
Abstract
The receptor binding domain (RBD) of the Spike protein from SARS-CoV-2 is a promising candidate to develop effective COVID-19 vaccines since it can induce potent neutralizing antibodies. We have previously reported the highly efficient production of RBD in Pichia pastoris, which is structurally similar to the same protein produced in mammalian HEK-293T cells. In this work we designed an RBD multimer with the purpose of increasing its immunogenicity. We produced multimeric particles by a transpeptidation reaction between RBD expressed in P. pastoris and Lumazine Synthase from Brucella abortus (BLS), which is a highly immunogenic and very stable decameric 170 kDa protein. Such particles were used to vaccinate mice with two doses 30 days apart. When the particles ratio of RBD to BLS units was high (6–7 RBD molecules per BLS decamer in average), the humoral immune response was significantly higher than that elicited by RBD alone or by RBD-BLS particles with a lower RBD to BLS ratio (1–2 RBD molecules per BLS decamer). Remarkably, multimeric particles with a high number of RBD copies elicited a high titer of neutralizing IgGs. These results indicate that multimeric particles composed of RBD covalent coupled to BLS possess an advantageous architecture for antigen presentation to the immune system, and therefore enhancing RBD immunogenicity. Thus, multimeric RBD-BLS particles are promising candidates for a protein-based vaccine.
Similar content being viewed by others

Introduction
The coronavirus SARS-CoV-2, which was detected about 1 year ago in Wuhan, Republic of China in December 2019, has produced over 4.88 million deaths by October 2021 (https://ourworldindata.org/covid-deaths). Since the discovery of the new strain, several vaccines have been developed (https://covid19.trackvaccines.org/vaccines/), based on inactivated viruses, DNA, RNA and/or proteins1,2,3. Furthermore, multivalent antigens that mimic antigen presentation by viral particles have been shown to induce a stronger immune response than monomeric antigens4, thus allowing immunization with lower doses of antigen, as well as the combination of different antigen variants. This approach would allow simultaneous immunization against the multipleSARS-CoV-2 lineages that have unfortunately emerged around the world5.
The receptor binding domain (RBD) of SARS-CoV-2 Spike protein is responsible for the interaction of the virus with the receptor Angiotensin-Converting Enzyme 2 (ACE2)6, which allows the penetration of the virus into the cells. Therefore, RBD constitutes a suitable target to develop diagnostic and therapeutic tools, as well as vaccines. It has been found that several neutralizing antibodies produced upon SARS-CoV-2 infection are directed towards RBD7,8. The structure of the complex formed by SARS-CoV-2 RBD and human ACE2 was recently determined9. Unlike SARS-CoV-1, several residue changes in SARS-CoV-2 RBD stabilize two virus-binding hotspots at the RBD/ACE2 interface, thus increasing its affinity for ACE29.
SARS-CoV-2 Spike’s RBD is a challenging protein to produce in heterologous systems because it has nine cysteine residues (eight of which form disulfide bonds), a complex topology and two N-glycosylation sites10,11. For these reasons, RBD is often expressed in mammalian cells9,12. Nevertheless, our AntiCovid consortium was able to produce RBD at high yields in Pichia pastoris, with a stability, biophysical properties and immunogenicity similar to those of RBD produced in mammalian HEK-2913T cells12. Remarkably, the production of RBD in yeast cells is highly cost effective, and the fermentation bioprocess can be easily scaled up under Good Manufacturing Practice conditions. In pilot-scale experiments in a bioreactor we attained yields of 180 mg L−1 of ~ 90% of pure RBD in P. pastoris, which is currently used as an antigen for serological test kits, and as an immunogen. RBD may be used to produce neutralizing antibodies at large scale in animal systems such as egg yolk (IgY)13,14 or in horses15.
While RBD is immunogenic per se, further enhancement of its immunogenicity should allow the development of a new generation of subunit vaccines, which ideally should be capable of simultaneously providing immunity against the different emerging RBD variants. A possible strategy to achieve this goal is coupling RBD to an immunogenic carrier protein. Lumazine synthase (LS, EC 2.5.1.78) from Brucella abortus (BLS) is a highly immunogenic and very stable decameric protein ideally suited to function as an immunogenic carrier. Five BLS subunits (17 kDa each) form a pentamer, and two pentamers a very stable decamer, which can be decorated with any protein whose antigenicity needs to be increased by coupling it to the N-terminus of BLS16,17,18. Moreover, BLS activates dendritic cells in vitro, increasing the levels of co-stimulatory molecules and the secretion of proinflammatory cytokines, and also recruits dendritic cells in vivo, in both cases in a TLR4-dependent manner19. In addition, BLS chimeras are extremely effective to rapidly activate specific CD8+ lymphocytes, and to induce significant cytotoxic activity20. While other carrier proteins have been non-covalently attached to target proteins through a pair of interacting proteins (e.g. X-dockerin-cohesin-Y21 where X and Y are proteins expressed as a fusion protein with the dockerin and cohesin domains, respectively), such non-covalent interactions might be relatively weak particularly for immunization purposes, which normally require the use of strong adjuvants. The translational fusion of target proteins and peptides with BLS has allowed researchers to overcome this problem22. However, given that BLS fusions are usually expressed at high yields in Escherichia coli, this strategy may be not adequate for proteins with a complex structure and high disulfide bonds content, and/or that require post translational modifications, as it is the case of RBD, which cannot be properly expressed in E. coli12. This problem could be circumvented through the covalent coupling of RBD and BLS in vitro, in their native state, using the Sortase A enzyme.
Sortase A and its variants can efficiently catalyze a transpeptidation reaction that creates a covalent link between two native proteins or peptides23. This reaction requires the N-terminal protein to be covalently linked to contain a specific signal located in its C-terminal stretch: ProteinN-Leu-Pro-X-Thr-Gly (Fig. 1). This site should be preferably in an unstructured context, and the Gly residue is usually followed by a histidine tag to facilitate its purification. The C-terminal protein to be covalently linked should contain three Gly amino acid residues in its N-terminal region (Gly-Gly-Gly-ProteinC). After the Sortase A transpeptidation reaction, both subunits are joined to produce the covalently-linked product ProteinN-Leu-Pro-X-Thr-Gly-Gly-Gly-ProteinC (Fig. 1). Thus, the Sortase A-mediated transpeptidation reaction can couple preassembled oligomeric carriers, such as BLS or virus-like particles (VLPs), to an immunogen expressed in a different system24. An additional advantage of Sortase A-mediated coupling is that the scar of the transpeptidation reaction in the protein product is only seven residues, of which three are glycines.

The strategy: Sortase A-mediated transpeptidation reaction. (A) The reaction is exemplified by the coupling of a BLS subunit (red) and the SARS-Co-V2 RBD domain (green). The enzymatic intermediary form (RBD covalent bound to Sortase A) is shown, as well as its subsequent hydrolysis to yield the by-product RBD-Leu-Pro-Glu-Thr. The latter cannot be reused in the coupling reaction. The reaction catalyzed by TEV protease to yield Gly-Gly-Gly-BLS, the substrate of Sortase A is also shown. (B) A hypothetical reaction product with a multiplicity of 10:10 (RBD10/BLS10) is shown.
In this work, we produced a multimeric RBD-BLS by a Sortase A-mediated transpeptidation covalent coupling reaction between RBD expressed in P. pastoris and BLS produced in E. coli, with the purpose of increasing RBD immunogenicity. RBD-BLS induced a strong humoral response against SARS-CoV-2 RBD. Remarkably, the RBD antigen produced a significantly higher humoral response when presented to the immune system as an RBD-BLS multimer with a high number of RBD molecules per particle (high multiplicity) than as a monomer, or as low multiplicity particles RBD-BLS. Thus, the RBD-BLS multimer is a potential vaccine candidate, which can also be used to produce neutralizing antibodies in animal systems. This platform could be also used to simultaneously present several proteins to the immune system, such as different RBD variants and other domains of the spike protein.
