
Abstract
The main protease (MPro) of SARS-CoV-2 plays a crucial role in viral replication and is a prime target for therapeutic interventions. Phytochemicals, known for their antiviral properties, have been previously identified as potential MPro inhibitors in several in silico studies. However, the efficacy of these remains in question owing to the inherent flexibility of the MPro binding site, posing challenges in selecting suitable protein structures for virtual screening. In this study, we conducted an extensive analysis of the MPro binding pocket, utilizing molecular dynamics (MD) simulations, principal component analysis (PCA) and free energy landscape (FEL) to explore its conformational diversity. Based on pocket volume and shape-based clustering, five representative protein conformations were selected for virtual screening. Virtual screening of a library of ~ 48,000 phytochemicals suggested 39 phytochemicals as potential MPro inhibitors. Based on subsequent MM-GBSA binding energy calculations and ADMET property predictions, five compounds were advanced to cell-based viral replication inhibition assays, with three compounds (demethoxycurcumin, shikonin, and withaferin A) exhibiting significant (EC50 < 10 μm) inhibition of SARS-CoV-2 replication. Our study provides an understanding of the binding interactions between these phytochemicals and MPro, contributing significantly to the identification of promising MPro inhibitors. Furthermore, beyond its impact on therapeutic development against SARS-CoV-2, this research highlights a crucial role of proper nutrition in the fight against viral infections.
Similar content being viewed by others

Introduction
As the world marks four years since the onset of the COVID-19 pandemic, we continue to grapple with the devastating impact of this global health crisis caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), first reported in Wuhan, China, in late 20191,2. As per the latest data from World Health Organization, (https://data.who.int/dashboards/covid19/cases?n=c, as of 22 January 2024), the global death toll from COVID-19 has now exceeded 7 million, and the disease continues to claim hundreds of lives daily. The onset of 2024 marked a deadly surge in COVID-19 cases, with over 1 million new cases and above 8000 deaths reported in the past month alone, highlighting the persistent threat posed by the virus. Although there are several highly efficient vaccines authorized for SARS-CoV-2 in the USA and other nations, the number of approved small-molecule COVID-19 drugs remain limited to remdesivir (e.g., Veklury), baricitinib (e.g., Olumiant), and nirmatrelvir/ritonavir (e.g., Paxlovid). Other approved medications, such as hydroxychloroquine and ivermectin, gained considerable attention based on initial in vitro and clinical evidence. However, these were rendered ineffective in subsequent clinical trials3,4.
In the pursuit of effective drug development strategies against SARS-CoV-2, researchers are exploring a diverse range of approaches, including investigating plant-derived compounds that have a rich history of use in traditional medicine for combating viral infections5,6,7,8,9,10. Phytochemicals, chemicals synthesized by plants demonstrate vast scaffold diversity and structural complexity, which enable these molecules to interact with high specificity to a wide range of biological macromolecules. Unique chemical features in phytochemicals can provide distinct advantages for their effectiveness as interventions. For instance, high rigidity due to a large number of chiral centers, often enable phytochemicals to disrupt protein-protein interactions11. However, identifying the bioactive component within plant extracts is a cumbersome process. Usually, plant-derived natural product screens involve a library of botanical extracts containing complex mixtures of molecules; this complexity is often unsuitable for traditional target-based efficacy studies12. Recognizing this challenge, application of cheminformatics has emerged as a highly promising strategy. Computational techniques offer a powerful toolset for identifying potential molecular targets, synergistic effects, partial agonist/antagonist activities, off-target interactions, and the intricate mechanisms13 of action associated with phytochemicals in extracts. Thereby, adding a layer of convenience in facilitating experimental validation and potentially streamlining the process of discovering novel therapeutics.
The application of cheminformatics to pinpoint active phytochemicals within complex botanical blends hold immense potential for advancing nutritional product development. This newfound insight could guide designing extraction strategies to ensure the presence and abundance of key bioactive markers within the final extracts. Large phytochemical databases, combined with cutting-edge computational methods, offer a robust toolkit for achieving this, ultimately fine-tuning the production of nutritionally efficacious products.
The protein MPro is a homodimer of two protomers, with each protomer comprising domain I (residues 8-101), domain II (residues 102–184), and domain III (residues 201–303)17,18,19,20,21 (Fig. 1). A long loop (residues 185–200) connects domains I and II with III. Domains I and II form an active site consisting of S1’, S1, S2, and S4 subsites22. The catalytic residues His41 and Cys145 form a portion of S2 and S1 pockets, respectively. His41 forms a crucial hydrogen bond with a water molecule, facilitating interactions with the side chains of Asp187 and His16423. Asp187 is further stabilized through a salt-bridge to Arg4023. This intricate arrangement allows His41 to function as a base, extracting a proton from the catalytic Cys145 side chain and activating it for a nucleophilic attack and cleavage of the overlapping polyproteins pp1a and pp1ab into 16 non-structural proteins. This cleavage releases the functional polypeptide for viral replication and transcription15,16.

Structure of SARS-CoV-2 MPro. [Left] Cartoon representation of one protomer showing three domains: Domain I (blue, residues 8-101), Domain II (yellow, residues 102–184), and Domain III (green, residues 201–303). The catalytic residues His41 and Cys145 are shown as sticks. [Right] Surface representation of the substrate-binding pocket with key subsites (S1, S1’, S2, and S4) that serve as binding locations for inhibitors.
The pursuit of MPro inhibitors, whether synthetic or plant-derived, as potential antiviral agents against the virus, has been the focus of extensive research. One of the primary challenges in targeting MPro stems from the inherent plasticity of its active site. Molecular dynamics (MD) simulations of SARS-CoV-2 and SARS-CoV MPro proteins have identified differences in shape and size24,25, of the binding site, despite the active site residues being identical. This conformational flexibility arises largely from 12 distant residues that differ between SARS-CoV-2 MPro and SARS-CoV MPro26; SARS-CoV-2 MPro exhibit increased structural flexibility and plasticity27. These remote residues play a pivotal role in the dynamic plasticity of the binding site, exerting substantial influence on inhibitor binding affinity and specificity. Thus, delineating the conformational changes of the binding site is important for selecting suitable target structures for virtual screening. Therefore, prior to virtual screening, we sampled ensembles of binding site pockets produced from MD simulations and compared pocket volumes and shapes. These comparisons identified novel, pharmacologically relevant binding-pocket conformations (that may not be readily apparent in experimental structures) for virtual screening.
Considerable efforts have been directed towards the development of inhibitors targeting the SARS-CoV-2 MPro, with a particular focus on substrate-based inhibitors. These inhibitors form covalent bonds with the nucleophilic thiolate of the catalytically active Cys145 residue of protein28a strategy commonly employed in inhibiting various viral and non-viral proteases. Despite their efficacy in blocking the protein’s binding site, covalent inhibitors raise safety concerns due to potential off-target effects and prolonged effects29,30. Interestingly, certain noncovalent inhibitors, such as 23R, have shown high selectivity compared to their covalent counterparts like GC376, making them promising candidates31. In our study, we employed structure-based virtual screening of phytochemical libraries against diverse binding site pockets of MPro to identify potential noncovalent phytochemical inhibitors. Specifically, we utilized the CMAUP14 database, which contains approximately 48,000 small-molecule phytochemicals, representing the largest library of plant-derived molecules. During our in-silico screening process, we prioritized well-characterized phytochemicals to ensure precise molecular characterization and enhance the translational potential of our findings.
Results
Stability analysis of MPro protein during MD simulation
To evaluate the structural stability and dynamic behavior of the apo form of MPro during the 300 ns molecular dynamics (MD) simulation, we analyzed the root-mean-square deviation (RMSD) of the protein at both the global and domain-specific levels. As shown in Fig. 2A, the global RMSD of the Cα atoms, calculated relative to both the initial simulation frame and the crystallographic structure, exhibited a three-phase trend over the 300 ns simulation. During the first 100 ns, the RMSD gradually increased, reaching a local maximum of approximately 0.25–0.28 nm. A sharp decrease in RMSD was observed between 100 and 120 ns, with values dropping as low as ~ 0.11–0.12 nm for the first frame reference and ~ 0.13–0.14 nm for the crystal structure reference. Following this transition, the RMSD gradually increased and stabilized, fluctuating within a range of approximately 0.17–0.23 nm for the remainder of the trajectory. The close overlap between the two RMSD profiles indicates that the overall structure of MPro was maintained after the initial relaxation phase.


