
Abstract
DNA methylation and histone modifications encode epigenetic information. Recently, major progress was made to measure either mark at a single-cell resolution; however, a method for simultaneous detection is lacking, preventing study of their interactions. Here, to bridge this gap, we developed scEpi2-seq. Our technique provides a readout of histone modifications and DNA methylation at the single-cell and single-molecule level. Application in a cell line with the FUCCI cell cycle reporter system reveals how DNA methylation maintenance is influenced by the local chromatin context. In addition, profiling of H3K27me3 and DNA methylation in the mouse intestine yields insights into epigenetic interactions during cell type specification. Differentially methylated regions also demonstrated independent cell-type regulation in addition to H3K27me3 regulation, which reinforces that CpG methylation acts as an additional layer of control in facultative heterochromatin.
Similar content being viewed by others


Main
DNA methylation (5-methylcytosine; 5mC) and histone modifications are the two major, heritable epigenetic marks. Pioneering work has established single-cell 5mC profiling by bisulfite sequencing (scBS-seq)1,2. In addition, several methods are capable of single-cell histone modification profiling where specific antibodies tether either protein A-micrococcal nuclease (scChIC) or protein A-Tn5 transposase (scCUT&TAG) to modified nucleosomes3,4,5. Histone modifications and DNA methylation interact in several ways, such as recruitment of DNMTs6 to H3K36me3 or binding of H3K9me3 by UHRF1 (ref. 7); however, there is currently no technique that is capable of capturing these interactions in single cells. To bridge this gap, we developed single-cell Epi2-seq (scEpi2-seq), which achieves joint readout of histone modifications and DNA methylation in single cells. Our method yields high-quality data and provides valuable insights into the dynamics of epigenetic interactions.
Results
Single-cell Epi2-seq detects histone modifications and CpG methylation in single cells
Based on sortChIC8, scEpi2-seq leverages TET-assisted pyridine borane sequencing (TAPS9) for multi-omic readout of DNA methylation and histone modifications in single cells (Fig. 1a). After isolation and permeabilization, a pA–MNase fusion protein is tethered to specific histone modifications using antibodies. Single cells are sorted into 384-well plates by fluorescence-activated cell sorting (FACS). Next, MNase digestion is initiated by adding the essential cofactor Ca2+. The resulting fragments are repaired and A-tailed. Next, adaptors containing a single-cell barcode, unique molecular identifier (UMI), T7 promoter and Illumina handle are ligated to these fragments. Material from a 384-well plate is collected and subjected to TAPS. Unlike bisulfite-based approaches, TAPS converts methylated cytosine (5mC) to uracil, leaving barcoded single-cell adaptors intact. Library preparation includes in vitro transcription (IVT), reverse transcription and PCR. Following paired-end sequencing, multiple pieces of information are extracted from each read. Mapping genomic locations reveals position of modified histones, and C-to-T base conversions identify methylated cytosines. Also, nucleosome spacing can be inferred from distances between the start of sequencing reads (Fig. 1a). In addition, reads originating from IVT and PCR duplicates are used to correct for sequencing errors (Extended Data Fig. 1a). Like bisulfite sequencing, TAPS does not distinguish between 5hmC/5mC; however, HPLC–MS analyses indicate that 5hmC is approximately 30-fold less abundant than 5mC in most tissues10. Thus, the majority of the signal detected by TAPS and bisulfite sequencing originates from 5mC.

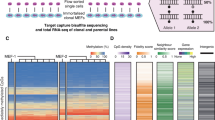
a, Overview of scEpi2-Seq workflow. Figure created with BioRender.com. b, Single-cell heatmaps of cut sites across three different histone modifications. Signals are aggregated per mark (top). c,d, Single-cell heatmap and line chart of distance between MNase cut sites with oscillations related to nucleosome positioning. Line indicates a LOESS smoothened curve of the underlying data points and error bands indicates the s.e. of the LOESS fit. e, Single-cell heatmaps of DNA methylation across three different histone modifications. Signals are aggregated per mark (top). f, Average methylation per mark with respect to distance from cut site.
To validate scEpi2-seq, we performed multi-omic profiling of DNA methylation histone modification in K562 cells. In total, we processed 2,660 cells across three histone marks (H3K9me3, H3K27me3 and H3K36me3). These libraries display high rates of cell barcode retrieval, high mappability and low mismatch rates (Extended Data Fig. 1b–d). We used in vitro CpG methylated spike-ins to assess TAPS conversion and found high C-to-T conversion rates (~95%, Extended Data Fig. 1e). Overall, we observe above average unique reads per cell, fraction reads in peaks and correlations to ENCODE ChIP-seq data compared to other single-cell chromatin methods (Extended Data Fig. 1f). We selected high-quality cells based on the number of unique reads and average methylation level per cell, which resulted in 60.2–77.9% cells passing quality control (QC) (Extended Data Fig. 2a). In the remaining cells (n = 1,981), we detect over 50,000 CpGs per single cell (Extended Data Fig. 2b). Next, we quantified the specificity of the single-cell histone measurements by calculating the fraction of reads in peaks (FRiP) for each cell (identified by MACS3 (ref. 11)). Average FRiP ranged between 0.72–0.88, depending on the antibody (Extended Data Fig. 2c). We detected orders of magnitude fewer reads in empty negative control wells (Extended Data Fig. 2d), underlining the high sensitivity and specificity of chromatin modality of scEpi2-seq data. Pseudobulk comparisons of our measurements and previously published data, including sortChIC8 and ENCODE ChIP-seq12 reveal high correlations (Extended Data Fig. 2h,i). Single-cell tracks display generally non-overlapping histone modifications as expected (Fig. 1b and Extended Data Fig. 2j). This observation is readily observed at higher resolution (200 bp; Extended Data Fig. 2f) and quantification further supports the mutually exclusive pattern. As a consequence, data across cells and modifications yield a near genome-wide CpG coverage (~80%; Extended Data Fig. 2e). These distributions are also observed at the PRAME gene locus, a highly expressed gene in K562 cells13 (300 kb; Extended Data Fig. 2j) and showed high levels of H3K36me3 demarcating the transcribed region, whereas the surrounding genome is decorated by the repressive marks H3K27me3 and H3K9me3. Next, we extracted distances between MNase cut sites from read mapping positions. Single-cell heatmaps (Fig. 1c) and data aggregated per histone mark (Fig. 1d) revealed the expected oscillatory patterns related to nucleosome spacing14. These oscillations disappear for H3K36me3 at distances larger than 1 kb, whereas they persist for H3K27me3 and H3K9me3, potentially related to the increased nucleosome dynamics during transcription15.
Next, we assessed 5mC levels in different chromatin contexts. First, we compared available ENCODE whole-genome bisulfite sequencing (WGBS) data to our scEpi2-seq split by chromatin domain for K562 cells. We observe that regions covered by the repressive histone marks H3K27me3 and H3K9me3 showed much lower methylation levels as compared to H3K36me3 (Extended Data Fig. 3a). These results are in agreement with 5mC levels per cell as measured with TAPS (Extended Data Fig. 2a). In addition, we compared in silico bulk measurements of DNA methylation levels (β values) to ENCODE WGBS (Extended Data Fig. 3b). These analyses revealed a high correlation between CpG methylation for 10-kb bins across the genome for K562, but not for HepG2, H1 or GM12878 cells (Extended Data Fig. 3b). At the single-CpG level, correlations exceed 0.8 (Pearson’s r; Extended Data Fig. 3c), indicating high data quality. Similar to our chromatin profiles, we display single-cell methylation tracks for a large region of chromosome 2 and observe high methylation for H3K36me3 and lower for H3K27me3 and H3K9me3 (Fig. 1e and Extended Data Fig. 3d). Plotting average methylation with respect to the cut site also shows much lower 5mC levels for H3K27me3 and H3K9me3 (8–10%) compared to H3K36me3 (50%) (Fig. 1e,f). This is in line with our analysis using WGBS data (Extended Data Fig. 3a,d) and with previous reports of higher methylation in gene bodies16. Taken together, scEpi2-seq simultaneously measures DNA methylation and histone methylation in the same single cell and shows high correlations with single-omics reference data.