Results
Production of Sortase A-substrates: BLS in E. coli and RBD in Pichia pastoris
In order to obtain a multimeric carrier for RBD, we first produced an oligomeric form of BLS in E. coli. For this purpose we synthesized and sequenced a gene encoding a BLS carrier protein variant which included in its N-terminal region a TEV protease cleavage site (in italics) followed by a Gly-tag (Met-Glu-Asn-Leu-Tyr-Phe-Gln-Gly-Gly-Gly-BLS) (Fig. 1). This strategy allows the removal of the N-terminal Met after a quantitative digestion with the TEV protease, thus producing a polypeptide carrying an N-terminal Gly-Gly-Gly required for the Sortase A-mediated transpeptidation reaction. Following BLS expression in E. coli, the protein was purified in two steps, by anion exchange chromatography (Q-Sepharose) followed by size exclusion chromatography (SEC-HPLC, Superdex S200). The protein was obtained with a high yield (100 mg L−1, > 98% purity), and its identity was verified after purification by mass spectrometry. BLS was then digested with TEV protease to generate the N-terminal Gly-Gly-Gly motif so that it can function as a Sortase A substrate. The digestion was confirmed by SDS-PAGE and mass spectrometry (Figs. S1, S2), which showed the expected “activated” multimeric carrier Gly-Gly-Gly-BLS. It is worth mentioning that the TEV digestion of BLS was complete.
To generate the second substrate of the coupling reaction, an RBD variant including a Sortase A recognition signal (Leu-Pro-Glu-Thr-Gly-) followed by a (His)6-tag at its C-terminus was produced in P. pastoris (RBD-Leu-Pro-Glu-Thr-Gly-(His)6) and obtained from culture supernatants with a high yield (180 mg L−1, > 90% purity). We have previously shown that this protein is conformationally stable and drives a significant immune response in mice when administered in a formulation with adjuvants25. We assessed the tertiary structure of the purified RBD through near-UV-CD spectroscopy. The results are compatible with an asymmetric and rigid environment of the aromatic side chains, indicating a native packing (Fig. 2A,B). Moreover, the protein does not exhibit significant aggregation, as inferred from the SEC-HPLC profiles and the absence of species with low elution volumes (Fig. 2C). In addition, as previously observed25, SEC-HPLC analysis revealed the existence of two different species (differing in their glycosylation patterns) with elution times compatible with molecular masses between 45 and 20 kDa.

RBD expressed in Pichia pastoris is properly folded and does not aggregate. Near-UV circular dichroism spectra of purified RBD: high tension voltage (A) and Near-UV CD spectra (B). The protein concentration was 30 µM, in 20 mM Tris–HCl, 100 mM NaCl, pH 7.4. (C) SEC-HPLC of RBD elution profiles (monitored at 280 nm) corresponding to two different batches (R4 and S4) of independently expressed and purified RBD, and of molecular mass markers (158, 44, 17 and 1.35 kDa).
Native Sortase A mediated coupling of RBD to a fluorescent peptide
To evaluate the ability of recombinant RBD-Leu-Pro-Glu-Thr-Gly-(His)6 to act as a Sortase A substrate, we first carried out a preliminary reaction using the fluorescent peptide probe Gly-Gly-Gly-Ser-{Lys-(TMR)} as a substrate instead of BLS. This peptide contains a covalently bound tetramethylrhodamine (TMR) label. The product of this reaction was the covalently coupled RBD-Leu-Pro-Glu-Thr-Gly-Gly-Gly-Gly-Ser-(Lys-TMR) (RBD-TMR).
After the coupling reaction and SEC (G25) separation, nearly 17% of input RBD was labeled with the fluorescent probe as inferred from UV and visible spectra analysis (Fig. 3A,B). The unreacted RBD retained the (His)6 tag, and could therefore be separated and recovered by addition of NTA resin to the reaction mix followed by centrifugation. This procedure produced a final yield of ~ 50% labelled RBD. Moreover, SEC-HPLC analysis, monitored at 554 nm, which allows detection of the TMR label, revealed the presence of the reaction product. However, no free fluorescent peptide was observed in the elution profile (Fig. 3C), indicating that the G25 matrix was capable of separating the residual free peptide. The analysis of the reaction product did not show peaks at short elution times, suggesting that RBD-TMR was not prone to aggregation after the covalent coupling of the probe. These results showed that Sortase A was functional, and it was possible to carry out the covalent coupling between recombinant RBD and a Gly-Gly-Gly-tagged substrate.

Covalent Coupling of RBD to a Fluorescent Probe Mediated by Sortase A. (A) UV and (B) visible absorption spectra of the free fluorescent probe (blue) and the coupling reaction product RBD-TMR (green) after a G25 column separation. The RBD spectrum is also shown as a reference (red) (C) SEC-HPLC elution profiles (Superose 6 column) of RBD (monitored at 280 nm, red), TMR (monitored at 554 nm, blue) and RBD-TMR (monitored at 554 nm, green) after coupling and G25 separation.
Functionality of recombinant RBD
In addition to the conformational analysis, we also assessed the functionality of purified RBD-TMR. Therefore, we tested its ability to bind the RBD receptor ACE2. For this purpose, we incubated a mix of (a) the labeled protein (RBD-TMR), (b) a soluble version of ACE2 with a C-terminal (His)6 tag, and (c) agarose-NTA-Ni2+resin (Fig. 4A) during 30 min. Next, we separated the ternary complex ACE2/RBD-TMR/agarose-NTA-Ni2+ by centrifugation. Since the matrix agarose-NTA-Ni2+ interacts with the (His)6 tag that it is present only in the C-terminal of ACE2 (and not in RBD-TMR), the centrifugation pulls down the resin bound to ACE2, which is in turn bound to RBD-TMR. This causes a significant decrease of the fluorescence intensity of the supernatant (570–580 nm, Fig. 4B). Elution of the complex from the NTA matrix by addition of 300 mM imidazole caused a strong fluorescence increase in the soluble fraction, further indicating that (His)6-tagged ACE2 forms a complex with RBD-TMR (Fig. 4C). We concluded that RBD-TMR was capable of interacting with the ACE2 soluble domain, indicating that the structural features relevant for the interaction are preserved in recombinant RBD produced in P. pastoris, and that the conditions employed to perform the transpeptidation reaction to produce RBD-TMR did not affect its tertiary structure.

Evaluation of RBD-TMR Binding to ACE2. (A) RBD-TMR pull down and elution from the matrix. (His)6-tagged ACE2 was immobilized in an NTA matrix (300 μL of 0.9 μM ACE2 + 75 μL of agarose-NTA-Ni2+ resin). A control incubation was performed omitting ACE2. Samples were centrifuged and washed three times and subsequently incubated in the presence of 1 μM RBD-TMR for 30 min under mild agitation. Created with BioRender.com. (B) After centrifugation, the supernatant was transferred to a fresh tube and analyzed by fluorescence spectroscopy. (C) The matrix was washed three times and eluted with 300 μL of 20 mM Tris–HCl pH 8.0, 150 mM NaCl and 250 mM imidazole. The sample was centrifuged, and the supernatant was analyzed by fluorescence spectroscopy. In (B,C) black lines correspond to the incubation in the presence of ACE2, while red lines correspond to the controls without ACE2. The excitation wavelength was 550 nm, the bandwidths for both excitation and emission were of 5 nm. All the measurements were made at 25 °C.
