
Abstract
Chronic heart failure (CHF) can induce chronic kidney disease (CKD), called type 2 cardio-renal syndrome (CRS2). The mechanism is not completely clear, and there is a lack of early warning biomarkers of CKD in the context of CHF. Two CKD, one CHF-PBMC and four CHF-cardiac tissue expression profile datasets were obtained from GEO database. Differential expression analysis and WGCNA were used to detect CKD key genes and CHF-related secreted proteins. Protein–protein interaction (PPI), functional enrichment, and cMAP analysis reveal potential mechanisms and drugs of CHF-related CKD. Five machine learning algorithms were used to screen candidate biomarkers, construct a diagnostic nomogram for CKD and validate it in two external cohorts. Clinical serum samples were collected in our hospital to evaluate the correlation and diagnostic value of biomarkers and CKD. 225 CKD key genes and 316 CHF-related secreted proteins were identified. Four key subgroups, including 204 genes, were identified as CRS2-related pathogenic genes by PPI analysis. Enrichment analysis revealed that the identified subgroups exhibited significant enrichment in cytokine action, immune responses, and inflammatory processes. The cMAP analysis highlighted metiradone as a drug with greater potential for therapeutic intervention for CRS2. Utilizing five machine learning algorithms, three hub genes (CD48, COL3A1, LOXL1) were pinpointed as potential biomarkers for CKD, and a nomogram model was constructed. Receiver operating characteristic analysis demonstrated that the nomogram’s area under the curve (AUC) exceeded 0.80 in both the CKD combined dataset and two external cohorts. In addition, the three biomarkers were significantly correlated with the glomerular filtration rate, and the AUC of the model predicting disease progression was 0.944. Furthermore, analysis of immune cell infiltration indicated a correlation between the three biomarkers and the infiltration fraction of macrophages, neutrophils, and other immune cells in CKD. Our clinical cohort validated the expression patterns of three biomarkers in serum, and the diagnostic model achieved an AUC of 0.876. CHF has the potential to facilitate the progression of CKD via the release of cardiac and PBMC secreted proteins. Furthermore, CD48, COL3A1, and LOXL1 have been identified as potential biomarkers for the detection of CKD.
Similar content being viewed by others

Introduction
The intricate and extensive connection between the heart and kidneys has been widely recognized for decades. In 2008, Professor Ronco introduced the concept of cardiorenal syndrome (CRS), defining it as the acute or chronic dysfunction of one organ leading to a similar dysfunction in the other, which is further categorized into 5 subtypes1. Among these subtypes, type 2 CRS (CRS2) refers to chronic kidney disease (CKD) resulting from chronic heart failure (CHF). Research indicates that up to 40–60% of patients with CHF also suffer from CKD2,3, and the mortality rate for those with both conditions is more than double compared to those with only CHF4. However, due to the insidious onset and prolonged course of CKD, along with a lack of early warning biomakers, clinical attention is often only directed towards significant decreases in glomerular filtration rate or increases in creatinine levels. Therefore, there is value in investigating methods for predicting the occurrence of CKD in CHF patients.
The approach of investigating the pathological mechanism to identify early biomarkers of disease is commonly employed, yet the precise understanding of how CHF contributes to CKD remains incomplete. Initially, it was widely believed that hemodynamic issues were dominant; however, findings from the ESCAPE (Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan) trial revealed no association between hemodynamic variables assessed through pulmonary artery catheterization and serum creatinine levels in 194 patients5. This suggests that there are alternative pathways for heart-kidney communication beyond renal injury resulting from inadequate perfusion. The activation of the renin–angiotensin–aldosterone system (RAAS), oxidative stress, and Inflammatory factor are currently recognized as humoral mechanisms contributing to renal injury6. Furthermore, related substances have been identified as potential disease biomarkers. For instance, arginine vasopressin (AVP) released in response to RASS activation can lead to water retention and hasten the progression of CKD; elevated plasma levels of AVP have been linked to adverse outcomes in congestive CHF7. Patients with CHF are in a proinflammatory state, characterized by the involvement of tumor necrosis factor-α, interleukin-1 (IL-1) and IL-6 family as the most crucial cytokines in cardiac inflammation8. These cytokines also play a role in kidney injury and are closely associated with the prognosis of CKD6.
This evidence suggests that secreted proteins may be partially involved in the pathological mechanism of CRS2. The ability to secrete extra-cellular into the circulation gives secreted proteins an inherent advantage as biomarkers. However, due to limited understanding of secreted proteins, there have been few studies searching for disease markers from this group of proteins.
In recent years, The Human Protein Atlas database has continuously updated the set of genes encoding secreted proteins, and currently predicts that there are over 3900 genes encoding secreted proteins. Therefore, this study utilized a series of bioinformatics methods to analyze the potential mechanisms of CRS2 based on secreted proteins and employed five machine learning (ML) algorithms to identify potential biomarkers. Additionally, a CKD prediction model was established using these biomarkers and validated in external cohorts and clinical data.
Materials and methods
Expression data collection and processing
Two microarray datasets of CKD, namely GSE32591 and GSE66494, were accessed from the GEO database9. Additionally, raw expression profile datasets of heart tissues (GSE1145, GSE5406, GSE21610, and GSE141910) and peripheral blood mononuclear cells (PBMC) (GSE59867) from patients with CHF were also retrieved from the GEO database. The integration of these expression profiles was accomplished using the “ComBat” function within the “SVA” package in R software (version 4.3.2, https://www.r-project.org/).