H3K9me3, H3K27me3 and H3K36me3 chromatin and CpG methylation scEpi2-seq multi-omics validation on RPE-1 hTERT FUCCI cells
To validate K562 findings, we generated additional scEpi2-seq data for the RPE-1 hTERT cell line. In total, we profiled 3,420 single cells across three histone modifications (H3K9me3, H3K27me3 and H3K36me3). Similar to K562, we find that our negative control wells contain orders of magnitude fewer reads compared to positive wells (Extended Data Fig. 4a). To estimate the expected per-cell methylation levels, we intersected the peaks identified from the ChIC portion of our data with bulk WGBS measurements. While genome-wide methylation levels were higher than K562 cells, differences between histone marks showed similar behavior. 5mC levels were low to intermediate in regions covered by repressive histone marks (H3K9me3 and H3K27me3) and high in regions covered by H3K36me3 (Extended Data Fig. 4e). While plotting ChIC counts and per-cell 5mC levels, we noted two clusters among the cells with high numbers of unique cut sites. In addition to the population that displayed similar average 5mC levels in accordance with the estimates from intersecting ChIC peaks and WGBS (Extended Data Figure 4b), we identified a second, smaller population for all histone marks (Extended Data Figure 4b). These cells exhibited lower FRiP and per-cell 5mC levels closer to the genome-wide average. This effect likely results from excessive MNase activity. Our filtering effectively removed cells with low numbers of unique cut sites and those with excessive MNase activity. Overall, 35.4–40.6% of cells passed our thresholds and 1,716 single-cell profiles were available for downstream analyses (Extended Data Fig. 4b). Similar to K562 cells, FRiP values were high for RPE-1 hTERT cells passing QC (0.83–0.88; Extended Data Fig. 4c). The median number of CpGs detected exceeded 50,000 for all histone modifications (Extended Data Fig. 4d). The mutually exclusive patterns of H3K36me3, H3K27me3 and H3K9me3 were reproducible in RPE-1 hTERT cells (Extended Data Fig. 4f,j). Nucleosome positioning followed the same trend as previously observed in K562 cells, where H3K27me3 is more regularly spaced over longer genomic distances compared to H3K36me3 chromatin (Extended Data Fig. 4g). Again, genome-wide comparisons between WGBS and TAPS methylation showed excellent correlation (Extended Data Fig. 4h; Pearson correlation >0.9; P < 2.2 × 1016). CpG methylation levels as function of distance to the MNase cut site aligned well with per-cell 5mC levels (Extended Data Fig. 4i). Taken together, scEpi2-seq showed similar performance in RPE-1 cells with respect to single-cell metrics and correlation with reference data.
Cell-cycle-dependent DNA methylation dynamics are primarily driven by DNA replication
To study the interaction of epigenetic marks during cell cycle progression, we took advantage of the FUCCI cell cycle reporter system17 in the RPE-1 hTERT cells. Intensities of the FUCCI reporters are recorded for each single cell during sorting, allowing a measurement of the cell cycle stage of each cell. To visualize CpG dynamics across the cell cycle, we plot the average CpG methylation for each cell and fluorescent intensities of FUCCI markers (S/G2 marker, mAG-Geminin, x axis and G1 marker, Cdt1-mKO, y axis; Extended Data Fig. 5d). Upon visual inspection, we observed slight alterations over the cell cycle; however, these seemed to be obscured by outlier cells. To visualize changes over the cell cycle, we performed a rolling window smoothing. First, a progression over the cell cycle (integrated cell cycle progression) was converted into pseudo-time to FUCCI intensities using Wanderlust18 (Extended Data Fig. 5a–c). Next, we used integrated cell cycle progression to perform rolling window smoothing for CpG methylation during cell cycle (Extended Data Fig. 5d,e). Using this smoothing, we observed that CpG methylation over cell cycle progression exhibited a transient decrease during S phase for each histone modification (Fig. 2a). CpG methylation seems to reach its minimum during S phase (high hGeminin-mAG and low hCdt1-mKO2 levels) for H3K27me3- and H3K36me3-positive regions. In contrast, CpG methylation reached a minimum during G1 phase (low hGeminin-mAG and low hCdt1-mKO2 levels) for H3K9me3-covered regions. To improve resolution with respect to genomic replication timing, we integrated scEpi2-seq data with single-cell EdU-seq data (scEdU-seq19) for the same cell line (Fig. 2b and Methods). Using this integration, we are able to assess CpG methylation and histone methylation with respect to two time-resolved axes; cell cycle progression extracted from the FUCCI reporters and DNA replication timing inferred from the scEdU-seq experiment (Fig. 2c). As previously described, genomic distribution of histone marks is tightly correlated with replication timing20,21. H3K36me3 and H3K9me3 are enriched for early and late-replicating regions, respectively, whereas H3K27me3 is broadly distributed during mid-replication timing domains (Fig. 2c). We found that maintenance methylation extends into G1 phase within late-replicating domains, both for regions marked with H3K27me3 and H3K9me3 (Fig. 2c). The association between replication-coupled methylation loss and replication timing is also apparent when both modalities are visualized along the genome (Fig. 2d). Taken together, our data imply that the observed cell-cycle-dependent DNA methylation dynamics are primarily driven by DNA replication. The decrease in DNA methylation is subtle (a few percent), suggesting that DNA methylation maintenance is overall efficient. Recent data from the Groth and Hajkova laboratories revealed that DNA methylation does not immediately recover with the forks and is not even fully recovered in the subsequent mitosis22,23. In fact, replication-coupled maintenance is followed by a slower, replication-uncoupled phase of methylation maintenance23. This slower phase is facilitated by direct interaction of UHRF1 with H3K9me3 (ref. 7). In line with these observations, our data show that H3K9me3 chromatin does not fully recover CpG methylation within a single cell cycle. This results in a protracted, slower phase of re-methylation that extends into G1 phase for late-replicating domains (reviewed by Unoki24).

a, Scatter-plot of cell cycle reporter intensities for RPE-1 FUCCI cells. Color indicates neighbor-averaged methylation per cell. b, Heatmap of scEdU-seq signal intensity for a representative region of chromosome 1. Single cells (y axis) are ordered by S-Phase progression. c, Heatmap of normalized methylation across the cell cycle (left) and histogram of histone modification signal intensities (right). Rows are ordered by genomic replication timing (RT). d, Heatmap of normalized methylation for a representative region on chromosome 1. Rows are ordered based on cell-cycle progression. Data for most prevalent histone modification per bin are used to compute average methylation. Dashed line shows replication timing for the same region inferred from scEdU-seq.
Nucleosome occupancy direct DNA methylation maintenance following DNA replication
To assess the interplay of nucleosome occupancy and methylation maintenance, we utilized the single-molecule aspect of scEpi2-seq data. The start of sequencing reads coincides with MNase cut sites within linker DNA25, which is reflected in the oscillatory distribution of cut site distances (Fig. 1d and Extended Data Fig. 4g). First, we assessed DNA methylation with respect to the distance from an MNase cut over integrated cell cycle progression (Fig. 3a). We observed that the decrease in DNA methylation during S phase (x axis) was correlated with the distance from the cut site (y axis). We chose three representative distances to probe the correlation of DNA methylation maintenance and nucleosome occupancy. While distances of 90 and 270 bp correspond to the midpoints of the first and second nucleosome from the cut site, a distance of 180 bp reflects linker DNA. Additional analyses reveal that the decrease in methylation is more pronounced in regions that are covered by nucleosomes, especially for H3K27me3 and H3K9me3 (Fig. 3b). This effect became even more apparent when we investigated the absolute methylation levels with respect to cut site distance (Fig. 3c). We observe that levels at the nucleosome depleted region (180 bp, green line) fluctuate less throughout cell cycle progression and behave differently from the regions that are covered by nucleosomes. This suggests that parts of the genome that are bound by nucleosomes display altered kinetics of DNA methylation recovery after DNA replication. These dynamics are also visible upon normalization of CpG methylation (Fig. 3d; z-scoring CpG methylation per chromatin mark). In line with this observation, CpG methylation on DNA wrapped around nucleosomes was altered by a maximum of 12% compared to 4% in linker regions (Fig. 3e). In addition, these dynamics were highly reproducible between H3K27me3 and H3K9me3. For H3K36me3, the overall drop was much less pronounced and less clearly associated with nucleosome occupancy. This strongly suggests that high turnover of histones during RNA polymerase II transcription enables faster recovery of CpG methylation levels following DNA replication. Taken together, these findings suggest that in addition to replication timing, nucleosome occupancy has a major influence on DNA methylation maintenance.

a, Overview of single-molecule analysis. Average methylation values are computed with respect to the start of the read, which is the MNase cut site within linker DNA. Figure created with BioRender.com. b, Heatmap of normalized methylation across the cell cycle at different distances from MNase cut site. c, Line charts of average methylation change across the cell cycle for three representative cut site distances. While distances of 90 and 270 bp represent nucleosome-covered regions, 180 bp consists of the linker DNA region. d, Relative change of CpG methylation (y axis, percent point from total) as a function of distance from the MNase cut, which are colored and split by chromatin domain. The line indicates LOESS smoothened representation of underlying data points and error band indicates the s.e. of the fit.
