
Abstract
The role and detailed mechanisms of lncRNAs in idiopathic pulmonary fibrosis (IPF) are not fully understood. qPCR was conducted to verify lncRNA FEZF1-AS1 expression in the transforming growth factor-beta 1 (TGF-β1)-stimulated human lung fibroblasts (HLF) and A549. The EMT-related proteins were performed by western blotting. Cell proliferation, migration, and transition were detected by CCK-8, colony formation, wound-healing and transwell assays. A dual-luciferase reporter assay was conducted to validate the target relationship of FEZF1-AS1 and miR-200c-3p. FEZF1-AS1 is highly expressed in the fibrotic A549 and HLF. in vitro experiments revealed that FEZF1-AS1 facilitates cell proliferation, migration and invasion. Knockdown of FEZF1-AS1 attenuated TGF-b1-induced fibrogenesis both in vitro. Moreover, silencing FEZF1-AS1 inhibited fibrogenesis through modulation of miR-200c-3p. In addition, inhibition of miR-200c-3p promoted fibrogenesis by regulation of Zinc finger E-box binding homeobox 1 (ZEB1). Mechanistically, FEZF1-AS1 promoted lung fibrosis by acting as a competing endogenous RNA (ceRNA) for miR-200c-3p. FEZF1-AS1 silencing increased the expression and activity of miR-200c-3p to inhibit ZEB1 and alleviate lung fibrogenesis in A549 and HLF. In addition, our study showed that FEZF1-AS1 can regulate enhancer of zeste homolog2 (EZH2) to upregulate fibrosis-related proteins and promote lung fibrosis. In summary, the results of our study revealed the pulmonary fibrogenic effect of FEZF1-AS1 in cellular experiments, demonstrating the potential roles and mechanisms of the FEZF1-AS1/miR-200c-3p/ZEB1 and FEZF1-AS1/EZH2 pathways, which provides a novel and potential therapeutic target to lung fibrosis.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive interstitial lung disease of unknown etiology, marked by epithelial injury and lung fibroblasts aberrant activation which caused excessive deposition of extracellular matrix (ECM) proteins, leading to irreversible fibrosis and destruction of lung architecture1. The growing evidence supports that the prevailing pathogenesis of IPF contains epithelial-mesenchymal transition (EMT) and fibroblast to myofibroblast transition following continued epithelial injury and aberrant fibroblast activation. Patients with IPF often manifested as progressive dyspnea, deterioration in lung functions, which then develops into respiratory failure and eventually death if left unattended2. Currently, Pirfenidone and Nintedanib are two of the drugs recommended for IPF, though both drugs have limited efficacy in slowing disease progression or improving the patients’ quality of life3. Lung transplant remains as the only surgical option in the management of advanced-stage of IPF, in which this surgery is often accompanied with complications that result in significant mortality rate. Thus, there are urgent needs in the in-depth study and exploration of molecular mechanisms that are associated with IPF, as a pivotal step in achieving breakthroughs in the treatment of such disease.
long non-coding RNAs (lncRNAs) are a type of non-coding RNA transcripts greater than 200 nucleotides in length without protein-coding function that can modulate the expression of target genes through diverse cellular mechanisms containing signal, guide, decoy, and scaffold4. As such, Emerging evidence has indicated that lncRNAs play vital roles in the pathogenesis of multiple diseases, including cancer, cardiovascular disorders, myocardial and liver fibrogenesis, et cetera, by regulating RNA-protein interaction, DNA-protein interaction, protein-protein interaction, and serving as a sponge for microRNAs (miRNAs)5,6,7,8. EMT has been highlighted as one of the main pathogenic mechanisms for the onset of IPF, in which numerous lncRNAs, such as TINCR9, RP11-390F4.310, NR2F1-AS111, lncRNA-ATB12, lncRNA H1913, and ERLR14 are closely related to the mechanism of EMT. Specifically, TINCR was reported to promote epithelial-mesenchymal transition and trastuzumab resistance by targeting miR-125b in breast cancer9. Peng et al. revealed that lncRNA RP11-390F4.3 play critical role in hypoxia-induced EMT and metastasis through activating of multiple EMT regulators10. ERLR, mainly located in the cytoplasm, overexpressing ERLR in retinal pigment epithelial cells directly triggered TGF-β1-induced EMT and is involved in proliferative vitreoretinopathy14. Although studies have suggested a strong relationship between lnc-RNAs and EMT, the detailed mechanisms between how lnc-RNAs are linked to IPF, via the pathogenetic process of EMT remains unclear.
The results from our study indicated that lncRNA FEZ family Zinc Finger 1-Antisense RNA 1 (FEZF1-AS1), also called AK057037 or LOC154860, as a newly discovered lncRNA, is significantly expressed in IPF, in vitro. FEZF1-AS1 is located on the opposite strand of gene FEZF1 and mapped to chromosome 7q31.32, with a length of 2653 bp. FEZF1-AS1 encodes a 2,564 bp transcript and contains three splicing variants (FEZF1-AS1-201, FEZF1-AS1-202, FEZF1-AS1-203) and seven “exons”15. However, the biological functions and molecular mechanisms of FEZF1-AS1 in IPF are completely undefined. Therefore, we further explored the potential molecular mechanism of FEZF1-AS1 in pulmonary fibrosis. The results demonstrated that FEZF1-AS1 can facilitate cell proliferation, promote cell migration and invasion, and silencing FEZF1-AS1 alleviated IPF by modulating the FEZF1-AS1/miR-200c-3p/zinc finger E-box binding homeobox 1 (ZEB1) pathway and FEZF1-AS1/enhancer of zeste homolog2 (EZH2) pathway. Our study not only provides new insights into the pathophysiological mechanisms of IPF but also offers a potential therapeutic strategy for treating this disease.
Materials and methods
Cell culture and transfection
A549 cells were obtained from Shanghai Fuheng Biotech Co., Ltd. (Shanghai, China), human lung fibroblasts (HLF) were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China), which were maintained in DMEM/F12 (SH30023.01, HyClone, USA) containing 10% foetal bovine serum (FBS, cat:10099141, GIBCO, USA) at 37 °C with 5% CO2. HLF and A549 cells were treated with TGF-β1 (10 ng/mL; CA59, Jinan Biotech, Co., Ltd, Shanghai, China) for 48 h before subsequent experiments. For FEZF1-AS1 knockdown, shRNAs targeting FEZF1-AS1 (sh-FEZF1-AS1), negative control shRNA (sh-NC), miR-200c-3p inhibitor, and its negative control were purchased from (Guangzhou RiboBio Co., Ltd, Guangzhou, China). MiR-200c-3p inhibitor is a specially modified miRNA inhibitor synthesized by chemical synthesis, which can specifically bind with mature miR-200c-3p to inhibit its expression. The miR-200c-3p inhibitor (100 pmol) were transfected into the cells using Lipo6000™ Transfection Reagent (C0526-1.5 ml, Beyotime Biotech, Co., Ltd, Shanghai, China). For EZH2 overexpression, the plasmid containing full-length EZH2 cDNA was obtained from Youbao Biotech, Co., Ltd. (Hunan, China). HLF and A549 cells transfection using Lipo6000™ was conducted according to a standard protocol.
