
Abstract
This work investigates the anti-trypanosomal activities of ten thiohydantoin derivatives against the parasite Trypanosoma cruzi. Compounds with aliphatic chains (THD1, THD3, and THD5) exhibited the most promising IC50 against the epimastigote form of T. cruzi. Also, it showed lower cytotoxicity to mammalian cells. THD3 and THD5 (IC50 = 72.4 µg/mL and 115 µg/mL) presented great activity against trypomastigote and amastigote forms (IC50 = 47.7 µg/mL and 34.1 µg/mL). THD5 had high selectivity index (SI = 15.1) against the amastigote form. The molecular docking and molecular dynamics simulations were performed to understand the interaction between the THD and the important target CYP51 enzyme essential to T. cruzi. THD3 and THD5 were found to have strong interactions within the hydrophobic channel of CYP51 due to their aliphatic side chains, leading to favorable binding free energies. Despite the possibility of cross-reactivity between THD5 and human CYP2C9, the results indicate low identity and similarity between the homolog enzymes and possible selectivity of THD5 for the protozoan one, suggesting that these compounds could inhibit sterol biosynthesis, crucial for the parasite’s survival. These findings indicate that THD3 and THD5 are promising hits for the development of Chagas disease treatments. To fully validate this potential, carrying out enzymatic and other in vitro and in vivo assays is essential in the future.
Similar content being viewed by others

Introduction
Chagas disease (CD) was discovered 112 years ago by Carlos Chagas and is caused by the protozoan Trypanosoma cruzi1. It is a Neglected Tropical Disease (NTD’s) listed by the World Health Organization (WHO), endemic in 21 countries, mainly in Latin America, which has an annual incidence of 30,000 cases. CD affects approximately 6 million people worldwide, causing about 12,000 deaths per year, instigating profound social and economic impacts, and remaining an important public health concern2.
Human infection occurs through the transmission of T. cruzi by hematophagous reduviid vectors. Alternatively, the parasite can be transmitted to mammalian hosts by organ transplant, blood transfer, ingesting contaminated food, or vertically from mother to newborn3. The clinical course of CD usually consists of acute (generally asymptomatic) and chronic phases. The life cycle of T. cruzi involves the following main morphological stages: the replicative epimastigotes and non-replicative metacyclic trypomastigotes (insect vector), the replicative intracellular amastigotes, and non-replicative blood trypomastigotes (mammalian host)4.
Since 1960–1970, the treatment of CD has been restricted to only two nitroheterocyclic drugs, Nifurtimox and Benznidazole. Both drugs have several limitations, such as prolonged treatment durations, lack of efficacy in the chronic phase of the disease, a wide range of side effects, or mutagenic potential that lead to treatment discontinuation by the patients1,5. Thus, efforts to find new antitrypanosomal agents are still needed. The concept of molecular mechanism of action of bioactive substances has been a critical strategy in drug design. These studies can be investigated using computational tools such as molecular docking to evaluate ligand binding orientation and affinity with important targets6.
The 14α-demethylase (CYP51) (EC 1.14.14.154), a cytochrome P450 enzyme, is responsible for the oxidative removal of the 14α-methyl group from sterol precursors in the sterol biosynthetic pathway. The CYP51 is expressed in all stages of the parasite’s life cycle and regulated in replicative forms. The products resulting from the sterol biosynthetic pathway are required for membrane integrity and function as cell viability and proliferation in protozoa, thus have been considered a viable target against T. cruzi over the years4,7,8.
Heterocycles are privileged scaffolds, reported by their several biological activities9, including the thiohydantoins, which are the subject of many studies in drug discovery due to their simple synthesis10 and activities as antibacterial11, anticancer12, antioxidant13, and antiparasitic14. Our research group is particularly interested in developing new drug candidates against neglected diseases. Recently, we described the antitrypanosomal activity of benzoylthioureas15 and 1H-pyrazolo[3,4-b]pyridine16 against T. cruzi, besides the antileishmanial effect of benzoylguanidines17 and thiohydantoins against Leishmania amazonensis18.
Hence, those promising results encouraged us to investigate the antitrypanosomal potential of a series of thiohydantoins derived from L-amino acids against epimastigote, amastigote, and trypomastigote forms of T. cruzi aiming to establish a structure-activity relationship (SAR) study by substitution of aliphatic, aromatic, or polar groups introduced on side chains of the thiohydantoin ring. Following that, the three most promising compounds were chosen for investigations into their potential molecular mechanisms of action. This involved using molecular docking and molecular dynamics simulations on the target enzyme, CYP51, which is crucial for the survival and propagation of the protozoan parasite. Finally, the selectivity of CYP51 was studied and compared to human CYPs, specifically examining the isoform CYP2C9 using molecular modeling techniques (Fig. 1).

The experimental approach to evaluate the antitrypanosomal potential of thiohydantoins (THD1-10).
Results and discussion
Thiohydantoins (THD1-10) were previously synthesized and reported by our research group and fully characterized by assignments of1H and13C NMR spectra11. A preliminary SAR study was conducted to identify critical structural features by modifying the aliphatic, aromatic, or polar groups introduced on the side chains at the C5 position of the thiohydantoin ring. Therefore, we conducted an antitrypanosomal activity screening of ten thiohydantoins by evaluating their inhibitory effects on the growth of epimastigote forms of T. cruzi (IC50epi) Y-strain, determination of cytotoxicity (CC50) to LLCMK2 cells and selectivity index (SI).
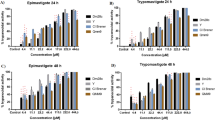
All thiohydantoins can inhibit the growth of epimastigotes forms at non-toxic concentrations to LLC-MK2 cells in vitro; except for compound THD10, all the other (THD1-9) demonstrated lower cytotoxicity than the standard drug benznidazole (BZN). The IC50epi and CC50 values of the compounds ranged from 12.8 to 153 µg/mL and 107 to 917 µg/mL, respectively (Table 1).