Native Sortase A-mediated coupling of RBD to BLS
Given that recombinant RBD proved to be a good Sortase A substrate, we attempted the coupling of RBD-Leu-Pro-Glu-Thr-Gly-(His)6 and TEV-digested Met-Glu-Asn-Leu-Tyr-Phe-Gln-Gly-Gly-Gly-BLS. The expected product was the covalently coupled RBD-Leu-Pro-Glu-Thr-Gly-Gly-Gly-Gly-Ser-Gly-Ser-Gly-BLS (RBD-BLS in Fig. 1). The SEC-HPLC profiles (Superose 6 column) corresponding to a BLS multimer, RBD alone and the reaction product are shown in Fig. 5A. The product of the covalent coupling mediated by Sortase A exhibited an elution time compatible with an increment in the hydrodynamic radius of the assembly compared to that observed for the decameric BLS (27 and 31.5 min, respectively). The unreacted BLS decamer and RBD are also clearly distinguishable in the SEC profile.

Sortase A-mediated covalent coupling of RBD and BLS. (A) SEC-HPLC profiles correspond to Gly-Gly-Gly-BLS (black), RBD (red), the control reaction mixture without Sortase A (violet) and the product of the reaction catalyzed by Sortase A, (green, lower panel). An equivalent volume of a 10 mM EDTA, 10 mM CaCl2 solution was loaded as a control (magenta, lower pane). Decameric BLS has a molecular weight of approximately 170 kDa, and each RBD subunit adds approximately 26 kDa or 40 kDa (excluding or including glycosylation, respectively). The observed multiplicity was ~ 6.6 (RBD6.6/BLS10) with a yield of ~ 30% relative to the initial RBD mass. The RBD-BLS oligomer eluted at ~ 25 min. The peak observed around 38–43 min corresponds to EDTA-Ca2+. Since Sortase A requires 10 mM CaCl2 for the activation of its catalytic activity, 10 mM EDTA was added to stop the reaction, which was carried out during 240 min at 4 °C. (B) SDS-PAGE analysis of Sortase A-mediated RBD-BLS coupling. To facilitate the detection of the Sortase A reaction products, the protein samples were treated or not with endoglycosidase H. Lanes 1–2: RBD + BLS without Sortase A, deglycosylated (lane 1), or not (lane 2), Lanes 3–4: RBD + BLS in the presence of Sortase A, deglycosylated (lane 3) or not (lane 4). Full Image of the SDS-PAGE is in Supplementary Material, Figure S5.
Since BLS is a decameric protein, the transpeptidation products are heterogeneous in non-saturating conditions. Different multiplicities of RBD molecules per decamer (RBDn/BLS10) can be obtained depending on the ratio of substrates used in the Sortase A reaction. We used low RBD and high BLS concentrations to obtain low multiplicities (between 1 and 2 of RBD copies per multimer, Fig. S3, referred as RBD-BLS LM), whereas the higher multiplicities (referred as RBD-BLS HM) were obtained with higher RBD/BLS ratios (Fig. S3, Fig. 5A).
SEC-HPLC and SDS-PAGE analysis of BLS, RBD, and RBD-BLS present in the reaction mixture before and after the addition of Sortase A allowed us to quantify the yield of the coupling reaction and the multiplicity of the products (Fig. S4), that is the average number of RBD copies per RBD-BLS particle.
The presence of heterogeneous sizes of glycan moieties in RBD resulted in a high heterogeneity which complicates the analysis. Therefore, we trimmed the glycans with endoglycosidase H and resolved the deglycosylated products of the coupling reaction by SDS-PAGE (Fig. 5B, compare lanes 1 and 3 to 2 and 4). The sharp band that migrates close to the 40 kDa marker in lane 3 corresponds to the covalently coupled RBD-BLS (26 kDa of deglycosylated RBD + 17 kDa of monomeric BLS, 43 kDa, Lane 1). It should be noted that a large amount of RBD still remains uncoupled (blue arrow in Lane 3), therefore the efficiency of the reaction still needs to be improved. Additionally, a byproduct (around 15–18 kDa) was observed that still needs to be identified (lanes 3 and 4).
In batch conditions, in the presence of twofold molar excess of RBD compared to BLS, we obtained yields of 10–20% RBD coupling with respect to the total amount of RBD used, leading to complexes with average multiplicities between 2 and 4 (RBD2/BLS10 and RBD4/BLS10, respectively) in preliminary experiments. However, the incorporation of a concentration step (with a 10 kDa cutoff Centricon centrifugal filter unit) allowed us to enhance the reaction yield, obtaining higher multiplicities (between RBD6./BLS10 and RBD7./BLS10, RBD-BLS HM, with a ~ 30% yield relative to the initial RBD mass). A further increase in the coupling of RBD can be achieved by increasing BLS concentration, albeit at the expense of decreasing the multiplicity of the final product. Saturation of BLS with RBD was difficult to achieve, due to the intrinsic equilibrium of the coupling reaction.
In summary, we succeeded in coupling the eukaryotic-produced RBD antigen with the E. coli expressed decameric carrier BLS under native conditions. In addition, the possibility of obtaining RBD-BLS with different multiplicities allowed us to study the effect of this parameter on the elicitation of the immune response.
Once purified by SEC, the coupling product was found to be stable after freezing and subsequent thawing, and did not show any significant aggregation tendency. In addition, the product could be easily concentrated, and did not show any apparent interaction with the concentrator membranes (data not shown). Importantly, the coupling products were also separated from the Sortase A enzyme by SEC. This is an essential step to avoid reverse transpeptidation and RBD-Sortase A hydrolysis (that would result in a RBDh product that can not be recycled by the enzyme)26, which would reduce the global yield of the reaction.
The immunogenicity of RBD is increased by coupling to a BLS multimer
To assess whether RBD coupled to BLS can improve the immunogenicity of RBD, we immunized mice with two doses 30 days apart of 15 μg containing RBD coupled or uncoupled to BLS at different multiplicities, i.e. RBD-BLS HM RBD-BLS LM, and monomeric RBD (Table 1, “Materials and methods”). RBD and RBD-BLS-HM formulations were adjuvanted or not with Al(OH)3.
The multimeric RBD-BLS HM formulation (RBD6.6/BLS10) elicited a strong humoral response with high anti-RBD IgG titers (82,000 ± 28,000), which increased an additional ~ 7-fold when adjuvanted with Al(OH)3 (553,000 ± 366,000) (Fig. 6). By contrast, monomeric RBD did not elicit a significant anti-RBD IgG specific antibody response even in the presence of Al(OH)3. Remarkably, a formulation containing 40 μg of monomeric RBD per dose in the presence of Al(OH)3) did not result in a significant increase of the IgG specific antibody response (data not shown). The RBD + BLS group in which mice were immunized with BLS together with monomeric RBD (i.e. not covalently coupled), each at the same concentration as used to immunize the RBD-BLS HM group, did not show any significant difference compared to the group immunized with RBD alone. This result indicates that BLS needs to be covalently coupled to the RBD to enhance its immunological response. Furthermore, the multiplicity of RBD-BLS multimers turned out to be a key variable, since a high multiplicity elicited strong immune response, while a low multiplicity did not (RBD-BLS LM group, between 1 and 2 copies of RBD per multimer). It is worth mentioning that the RBD-BLS LM formulation included the same mass of RBD (15 µg/dose), but a significant higher mass of BLS than the other groups (70 µg BLS/dose for RBD-BLS LM vs. 17 µg BLS/dose for RBD-BLS HM and RBD + BLS groups, Table 1).