Western blot analysis
Protein samples from cultured A549 and fibroblasts were extracted and lysed for 30 min on ice in RIPA buffer (P0013, Beyotime Biotech, Co., Ltd, Shanghai, China) containing protease inhibitor, then centrifuged at 12,000 rpm for 5 min at 4°C. Protein concentrations were quantified using a BCA protein assay kit (P0011, Beyotime Biotech, Co., Ltd, Shanghai, China). Total protein was separated by standard SDS PAGE, transferred onto polyvinylidene fluoride (PVDF) membranes(Millipore, Billerica, MA, USA). The membranes were then blocked with 5% non-fat milk and washed with Tris-buffered saline and Tween (TBST) buffer before incubation overnight with the primary antibodies: E-cadherin (1:5000, 20874-1-AP, proteintech, Rosemont, IL, USA), N-cadherin (1:2000, 22018-1-AP, proteintech, Rosemont, IL, USA), Vimentin (1:2000, 10366-1-AP, proteintech, Rosemont, IL, USA), α-SMA (1:1000, 14395-1-AP, proteintech, Rosemont, IL, USA), Collagen I (1:1000, 14695-1-AP, proteintech, Rosemont, IL, USA), ZEB1 (1:500, 21544-1-AP, proteintech, Rosemont, IL, USA), EZH2 (1:1000, 21800-1-AP, proteintech, Rosemont, IL, USA). GAPDH (1:5000; 6004-1-1 g, Proteintech, Rosemont, IL, USA) was used as an internal control. The enhanced chemiluminescence assays were applied for detecting the immunoreactivity.
qRT-PCR
The procedures were conducted according to manufacturer’s instructions. Total RNA was extracted from cultured A549 and fibroblasts using the TRIzol-chloroform-isopropyl alcohol procedure. Total RNA was extracted using RNAsimple Total RNA Kit (yuduobio, Co., Ltd., Shanghai, China) and miRcute miRNA Isolation Kit (Tiangen Biotech, Co., Ltd, Beijing, China). First-strand cDNA synthesis was using FastKing gDNA Dispelling RT SuperMix (Tiangen Biotech, Co., Ltd, Beijing, China) and miRcute Plus miRNA First-Strand cDNA Kit (Tiangen Biotech, Co., Ltd, Beijing, China). Quantitative real-time PCR (qRT-PCR) was condncted using the SYBR-Green PCR kit (TransGen Biotech, Co., Ltd, Beijing, China). GADPH or U6 were applied as internal control. The miR-200c-3p primer sequence was performed from Ruibo Biotech, Co., Ltd. (Guangzhou, China). The primer sequences used in this study are shown in supplementary material: Table S1.
Cell counting Kit-8 (CCK-8) cell viability assay
A549 and human lung fibroblasts (HLF) cells proliferation were detected by CCK-8 assay. Cells with various treatments were seeded into 96-well plates (4000 cells per well). When the cells cultured for indicated times, 10 µl CCK-8 (DoJinDo, Kyushu Island, Japan) was added into each well and incubated in cell incubator for 4 h. After that, the OD value at 450 nm was measured on a microplate reader (MULTISKAN FC, ThermoFisher, USA).
Scratch wound-healing assay
A549 and HLF cells seeded on 6-well plates and cultured at normal cultural conditions. When the cells reach 100% confluence, artificial wounds were created with a sterile 10 µL Eppen-dorf tip. The cells were treated with or without TGF-β1 (10 ng/ml) and transfected with or without modifications. Then, the cells were fixed with paraformaldehyde and the images were taken at different time points using a fluorescence microscope (IX71, OLYMPUS, Germany).
Transwell assay
A549 and HLF cells (1 × 104) with various treatments were seeded in the medium without Foetal Bovine Serum (FBS) in upper Transwell chamber. Migration-inducing medium with TGF-β1 (20 ng/ml) was added into the bottom Transwell chambers. After 48 h, the cells were subjected to fixation with 4% paraformaldehyde and stained with 2% crystal violet (Wuhan Guge Biotech, Co., Ltd, Wuhan, China). The images of migratory cells were photographed using a fluorescence microscope (IX71, OLYMPUS, Germany). Each experiment was performed in triplicates.
Colony formation assay
To determine the HLF and A549 capacity of migration, the colony formation assay was performed. Cells with various treatments were seeded into 6-well plates (1500 cells per well) and cultured in DMEM/F12 containing 10% FBS for 10–14 days. The colonies were subjected to fixation with 4% paraformaldehyde and stained with crystal violet. A fluorescence microscope was used to capture images of the culture dish to better visualize the cell colonies. The captured images were then analyzed, and colonies were counted using ImageJ software to ensure data accuracy and to minimize bias.
RNA fluorescence in situ hybridization (FISH)
To clarify the subcellular localization of FEZF1-AS1, this study employed RNA fluorescence in situ hybridization (FISH) to label cells and analyze images. The RNA FISH assay was conducted with a Fluorescent In Situ Hybridization Kit (BW2521-01, BaiweiBio, Guangzhou, China). HLF or A549 cells with related treatment were fixed using 4% formaldehyde/5% acetic acid for 30 min followed by three washes with cold 1 × PBS. Then, the fixed cells were added to pre-cooled permeabilization solution and allowed to stand at room temperature for 5 min, followed by three washes with cold 1 × PBS. The cells were then pre-hybridized at 37 °C for 30 min followed by incubation with lncRNA FISH Probe Mix or negative control Probe Mix for hybridization at 37 °C overnight. Following that, the cells were washed three times and stained the nucleus with DAPI (C1006, Beyotime Biotech, Co., Ltd, Shanghai, China). The lncRNA FEZF1-AS1 probe was labeled with FAM and the nuclei were stained with DAPI. The FAM channel displayed green fluorescent images and the DAPI channel displayed blue fluorescent images. Images were obtained using a laser scanning confocal microscope (Nikon, Japan). The FAM fluorescence intensity in both the nucleus and cytoplasm was quantified using ImageJ. DAPI staining was employed to delineate the nuclear region. For each cell type, 20 cells were quantified, and the experiment was conducted in triplicate. The sequences of the lncRNA FEZF1-AS1 probe and negative control probe are shown in supplementary material: Table S2.
Luciferase reporter assay
The 3ʹ-UTR of FEZF1-AS1 contain conserved miR-200c-3p binding sites, which were predicted through the lncRNA database (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid). We downloaded high-resolution UCSC Genome Browser illustrations covering all known isoforms of the FEZF1-AS1, which were obtained from the GENCODE database, and highlighted the 3’-UTR fragment utilized for the luciferase assay. This fragment is located in the 3’-UTR region of the major isoforms, ensuring that the results of our experiments hold broad biological relevance. Full-length 3′-UTR of FEZF1-AS1 gene and the fragment including of assumed miR-200c-3p binding site was amplified from human genomic DNA and then cloned into the Sall and Sacl downstream sites of the luciferase gene in the pmirGLO vector. Similarly, the fragment of lncRNA FEZF1-AS1 3′-UTR mutant was also amplified and cloned into the pmirGLO control vector at the same sites. The fragment lncRNA FEZF1-AS1 3′-UTR mutant was constructed through mutating the predicted miR-200c-3p binding sites. Site-directed mutagenesis technology was applied to introduce mutations and specific primers that contained base substitutions at the mutation site that were complementary to the target mutation site were also designed. Hieff Mut™ Multi Site-Directed Mutagenesis Kit (11004ES10, Yeasen Biotech, Co., Ltd, Shanghai, China) was applied and experiments were conducted according to the standard operating procedures provided by the manufacturer. A549 cells were co-transfected with miR-200c-3p mimics, miR-200c-3p inhibitor and the 3ʹ-UTR luciferase vector via Lipo6000™. The miR-200c-3p mimics were double-stranded small molecule RNA oligonucleotides based on mature miR-200c-3p sequence, which was designed to mimic endogenous mature miR-200c-3p. MiR-200c-3p inhibitor is chemically modified single-stranded RNA molecule that can competitively bind to and inhibit the function of specific endogenous mature miR-200c-3p. After transfecting for 48 h, luciferase activity was measured using the dual-luciferase reporter gene assay kit (Yeasen Biotech, Co., Ltd, Shanghai, China) according to the manufacturer’s protocols.