In general, thiohydantoins without substituents (THD1) or with aliphatic chains at the C5 position, such as THD3 and THD5, showed the best IC50epi values, except for THD2 and THD4. On the other hand, the presence of phenyl rings (THD8 and THD9) or polar groups like (CH2)2SCH3 (THD6), and pyrrolidine (THD7) reduced activity. Among the aliphatic subset (THD1-5), the chain length or extension could be considered a critical feature for the anti-epimastigote effect since more extended or bulky chains, such as substituted with isopropyl group (THD4) showed lowest activities in comparison with other aliphatic ones (Table 1). The presence of aromatic rings did not contribute to the anti-epimastigote activity. THD8 and THD9 showed lower IC50epi values than aliphatic ones, especially compound THD10, an indole-substituted compound, which presents the worst activity (IC50epi = 153 µg/mL). It is important to notice that unsubstituted derivative THD1 showed the best potential of the series (IC50epi = 12.8 µM), showing that the thiohydantoin ring itself could be essential for anti-trypanosome activity (Table 1).
Furthermore, most thiohydantoins (THD1-9) were more selective to the epimastigote forms of T. cruzi than to the mammalian cells, which can be demonstrated by SI values that ranged between 4.93 and 38.6, except for THD10 (SI = 0.69), which was notable that the introduction of indole ring at C5-position leads to the increasing of cytotoxicity. Among all thiohydantoins, the most promising selective compounds were THD1, THD3, and THD5, which showed SI values > 1020. In this way, aligned with its, these derivatives, with IC50epi values 12.8, 19.9, and 18.8 µg/mL, respectively, were selected as hits and subjected to assays against both trypomastigote and amastigote forms of the parasite (Table 2).
Our findings demonstrated that although the compounds are not more effective against the parasite than the BZN standard, THD1 specifically inhibits T. cruzi, exhibiting the most potent activity against epimastigotes with great SI value (IC50epi = 12.8 µg/mL, SI = 38.6). Still, it can inhibit the amastigote form (Table 2, IC50ama = 73.1 µg/mL, SI = 6.74). The compounds THD3 and THD5 showed anti-trypomastigote and anti-amastigote activity. The IC50 values for THD3 were 72.4 µg/mL for trypomastigotes and 47.7 µg/mL for amastigotes. For THD5, the IC50 values were 115 µg/mL for trypomastigotes and 34.1 µg/mL for amastigotes (Table 2). THD3 had the best selectivity index of 5.8 compared to THD5, with an SI of 4.5 for trypomastigote forms. However, for amastigote results, THD5 demonstrated the best SI value of 15.1 compared to THD3, with an SI of 8.82.
Nitrogen synthesis is crucial for the metabolism of T. cruzi, as it produces essential biomolecules required for the parasite’s survival, replication, and pathogenicity19. Several enzymes and cofactors that T. cruzi relies on for metabolic processes contain nitrogen20. Nitrogen synthesis inhibitors are most effective against replicative forms (epimastigotes and amastigotes) of T. cruzi21. On the other hand, trypomastigote forms are less responsive to these inhibitors21. This is because trypomastigotes, which are non-replicative forms, rely less on active biosynthetic processes. In contrast, amastigotes, replicative forms within mammalian host cells, depend on a high rate of nucleotide and protein synthesis. Therefore, they are more susceptible to inhibitors that disrupt these pathways22.
In previous research conducted by our team, we found that thiohydantoins do not increase the production of NO (nitric oxide) in macrophages infected with L. amazonensis, another type of trypanosomatid species23. In this work, our findings demonstrated that THD1 specifically inhibits epimastigote and amastigote of T. cruzi, and it does not affect the viability of trypomastigotes, suggesting that THD1 may function as a nitrogen synthesis inhibitor.
The development of drugs for treating CD should focus on the specific objective of the study and the phase of the disease being targeted. The amastigote form of T. cruzi, found in the mammalian host tissues, is particularly important because it causes damage to the tissues and is the main replicative form in the host. This is especially significant given that CD is typically asymptomatic and is often detected late24.
The CYP51 enzyme plays a key role in the biosynthesis pathway of sterols, which are essential for the cell membrane of T. cruzi. The expression of CYP51 is higher in the amastigote, parasitic forms that actively replicate within the host cells and thus require high levels of sterols to form and maintain its cell membranes25,26. Based on our outstanding results, including in the amastigote forms, we carried out molecular docking simulations of the three selected active compounds (THD1, THD3, and THD5) by the in vitro studies in enzymatic target CYP5127,28. We aimed to search for information about possible mechanisms of action for antitrypanosomal activity.
The GOLD program successfully reproduced the crystal conformation of ligand N26 (Nalpha-(2-fluoro-4-{4-[4-(trifluoromethyl)phenyl]piperazin-1-yl}benzoyl)-N-pyridin-4-yl-D-tryptophanamide) with root mean square deviation (RMSD) values of 0.89 Å for CYP51, using the ASP score function (Figure S1). This redocking validates our previous article with CYP5129, as the RMSD value was less than 2 Å, supporting the reliability of the docking procedure30.