Structural stability and flexibility analysis of SARS-CoV-2 MPro during 300 ns MD simulation. (A) Global RMSD of Cα atoms compared to initial frame and crystal structure, showing distinct conformational phases. (B) Domain-specific RMSD revealing differential flexibility: Domain II remains stable, Domain I shows moderate flexibility, Domain III exhibits pronounced conformational changes, and pocket residues display temporal correlation with Domain III movements. (C) RMSF analysis comparing backbone and sidechain atom flexibility.
To delve deeper into the protein’s stability profile, we investigated the RMSD of Cα atoms of each domain (Fig. 2B). This domain-specific analysis provides a detailed view of the differential stability across the protein’s structural regions. Domain II exhibits stability throughout the entire trajectory, maintaining low RMSD values typically below 0.13 nm while Domain I shows intermediate flexibility, with RMSD values generally ranging between ~ 0.10–0.18 nm. In contrast, Domain III demonstrates pronounced conformational flexibility, particularly during two distinct periods (75–100 ns and beyond 225 ns), where RMSD values exceed 0.30 nm. These heightened fluctuations in Domain III represent significant conformational transitions that may be relevant to the protein’s function.
The pocket residues exhibit a distinct RMSD profile that generally parallels the motion of Domain I—where several of these residues are located—with subtle deviations observed during specific intervals, particularly within the first 50 ns and around 250 ns. This suggests that local conformational changes influence the binding site independently of broader domain motions. Further analysis of conformational dynamics reveals a nuanced relationship between Domain III and the protein’s binding pocket. Domain III shows peak flexibility between 75-100ns, coinciding with the onset of pocket residue stabilization that persists until ~ 220ns. After 220ns, both regions initially show increased fluctuations, but pocket residues stabilize around 260-270ns while Domain III continues to fluctuate. These observations reveal a complex allosteric relationship where Domain III appears to trigger binding pocket stabilization, though additional factors likely regulate pocket dynamics independently of Domain III’s state.
To gain insights into the flexibility of the residues during simulations, the RMSF (Root Mean Square Fluctuations) analyses of the backbone and sidechain atoms of MPro protein was subsequently performed. As shown in Fig. 2C, a significant difference in RMSF values was observed between the backbone and sidechain atoms. The sidechain atoms showed fluctuations approximately between 0.10 and 0.40 nm, while the backbone atoms displayed more limited fluctuations in the range of 0.05 to 0.20 nm.
Domain III showed the greatest side chain flexibility across its structure, supporting our RMSD findings that indicated substantial conformational changes in this domain during the simulation. Domain I also exhibited considerable side chain mobility, especially between residues 40–70 – a region containing the first catalytic residue, His41, as well as several residues contributing to the S2 and S4 binding pockets. Among these, the highest fluctuations were observed for Thr24-25, Glu47, and Tyr54. In contrast, Domain II displayed moderate side chain flexibility, with several distinct peaks observed along its sequence, including notable fluctuations around residues 140–160—a region that contains the second catalytic residue, Cys145, as well as residues forming the S1 and S2 pockets. The RMSF plot revealed that side chain atoms of most pocket-forming residues in Domain II showed relatively low flexibility, except for Asn142. This limited flexibility is likely due to the formation of β sheets, as previously reported35. The loop region between Domains II and III showed variable flexibility, with prominent peaks corresponding to Arg188, Gln189, Thr190 and Ala191. RMSF values for these residues ranged from 0.20 nm to 0.40 nm. Notably, Asn142, Arg188, and Gln189 have previously been reported32,33,34 to play a crucial role in adapting to different ligand structures and sizes, facilitating the recognition and binding of various inhibitors. Interestingly, Tyr154 located at the distal side of the binding site showed the highest fluctuations with an RMSF value of 0.35 nm. This underscores the pivotal role of specific flexible residues in dictating the flexibility of MPro needed for ligand binding.
Collectively, our stability analyses show that MPro is a dynamic system with domain-specific mobility, suggesting potential allosteric communication between Domain III and the binding pocket. The observed flexibility across domains and key pocket residues indicates that MPro relies on conformational plasticity rather than rigidity to perform its proteolytic function and bind diverse ligands.
Principal component analysis and free energy landscape
To elucidate the essential dynamics governing the function of MPro, we performed Principal Component Analysis (PCA) on the 300 ns molecular dynamics trajectory. PCA helps us identify the most important movements within the protein structure that contribute to its functional states. Figure 3A displays the time-colored trajectory illustrating progressive transitions through different regions of conformational space. The distribution reveals multiple populated regions in the PC1-PC2 subspace, suggesting the protein samples a diverse ensemble of structures that may represent functionally relevant states. The first two principal components captured a substantial portion (38.6%) of the overall variance (Supplementary Fig. S1A), with PC1 accounting for 21.0% and PC2 contributing 17.6% of the total motion.

Conformational analysis of SARS-CoV-2 MPro. (A) PCA projection onto the first two principal components (PC1 and PC2) showing major protein motions during the 300 ns simulation, with points colored by simulation time progression (blue→purple). (B) [Left] FEL revealing three major conformational states (Basins 1–3), where deeper blue regions represent energetically favorable conformations. [Right] Structural overlay of representative conformations from each basin (Basin 1: steel blue, Basin 2: plum, Basin 3: golden), highlighting regions with significant conformational differences.
Free Energy Landscape (FEL) analysis, projected onto PC1 and PC2, was used to quantify the free energy barriers between conformational basins and to provide a clearer picture of the system’s functional dynamics. In general, the FEL shows us how much energy it takes for MPro to transition between different shapes or states. Our analysis reveals a rugged energy surface with three well-defined basins corresponding to distinct conformational states (Fig. 3B, left). The global minimum (Basin 1) represents the dominant ensemble with subtle substates, while Basin 2 (intermediate transition state-like) and Basin 3 (higher-energy metastable) underscore the conformational plasticity of the system. The estimated energy barriers between Basin 1 and Basin 2 (~ 3–6 kcal/mol) were relatively low, while transitions between Basin 3 and the other basins encounter moderately higher barriers (~ 6–12 kcal/mol), suggesting accessible conformational exchanges between the metastable states, facilitating functional adaptability. The bifurcated nature of the global minimum highlights a finely tuned energy landscape, allowing MPro to maintain stability while permitting subtle structural variation.
To connect the energetic insights with structural features and our previous findings on domain-specific dynamics, we examined representative structures from each energy basin. The conformations shown in Fig. 3B (right panel) highlight that transitions primarily involve rearrangements in loop regions that form or surround the binding pockets. Such shifts may be significant, as they could influence how the protein engages with potential inhibitors. Importantly, residues 46–50 in the lowest-energy basin (Basin 1, shown in steel blue) displayed a short helical segment emerging within a previously disordered loop, indicating stabilization of secondary structure in this favorable energetic state. Structural variations near the catalytic regions are consistent with our earlier observations of domain-specific mobility and point to potential effects on ligand accessibility and binding site flexibility.
Residue contribution analysis (Supplementary Fig. S1B) identified key regions driving the observed conformational transitions. Along PC1, most residues exhibited moderate displacements (~ 1.5 Å), while PC2 revealed more substantial shifts (~ 2.5 Å). The largest movements in both components were seen in residues within or near the binding pocket, notably Asn53–Leu57, Gly138–Ser139, Tyr154. Additionally, several residues in Domain III showed high mobility along PC2, reinforcing their strategic location and potential role in substrate recognition and binding site plasticity, which is consistent with our earlier observations of allosteric communication between Domain III and the active site.