Profiling of mouse intestinal cells reveals cell-type-specific differential DNA methylation within H3K27me3 regions
After exploring CpG methylation in specific chromatin domains in a relatively homogeneous cells, we set out to assess the functionality of scEpi2-seq in a complex tissue. Given the critical roles of DNA demethylation during intestinal differentiation26 and PRC2-dependent repression in intestinal stem cell maintenance27,28,29,30, we generated H3K27me3-targeted scEpi2-seq data for single cells isolated from the small intestine of mice. Cells from the proximal, distal and middle portion of the small intestine were isolated from three mice (Fig. 4a) to reduce confounding effects on H3K27me3 deposition patterns along the anteroposterior axis31. Cells from each intestinal fraction were fluorescently labeled, pooled, sorted and their fluorescence recorded during the single-cell sort (Extended Data Fig. 6a). Following QC, we retain 3,123 mouse intestinal cells. Signac32 and MethSCAn33 were utilized to perform dimensionality reduction and clustering for H3K27me3 patterns and CpG methylation, respectively. We observed several clusters in both H3K27me3 and CpG methylation data, which are split between cells from proximal and middle versus distal regions (Fig. 4b). To identify the cell types within these clusters, we performed marker gene detection on the H3K27me3 signal. Given that H3K27me3 marks PRC2-repressend facultative heterochromatin, histone counts are inversely correlated to messenger RNA expression34 (Extended Data Fig. 6b). This analysis revealed three clusters; absorptive (for example, Pou2f3, Tcf4, Ephb3, Foxa1 and Foxa2), secretory (for example, Nptx1, Nkx2-2, and Fev) and immune (for example, Ikzf3, Pax5 and Pou2f2) cells. The absorptive and secretory cells are present in all anteroposterior fractions, whereas immune cells are enriched in the distal fraction (Fig. 4b,c, Extended Data Fig. 6b and Supplementary Table 4). This is in line with the increase of lymphoid follicles called Peyer’s patches toward the distal part of the small intestinal tract35.

a, Overview of the intestinal samples, fluorescent labeling and computational analyses for scEpi2-seq intestinal dataset. b, Uniform Manifold Approximation and Projections (UMAPs) generated from 3,123 cells from the individual modalities; H3K27me3 (left) and CpG methylation (right). Cells are colored based on the position along the small intestinal tract. The dashed lines in the right plot indicate two 5mC clusters retrieved from Leiden clustering on the residuals on the variable methylated regions (MethSCAn) c, Same as in b, but cells are colored based on coarse clustering of the H3K27me3 data. d, Same as in b, but cells are colored based on refined clustering of the coarse H3K27me3 cluster in c. e, The number of unique H3K27me3 reads per single cell split by the subclusters based on the H3K27me3 signal from three individual mice. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5 × IQR. f, The percentage of CpG methylation on H3K27me3 ChIC fragments per single cell split by the subclusters based on the H3K27me3 signal derived from three individual mice. The boxplot is defined by the median ± IQR and whiskers represent 1.5 × IQR. g, LOESS smoothed line plot of the pairwise distances between MNase cut sites originating from all cells assigned to either absorptive, secretory or immune cells, which depict oscillations related to nucleosome positioning. Line indicates a LOESS smoothened curve of the underlying data points and error bands indicates the s.e. of the LOESS fit. h,i, Plots depicting DMRs in close proximity to Nkx2-3 (h) and Nr2f2 (i) genes, where the blue dashed line indicates the DMR identified by MethSCAn. The top is a line plot with the total number of CpG covered in a 5-kb bin for all cells in either of the 5mC clusters. The bottom two plots depict the average CpG methylation (%) over a 100-bp bin for both 5mC clusters. j, Heatmap containing DMRs (x axis) in each H3K27me3 subcluster (y axis) with the colors depicting the percentage of CpG methylation. The DMRs and subclusters were hierarchically clustered based on minimizing the variance (Ward’s method).
Next, we investigated the H3K27me3 chromatin domain distribution between these cell types. Upon visual inspection, we observed that most of the signals were similar between cell types with a few domains present in cell-specific clusters (Extended Data Fig. 6c). We previously observed that CpG methylation levels are relatively stable within a H3K27me3 domain in RPE-1 hTERT and K562 cells (Fig. 1f and Extended Data Fig. 4i). We observed a similar pattern for CpG methylation as a function of the distance to the H3K27me3 cut site in mouse intestinal cell types (Extended Data Fig. 6d). In addition, we found that 5mC levels within H3K27me3 domains are primary cells (~65%) compared to tissue culture cell lines (~10% for K562 and ~35% for RPE-1 hTERT). These data suggest that 5mC levels are possibly eroded in cell lines due to cell divisions, which has been previously described for heterochromatin36,37. Last, we observed unaltered nucleosome occupancy patterns within the H3K27me3 domains between absorptive, secretory or immune cells (Fig. 4g and Extended Data Fig. 6e). This observation suggests that nucleosome periodicity patterns are divergent between chromatin states, but not between cell types within the same chromatin state.
To achieve higher cell type resolution, dimensionality reduction and clustering were performed again for each cluster separately (absorptive, secretory and immune). First, we found that the coarse absorptive cluster split into two very similar enterocyte clusters (Fig. 4d). Second, the coarse secretory cluster split into two subclusters; enteroendocrine cells (EECs) and goblet cells, which we determine based on the mutual exclusive H3K27me3 signal on Pax6 for goblet cells and Gfi1 for EECs (Fig. 4d and Extended Data Fig. 6f,g). Last, we found three subclusters in the coarse immune cluster representing B cells, T cells and myeloid cells, which we identified based on the H3K27me3 signal on Lyn (Extended Data Fig. 6h; T cells, not B cells or myeloid), Syk (Extended Data Fig. 6i; T cells, not B cells or myeloid) and Il10ra (Extended Data Fig. 6j; myeloid, not T cells or B cells). Using these refined cell types, we investigated the levels of H3K27me3 as well as CpG methylation (Fig. 4e,f). We observed the lowest H3K27me3 levels in enterocytes, which also have the lowest CpG methylation levels. As for secretory cells, we observed similar levels of H3K27me3 but higher CpG methylation in goblet cells compared to EECs. For immune cells, we saw the lowest levels of H3K27me3 levels in B cells compared to T cells or myeloid cells, but these displayed the highest levels of CpG methylation. These diverging trends for CpG methylation and H3K27me3 levels in immune cells suggest interactions between these two repressive epigenomic regulatory layers.
Differentially methylated regions (DMRs) are closely spaced CpG sites that exhibit methylation variability across cell types, disease states and even individuals38,39. Their identification is crucial for understanding epigenetic landscapes and gene regulation. First, we set out to identify cell clusters based on the CpG profiling of the mouse intestine. We utilized MethSCAn, leveraging its performance with minimal coverage per cell, making it ideal for capturing CpG heterogeneity in single-cell chromatin libraries, which generally display low coverage. We identified two CpG clusters in our intestinal dataset (Extended Data Fig. 6k–m and Fig. 4b–d), corresponding to the immune and absorptive/secretory H3K27me3 clusters. Next, we identified DMRs within our mouse intestinal data using MethSCAn (Methods and Extended Data Fig. 6n). These data demonstrate that, even though H3K27me3 decorates these regions at similar levels, CpG methylation displays notably different levels between epithelial and hematopoietic lineages (Fig. 4h–j). These contrasting levels in CpG methylation demonstrate that, on top of H3K27me3 regulation, CpG methylation functions as an additional regulatory layer. Closer inspection of the DMRs between the two 5mC clusters reveals that all DMRs are associated with at least a single gene and that the majority of DMRs are located within 50 kb of transcription start sites, which suggest a large role in the cis-regulatory function of these DMRs. Moreover, pathway enrichment analysis of DMRs with GREAT40 revealed an increase for genes present in transcription factor complexes (q-value, 3.82 × 10−8), which display RNA polymerase II transcription factory activity (q-value, 1.85 × 10−23) and sequence specific DNA binding (q-value, 3.47 × 10−45). We next visualized the top 25 DMRs identified by MethSCAn in all pairwise comparisons of H3K27me3 subclusters and 5mC clusters (Fig. 4j). Following hierarchical clustering, we recovered the separation between immune and intestinal (absorptive and secretory) cells. Within these groups, we observed that T cells and B cells are more similar than myeloid cells. At the same time, we found that cells from the epithelial 2 cluster are more similar to goblet cells compared to epithelial 1 and enteroendocrine cells, which contrasts with our observation using only the H3K27me3 data (Fig. 4b–d) demonstrating the added value of the multi-omic measurements. Using scEpi2-seq, we profiled both CpG methylation and H3K27me3 from the same single cells in the mouse small intestine, enabling joint analysis of two repressive epigenetic layers across cell types and anatomical regions. We identified major epithelial and immune lineages and resolved distinct subtypes, observing coordinated and divergent patterns of methylation and H3K27me3 between lineages. Notably, DMRs revealed lineage-specific epigenetic regulation not captured by H3K27me3 alone, highlighting CpG methylation as an additive regulatory layer in gene regulation within facultative heterochromatin.