The molecular docking simulations of THD1, THD3, and THD5 (Fig. 2a-c) revealed distinct interaction profiles within the catalytic pocket. THD1, despite its proximity to the Fe-heme at 3.0 Å, cannot interact with any residues in the catalytic pocket (Fig. 2a). In contrast, THD3 did not exhibit the proximity of the sulfur atom for interaction with Fe but performed H-bond with catalytic Tyr116 through the NH group of the thiohydantoin ring. Additionally, THD3 is involved in alkyl interactions between its side chain methyl group with Ala291 and Leu356 and pi-alkyl interaction with porphyrin rings of the heme cofactor. Hydrophobic interactions by Pi-Pi Stacked type between thiohydantoin and porphyrin rings were also observed (Fig. 2b). The extended side chain (isobutyl group) of THD5 allowed the Pi-alkyl interaction with Phe110 and with a methyl group of a porphyrin ring from heme. Furthermore, the binding mode of THD5 contributed to the H-bond of the carbonyl group from the thiohydantoin ring, as well as S-Fe coordination at 2.5 Å (Fig. 2c).

Binding mode and molecular interactions of thiohydantoins (balls and sticks in green color) with CYP51. (A) THD1; (B) THD3; (C) THD5. Interacting amino acid residues are represented by sticks in gray color and cofactor HEME in yellow color. The dashed yellow lines represent the H-bond, orange lines Pi-alkyl, light green lines Pi-Pi Stacked, and gray ones metal interaction.
Interestingly, in this case, the longer side chain of THD3 and THD5 seems critical to binding and interactions compared to unsubstituted thiohydantoin itself. These results support the findings of the in vitro assays. The unsubstituted thiohydantoin THD1 was the most effective compound for the epimastigote forms. Meanwhile, for the amastigote forms, compounds THD3 and THD5 showed significant selectivity (SI > 8 and 15). CYP51 is more expressed in these forms, indicating that our molecular docking results revealed interesting interactions of these two compounds in inhibiting this molecular target.
The cytochrome P450 superfamily plays a crucial role in the mammalian host, metabolizing over 90% of the many drugs31. Predicting how a substance interacts with human CYPs can be helpful. If a substance is a CYP substrate, it may have low oral bioavailability due to pre-systemic metabolism. Conversely, if it acts as an inhibitor, it can affect the plasma concentration of co-administered drugs metabolized by one of the isoforms, potentially leading to increased toxicity and a longer half-life31. Based on this, we predict in silico the possibility of THD1, THD3, and THD5 interacting with isoforms of human CYPs using ADMETlab 2.0 platform32 (Table 3).
Among the compounds, only THD5 was predicted to act as a CYP2C9 substrate, an enzyme responsible for the metabolism of endogenous substrates (e.g., steroids) and xenobiotics (e.g., drugs)33. We performed molecular docking simulations of THD5 against human CYP2C9. The protocol was validated by redocking of co-crystallized inhibitor TCA007 (ethyl {2-[([1,3]thiazolo[4,5-c]pyridine-2-carbonyl)amino]thiophene-3-carbonyl}carbamate), yielding an RMSD of 0.15 Å, using ChemPLP scoring function (Figure S2).
In contrast to its behavior with CYP51, THD5 did not demonstrate the proximity of the sulfur atom from the thiohydantoin ring to Fe’s interaction in the CYP2C9 complex. However, it exhibited Pi-alkyl interaction with Phe100 (Val102 in CYP51), Phe114 (Tyr116 in CYP51), and Leu366 (Leu356 in CYP51) via its side chain (isobutyl group) (Fig. 3).

Binding mode and molecular interactions of THD5 (balls and sticks in green color) with CYP2C9. Interacting amino acid residues are represented by sticks in gray color and cofactor HEME in yellow color. The dashed orange lines represent the Pi-alkyl interaction.
To access the potential for cross-reactivity due to the high structural identity between the CYP51 and CYP2C9, given that these enzymes are homologs, we compared its amino acid sequences through structural alignment between the human and the protozoa enzymes. The sequence alignment demonstrated a low percentage of 17.4% identity and 32.9% similarity between CYP51 and CYP2C9 sequences (Figure S3). Furthermore, in the case of residues present in the catalytic cavity, such as Met106, Phe110, Tyr116, and Met358, in the sequence of human CYP2C9 correspond respectively to alanine, asparagine, valine, and threonine and are not similar in interaction aspects. Only the Leu356 amino acid is identical in CYP2C9 (Leu336), and the Tyr103 is substituted in human CYP for phenylalanine (Figure S3).
Although the predictions suggested that THD5 could function as a CYP2C9 substrate and molecular docking revealed interactions with the target, the low sequence identity and similarity between the two CYPs suggest that cross-reactivity might not be a significant issue if this potential hit is used to treat Chagas disease.
Based on the molecular docking results, we conducted molecular dynamics (MD) simulations of THD1, THD3, and THD5 complexes with CYP51 and THD5-CYP2C9 to access the potential persistence of interactions and to observe the behavior of derivatives in an aqueous system.
We observed a consistent pattern in the RMSD analysis of the ligands. The THD compounds showed an average variation profile of about 4 Å, indicating stability throughout the 200 ns of MD simulation. Despite the lower RMSD value, the most significant standard deviation was observed for the THD1-CYP51 complex with 3.40 ± 1.55 Å (Fig. 4A), compared to THD3-CYP51, which had an RMSD of 3.87 ± 0.75 Å (Fig. 4B). The THD5-CYP51 complex showed minor fluctuation in its binding mode, with an RMSD of 5.15 ± 0.32 Å throughout the entire MD simulation period (Fig. 4C).

RMSD analysis of derivatives THD1 (A), THD3 (B), and THD5 (C) on CYP51 complexes. Cα-RMSD analysis of CYP51 enzyme in the presence of ligands (D). RMDS analysis of THD5 and Cα-atoms of the protein CYP2C9 (E).
The stability of each complex was thoroughly assessed by analyzing the trajectories based on protein Cα-atoms shifts (Fig. 4D). The CYP51 complexes showed an average RMSD variation profile of approximately 4 Å, remaining stable throughout the 200 ns of MD simulation. Specifically, RMSD values were 4.18 ± 0.68 Å, 3.66 ± 0.57 Å, and 3.76 ± 0.74 Å in the presence of THD1, THD3, and THD5, respectively with low standard deviation (Fig. 4D).