Pocket dynamics analysis
The significant conformational heterogeneity revealed by PCA and FEL analyses, particularly in residues shaping the binding pocket, emphasized the need to consider structural diversity when selecting conformations for docking-based virtual screening. To further investigate how binding site geometry varies across these states, we performed a pocket dynamics analysis using volume-based clustering. POVME36,37 was used to compute the binding site volumes from 60 frames, sampled every 5 ns during the 300 ns MPro simulation. These frames were sampled at 5 ns intervals during the 300 ns simulation. Active site pocket volumes ranged from 190 to 498 Å3with surface areas varying between 188 and 380 Å2 during the MD simulations. The active site volumes in our study were slightly greater than previously reported38,39 likely due to the expansion of subsite pockets and the formation of new pockets within the active site.
The POVME clustering workflow classified the binding sites within the sampled frames into five clusters, each representing frequently observed pocket shapes. The representative structure from each cluster is shown in Fig. 4. Differences between the binding site shape in each cluster originate from the opening or closing of regions that constitute the active site. For Clusters 1 through 4, a gradual widening of the S1 pocket is observed; however, this widening coincides with a concurrent reduction in the pocket’s depth, resulting in a shallower S1 pocket configuration (shown in Fig. 4A-D). In Cluster 5, the shape and size of the S1 subsite is decreased significantly compared to the other clusters (shown in Fig. 4E). The S2 subsite, on the other hand, maintained its small size in all conformations, with a portion of it being deeply embedded and thus inaccessible for ligand binding. New subsites, termed the S2’ subsite, emerged adjacent to the S1’ subsite in structures corresponding to Clusters 1, 3, 4, and 5 (shown in Fig. 4A, C-E). The S1’ pocket showed the least divergence when compared to the average pocket geometry, retaining a consistent shape in Clusters 1–3 but appearing markedly smaller in Clusters 4 and 5. Among all subsites, S4 exhibited the most pronounced dissimilarity in shape across all five clusters, highlighting its structural plasticity.

Binding pocket conformations of SARS-CoV-2 MPro identified through volume-based clustering. (A-E) Surface representations of representative structures from five distinct clusters (Clusters 1–5), showing the variable geometry of the binding site. The catalytic residues His41 (pink) and Cys145 (green) are highlighted, with binding subsites (S1, S1’, S2, S2’, S4) labeled in red.
Molecular docking
The CMAUP database14 was screened to identify phytochemicals capable of binding to each of the five active site conformations of MPro. We identified nearly 500 phytochemicals with docking scores above − 7.5 kcal/mol across five protein conformations. This criteria for the docking score cutoff was based on the docking scores observed for reported small-molecule MPro inhibitors40,41,42. Among the conformations, Cluster 3 emerged as the most favorable binding site, with approximately 200 compounds scoring above the set threshold. Whereas Cluster 5 was the least favorable with, only 25 compounds meeting the criterion.
Table 1 lists the 20 phytochemicals with high docking scores across all five conformations. Additional details of the phytochemicals, including structure, PubChem ID, and the plant sources are listed in Supplementary Table 1. From these phytochemicals, we identified four aglycones and sixteen glycosides. Aglycones are of particular interest as they typically harbor the pharmacological or biological moiety, whereas the glycosidic bonds primarily affect properties like solubility, stability, and bioavailability.
The four identified aglycones were shimobashiric acid C (SAC), salvianolic acid L (SAL), AHDPH, and shikonin. Figure 5 shows their binding modes in their most favorable protein conformations. SAC, SAL, and AHDPH bound most strongly to Cluster 3 conformation (scores of -10.5, -13.3, and − 11.6 kcal/mol, respectively), while shikonin preferred Cluster 5 (score − 9.5 kcal/mol).

Binding modes of top-scoring non-glycoside phytochemicals with SARS-CoV-2 MPro. Left panels (A, C,E, G) show 3D surface representations of the binding pocket with compounds in stick format; right panels (B, D,F, H) display 2D interaction diagrams. Binding subsites (S1, S1’, S2, S2’, S4) are labeled in red. Compounds shown: (A-B) SAC binding to Cluster 3 conformation, (C-D) SAL binding to Cluster 3 conformation, (E-F) AHDPH binding to Cluster 3 conformation, and (G-H) Shikonin binding to Cluster 5 conformation.
For SAC, a complex molecule with a cyclobutane core and multiple hydroxyphenyl groups, one hydroxyphenyl group fits in the S2 while the others interact with the peripheral residues of the S1, S4, and newly formed S2’ pocket (Fig. 5A-B). The key interactions of SAC include a pi-pi interaction with the catalytic residue, His41 and hydrogen bonds with several residues including Cys44, Glu166, Asp187, Gly143, Ser144, Cys145, and Gln189.
SAL consists of a naphthalene core with multiple 3,4-dihydroxyphenyl groups, and a 3-(3,4-dihydroxyphenyl) propanoic acid moiety connected via a carboxyethyl linker. As depicted in Fig. 5C-D the dihydroxyphenyl groups occupy the S1’, S1, and S4 pockets, while the naphthalene ring interacts with S2’ pocket. The compound forms six hydrogen bonds with protein residues, including Cys44, Gly143, Cys145, His164, Glu166, Arg188, and Gln192. In the case of AHDPH, S1 and S2 pockets remain unoccupied, while S1’ and S4 accommodate its dihydroxyphenyl groups. Key interactions include hydrogen bonds with four residues, Thr26, Arg188, Gln192 and Glu 166 (Fig. 5E-F).
For shikonin, the naphthalene ring fits the narrow S4 pocket, with its hydroxy groups interacting with charged pocket residues—Glu166, Asp187, Arg188, and Gln189, while its pentyl chain interacts with the hydrophobic residues forming the S2 pocket (Fig. 5G-H).
Considering the metabolic cleavage of glycosides to aglycones in the body, we also investigated their corresponding metabolites (Table 2). These metabolites (aglycones) exhibited moderate binding with most preferring specific comformations, except for quercetin that showed good binding to all conformations with a docking score >= 7.0 kcal/mol. Several aglycones, including dihydrocaffeic acid, brevifolincarboxylic acid, DDHH, pinoresinol, kaempferol, luteolin, isoharmnetin, pectolinarigenin, and secoisolariciresinol preferentially bound to at least two conformations with a docking score > = 7.0 kcal/mol. Hesperetin and eriodictyol bounded favorably with the first conformation, while hydroxytyrosol preferred the third conformation. Gallic acid, and caffeic acid exhibited a low docking score across all conformations compared to its glycoside. Some of these aglycones have been previously reported for their anti-SARS-CoV-2 activity43,44,45,46,47.
In addition to examining the top 20 phytochemicals and their respective metabolites, we extended our analysis to include four phytochemicals that exhibited high docking scores for at least one protein conformation. These included cynarin, demethoxycurcumin, hexahydrocurcumin, and withaferin A (Table 3).
Cynarin scored well with Cluster 1 and 3, where its docking scores exceeded − 9.0 kcal/mol. As shown in Fig. 6A-B, the dihydrocyclohexane core of the molecule occupies the S1 pocket, the carboxylic acid group interacts with S1’ site, and one of the two hydroxyphenyl groups occupies the S4 pocket. The other hydroxyphenyl group, although not positioned within the S1’ pocket, interacts with the residues forming the pocket. The key interactions include Thr26, Asn142, Gly143, Glu166, and Thr190.

Binding interactions of SARS-CoV-2 MPro and phytochemicals with high docking scores in at least one protein conformation. Left panels (A,C,E,G) show 3D surface representations of the binding pocket with compounds in stick format; right panels (B, D,F, H) display 2D interaction diagrams. Binding subsites (S1, S1’, S2, S4) are labeled in red. Compounds shown: (A-B) Cynarin binding to Cluster 1conformation, (C-D) Demethoxycurcumin binding to Cluster 2 conformation, (E-F) Hexahydrocurcumin binding to Cluster 3 conformation, and (G-H) Withaferin A binding to Cluster 5 conformation.
Demethoxycurcumin exhibits a docking score of approximately − 8.0 kcal/mol when binding to the representative protein conformation of Cluster 2, the highest among other protein conformations. As shown in Fig. 6C-D, the hydroxyphenyl group occupies the region between the S2 and S1’ pockets, with the S2 pocket fits the hepta-1,6-diene-3,5-dione bridge. The hydroxy-3-methoxy group of the phytochemical effectively occupies the S1 pocket. This binding mode forms hydrogen bonds with Thr26, Leu141, Ser144, and Gly143.