Differentially expressed genes (DEGs) analysis
The CKD combined dataset and CHF-related datasets underwent preprocessing steps including background correction, normalization, and gene symbol conversion. Subsequently, differentially expressed genes (DEGs) in the CKD and CHF datasets were identified using the “Limma” package in R software. DEGs were screened based on the criteria of adjusted p-value < 0.05 and fold change > 1.2. Following this, the expression patterns of the DEGs were visually represented through the utilization of the “ggplot2” package for volcano plots and the “pheatmap” package for heatmaps in R, respectively.
Weighted gene co-expression network analysis (WGCNA) and significant modules filtering
The application of WGCNA was employed to unveil gene association patterns across diverse samples and to pinpoint potential biomarker genes or therapeutic targets. The interaction between gene sets and their relationship with phenotypes played a crucial role in the selection process. The initial step involved filtering out genes with a median absolute deviation of 0 from each sample. Subsequently, the “goodSamplesGenes” function was employed in Step 2 to detect missing values, and samples surpassing a cutHeight threshold of 20,000 were excluded as outliers. In Step 3, an optimal soft threshold of 5 is determined using cex1 = 0.85, which aids in the establishment of a scale-free co-expression gene network. Subsequent to gene clustering, modules are acquired and similar ones are consolidated based on MEDissThres = 0.25 criteria. Step 4 involves the creation of a heat map to demonstrate module-trait relationships, while Step 5 includes the computation of module membership (MM) and gene significance (GS) values and the generation of scatter plots to depict MM-GS correlations for each module10.
Secreted proteins access
The Human Protein Atlas database facilitated the retrieval of secreted proteins11. A total of 3947 genes encoding secreted proteins were obtained from the category of “SPOCTOPUS predicted secreted proteins”.
The establishment of protein–protein interaction (PPI) network
The study investigated the interactions between CHF-related secreted proteins and CKD key genes, resulting in the construction of a protein–protein interaction (PPI) network. This network was created by integrating data from the STRING database12 using a confidence score threshold of 0.4. Cytoscape software (version 3.9.0, https://cytoscape.org/) was used to visualize the PPI network. Furthermore, an additional procedure entailed the utilization of the Cytoscape plug-in, MCODE, to identify significant subsets within the network. Subsets scoring above 10 were designated as CRS2-related pathogenic genes for subsequent analytical investigations.
Functional enrichment analysis
The investigation of the biological function and mechanistic foundations of CRS2-related pathogenic genes involved the utilization of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis13. These genes were inputted into the SangerBox14 for this purpose. Statistical significance in enrichment was assessed with a threshold set at adjusted p < 0.05. Additionally, the results of the functional enrichment analysis were visually depicted using a lollipop chart.
Connectivity map (cMAP) analysis
The cMAP15 functions as a database of gene expression profiles that employs the analysis of gene expression patterns to unveil associations between genes, diseases, and small molecule compounds. The inclusion of upregulated genes associated with CRS2-related pathogenic genes was conducted, with these genes subsequently integrated into the cMAP online repository to identify potential small-molecule drugs for the treatment of CKD. Ultimately, the research successfully identified the top ten compounds with the highest enrichment scores.
ML algorithms
In this study, a combination of five ML algorithms, namely random forest (RF), eXtreme gradient boosting (XGB), support vector machine (SVM), generalized linear model (GLM), and the least absolute shrinkage and selection operator (LASSO), were utilized for the identification of candidate biomarkers and development of a diagnostic model for CKD. First, the CKD combined dataset was randomly split according to a 6:4 ratio: 60% was the training set, and the other 40% was the test set. Next, LASSO regression was employed for feature dimensionality reduction on the CKD combined dataset. The aforementioned five ML algorithms were implemented utilizing the “glmnet,” “caret,” “randomForest,” “kernlab,” and “xgboost” packages within the R software. The candidate genes identified by the five algorithms were compared, and those genes present in the intersection were designated as hub genes for the development of CHF-related CKD diagnostic models.
The establishment and evaluation of diagnostic nomogram model
The nomogram was establishment using the three hub genes with the assistance of the “rms” package. Diagnostic performance for CKD was assessed through the generation of receiver operating characteristic (ROC) curves for each hub gene and the nomogram. Calibration curves and decision curve analysis (DCA) were utilized to evaluate the predictive efficiency of the nomogram in heart failure (HF) comorbid with CKD.
External verification of hub genes expression pattern and diagnostic efficacy
Two independent datasets (GSE180394 and GSE104954) containing CKD cases and control were obtained from the GEO database. The expression patterns of the hub genes in external datasets were analyzed by the violin plot, while the diagnostic efficacy of both the hub genes and nomogram model were assessed using ROC analysis.
Correlation analysis of hub genes with CKD severity and disease progression
A dataset (GSE137570) containing clinical information such as age, gender, estimated glomerular filtration rate (eGFR), degree of renal tubulointerstitial fibrosis (TIF), and disease progression of CKD patients was obtained from the GEO database. Sangerbox database was used to draw the scatter map of the correlation between hub genes and various clinical features, and ROC was used to analyze the predictive efficacy of hub genes and model on eGFR, TIF and CKD progression.
Immune infiltration measurement
ssGSEA (Single Sample Enrichment Analysis) algorithm was used to estimate the immune cell and immune function enrichment scores of each sample to represent the relative infiltration abundance by means of the “GSVA” package. The box plot, generated using the “ggplot2” package, was employed to present these significant differences. Following that, Spearman’s rank correlation coefficient was employed to present the correlation of the expression of biomarkers with the quantity of infiltrated immune cells and immune functions.