About the THD5 derivative within CYP2C9, we observed an RMSD value of 5.30 ± 0.58 Å (Fig. 4E), similar to theTHD5-CYP51. However, the RMSD for Cα-atoms of CYP2C9 was substantially higher at 8.84 ± 1.14 Å, indicating less stability in the complex formation (Fig. 4E).
To further assess the impact of fluctuations on residues, we calculated the root mean of square fluctuations (RMSF) during the 200 ns of MD. Our analysis revealed significant fluctuations greater than 3.1 for amino acids between Arg400-His420 (yellow) close to the heme cavity and regions of Leu200-Ser226 (red), Arg248-Tyr258 (blue) at 6.3 Å in the presence of THD1, THD3, and THD5 ligands to CYP51, as shown in Fig. 5A. These fluctuations were minimal for CYP2C9 in the presence of THD5, with RMSF values lower than 4 Å, except by the region between Gly400-Phe410 exhibiting 5.5 Å, indicating overall stability for the entire protein (Fig. 5B).

(A) RMSF analysis of derivatives THD1, THD3, and THD5 on CYP51 complexes; 3D structure highlighting the significant fluctuations observed: Leu200-Ser226 (red), Arg248-Tyr258 (blue), and Arg400-His420 (yellow). (B) RMSF analysis of derivative THD5 on CYP2C9 complex.
The hydrogen bonding analysis in the CYP51 complexes revealed that THD1 formed a hydrogen bond between the NH group and the catalytic Tyr116 residue, with a low persistence of 9% (Fig. 6A). In the case of THD3, a similar docking pose was observed, forming a hydrogen bond with Tyr116 exhibiting a lifetime of 21.7% (Fig. 6B). Additionally, THD5 demonstrated an H-bond with the same residue as Tyr116 and a high persistence of 47%, as well as another H-bond with Ala287, with a persistence of 21.6% of the simulation time (Fig. 6C). In the THD5-CYP2C9 complex, the observed interaction was with the Thr301 residue, located slightly further from the heme cofactor binding site, with a low persistence of 17.9% (Fig. 6D).
Our molecular dynamics results reinforce that the three THD compounds, due to the donor potential of the NH group from the thiohydantoin ring, could interact with Tyr116, which could be prejudicial to potentially impacting the catalytic activity of enzyme.

THD-CYP51 complexes in the representative pose interactions of MD: THD1 (A), THD3 (B), and THD5 (C). THD5-CYP2C9 complex in the representative pose interaction of MD (D). The H-bonds between ligands (sticks and balls in green) and residues (sticks in yellow color) are represented by dashed yellow lines.
CYP51 from T. cruzi is reported to have a hydrophobic channel, with amino acid residues in the active site offering a favorable binding patch for the esterified sterols34. Therefore, it is essential for compounds designed to inhibit CYP51 to possess groups that can act as both hydrogen bond acceptors or donors and hydrophobic characteristics that can interact with this activity cavity. Notably, the hydrophobic residues Tyr103, Met106, Phe110, and Tyr116 are reported in the literature to form an electron-rich cluster that supports the conformation adopted by the heme cofactor29,35.
As previously mentioned, the minor potential of THD1 in performing interactions was expected due to the observed need for an extended carbon side chain, as seen in THD3 and THD5, which can interact particularly well with the hydrophobic residues of the cavity.
When analyzing the surfaces of the heme cofactor cavity and the key hydrophobic residues required for its active conformation, a channel formation is evident, allowing substrates or designed inhibitors to enter and bind to the molecular target (Fig. 7A-C). For THD1, a relatively small molecule, its volume is insufficient to interact with both regions effectively, thereby limiting its ability to facilitate favorable interactions for catalytic inhibition (Fig. 7A). In contrast, the derivatives THD3 (Fig. 7B) and THD5 (Fig. 7C) have aliphatic side chains capable of accessing these surfaces and creating hydrophobic interactions with the amino acids, while the thiohydantoin ring located between the cavities can form hydrogen bonds, enhancing their inhibitory potential.

Surfaces of cofactor heme (in blue) and hydrophobic residues Tyr103, Met106, Phe110, Tyr116, Leu357, and Met358 (in yellow) in the presence of ligands (in green) (A) THD1, (B) THD3, and (C) THD5.
Another essential factor to investigate is the potential coordination with the Fe metal of the enzyme cofactor. Heterocyclic compounds can coordinate with heme, replacing the water in the iron coordination sphere and affecting substrate binding and metabolism36.
In the complexes formed with the CYP enzymes, the sulfur atom from thiohydantoin and the Fe atom from heme is in proximity during the initial nanoseconds of the simulation. For the THD1-CYP51 (Fig. 8A) and THD3-CYP51 (Fig. 8B) complexes, the smallest distance between these two atoms occurs between 0 and 30 ns, with values of 4.62 ± 1.4 and 9.13 ± 1.0 Å, respectively. Subsequently, this distance increases to 7.95 ± 2.8 Å for THD1 and decreases to 7.83 ± 1.8 Å for THD3. For the THD5-CYP51 complex, this distance is more significant in the first 13 ns of simulation, measuring 8.71 ± 3.4 Å, and remains for the rest of the simulation period with an average distance of 8.97 ± 0.4 Å (Fig. 8C).

Distance (Å) between S atom from thiohydantoin ring and Fe atom from heme in complexes: (A) THD1-CYP51, (B) THD3-CYP51, and (C) THD5-CYP51 and THD5-CYP2C9.