RBD-specific humoral immune response in mice vaccinated with monomeric RBD and RBD-BLS multimers. (A) Immunization scheme and blood collection. BALB/c mice (n = 5) were vaccinated with two doses of the formulations 30 days apart subcutaneously, and sera were obtained 20 days after the boost. Created with BioRender.com. (B) Evaluation of anti-RBD IgG antibody titers. Serial dilutions of sera samples were dispensed in a multi-well plate and RBD-specific IgG titers were determined by ELISA at day 50. The addition or not of 500 μg of Al(OH)3 adjuvant is indicated. The results are expressed as mean endpoint titers. The RBD + BLS group wasco-immunized with BLS and RBD at the same concentration as used in the RBD-BLS HM group, but not coupled. Control and PreIS correspond to normal mouse and preimmune sera, respectively. The S.E.M. is indicated by vertical lines. Differences were considered statistically significant when p < 0.05 (*, Student’s t test). Black dots show the individual values corresponding to the RBD-specific IgG titer.
These results clearly show that coupling of RBD to BLS at high multiplicity strongly enhances RBD immunogenicity, which may be due at least in part to the spatial arrangement of the RBD subunits in the resulting particles (Fig. 1B).
Immunization with RBD-BLS elicit antibodies with neutralizing activity against a SARS-CoV-2 S pseudotyped virus transduction
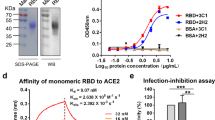
To analyze the neutralizing activity of immune sera elicited by RBD-BLS immunization, in vitro neutralization assays using SARS-CoV-2 S pseudotyped lentiviruses were performed27. Serial dilutions of sera were made in the DMEM medium and incubated with equal amounts of pseudotyped virus, then added to HEK-293T cells previously transfected with ACE2 and TMPRSS2 protease. While pseudotyped virus particles were capable of delivering GFP-encoding RNA into the cells in the presence of preimmune serum, immune sera prevented transduction, shown as a percentage of inhibition, in a concentration-dependent manner. Sera from mice immunized with RBD-BLS HM (Table 1), strongly reduced the number of GFP-expressing cells (Fig. 7). Remarkably, immunizations with RBD-BLS HM administered in the presence or absence of Al(OH)3 adjuvant resulted in high neutralizing antibody titers (Fig. 7, IC50 values of 1816, with adjuvant, and 1503, without adjuvant), as judged by the significant reduction observed in pseudotyped virus transduction. By contrast, sera from mice immunized with Pichia pastoris-expressed RBD domain plus BLS (not covalently coupled) resulted in significantly lower neutralizing antibody titers (IC50 = 17, Table 1, RBD + BLS). Altogether these results show that RBD-BLS HM is a very promising candidate both to induce neutralizing antibodies in animal models.

SARS-CoV-2 S pseudotyped virus neutralization assay. (A) Scheme of the assay. HEK-293T cells transfected with ACE2 and TMPRSS2 protease were transduced with a SARS-CoV-2 S pseudotyped lentivirus carrying a GFP-encoding mRNA in the presence of different dilutions of mouse sera. Forty eight hours later, cells were observed under the microscope. Created with BioRender.com. (B) The number of GFP positive cells for each serial dilution was determined. The serum antibody dilution causing a 50% reduction of GFP positive cells (IC50) compared to control “virus only” treated cells was calculated using Graphpad Prism. PreIS corresponds to the preimmune sera.
Discussion
A year ago, just before SARS-CoV-2 was first detected in Argentina, a group of researchers from different institutions and with different relevant expertises joined forces to help fight the ongoing pandemic. Our work was aimed at producing the RBD domain of SARS-CoV-2 spike protein, as it is a useful tool for the serological detection of infected patients, and has the potential to be used as antigen to develop a low cost vaccine. So far we have successfully produced RBD in different expression systems, among them in the yeast P. pastoris, which showed biophysical properties similar to that of RBD expressed in mammalian cells25.
In this work RBD was covalently coupled to a multimeric carrier with the aim of improving its immunogenicity to develop a novel vaccine candidate. As a carrier we used the multimeric lumazine synthase protein from Brucella abortus (BLS), which has been previously shown to be a good antigen carrier for systemic immunization and as mucosal-adjuvant, which makes it a good candidate to be used for oral subunit vaccines17. We have improved the efficiency of the RBD-BLS coupling reaction to scale up the production and purification of the RBD-BLS oligomer, obtaining different multiplicities (1–6) and compared the immunogenicity of RBD-BLS vs. RBD in mice.
Our results showed that the RBD-BLS multimer can elicit an increased immune response compared to that of monomeric RBD, even in the absence of an adjuvant. This increase was dependent on the covalent coupling of RBD to BLS, as the co-administration of RBD with BLS (i.e. not covalently coupled) did not induce an equivalent immune response. We also observed that the multiplicity of RBD coupled to BLS was an important factor in the enhancement of the immunological response. In immunological studies we observed that RBD coupled to BLS at a high multiplicity (6–7 RBD copies per BLS decamer) is remarkably more immunogenic than monomeric RBD, or than RBD coupled to BLS at a low multiplicity (1–2 RBD copies per BLS decamer). A significant increase in the anti-RBD humoral immune response was observed when mice were immunized with RBD-BLS HM as compared to monomeric RBD. By contrast, no significant humoral immune response was elicited by monomeric RBD in the presence or absence of Al(OH)3, or by the addition of BLS to RBD (without covalent coupling).
Whereas BLS is highly resistant to urea-induced unfolding, its decameric structure is sensitive to low pH (pH 4.0–5.5), or to moderate guanidinium chloride concentrations (2.0 M)28, which cause the dissociation of the decameric forms to pentamers. In addition, BLS engineered variants exist with mutations at the pentamer-pentamer interface that allow the production of polarized heteropentameric decamers29. This makes BLS a very malleable protein, which might allow, for example, to include different immunogens in a single protein particle. BLS oligomers carrying different coupled proteins could be disassembled by incubation at low pH, mixed and reassembled so as to generate a diversity of homo and hetero decamers.
It is interesting that RBD-BLS (HM) induced significant antibody response even without adjuvant. It has previously been reported that BLS induces the activation of dendritic cells in a TLR4 dependent manner in the absence of adjuvants. Also the ability of several BLS chimeras to activate dendritic cells have been tested20. As a Toll-like receptor (TLR) agonist, it has been shown that BLS can stimulate both innate and adaptive immune responses, thereby improving vaccine efficacy17,20. Thus RBD-BLS particles, alone or in combination with adjuvants, have the potential to boost T- and B-cell responses against SARS-CoV-2, through mechanisms that deserve further exploration. A detailed analysis of the profiles of immunoglobulins elicited, cellular response and challenge experiments are yet to be performed to assert a long-term protection against Sars-CoV-2 infection.
The idea of coupling RBD to a multimeric protein to produce a stronger immunogen has been considered by other groups. Some molecular platforms have been proposed to multimerize CoV antigens. A very interesting recent work reports the production of particles displaying regions of the Middle East respiratory syndrome coronavirus (MERS-CoV) spike protein, by coupling its RBD domain to a multimeric lumazine synthase (LS), using the SpyTag/SpyCatcher system30, in which the peptide SpyTag that includes 13 amino acids reacts with the 12.3 kDa-SpyCatcher protein31,32. The SpyTag/SpyCacther system was recently used for the construction of a ferritin nanoparticle-based SARS-CoV-2 RBD vaccine33. A similar strategy has been recently developed by Zhang and coworkers for SARS-CoV-2 spike protein using both Aquifex aeolicus LS and Helicobacter pylori ferritin as nanoparticle scaffolds34. They coupled a trimeric version of the antigen to LS, which caused such nanoparticles to elicit significantly higher neutralizing antibody responses than the spike protein alone. New strategies to develop novel vaccines against SARS-CoV2 also include self-assembling fusion proteins. For example, Powell and coworkers have recently prepared in Expi293F mammalian cells a fusion protein containing the spike ectodomain followed by H. pylori ferritin, a 19 kDa protein that self-assembles into a 24-subunit nanoparticle35. Of note, such nanoparticles display eight copies of a trimeric antigen on the surface, as previously used for haemagglutinin from H1N1 virus36. The authors showed that multivalent presentation of spike trimers on ferritin can notably increase elicitation of neutralizing antibodies in mice after a single dose.