Hexahydrocurcumin had its highest binding score (~ 10.0 kcal/mol) when interacting with the representative conformation of Cluster 3. As shown in Fig. 6E-F, one hydroxymethoxyphenyl groups of the phytochemical occupies the S2-S4 pocket, interacting with Glu166, Asp187, and Thr190. The second hydroxymethoxyphenyl group fits into the S1 pocket, interacting with Gly143 and Cys145. Interestingly, despite the structural similarity between the two curcumin derivatives, their predicted affinities toward specific protein conformations and their adopted binding orientations are distinct.
Withaferin A preferred the representative structure of Cluster 5 conformation, binding with a docking score of -8.4 kcal/mol. The dihydropyran ring and the hydroxymethyl substituent of the molecule occupy the S4 pocket, while the main cyclic structure of withaferin A (oxapentacyclooctadec-4-en-3-one) interacts with the S2 and S1’ sites of the binding pocket. Crucial interactions included residuesThr26, Asn119, and Glu166. (Fig. 6G-H).
MM-GBSA prediction
To improve the accuracy of our inhibitor binding predictions, the initially identified 39 phytochemicals were rescored based on binding energies calculated with Prime/MM-GBSA. For these binding energy calculations, the protein-ligand conformation with the highest docking score was selected. Table 4 summarizes the MM-GBSA energies for the selected 20 phytochemicals (Table 1), their corresponding aglycones (Table 2), and phytochemicals exhibiting strong docking scores for at least one protein conformation (Table 3), a total of 39 phytochemicals. The MM-GBSA energy calculations revealed that the binding mode of all the phytochemicals was primarily driven by negative ΔG values for ΔGcoulomb, ΔGhbond, ΔGlipo, ΔGpacking, and ΔGvdw. These scores indicate the presence of attractive Coulombic interactions, hydrogen bonding, and strong hydrophobic interactions, including lipophilic and van der Waals forces. However, the positive ΔGcovalent and ΔGsolv_GB scores suggest that covalent bond formation and binding of the phytochemicals in an aqueous environment are energetically unfavorable.
Among the selected phytochemicals, Forsythiaside had the best calculated binding energy, ΔGbind = -82.2 kcal/mol. Luteolin 7-rutinoside ranked second with a ΔGbind value of -80.2 kcal/mol, followed by quercetin 3-glucoside 2”-gallate and hyperin 6’-[glucosyl-(1 > 3)-rhamnoside] with a ΔGbind value of approximately − 76.0 kcal/mol. All of these molecules also showcased high docking scores above − 12.0. kcal/mol.
Pinoresinol, one of the metabolites of forsythiaside exhibited the highest binding energy of ~ -59.0 kcal/mol among all the aglycones studied. It also showcased a significant docking score of -8.7 kcal/mol. Secoisolariciresinol was the second-best aglycone with a binding energy of -53.8 kcal/mol, followed by kaempferol. Overall, the binding energy calculations aligned well with the computed docking scores of top 20 phytochemicals (Table 1), except for SAC and kaempferol 3-(3R-glucosylrutinoside), which deviated with relatively low binding energies (ΔGbind < -40 kcal/mol), despite securing high docking scores across all protein conformations. Cynarin, while displaying a significantly high docking score for the first protein conformation, exhibited a low binding energy value (ΔGbind = -36.1 kcal/mol). Additionally, the aglycones luteolin and quercetin demonstrated lower binding energies (ΔGbind = -39.4 and − 36.4 kcal/mol, respectively), although they showcased moderate docking scores in at least one protein conformation.
We studied the protein-ligand interaction analysis of the top ten phytochemicals based on docking scores and MM-GBSA energies. The heatmap illustrated in Fig. 7 highlights the significance of specific binding site residues, including His41, Asn142, Gly143, Cys145, Met165, Glu166, Arg188, and Glu189, in stabilizing the phytochemicals within the binding site. These residues formed van der Waals and Coulombic interactions with at least five out of the ten phytochemicals, emphasizing their crucial role in ligand binding.

Interaction energy heatmaps between MPro residues and top ten phytochemicals. (A) Van der Waals interaction energies (green scale) and (B) Coulombic interaction energies (red scale). Values shown are in kcal/mol, with more negative values (darker colors) indicating stronger interactions. Several residues, including His41, Gly143, and Glu166, form strong interactions with multiple phytochemicals.
Bioavailability prediction
To gain insights into the bioavailability of the selected phytochemicals, we conducted an in silico ADMET study using ADMET Predictor by Simulations Plus48. This calculation generates a ADMET Risk score that reflects potential compound liabilities based on 20 rules within three Risk models (Absn_Risk, CYP_Risk, and Tox_Risk) of their ADMET Predictor. Additionally, the ADMET Risk score models pharmacokinetic properties termed fraction unbound (fu) and high-steady-state volume of distribution (Vd). Each Risk score is paired with a mnemonic Code list that identifies the rules that have been violated. Of the 39 phytochemicals predicted to bind strongly to MPro, ten had ADMET Risk scores of < = 1 (Table 5). On the other hand, six phytochemicals, including 1,3,6-Tri-O-galloyl-beta-D-glucose, 2-acetylacteoside, quercetin 3-glucoside2’’ -gallate, balanophotannin E, SAL, and SAC had problematic ADEMT Risk scores exceeding 6.
We calculated absorption and bioavailability characteristics of these phytochemicals using three human clearance models (liver microsomes, hepatocytes, and systemic) at a dose of 100 mg for an immediate release tablet and observed significant variability in the calculated pharmacokinetics parameters (Table 5). Out of 39 phytochemicals examined, 20 exhibited high absorption (%Fa) and minimal metabolism or elimination in the liver during the first-pass effect (%Fb). A slight decrease in %Fb values were observed in the hepatocytes for some of these phytochemicals, suggesting that a portion of the compound is subject to metabolism.
In addition to absorption and bioavailability estimations, we calculated phytochemical plasma half-life (T-half) in humans and cell permeability (Madin-Darby canine kidney; MDCK), the latter serving as an estimate for intestinal epithelial absorption (Table 5). Demethoxycurcumin, shikonin, SAC, and withaferin, demonstrated an extended plasma half-life, surpassing 7 h. Phytochemicals with low ADMET risks also exhibited high MDCK permeability, except for secoisolariciresinol and DDHH.
Cytotoxicity and viral replication assay
We performed cell-based antiviral assay to evaluate the inhibitory effects of five highly performing phytochemicals (demethoxycurcumin, hydroxytyrosol, kaempferol, shikonin, and withaferin A) on SARS-CoV-2 replication. These compounds were selected based on their high overall performance in terms of docking score (above − 7.5 kcal/mol for at least one protein conformation), MM-GBSA binding energy (ΔGbind > -50 kcal/mol), and ADMET properties. Glycosides were excluded from the viral replication assay as their metabolic modification in the body would render the results less relevant. Although pinoresinol and hexahydrocurcumin fulfilled the criteria for top-performing molecules, meeting the specified factors, we opted not to include them in the study due to their current unavailability for immediate testing.
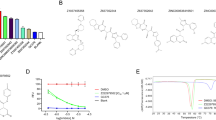
Viral assay demonstrated the SARS-CoV-2 replication inhibitory activity of three compounds, demethoxycurcumin (EC50 = ~ 8.8 µM), shikonin (EC50 = ~ 10 µM), and withaferin (EC50 = ~ 2.8 µM) (Fig. 8). In contrast to shikonin and withaferin, demethoxycurcumin showed no apparent cytotoxicity. These phytochemicals exhibit desirable physicochemical properties essential for drug development.

Antiviral activity and cytotoxicity of phytochemicals against SARS-CoV-2. (A) Dose-response curve for Remdesivir (control), showing viral inhibition (blue) and cell viability (red), EC50 = 3 µM. (B) Evaluation of Shikonin (EC50 = 10 µM), Demethoxycurcumin (EC50 = 8.8 µM), and Withaferin A (EC50 = 2.8 µM). FA denotes the fraction of cells affected; GRI represents the predicted compound response at an infinite concentration.