Discussion
Here, we introduce scEpi2-seq, an addition to the growing single-cell multi-omic toolbox. Recent advances have enabled researchers to measure 5mC or histone modifications in thousands to hundreds of thousands of cells41; however, bisulfite treatment is destructive, causes DNA breakage and is challenging to combine with histone modification assays, which retrieve molecules of interest by ligation of adaptors. Taking into account these limitations, we developed scEpi2-seq to study 5mC and the surrounding histone landscape. Combining sortChIC with TAPS conversion allowed multi-omic measurements, accurately profiling histone modifications and DNA methylation at the same time in single cells. In-depth validation of the histone and 5mC portions of our K562 data revealed high specificity as well as high numbers of unique cuts per cell. Comparison with reference data revealed high correlations with single-omics bulk measurements as well as the single-cell methods scCUT&RUN or scCUT&TAG4,5 for H3K36me3, H3k27me3 and H3k9me3 histone marks. Our comparison indicates that Tn5-based methods usually offer higher throughputs, albeit at the expense of the number of unique fragments per cell. Also, MNase digestion offers improved resolution for heterochromatic regions8; however, scCUT&TAG is preferred for atlas-scale endeavors where high throughput is essential to capture rare cell types. We showcase the utility of plate-based assays and flow cytometry by resolving reporter constructs (cell cycle progression in RPE-1 cells) as well as dyes introduced during sample preparation (to label anteroposterior axis in the mouse intestine). Of note, scEpi2-seq provides additional information on CpG DNA methylation allowing the exploration of the coupling between the two major epigenetic marks: histone modifications and DNA methylation.
Employing scEpi2-seq and scEdU-seq in RPE-1 FUCCI cells allowed us to assess the interplay of methylation maintenance, histone modifications and DNA replication timing across the cell cycle. Theoretic models about the heritability of 5mC patterns existed as early as 1975 (ref. 42). Murine DNMT1 was first cloned in 1988 (ref. 43) and subsequently recognized as the main enzyme responsible for maintenance methylation (reviewed by Mattei et al.44). A multitude of binding partners have been characterized, most notably PCNA and UHRF1, which recruit DNMT1 to the replication fork. Publications on the kinetics of maintenance find the process to be overall efficient; however, sequencing-based methods report faster re-methylation rates23,45,46 as opposed to mass spectrometry measurements22. While the precise kinetics are a matter of active research, the publications above agree on the fact that methylation extends into G1 phase for some regions. Regions with slower maintenance tend to be late replicating, heterochromatic and associated with the nuclear lamina47. Direct interactions between G9a/DNMT1 (ref. 48) and H3K9me3/UHRF1 (ref. 7) might serve as mechanisms to target the maintenance methylation machinery. Notably, increased cell turnover leads to erosion of DNA methylation in late-replicating regions. This has been observed in cancer49,50 and extended cell culture51. Using scEpi2-seq, we confirm that maintenance methylation extends into G1 phase for late-replicating regions22. Heterochromatic regions tend to replicate late and as such, the effect is most obvious in our H3K9me3 dataset; however, we note that late-replicating regions decorated with H3K27me3 show similar dynamics. Thus, protracted maintenance methylation seems to be driven more by replication timing than by a specific histone mark. In vitro studies have shown that nucleosome occupancy can interfere with the binding of DNMT1 (ref. 52) and DNMT3a/b53 to DNA. In line with these findings, nucleosome-covered regions showed a more pronounced drop in methylation during S phase. We hypothesize that nucleosomes block DNMT1 from interaction with DNA. This study provides direct evidence for slower 5mC maintenance in nucleosome-covered regions. Taken together, scEpi2-seq provides new insights into the interplay of chromatin structure and epigenetic inheritance.
Finally, we profiled H3K27me3 and 5mC along the anteroposterior axis of the mouse small intestinal tract. We find a variety of cell types with distinct H3K27me3 profiles. In addition, we observed that CpG methylation separates the hematopoietic lineage from the epithelial lineage. Moreover, immune cells show high CpG methylation and variable H3K27me3 levels, whereas epithelial cells are more distinct in both epigenetic marks. This points to distinct repressive strategies between immune and epithelial lineage. These data suggest that H3K27me3 methylation levels operate as an independent regulatory mechanism from CpG methylation. In addition, we observe differences in the epithelial cell subtypes, identified with H3K27me3, which show strong separation based on the anteroposterior axis. This could potentially indicate an effect of the position along the intestinal tract to shape differentiation trajectories, which has been previously described54,55. Our data suggest that chromatin states may contribute to cell fate decisions. Conversely, we do not observe a difference in nucleosome occupancy periodicity across cell types within H3K27me3 regions. This observation suggests that chromatin structure is dictated by epigenomic state rather than cell identity. In addition, we observe many DMRs between immune and intestinal cells that are located upstream of the gene bodies and that these genes are often transcription factors with known function in differentiation trajectories (for example Nr2f2 and Pou2f3). This suggests that in addition to H3K27me3, these transcription factors need to be additionally repressed by CpG methylation to fully block illegitimate expression. Taken together, these observations in the mouse small intestine warrant co-acquisition of both histone marks and CpG methylation status with scEpi2-seq as these provide a more extensive view on epigenome regulation in complex tissues.
Methods
The manuscript is accompanied by a detailed, step-by-step version of the protocol, which can be accessed at https://www.protocols.io/view/single-cell-epi2-seq-cqk7vuzn.
Cell culture
K562 cells (CCL-243, ATCC) were cultured in RPMI 1640 GlutaMAX medium (Gibco), supplemented with 5% FBS and pen–strep. hTERT RPE-1 FUCCI cells (a kind gift from the R.H. Medema laboratory) were grown in adherent culture with DMEM/F12 GlutaMAX medium (Gibco) supplemented with pen–strep and 10% FBS. TrypLE and PBS were used to passage RPE-1 hTERT FUCCI cells.
Mice
Our research complies with all relevant ethical guidelines. Experimental procedures were approved by the Dier Experimenten Commissie (DEC) of the Koninklijke Nederlandse Akademie van Wetenschappen (KNAW) and performed according to the guidelines. The mice that were used were the C57BL/6 strain. Animals used in the experiments were between 8 and 22 weeks of age. Both male and female mice were used for the experiments. Mice were housed in open housing with a 14–10-h light–dark cycle at 24 °C and 45–70% humidity with food and water ad libitum. Three individual mice were used, the proximal, middle and distal part of the intestines from a single individual were pooled following the generation of single cell suspension, fixation and CellTrace labeling (Invitrogen, according to the manufacturer’s protocol); randomization and blinding were not performed.
Single-cell histone profiling
Buffers
scChIC required three wash buffers. Wash buffer 1 (WB1) contained 20 mM HEPES, 150 mM NaCl, 0.5 mM spermidine, 0.05% Tween-20, 2 mM EDTA and one cOmplete Protease Inhibitor tablet (per 50 ml). Wash buffer 2 (WB2) had the same composition as WB1 but omitting EDTA. Wash buffer 3 (WB3) had the same composition as WB1 but lacking EDTA and protease inhibitor.
Fixation and storage
All steps were performed in Protein LoBind tubes. Cells were collected and washed twice with PBS and fixed in 1 ml 70% ice-cold ethanol per 106 cells for 2 h at −20 °C. If not processed directly, cells were stored at −80 °C in WB1 with 10% dimethylsulfoxide (DMSO).
Antibody and pA–MNase binding
Protein A–MNase fusion protein (pA–MNase) was expressed in bacteria and purified as outlined in Zeller et al.8. Cells were thawed, washed twice with WB1 and resuspended in 500 µl WB1. Antibody incubation was performed overnight at 4 °C with gentle agitation. Cells were washed once and resuspended in 500 µl of WB2. pA–MNase (3 ng ml−1) and Hoechst 34580 (5 µg ml−1) were added to each sample, followed by incubation for 1 h at 4 °C with gentle agitation. Cells were washed twice with WB2, resuspended in 500 µl of WB3, filtered through a 70-µm strainer and transferred to FACS tubes. Antibodies used in this study were anti-H3K9me3 (Abcam, ab8898, 1:100 dilution), anti-H3K27me3 (NEB, C36B11, 1:200 dilution) and anti-H3K36me3 (Thermo Fisher, MA5-24687, 1:2,000 dilution).
Fluorescence-assisted cell sorting
First, 10 µl of sterile filtered mineral oil was added to each well of 384-well hard-shell plate. Cells were sorted into 384-well plates on a BD Influx. Hoechst signal was used to select for K562 cells in G1 phase. Four gates were used for RPE-1 FUCCI cells to sample evenly from G1, S and G2 phases. Four to eight wells were left empty as controls in all plates. After sorting, cells were spun down for 1 min at 2,000g.
Processing of single-cell plates
All dispensions below were performed using an Innovadyne Nanodrop II, after which plates were sealed with covers and spun for 1 min at 2,000g.
pA–MNase activation and Proteinase K digest
MNase digestion was initiated by adding 100 nl of WB3 supplemented with 2 mM CaCl2 to each well and incubated for 30 min at 4 °C. Digestion was stopped by dispensing 100 nl of the following solution (final concentrations): H2O, 40 mM EGTA, 1.5% NP-40 and 2 mg ml−1 Proteinase K and incubated for 20 min at 4 °C; 6 h at 65 °C; and 2 min at 80 °C.
Blunting
A total of 100 nl of the following mix was added to each well: 2 nl Klenow, large fragment (NEB); 2 nl T4 PNK (NEB); 5 nl dNTP solution (Promega); 30 nl 10 mM ATP; 30 nl 10× PNK Buffer (NEB); 10 nl 25 mM MgCl2; 5 nl 50% PEG8000; 1.5 nl 20 mg ml−1 BSA (NEB); 14.5 nl H2O and incubated 30 min at 37 °C; 20 min at 75 °C.