Several technologies are being applied for SARS-CoV-2 vaccines development. These include mRNA, non-replicating viral vectors, protein subunit and inactivated virus vaccines technologies, which have demonstrated to be safe and efficient with reported efficacy between 50 and 95%37. The use of mRNA and adenovirus is relatively new compared with that of protein-based vaccines, which is a long-time tested technology. Clinical trials for SARS-COV2 RBD-based protein subunit vaccines demonstrated them to be safe, and to elicit acceptable immune responses. Moreover, they do not require very low storage temperatures, which may represent an important limitation for developing countries with limited resources to keep cold-chain supplies. However, they often fail to elicit potent and long-lasting immune responses, and need to be formulated with potent adjuvants. Multivalent antigens coupled to nanoparticles have proven to have higher efficacy at a lower dose than traditional protein-subunit vaccines. Here we demonstrated that conjugation of RBD to BLS at high multiplicity is able to significantly increase the immunogenicity of RBD, even in the absence of adjuvants. Compared with other multivalent antigens, the sortase A-mediated transpeptidation coupling used to produce RBD-BLS leaves only a minimal protein scar of only seven residues, of which three are glycines. Multi-antigen vaccines might be better for protection against virus variants. The modular design of the RBD-BLS nanoparticle would allow the easy delivery of new antigen-variants combinated in the same particle. Even though this is a promising strategy, the yield of the coupling reaction is still limited and affects the multiplicity and homogeneity of the product. Lowering the complexity of the production process and increasing the yield of the conjugation reaction would be necessary for scaling up the antigen production. In this context, the strategy presented here involving RBD-BLS particles may be considered as a new useful tool against SARS-CoV-2. The platform is ductile enough to incorporate new variants like E484K, which has been associated with escape from neutralizing antibodies38,39,40 or N501Y, which is associated with increased binding specificity and faster-growing lineages41.
Materials and methods
SARS-CoV-2 RBD protein expression and purification
The receptor binding domain of spike protein from SARS-CoV-2 (RBD) was expressed in Pichia pastoris. The RBD coding sequence (spike amino acid residues 319–537) was fused to the Saccharomyces cerevisiae alpha factor secretion signal (N-terminal) followed by a C-terminal Sortase A recognition sequence and a His6 tag (C-terminal) given the C-terminal sequence RBD-Leu-Pro-Glu-Thr-Gly-(His)6 as previously reported25. The expression was carried out in a bioreactor as indicated, and the protein was purified with a yield of 150 mg L−1 of culture supernatant25. RBD concentration was determined by UV spectrophotometry using an absorption coefficient of 33,850 M−1 cm−1.
BLS expression and purification
A gene encoding a variant of lumazine synthase of Brucella abortus (BLS) including an N-terminal TEV site followed by a Gly-Gly-Gly sequence and a Gly-Ser-GLy-Ser-GLy spacer (Met-Glu-Asn-Leu-Tyr-Phe-Gln-Gly-Gly-Gly-Gly-Ser-GLy-Ser-Gly-BLS) was assembled and cloned in the pET11a vector (Novagen). The construct was checked by sequencing. The protein was expressed in E. coli BL21(DE3). Cells were grown in Luria Bertani broth at 37 ºC to OD600 1.0, and protein expression was induced by 1 mM IPTG. Expression was carried out at 28 ºC overnight. The cells were harvested by centrifugation at 6000 rpm for 20 min and freezed at − 20 ºC until use. The cell pellet was resuspended in 20 mL of 20 mM Tris–HCl, pH 7.4 and gently sonicated in an ice-water bath. The sample was centrifuged at 10,000 rpm for 30 min, and the soluble fraction was transferred to a fresh tube. The protein was purified in two steps. First the soluble fraction was loaded onto an anionic exchange chromatographic column (Q-Sepharose, given that the theoretical pI value of BLS is 6.4). The fractions containing BLS, as judged by SDS-PAGE analysis, were pooled and loaded onto a size exclusion chromatographic column (Superdex S200). Fractions containing BLS were pooled and the purified protein was analyzed by mass spectrometry and SDS-PAGE. BLS concentration was determined by UV spectrophotometry using an absorption coefficient of 18,405 M−1 cm−1.
ACE2 expression and purification
The expression plasmid pcDNA3-sACE2(WT)-8his, which includes the human ACE2 soluble domain (residues 1–615) coding gene, followed by a Gly-Ser-Gly linker and eight His residues for purification, was transfected in HEK-293T mammalian cells. Cells were grown in high glucose (4.5 g L−1) Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Natocor), 110 mg L−1 of sodium pyruvate (Thermo Fisher Scientific) and penicillin/streptomycin (100 units mL−1 and 100 μg mL−1 respectively, Thermo Fisher Scientific) in a 37 °C incubator under 5% CO2. Cells were plated (2 × 107 cells per 150 mm plate) and grown for 24 h before transfection with Polyethylenimine (PEI, Sigma). After 72 h, the cell culture medium was centrifuged twice at 12,000×g for 20 min at 4 °C. The supernatant pH was adjusted to pH 8.0 with an equilibration buffer (50 mM sodium phosphate, 300 mM NaCl and 20 mM imidazole, pH 8.0). Soluble ACE2 was purified using a previously equilibrated Ni 2+-NTA-agarose column. The protein was eluted by increasing imidazole concentrations (prepared in the equilibration buffer). Fractions containing the ACE2, as judged by the SDS-PAGE analysis, were pooled and dialyzed against a buffer without imidazole (20 mM sodium phosphate, 150 mM NaCl, pH 7.4). pcDNA3-sACE2(WT)-8his was a gift from Erik Procko (Addgene plasmid # 149268; http://n2t.net/addgene:149268; RRID:Addgene_149268)42, and the HEK-293T mammalian cell line was kindly provided by Xavier Saelens (VIB-University of Ghent, Belgium). ACE2 concentration was determined by UV spectrophotometry using an absorption coefficient of 157,220 M−1 cm−1.
TEV protease expression and purification
Tobacco Etch Virus protease (TEV) was a gift from Helena Berglund (Addgene plasmid # 125194; http://n2t.net/addgene:125194; RRID:Addgene_125194)43. Briefly, E coli Rosetta (DE3) cells (starting with a 1/20 dilution of an overnight pre-culture) were grown in Luria Bertani media at 37 °C up to OD600nm of 0.6. TEV was induced (for 4 h at 37 °C) by the addition of 0.5 mM IPTG. Cells were centrifuged, resuspended, and sonicated in 20 mL of lysis buffer (50 mM phosphate pH 8.0, 150 mM NaCl, 20 mM Imidazole, 10% glycerol). The sample was subsequently centrifuged, and the soluble fraction loaded in a NTA-Ni2+ agarose column previously equilibrated with the lysis buffer. A washing step with 10 column volumes was performed, and the protein was eluted with 2.5 volumes of elution buffer (50 mM phosphate pH 8.0, 150 mM NaCl, 250 mM Imidazole, 10% glycerol). Finally, to remove imidazole, the protein was subjected to dialysis and stored at − 80 °C in small fractions.