It is interesting to note that in the case of the THD5 complex with CYP2C9, the orientation of the thiohydantoin nucleus is contrary to that observed for the CYP51 complexes, resulting in a significantly greater distance between the S and Fe atoms throughout the simulation, 14.30 ± 1.9 Å (0 to 14 ns) and 10.69 ± 0.6 Å (14–200 ns). This considerable distance suggests that coordination is unlikely from the beginning of the complex formation (Fig. 8C). In general, only THD1 could coordinate with Fe at the start of the ligand-enzyme complex formation, but this coordination was brief. This observation reinforces that for THD3 and THD5, the interactions involving the aliphatic side chain are predominant in the interaction with the target.
Finally, we calculated the free binding energy (ΔGb) for all complexes evaluated using the MMPBSA method from the representative frames during the stability period of the complexes. Throughout complex formation, the solvent-accessible surface area of all THD appeared energetically favorable due to the interactions at the binding site, with similar ΔEsasa values ranging from − 2.0 to -2.7 kcal/mol (Table 4). The CYP51 enzyme possesses a large and hydrophobic active site that facilitates strong interactions with compounds such as THD3 and THD5, as evidenced by their ΔEvdw values of -19.7 and − 14.3 kcal/mol, respectively (Table 4). In contrast, despite THD1 having an ΔEvdw = -16.3 kcal/mol, the solvation energy analysis indicates a higher energetic cost (ΔEsolv = 13.8 kcal/mol) for the THD1 ligand to access the CYP51 active site due to its hydrophobic environment, which is reflected in its less favorable ΔGb value of -2.30 kcal/mol (Table 4).
In our analysis of molecular dynamics simulations, we observed that compounds THD3 and THD5, when in complex with CYP51, demonstrated enhanced interactions with the hydrophobic channel. Moreover, these compounds exhibited no significant increase in energetic cost for desolvation and presented the most favorable ΔGb values of -12.1 and − 12.8 kcal/mol, respectively. In contrast, in the complex involving THD5 and CYP2C9, the ΔEsolv indicates a higher energy cost (12.5 kcal/mol) compared to CYP51 (7.68 kcal/mol). This difference arises because THD5 binds at a greater distance from the hydrophobic channel to carry out interactions, as visually depicted in Fig. 6D. Despite this, THD5 forms a stable complex with ΔGb = -11.3 kcal/mol (Table 4).
Based on the molecular dynamics analysis, it is suggested that THD3 and THD5 can bind to the active site of CYP51 with a likely selectivity of THD5 for the parasite enzyme over the human CYP2C9. This finding aligns with the in vitro assay results, which indicate that these two compounds are the most active within the series evaluated. They also act on the amastigote form of T. cruzi, where this enzyme is expressed in more significant quantities.
Experimental section
Animals and ethics committee
This study protocol was approved by the Ethics in Animal Utilization Committee (CEUA) of the Universidade Estadual de Londrina (UEL), Paraná, Brazil (CEUA/UEL number 051.2023), according to the Brazilian federal law (11.794/2008, decreto nº 6.899/2009). Furthermore, the animals were kept in a controlled and disinfected environment, with access to sterile water and a commercial rodent diet (Nuvilab-CR1, Quimtia-Nuvital, Colombo, Paraná, Brazil) ad libitum, and cage floor wood shavings, in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences, USA. The experiment involving animals follows the recommendations described in the ARRIVE guidelines.
Trypanosoma cruzi (Y strain)
Epimastigote form
The epimastigote forms of T. cruzi (Y strain) were cultivated in LIT (Liver Infusion Tryptone) medium supplemented with 10% fetal bovine serum (FBS) (Gibco®, Invitrogen) at 28° C. Subcultures were performed every 72 h, following the protocol of Camargo et al. (1964)37.
Effect of thiohydantoin derivatives (THD1-10) on growth of epimastigote forms
A suspension of 106 parasites/mL was seeded in 24-well plates containing LIT with 10% FBS medium and increasing concentrations of the tested THD (50, 100, 200, and 400 µg/mL) and standard BZN concentrations of 100 to 0.78 µg/mL. The plate was incubated at 28° C for 72 h. Cultures of untreated parasites cultivated in LIT medium were used as controls. After 72 h of incubation, the number of parasites was quantified by counting in a Neubauer chamber, and the IC50 (concentration capable of inhibiting 50% of parasite growth) was obtained38.
Trypomastigote forms
The trypomastigote forms were obtained from supernatants of infected LLC-MK2 cell line (renal epithelial cells of Macaca mulatta, CCL-7 ATCC, Sigma-Aldrich, Brazil) cultures infected with T. cruzi (Y strain) at the peak of parasitemia. The epithelial cells were cultivated in DMEM with 10% FBS at 37 °C and 5% CO2, and at the confluence, the monolayer was inoculated with blood from Swiss mice infected with T. cruzi (Y strain). The infected LLC-MK2 cells were incubated at 37 °C and 5% CO2 until the trypomastigote forms were released in the supernatant.
Effect on the viability of trypomastigote forms
For the assay, 107/mL trypomastigote forms were incubated with the THD concentrations of 200, 100, 50, and 25 µg/mL and standard BZN concentrations of 100 to 0.78 µg/mL at 37 °C, 5% CO2 for 24 h. The viability of the parasites was analysed by counting in a Neubauer chamber39 and the results obtained were used to calculate the IC50trp (concentration capable of inhibiting 50% of viability of trypomastigotes).
Effect on the proliferation of intracellular amastigotes
LLC-MK2 cells were seeded in a 24-well plate with glass coverslips (13 mm) and after cell adhesion, they were infected with trypomastigote forms of T. cruzi (MOI 5:1) for 18 h. After the infection, the wells were washed with PBS to remove extracellular parasites. The cells were subjected to treatments at concentrations of 100, 50, and 25 µg/mL (THD1 and THD3) and 200, 100, and 50 µg/mL (THD5) and standard BZN concentrations of 100 to 0.78 µg/mL, incubated for 48 h at 37 °C, 5% CO2. After incubation, the cells were fixed with methanol stained with Giemsa, and the coverslips were mounted on glass slides using Permont. A total of 400 cells were counted under light microscope. The infection index was determined by the % of infected cells x the average number of parasites per infected cell, according to the protocol described by Barrias et al., (2010)40 with modifications.