All of the three compounds adhere to Lipinski’s rule of five, a guideline for orally bioavailable drug development. Specifically, their molecular weights are under 500 g/mol, their Log P values are below 4, they possess five or fewer hydrogen bond acceptors with three or less hydrogen bond donors. These compounds are soluble in organic solvents. In DMSO, the solubility of demethoxycurcumin, shikonin, and withaferin A is 10.0, 11.0, and 5.0 mg/ml, respectively49,50,51. Shikonin and withaferin A are sparingly soluble in aqueous buffer. Additionally, the stability of these compounds exceeds 2 years under appropriate storage conditions, specifically when stored at -20 °C49,50,51. Taken together, the bioactivity and favorable chemical properties of these compounds make them promising candidates for further drug development initiatives.
The inhibitory effects of these three compounds against MPro were previously speculated52,53,54,55,56,57,58,59however, the referenced studies primarily relied on computational or enzyme-based assays. Our study presents a robust cell-based antiviral data, providing a more direct and clinically relevant perspective on the inhibitory potential of these compounds against SARS-CoV-2 replication.
While our findings contribute to the ongoing discussion on effective viral inhibition potential of demethoxycurcumin, shikonin, and withaferin A, we also recognize the necessity for further research to elucidate the intricate mechanisms underlying this inhibition. Our work adds valuable insights to this evolving dialogue, underscoring the importance of continued investigation in this area.
Discussion
Plant-derived natural product reservoir harbors a distinctive wealth of ‘bioactive’ molecules, spanning broader chemical diversity than synthetic libraries60. Interestingly, phytochemicals are structurally optimized by evolution, to serve specific biological functions. Among these functions is the regulation of endogenous defense mechanisms against microorganisms such as viruses and bacteria61. Additionally, the utilization of phytochemicals in traditional medicine could offer valuable insights into their effectiveness and safety. These characteristics elevates the importance of phytochemicals for antiviral and antibacterial interventions, revealing fresh avenues for exploration. Our study aimed to identify phytochemicals capable of inhibiting SARS-CoV-2 MPro protein, using a phytochemical database comprising ~ 48,000 compounds. In the current literature landscape of studies on virtual screening for SARS-CoV-2 phytochemical inhibitors, researchers predominantly rely on relatively small molecule libraries, typically comprising hundreds or thousands of compounds62,63or concentrate on a limited number of medicinal plants64,65,66 that are traditionally known for its antiviral activity. Therefore, the utilization of significantly large database widens the scope of our investigation and markedly enhances the probability of identifying novel MPro inhibitors.
MPro is a challenging target due to its inherent plasticity mediated by allosteric residues. The high flexibility of MPro alters the characteristics of its binding pocket subsites, leading to diverse packing modes for inhibitors. This variation is evident from over 600 experimental structures of MPro deposited in the protein data bank67. Our PCA and FEL investigations provided quantitative evidence of this conformational plasticity, revealing that MPro explores a diverse ensemble of structural states during simulation. Structural comparison of representative conformations from different energy basins showed that variations predominantly occur within loop regions proximal to the binding pocket. Importantly, the catalytically important residues in these regions exhibited significant spatial displacements, directly influencing pocket geometry and ligand-binding capacity. To investigate the ensemble of binding site conformations adopted by the protein we integrated MD simulations with pocket shape-based clustering approach for conducting virtual screening. Since performing docking on MD simulations generated hundreds of thousands of protein conformations is computationally demanding, employing pocket-based filtering allows the selection of representative protein structures that capture the full range of conformational diversity observed in the binding site.
While numerous in silico studies on MPro inhibitors have been conducted, our study stands out as one of the few that not only investigated the ensemble of binding site conformations but also conducted in-depth analysis of the changes in the binding site geometry. This approach yielded novel insights into the restructuring of binding site pockets during MD simulations. For example, we observed a dual pattern in the binding site pockets: the S2 and S4 pockets exhibited shifts, transitioning from two distinct pockets to a single, larger pocket, while simultaneously, the area and volume of the S4 pocket increased. Additionally, we noted the opening of the S2 pocket and the formation of new accessible subsites. Some of the structural features of the binding sites observed in our analysis have been captured in experimental structures of M[Pro68–70. Our study also corroborated two crucial features: the conformational changes in the S2 pocket and the formation of distinct subsites other than S1, S1’, S2, and S4, which have been previously studied71,72. This underscores the importance of considering binding site dynamics in drug design approaches, as these conformational changes can significantly impact inhibitor binding and efficacy.
While our analysis provided valuable insights, a potential avenue for further enhancement would be to extend the length of the MD simulation and expand the number of protein conformations used for pocket-based clustering. Enriching the dataset could yield more comprehensive understanding of the intricate dynamics governing the MPro binding site, thereby facilitating an improved selection of potential drug candidates.
In line with the variations observed during pocket-based clustering analysis, virtual screening against the representative structures yielded a striking divergence in the landscape of high-performing phytochemicals; around 1% of the phytochemicals in the dataset showed potential binding across all examined MPro pocket conformers. A majority of molecules with high docking scores belonged to the class of flavonoid glycosides. This observation was consistent with previous reports73 on insilico studies of natural products, although the structural basis for their potency compared to other phytochemical classes remain unclear. In this study, we assessed 20 phytochemicals with high docking scores across all protein conformations and four phytochemicals that displayed high docking scores for at least one conformation. Some of the phytochemicals identified in our study, such as luteolin, quercetin, pectolinarin, and pinoresinol have been extensively studied for its MPro inhibitory bioactivity through structure-based studies74,75,76,77. Whereas phytochemicals previously reported to exhibit high docking scores for MPro, such as myricetin78epicatechin gallate79glycyrrhizic acid80chlorogenic acid81and naringenin82did not perform well in our studies. This discrepancy could be attributed to the database utilized in our study, which included a significantly larger number of molecules. It’s possible that our expanded database contains molecules better suited to efficiently bind MPro, leading to difference. Interestingly, a prior in silico analysis83 that utilized the same database to screen for MPro inhibitors also did not identify these phytochemicals. However, there was a difference in high-performing molecules. While our investigation predominantly identified flavonoids and their subgroups as high-performing molecules, the previous study predominantly highlighted terpenoids and their derivatives as MPro inhibitors. This variation may stem from our consideration of five different conformations of the protein, capturing the full range of conformational diversity observed in the binding site.
Given the likelihood of metabolic transformations of glycosides within the body, we examined the binding capabilities of the aglycones of these glycosides. This study showed a consistent superiority of glycosides over their aglycone counterparts in terms of docking scores, signaling a pivotal role for glycosylation in increasing the score of binding calculations. Moreover, this observation accentuates the importance of investigating phytochemical metabolites to ensure efficacy, especially in the case where parent compounds that are prone to biotransformation. By examining metabolite-protein interactions, the likelihood of overlooking potential therapeutic benefits originating from these modified forms can be minimized, concurrently highlighting potential reductions in bioactivity among the resultant metabolites.
Among all the phytochemicals identified in the virtual screening process, seven are reported here for the first time for their MPro binding tendency. These include hyperin 6’-[glucosyl-(1 > 3)-rhamnoside], balanophotannin E, plantagineoside C, SAC, AHDPH, brevifolincarboxylic acid, and secoisolariciresinol. However, the ADME profiles and bioavailability of these phytochemicals generally fall within the moderate to poor range, except for AHDPH, brevifolincarboxylic acid and secoisolariciresinol, which exhibit more favorable attributes. For forthcoming investigations, it would be interesting to explore the impact of these molecules on MPro inhibition and SARS-CoV-2 replication. This exploration could uncover their significance in combating the virus and could potentially yield novel insights for future therapeutic approaches. Consistent with previous computational and enzyme-based studies52,53,54,55,56,57,58,59three molecules—demethoxycurcumin, shikonin, and withaferin A—demonstrated high MPro inhibition activity in our docking analysis and were further experimentally validated for their inhibitory bioactivity against SARS-CoV-2 replication. By focusing on pure, well-characterized phytochemicals, we addressed the critical challenge of identifying bioactive components within complex plant extracts, demonstrating a more precise approach to natural product-based drug discovery.
