microscopy Data Bank under accession codes EMD-50034 and EMD-50035. The TEM micrographs, tomograms and all other data are available from the corresponding author(s) on reasonable request. Source data are provided with this paper.
References
Zhang, Z. et al. Structure of SARS-CoV-2 membrane protein essential for virus assembly. Nat. Commun. 13, 4399 (2022).
Neuman, B. W. et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 174, 11–22 (2011).
Bracquemond, D. & Muriaux, D. Betacoronavirus assembly: clues and perspectives for elucidating SARS-CoV-2 particle formation and egress. mBio 12, e0237121 (2021).
de Wit, E., van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14, 523–534 (2016).
Our World in Data. Estimated cumulative excess deaths per 100,000 people during COVID-19, Jun17, 2024. https://ourworldindata.org/explorers/coronavirus-data-explorer.
World Health Organization. Post COVID-19 condition (long COVID). https://www.who.int/europe/news-room/fact-sheets/item/post-covid-19-condition (2022).
Halford, B. The path to paxlovid. ACS Cent. Sci. 8, 405–407 (2022).
Hammond, J. et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 386, 1397–1408 (2022).
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Kabinger, F. et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 28, 740–746 (2021).
Toussi, S. S., Hammond, J. L., Gerstenberger, B. S. & Anderson, A. S. Therapeutics for COVID-19. Nat. Microbiol. 8, 771–786 (2023).
Mackman, R. L. et al. Discovery of GS-5245 (obeldesivir), an oral prodrug of nucleoside GS-441524 that exhibits antiviral efficacy in SARS-CoV-2-infected African green monkeys. J. Med. Chem. 66, 11701–11717 (2023).
Gao, M. et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 (2010).
Kaptein, S. J. F. et al. A pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction. Nature 598, 504–509 (2021).
Goethals, O. et al. Blocking NS3–NS4B interaction inhibits dengue virus in non-human primates. Nature 615, 678–686 (2023).
Steiner, S. et al. SARS-CoV-2 biology and host interactions. Nat. Rev. Microbiol. 22, 206–225 (2024).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Mandala, V. S. et al. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 27, 1202–1208 (2020).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738 (2020).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226 (2008).
Snijder, E. J. et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80, 5927–5940 (2006).
Cortese, M. et al. Integrative imaging reveals SARS-CoV-2-induced reshaping of subcellular morphologies. Cell Host Microbe 28, 853–866 (2020).
Wolff, G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398 (2020).
Arndt, A. L., Larson, B. J. & Hogue, B. G. A conserved domain in the coronavirus membrane protein tail is important for virus assembly. J. Virol. 84, 11418–11428 (2010).
Sergio, M. C., Ricciardi, S., Guarino, A. M., Giaquinto, L. & De Matteis, M. A. Membrane remodeling and trafficking piloted by SARS-CoV-2. Trends Cell Biol. 34, 785–800 (2024).
Lu, S. et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 12, 502 (2021).
Boson, B. et al. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 296, 100111 (2021).
Chiu, W. et al. Development and optimization of a high-throughput screening assay for in vitro anti-SARS-CoV-2 activity: evaluation of 5676 phase 1 passed structures. J. Med. Virol. 94, 3101–3111 (2022).
Wang, A. Q. et al. Preclinical pharmacokinetics and in vitro properties of GS-441524, a potential oral drug candidate for COVID-19 treatment. Front. Pharmacol. 13, 918083 (2022).
Jochmans, D. et al. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. mBio 14, e0281522 (2023).
Abdelnabi, R. et al. A SCID mouse model to evaluate the efficacy of antivirals against SARS-CoV-2 infection. J. Virol. 96, e0075822 (2022).
Van Damme, E. et al. A small-molecule SARS-CoV-2 inhibitor targeting the membrane protein. Nature https://doi.org/10.1038/s41586-025-08651-6 (2025).
Koch, J. et al. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 40, e107821 (2021).
Dolan, K. A. et al. Structure of SARS-CoV-2 M protein in lipid nanodiscs. eLife 11, e81702 (2022).
de Haan, C. A., Vennema, H. & Rottier, P. J. Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J. Virol. 74, 4967–4978 (2000).
Ujike, M. & Taguchi, F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses 7, 1700–1725 (2015).
Dutta, M. et al. Direct lipid interactions control SARS-CoV-2 M protein conformational dynamics and virus assembly. Preprint at bioRxiv https://doi.org/10.1101/2024.11.04.620124 (2024).