In silico prediction of human CYPs substrate or inhibitor
The in silico pharmacokinetic metabolism of human CYPs: CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 of the compounds THD1, THD3, and THD5 were assessed using ADMETlab 2.0 platform (https://admetmesh.scbdd.com/).
Sequence alignment between CYP51 from T. cruzi and human CYP2C9
The sequence alignment was performed with the T-Coffee server (http://tcoffee.crg.cat/)41 using the UniProt code Q7Z1V1 to CYP51 from T. cruzi and P11712 to human CYP2C9.
The identity of the sequence pair was evaluated using the Sequence Manipulation Suite (SMS) server (https://www.bioinformatics.org/sms2/ident_sim.html)42.
Molecular docking
The molecular docking was carried out using GOLD (Genetic Optimization for Ligand Docking) v. 2020.1, applying the score functions GoldScore43, ChemScore44, ASP45, and ChemPLP46. The X-ray crystal structure of CYP51 (PDB ID: 4C27, resolution: 1.95 Å)47 and CYP2C9 (PDB ID: 5W0C, resolution 2.0 Å)48 are available in the Protein Data Bank (PDB) server (https://www.rcsb.org)49. The hydrogen atoms were added to the proteins based on ionization inferred by the program, the number of genetic operations in each run, and other parameters were set as default. The binding sites were defined within a 12 Å radius. Also, the ligands were subjected to 50 iterative runs. The grid box size dimensions were prepared according to the protocol previously described by our research group16 considering the following coordinates: CYP51 was centered between iron ion on HEME cofactor at x: -5.208, y: 11.497, z: -18.258. CYP2C9 was centered between iron ion on the HEME cofactor at x: -24.2428, y: 59.7815, z: 1.46993.
The 3D structures of thiohydantoins derivatives (THD1, THD3, and THD5) were built up in ChemDraw, and then, the geometry optimization was performed using the MM2 force field implemented on ChemBio3D v.12.0 (PerkinElmer Informatics)50. The best-scored pose for which docking results were considered by RMSD calculation of each function applicable. The intermolecular interaction analyses and figures were generated by the PyMOL program v. 2.551.
Molecular dynamics
The molecular dynamics simulations were performed using the GROMACS version 5.1.4 and 2019 package52,53, applying the CHARMM36 all-atom (AA) force field 55 and TIP3P water model with the protein-ligand poses (complexes) from molecular docking. The atom’s order was corrected by sort_mol2_bonds.pl Python script and ligand topology acquired through the CHARMM General Force Field for organic molecules (CGenFF) server (https://cgenff.umaryland.edu/initguess/).
The missing residues of proteins were completed with the CHARMM-GUI server manipulator (https://www.charmm-gui.org/?doc=input/pdbreader). The ionization states of the protein’s residues were adjusted to pH 7.4 using the pdb2gmx Python script. Each complex was centered inside a cubic periodic box 5.696 × 4.481 × 5.023 nm, volume = 1025.7 nm³, neutralized with Cl− ion to CYP51 and 6.465 × 4.757 × 4.014 nm, volume = 987.63 nm³, neutralized with four Cl− ions to CYP2C9, and solvated with water TIP3P type.
Systems were submitted to energy minimization, two equilibration steps (V-rescale thermostat and Parrinello-Rahman barostat), considering volume, and temperature constant (NVT ensemble), and then considering the system as isothermal-isobaric (NPT ensemble) following the protocol of our research group54. Hydrogen atoms were frozen using the LINCS algorithm, and the long-distance electrostatic interactions using the PME algorithm with a cut-off radius of 1 nm were applied to the van der Waals and Coulomb interactions. After the thermalization step, the molecular dynamics (MD) simulations were performed during 200 ns, using 2 fs integration time and 20 Å of cut-off radius to the long-distance interactions.
The CYP51 and CYP2C9 ligand complexes in aqueous systems were evaluated by RMSD and RMSF, the hydrogen bonding (H-bond) and distance between atoms by gmx rms, gmx rmsf, gmx hbond, and gmx distance, respectively, package of GROMACS 2019. The H-bonds frequency was calculated using the hbmap2grace package55, and the graphs were plotted using the XMGRACE 5.1.19 program56.
The binding free energy (ΔGb) was calculated using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method through the module added to the GROMACS 5.1.4. Figures were constructed using the Visual Molecular Dynamics (VMD) program (http://www.ks.uiuc.edu/Research/vmd/).
Conclusions
The results indicate that thiohydantoin derivatives (THD1-10) have promising activity against Trypanosoma cruzi. Among the compounds, THD3 and THD5 stood out for their IC50 = 72.4 µg/mL (SI = 5.8) and 115 µg/mL (SI = 4.5) in the trypomastigote or amastigote form IC50 = 47.7 µg/mL (SI = 8.82) and IC50 = 34.1 µg/mL (SI = 15.1), showing effective and less toxicity to mammalian host cells.
Molecular modeling studies suggest that the interaction of two compounds, THD3 and THD5, with the CYP51 enzyme, essential for sterol biosynthesis in the parasite, may be responsible for the antitrypanosomal activity observed. These derivatives have aliphatic side chains capable of accessing the surfaces between the heme cofactor and site activity cavity, creating hydrophobic interactions with the key amino acids. In contrast, the thiohydantoin ring binding between the cavities can form hydrogen bonds, enhancing their inhibitory potential. Besides the possibility of cross-reactivity of THD5 due to the interaction with the human CYP2C9, our results indicated low identity and similarity between the two homolog enzymes and fewer interactions of THD5 with human CYP compared to protozoan, consolidating the viability of the compounds as potential candidates for developing new treatments against Chagas Disease. However, to truly validate this potential, it is essential to carry out enzymatic and other in vitro and in vivo assays.