While our computational framework has yielded valuable insights into potential MPro inhibitors, we acknowledge its inherent limitations in fully capturing the biological complexity of physiological systems. In silico models, by design, simplify key processes such as metabolism, tissue distribution, off-target interactions, and immune modulation. For example, predicted binding affinities may be altered by serum protein binding, enzymatic degradation, or membrane transport—factors not fully accounted for in our simulations. Furthermore, the dynamic interplay between inhibitors and the conformational ensemble of MPro within a host cellular context introduces additional layers of complexity.
Despite these limitations, integrating computational predictions with in vitro validation strengthens confidence in our findings. Notably, the best-performing phytochemicals—demethoxycurcumin, shikonin, and withaferin A—have established safety profiles and a history of traditional use, providing a meaningful biological context. Although some cytotoxicity was observed at higher concentrations, these data offer a promising starting point for future optimization. Moving forward, in vivo studies will be critical to confirm efficacy and pharmacokinetics, and to bridge the gap between bench and bedside. Collectively, this integrative approach enhances both the efficiency and translational potential of our natural product-based antiviral discovery pipeline.
Methods
Molecular dynamics simulation
The protein structure of MPro protein was obtained from the Protein Data Bank84 (PDB code: 5R80). Two independent molecular dynamics simulations of the ligand unbound protein were carried out utilizing the GROMACS 5.0.7 software85. The force field used for the protein was CHARMM3686. The models were immersed in a dodecahedron box with dimensions 5.0777 × 5.069 × 3.672 nm, containing TIP3P87 water molecules. Subsequently, two chloride ions, twelve, and four sodium ions were added to model 1, 2, and 3, respectively to achieve neutrality of the system. Periodic boundary conditions and a 2 fs time step were employed for each simulation. Particle Mesh Ewald88 (PME) was employed to treat long-range electrostatic interactions, and a cutoff of 0.9 nm was used for non-bonded interactions. Combination of steepest descent and conjugate gradient methods of energy minimization (EM) were used to remove steric clashes and to minimize the interaction forces. EM was achieved in four steps89. In Step 1 the protein and ligand were restrained, in Step 2 the protein heavy atoms and ligand were restrained, in Step 3 the protein main chain was restrained and in Step 4, unrestrained EM was performed. Unrestrained minimization was carried out until convergence where the maximum atomic force was < 100 kJ/mol–nm. The minimized structures were first gradually heated to 300 K in the NVT ensemble for 20ps using a position restraint of 1000 kJ/mol-nm constant force on the proteins. The systems were equilibrated under NPT (isobaric-isothermal) ensemble by 200ps by gradually decreasing the position restraint force to 700, 400, and then to 100 and finally to 10 kJ/mol–nm and then equilibrated for an additional 2 ns without any restraints. A production run for 300 ns were carried out under the NPT ensemble. The temperature was maintained at 300 K with the Berendsen weak coupling method90. Bond lengths were constrained using the linear constraint solver (LINCS) algorithm91 and the van der Waals cutoff was at 1.4 nm. The trajectories were sampled every 10ps for analysis in production dynamics. Molecular visualization and analysis were performed using VMD92 and UCSF Chimera package93.
Principal component analysis and free energy landscape
PCA was performed using GROMACS85 to characterize the dominant motions of apo MPro during the 300 ns molecular dynamics trajectory. The trajectory was first RMS-fitted to the backbone atoms of the reference structure to remove global translational and rotational motions. A covariance matrix of atomic positional fluctuations was computed using gmx covar, focusing on Cα atoms. The resulting eigenvectors and eigenvalues were analyzed using gmx anaeig, and the first two principal components (PC1 and PC2), which captured the largest variance in atomic displacements, were selected for further analysis.
To explore the conformational landscape, a two-dimensional FEL was generated based on the projection along PC1 and PC2. The gmx sham utility was employed to construct a 2D histogram and compute the relative free energy surface by applying Boltzmann inversion:
where ΔG is the free energy, kB is the Boltzmann constant, T is the temperature, and P is the normalized probability of each bin in the PC1–PC2 space. The FEL enabled identification of distinct conformational basins, and representative structures from local energy minima were extracted for structural comparison and downstream analyses.
Pocket volume analysis
POVME 3 36 software was utilized for characterizing pocket volumes and shapes of ensembles of binding pockets from molecular dynamics simulation.
The volume of the active site is calculated by first defining the binding pocket region. The coordinates of Cβ atom of residue 165 located at the center of the active site was considered as the point of inclusion with 8 Å radius. Subsequently, the user-specified inclusion region encompassing all the binding-pocket conformations of the trajectory is filled with equidistant points spaced at 1.0 Å. The program then removes all the points that are close to the receptor alpha carbon atoms and leaves the points that are positioned within the pockets. Two algorithms, gift wrapping, and Akl-Toussaint heuristic are used to define the convex hull of receptor atoms near the inclusion sphere; any points outside the convex hull are then removed. Lastly, to remove the isolated patches of points lying outside the primary pockets or the pockets not contiguous with the primary pockets are removed. POVME scripts (binding_site_overlap.py and cluster.py) were used to calculate the similarity of the pockets from all protein conformations, to measure the binding site overlap, and to perform hierarchical clustering.
Structure-based virtual screening
Protein and ligand Preparation
The representative protein structures derived post clustering was prepared for docking utilizing the “Protein preparation wizard” tool in Schrödinger suite94. The protocol included, the removal of water molecules and cofactors, rectifying misidentified elements, introducing hydrogen atoms, determining bond orders, and optimizing hydrogen bonds. Hydrogen bond assignment was done using PROPK at pH 7.0, the orientation of hydroxyl groups, side chain amide groups of Asn and Gln, and the charge state of His residues were optimized. The protein structures were minimized to an RMSD limit from the starting structure of 0.3 Å using the OPLS3 force field95. The prepared proteins were subsequently utilized for grid creation using the “Receptor Grid Generation” panel within the Glide module of the Schrödinger suite96. Binding site was defined by selecting key residues located at the subsites.
The library of phytochemicals was prepared using Schrödinger’s Maestro LigPrep tool97which involved applying the OPLS4 force field, optimizing the structures, and incorporating hydrogen atoms. Additionally, Epik, integrated into the process, was utilized to assign probable protonation states within a pH range of 7 ± 2 and determine tautomeric forms for each compound.
Molecular docking
Virtual screening was performed using Glide program96. A ligand-flexible docking of prepared ligands to the binding sites of five protein conformations was performed at two different levels, standard precision (SP) and extra precision (XP) using Virtual Screening Workflow protocol of GLIDE.
MM-GBSA prediction
The examination of the protein and ligand complexes’ free binding energies was carried out by utilizing the MM-GBSA (Molecular Mechanics, the Generalized Born model, and Solvent Accessibility) tool of Schrödinger. To determine the optimal binding energy the Prime module within the Schrödinger software was employed. Solvation model VSGB 2.098 and OPLS-AA force field95 was used for the calculations. Additionally, the protocol incorporates physics-based modifications catering to π-π interactions, hydrophobic interactions, and self-contact interactions involving hydrogen bonding.
ADMET and bioavailability prediction
The SMILES (Simplified Molecular-Input Line-Entry System) strings for phytochemicals were imported into ADMET Predictor v11.048. Physicochemical and pharmacokinetic properties were predicted using quantitative structure-activity relationship (QSAR) models within AP11.0. For a comprehensive understanding of the model specifications, validation, and performance, please refer to the AP11.0 user manual and relevant publications99,100.
Cytoxicity assay
The compounds selected for cytotoxicity and viral replication testing were carefully selected based on its purity and reliability from reputable suppliers. Hydroxytyrosol (purity: 98.78%), shikonin (purity: 99.79%), and demethoxycurcumin (purity: >= 99.0%) were procured from MedChemExpress (https://www.medchemexpress.com).
Kaempferol (purity: 99.7%) and withaferin A (purity: 96.0%) were sourced from ChromaDex (https://www.chromadex.com).