Patients’ samples collection
Serum samples from healthy controls and CHF patients with or without CKD were collected from Shaoxing Second Hospital. All CKD patients had never used dialysis-related treatments such as peritoneal dialysis or hemodialysis. Patients with diabetes, infectious nephropathy, drug or allergic nephropathy, systemic lupus erythematosus and other immune system diseases were excluded. The clinical characteristics of our cohort are shown in Table 1. The protocol of human samples has been approved by the Ethics Committee of Shaoxing Second Hospital (Ethics batch number: 2023081), and all participants provided written informed consent. In addition, ELISA kit (Elabscience, Wuhan, China) was used to detect serum COL3A1 levels, and ELISA kits (Animaluni, Shanghai, China) were used to detect serum CD48 and LOXL1 levels, respectively, according to the manufacturer’s protocols.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 9.5.1 (GraphPad Software Inc., San Diego, CA, USA, https://www.graphpad.com/) in this study. Measurement data were presented as mean ± standard deviation, while counting data were expressed as quantity (percent). Inter-group comparisons of measurement data were performed using unpaired Student’s t-test or ANOVA, and counting data were compared using the Chi-square test. Statistical significance was defined as p < 0.05.
Result
Identification of DEGs and WGCNA key modules in CKD
The bioinformatics analysis methodology is depicted in Fig. 1. Following batch correction, the CKD combined dataset consisted of 85 samples from the CKD group and 23 samples from the control group. Figure 2A–D demonstrates that the elimination of batch effects resulted in a significant decrease in disparities between the two datasets. Differential Expression Analysis (DEA) between the CKD and control samples revealed a total of 3252 DEGs, with 1721 genes showing upregulation and 1531 genes displaying downregulation. The expression features of these DEGs in the CKD combined dataset was illustrated using a volcano plot and heatmap (Fig. 2E, F). In order to further investigate the identification of CKD key genes, a WGCNA was conducted to identify gene modules that are highly correlated with CKD samples. The selection of a soft-thresholding power of 3 was based on considerations of scale independence and average connectivity. This power was utilized to generate a total of 8 modules, and the clustering of module eigengenes can be visualized in Fig. 2G. Furthermore, this study conducted an investigation into the relationship between CKD and 8 gene modules, as shown in Fig. 2H. The analysis revealed that the pink module had the strongest positive correlation with CKD, consisting of 83 genes with a p-value of 3e−5. The pink and blue modules, which demonstrated statistical significance, were identified as key modules for subsequent analyses. To identify CKD key genes, an intersection analysis was conducted between DEGs and genes from the key modules identified through WGCNA. This analysis identified a total of 225 CKD key genes for further investigation, as illustrated in Fig. 2I.

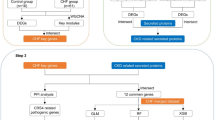
Graphical representation detailing the design flow of this research.

Identification of DEGs and WGCNA key modules in CKD. (A,B) Gene expression density map before (A) and after (B) batch correction of CKD datasets. (C,D) PCA plots before (C) and after (D) batch correction of CKD datasets. (E) Volcano plot illustrating the DEGs in the CKD combined dataset. In the visual representation, genes that are upregulated are symbolized by red dots, whereas genes that are downregulated are symbolized by blue dots. (F) Heatmap featuring the top 30 upregulated and top 30 downregulated DEGs in the CKD combined dataset. (G) Gene dendrogram displaying the cleaved gene modules. (H) Heatmap highlighting the associations of MEs with CKD status, with blue and pink modules exhibiting significant correlations with CKD (p < 0.05). (I) Venn diagram illustrating the identification of 225 CKD key genes through the intersection of key module with DEGs. DEG, differentially expressed gene; WGCNA, weighted gene co-expression network analysis; CKD, chronic kidney disease; ME, module eigengene.
Identification of differentially expressed secreted proteins in CHF
Following batch correction, the CHF combined dataset, consisting of 288 samples from the CHF group and 201 samples from the control group, was acquired. The elimination of batch effects resulted in a significant decrease in discrepancies observed among the four datasets, as demonstrated in Fig. 3A–D. Through the use of volcano plots and heatmaps, 700 DEGs were identified in heart tissue (Fig. 3E and F) of CHF individuals, and 367 DEGs in PBMC (Fig. 3H and I). Given the consideration that the development of CKD may be primarily facilitated by the release of secreted proteins in individuals with CHF, the CHF-related secreted proteins were subsequently acquired by combining the differentially expressed secreted proteins from CHF heart tissues (Fig. 3G) and PBMC datasets (Fig. 3J).

Identification of differentially expressed secreted proteins in CHF. (A,B) Gene expression box plot before (A) and after (B) batch correction of CHF heart datasets. (C,D) PCA plots before (C) and after (D) batch correction of CHF heart datasets. (E,F) Volcano plot (E) and heatmap (F) show DEGs in the CHF combined dataset-Heart. (G) The Venn diagram show that 256 CHF-related cardiac secreted proteins were identified by the intersection of DEGs and secreted protein gene sets. (H,I) Volcano plot (H) and heatmap (I) show DEGs in the CHF dataset-PBMC. (J) The Venn diagram show that 69 CHF-related PBMC derived secreted proteins were identified by the intersection of DEGs and secreted protein gene sets.
PPI, functional enrichment and drug screening of the CRS2-related pathogenic genes
To elucidate the potential pathogenic genes and underlying mechanisms in CRS2, the interaction between CHF-related secreted proteins and CKD key genes was investigated using the STRING database. Utilizing the MCODE analysis plugin, four significant modules were identified, highlighting 204 genes as potentially CRS2-related pathogenic genes. The examination of GO terms related to biological processes (BP) revealed that CRS2-related pathogenic genes were significantly enriched in processes such as “extracellular matrix organization” and “chemokine-mediated signaling pathway” (Fig. 4A). Additionally, the molecular function (MF) analysis indicated a strong correlation between the pathogenic genes and functions such as “extracellular matrix structural constituent” and “cytokine activity” (Fig. 4A). The results of the KEGG pathway analysis revealed strong correlations between the CRS2-related pathogenic genes and important pathways such as “cytokine-cytokine receptor interaction” and “cell adhesion molecules”. Furthermore, these genes showed enrichment in immune-related pathways, including “antigen processing and presentation”, “T helper type 17 (Th17) cell differentiation”, and “natural killer cell mediated cytotoxicity” as depicted in Fig. 4B. Additionally, an analysis of drug screening within the cMAP database revealed that the top ten compounds with the most negative scores, including metyrapone, albendazole, PKCbeta-inhibitor, XMD-1150, phensuximide, KIN001-220, acamprosate, isoliquiritigenin, diazepam, and estrone, have been identified as potential pharmacological agents for the treatment of CRS2 (Fig. 4C). The depiction of the specific pathways targeted by these compounds is illustrated in Fig. 4D.