Data availability
The authors confirm all data generated and analyzed during this study are available in the article and the supplementary information.
References
Guedes-da-Silva, F. H. et al. Antitrypanosomal activity of sterol 14␣- demethylase (CYP51) inhibitors VNI and VFV in the Swiss mouse models of chagas disease induced by the Trypanosoma cruzi Y strain. Antimicrob. Agents Chemother. 61, 1–9 (2017).
Pan American Health Organization. Chagas Disease. https://www.paho.org/en/topics/chagas-disease (2021).
Espinosa, R. et al. Synthesis and evaluation of the in vitro and in vivo antitrypanosomal activity of 2-styrylquinolines. Heliyon 7 (2021).
Franco, C. H. et al. Novel structural CYP51 mutation in Trypanosoma cruzi associated with multidrug resistance to CYP51 inhibitors and reduced infectivity. Int. J. Parasitol. Drugs Drug Resist. 13, 107–120 (2020).
Shaykoon, M. S. et al. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1,2,4-triazole and 1,3,4-oxadiazole derivatives. Bioorg. Chem. 100, 103933 (2020).
Aliebrahimi, S., Montasser Kouhsari, S., Ostad, S. N., Arab, S. S. & Karami, L. Identification of Phytochemicals Targeting c-Met kinase domain using Consensus Docking and Molecular Dynamics Simulation studies. Cell. Biochem. Biophys. 76, 135–145 (2018).
Riley, J. et al. Development of a fluorescence-based Trypanosoma cruzi CYP51 inhibition assay for effective compound triaging in drug discovery programmes for chagas disease. PLoS Negl. Trop. Dis. 9, 1–12 (2015).
Doyle, P. S. et al. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 54, 2480–2488 (2010).
Kalaria, P. N., Karad, S. C. & Raval, D. K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 158, 917–936 (2018).
Wang, Z. D., Sheikh, S. O. & Zhang, Y. A simple synthesis of 2-thiohydantoins. Molecules 11, 739–750 (2006).
de Carvalho, P. G. C. et al. Synthesis and antimicrobial activity of thiohydantoins obtained from L-amino acids. Lett. Drug Des. Discov. 17, 94–102 (2018).
Cho, S. H., Kim, S. H. & Shin, D. Recent applications of hydantoin and thiohydantoin in medicinal chemistry. Eur. J. Med. Chem. 164, 517–545 (2019).
Aichouchebouzroura, S. et al. Antioxidant and anti tumoral activities of hydrazylpyrrolidine 2, 5 dione substituted and 2-thioxo imidazolidine 4-one. Int. J. Pharm. Chem. Biol. Sci. 4, 447–452 (2014).
Buchynskyy, A. et al. 1-Benzyl-3-aryl-2-thiohydantoin derivatives as new anti-trypanosoma brucei agents: SAR and in vivo efficacy. ACS Med. Chem. Lett. 8, 886–891 (2017).
Pereira, P. M. L. et al. In vitro evaluation of antitrypanosomal activity and molecular docking of benzoylthioureas. Parasitol. Int. 80, 102225 (2021).
da Silva Lima, C. H. et al. Anti-Trypanosoma cruzi activity and molecular docking studies of 1Hpyrazolo[ 3, 4-b]pyridine derivatives. Lett. Drug Des. Discov. 17, 184–191 (2019).
Santiago-silva, K. M. et al. Exploring the antileishmanial activity of N, N - disubstituted-benzoylguanidines : synthesis and molecular modeling studies. J. Biomol. Struct. Dyn. 0, 1–16 (2021).
Camargo, P. G. et al. Thiohydantoins as anti-leishmanial agents: n vitro biological evaluation and multi-target investigation by molecular docking studies. J. Biomol. Struct. Dyn. 0, 1–10 (2020).
Silber, A., Colli, W., Ulrich, H., Manso Alves, M. & Pereira, C. Amino acid metabolic routes in Trypanosoma cruzi: possible therapeutic targets against chagas; disease. Curr. Drug Target. Infect. Disord. 5, 53–64 (2005).
Márquez, V. E. et al. Redox metabolism in Trypanosoma cruzi. Biochemical characterization of dithiol glutaredoxin dependent cellular pathways. Biochimie 106, 56–67 (2014).
Urbina, J. A. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 115, 55–68 (2010).
Bringaud, F., Rivière, L. & Coustou, V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol. Biochem. Parasitol. 149, 1–9 (2006).
Taciane da Silva et al. Investigation of the antileishmanial activity and mechanisms of action of acetyl-thiohydantoins. Chem. Biol. Interact. 351, 109690 (2021).
Franco-Paredes, C. American trypanosomiasis: chagas disease. Chagas disease. Manson’s Trop. Dis. Twenty-Third Ed. 622–630. https://doi.org/10.1016/B978-0-7020-5101-2.00047-9 (2013).
Jun, Y. C. William. Structure based design of CYP51 inhibitors. Curr. Top. Med. Chem. 17, 30–39 (2017).
Friggeri, L. et al. Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α-demethylase: two regions of the enzyme molecule potentiate its inhibition. J. Med. Chem. 57, 6704–6717 (2014).
Sykes, M. L. & Avery, V. M. 3-Pyridyl inhibitors with novel activity against Trypanosoma cruzi reveal in vitro profiles can aid prediction of putative cytochrome P450 inhibition. Sci. Rep. 8, 1–12 (2018).