A-tailing
Then, 150 nl of the following mix was added to each well (volumes per well): 1 nl AmpliTaq360 DNA Polymerase (Applied Biosystems); 1 nl 100 mM dATP; 25 nl 2 M KCl (Invitrogen); 10 nl PEG8000 50%; 1 nl 20 mg ml−1 BSA; 112 nl H2O and incubated for 15 min at 37 °C; 10 min at 72 °C.
Adaptor dispension
To each well, 50 nl of 5 µM barcoded adaptor was added using a Mosquito HTS Nanolitre Liquid handler (ttplabtech). Adaptor sequences are provided in Supplementary Table 1.
Adaptor ligation
Then, 150 nl of the following mix was added to each well: 25 nl T4 Ligase at 400,000 U ml−1 (NEB); 3 nl 1 M MgCl2; 45 nl 0.1 M dithiothreitol (DTT); 20 nl 10 mM ATP; 5 nl 50% PEG8000; 1 nl 20 mg ml−1 BSA; 51 nl H2O and incubated for 20 min at 4 °C; 16 h at 16 °C; 10 min at 65 °C.
Pooling of plates
Plates were inverted and placed in VBLOK200 reservoir (Click-Bio) pre-coated with mineral oil and spun for 2 min at 500g and the liquid phase was transferred to 1.5 ml Eppendorf tubes. Next, DNA was purified by incubating for 10 min with 0.8× Ampure XP bead cleanup (Beckman Coulter, pre-diluted 1:4 in bead binding buffer; 1 M NaCl, 20% PEG8000, 20 mM Tris, pH 8.0 and 1 mM EDTA). Beads were pelleted on a magnet, washed twice with 80% ethanol, air dried and resuspended in 19 µl H2O and transferred to a 0.5-ml Eppendorf tube. The material was stored at −20 °C until further processing.
TAPS methylation profiling
TET1 enzyme production
mTET1CD was expressed as outlined by Liu and colleagues9. In brief, FLAG-tagged mTET1CD was expressed in Expi293F cells. After lysis, mTET1CD–FLAG was bound to Anti-Flag M2 Affinity Gel (Sigma) and purified on gravity chromatography columns according to the manufacturer’s specifications. Protein was concentrated on Amicon Ultra-4 Centrifugal Filter units followed by buffer exchange with Bio-Spin P-30 Gel Columns. Protein was stored at −80 °C in 20 mM HEPES, pH 8.0, 150 mM NaCl, 1 mM DTT and 30% glycerol.
Preparation of fully methylated lambda phage spike-in
Lambda phage DNA was methylated using M.SssI (NEB) using the following reaction: 5 µl NEB Buffer 2, 1 µl 32 mM S-adenosylmethionine, 0.5 µl M.SssI (4,000 U ml−1), 1 µg of lambda phage DNA and H2O (to 50 µl). After incubation at 37 °C for 2 h, additional SAM (1 µl) and M.SssI (0.5 µl) were supplemented and incubated for 2 h at 37 °C. DNA was purified using a 1:1 bead-to-sample ratio Ampure XP SPRI cleanup and eluted in 20 µl nuclease-free water. This methylation reaction was repeated once using the previously methylated DNA as input, again supplementing with SAM. A second SPRI cleanup was performed, eluted in 20 µl nuclease-free water and diluted to 1 pg µl−1.
TAPS conversion and cleanup
Reaction buffer for TAPS consists of: 167 mM HEPES; 333 mM NaCl (Invitrogen); 3.3 mM α-ketoglutarate; 6.67mM l-ascorbic acid; 4 mM ATP; 8.33 mM DTT. The following reaction was assembled on ice: 19 µl of pooled material, 1 µl of methylated lambda spike-in libraries, 15 µl of TAPS reaction buffer, 3.33 µl of 1.5 mM Fe2+ solution and 12 µl of mTET1CD. Samples were incubated for 80 min at 37 °C. Then, 1 µl of 20 mg ml−1 Proteinase K was added per reaction, followed by incubation for 15 min at 55 °C. Next, samples were cleaned up with 2× volumes of Ampure XP DNA beads and eluted in 19.67 µl of nuclease-free H2O. The above reaction, Proteinase K digest and bead cleanup were repeated and eluted in 33.75 µl H2O. Next, 10 µl of 3 M NaAc (pH 4.3) and 6.25 µl of 10 M pyridine borane solution were added to the libraries. Samples were incubated for 16 h in a thermal shaker (37 °C at 850 rpm). After pyridine borane incubation, reactions were subjected to oligonucleotide clean and concentrator columns (Zymo) according to the manufacturer’s protocol and volumes were reduced to 9.6 µl using SpeedVac.
Sequencing library preparation
In vitro transcription
TAPS-converted libraries were subjected to IVT by adding 14.4 µl of IVT reaction mix (MEGAscript T7 Transcription kit) and incubated for 14 h at 37 °C. Next, 6 µl of H2O and 3 µl of Turbo DNase were added and samples incubated for 15 min at 37 °C to digest template DNA. Amplified RNA (aRNA) was fragmented by adding 7.88 µl of fragmentation buffer (200 mM Tris-acetate; 500 mM KaOAc; 150 mM MgOAc) followed by incubation for 90 s at 94 °C. Samples were immediately chilled on ice and 4.13 µl of 0.5 M EDTA, pH 8. Then, aRNA was cleaned with 0.8× RNAClean XP bead cleanup and eluted in 6 µl of H2O.
Reverse transcription and library amplification
After QC, 5 µl of aRNA was combined with 0.5 µl of 10 mM dNTP solution and 1 µl of random hexamer RT primer 20 µM (Supplementary Table 2). Samples were heated to 65 °C for 5 min and then immediately chilled on ice. Then, 6.5 µl of primed sample was combined with 2 µl First Strand Buffer, 1 µl 0.1 M DTT, 0.5 µl of SuperScriptII and 0.5 µl of RNaseOUT and incubated for 10 min at 25 °C; and 60 min at 42 °C. Then, 2 µl of barcoded RPIx primer (sequences in Supplementary Table 3) was added to each sample. Library PCR was performed by adding 11 µl nuclease-free H2O, 25 µl of NEBNext Ultra II Q5 Master Mix (NEB) and 2 µl of 10 µM RP1 primer (Supplementary Table 2). Samples were amplified with 10–13 cycles of PCR, dependent on histone modification. PCR settings: 30 s at 98 °C; 10 s at 98 °C, 30 s at 60 °C, 30 s at 72 °C for 10–13 cycles and 10 min at 72 °C. Amplified DNA was cleaned with two subsequent 0.8× AMPure XP bead cleanups and eluted in 15 µl of nuclease-free H2O. Concentration and size distribution of the final libraries were measured on a Qubit (Invitrogen) and Bioanalyzer (Agilent), respectively. Samples were pooled and sequenced on the Illumina NextSeq2000 platform (2 × 150 bp) according to the manufacturer’s specifications.
Data analysis
Trimming, demultiplexing, mapping and deduplication
The scChIC mapping and counting workflow is described by Zeller et al.8, which we further expand to include the steps required for molecule consensus and methylation calling. Sequenced reads were demultiplexed using SingleCellMultiOmics demux.py and adaptor sequences were removed using Cutadapt. A custom reference was prepared by combining the following assemblies: Human Ensembl assembly v.97 (hg38), Lambda phage (acc. J02459.1), Cutibacterium acnes (acc. KPA171202) and Escherichia coli strain RHB09-C15. hg38 was used for mapping of the (human) cell lines K562 and RPE-1 and lambda phage to detect methylated spike-ins. Bacterial genomes were added to detect contaminants. Similarly, mouse intestinal samples were aligned to Mouse Ensembl assembly v.97 (mm10), Lambda phage (acc. J02459.1), C. acnes (acc. KPA171202) and E. coli strain RHB09-C15. Trimmed reads were mapped paired-end using bwa-mem with default parameters. Mapped reads were filtered and deduplicated using SingleCellMultiOmics (bamtagmultiome.py).
Molecule consensus calling
In the molecule consensus calling step, the information from reads derived from the same original DNA-template is aggregated (Extended Data Fig. 1a). Reads containing the same UMI, starting coordinate, strand and haplotype were aggregated into a single molecule. Paired-end reads are merged, for positions with overlapping read pairs the base call with the highest Phred score is selected and lower than 15 are discarded. For dovetailing alignments, the overlapping segments are not considered. For each covered position, the most common base is selected using majority voting over all reads (IVT/PCR duplicates) that cover a location, resulting in a base call for each covered position of the consensus molecule. Ties are resolved by inserting an ambiguous base call (N).
Methylation calling
Methylation calling was performed on the generated consensus base calls. For every covered cytosine of the consensus molecule, a C is considered unmethylated, a T is considered methylated and other nucleotides result in an ambiguous methylation call. To avoid incorrect methylation calls due to the presence of single-nucleotide variants, genomic locations with known C-to-T variants were masked.