Statistical analysis
All data from at least three independent experiments are presented as mean ± standard deviation (SD). Statistical analyses were performed using the SPSS 23.0 and GraphPad Prism 8.0. Unpaired Student’s t test was applied for two groups comparison, and one-way ANOVA was applied for multiple comparisons. P < 0.05 were considered statistically significant.
Results
LncRNA FEZF1-AS1 contributes to pulmonary fibrogenesis in vitro
Genomic locus of LncRNA FEZF1-AS1 obtained from the UCSC genome browser (Fig. 1A). The prevailing pathogenesis of IPF involves EMT and fibroblast to myofibroblast transition. Therefore, A549 (epithelial cell) and HLF (fibroblast cell) were exposed to transforming growth factor (TGF)-β1 in this study. In order to accurately explore the role of FEZF1-AS1 in PF, we constructed cell models via treatment with 10 ng/mL TGF-β1 for 48 h. In order to accurately explore the role of FEZF1-AS1 in PF, we also designed a TGF-β1 treatment group (TGF-β1 group) where cells were treated with 10 ng/mL TGF-β1 for 48 h to serve as a baseline for assessing TGF-β1 effects independently of any shRNA interference. The results from our study indicated that FEZF1-AS1 was significantly up-regulated in both types of TGF-β1 treated cells (Fig. 1B). A549 and HLF cells were transfected with sh-NC or sh-FEZF1-AS1, i.e., sh-1, sh-2, and sh-3. The results of qPCR indicated sh-1 in A549 and HLF are the most efficient short hairpin RNAs (shRNAs) in FEZF1-AS1 knockdown (Fig. 1C), and thus, they were selected to represent FEZF1-AS1 knockdown. As shown in Fig. 1D, transfected with sh-FEZF1-AS1 (sh-1) inhibited the expression of FEZF1-AS1 in TGF-β1-treated HLF and A549.

Silencing FEZF1-AS1 abrogates TGF-β1-induced HLF and A549 cell proliferation, migration and invasion in vitro. (A) Genomic locus of LncRNA FEZF1-AS1. (B) qRT-PCR analysis of the expression of FEZF1-AS1 in cultured HLF and A549 treated with TGF-β1. (C) The efficiency of FEZF1-AS1 knockout vector transfection was assessed via qPCR. (D) qPCR analysis of FEZF1-AS1 after treated with TGF-β1 with or without FEZF1-AS1 knockdown. (E) CCK8 assay was used to test the effect of FEZF1-AS1 knockdown on TGF-β1-treated HLF and A549 proliferation. (F) Colony formation assay (seeded at 6-well plate) indicated the effect of FEZF1-AS1 inhibition on TGF-β1-induced HLF and A549 proliferation. (G) Wound-healing assay was used to evaluate the migration of TGF-β1-treated HLF and A549 after FEZF1-AS1 depletion. (H) Representative images of transwell invasion assay (left) and quantification of cell invasion ability (right) for FEZF1-AS1’s role in TGF-β1-treated HLF and A549 cells. Control, HLF and A549 cell; sh-NC, TGF-β1-treated HLF and A549 cells transfected with a non-targeting shRNA sequence (sh-NC), serving as a control for FEZF1-AS1 knockdown; sh-1, TGF-β1-treated HLF and A549 cell with FEZF1-AS1 knockdown (D-H). All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
To identify whether FEZF1-AS1 accelerates cell growth in HLF and A549 cells, CCK8 assay and colony formation assays were utilized. The cell proliferation ability of the sh-NC group was higher than control group, indicated that TGF-β1 enhanced the ability of cell proliferation, whereas the knockdown of FEZF1-AS1 using shRNA attenuated TGF-β1-induced cell proliferation, in contrast to the sh-NC group (Fig. 1E and F, Fig. S1A). These findings indicated that FEZF1-AS1 considerably accelerates TGF-β1-treated HLF and A549 growth.
Furthermore, we explored the effectiveness of FEZF1-AS1 in HLF and A549 cell migration and invasion. The results of sh-NC group compared with the control group demonstrated that TGF-β1 promoted cell migration and invasion ability, however inhibition of FEZF1-AS1 suppressed TGF-β1-induced cell migration and invasion (Fig. 1G and H, Fig. S1B). In summary, these results demonstrated that cell migration and invasion were significantly promoted by FEZF1-AS1 in PF.
Silencing FEZF1-AS1 mitigated TGF-β1-induced the changes of fibrosis-related proteins
The progressive pulmonary fibrosis is associated with epithelial-mesenchymal transition (EMT) and fibroblast to myofibroblast transition. Therefore, the fibrosis-related proteins include EMT-related proteins and the proteins associated with fibroblast-myofibroblast transition. This process of EMT is characterized by inhibition of epithelial marker called E-cadherin, as well as up-regulation of mesenchymal markers named N-cadherin and vimentin, resulting in the loss of epithelial phenotype and acquisition of mesenchymal characteristics. This is indicated in the findings from our study, as shown in Fig. 2A, the expression of E-cadherin was downregulated, whilst the expression of N-cadherin and vimentin were upregulated in the TGF-β1-treated A549 cells. The fibroblast-myofibroblast transition related proteins include α-SMA and vimentin, which are two major markers of fibroblast-myofibroblast transition. In addition, collagen I as a fibrosis marker is another key fibrosis-related protein. As illustrated in Fig. 2B, the expression of α-SMA, vimentin and collagen I were enhanced in the TGF-β1-treated HLF cells. We further investigated the role of FEZF1-AS1 on the fibrosis-related proteins. Our results indicated that the TGF-β1-treated up-regulation of N-cadherin and vimentin, and inhibition of E-cadherin in A549 cells were attenuated by silencing FEZF1-AS1 (Fig. 2C). Consistent with these results, we also found that knockdown of FEZF1-AS1 using shRNA eliminated TGF-β1-induced fibroblast-myofibroblast transition in HLF (Fig. 2D). In summary, these results confirmed that silencing FEZF1-AS1 could significantly improve fibrosis by altering the expression level of fibrosis-related proteins induced by TGF-β1. To clarify the molecular mechanism, we first evaluated the subcellular localization of FEZF1-AS1. FISH was executed to identify the localization of FEZF1-AS1 in cells. In the fluorescence microscopy images using FAM dye, fluorescence signals were clearly observed expressing in both the cytoplasm and the nucleus. Specifically, the fluorescence is slightly concentrated in the nuclear regions, which may be attributed to cellular structures. The yellow arrows in the images indicate representative nuclear regions, while the green arrows point to representative cytoplasmic areas. We conducted subcellular localization quantification of FEZF1-AS1 in A549 and HLF cells. The results demonstrated significant fluorescence expression not only in the nucleus but also in the cytoplasm, with a higher fluorescence intensity observed in the nucleus compared to the cytoplasm (Fig. 2E). This is consistent with our expectations and further supports the localization of FEZF1-AS1 in both the cytoplasm and nucleus. Therefore, we hypothesize that FEZF1-AS1 may play a role in the pathogenesis of IPF through both nuclear and cytoplasmic pathways.