De Corte, B. L. From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepin-2(1H)-one (TIBO) to etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 48, 1689–1696 (2005).
Battles, M. B. et al. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 12, 87–93 (2016).
Ivens, T. et al. Development of a homogeneous screening assay for automated detection of antiviral agents active against severe acute respiratory syndrome-associated coronavirus. J. Virol. Methods 129, 56–63 (2005).
Chiu, W. et al. Development of a robust and convenient dual-reporter high-throughput screening assay for SARS-CoV-2 antiviral drug discovery. Antiviral Res. 210, 105506 (2023).
Ogando, N. S. et al. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J. Virol. 94, e01246-20 (2020).
Boudewijns, R. et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 11, 5838 (2020).
Thi Nhu Thao, T. et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 582, 561–565 (2020).
van der Hoek, L. et al. Identification of a new human coronavirus. Nat. Med. 10, 368–373 (2004).
Pensaert, M. B. & de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 58, 243–247 (1978).
Abdelnabi, R. et al. Nirmatrelvir-resistant SARS-CoV-2 is efficiently transmitted in female Syrian hamsters and retains partial susceptibility to treatment. Nat. Commun. 14, 2124 (2023).
Tischer, B. K., Smith, G. A. & Osterrieder, N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634, 421–430 (2010).
Thiel, V. et al. Multigene RNA vector based on coronavirus transcription. J. Virol. 77, 9790–9798 (2003).
Lamers, M. M. et al. Human airway cells prevent SARS-CoV-2 multibasic cleavage site cell culture adaptation. eLife 10, e66815 (2021).
Kurhade, C., Xie, X. & Shi, P.-Y. Reverse genetic systems of SARS-CoV-2 for antiviral research. Antiviral Res. 210, 105486 (2023).
Do, T. N. D. et al. A robust SARS-CoV-2 replication model in primary human epithelial cells at the air liquid interface to assess antiviral agents. Antiviral Res. 192, 105122 (2021).
Nakabayashi, H., Taketa, K., Miyano, K., Yamane, T. & Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42, 3858–3863 (1982).
Desmarets, L. et al. The KxGxYR and DxE motifs in the C-tail of the Middle East respiratory syndrome coronavirus membrane protein are crucial for infectious virus assembly. Cell. Mol. Life Sci. 80, 353 (2023).
Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y. & Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Mastronarde, D. N. & Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 197, 102–113 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF Chimera−a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kidmose, R. T. et al. Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 (2009).
Agirre, J. et al. Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 (2015).
Dialpuri, J. S. et al. Analysis and validation of overall N-glycan conformation in Privateer. Acta Crystallogr. D Struct. Biol. 79, 462–472 (2023).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020).
Chen, K. Y. et al. A highly sensitive cell-based luciferase assay for high-throughput automated screening of SARS-CoV-2 nsp5/3CLpro inhibitors. Antiviral Res. 201, 105272 (2022).
Eydoux, C. et al. A fluorescence-based high throughput-screening assay for the SARS-CoV RNA synthesis complex. J. Virol. Methods 288, 114013 (2021).
Campagnola, G., Govindarajan, V., Pelletier, A., Canard, B. & Peersen, O. B. The SARS-CoV nsp12 polymerase active site is tuned for large-genome replication. J Virol. 96, e0067122 (2022).
Zhu, L., George, S., Schmidt, M. F., Al-Gharabli, S. I., Rademann, J. & Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 92, 204–212 (2011).
Bouvet, M. et al. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 6, e1000863 (2010).
Ianevski, A., He, L., Aittokallio, T. & Tang, J. SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics 33, 2413–2415 (2017).