Functional enrichment analysis and drug screening of the CRS2-related pathogenic genes. (A) Lollipop plot highlighting the results of GO enrichment analysis covering BP, CC, and MF of CRS2-related pathogenic genes. (B) Lollipop plot presenting the KEGG results pertaining to CRS2-related pathogenic genes. (C) Heatmap presenting the top 10 compounds with the most significantly negative enrichment scores as indicated by the cMAP analysis. (D) The description of those top10 compounds. CRS2, Cardio-renal syndrome type 2. GO, gene ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclo-pedia of Genes and Genomes; cMAP, connectivity map.
Screening of biomarkers and establishment of diagnostic models for HF-related CKD
Due to the potential significant role of commonly differentially expressed secreted proteins in CHF and CKD, 24 common genes were identified at the intersection of CHF-related secreted proteins and up-regulated CKD key genes. These genes were then employed in the construction of a diagnostic model for CKD, which has the potential to differentiate between CHF patients with and without CKD (Fig. 5A). In order to enhance the identification of diagnostic biomarkers, four ML algorithms, specifically GLM, SVM, RF, and XGB, were utilized to rank the 24 common genes based on their significance as variable indicators (Fig. 5B–D). Furthermore, the implementation of the LASSO regression algorithm led to the discovery of 15 promising candidate genes from the initial set of 24 common genes (Fig. 5E, F). It is noteworthy that, after identifying the common intersection among the candidate genes generated by the five ML algorithms, only three hub genes—CD48, collagen type III alpha 1 chain (COL3A1), and lysyl oxidase like 1 (LOXL1)—were consistently present in the intersection across all five subsets (Fig. 5G).

Screening of potential diagnostic biomarkers for CHF-related CKD. (A) Venn diagram displaying the 24 intersecting genes among CKD-related secreted proteins, upregulated CKD DEGs, and CKD key module genes. (B) Boxplot presenting the visualization of the residuals of four ML algorithms (RF, SVM, XGB, and GLM). (C) Top 10 gene feature importance scores from these four ML algorithms. (D) ROC analysis for results of four ML algorithms. (E,F) The minimum (E) and lambda values (F) of diagnostic biomarkers (n = 15) as reflected by the LASSO logistic regression. (G) Venn diagram revealing three common genes identified by all five ML algorithms, signifying them as hub genes in the context of CHF comorbid with CKD. CKD, chronic kidney disease; CHF, chronic heart failure; ML, machine learning; DEGs, differentially ex-pressed genes; RF, random forest; SVM, support vector machine; XGB, eXtreme Gradient Boosting; GLM, generalized linear model; LASSO, least absolute shrinkage and selection operator; ROC, receiver operating characteristic.
As depicted in Fig. 6A–C, the expression levels of the three hub genes were found to be significantly elevated in the CKD group compared to the control group. In order to improve diagnostic accuracy and predictive capacity, a nomogram was developed that incorporates these three hub genes (Fig. 6D). The analysis of calibration curves unveiled a close resemblance between the predicted probabilities generated by the established nomogram diagnostic model and those of the ideal model (Fig. 6E). Additionally, the evaluation of the nomogram’s performance was conducted using DCA, indicating that the nomogram model may provide benefits in the identification of CKD (Fig. 6F). In ROC analysis, the Area Under the Curve (AUC) values of the three hub genes exceeded 0.75, with the nomogram exhibiting a higher AUC value (0.82) compared to each individual hub gene (Fig. 6G, H).

Establishment and evaluation of diagnostic model. (A–C) Violin plot of CD48 (A), COL3A1 (B), and LOXL1 (C) gene expression in the CKD combined dataset. (D) Construction of a nomogram by considering the diagnostic biomarkers. (E) Calibration curve of the predictive performance of the nomogram model in CKD. The solid curve labeled “Bias-corrected” closely aligns with the dashed line labeled “Ideal” indicating the relative reliability of the results. (F) DCA for the performance of the nomogram model. The black line labeled as “None” represents the net benefit when assuming no CKD patients, the grey line labeled as “All” indicates the net benefit when assuming all patients have CKD, and the red line labeled as “Nomogram model” represents the net benefit based on the diagnostic predictions of nomogram model for CKD. (G,H) ROC curve for the diagnostic performance of each candidate biomarker (G) and the nomogram model (H) constructed for CKD in the CKD combined dataset. CKD, chronic kidney disease; DCA, decision analysis curve; ROC, receiver operating characteristic.
External validation of expression patterns and diagnostic efficacy of 3 hub genes
The datasets GSE180394 and GSE104954 each consisted of 9 controls, 44 patients with CKD, and 18 controls, 35 patients with CKD, respectively. The violin plots illustrated a significant upregulation of hub genes in the CKD group of both GSE180394 (Fig. 7A–C) and GSE104954 (Fig. 7D–F). ROC analysis demonstrated that the three hub genes exhibited good diagnostic performance for CKD in both external datasets (Fig. 7G, I), and the nomogram model displayed excellent diagnostic accuracy with AUC values of 0.970 and 0.929, respectively (Fig. 7H, J).