Silencing FEZF1-AS1 mitigated fibrosis-related proteins changes induced by TGF-β1 and the localization of FEZF1-AS1. (A) Western blot analysis of EMT-related proteins in TGF-β1-treated A549. (B) Western blot analysis of the proteins associated with fibroblast-myofibroblast transition in TGF-β1-treated HLF. (C) Western blot analysis of EMT-related proteins in TGF-β1-treated A549 cells after FEZF1-AS1 silencing. (D) Western blot analysis of the proteins associated with fibroblast-myofibroblast transition in TGF-β1-treated HLF after FEZF1-AS1 silencing. (E) The fluorescent microscopic images of A549 and HLF cells show the distribution of the FAM dye in the cytoplasm and nucleus. Yellow arrows indicate nuclear regions, while green arrows indicate the cytoplasmic region. Fluorescent signals are expressed in both regions, indicating that FEZF1-AS1 is localized in both cytoplasm and nucleus. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
Silencing lncRNA FEZF1-AS1 inhibited TGF-β1-induced lung fibrogenesis through activation of miR-200c-3p
The mechanism on how lung fibrosis can be alleviated by silencing FEZF1-AS1 was further explored. The accumulating evidence has suggested that lncRNAs are able to bind to miRNAs competitively, which then inhibit the activity of miRNAs as competing endogenous RNAs (ceRNAs). To determine the target miRNA which mediates the antifibrotic effect and has potential binding sites for FEZF1-AS1, bioinformatics analysis was conducted using the lncRNA database (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid). Our study hypothesized that FEZF1-AS1 contributes to lung fibrosis through modulation of miR-200c-3p. In order to support this hypothesis, we transfected sh-FEZF1-AS1 into HLF and A549 cells to examine the effect of FEZF1-AS1 on fibrogenesis and to determine whether miR-200c-3p mediates this effect. For detecting cell proliferation, migration, and transition, we performed the CCK8 assay, wound-healing assay, and transwell invasion assay. The results indicated inhibition of FEZF1-AS1 had mitigated the TGF-β1-treated cell proliferation, migration, and transition, however, these effects were nearly reversed by miR-200c-3p inhibition (Fig. 3A and C, Fig. S2A-S2B). Furthermore, this study also found that knockdown of FEZF1-AS1 inhibited TGF-β1-treated EMT-related proteins production in A549 cells, whereas these effects were nearly alleviated by miR-200c-3p inhibition (Fig. 3D). Consistent with these results, FEZF1-AS1 knockdown also inhibited TGF-β1-treated the proteins production associated with fibroblast-myofibroblast transition in HLF cells, whereas these effects were nearly alleviated by miR-200c-3p inhibition (Fig. 3E). Therefore, our data demonstrated that silencing FEZF1-AS1 leads to inhibition of TGF-β1-treated lung fibrogenesis through activation of miR-200c-3p.

Silencing lncRNA FEZF1-AS1 inhibited TGF-β1-induced lung fibrogenesis through activation of miR-200c-3p. (A) CCK8 assay indicated the effect of FEZF1-AS1 knockdown and miR-200c-3p inhibition on TGF-β1-treated HLF and A549 proliferation. (B) Wound-healing assay evaluated the migration of TGF-β1-treated HLF and A549 cells after transfected with sh-FEZF1-AS1 and miR-200c-3p inhibitor. (C) Transwell invasion assay for the evaluation of invasion ability after transfected with sh-FEZF1-AS1 and miR-200c-3p inhibitor in TGF-β1-treated HLF and A549 cells. (D) Western blot analysis of EMT-related proteins in TGF-β1-treated A549 cells after transfected with sh-FEZF1-AS1 and miR-200c-3p inhibitor. (E) Western blot analysis of the proteins associated with fibroblast-myofibroblast transition in TGF-β1-treated HLF cells after transfected with sh-FEZF1-AS1 and miR-200c-3p inhibitor. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
FEZF1-AS1 regulates the expression and activity of miR-200c-3p by acting as a ceRNA
Growing evidence supports that lncRNAs may function as ceRNA by binding to miRNAs to regulate their biological effects. To explore the effect of FEZF1-AS1 on miR-200c-3p, the expression level of miR-200c-3p in IPF was examined. The expression of miR-200c-3p was found to be down-regulated in TGF-β1-treated HLF and A549 cells (Fig. 4A). Meanwhile, Fig. 4B illustrated sh-FEZF1-AS1 increased the expression of miR-200c-3p in TGF-β1- induced HLF and A549 cells (Fig. 4B).

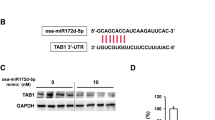
FEZF1-AS1 regulates the expression and activity of miR-200c-3p by acting as a ceRNA. (A) qRT-PCR analysis of miR-200c-3p expression in TGF-β1 treated A549 and HLF cells (10 ng/ml) for 48 h. (B) qRT-PCR analysis of miR-200c-3p expression after FEZF1-AS1 inhibition in TGF-β1-treated HLF and A549. (C) Sequence alignment of miR-200c-3p with the predicted binding sites within the wild-type or mutated FEZF1-AS1 regions. (D) Luciferase reporter activity of vectors carrying the luciferase gene and a fragment of FEZF1-AS1 containing wild-type or mutated miR-200c-3p binding site. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
In order to obtain more direct evidence of the interaction between FEZF1-AS1 and miR-200c-3p, a dual-luciferase reporter assay was conducted to further determine whether FEZF1-AS1 is a practical target for miR-200c-3p. Luciferase vectors carrying FEZF1-AS1 fragments containing wild-type (FEZF1-AS1-WT) and mutated (FEZF1-AS1-MUT) binding sites for miR-200c-3p were constructed respectively (Fig. 4C). We also provided a detailed scheme showing the region of the transcript that is used in the luciferase assay. To ensure that the 3’-UTR region we utilized is conserved and representative across all known isoforms of the lncRNA FEZF1-AS1, we conducted a detailed annotation analysis of FEZF1-AS1 using the UCSC Genome Browser (Fig. S3A). Figure S3A illustrates the location of the FEZF1-AS1 transcript and its 3’-UTR region. In the figure, the 3’-UTR region of the FEZF1-AS1 transcript is clearly delineated within the q31.32 (122306984–122307283) of chromosome 7, with the specific transcript used in this study highlighted within a red box. The precise definition of the 3’-UTR position of this transcript provides a critical foundation for further investigation into the role of FEZF1-AS1 in gene expression and regulation. This information is significant on multiple levels. First, it offers a clear localization for elucidating the interactions between FEZF1-AS1 and other genes. Second, by explicitly identifying the specific transcript utilized in the research, it ensures the accuracy of the experiments and the reliability of the results. Finally, these data support our hypotheses regarding the role of FEZF1-AS1 in the pathogenic mechanisms of IPF. As shown in Fig. 4D, transfection with the miR-200c-3p mimic decreased and miR-200c-3p inhibitor increased the luciferase activity of FEZF1-AS1, while no significant difference was observed in the FEZF1-AS1-Mut group. Those results indicated that miR-200c-3p can directly bind to FEZF1-AS1 at the recognized sites.