Whole-genome bisulfite sequencing
Bulk genomic DNA was extracted from RPE-1 cells using the Monarch Genomic DNA Purification kit (NEB). DNA (10 ng) was bisulfite converted and libraries were prepared using the Pico Methyl-Seq Library Kit (Zymo) according to the manufacturer’s instructions. Libraries were sequenced on a NextSeq500 (2 × 150bp). Fastq files were derived from raw sequencing data by demultiplexing with bcl2fastq. Subsequently, data were trimmed using Cutadapt and mapped and deduplicated and coverage plus average methylation status for every CpG were exported using BisMark.
Calculation of conversion rates from methylated spike-in
To determine CG dinucleotide conversion efficiencies in methylated lambda phage spike-ins, mapped BAM files were analyzed using an estimate_conversion.py. Each read group, nucleotide calls covering CG sites were aggregated and majority-vote consensus sequences were determined. Sites were classified as unconverted (CG) or converted (TG or CA), and counts were recorded for each position. The final output included genomic coordinates, conversion counts and trinucleotide context.
Extraction of sequencing statistics
Basic sequencing statistics were extracted from BAM files using the command libraryStatistics.py from the SingleCellMultiOmics package with default parameters.
Calculation of mismatch rates
Mismatch rates were computed from files generated with ‘samtools mpileup’. Specifically, we used a subset of libraries from the RPE-1 dataset for which unconverted data were available. Mismatches were calculated as the difference between total coverage and matches, and mismatch rates were expressed as the fraction of mismatches over total coverage. Each position was assigned a trinucleotide context (±1-bp window) and sites containing ambiguous bases (‘N’) were excluded. Mismatch rates were then aggregated by library and sequence context to quantify base-specific error profiles. Of note, all sequence contexts containing CG dinucleotides were filtered to account for increased mismatch rates due to the conversion in TAPS libraries.
Calculation of fraction reads in peaks
For each dataset, histone modification-specific peaks were identified using MACS11 with broad peak calling enabled and without local lambda background correction. For each library, cell-level BAM files were created using a custom Python script (split_bam_by_cellbarcode.py, available on GitHub). Next, reads from each BAM file were intersected with their corresponding peak regions using countOverlaps from the GenomicRanges package. The FRiP score was computed as the fraction of reads falling within peak regions over the total number of aligned reads.
Overlap of histone modification peaks with DNA methylation
Histone modification peaks extracted from scEpi2-seq data were intersected with WGBS acquired from ENCODE (K562 cells) or generated as part of this manuscript (RPE-1). CpGs overlapping histone modification peaks were identified using ‘GenomicRanges::findOverlaps’. Next, average CpG methylation (β value), number of CpGs and coverage per peak were calculated for each histone modification. Peaks with >50 reads total coverage and >10 CpGs were retained for visualization.
Comparison of scEpi2-seq and whole-genome bisulfite sequencing data
scEpi2-seq data for K562 cells were binned into 10-kb windows for cells passing QC. WGBS data for K562 and three additional cell lines (GM12878, H1 and HepG2) was acquired from ENCODE and also binned at 10-kb resolution and analyzed using Pearson’s correlation coefficient. For single CpG resolution, the maximum correlation attainable (referred to as ‘simulated’ in Extended Data Fig. 3c) was calculated as follows: First, WGBS data were filtered for a coverage of at least 10×. Methylation values were generated from WGBS data using binomial sampling with empirically determined parameters for TAPS conversion efficiency (α = 0.875, extracted from lambda phage spike-ins) and false-positive rate (β = 0.0023, from original TAPS publication9). Coverage levels were sampled from the empirical scEpi2-seq distribution. Pearson correlations were computed between actual and simulated methylation, as well as between matched CpG methylation values from scEpi2-seq and WGBS across a range of coverage thresholds.
Comparison of scEpi2-seq, single-cell ChIC and ChIP
Read counts for K562 cells were aggregated per histone modification. Data were binned into non-overlapping regions of 50 kb for comparison with ChIC and ChIP data, and Spearman correlation coefficients were computed across matched bins.
Preprocessing of single-cell chromatin data for analysis with Signac and Seurat
BAM files were processed including mapping and tagging as outlined above using custom software (SingleCellMultiOmics package). BAM files were converted to fragment files using sinto with cell barcodes extracted from the ‘bc’ BAM tag. The resulting files were sorted, compressed with bgzip and indexed using tabix. Processed fragment files were imported into R using Signac/Seurat’s FragmentObject function and summarized in 50-kb genomic bins. Chromatin assays were created with a minimum cell (n = 10) and feature (n = 1,000) threshold and merged into a single Seurat object. Finally, dimensionality reduction was performed using TF-IDF normalization, singular value decomposition (SVD) and UMAP.
Integration of scEpi2-seq and scEdU-seq data
To integrate scEpi2-seq and scEdU-seq datasets, we used the FUCCI fluorescent intensities to compute a pseudo-time across the cell cycle (integrated cell cycle progression) using the Wanderlust algorithm for both scEpi2-seq dataset (Extended Data Fig. 5e). Next, we aligned the scEdU-seq S phase progression19 to the integrated cell cycle progression by aligning the start of S phase progression with cells starting to accumulating mAG-Geminin (integrated cell cycle progression of 0.4) and aligning the end of S phase progression with cells fully losing the mKO-Cdt1 (integrated cell cycle progression of 0.8), which we extracted from our previously published scEdU-seq data.
Normalization of histone mark densities
To normalize histone mark density over S phase progression, the densities for the chromatin mark were split by histone modification and subsequently maximum normalized to the highest replication timing bin (highest replication timing bin becomes 1.0).
Signac workflow to identify cell types using H3K27me3 ChIC from mouse intestine
Signac was used within Seurat to create a chromatin assay, filtering cells with fewer than 103 unique reads. A gene activity matrix was computed and principal-component analysis was performed, after which the first component was removed as it correlated with sequencing depth. Next, dimensionality reduction was performed using latent semantic indexing, followed by UMAP visualization and clustering. The coarse clustering resulted in three large clusters that were subsequently split and reanalyzed to yield seven subclusters. Regions with differential abundance in H3K27me3 were identified by using the FindMarker function and visualized using the CoveragePlot function.
MethSCAn workflow to identify differentially methylated regions across cell types from mouse intestine
Methylation calls from single mouse intestinal cells were extracted as previously described by the tapsTabulator function. These were converted into a single-cell BisMark input format and subjected to the standard MethSCAn workflow. Next, we detected differentially methylated regions (DMRs) between the 5mC cluster 1 and 5mC cluster 2 or all pairwise H3K27me3 celltypes and filtered them for at least 25 CpGs covered in both conditions as well as a P value < 0.001. The top 25 DMRs (before filtering) from each comparison were used to generate a heatmap displaying CpG methylation percentage per DMRs over all subclusters, both were hierarchically clustered using ward.D2 to minimize the variance within clusters.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated for this study have been deposited at the Gene Expression Omnibus (accession code GSE232637). Data downloaded from ENCODE are listed in the GitHub repository accompanying this publication (https://github.com/cgeisenberger/taps-manuscript). Single-cell ChIC data were downloaded from the Gene Expression Omnibus (accession code GSE164779). Whole-genome bisulfite data for K562 (ENCFF867JRG and ENCFF721JMB), HepG2 (ENCFF817LMT and ENCFF453UDK), H1 (ENCFF573YXL and ENCFF434CNG) and GM12878 (ENCFF614QHA and ENCFF570TIL) were downloaded from ENCODE. Bulk ChIP-seq data from K562 for H3K27me3 (ENCSR000EWB), H3K36me3 (ENCSR000DWB) and H3K9me3 (ENCSR000APE) were downloaded from ENCODE.
Code availability
Processing of sequencing data up to the generation of count and methylation tables was performed with the software package SingleCellMultiOmics, which can be accessed at https://github.com/BuysDB/SingleCellMultiOmics. Code for downstream analysis and generation of figures can be accessed at https://github.com/cgeisenberger/taps-manuscript.
Change history
14 November 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41592-025-02979-7
References
Smallwood, S. A. et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820 (2014).
Luo, C. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017).
Hainer, S. J., Bošković, A., McCannell, K. N., Rando, O. J. & Fazzio, T. G. Profiling of pluripotency factors in single cells and early embryos. Cell 177, 1319–1329.e11 (2019).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Ku, W. L. et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 16, 323–325 (2019).
Yano, S. et al. Histone H3K36me2 and H3K36me3 form a chromatin platform essential for DNMT3A-dependent DNA methylation in mouse oocytes. Nat. Commun. 13, 4440 (2022).
Rothbart, S. B. et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 19, 1155–1160 (2012).
Zeller, P. et al. Single-cell sortChIC identifies hierarchical chromatin dynamics during hematopoiesis. Nat. Genet. 55, 333–345 (2023).
Liu, Y. et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 37, 424–429 (2019).
Globisch, D. et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5, e15367 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, 1–9 (2008).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
Oehler, V. G. et al. The preferentially expressed antigen in melanoma (PRAME) inhibits myeloid differentiation in normal hematopoietic and leukemic progenitor cells. Blood 114, 3299–3308 (2009).