External validation of expression patterns and diagnostic efficacy of 3 hub genes. (A–C) Violin plot of LOXL1 (A), COL3A1 (B), and CD48 (C) gene expression in GSE180394 dataset. (D–F) Violin plot of LOXL1 (D), COL3A1 (E), and CD48 (F) gene expression in GSE104954 dataset. (G–J) ROC curve demonstrating the diagnostic performance of each candidate biomarker and the nomogram model for CKD in two external datasets, GSE180394 (G,H) and GSE104954 (I,J). ROC, receiver operating characteristic; CKD, chronic kidney disease.
Correlation analysis of 3 hub genes with CKD severity and disease progression
The GSE137570 dataset includes age, sex, eGFR, and TIF levels for 24 CKD patients (cohort1), as well as age, sex, and disease progression data for an additional 17 CKD patients (cohort2). In the two cohorts, the expression levels of the three hub genes were not significantly correlated with age and gender (Figs. 8A–F and 9A–F). In cohort1, the expression levels of 3 hub genes were significantly negatively correlated with eGFR levels and significantly positively correlated with TIF levels (Fig. 8G–L). ROC analysis showed that the predictive efficiency for eGFR (< 45 ml/min) and TIF (> 50%) reached 0.832 and 0.964, respectively (Fig. 8M–P). In cohort 2, the expression levels of 3 hub genes were significantly positively correlated with disease progression (Fig. 9G–I). ROC analysis showed that the AUC of the 3 hub genes predicting disease progression was higher than 0.85 (Fig. 9J), and the AUC of the model reached 0.944 (Fig. 9K).

Correlation analysis of 3 hub genes with CKD severity. (A–C) Correlation scatter plots of LOXL1 (A), COL3A1 (B), and CD48 (C) expression with age, respectively. (D–F) Correlation scatter plots of LOXL1 (D), COL3A1 (E), and CD48 (F) expression with gender, respectively. (G–I) Correlation scatter plots of LOXL1 (G), COL3A1 (H), and CD48 (I) expression with gender, respectively. (J–L) Correlation scatter plots of LOXL1 (J), COL3A1 (K), and CD48 (L) expression with gender, respectively. (M,N) ROC curve demonstrating the diagnostic performance of each candidate biomarker and the nomogram model for GFR in GSE137570 dataset cohort1. (O,P) ROC curve demonstrating the diagnostic performance of each candidate biomarker and the nomogram model for TIF in GSE137570 dataset cohort1. CKD, chronic kidney disease; GFR, glomerular filtration rate; TIF, tubulointerstitial fibrosis; ROC, receiver operating characteristic.

Correlation analysis of 3 hub genes with CKD progression. (A–C) Correlation scatter plots of LOXL1 (A), COL3A1 (B), and CD48 (C) expression with age, respectively. (D–F) Correlation scatter plots of LOXL1 (D), COL3A1 (E), and CD48 (F) expression with gender, respectively. (G–I) Correlation scatter plots of LOXL1 (G), COL3A1 (H), and CD48 (I) expression with CKD progression, respectively. (J,K) ROC curve demonstrating the diagnostic performance of each candidate biomarker and the nomogram model for CKD progression in GSE137570 dataset cohort2. CKD, chronic kidney disease; ROC, receiver operating characteristic.
Immune cell infiltration of CKD
A close association was unraveled between immune processes and the pathway analysis of CRS2-related pathogenic genes. The characteristics of 16 subpopulations of immune cells and 13 types of immune functions were derived using the ssGSEA algorithm. Higher proportions of B cells, macrophages, neutrophils Th2 cells and regulatory T cells were observed in CKD in contrast to the control group (Fig. 10A). Immune functions analysis showed that cytolytic activity, inflammation promoting and parainflammatory response were activated in CKD group (Fig. 10B). Notably, as displayed in Fig. 10C and D, the hub genes showed significant correlation to several immune cells and immune functions in CKD.

Immune cell infiltration of CKD. (A) The box plot showing the differential comparison of the infiltration score of 16 immune cells between CKD and control groups. (B) The box plot showing the differential comparison of the infiltration score of 13 immune functions between CKD and control groups. The heatmap reveals the correlation of infiltration of 16 immune cells. (C) The correlation heatmap represents the correlation between differentially infiltrated immune cells and 3 hub genes. (D) The correlation heatmap represents the correlation between differentially infiltrated immune functions and 3 hub genes. CKD, chronic kidney disease.
Validation of 3 biomarkers and diagnostic model in clinical serum samples
To further validate the clinical value of the 3 biomarkers, clinical serum samples were gathered to measure protein concentrations. As shown in Fig. 11A–C, compared with the healthy control group, the concentrations of the 3 biomarkers in the CHF with CKD group were all significantly increased, and the concentrations of CD48 and COL3A1 were significantly increased compared with the CHF without CKD group, while the differences in LOXL1 concentrations were not statistically significant. A nomogram containing these three biomarkers is shown in Fig. 11D, and analysis of calibration curves shows that the prediction probability generated by the established nomogram diagnosis model is similar to that of the ideal model (Fig. 11E). In addition, the performance of the nomogram was evaluated using DCA, showing that the nomogram model can provide benefits in the identification of CKD (Fig. 11F). In ROC analysis, the AUC values of the 3 biomarkers all exceeded 0.75 (Fig. 11G), and the AUC values of the nomogram model reached 0.876(Fig. 11H).