Inhibition of miR-200c-3p promotes TGF-β1-driven fibrogenesis in A549 and HLF cells
This study hypothesized that reduced expression of miR-200c-3p promotes fibrogenesis in TGF-β1 treated HLF and A549 cells. To explore whether miR-200c-3p plays a negative role in lung fibrosis, miR-200c-3p inhibitor was transfected with TGF-β1-treated and non TGF-β1-treated A549 and HLF cells respectively (10 ng/ml) and were incubated for 48 h. As illustrated in Fig. 5A and D, Fig. S4A and Fig. S4B, TGF-β1 enhanced the ability of cell proliferation, migration and invasion, whereas reduced expression of miR-200c-3p further promoted the TGF-β1-treated cell proliferation, migration and invasion in A549 and HLF cells. Besides, our results also indicated that inhibition of miR-200c-3p contributed to TGF-β1-treated EMT in A549 cells, as well as up-regulation of N-cadherin and vimentin, and down-regulation of E-cadherin (Fig. 5E). Furthermore, we also found that knockdown of miR-200c-3p promoted TGF-β1-induced up-regulation of the related protein markers with fibroblast-myofibroblast transition in HLF cells, including α-SMA, vimentin and collagen I (Fig. 5F). These results suggested inhibition of miR-200c-3p promotes lung fibrogenesis.

Inhibition of miR-200c-3p promotes TGF-β1-driven fibrogenesis in A549 and HLF cells. (A) CCK8 assay demonstrated the effect of miR-200c-3p inhibition on TGF-β1-induced A549 and HLF cells proliferation. (B) Colony formation assay (seeded at 6-well plate) indicated the effect of miR-200c-3p inhibition on TGF-β1-induced proliferation in A549 and HLF cells. (C) Scratch assay demonstrated the effect of miR-200c-3p inhibition on TGF-β1-induced migration in A549 and HLF cells. (D) Transwell invasion assay evaluated the invasiveness of TGF-β1-treated A549 and HLF cells after miR-200c-3p inhibition. (E) Western blot analysis of fibrosis-related proteins in TGF-β1-treated A549 cells after miR-200c-3p inhibition. (F) Western blot analysis of fibrosis-related proteins in TGF-β1-treated HLF after miR-200c-3p inhibition. Control, HLF and A549 cells; Negative Control, TGF-β1-treated HLF and A549 cells without miR-200c-3p inhibition; miR-200c-3p inhibitor, TGF-β1-treated HLF and A549 cells with miR-200c-3p inhibition (A-D). All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
FEZF1-AS1’s effects of fibrogenesis are partly attributed to sponging miR-200c-3p, and then triggering ZEB1
In order to further investigate the anti-fibrotic mechanisms of miR-200c-3p, the TargetScan database was used to identify potential targets of miR-200c-3p, in which ZEB1 3’-UTR contained perfect matching sites for miR-200c-3p. Among human, mouse and rat species, it was broadly conserved. Accumulating evidence has indicated that ZEB1 promotes lung fibrosis via the pathogenic mechanisms of EMT and myofibroblast differentiation16,17. Thus, we first explored the interaction between ZEB1 and miR-200c-3p, the results suggested that ZEB1 expression was up-regulated in TGF-β1-induced HLF and A549 cells (Fig. 6A). We then investigated the relationship between ZEB1 and FEZF1-AS1, our study found that FEZF1-AS1 suppression attenuated the expression of ZEB1 in TGF-β1-treated HLF and A549 cells (Fig. 6B). Meanwhile, we found that inhibition of miR-200c-3p promoted the expression of ZEB1 in TGF-β1-treated A549 and HLF cells (Fig. 6C). The results demonstrated that miR-200c-3p directly suppressed the expression of ZEB1. On the other hand, ZEB1 which was suppressed by sh-FEZF1-AS1 was up-regulated by co-transfection with miR-200c-3p inhibitor (Fig. 6D and E). In summary, this study suggested that FEZF1-AS1 regulates ZEB1 by directly adsorbing miR-200c-3p. FEZF1-AS1’s efficiency of fibrogenesis was partially attributed to sponging miR-200c-3p which in turn triggered ZEB1 in IPF.

FEZF1-AS1’s effects of fibrogenesis are partly attributed to sponging miR-200c-3p, and then triggering ZEB1. (A) qRT-PCR analysis of ZEB1 expression in cultured HLF and A549 cells that responded to TGF-β1 (10 ng/ml) for 48 h. (B) Western blot analysis of the expression of ZEB1 after FEZF1-AS1 knockdown in TGF-β1-treated HLF and A549 cells. (C) Western blot analysis was performed to detect ZEB1 protein levels in TGF-β1-treated A549 and HLF after transfected with miR-200b-3p inhibitor. (D, E) Western blot analysis of expression of ZEB1 after transfected with sh-FEZF1-AS1 and miR-200c-3p inhibitor. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
The association between lncRNA FEZF1-AS1 and the expression of EZH2 in PF
The recent studies proposed that an abnormally expressed lncRNA was involved in the development and progression of PF by regulating a downstream gene. Recent studies have shown that lncRNAs regulates the expression of EZH2, which exhibit pulmonary fibrosis by mediating EMT and fibroblast-to-myofibroblast transition18,19,20. To investigate whether EZH2 is a downstream gene of lncRNA FEZF1-AS1 in PF, detection of EZH2 expression in PF was first carried out in this study. Our study found that EZH2 was effectively up-regulated at both mRNA (Fig. 7A) and protein level (Fig. 7B) in TGF-β1-induced HLF and A549 cells. Meanwhile, the results demonstrated that knockdown of FEZF1-AS1 down-regulated the expression of EZH2 at both mRNA (Fig. 7C) and protein level (Fig. 7D) in TGF-β1-treated HLF and A549 cells. However, overexpression of EZH2 had no effect on the expression level of FEZF1-AS1 (Fig. S5B). Taken together, current results suggest a potential association between lncRNA FEZF1-AS1 and the expression of EZH2, which indicated that lncRNA FEZF1-AS1 and EZH2 may play important roles in regulating the progression of PF. However, it remains challenging to determine the precise sequence of events and to differentiate between direct and indirect effects at this stage.