Sortase A expression and purification
The Sortase A pentamutant (eSrtA) in pET29 was a gift from David Liu (Addgene plasmid # 75144; http://n2t.net/addgene:75144; RRID:Addgene_75144)44. Briefly, E. coli BL21 (DE3) cells (starting with a 1/40 dilution of an overnight pre-culture) were grown in Terrific Broth at 37 °C up to OD600nm of 0.6. Sortase A expression was induced for 5 h at 37 °C by the addition of 1 mM IPTG. Cells were centrifuged, resuspended, and sonicated in 35 mL of lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 20 mM Imidazole, 10% glycerol). After that the sample was centrifuged and the soluble fraction loaded in NTA-Ni2+ agarose previously equilibrated with the lysis buffer. A washing step with 10 column volumes was performed and the elution was carried out with 2.5 volumes of elution buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 350 mM Imidazole, 10% glycerol). Finally, imidazole was removed and the protein was concentrated using a Centricon (Merck Millipore) and preserved at − 80 °C in small fractions at 2 mM concentration.
ESI MS for intact mass analysis
The protein samples were analyzed on an LC − ESI–MS platform consisting of a Surveyor MS pump system (C4 column, Higgins Analytical Proto 300 5 µm RS-0301-W045, 30 mm × 1.0 mm) coupled to an electrospray mass spectrometer (Thermo Finnigan LCQ duo, equipped with an ion trap mass analyzer). The gradient was 5% solvent B (96% acetonitrile, 2% acetic acid) in solvent A (2% acetonitrile, 2% acetic acid) for 2 min, 5–100% solvent B in solvent A in 4 min, and 100% solvent B for 8 min at a flow rate of 40 μL min−1. The ESI spectra was deconvoluted to the zero-charge domain by the ProMass Deconvolution Software (Novatia) using a standard parameter set: average mass type, mass tolerance 0.02%, minimum tolerance 2 Da and input m/z range 500–2000 units.
Circular dichroism spectroscopy
CD spectra measurements were carried out at 20 °C with a Jasco J-815 spectropolarimeter. Near-UV CD spectra were collected using cells with path lengths 1.0 cm. High tension voltage was registered simultaneously. Data was acquired at a scan speed of 20 nm min−1 (five scans were averaged, scans corresponding to buffer solution were averaged and subtracted from the spectra). Values of ellipticity were converted to molar ellipticity.
Fluorescent probe
The tetramethylrhodamine (TMR) labeled peptide with sequence Gly-Gly-GLy-Ser-{Lys-(TMR)}was purchased from Genscript. Its mass, as determined by mass spectrometry analysis, and excitation and emission spectra were compatible with the predicted theoretical values. An absorption coefficient of 87,500 M−1 cm−1 at 560 nm was used to calculate the TMR concentration in solution. To evaluate the transpeptidation efficiency, a spectrum of free TMR was subtracted from the spectrum of protein in order to correct the 280 nm absorbance value of the protein solution.
Transpeptidation and multiplicity
The transpeptidation reaction was performed by the Sortase A enzyme. To obtain a product with high multiplicity (6:10, RBD:BLS) 70 and 35 µM of RBD and BLS respectively were used. For lower ratios (approximately 2:10, RBD:BLS), 70 and 350 µM of RBD and BLS respectively were used. The reaction was started by the addition of Sortase A (0.4 µM final concentration from a 2 mM stock solution preserved at − 80 °C). The reaction was performed in a 20 mM Tris–HCl, 150 mM NaCl, pH 7.4, 10 mM CaCl2 buffer. CaCl2 allowed the activation of Sortase A, therefore the reaction was stopped by the addition of 10 mM EDTA. The reaction previously treated or not with 5 mU of endoglycosidase H during 1 h at 37 °C was analyzed by SDS-PAGE and by SEC-HPLC. In order to improve the efficiency and the irreversibility of the transpeptidation reaction, one of the reaction products, the short peptide Gly-His-His-His-His-His-His splitted by Sortase A from the C-terminal of RBD (Fig. 1 for reference), was extracted from the mixture. For this purpose, we performed the reaction in a concentrator of 10 kDa cutoff under centrifugation at 3000 rpm at 4 °C. Control experiments were performed without centrifugation. All samples were kept at 4 °C.
Hydrodynamic behavior of the proteins
Size exclusion chromatography (SEC-HPLC) was carried out using a Superose-6 column (GE Healthcare). The protein concentration was 10–30 μM, a volume of 50 μL was typically injected, and the running buffer was 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0. The experiment was carried out at room temperature (∼ 25 °C) at a 0.5 mL min−1 flow rate. A JASCO HPLC instrument was used. It was equipped with an automatic injector, a quaternary pump and a UV–VIS UV-2075 (the elution was monitored at 280 nm).
Interaction between recombinant RBD and ACE2
The interaction between the covalently coupled product RBD-TMR and the ACE2 soluble domain was studied taking advantage of the presence of a (His)6 tag in the C terminus of ACE2, which is not present in RBD-TMR. ACE2 (1.0 μM) and RBD-TMR (1.0 μM) were incubated during 30 min, with gentle agitation in darkness in the presence of NTA-Ni2+ in 20 mM sodium phosphate, 150 mM NaCl, pH 7.40. The complex was separated from free RBD-TMR by centrifugation (1 min at maximum speed in a microcentrifuge). The fluorescence after the incubation was measured using an excitation wavelength of 560 nm, the emission monitored between 570 and 670 nm, a 4 nm bandwidth was used for both excitation and emission. Measurements were performed in a JASCO spectrofluorometer equipped with a thermostated cell holder connected to a circulating water bath set at 25 °C. A path length cell of 0.3 cm was used. Additionally, the complex bound to the NTA matrix was eluted by the addition of imidazole and the fluorescence of the supernatant was analyzed in the same fashion.
Endotoxin remotion and detection
Lipopolysaccharide (LPS)45 was removed from the protein preparations by an overnight incubation (4 °C) of the proteins with polymyxin B matrix followed by a second incubation (2–3 h) at room temperature. The matrix was previously equilibrated in 20 mM Tris–HCl, 100 mM NaCl, pH 7.0 buffer solution. We measured the LPS content of purified RBD-BLS preparations as previously described19. The endotoxin concentration in the protein samples was determined by Cassará Fundation Laboratories. Briefly, all protein samples were brought to a final volume of 200 μL and the LPS content was measured using a commercial kit from Charles River Endosafe (R160), based on the Limulus amebocyte lysate (LAL) assay chromogenic method, with a sensitivity of 0.015 EU/mL. No LPS was detected in the RBD-BLS preparation following endotoxin removal.
Immunization protocols
Mice immunization was carried out by experts from the High Level Technological Service from CONICET (STAN No. 4482), under guidelines from the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL). BALB/c mice were obtained from the animal facility of the Faculty of Veterinary Sciences, University of La Plata (Argentina), and housed at the animal facility of the Instituto de Ciencia y Tecnología Dr. César Milstein, Fundación Pablo Cassará. Six groups of five Females mice (6–8 week-old) (n = 5) were immunized subcutaneously with the same amount of RBD protein (15 µg) coupled or not to BLS in the presence or absence of 500 µg aluminum hydroxide (Al(OH)3). Two different BLS coupling formulations were obtained (Table 1): RBD6.6/BLS10 (high multiplicity, RBD-BLS HM) and RBD1.5/BLS10 (low multiplicity, RBD-BLS LM). Additionally, uncoupled RBD + BLS formulation (RBD + BLS, Table 1) was tested with the same equimolar conditions used for the RBD-BLS HM group. Pre-immune sera also were collected before starting the immunization. Mice received two immunizations with the same dose at days 0 (antigen prime) and 30 (antigen boost). Additional control animals were injected with 500 µg Al(OH)3 per mouse with the same immunization schedule.
Blood samples were obtained at 20 days post-second immunization by venipuncture from the facial vein. After coagulation at room temperature for 1–2 h, blood samples were spun in a centrifuge at 3000 rpm/min for 10 min at 4 °C. The upper serum layer was collected and stored at − 20 °C.