Acknowledgements
We thank C. De Keyzer, L. Bervoets, S. Hendrickx, K. Abbasi, R. Van Berwaer, J. Vandeput, J. Zevenhoven, W. Noteborn, B. Vinayagam, S. Cumps and J. Meerkens for technical assistance; V. Vergote and E. Heylen for the operational management of the animal and 3CAPS facility at KU Leuven; A. Calleja and D. Marchand for assistance with analytical chemistry (quality control and preparative purifications); A. Kilonda for organic-chemistry activities; L. Steegmans for compound plating; L. Watters for support with data management; and K. Castermans for project management support; C. Drosten and the European Virus Archive for providing the SARS-CoV-2 BavPat1/2020 isolate; the Electron Microscopy Core Facility (EMCF) at the European Molecular Biology Laboratory, in particular M. Schorb for technical support; the EMCF at Heidelberg University for assistance with electron-microscopy analyses, especially U. Haselmann for assistance with sample preparation (the EMCF of Heidelberg University is a Deutsche Forschungsgemeinschaft (DFG)-registered research infrastructure, RI 00565); L. Rouilly, A. Delpal, J.-C. Guillemot, B. Selisko, F. Ferron and A. Shannon for help with the Nsp5, Nsp3, Nsp14, Nsp10/Nsp16 and RdRp assays. Funding: KU Leuven, CISTIM, Leiden University Medical Center, Utrecht University, Jagiellonian University, Aix-Marseille University, CNRS and University of Lübeck acknowledge funding through the CARE project from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement 101005077; the JU receives support from the European Union’s Horizon 2020 research and innovation program, EFPIA and the Bill & Melinda Gates Foundation, the Global Health Drug Discovery Institute and the University of Dundee; the content of this publication reflects the author’s view and the JU is not responsible for any use that may be made of the information it contains; KU Leuven, CISTIM, Leiden University Medical Center, Utrecht University, Aix-Marseille University, CNRS and University of Lübeck acknowledge funding through the European Union’s Horizon 2020 research and innovation program under grant agreements 101003627 (SCORE project), 101137229 PanViPrep and 101102733 DURABLE; KU Leuven also acknowledges funding from the Covid-19 Fund from KU Leuven/UZ Leuven. Part of this research work was performed using the Caps-It research infrastructure (project ZW13-02), which was financially supported by the Hercules Foundation and the Rega Foundation, KU Leuven. KU Leuven also acknowledge funding by the Belgian federal government for the VirusBank Platform and funding from National Institutes of Health grant R01AI177512-01 and the Bill & Melinda Gates Foundation under grant agreement INV-006366. Work at Utrecht University is supported by the Netherlands Electron Microscopy Infrastructure), project 184.034.014 of the National Roadmap for Large-Scale Research Infrastructure of the Dutch Research Council (NWO) and by the Beijerinck Premium of the M. W. Beijerinck-Virology Fund, Royal Netherlands Academy of Arts and Sciences (KNAW), awarded to D.L.H. Work at Heidelberg University was supported by the DFG, project 240245660- SFB 1129 and project 272983813- TRR 179, by the Project Virological and immunological determinants of COVID-19 pathogenesis − lessons to get prepared for future pandemics (KA1-Co-02 COVIPA), a grant from the Helmholtz Association’s Initiative and Networking Fund and by the German Center for Infection Research (DZIF), TTU 05.712, and by the CoCID project (101017116) funded within the European Union Research and Innovation act. Y.S. was supported by the Hector Fellow Academy. Work at the University of Tokyo is partly supported by JSPS KAKENHI (grant 22K15046 (to Z.Z.) and 22H02556 (U.O.). Work at Kyoto University is partly supported by JSPS KAKENHI (grant JP23H02724 to N.N.), the Joint Usage/Research Center Program of the Institute for Life and Medical Sciences at Kyoto University (N.N.), the AMED Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS, JP23ama121007). Work at the Center for Infection and Immunity of Lille is supported by the French National Research Agency (grant ANR-22-CE15-0030-01).