Interactions between lncRNA FEZF1-AS1 and EZH2. (A) The mRNA level of EZH2 in TGF-β1-treated HLF and A549 cells by qRT-PCR analysis. (B) The protein level of EZH2 in TGF-β1-treated HLF and A549 cells by western blot analysis. (C) qRT-PCR analysis of the expression of EZH2 after FEZF1-AS1 knockdown in TGF-β1-treated HLF and A549 cells. (D) The protein level of EZH2 after FEZF1-AS1 knockdown in TGF-β1-treated HLF and A549 cells. (E) Overview of involvement of FEZF1-AS1 in IPF. Schematic representation of proposed model for FEZF1-AS1, acting as a ceRNA for miR-200c-3p to up-regulate ZEB1 expression and upregulating EZH2 expression to promote EMT and fibroblast-myofibroblast transition, leading to lung fibrosis. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
EZH2 overexpression reversed the effect of FEZF1-AS1 knockdown on PF in TGF-β1-stimulated HLF and A549 cells
Next, we further to explore the role of the lncRNA FEZF1-AS1/EZH2 axis in the progression of PF, the role of EZH2 on FEZF1-AS1 knockdown in the inhibition fibrogenesis in TGF-β1-treated A549 and HLF cells was explored, the expression of EZH2 was up-regulated and genetic rescue experiments were performed using the following combinations: sh-FEZF1-AS1 + OE-NC group and sh-FEZF1-AS1 + OE-EZH2 group. The western blot analysis results indicated that EZH2 OE significantly increases the expression of EZH2 (Fig. 8D). Furthermore, FEZF1-AS1 knockdown suppressed the TGF-β1-induced cell proliferation, migration, and transition, whereas EZH2 overexpression effectively alleviated these effects (Fig. 8A and C, Fig. S6A-S6B). Therefore, silencing FEZF1-AS1 reduced the protein level of the EMT-related proteins including E-cadherin, N-cadherin, and vimentin in TGF-β1-induced A549 cells, in which such effects were nearly reversed by overexpression of EZH2 (Fig. 8D), along with the expression of α-SMA, vimentin, and collagen I in the TGF-β1-treated HLF cells as analyzed by western blot (Fig. 8D). Overall, knockdown of lncRNA FEZF1-AS1 significantly attenuated TGF-β1-induced PF by regulating EZH2.

EZH2 overexpression reversed the effect of FEZF1-AS1 knockdown on lung fibrosis in TGF-β1-stimulated HLF and A549 cells. (A) CCK8 assay evaluated the function of FEZF1-AS1 knockdown on TGF-β1-treated HLF and A549 cells proliferation which were simultaneously treated with up-regulated EZH2. (B) Wound-healing assay indicated the effect of FEZF1-AS1 knockdown together with EZH2 overexpression on TGF-β1-treated HLF and A549 migration. (C) Transwell invasion assay evaluated the invasiveness of TGF-β1-treated HLF and A549 cells after transfected with sh-FEZF1-AS1 and up-regulated EZH2. (D) Western blot analysis of fibrosis-related proteins and EZH2 expression in TGF-β1-treated A549 and HLF cells after silencing FEZF1-AS1 and up-regulation of EZH2. All data were presented as mean ± SD. n = 3 independent experiments. *p < 0.05.
Discussion
To date, the role and mechanism of LncRNA FEZF1-AS1 in pulmonary fibrosis remains unreported. The FISH results in our study indicate that FEZF1-AS1 is expressed in both the nucleus and the cytoplasm. The strong fluorescent signal in the nucleus suggests that it may play a role in the regulation of gene expression. The expression in the cytoplasm implies its potential involvement in signaling pathways and post-transcriptional regulation. In this study, we discovered that FEZF1-AS1 promotes the development of lung fibrosis by the FEZF1-AS1/miR-200c-3p/ZEB1 pathway and FEZF1-AS1/EZH2 pathway. Our study revealed that FEZF1-AS1 contributes to the pathogenic development of EMT and fibroblast-myofibroblast transition by acting as a miR-200c-3p sponge and upregulating EZH2 expression (Fig. 7E). We also found out that inhibition of miR-200c-3p led to fibrosis in TGF-β1-treated A549 and HLF cells by targeting ZEB1, resulting in dysregulation of pulmonary fibrosis-associated proteins. Besides, the results suggested that silencing FEZF1-AS1 resulted in alleviation in the EMT and fibroblast to myofibroblast transition via regulation of the ZEB1, in which FEZF1-AS1 act as a ceRNA for miR-200c-3p. This is consistent with previous studies, which have found that FEZF1-AS1 regulates miRNAs, such as miR-30a and miR-107, through a sponge mechanism21,22. The results, combined with LncRNA Atlas data, may further support the conclusion that FEZF1-AS1 influences gene expression in the cytoplasm through its sponge effect. ZEB1 as a member of the dizinc finger protein family, is involved in the signaling pathway of the TGF-β1. Recent studies have shown that ZEB1 is mainly focused on its role in promoting EMT and fibroblast to myofibroblast transition23,24. These findings are consistent with our results. Additionally, we discovered that FEZF1-AS1 significantly promoted TGF-β1-induced lung fibrosis by regulating EZH2. Collectively, our findings suggest that FEZF1-AS1 may be a novel and potential therapeutic target for lung fibrosis.
It is now increasingly proven that LncRNAs play essential roles in IPF. Recent researches reported that LINC00941/lncIAPF accelerate lung fibrosis by promoting fibroblast to myofibroblast transition via blocking autophagy depending on ELAVL1/HuR18. lnc-SNHG1 promotes fibroblast-to-myofibroblast transition during the development of lung fibrosis, in which the process was induced by silica particles exposure via sponging miR-326 and facilitating the expression of SP125. Meanwhile, by acting as a ceRNA for miR-450b-5p, Hoxaas3 also promoted fibroblast-myofibroblast transition via up-regulation of Runx1 expression, hence contributing to the development of IPF26. Previous studies suggested that FEZF1-AS1 is highly expressed and facilitates cell proliferation, migration and invasion in colorectal carcinoma, gastric cancer, lung adenocarcinoma and other human malignancies, whilst, silencing FEZF1-AS1 resulted in down-regulation of EMT markers such as N-cadherin and vimentin, as well as up-regulation of epithelial marker E-cadherin27. However, the roles of FEZF1-AS1 in pulmonary fibrosis (PF) remain unclear. Our study discovered that FEZF1-AS1 level is significantly upregulated in TGF-β1-treated A549 and HLF cells. In addition, we found that FEZF1-AS1 facilitated the proliferation, migration and invasion of TGF-β1-treated A549 and HLF cells, as well as promoted the expression of fibrosis-related proteins. By acting as a ceRNA, one lncRNA can bind and regulate the expression and activity of miRNAs that participate in the progression of diseases. Therefore, we conducted bioinformatics analysis using the bibiserv database to explore the target miRNA. The results indicated that miR-200c-3p harbors the potential binding sites for FEZF1-AS1.
Increasing evidence has suggested that miRNAs play crucial roles in pulmonary fibrosis, encompassing their roles in disease initiation, progression, and their potential as diagnostic biomarkers and therapeutic targets28,29. For example, Guiot et al. found that the overexpression of miR-142-3p in HLF and alveolar epithelial cells could reduce the expression of profibrotic genes. Furthermore, macrophage-derived exosomes fight against the progression of IPF through the delivery of antifibrotic miR-142-3p30. Chen et al. reported that down-regulation of miR-15a resulted in the activation of fibroblast via inhibition of YAP1, whereas overexpression of miR-15a exerted the opposite effect31. Many studies have illustrated the protective role of miR-200c-3p in a variety of diseases, such as breast cancer32,33, cardiovascular disease34,35,36, and gastric cancer37,38. In addition, a recent study reported that miR-200c-3p promotes endothelial-to-mesenchymal transition (EndoMT) in artery bypass grafts by regulating fermitin family member 2 (FERM2)39. However, the role of miR-200c-3p in pulmonary fibrosis remains largely unclear. In the present study, we observed miR-200c-3p is down-regulated in TGF-β1-treated A549 and HLF cells. Furthermore, our study indicated that inhibition of miR-200c-3p promoted the EMT and fibroblast to myofibroblast transition in pulmonary fibrosis, suggesting miR-200c-3p exerted an anti-fibrotic effect in pulmonary fibrosis. However, the down-stream regulator for miR-200c-3p in PF is not yet clear. We applied the TargetScan database and found that ZEB1 3’-UTR contained perfect matching sites for miR-200c-3p.