Identification of serum antibody against protein RBD in mice using an ELISA assay
The antibody response against RBD and RBD-BLS was measured using a standard ELISA procedure. Briefly, flat-bottom 96-well plates (Thermo Scientific NUNC-MaxiSorp) were coated with recombinant RBD protein produced in P. pastoris at a final concentration of 1 µg/mL (100 µL/well) in phosphate-buffered saline (PBS) coating buffer (pH 7.4) at 4 °C overnight. After blocking (8% nonfat dry milk PBS for 2 h at 37 °C) the plates were washed 5 times with PBS containing 0.05% Tween 20 (PBST). Serially diluted mouse sera were incubated at 37 °C for 90 min in PBS containing 1% non-fat dry milk (blocking solution), and plates were subsequently washed with PBST. For total specific IgG determination, IgG horseradish peroxidase (HRP)-conjugated antibody (DAKO P0447) was diluted 1/1000 in blocking solution and added to the wells. After incubation for 1 h at 37 °C, plates were washed five times with PBST and developed with 3,3′,5,5′-tetramethylbiphenyldiamine (TMB) for 15 min. The reaction was stopped with 50 µL/well of 1 M H2SO4 (stop solution). The absorbance was measured in a microplate reader (Thermo Multiscan FC ELISA) at 450 nm.
The antibody titer was determined as the inverse of the last dilution that was considered positive, with a cut-off value defined as absorbance at 450 nm of 0.20, which was twice as high as that from a pool of normal mouse sera (from 30 unimmunized animals). Statistical significance was evaluated by the Student’s t test, using a logarithmic transformation of the ELISA titers. Differences were considered significant if p < 0.05.
Pseudotyped lentivirus neutralization assay
SARS-CoV-2 S-Pseudotyped lentivirus27,46 were produced by co-transfection of HEK-293T cells with plasmids bearing a GFP reporter gene (pLB was a gift from Stephan Kissler, Addgene plasmid #11619; http://n2t.net/addgene:11619; RRID:Addgene_11619), a lentivirus backbone (VRC5602, NIH), and the Spike protein (VRC7475_2019-nCoV-S-WT, NIH). Briefly, HEK-293T cells (2 × 107) were seeded in a 150-mm tissue culture dish in DMEM medium containing 10% FBS. The next day, cells were transfected with 10 μg of VRC5602, 5 μg pLB-GFP, and 3 μg of VRC7475_2019-nCoV-S-WT in OptiMEM medium using PEI in a 1:3 DNA:PEI ratio. Twenty four hours later, transfection efficiency, indicated as GFP fluorescence, was checked under a fluorescent microscope. Pseudotyped lentivirus supernatants were harvested 48 h post-transfection and stored at 4 °C, fresh media (DMEM + 5% FBS) was added to the plates. After 48 h, combined supernatants were clarified by centrifugation for 10 min at 3000 rpm to pellet the residual cells. The clarified supernatant was centrifuged for 5 h at 10,000 rpm. The pellet was resuspended in a storage medium (OptiMEM + 6% Sucrose) and aliquots were frozen at − 80 °C until use.
Pseudotyped lentivirus titers were measured by transducing HEK-293T cells previously seeded in 96-well plates (2 × 104 cells/well) and transiently transfected with 100 ng of ACE2 (NIH) and 10 ng of TMPRSS2 protease (NIH) per well. The pseudotyped virus stock (concentrated supernatant) was serially diluted in assay medium (DMEM + 2.5% FBS), incubated for 2 h at 37 °C and added to the transfected cells. Virus titers were calculated by counting the GFP positive cells using an automated counting tool in ImageJ (NIH), a titer of 200–225 GFP positive cells/field using 100× magnification was used for the assay.
Neutralization assays were performed on HEK-293T cells transiently transfected 24 h before transduction with ACE2 and TMPRSS2 protease genes. Fifty microliters of serial fivefold diluted heat-inactivated serum (56 °C for 45 min), starting dilution of 1:100, were prepared in assay medium and incubated for 2 h with an equal volume of titrated pseudotyped lentivirus. Pseudovirus and sera dilution mixtures (100 μL) were then added onto 96-well plates containing 2 × 104 cells/well. Pseudovirus infectivity was scored 48 h later and images were obtained using an inverted microscope (IX-71 OLYMPUS), and analyzed with the Micro-Manager Open Source Microscopy Software. Sera antibody neutralization titers were calculated by a nonlinear regression curve fit using GraphPad Prism software Inc. (La Jolla, CA, USA). Half maximal inhibitory concentration (IC50), corresponding to the serum antibody dilution causing a 50% reduction of GFP positive cells compared to control “virus only” treated cells, was determined using the same software according to Ferrara and Temperton47.
Ethical approval
All experimental protocols were carried out by experts from the High-Level Technical Service of the National Research Council (CONICET, Argentina) (STAN #4482), under ISO9001 guidelines. The protocol was approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL). The study was carried out in compliance with the ARRIVE guidelines.
Code availability
UniProt Accession IDs: UniProtKB-P0DTC2 (SPIKE_SARS2); Spike glycoprotein from SARS-CoV-2. UniProtKB-Q2YKV1 (RISB2_BRUA2); 6,7-dimethyl-8-ribityllumazine synthase 2. UniProtKB-Q9BYF1 (ACE2_HUMAN); human angiotensin-converting enzyme 2.
Change history
25 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-05984-4
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- BLS:
-
Lumazine synthase from Brucella abortus
- CD:
-
Circular dichroism
- CoV:
-
Coronavirus
- EDTA:
-
Ethylenediaminetetraacetic acid
- ELISA:
-
Enzyme-linked immunosorbent assay
- ESI–MS:
-
Electrospray ionization mass spectrometry
- GFP:
-
Green fluorescent protein
- HM:
-
High multiplicity
- HPLC:
-
High-performance liquid chromatography
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside, Isopropyl β-D-thiogalactoside
- LM:
-
Low multiplicity
- LPS:
-
Lipopolysaccharide
- LS:
-
Lumazine synthase
- NTA-Ni2+:
-
Nickel-charged nitrilotriacetic acid affinity resin
- PBS:
-
Phosphate-buffered saline
- PDB:
-
Protein Data Bank
- PEI:
-
Polyethylenimine
- RBD:
-
Receptor binding domain
- RBD-BLS:
-
The multimeric particle constituted by RBD covalently coupled to the BLS decamer
- RBD-TMR:
-
RBD covalently coupled to tetramethylrhodamine
- SARS-CoV-1:
-
Severe acute respiratory syndrome coronavirus 1
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SDS-PAGE:
-
Polyacrylamide gel electrophoresis
- SEC:
-
Size exclusion chromatography
- TEV:
-
Tobacco Etch Virus protease
- TMR:
-
Tetramethylrhodamine
- TFA:
-
Trifluoroacetic acid
- SDS-PAGE:
-
SDS polyacrylamide gel electrophoresis
References
Krammer, F. SARS-CoV-2 vaccines in development. Nature 586, 516–527 (2020).
Sharma, O., Sultan, A. A., Ding, H. & Triggle, C. R. A review of the progress and challenges of developing a vaccine for COVID-19. Front. Immunol. 11, 2413 (2020).
Chen, W.-H. et al. Genetic modification to design a stable yeast-expressed recombinant SARS-CoV-2 receptor binding domain as a COVID-19 vaccine candidate. Biochim. Biophys. Acta BBA-Gen. Subj. 20, 129893 (2021).
Cohen, A. A. et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 371, 735–741 (2021).
da Silva Francisco Jr, R. et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 20, 198345 (2021).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Brouwer, P. J. et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020).
Shi, R. et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333 (2020).
Shajahan, A., Supekar, N. T., Gleinich, A. S. & Azadi, P. Deducing the N-and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 30, 981–988 (2020).
Tai, W. et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 17, 613–620 (2020).