Author information
Authors and Affiliations
Contributions
M.L., D.J. and J.N. planned, coordinated and performed (M.L. and D.J.) the experimental virology work. R.A. planned and coordinated the animal experiments. P.L., W.C. and S.D.J. planned and coordinated work at the Caps-It infrastructure. N.C., K.D., T.F., T.V.B., J.R. and J.S. did experimental virology work. A.S. reverse engineered rBF.7-WT, M(M91K), M(S99A), M(N117K) and M(P132S), and L.N. supervised this. D.B. and J.-C.V. planned, coordinated and executed the medicinal chemistry work at CISTIM. M.K. and P.A. did organic synthesis. H.K. coordinated the primary high-content screen. A.M. coordinated and guided the medicinal chemistry, ADME and PK activities at CISTIM. S.Boland performed modelling. I.V. performed binding studies with purified M protein, J.W. provided project-management support. P.C. coordinated and guided overall drug discovery-related aspects around the discovery of CIM-834. L.D. performed VLP assays. Supervised by S.Belouzard and J.D. D.L.H. and O.J.D.-A. planned, coordinated and executed the cryo-EM work. O.J.D.A. performed pseudotype assays, supervised by B.-J.B. and F.V.K. Z.Z., U.O. and T.S. prepared recombinant SARS-CoV-2 M protein. N.N. generated Fab fragments. G.M. and Y.S. executed the EM work, supervised by R.B. B.W.B. and G.J.M.G. reverse engineered and characterized rWuhan-WT and M(P132S), supervised by E.J.S. P.W. did experiments with MERS-CoV and SARS-CoV, supervised by M.J.v.H. A.Z. performed antiviral assays with HCoV-NL63, HCoV-OC43, PEDV and FIPV. K.O. developed the A549 cell line overexpressing TMPRSS2 and DPP-4, and did the antiviral assay with MERS-CoV, supervised by K.P. S.J. did Mpro enzymatic assays, supervised by R.H. C.E. and E.D. performed Nsp5, Nsp3, Nsp14, Nsp10/Nsp16 and RdRp assays, supervised by B.C. B.W. did the lung histology analysis. Joint efforts of different institutes: J.N. and P.C. designed and initiated the project. J.N., P.C., A.M. and P.L. secured funding. M.L., D.J., D.L.H., O.J.D.-A., L.D., G.M. and R.B. prepared the figures. M.L., D.J. and J.N. wrote the manuscript with important contributions from D.L.H., O.J.D.-A., L.D., G.M. and R.B. and with comments from all authors.
Corresponding author
Ethics declarations
Competing interests
KU Leuven has filed a patent application claiming the discovery of a class of antiviral molecules to which CIM-834 belongs as SARS-CoV-2 replication inhibitors. The authors declare no other competing interests.
Peer review
Peer review information
Nature thanks Yunjeong Kim, Shengyong Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Microsomal stability, mouse and hamster PK parameters, CYP inhibition and in vivo efficacy of CIM-834.
a, Liver microsomal stability was determined as described in the materials and methods. For PK studies, CIM-834 was formulated as a solution in propylene glycol/Tween80/pH 5 citrate buffer (14/1/85) and administered to male CD-1 mice at 10 or 100 mg/kg, or administered to female hamsters at 100 mg/kg after a pre-treatment of ritonavir as a solution formulated in EtOH/propylene glycol/water (43/27/30) at 50 mg/kg. RoA: route of administration. CL, clearance; Vss, steady-state volume of distribution; Cmax, maximum concentration; AUClast, area under the curve up to the last measurable concentration. F was calculated based on AUClast iv and po and corrected for dose. ND: not determined. b, IC50 of CIM-834 on different CYP enzymes. c, SCID mice were intranasally infected with 105 TCID50 SARS-CoV-2 B.1.351 and treated orally once (QD) or twice (BID) daily with CIM-834 or nirmatrelvir. No ritonavir was used in either of the treated groups. Treatment was initiated just before infection. Lung infectious titres, vRNA and body weight change were determined 3 days post-infection (Kruskal-Wallis test and Dunn’s comparison, median values (infectious virus and vRNA) or mean ± s.d, (weight change), n = 2 biologically independent experiments with 6 mice in each group/experiment).
Extended Data Fig. 2 CIM-834 blocks coronavirus particle formation.
a, Setup of the time-of-drug-addition assay (TOA). b,c, Ten hr p.i. intracellular vRNA b, and infectious viral particles c, were quantified (mean ± s.d., n = 3 biologically independent experiments, one-way ANOVA followed by Dunnett’s multiple comparisons with the DMSO control). d, TOA assay using VeroE6-H2B-mCherry cells and rSARS-CoV-2-mNeonGreen. e, Quantification of %infected cells (mNeonGreen+ and mCherry+ VeroE6 cells) relative to the DMSO control (mean ± s.d., n = 6 biologically independent experiments). f, Virus kinetics experiment with the same setup (compounds added at 0 h p.i.) and quantification of infected cells at different time points. In a-f, hydroxychloroquine was tested at 10 µM, CIM-834 and nirmatrelvir at 1 µM. g, CIM-834 does not influence the co-localization of M and TGN46, a marker for the trans-Golgi network. Data (mean ± s.d.) from two independent experiments. h, TEM (top) and tomographic reconstruction (bottom) of cytoplasmic regions of DMSO- or CIM-834(1 µM)-treated SARS-CoV-2 infected cells (10 h p.i.). a1, a2, Details of boxed regions highlighted in Fig. 4g, showing additional examples of assembly sites located both at the center of the DMV cluster and at its periphery, in proximity of the terminal region of Golgi cisternae (G). m, mitochondrion. b1, b2: Magnified regions from Fig. 4g, showing concentric stacks of membranes (orange dashed lines) detected in CIM-834 treated cells and their proximity to DMVs (*). c1,c2: Slices from tomographic acquisition of DMSO (c1) and CIM-834-treated cells (c2), highlighting the regions magnified respectively below (d, f) or in Fig. 4h (c, e). Section thickness: 70 nm (a1,a2,b1,b2). Tomographic section thickness: 0.78 nm (c1,c2). i, Inhibition of the activities of purified, recombinant enzymes assayed in vitro in the presence of CIM-834 and appropriate reference compounds.