Calvet, C. M. et al. 4-Aminopyridyl-based CYP51 inhibitors as anti-Trypanosoma cruzi drug leads with improved pharmacokinetic profile and in vivo potency. J. Med. Chem. 57, 6989–7005 (2014).
Flores-Junior, L. A., dos Santos, E., Muri, E. M., Lima, C. H. & Dias, L. R. Putative inhibitor of TcCYP51 from a library of approved drugs: a virtual screening study. J. Braz Chem. Soc. https://doi.org/10.21577/0103-5053.20240064 (2024).
Hevener, K. E. et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 49, 444–460 (2009).
Nelson, D. R. et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6, 1–42 (1996).
Xiong, G. et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49, W5–W14 (2021).
Zhou, S. F., Liu, J. P. & Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 41, 89–295 (2009).
Warfield, J., Setzer, W. N. & Ogungbe, I. V. Interactions of antiparasitic sterols with sterol 14α-demethylase (CYP51) of human pathogens. Springerplus 3, 679 (2014).
Vieira, D. F. et al. Binding mode and potency of N -indolyloxopyridinyl-4-aminopropanyl-based inhibitors targeting Trypanosoma Cruzi CYP51. J. Med. Chem. 57, 10162–10175 (2014).
Lepesheva, G. I., Villalta, F. & Waterman, M. R. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv. Parasitol. 75, 65–87 (2011).
CAMARGO, E. P. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop. Sao Paulo 6, 93–100 (1964).
Contreras Lancheros, C. A. et al. Selective antiprotozoal activity of nitric oxide-releasing chitosan nanoparticles against Trypanosoma cruzi: toxicity and mechanisms of action. Curr. Pharm. Des. 24, 830–839 (2018).
Izumi, E., Ueda-Nakamura, T., Veiga, V. F., Pinto, A. C. & Nakamura, C.V. Terpenes from copaifera demonstrated in vitro antiparasitic and synergic activity. J. Med. Chem. 55, 2994–3001 (2012).
Barrias, E. S., Reignault, L. C., De Souza, W. & Carvalho, T. M. U. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One 5, e7764 (2010).
Notredame, C., Higgins, D. G. & Heringa, J. T-coffee: a novel method for fast and accurate multiple sequence alignment 1 1Edited by J. Thornton. J. Mol. Biol. 302, 205–217 (2000).
Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 (2000).
Verdonk, M. L., Cole, J. C., Hartshorn, M. J., Murray, C. W. & Taylor, R. D. Improved protein–ligand docking using GOLD. PROTEINS Struct. Funct. Genet. 52, 609–623 (2003).
Eldridge, M. D., Murray, C. W., Auton, T. R., Paolini, G. V. & Mee, R. P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 11, 425–445 (1997).
Mooij, W. T. M. & Verdonk, M. L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Genet. 61, 272–287 (2005).
Korb, O., Stützle, T. & Exner, T. E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 1, 115–134 (2007).
Wiggers, H. J. et al. Non-peptidic Cruzain inhibitors with trypanocidal activity discovered by virtual screening and in vitro assay. PLoS Negl. Trop. Dis. 7 (2013).
Liu, R. et al. Determinants of the inhibition of DprE1 and CYP2C9 by antitubercular thiophenes. Angew. Chem. Int. Ed. 56, 13011–13015 (2017).
Berman, H. M. et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 58, 899–907 (2002).
Evans, D. A. History of the Harvard ChemDraw project. Angew. Chem. Int. Ed. 53, 11140–11145 (2014).
DeLano, W. L. & Pymol An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92 (2002).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Camargo, P. G., dos Santos, C. R., Girão Albuquerque, M., Rangel Rodrigues, C. & Lima, C. H. da S. Py-CoMFA, docking, and molecular dynamics simulations of Leishmania (L.) amazonensis arginase inhibitors. Sci. Rep. 14, 11575 (2024).
Gomes, D. E. B., da Silva, A. W., Lins, R. D. & Pascutti, P. G. & A., S. HbMap2Grace. Software for mapping the hydrogen bond frequency. Lab. Mol. Model. Dyn. (2009).
Turner, P. J. & XMGRACE Version 5.1.19. Center for Coastal and Land-Margin Research (Oregon Graduate Institute of Science and Technology, 2005).
Acknowledgements
The authors would like to thank the Centro Nacional de Processamento de Alto Desempenho em São Paulo (CENAPAD-SP) for the resources for the Molecular Dynamics simulations.
Funding
This research was funded by the Brazilian governmental agencies: FAPERJ (“Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro”) (SEI-260003/019723/2022, SEI-260003/007043/2022, SEI-260003/003788/2022), CAPES (“Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior” - Funding Code 001), and CNPq (“Conselho Nacional de Desenvolvimento Científico e Tecnológico”).
Author information
Authors and Affiliations
Contributions
P.G.C. Conceptualization, methodology, formal analysis, investigation, writing. H.T.S. Conceptualization, methodology, formal analysis, investigation.P.M.L.P Methodology, formal analysis, investigation. M.L.S. Conceptualization, methodology, formal analysis.F.M.J. Supervision.M. G. A. Review and editing, supervision.C. R. R. Project administration, supervision, software.S.F.Y.O. Project administration, supervision.C. H. S. L. Review and editing, project administration, supervision, software.M.L.F.B. Review and editing, project administration, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Camargo, P.G., Suzukawa, H.T., Pereira, P.M.L. et al. In vitro assays identified thiohydantoins with anti-trypanosomatid activity and molecular modelling studies indicated possible selective CYP51 inhibition. Sci Rep 15, 465 (2025). https://doi.org/10.1038/s41598-024-84697-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-84697-2
Keywords
This article is cited by
-
Synthesis and in vitro antitrypanosomatid efficacy of 5-benzylidene-2-thiohydantoin esters
Medicinal Chemistry Research (2025)