Validation of 3 biomarkers and diagnostic model in clinical serum samples. (A–C) Histogram of CD48 (A), COL3A1 (B), and LOXL1 (C) concentrations in clinical serum samples. (D) Construction of a nomogram by considering the diagnostic biomarkers. (E) Calibration curve of the predictive performance of the nomogram model for CKD. The solid curve labeled “Bias-corrected” closely aligns with the dashed line labeled “Ideal” indicating the relative reliability of the results. (F) DCA for the performance of the nomogram model. The black line labeled as “None” represents the net benefit when assuming no CKD patients, the grey line labeled as “All” indicates the net benefit when assuming all patients have CKD, and the red line labeled as “Nomogram model” represents the net benefit based on the diagnostic predictions of nomogram model for CKD. (G,H) ROC curve for the diagnostic performance of each biomarker (G) and the nomogram model (H) constructed for CKD in clinical serum samples. CKD, chronic kidney disease; DCA, decision analysis curve; ROC, receiver operating characteristic.
Discussion
This study has elucidated the relationship between CHF and subsequent CKD through a series of bioinformatics analysis methods. The potential humoral mechanisms of CRS2 may include the binding of cell adhesion molecules, multiple immune processes, and cytokine-related signaling pathways. The utilization of ML algorithms has facilitated the development of a diagnostic nomogram model that integrates three hub genes (CD48, COL3A1, and LOXL1) to predict the risk of CKD. ROC analysis demonstrated that these three hub genes and models were highly effective in predicting CKD. Furthermore, two external GEO datasets confirmed the consistency of the up-regulated expression patterns of CD48, COL3A1, and LOXL1 with CKD combined dataset. Based on clinical serum data, the predictive value of these three hub genes as biomarkers for CKD in patients with CHF was reaffirmed.
The main pathological features of CRS2 renal injury include glomerular sclerosis and tubular cell swelling, necrosis, and subsequent tubule interstitial fibrosis. The pathogenesis of CRS2 is complex, and current studies have shown that CHF leads to hemodynamic changes that lead to the activation and release of neurotransmitters in the sympathetic nervous system and the RAAS, which further leads to decreased renal perfusion pressure and ultimately to the development of CKD16. In this study, the pathological mechanism of CRS2 was explored from the perspective of secreted proteins. In the GO enrichment analysis of CRS2-related pathological genes, the top item in BP ranking was “extracellular matrix organization”, and the top items in CC and MF were also related to extracellular matrix. In addition, other top-ranking items, including “positive regulation of cell adhesion” and “neutrophil migration”, have also been shown to be major contributors to renal fibrosis17. KEGG enrichment analysis identified fibrosis-related pathways like CAM and ECM receptor interaction, along with multiple cytokine and chemokine-related pathways. This suggests that CHF may impact the kidney through cytokines secreted by the heart and PBMCs, leading to tubulointerstitial fibrosis and impaired renal function.
CKD is widely recognized as a collection of chronic inflammatory conditions characterized by immune cell dysfunction18. Analysis of the CKD combined dataset using ssGSEA identified notable alterations in immune cell populations and immune function. Specifically, the analysis revealed a significant increase in scores related to inflammation-promoting, para-inflammation, interferon response, and HLA expression in the CKD group, aligning with the persistent inflammatory milieu characteristic of CKD. Additional examination of immune cell infiltration demonstrated an increase in the infiltration fraction of B cells, macrophages, neutrophils, TH2 cells, Treg, and other cells within the CKD group. Notably, analysis of functional enrichment of PPI key subsets also revealed a diverse array of immune cells and associated dysfunction. GO analysis identified enriched items such as “leukocyte migration” and “neutrophil migration”, while KEGG analysis highlighted immune cell related pathways including “TH17 cell differentiation”, “TH1 and TH2 cell differentiation”, and “natural killer cell mediated cytotoxicity”. Neutrophils are the main inflammatory cells in circulation, and animal models have found that there is significant neutrophilic infiltration in kidney injured tissues, and reducing neutrophilic infiltration can delay the development of CKD19. Early studies have suggested that circulating neutrophil count is associated with the progression of CKD, and in recent years, several studies have further confirmed this phenomenon20. A clinical study of 938 CKD patients found that baseline neutrophil/lymphocyte ratio (NLR) was associated with an increased risk of end-stage renal disease in pre-dialysis patients with stage 4 CKD21. Another study involving 3252 diabetic patients further showed that neutrophil count was an independent risk factor for CKD progression (OR 2.556, 95% CI 1.11–5.88)22. TH17 cells are pro-inflammatory T cells derived from CD4T cells, and IL-17 is its signature cytokine23. Previous studies have found that TH17 cells are rapidly activated in ischemia–reperfusion induced AKI, and continuously activated TH17 cells can induce significant renal fibrosis by activating the IL-17 signaling pathway, promoting the transition from acute kidney injury (AKI) to CKD, and early application of IL-17 antagonists can reduce the degree of renal fibrosis24. IL-17 may also contribute to CKD-related renal fibrosis by recruiting neutrophils, making it a potential target for CKD treatment. Various experimental models of kidney injury, such as glomerulonephritis and lupus nephritis, have demonstrated an elevation in circulating Th17 cells and TH17-associated cytokines25. Furthermore, a mendelian randomization analysis investigating the causal link between 91 inflammatory proteins and CKD revealed that elevated levels of IL-17 were correlated with an augmented risk of CKD26. These findings indicate that CHF-related secreted proteins may facilitate the onset or advancement of CKD via immune mechanisms like neutrophil migration and TH17 cell activation.