Remarkably, ZEB1 has been discovered to be a pivotal transcription factor in the process of EMT. Yao et al. demonstrated that ZEB1 mediated EMT in human alveolar epithelial type II (ATII) cells augments TGF-β1-induced profibrogenic responses. Besides, the paracrine signaling between RAS-activated ATII cells and fibroblasts augmented fibroblast recruitment was associated with ZEB1 tissue plasminogen activator axis17. Shao et al. illustrated that angiotensin (1–7) suppressed the expression of ZEB1 via disruption of TGF-β1-Smad signaling pathway which possessed protective effects against IPF40. By activating ZEB1, CTBP1 promotes the activation of lung fibroblasts, thereby causing excessive deposition of the extracellular matrix which aggravates IPF41. However, the up-stream regulator for ZEB1 in IPF remains unclear. Our findings demonstrated that miR-200c-3p suppressed the expression of ZEB1 and FEZF1-AS1 suppression attenuated the expression of ZEB1. Meanwhile, sh-FEZF1-AS1 induced downregulation of ZEB1 was reversed by co-transfection with miR-200c-3p inhibitor. These results suggested that ZEB1 acts as one of the targets in FEZF1-AS1/miR-200c-3p pathway and possesses mediation in anti-fibrotic effect of miR-200c-3p. And the results are consistent with previous researches, which have shown that miR-200c-3p promotes cell proliferation, migration, EMT, and fibroblast activation by targeting ZEB1 in different diseases23,42,43.
Different cellular localization may lead to multiple mechanisms by which lncRNAs exert their functions. FISH experiments showed that FEZF1-AS1 is primarily located in the nucleus but is also distributed in the cytoplasm of A549 and HLF cells. This finding suggests that FEZF1-AS1 could be an essential regulatory player in the nucleus and cytoplasm of A549 and HLF cells. LncRNAs could regulates EZH2 transcription in the nucleus and then exhibit oncogenic roles by mediating EMT19,20. Highly up-regulated expression of LINC00941 promotes fibroblast-to-myofibroblast transition, as well as myofibroblast proliferation and migration by regulating the stability of EZH2, which ultimately accelerates pulmonary fibrosis18. But to our knowledge, the relationship between FEZF1-AS1 and EZH2, as well as its regulatory function in PF have not been reported. In this study, our results indicated a potential association between lncRNA FEZF1-AS1 and the expression of EZH2. Studies reported that inhibition of EZH2 attenuates pulmonary fibrosis via suppressing EMT and fibroblast-to-myofibroblast transition. Consistent with the previous studies, our findings indicate that the overexpression of EZH2 accelerates processes such as cell proliferation, migration, invasion, EMT, and fibroblast to myofibroblast transition. Additionally, it diminishes the protective effects of FEZF1-AS1 knockdown against fibrosis. Current results suggest a potential association between lncRNA FEZF1-AS1 and the expression of EZH2. However, it remains challenging to determine the precise sequence of events and to differentiate between direct and indirect effects at this stage.
Taken together, the findings from our study proposed that FEZF1-AS1 is up-regulated in TGF-β1-treated A549 and HLF cells. Also, the results have consolidated the fact that FEZF1-AS1 facilitates cell proliferation, migration and invasion in IPF. Our findings have also highlighted that FEZF1-AS1 partially mediates fibrogenic effects by acting as a ceRNA for miR-200c-3p thereby increasing the expression level of ZEB1, in which FEZF1-AS1 also regulates EZH2 expression, all of which contributing to the aggravation of IPF. In conclusion, the results from this study demonstrated a potential mechanism that underlies the fibrogenic effect of FEZF1-AS1 in IPF, suggesting that FEZF1-AS1 as a potential novel biomarker and valuable therapeutic target in the case of IPF. However, it is important to acknowledge that our studies had its limitations. Firstly, due to limitations in resources and time, we have not been able to conduct relevant clinical trials or alternative animal model experiments. Secondly, the mechanism that underlies the association between lncRNA FEZF1-AS1 and the expression of EZH2 is not yet explained. The relationship between lncRNA, EZH2 and potential genomic sites requires further investigations. Additionally, our research primarily focused on a specific 3’-UTR region, and future studies could explore the functional differences between different isoforms and their potential applications in cancer research. Finally, the generalizability of the experimental conditions and results in this study may be limited. The functions and mechanisms of FEZF1-AS1 could vary across different cell types or tissue environments. Therefore, further research is necessary to validate these findings under a variety of experimental conditions.
Furthermore, it is important to note that this study has some limitations regarding the FISH experiments. First, although the FAM dye demonstrated efficiency and specificity under our experimental conditions, the STELLARIS guidelines (https://www.biosearchtech.com/support/education/stellaris-rna-fish/dyes-and-modifications-for-stellaris) indicate that its application may be limited when using fewer than 35 probes. This could mean that some signals might be affected by autofluorescence. To mitigate this limitation, we conducted multiple control and repeat experiments to confirm the specificity and reliability of the signals; however, future studies could consider utilizing other more suitable fluorescent dyes to further validate our results. Furthermore, future research should aim to optimize the signal quantification methods and report larger sample sizes of cells to ensure the comprehensiveness and reproducibility of the results. Additionally, due to the constraints of our research timeline, our negative control did not include cells depleted of lncRNA. A more appropriate control approach should include such a depletion treatment to accurately assess the specificity of the target RNA signals. Including this control in future studies will be essential to enhance the reliability of the findings.
Data availability
All data in the manuscript is available through the responsible corresponding author.
References
Spagnolo, P. et al. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 222, 107798 (2021).
Lederer, D. J. & Martinez, F. J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378(19), 1811–1823 (2018).
Raghu, G. et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 205(9), e18–e47 (2022).
Yang, M. et al. lncRNAfunc: a knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 50(D1), D1295–d1306 (2022).
Tang, R. et al. A Roadmap for fixing the heart: RNA Regulatory Networks in Cardiac Disease. Mol. Ther. Nucleic Acids. 20, 673–686 (2020).
Chen, T. et al. LncRNA Airn maintains LSEC differentiation to alleviate liver fibrosis via the KLF2-eNOS-sGC pathway. BMC Med. 20(1), 335 (2022).
Liu, C. Y. et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 9(1), 29 (2018).
Statello, L. et al. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 22(2), 96–118 (2021).
Dong, H. et al. Activation of LncRNA TINCR by H3K27 acetylation promotes Trastuzumab resistance and epithelial-mesenchymal transition by targeting MicroRNA-125b in breast Cancer. Mol. Cancer. 18(1), 3 (2019).
Peng, P. H. et al. Hypoxia-induced lncRNA RP11-390F4.3 promotes epithelial-mesenchymal transition (EMT) and metastasis through upregulating EMT regulators. Cancer Lett. 483, 35–45 (2020).
Li, D. et al. The EMT-induced lncRNA NR2F1-AS1 positively modulates NR2F1 expression and drives gastric cancer via miR-29a-3p/VAMP7 axis.Cell Death Dis. 13(1), 84 (2022).
Li, J. et al. LncRNA-ATB: an indispensable cancer-related long noncoding RNA. Cell. Prolif. 50(6), e12381 (2017).