Extended Data Fig. 3 Tomographic analysis of SARS-CoV-2 infected A549ACE2+TMPRSS2 cells (10 h p.i.).
a, Example electron micrograph of a high-pressure frozen and freeze substituted DMSO-treated infected cell. Yellow arrowheads indicate assembling virions and black stars (*) indicate DMVs. b,c, Detail of the regions boxed in panel a, showing a strong contrast of the assembling viral ribonucleoprotein at progressive stages of assembly. The tomographic slice in panel b corresponds to a diagonally resliced tomographic volume capturing a view of three adjacent assembly sites. Three-dimensional segmentations of the progressively assembling virions are shown on the right of the tomographic sections. Light blue volumes represent DMVs, light green the ERGIC/Golgi membranes containing the virions. The virions’ color coding indicates stages of budding: early (yellow), intermediate (orange) and complete (red). Viral RNPs are represented in purple. d,e, Further examples of virions in very early and late stages, respectively taken from the tomographic volume in a at a z height not visible in the overview. f, Tomographic slice through an infected and CIM-834 (1 µM)-treated cell showing complete absence of virions and assembly events, despite a high abundance of DMVs of similar size and distribution as observed in DMSO-treated cells. Darkened, elongated mitochondria (m) are present in the area surrounding the DMVs. g, Segmentation of the volume displayed in f, showing the spatial relationship of DMVs (light blue) and mitochondria (pink), confirming the absence of assembly sites throughout the volume. h, i Magnified tomographic sections of the boxed regions in panel f. Tomographic section thickness: 0.78 nm (a,f), average of 5 × 0.78 nm thick slices (b,c,h,i).
Extended Data Fig. 4 CIM-834 binds directly to SARS-CoV-2 M protein.
a, Offline affinity selection mass spectrometry (AS-MS) of 5 µM M protein with 5 µM CIM-834 (blue) or buffer control (purple), (mean ± s.d., n = 3 independent experiments). b, Example of extracted ion chromatogram (EIC) for CIM-834 with 10 ppm mass tolerance with M protein (blue) and buffer control (purple). c, MS spectrum corresponding to the EIC peak for CIM-834 with M protein (blue).
Extended Data Fig. 5 Single-particle cryo-EM data processing pipeline for the FabE complex.
Schematic representation of the single-particle cryo-EM data processing pipeline for the FabE complex. Major steps include particle picking, 2D classification, initial model generation, 3D classification, refinement, and map sharpening. The final reconstruction yielded a 3.3 Å resolution map. Additional details are described in the single-particle image processing section of the materials and methods. CTF: contrast transfer function.
Extended Data Fig. 6 Single-particle cryo-EM data processing pipeline for the FabB complex.
Schematic representation of the single-particle cryo-EM data processing pipeline for the FabE complex. Major steps include particle picking, 2D classification, initial model generation, 3D classification, refinement, and map sharpening. The final reconstruction yielded a 3.2 Å resolution map. Additional details are described in the single-particle image processing section of the materials and methods. CTF: contrast transfer function.
Extended Data Fig. 7 Single-particle cryo-EM data processing for FabE and FabB complexes.
a, Representative motion-corrected electron micrograph of FabE complex embedded in vitreous ice. Scale bar = 20 nm. b, Representative reference-free 2D class averages. c, Gold-standard Fourier shell correlation (FSC) curve generated from the independent half maps contributing to the 3.3 Å global resolution density map of the FabE complex. d, Local resolution filtered EM density map for the refined FabE complex (left), and central-slice of the complex (right), coloured according to local resolution which was calculated in CryoSPARC. e, Representative motion-corrected electron micrograph of FabB complex embedded in vitreous ice. Scale bar = 20 nm. f, Representative reference-free 2D class averages. g, Gold-standard Fourier shell correlation (FSC) curve generated from the independent half maps contributing to the 3.2 Å global resolution density map of the FabB complex. h, Local resolution filtered EM density map for the refined FabB complex (left), and central-slice of the complex (right), coloured according to local resolution which was calculated in CryoSPARC. Location of the CIM-834 binding pocket is circled.