Kelly, T. K. et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506 (2012).
Ehara, H., Kujirai, T., Shirouzu, M., Kurumizaka, H. & Sekine, S.-I. Structural basis of nucleosome disassembly and reassembly by RNAPII elongation complex with FACT. Science 377, eabp9466 (2022).
Baubec, T. et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247 (2015).
Shaltiel, I. A. et al. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl Acad. Sci. USA 111, 7313–7318 (2014).
Bendall, S. C. et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014).
van den Berg, J. et al. Quantifying DNA replication speeds in single cells by scEdU-seq. Nat. Methods 21, 1175–1184 (2024).
Hansen, R. S. et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl Acad. Sci. USA 107, 139–144 (2010).
Pope, B. D. et al. Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405 (2014).
Stewart-Morgan, K. R. et al. Quantifying propagation of DNA methylation and hydroxymethylation with iDEMS. Nat. Cell Biol. 25, 183–193 (2023).
Ming, X. et al. Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging-associated methylome deterioration. Cell Res. 30, 980–996 (2020).
Unoki, M. Chromatin remodeling in replication-uncoupled maintenance DNA methylation and chromosome stability: Insights from ICF syndrome studies. Genes Cells 26, 349–359 (2021).
Muthurajan, U. et al. In vitro chromatin assembly: strategies and quality control. Methods Enzymol. 573, 3–41 (2016).
Ansari, I. et al. TET2 and TET3 loss disrupts small intestine differentiation and homeostasis. Nat. Commun. 14, 4005 (2023).
Koppens, M. A. J. et al. Deletion of polycomb repressive complex 2 from mouse intestine causes loss of stem cells. Gastroenterology 151, 684–697.e12 (2016).
Jadhav, U. et al. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165, 1389–1400 (2016).
Kim, T.-H. et al. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506, 511–515 (2014).
Chiacchiera, F., Rossi, A., Jammula, S., Zanotti, M. & Pasini, D. PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J. 35, 2301–2314 (2016).
Montavon, T. & Soshnikova, N. Hox gene regulation and timing in embryogenesis. Semin. Cell Dev. Biol. 34, 76–84 (2014).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Kremer, L. P. M. et al. Analyzing single-cell bisulfite sequencing data with MethSCAn. Nat. Methods 21, 1616–1623 (2024).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Cornes, J. S. Number, size, and distribution of Peyer’s patches in the human small intestine: part I The development of Peyer’s patches. Gut 6, 225–229 (1965).
Ehrlich, M. DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413 (2002).
Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008).
Reik, W., Dean, W. & Walter, J. Epigenetic reprogramming in mammalian development. Science 293, 1089–1093 (2001).
Rakyan, V. K., Down, T. A., Balding, D. J. & Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541 (2011).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Tian, W. et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science 382, eadf5357 (2023).
Holliday, R. & Pugh, J. E. DNA modification mechanisms and gene activity during development. Science 187, 226–232 (1975).
Bestor, T., Laudano, A., Mattaliano, R. & Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 203, 971–983 (1988).
Mattei, A. L., Bailly, N. & Meissner, A. DNA methylation: a historical perspective. Trends Genet. 38, 676–707 (2022).
Xu, C. & Corces, V. G. Nascent DNA methylome mapping reveals inheritance of hemimethylation at CTCF/cohesin sites. Science 359, 1166–1170 (2018).
Charlton, J. et al. Global delay in nascent strand DNA methylation. Nat. Struct. Mol. Biol. 25, 327–332 (2018).
Berman, B. P. et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 44, 40–46 (2011).
Estève, P.-O. et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20, 3089–3103 (2006).
Zhou, W. et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 50, 591–602 (2018).
Timp, W. et al. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 6, 61 (2014).
Endicott, J. L., Nolte, P. A., Shen, H. & Laird, P. W. Cell division drives DNA methylation loss in late-replicating domains in primary human cells. Nat. Commun. 13, 6659 (2022).
Schrader, A., Gross, T., Thalhammer, V. & Längst, G. Characterization of Dnmt1 binding and DNA methylation on nucleosomes and nucleosomal arrays. PLoS ONE 10, e0140076 (2015).
Felle, M. et al. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 39, 6956–6969 (2011).
Beumer, J. & Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 22, 39–53 (2021).
Zinina, V. V. et al. ID2 controls differentiation of enteroendocrine cells in mouse small intestine. Acta Physiol. 234, e13773 (2022).
Acknowledgements
We thank Utrecht Sequencing Facility (USEQ) in Utrecht for their excellent sequencing service and R. van der Linden for his support with FACS. We also thank all members of the van Oudenaarden laboratory and S.-W. Wang for valuable scientific discussions. This work was supported by a European Research Council Advanced grant (grant no. 101053581-scTranslatomics) Novo Nordisk Fonden Synergy Programme (0091873). This work is part of the Oncode Institute, which is partly financed by the Dutch Cancer Society. C.-X.S. was supported by the Ludwig Institute for Cancer Research and National Institute for Health and Care Research (NIHR) Oxford Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR or the UK Department of Health. In addition, we thank the Hubrecht Sorting Facility and the Utrecht Sequencing Facility, subsidized by the University Medical Center Utrecht, the Hubrecht Institute, Utrecht University and The Netherlands X-omics Initiative (NWO project 184.034.019).
Author information
Authors and Affiliations
Contributions
C.G., J.v.d.B. and A.v.O. conceived and designed the project. C.G. and J.v.d.B. developed the experimental scEpi2-seq protocol and performed experiments. J.v.d.B. generated scEdU-seq data. J.V.-L. generated WGBS data. Y.L., X.C. and C.S. developed the original TAPS protocol and provided reagents and feedback for establishing experimental procedures. A.L. processed and dissociated mouse small intestinal samples before single-cell sorting. B.d.B. wrote software to preprocess data with feedback from J.d.R. C.G., J.v.d.B., V.v.B. and A.v.O. analyzed data. All authors discussed and interpreted results. C.G. and J.v.d.B. wrote the manuscript with feedback from the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Goncalo Castelo-Branco, Yonglong Dang and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lei Tang, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Library statistics for scEpi2-seq libraries and comparison to other single-cell methods.
a. Overview of data extraction strategy for scEpi2-Seq data. Every fragment resulting from MNase digestion is ligated to an adaptor containing a UMI, single-cell barcode, Illumina handle and T7 promoter. Every fragment is uniquely labeled by cell barcode and UMI, and mapping positions of R1 and R2 can be used to distinguish between duplicates generated during in vitro transcription (IVT) or PCR. In addition, aggregating all sequencing reads derived from the same DNA fragment into a consensus molecule can mitigate sequencing errors and makes methylation estimates more robust. Created in BioRender (2025) https://BioRender.com/0dai1w3. b. Mapping, Demultiplexing (that is, reads with correct cell barcode) and Deduplication (that is, removed reads with same cell barcode, UMI and position in the genome per single cell) rates per plates (indicated as dots, n = 7 independent experiments) for all experiments in this study. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR c. Mapping rates from the RPE-1 hTERT plates, which were either TAPS converted or unconverted prior during library preparation. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR. d. Comparison of mismatch rates from the RPE-1 hTERT plates, which were either TAPS converted or unconverted prior during library preparation. Only nucleotides in a non-CpG context were included. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR e. Conversion efficiency of in vitro methylated lambda phage genome (spike-ins) after TAPS conversion. Each dot represents a single 384-plate (technical replicates) and results are grouped by sample of origin (n = 7 biological replicates over samples of origins). Inset displays the same data but with a selected range of conversion efficiency (that is, 85-100%). The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR f. Comparison between scEpi2-seq (this study) and other single- and multi-omic approaches for measuring histone modifications in single cells.
Extended Data Fig. 2 Application of scEpi2-seq in K562 cells.
a. Cells were quality-filtered based on the number of unique ChIC cut sites and DNA methylation. Scatter plots showing the number of unique cut sites (x-axis, log10 scale) and per-cell 5mC levels (y-axis) in the K562 dataset. b. Normalized density plot showing the number of CpGs detected per single K562 cell (x-axis, log10) stratified by histone mark (columns) and QC status (rows). c. Violin plot showing the fraction of reads in peaks (FRiP) for QC-filtered cells in the K562 data set (n = 2 biological replicates). Data are grouped for different modifications assessed with scEpi2-seq. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR d. Number of unique reads retrieved per single cell split by chromatin domain from wells containing a cell or a well left intentionally empty during sorting (n = 2 biological replicates). The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR. e. Percentage of the genome covered by histone peaks called from the ChiC portion of scEpi2-seq data from K562 cells. f. Ternary plot showing density of the number of cuts for three histone marks (that is, H3K36me3,H3K27me3 and H3K9me3) in 200 bp intervals across the genome. g. Observed over expected overlap between histone marks was compared to randomly sampled regions of the same size. Y-axis represents enrichment with respect to 1,000 resampled regions. Overlap between histone modifications was lower than would be expected by chance, demonstrating their overall mutually exclusive pattern. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR h. Correlation between in silico bulk measurements for histone modifications measured with scEpi2-seq and previously published single-cell sortChiC data (Zeller et al., 2023, H3K27me3, H3K9me3 and H3K36me3). Reads are counted and aggregated for non-overlapping 100 kb bins across the genome. Correlation (Spearman’s rho) ranged from 0.92 to 0.95. i. Correlation between in silico bulk measurements for histone modifications measured with scEpi2-seq and Encode ChIP (H3K27me3, H3K9me3 and H3K36me3) and previously published single-cell sortChiC data (H3K9me3). Reads are counted and aggregated for non-overlapping 50 kb bins across the genome. Correlation (Spearman’s rho) ranged from 0.82 to 0.95 for sortChiC and 0.63 to 0.95 for ENCODE. j. Coverage plot of Signac normalized pseudobulk H3K27me3, H3K36me3, and H3K9me3 signal in K562 cells for a region containing the PRAME gene (highly expressed in K562 cells).