Managing CKD in patients with CHF is a challenging topic, and one of the most significant contradictions lies in the use of diuretics. Diuretics are the most commonly used medication for rapid relief of CHF symptoms, but diuretics may exacerbate electrolyte imbalances, reduce the amount of fluid circulating, disrupt neurohormonal balance, and reduce kidney function27. Furthermore, research has demonstrated that ACEI/ARB can enhance long-term prognosis and decrease overall mortality rates in individuals with CHF28. However, the administration of ACEI/ARB may result in an initial deterioration of renal function29. The current study utilized cMAP drug sensitivity analysis, focusing on the up-regulated pathological genes associated with CKD in CHF patients, revealing metiradone as a potential therapeutic agent capable of reversing the gene alterations linked to CKD. Metiradone is an adrenal corticosteroid synthesis inhibitor, which can reduce the circulating concentration of glucocorticoid, inhibit the production of pro-inflammatory factors and monocyte accumulation30, suggesting that metiradone may have the value of treating CKD. In addition, a potential adverse effect of ACEI/ARB therapy for CHF is aldosterone escape, and it has been found that combining ACEI with aldosterone receptor antagonists can limit this effect and bring additional cardio-renal benefits27. Metiradone, as an inhibitor of corticosteroid synthesis, can also inhibit the synthesis of aldosterone and regulate the activity of the RAAS. These results suggest that metiradone may be a dual potential agent for treating CHF and preventing pathological changes in CKD.
The occurrence of CKD is long and insidious, and the increase of creatinine and the decrease of eGFR are the manifestations of renal function, but can not well reflect the early pathological changes of the kidney. In patients with CHF, the clinical manifestations of CKD, such as hypertension and edema, are easily mistaken for the manifestations of CHF, and it is difficult to immediately recognize the occurrence of CKD. Therefore, it is valuable to identify new early biomarkers of CKD in CHF patients. Three CKD biomarkers were identified using five ML algorithms and a diagnostic model was developed based on CHF-related secreted proteins and CKD key genes in this study. The diagnostic accuracy of these biomarkers was confirmed through tissue RNA assay in two external cohorts and serum ELISA assay in the clinical cohort of the study. Among them, COL3A1 is one of the classic markers of fibrosis and is commonly used in animal experiments to evaluate the degree of renal fibrosis31. Ruijing Zhang et al. proposed that CD48 may promote the overexpression of secreted proteins such as inflammatory factors in PBMC and renal tissue by mediating cell–cell interactions, thereby exacerbating the progression of CKD32. In addition, a recent study has shown that LOXL family proteins can be released extracellularly through exosomes in human tubular epithelial HK2 cells and participate in the occurrence of fibrosis33. Therefore, these three hub genes have the potential to serve as biomarkers for CRS2. However, CD48, COL3A1, and LOXL1 are not routine laboratory testing indicators in clinical practice, and larger clinical studies are needed to confirm their diagnostic and prognostic value and provide novel biomarkers for the early detection of CKD patients in clinical practice. Additionally, three biomarkers associated with eGFR and TIF were identified in external cohorts, suggesting a potential new indicator for assessing CKD severity.
The principal constraint of this study is the limited sample size of the clinical validation cohort, highlighting the need for larger clinical investigations to enhance the credibility of the correlation between the three biomarkers and the advancement of CKD in patients with CHF. Secondly, the absence of patient follow-up obscures the enduring prognostic significance of these three biomarkers for CHF patients with CKD. Furthermore, the CKD patients used in this study were non-dialysis patients, and further clinical cohort validation is required to determine the prognostic value of these three biomarkers in dialysis patients.
Data availability
The public datasets were downloaded and analyzed in this study including GEO data repository (accession numbers: GSE32591, GSE66494, GSE1145, GSE5406, GSE21610, and GSE141910).
Abbreviations
- CRS:
-
Cardiorenal syndrome
- CRS2:
-
Type 2 cardio-renal syndrome
- CKD:
-
Chronic kidney disease
- CHF:
-
Chronic heart failure
- RAAS:
-
Renin–angiotensin–aldosterone system
- AVP:
-
Arginine vasopressin
- IL:
-
Interleukin
- ML:
-
Machine learning
- MR:
-
Mendelian randomization
- SNP:
-
Single nucleotide polymorphism
- DEG:
-
Differentially expressed gene
- PCA:
-
Principal component analysis
- ME:
-
Module eigengene
- WGCNA:
-
Weighted gene co-expression network analysis
- MM:
-
Module membership
- GS:
-
Gene significance
- PBMC:
-
Peripheral blood mononuclear cells
- PPI:
-
Protein–protein interaction
- cMAP:
-
Connectivity map
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- BP:
-
Biological processes
- CC:
-
Cellular component
- MF:
-
Molecular function
- RF:
-
Random forest
- SVM:
-
Support vector machine
- XGB:
-
EXtreme gradient boosting
- GLM:
-
Generalized linear model
- LASSO:
-
Least absolute shrinkage and selection operator
- DCA:
-
Decision analysis curve
- ROC:
-
Receiver operating characteristic
- eGFR:
-
Estimated glomerular filtration rate
- TIF:
-
Tubulointerstitial fibrosis
- ssGSEA:
-
Single sample enrichment analysis
- ELISA:
-
Enzyme-linked immunosorbent assay
- TH17:
-
T helper type 17
- NLR:
-
Neutrophil/lymphocyte ratio
References
Ronco, C., Haapio, M., House, A. A., Anavekar, N. & Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 52(19), 1527–1539 (2008).
Hillege, H. L. et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 113(5), 671–678 (2006).
Heywood, J. T. et al. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: A report from the ADHERE database. J. Card. Fail. 13(6), 422–430 (2007).
Damman, K. et al. Renal impairment, worsening renal function, and outcome in patients with heart failure: An updated meta-analysis. Eur. Heart J. 35(7), 455–469 (2014).
Nohria, A. et al. Cardiorenal interactions. J. Am. Coll. Cardiol. 51(13), 1268–1274 (2008).