Vero cells were seeded using a multiDrop combi liquid dispenser (Thermo) into 384-well plates at a density of 500 cells/well suspended in 50 µL of media. Cells were allowed to recover and fully attach overnight (approximately 16 h), at which point library compounds were dispensed into wells using an Echo 550 acoustic dispenser (Labcyte). A total of six final concentrations where tested (50 µM, 25 µM, 12.5 µM, 6.25 µM, 3.125 µM, and 1.5625 µM) and wells were back filled with DMSO such that all wells contained a fixed ratio of DMSO. Compounds were incubated with cells for 1 h prior to addition of virus and then for an additional 24 h, then fixed with 10% formalin, permeabilized 0.1% Triton X-100, washed, and stained for SARS-CoV-2 N protein using a specific antibody (Sino Biological # MM05) and fluorescently labelled secondary antibody. Cells were counter stained with Hoechst 33,342 to detect cell nuclei, washed, and imaged with a Cytation 1 (Biotek) automated. Each image was then analyzed with a custom workflow in Cell Profiler (Broad Inst., Boston, MA) which involved counting of cell nuclei and infected cells. At least 4 replicates were used to construct dose response curves.
Statistics and data normalization
The growth rate index is calculated from cell counts using the following formula:
Where Xc is the observed cell count after drug exposure, X0 is the median cell count from a plate that was fixed at the time when the drug was added, and XDMSO is the median cell count of the on-plate DMSO control. The growth rate is then fit against the log10 molar concentration using a cascade of models (4 parameters logistic, 2 parameters logistic, and linear model). The model with the best overall fit, determined by minimizing the mean-squared-error, is then selected. Finally, compound effects are ranked using either potency (GR50, LD50) or the area under/over the curve (AUC_GRI, AOC_GR, and AOC_LD) calculated from the optimized fit.
Viral assay
As for the cytotoxicity assay, Vero cells were seeded into 384-well plates to a density of 70% per well. Cells were allowed to recover and fully attach overnight (approximately 16 h), at which point library compounds were added to cells. A total of ten final concentrations where tested ranging from 10 µM down to 20 nM. Compounds were incubated with cells for 1 h prior to addition of SARS-CoV-2 virus, Washington strain, and then for an additional 36 h. The cells were then fixed with 10% formalin, permeabilized 0.1% Triton X-100, washed, and stained for SARS-CoV-2 N protein using a specific antibody (Sino Biological # MM05) and fluorescently labelled secondary antibody. Cells were counter stained with Hoechst 33,342 to detect cell nuclei, washed, and imaged with a Cytation 1 (Biotek) automated. Each image was then analyzed with a custom workflow in Cell Profiler (Broad Inst., Boston, MA) which involved counting of cell nuclei and infected cells. At least 4 replicates were used to construct dose response curves.
Infected cells/total cell count ratios were calculated for each well. Then, each well was normalized to the average of the 14 negative control wells on each plate. These normalized ratios were input into GraphPad Prism (7.05), and curves were fitted to the data. A dose response curve ([Inhibitor] vs. response - Variable slope (four parameters); constrained to 0) was used. R2 > 0.80 was used as a threshold for a good fit of the dose-response curve. Total cell count was added to the right y-axis as a surrogate for potential cytotoxicity and dose-response curves were also run for total cell count following the same formula and constraint.
In Supplementary Table 2, the results are labeled accordingly: red, did not converge on a line of best fit; yellow, converged on a line of best fit but R2 < 0.80; green, converged on a line of best fit and R2 > 0.80.
Data availability
The data that support the findings of this study are available from the corresponding author, Khushboo Singh upon reasonable request.
References
Wu, F. et al. Author correction: A new coronavirus associated with human respiratory disease in China. Nature 580, E7 (2020).
Gorbalenya, A. E. et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-ncov and naming it SARS-COV-2. Nat. Microbiol. 5, 536–544 (2020).
Galan, L. E. et al. Phase 2 randomized study on chloroquine, hydroxychloroquine or Ivermectin in hospitalized patients with severe manifestations of SARS-COV-2 infection. Pathog Glob Health. 115, 235–242 (2021).
Vallejos, J. et al. Ivermectin to prevent hospitalizations in patients with covid-19 (IVERCOR-covid19) a randomized, double-blind, placebo-controlled trial. BMC Infect. Dis. 21, 635 (2021).
Dhawan, B. N. Anti-viral activity of Indian plants. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 82, 209–224 (2012).
M., H. C. C. et al. Phytochemical, antioxidant and microbial inhibitory effects of spondias mombin leaf and stem bark extracts. IOSR J. Pharm. Biol. Sci. 9, pp14–17 (2014).
Khanna, K. et al. Herbal Immune-boosters: substantial warriors of pandemiccovid-19 battle. Phytomedicine 85, 153361 (2021).
Ling, C. Traditional Chinese medicine is a resource for drug discovery against 2019 novel coronavirus (SARS-COV-2). J. Integr. Med. 18, 8788 (2020).
Remali, J. & Aizat, W. M. A review on plant bioactive compounds and their modes of action against coronavirus infection. Front. Pharmacol. 11, 589044 (2021).
Fei, J. et al. Contribution of traditional Chinese medicine combined with conventional Western medicine treatment for the novel coronavirus disease (COVID-19), current evidence with systematic review and meta‐analysis. Phytother. Res. 35, 5992–6009 (2021).
Lawson, A. D., MacCoss, M. & Heer, J. P. Importance of rigidity in designing small molecule drugs to tackle protein–protein interactions (ppis) through stabilization of desired conformers. J. Med. Chem. 61, 4283–4289 (2017).
Henrich, C. J. & Beutler, J. A. Matching the power of high throughput screening to the chemical diversity of natural products. Nat. Prod. Rep. 30, 1284 (2013).
Romano, J. D. & Tatonetti, N. P. Informatics and computational methods in natural product drug discovery: A review and perspectives. Front. Genet. 10, 368 (2019).
Zeng, X. et al. A database of collective molecular activities of useful plants. Nucleic Acids Res. CMAUP, 47, D1118–D1127 (2018).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable Bat origin. Nature 579, 270–273 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Anand, K. et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 21, 3213–3224 (2002).
Yang, H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. 100, 13190–13195 (2003).
Xue, X. et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 82, 2515–2527 (2008).
Ren, Z. et al. The newly emerged SARS-like coronavirus HCoV-EMC also has an achilles’ heel: current effective inhibitor targeting a 3 C-like protease. Protein Cell. 4, 248–250 (2013).
Wang, F. et al. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 6, 22677 (2016).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Kneller, D. W., Kovalevsky, A. & Coates, L. Structural plasticity of the SARS-COV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 11, 3202 (2020).
Bzówka, M. et al. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for Small-Molecule inhibitor design. Int. J. Mol. Sci. 21, 3099 (2020).
Gossen, J. et al. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from Activity-Based compound library screening guided by analysis of protein dynamics. ACS Pharmacol. Transl Sci. 4, 1079–1095 (2021).
Flynn, J. M. et al. Comprehensive fitness landscape of SARS-CoV-2 Mpro reveals insights into viral resistance mechanisms. Elife 11, e77433 (2022).
Estrada, E. Topological analysis of SARS CoV-2 main protease. Chaos 30, 061102 (2020).
Abranyi-Balogh, P. et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. 160, 94–107 (2018).
Singh, J., Peter, R. C., Baillie, T. A. & Whitty, A. The. Resurgence of covalent drugs. Nat. Rev. Drug Discov. 4, 307–317 (2011).
Bauer, R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today. 20, 1061–1073 (2015).
Kitamura, N. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 65, 2848–2865 (2022).
Bharadwaj, K. K. et al. Macrolactin A as a novel inhibitory agent for SARS-CoV-2 Mpro: bioinformatics approach. Appl. Biochem. Biotechnol. 193, 3371–3394 (2021).
Abdusalam, A. A. A. & Murugaiyah, V. Identification of potential inhibitors of 3CL protease of SARS-CoV-2 from ZINC database by molecular Docking-Based virtual screening. Front. Mol. Biosci. 7, 603037 (2020).
Gupta, A. et al. Structure-Based virtual screening and biochemical validation to discover a potential inhibitor of the SARS-CoV-2 main protease. ACS Omega. 5, 33151–33161 (2020).
Weng, Y. L. et al. Molecular dynamics and in Silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Sci. Rep. 11, 7429 (2021).
Durrant, J. D. et al. POVME 2.0: an enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput. 10, 5047–5056 (2014).
Wagner, J. R. et al. POVME 3.0: software for mapping binding pocket flexibility. J. Chem. Theory Comput. 13, 4584–4592 (2017).