Wei, S. et al. Chicken Egg Yolk Antibodies (IgYs) block the binding of multiple SARS-CoV-2 spike protein variants to human ACE2. Int. Immunopharmacol. 90, 107172 (2021).
Lu, Y. et al. Generation of chicken IgY against SARS-COV-2 spike protein and epitope mapping. J. Immunol. Res. 20, 20 (2020).
Zylberman, V. et al. Development of a hyperimmune equine serum therapy for COVID-19 in Argentina. (2020).
Hiriart, Y. et al. Characterization of structural and immunological properties of a fusion protein between flagellin from Salmonella and lumazine synthase from Brucella. Protein Sci. 26, 1049–1059 (2017).
Rosas, G. et al. Brucella spp. lumazine synthase: a novel adjuvant and antigen delivery system to effectively induce oral immunity. Microbes Infect. 8, 1277–1286 (2006).
Alfano, E. F. et al. Expression of the multimeric and highly immunogenic Brucella spp. lumazine synthase fused to bovine rotavirus VP8d as a scaffold for antigen production in tobacco chloroplasts. Front. Plant Sci. 6, 1170 (2015).
Berguer, P. M., Mundiñano, J., Piazzon, I. & Goldbaum, F. A. A polymeric bacterial protein activates dendritic cells via TLR4. J. Immunol. 176, 2366–2372 (2006).
Berguer, P. M. et al. A polymeric protein induces specific cytotoxicity in a TLR4 dependent manner in the absence of adjuvants. PLoS One 7, e45705 (2012).
Craig, S. J., Foong, F. C. & Nordon, R. Engineered proteins containing the cohesin and dockerin domains from Clostridium thermocellum provides a reversible, high affinity interaction for biotechnology applications. J. Biotechnol. 121, 165–173 (2006).
Craig, P. et al. Multiple display of a protein domain on a bacterial polymeric scaffold. Proteins Struct. Funct. Bioinform. 61, 1089–1100 (2005).
Pishesha, N., Ingram, J. R. & Ploegh, H. L. Sortase A: A model for transpeptidation and its biological applications. Annu. Rev. Cell Dev. Biol. 34, 163–188 (2018).
Tang, S., Xuan, B., Ye, X., Huang, Z. & Qian, Z. A modular vaccine development platform based on sortase-mediated site-specific tagging of antigens onto virus-like particles. Sci. Rep. 6, 1–9 (2016).
Arbeitman, C. R. et al. Structural and functional comparison of SARS-CoV-2-spike receptor binding domain produced in Pichia pastoris and mammalian cells. Sci. Rep. 10, 25 (2020).
Huang, X. et al. Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry 42, 11307–11315 (2003).
Tandon, R. et al. Effective screening of SARS-CoV-2 neutralizing antibodies in patient serum using lentivirus particles pseudotyped with SARS-CoV-2 spike glycoprotein. Sci. Rep. 10, 1–7 (2020).
Zylberman, V. et al. High order quaternary arrangement confers increased structural stability to Brucella sp. lumazine synthase. J. Biol. Chem. 279, 8093–8101 (2004).
Sosa, S. et al. Asymmetric bifunctional protein nanoparticles through redesign of self-assembly. Nanosc. Adv. 1, 1833–1846 (2019).
Okba, N. M. et al. Particulate multivalent presentation of the receptor binding domain induces protective immune responses against MERS-CoV. Emerg. Microbes Infect. 9, 1080–1091 (2020).
Zakeri, B. et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. 109, E690–E697 (2012).
Liu, Z. et al. A novel method for synthetic vaccine construction based on protein assembly. Sci. Rep. 4, 1–8 (2014).
Wang, W., Huang, B., Zhu, Y., Tan, W. & Zhu, M. Ferritin nanoparticle-based SARS-CoV-2 RBD vaccine induces a persistent antibody response and long-term memory in mice. Cell. Mol. Immunol. 18, 749–751 (2021).
Zhang, B. et al. A platform incorporating trimeric antigens into self-assembling nanoparticles reveals SARS-CoV-2-spike nanoparticles to elicit substantially higher neutralizing responses than spike alone. Sci. Rep. 10, 1–13 (2020).
Powell, A. E. et al. A single immunization with spike-functionalized ferritin vaccines elicits neutralizing antibody responses against SARS-CoV-2 in mice. ACS Cent. Sci. 20, 20 (2021).
Kanekiyo, M. et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 499, 102–106 (2013).
Tregoning, J. S., Flight, K. E., Higham, S. L., Wang, Z. & Pierce, B. F. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 20, 1–11 (2021).
Jangra, S. et al. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv 20, 20 (2021).
Andreano, E. et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv 20, 20 (2020).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57 (2021).
Gu, H. et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369, 1603–1607 (2020).
Chan, K. K. et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 369, 1261–1265 (2020).
Van Den Berg, S., Löfdahl, P. -Å., Härd, T. & Berglund, H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 121, 291–298 (2006).
Popp, M. W., Antos, J. M. & Ploegh, H. L. Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci. 56, 15–23 (2009).
Batista, P. de O. M. D. et al. Methods of endotoxin removal from biological preparations: a review. (2007).
Capcha, J. M. C. et al. Generation of SARS-CoV-2 Spike pseudotyped virus for viral entry and neutralization assays: A 1-week protocol. Front. Cardiovasc. Med. 7, 25 (2020).
Ferrara, F. & Temperton, N. Pseudotype neutralization assays: from laboratory bench to data analysis. Methods Protoc. 1, 8 (2018).
Acknowledgements
We thank LANAIS-PRO-EM for the support with mass spectrometry analysis of proteins and peptides, and Fundación Ciencias Exactas y Naturales from Universidad de Buenos Aires for their help. Also we would like to thank Dr. Fernando Goldbaum, Dra. Vanesa Zylberman and Dr. Santiago Sosa for providing us a BLS variant and helpful discussion concerning the use of BLS. We thank Dr. José María Delfino for providing us a microdialysis device for exploratory experiments. We thank Erik Procko for providing us with pcDNA3-sACE2(WT)-8his (Addgene plasmid #149268), Stephan Kissler for providing us with pLB (Addgene plasmid #11619) and David Liu for providing us with Sortase A pentamutant (Addgene plasmid #75144).
Funding
This study was supported by the Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (ANPCyT) (IP-COVID-19-234), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Universidad de Buenos Aires (UBA, PIDAE 2019) and Universidad Nacional de San Martín (UNSAM). We would like to thank the following Institutions for supporting M.F. Pavan and N.B.F. (CONICET), T.I.H. (ANPCyT) and M.F. Pignataro (F.A.R.A.).
Author information
Authors and Affiliations
Consortia
Contributions
All authors from the Argentinian AntiCovid Consortium (*) designed experiments, performed research, analyzed data and drafted the manuscript. All authors (listed in alphabetical order) contributed equally to this work. P. C. F. determined endotoxin contents in the protein samples prior to the formulation for immunizations.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in Figure 7 where a preliminary rendition was published.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Argentinian AntiCovid Consortium. Covalent coupling of Spike’s receptor binding domain to a multimeric carrier produces a high immune response against SARS-CoV-2. Sci Rep 12, 692 (2022). https://doi.org/10.1038/s41598-021-03675-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03675-0
This article is cited by
-
Development of a self-assembling multimeric Bann-RBD fusion protein in Pichia pastoris as a potential COVID-19 vaccine candidate
Scientific Reports (2025)
-
Improvement of Cellulomonas fimi endoglucanase CenA by multienzymatic display on a decameric structural scaffold
Applied Microbiology and Biotechnology (2023)
-
COVID-19 Vaccination Responses with Different Vaccine Platforms in Patients with Inborn Errors of Immunity
Journal of Clinical Immunology (2023)
-
Mutations in SARS-CoV-2 structural proteins: a global analysis
Virology Journal (2022)