Extended Data Fig. 8 Atomic model of SARS-CoV-2 M in complex with Fab-B and CIM-834.
a, Atomic model of the dimeric complex with residues coloured according to calculated B-factor. b, Zoomed-in view of the inhibitor binding site with the C1 symmetry (left) or C2 symmetry (right)-imposed EM density shown as a blue mesh. c, Map versus model FSC curves for the final refined model, generated in Phenix66. d, Two orthogonal views of the sharpened EM density for CIM-834 shown at a sigma level of 4.8 and 6.5. e, Two orthogonal views of the unsharpened EM density for CIM-834 shown at a sigma level of 4.6 and 5.8.
Extended Data Fig. 9 Amino acids in proximity of the CIM-834 binding pocket and their conservation in the M protein sequence of different human coronaviruses.
a-b, M protein chains in grey (dark grey = chain A, light grey= chain B). Bound compound displayed as surface. Green residues are within 5 Å of the bound compound. Blue residues are 5-10 Å from bound compound. c, Amino acid alignment of the M proteins of the seven human coronaviruses. Identical residues are shown as dots. Same colour coding as in a,b. d, Comparison of the overall conservation of the M protein sequence among different human coronaviruses versus conservation of the amino acids in the CIM-834 binding pocket (residues 10 Å and 5 Å from the bound compound). Conservation of the complete sequence of the main protease and its active site is provided for comparison. e, Mapping of the conservation rate (identity) of M residues on cryo-EM model (colour scale: red ≤20%, yellow 50%, grey ≥ 70%), based on an alignment of 100 coronaviral sequences. Sequences used for mapping were retrieved from a BLAST search against the M protein sequence, in the UniProtKB (www.uniprot.org).
Extended Data Fig. 10 Combinations of CIM-834 with GS-441524 or nirmatrelvir results in additive antiviral activity in vitro.
An antiviral assay was performed by infecting A549ACE2+TMPRSS2-mCherry with a SARS-CoV-2-mNeonGreen reporter virus as described previously41. Left, dose-response matrix for a, CIM-834 and GS-441524 or b, CIM-834 and nirmatrelvir representing average %inhibition of virus replication. Right, heat map of the delta scores (%) for the same combinations and ZIP analysis where δ = 0, δ > 0, and δ < 0 correspond to zero interaction, synergy, and antagonism, respectively. The overall zero interaction potency (ZIP) score represents the response beyond expectation (in %). In the range −10 < ZIP < 10, the compounds are likely to act in an additive manner, Score ≥10 indicate synergism. The results shown represent the means of two independent experiments for each combination. Data were then analysed with the SynergyFinder webtool80 based on zero interaction potency (ZIP) model.
Supplementary information
Supplementary Information
Supplementary Methods (compound synthesis and chemical characterization of CIM-834), Supplementary Fig. 1 (uncropped blots) and Supplementary Table 1.
Supplementary Video 1
Tomographic volume of SARS-CoV-2 A549ACE2+TMPRSS2-infected cells treated with DMSO. (Assembling) virions are indicated by the arrows. In the segmentation, ERGIC is shown in green, virions in yellow, microtubuli in blue and intermediate filaments in pink.
Supplementary Video 2
Tomographic volume of SARS-CoV-2 A549ACE2+TMPRSS2-infected cells treated with CIM-834. No assembling virions can be detected. DMVs are shown in cyan, membranes in orange.
Supplementary Video 3
The change between the short and long forms of the SARS-CoV-2 membrane protein.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Laporte, M., Jochmans, D., Bardiot, D. et al. A coronavirus assembly inhibitor that targets the viral membrane protein. Nature 640, 514–523 (2025). https://doi.org/10.1038/s41586-025-08773-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-08773-x
This article is cited by
-
Structural insights into MERS and SARS coronavirus membrane proteins
Communications Biology (2025)
-
A small-molecule SARS-CoV-2 inhibitor targeting the membrane protein
Nature (2025)