Zhou, W. et al. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR-200b/c and let-7b. Sci. Signal. 10(483), eaak9557 (2017).
Yang, S. et al. Long noncoding RNA ERLR mediates epithelial-mesenchymal transition of retinal pigment epithelial cells and promotes experimental proliferative vitreoretinopathy. Cell. Death Differ. 28(8), 2351–2366 (2021).
Gui, Z. et al. LncRNA FEZF1-AS1 promotes multi-drug resistance of gastric cancer cells via upregulating ATG5. Front. Cell. Dev. Biol. 9, 749129 (2021).
Yao, L. et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell. Death Differ. 26(5), 943–957 (2019).
Yao, L. et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 297(3), 101096 (2021).
Zhang, J. et al. ATF3 -activated accelerating effect of LINC00941/lncIAPF on fibroblast-to-myofibroblast differentiation by blocking autophagy depending on ELAVL1/HuR in pulmonary fibrosis. Autophagy. 18(11), 2636–2655 (2022).
Sun, C. C. et al. Long intergenic noncoding RNA 00511 acts as an oncogene in non-small-cell lung cancer by binding to EZH2 and suppressing p57. Mol. Ther. Nucleic Acids. 5(11), e385 (2016).
Fang, J., Sun, C. C. & Gong, C. Long noncoding RNA XIST acts as an oncogene in non-small cell lung cancer by epigenetically repressing KLF2 expression. Biochem. Biophys. Res. Commun. 478(2), 811–817 (2016).
Zhang, Z. et al. Long non-coding RNA FEZF1-AS1 promotes breast cancer stemness and tumorigenesis via targeting miR-30a/Nanog axis. J. Cell. Physiol. 233(11), 8630–8638 (2018).
Yao, J. et al. Long non-coding RNA FEZF1-AS1 promotes the proliferation and metastasis of hepatocellular carcinoma via targeting miR-107/Wnt/β-catenin axis. Aging (Albany NY). 13(10), 13726–13738 (2021).
Lai, Y. H. et al. Magnolol regulates miR-200c-3p to inhibit epithelial-mesenchymal transition and retinoblastoma progression by modulating the ZEB1/E-cadherin axis in vitro and in vivo. Phytomedicine. 110, 154597 (2023).
Krebs, A. M. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell. Biol. 19(5), 518–529 (2017).
Wu, Q. et al. Long non-coding RNA SNHG1 promotes fibroblast-to-myofibroblast transition during the development of pulmonary fibrosis induced by silica particles exposure. Ecotoxicol. Environ. Saf. 228, 112938 (2021).
Lin, S. et al. LncRNA Hoxaas3 promotes lung fibroblast activation and fibrosis by targeting miR-450b-5p to regulate Runx1. Cell. Death Dis. 11(8), 706 (2020).
Li, J. et al. The lncRNA FEZF1-AS1 promotes the progression of colorectal cancer through regulating OTX1 and targeting miR-30a-5p. Oncol. Res. 28(1), 51–63 (2020).
Kadota, T. et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. J. Extracell. Vesicles. 10(10), e12124 (2021).
Kabekkodu, S. P. et al. Clustered miRNAs and their role in biological functions and diseases. Biol. Rev. Camb. Philos. Soc. 93(4), 1955–1986 (2018).
Guiot, J. et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3. Thorax. 75 (10), 870–881 (2020).
Chen, Y. et al. YAP1/Twist promotes fibroblast activation and lung fibrosis that conferred by miR-15a loss in IPF. Cell. Death Differ. 26(9), 1832–1844 (2019).
Liu, Z. et al. Circular RNA hsa_circ_001783 regulates breast cancer progression via sponging miR-200c-3p.Cell Death Dis. 10(2), 55 (2019).
Zhang, D. D. et al. Phosphodiesterase 7B/microRNA-200c relationship regulates triple-negative breast cancer cell growth. Oncogene. 38 (7), 1106–1120 (2019).
Jiang, Y. et al. Upregulation of miR-200c-3p induced by NaF promotes endothelial apoptosis by activating Fas pathway. Environ. Pollut. 266 (Pt 1), 115089 (2020).
Vancheri, C. et al. Downregulation of circulating hsa-miR-200c-3p correlates with Dyslipidemia in patients with stable coronary artery disease. Int. J. Mol. Sci. 24(2), 1112 (2023).
Bakhashab, S. et al. Deciphering the role of miR-200c-3p in type 1 diabetes (subclinical Cardiovascular Disease) and its correlation with inflammation and vascular health. Int. J. Mol. Sci. 23(24), 15659 (2022).
Mao, X. et al. ELK4-mediated lncRNA SNHG22 promotes gastric cancer progression through interacting with EZH2 and regulating miR-200c-3p/Notch1 axis. Cell. Death Dis. 12(11), 957 (2021).
Zhang, C. et al. miRNAs derived from plasma small extracellular vesicles predict organo-tropic metastasis of gastric cancer. Gastric Cancer. 25(2), 360–374 (2022).
Chen, D. et al. miRNA-200c-3p promotes endothelial to mesenchymal transition and neointimal hyperplasia in artery bypass grafts. J. Pathol. 253(2), 209–224 (2021).
Shao, M. et al. Exogenous angiotensin (1–7) directly inhibits epithelial-mesenchymal transformation induced by transforming growth factor-β1 in alveolar epithelial cells. Biomed. Pharmacother. 117, 109193 (2019).
Li, X. et al. Toosendanin restrains idiopathic pulmonary fibrosis by inhibiting ZEB1/CTBP1 Interaction. Curr. Mol. Med. 24(1), 123–133 (2024).
Jiang, Y. et al. Exosomal miR-200c-3p negatively regulates the migraion and invasion of lipopolysaccharide (LPS)-stimulated colorectal cancer (CRC). BMC Mol. Cell. Biol. 21(1), 48 (2020).
Garrido-Cano, I. et al. Delivery of miR-200c-3p using tumor-targeted mesoporous silica nanoparticles for breast cancer therapy. ACS Appl. Mater. Interfaces. 15(32), 38323–38334 (2023).
Acknowledgements
Not applicable.
Funding
This study was funded by Clinical Research Center of Affiliated Hospital of Weifang Medical University, Shandong Province, China (2022wyfylcyj05), Weifang Municipal Health Commission Traditional Chinese Medicine Research Project (No.WFZYY2024-4-004), the National Natural Science Foundation of China (No. 82205079), and the Natural Science Foundation of Shandong Province, China (ZR2023QH061).
Author information
Authors and Affiliations
Contributions
Jun Man, Fusheng Gao and Mengmeng Liu designed the research; Jun Man, Mengmeng Liu, Yuxin Lai and Longfei Song performed cellular experiments; Mengmeng Liu, Jun Man and Yuxin Lai wrote the manuscript.
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent to participate
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, M., Song, L., Lai, Y. et al. LncRNA FEZF1-AS1 promotes pulmonary fibrosis via up-regulating EZH2 and targeting miR-200c-3p to regulate the ZEB1 pathway. Sci Rep 14, 26044 (2024). https://doi.org/10.1038/s41598-024-74570-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-74570-7
Keywords
This article is cited by
-
Repurposing disulfiram: targeting Zeb1 to attenuate paraquat-induced pulmonary fibrosis in rats
Journal of Molecular Histology (2025)