Kumar, U., Wettersten, N. & Garimella, P. S. Cardiorenal syndrome: Pathophysiology. Cardiol. Clin. 37(3), 251–265 (2019).
Francis, G. S. et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the studies of left ventricular dysfunction (SOLVD). Circulation 82(5), 1724–1729 (1990).
Zhang, H. & Dhalla, N. S. The role of pro-inflammatory cytokines in the pathogenesis of cardiovascular disease. Int. J. Mol. Sci. 25(2), 1082 (2024).
Gene Expression Omnibus. Available from: https://www.ncbi.nlm.nih.gov/geo/.
Zhu, E. et al. Screening of immune-related secretory proteins linking chronic kidney disease with calcific aortic valve disease based on comprehensive bioinformatics analysis and machine learning. J. Transl. Med. 21(1), 359 (2023).
The Human Protein Atlas. Available from: https://www.proteinatlas.org/.
STRING. Available from: https://cn.string-db.org/cgi/input?sessionId=bnpgMtpJVtsZ&input_page_show_search=off.
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
SangerBox. Available from: http://www.sangerbox.com/index.html.
Connectivity Map. Available from: https://clue.io/.
Xu, X. et al. Renal fibrosis in type 2 cardiorenal syndrome: An update on mechanisms and therapeutic opportunities. Biomed. Pharmacother. 164, 114901 (2023).
Hundae, A. & McCullough, P. A. Cardiac and renal fibrosis in chronic cardiorenal syndromes. Nephron Clin. Pract. 127(1–4), 106–112 (2014).
Sato, Y. & Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Renal Physiol. 315(6), F1501–F1512 (2018).
Leong, K. G. et al. Cyclophilin inhibition protects against experimental acute kidney injury and renal interstitial fibrosis. Int. J. Mol. Sci. 22(1), 271 (2020).
Chung, F. M., Tsai, J. C., Chang, D. M., Shin, S. J. & Lee, Y. J. Peripheral total and differential leukocyte count in diabetic nephropathy: The relationship of plasma leptin to leukocytosis. Diabetes Care 28(7), 1710–1717 (2005).
Yuan, Q. et al. Neutrophil-to-lymphocyte ratio and incident end-stage renal disease in Chinese patients with chronic kidney disease: Results from the Chinese cohort study of chronic kidney disease (C-STRIDE). J. Transl. Med. 17(1), 86 (2019).
Zhang, R. et al. Increased neutrophil count is associated with the development of chronic kidney disease in patients with diabetes. J. Diabetes 14(7), 442–454 (2022).
Noel, S. Orai1: CRACing the Th17 response in AKI. J. Clin. Invest. 129(11), 4583–4586 (2019).
Mehrotra, P., Sturek, M., Neyra, J. A. & Basile, D. P. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J. Clin. Invest. 129(11), 4951–4961 (2019).
Mehrotra, P. et al. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: Compensatory role of natural killer cells in athymic rats. Am. J. Physiol. Renal Physiol. 312(3), F385–F397 (2017).
Hu, Y., Hao, F., An, Q. & Jiang, W. Immune cell signatures and inflammatory mediators: Unraveling their genetic impact on chronic kidney disease through Mendelian randomization. Clin. Exp. Med. 24(1), 94 (2024).
Raina, R. et al. An update on the pathophysiology and treatment of cardiorenal syndrome. Cardiol. Res. 11(2), 76–88 (2020).
Ahmed, A. et al. Renin-angiotensin inhibition in systolic heart failure and chronic kidney disease. Am. J. Med. 125(4), 399–410 (2012).
Testani, J. M., Kimmel, S. E., Dries, D. L. & Coca, S. G. Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ. Heart Fail. 4(6), 685–691 (2011).
Niraula, A., Wang, Y., Godbout, J. P. & Sheridan, J. F. Corticosterone production during repeated social defeat causes monocyte mobilization from the bone marrow, glucocorticoid resistance, and neurovascular adhesion molecule expression. J. Neurosci. 38(9), 2328–2340 (2018).
Guo, J. et al. Relationship of clusterin with renal inflammation and fibrosis after the recovery phase of ischemia-reperfusion injury. BMC Nephrol. 17(1), 133 (2016).
Zhang, R. et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs—A novel multiple-hit model for disease progression. Redox Biol. 34, 101460 (2020).
Kamiya, T. et al. Inhibition of N-glycosylation by glucosamine hydrochloride inhibits TGF-beta1-induced LOXL2 secretion. J. Cell Biochem. 124(6), 797–807 (2023).
Funding
This research was supported by the National Nature Science Foundation of China (82300347, 81870255), Zhejiang Provincial Science Foundation of China (LY21H020001), Huadong Medicine Joint Funds of Zhejiang Provincial Natural Science Foundation of China (LHDMZ24H020001), Ningbo Municipal Science Foundation of China (2021J296), Ningbo Key Research and Development Program (2024Z210), and Ningbo Municipal Key Laboratory for Molecular Target Screening and Applications (2023-BZDS).
Author information
Authors and Affiliations
Contributions
Conceptualization: ZW, CZ, and JL; Methodology: JH, CZ, YS, and XJ; Investigation: ZW and CZ; Project Administration: JL and JZ; Writing—Original Draft Preparation: CZ; Writing—Review and Editing: CH and JL; Funding Acquisition: JL and YS. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Shaoxing Second Hospital.
Consent for publication
All authors have thoroughly reviewed the manuscript and consent to its publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, C., Wu, Z., Song, Y. et al. Exploring diagnostic biomarkers of type 2 cardio-renal syndrome based on secreted proteins and bioinformatics analysis. Sci Rep 14, 24612 (2024). https://doi.org/10.1038/s41598-024-75580-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-75580-1