Extended Data Fig. 3 Comparison of CpG methylation data.
a. Bar plot showing genome-wide single-cell CpG coverage in the K562 scEpi2-seq data set. b. Correlation between pseudobulk DNA methylation data extracted from the TAPS portion of K562 scEpi2-seq data and ENCODE WGBS data for different cell lines. Correlation is calculated for non-overlapping 10 kb bins with a coverage of at least 50 reads per bin. c. Correlation between the TAPS portion of K562 scEpi2-seq data and ENCODE WGBS at single-CpG resolution. X-axis represents different cutoffs (0 = no filtering, 1 = more than 1 read etc.). For comparison, WGBS data were downsampled to read depths as observed in our scEpi2-seq data. d. scEpi2-seq traces for different histone modifications (left) and corresponding CpG methylation (right) for K562 cells. The line plots contain comparisons between pseudobulk values (maximum normalized values for histone modifications and average CpG methylation for DNA methylation) from scEpi2-seq and corresponding measurements for bulk ENCODE data (ChIP for histone modifications and WGBS for CpG methylation).
Extended Data Fig. 4 Application of scEpi2-seq in RPE-1 hTERT FUCCI cells.
a. Number of unique reads retrieved per single cell split by chromatin domain from wells containing a cell or a well left intentionally empty during sorting (n = 2 biological replicates). The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR. b. Cells were quality-filtered based on the number of unique ChIC cut sites and DNA methylation. Top - Scatter plots showing the number of unique cut sites (x-axis, log10 scale) and per-cell 5mC levels (y-axis) in the RPE-1 data set. Similar to the top panel, but zoomed in showing only cells with higher numbers of unique ChiC counts ( > 8000 reads). Dots are colored by fraction reads in peaks (FRiP). c. Violin plot showing the fraction of reads in peaks (FRiP) for QC-filtered cells in the RPE-1 hTERT FUCCI data set (n = 2 biological replicates). Data are grouped for different modifications assessed with scEpi2-seq. The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR. d. Normalized density plot showing the number of CpGs detected per single RPE-1 hTERT FUCCI cells (x-axis, log10 scale stratified by histone mark (columns) and QC status (rows). e. Distribution of RPE-1 methylation levels (WGBS) within peaks called from scEpi2-seq data (top row). Also included are methylation levels for random regions with the same size distribution (bottom row). f. Heatmap with histone modification signal intensities (number of ChIC counts) of single RPE-1 cells (rows) for a representative region on chromosome 2. The genomic region is equivalent to the one presented in Fig. 1b. g. Single-cell heatmap for cut site spacing in RPE-1 cells for the 250 cells with highest unique coverage per chromatin mark and arranged by the unique read depth. The read pair correlation shows characteristic oscillations related to nucleosome spacing. h. Comparison of in silico bulk aggregates of relative methylation extracted from scEpi2-seq data and WGBS data of the same cell line in 25 kb bins across the genome. Correlation estimation was performed using Pearson correlation test with a correlation coefficent of 0.9 and the and a p-value < 2.2×10−16 i. Average CpG methylation in relation to distance from the cut site. Similar to K562 data, H3K36me3-decorated, actively transcribed regions show the highest average methylation. j. Coverage plot of Signac normalized pseudobulk H3K27me3, H3K36me3, and H3K9me3 signal in RPE-1 hTERT FUCCI cells for a region containing the FTH gene (highly expressed in RPE-1 hTERT cells).
Extended Data Fig. 5 Wanderlust computation of Integrated cell Cycle Progression and Nearest neighbor smoothing over Integrated Cell Cycle progression.
a. Fluorescent intensities of FUCCI markers (x-axis, S/G2 marker, mAG-Geminin and y-axis, G1 marker, Cdt1-mKO) and cells are colored based on Wanderlust ordering into Integrated Cell Cycle Progression for all cells acquired during the cell sorting process from all antibody conditions. b. Fluorescent intensities of FUCCI markers (x-axis, S/G2 marker, mAG-Geminin and y-axis, G1 marker, Cdt1-mKO) and cells are colored based on manual annotations of cell cycle phases for all cells acquired during the cell sorting process from all antibody conditions. c. DAPI intensity for cells from manually annotated cell cycle phases acquired during the cell sorting from all antibody conditions. d. CpG Methylation values (y-axis) per single cell over Integrated Cell Cycle Progression (x-axis). Each dot indicates the average CpG methylation within a single cell. The line is a rolling window smoothing for the average CpG methylation over 10 nearest neighbors. e. Fluorescent intensities of FUCCI markers (x-axis, S/G2 marker, mAG-Geminin and y-axis, G1 marker, Cdt1-mKO) and cells are colored based on CpG methylation values with each column representing Increasing number of nearest neighbor smoothing (that is, 1,2,5,10 plus 10 with winsorizing) split by chromatin domain per row (that is, H3K27me3, H3K36me3 and H3K9me3).
Extended Data Fig. 6 H3K27me3 scEpi2-seq for on mouse intestinal samples.
a. CellTrace labeling and visualization of intestinal fractions by flow cytometry. b. log2 fold changes of H3K27me3 signal on genes (each dot indicates a gene, color indicates adj. P-value, default likelihood ratio test using Signac) from 3 individual mice split by Absorptive, Secretory and Immune cells (x-axis, coarse Signac clusters, see Methods). Each facet contains genes associated with either Absorptive, Secretory or Immune cells (Supplementary Table 4). The boxplot is defined by the median ± interquartile range (IQR) and whiskers represent 1.5× IQR c. Heatmap with histone modification signal intensities (no. of ChIC counts) of single intestinal cells (rows) split by coarse Signac clusters (that is, Absorptive, Secretory and Immune) for a representative region on chromosome 1. d. Average CpG methylation (y-axis) in relation to distance from the cut site (x-axis). e. Single-cell heatmap for cut site spacing in intestinal cells split by coarse Signac cluster (that is, Absorptive, Secretory and Immune). The read pair correlation shows characteristic oscillations related to nucleosome spacing. f. Coverage plot of normalized H3K27me3 signal between Enteroendocrine cells and Goblet cells for a region containing the Gfi1 gene (expressed in Goblet cells). g. Coverage plot of normalized H3K27me3 signal between Enteroendocrine cells and Goblet cells for a region containing the Pax6 gene (expressed in EECs). h. Coverage plot of normalized H3K27me3 signal between T-cells, B-cells and Myeloid cells for a region containing the Lyn gene (expressed in B-cell and Myeloid cells). i. Coverage plot of normalized H3K27me3 signal between T-cells, B-cells and Myeloid cells for a region containing the Il10ra gene (expressed in B-cells and T-cells). j. Coverage plot of normalized H3K27me3 signal between T-cells, B-cells and Myeloid cells for a region containing the Syk gene (expressed in B-cells and Myeloid cells). k. Coverage of CpG per single cell (y-axis) versus average CpG methylation (x-axis) per single cell (each dot). Each dot is colored by quality control (pass/fail), cells that pass QC were used for downstream analysis. l. UMAPs generated from CpG methylation using MethSCAn. Cells are colored based on two 5mC clusters retrieved from Leiden clustering on the residuals on the variable methylated regions (see Methods). m. Fraction of 5mC cluster (y-axis) derived from Leiden clustering over the three individual mice (x-axis). n. Difference in CpG methylation (x-axis) and adj. P-value, y-axis) for each identified Differentially Methylated Regions (DMRs) by MethSCAn between 5mC cluster pseudobulks and all pairwise combination between pseudo-bulks from all H3K27me3 subclusters. Dots are colored by significant and non-significant DMRs derived using a two-sided t-test (CpG coverage pseudobulk > 25 within DMR an adj. P-value < 0.001).
Supplementary information
Supplementary Information
Supplementary Tables 1–4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Geisenberger, C., van den Berg, J., van Batenburg, V. et al. Single-cell multi-omic detection of DNA methylation and histone modifications reconstructs the dynamics of epigenomic maintenance. Nat Methods 22, 2042–2051 (2025). https://doi.org/10.1038/s41592-025-02847-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02847-4
This article is cited by
-
Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features
Nature Communications (2026)