Durdagi, S. et al. Near-physiological-temperature serial crystallography reveals conformations of SARS-CoV-2 main protease active site for improved drug repurposing. Structure 29, 1382–1396 (2021).
Sztain, T., Amaro, R. & McCammon, J. A. Elucidation of cryptic and allosteric pockets within the SARS-CoV-2 protease. J. Chem. Inf. Model. 61, 3495–3501 (2021).
Yang, X. et al. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular Docking. Front. Pharmacol. 13, 962863 (2022).
da Fonseca, A. M. et al. Screening of Potential Inhibitors Targeting the Main Protease Structure of SARS-CoV-2 via Molecular Docking, and Approach with Molecular Dynamics, RMSD, RMSF, H-Bond, SASA and MMGBSA. Mol. Biotechnol. Preprint at (2023). https://doi.org/10.1007/s12033-023-00831-x
Teli, D. M. et al. Fragment-based design of SARS-CoV-2 Mpro inhibitors. Struct. Chem. 33, 2155–2168 (2022).
Jamhour, R. M. A. Q. et al. Phytochemicals as a potential inhibitor of COVID-19: an In-Silico perspective. Russ J. Phys. Chem. 96, 1589–1597 (2022).
Agrawal, P. K. & Blunden, G. Phytochemicals against SARS-COV-2 infection. Nat. Prod. Commun. 18 (2023).
Biancatelli, R. M. L. C., Berrill, M., Catravas, J. & Marik, P. E. Quercetin and vitamin C: an experimental, synergistic therapy for the prevention and treatment of SARS-CoV-2 related disease (COVID-19). Front. Immunol. 11, 1451 (2020).
Mani, J. S. et al. Natural product-derived phytochemicals as potential agents against coronaviruses: a review. Virus Res. 284, 197989 (2020).
Issa, S. S. et al. The main protease of SARS-CoV-2 as a target for phytochemicals against coronavirus. Plants 11, 1862 (2022).
AP11.0. SimulationsPlus LLC, Lancaster, CA.
Cayman Chemical Company. Demethoxycurcumin. Item Number 10961. (2024). https://www.caymanchem.com/product/10961/demethoxycurcumin
Cayman Chemical Company. Shikonin. Item Number 14751. (2024). https://www.caymanchem.com/product/14751/shikonin
Cayman Chemical Company. Withaferin A. Item Number 11352. (2024). https://www.caymanchem.com/product/11352/withaferin-a
Alici, H., Tahtaci, H. & Demir, K. Design and various in Silico studies of the novel Curcumin derivatives as potential candidates against COVID-19 -associated main enzymes. Comput Biol. Chem (2022).
Mulu, A. et al. The impact of curcumin-derived polyphenols on the structure and flexibility COVID-19 main protease binding pocket: a molecular dynamics simulation study. PeerJ 9, e11590 (2021).
Khaerunnisa, S., Kurniawan, H., Awaluddin, R., Suhartati, S. & Soetjipto, S. Potential Inhibitor of COVID-19 Main Protease (Mpro) From Several Medicinal Plant Compounds by Molecular Docking Study. Preprint at (2020). https://doi.org/10.20944/preprints202003.0226.v1
Ghosh, A. et al. Structure-activity relationship (SAR) and molecular dynamics study of withaferin-A fragment derivatives as a potential therapeutic lead against the main protease (Mpro) of SARS-CoV-2. J. Mol. Model. 27, 97 (2021).
Chakraborty, S. et al. The natural products Withaferin A and withanone from the medicinal herb Withania somnifera are covalent inhibitors of the SARS-CoV-2 main protease. J. Nat. Prod. 85, 2340–2350 (2022).
Zhang, Y. et al. Structure-Based discovery and structural basis of a novel Broad-Spectrum natural product against the main protease of coronavirus. J. Virol. 96, e0125321 (2022).
Ma, C. et al. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl Sci. 3, 1265–1277 (2020).
Gupta, S. et al. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular Docking and simulation studies. J. Biomol. Struct. Dyn. 39, 4334–4345 (2021).
Lachance, H. et al. Charting, navigating, and populating natural product chemical space for drug discovery. J. Med. Chem. 55, 5989–6001 (2012).
Kaur, S. et al. How do plants defend themselves against pathogens-Biochemical mechanisms and genetic interventions. Physiol. Mol. Biol. Plants. 28, 485–504 (2022).
Sharma, P. J. Identification of natural inhibitors against Mpro of SARS-CoV-2 by molecular docking, molecular dynamics simulation, and MM/PBSA methods. J. Biomol. Struc T Dyn. 6, 2757–2768 (2022).
Hossain, A. Identification of medicinal plant-based phytochemicals as a potential inhibitor for SARS-CoV-2 main protease (Mpro) using molecular Docking and deep learning methods. Comput. Biol. Med. 157, 106785 (2023).
Gogoi, B. Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation. Mol. Divers. 3, 1963–1977 (2021).
Choe, J., Yong, H. P. & Xiang, N. Z. The efficacy of traditional medicinal plants in modulating the main protease of SARS-CoV-2 and cytokine storm. Chem. Biodivers. 11, e202200655 (2022).
Choudhary, P., Singh, T., Amod, A. & Singh, S. Evaluation of phytoconstituents of Tinospora cordifolia against K417N and N501Y mutant Spike glycoprotein and main protease of SARS-CoV-2- an in Silico study. J. Biomol. Struct. Dyn. 9, 4106–4123 (2023).
Protein Data Bank. (2023). https://www.rcsb.org. Retrieved 29 Aug 2023.
Zhang, L. & Hilgenfeld, R. Crystal structure of SARS-CoV-2 Mpro in complex with the activity-based probe, biotin-PEG(4)-Abu-Tle-Leu-Gln-vinylsulfone. https://doi.org/10.2210/pdb6Z2E/pdb (2020).
Dai, W., Zhang, B., Jiang, X. M., Su, H. & Li, J. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for the design of anti-SARS drugs. Science 300, 1763–1767 (2020).
Ferraz, W. R., Gomes, R. A., Al, S. N. & Trossini, G. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in Silico repurposing study. Future Med. Chem. 12, 1815–1828 (2020).
Kneller, D. W. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 50, 17365–17373 (2020).
Cherrak, S. A., Merzouk, H. & Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular Docking and simulation studies. PLoS One. 15, e0240653 (2020).
Harwansh, R. K. & Bahadur, S. Herbal medicines to fight against COVID-19: new battle with an old weapon. Curr. Pharm. Biotechnol. 2, 235–260 (2022).
Xu, H., Li, S. & Liu, J. Bioactive compounds from Haushi Baidu decoction possess both anti-viral and anti-inflammataory effects against COVID-19. Proc. Natl. Acad. Sci. USA 18, e2301775120 (2023).
Allam, A. E. .al.In silico study of natural compounds from sesame against COVID-19 by targeting Mpro, PLpro and RdRp. R Soc. Chem. 11, 22398 (2021).
Antonopoulou, I., Sapountzaki, E., Rova, U. & Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 20, 1306–1344 (2020).
Ul Qamar, M. T., Alqahtani, S. M., Alamri, M. A. & Chen, L. L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 10, 313–319 (2020).
Ngwe, T. M. M. Anti-SARS-CoV-2 activity of various PET-bottled Japanese green teas and tea compounds in vitro. Arch. Virol. 167, 1547–1557 (2022).
Zígolo, M. A., Goytia, M. R., Poma, H. R., Rajal, V. B. & Irazusta, V. P. Virtual screening of plant-derived compounds against SARS-CoV-2 viral proteins using computational tools. Sci. Total Environ. 781, 46400 (2021).
Su, H. X. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 41, 1167–1177 (2020).
Nguyen, T. T. H. The inhibitory effects of plant derivate polyphenols on the main protease of SARS coronavirus 2 and their Structure-Activity relationship. Molecules 26, 1924 (2021).
El-Ashrey, M. K., Bakr, R. O., Fayed, M. A. A., Refaey, R. H. & Nissan, Y. M. Pharmacophore based virtual screening for natural product database revealed possible inhibitors for SARS-COV-2 main protease. Virology 570, 18–28 (2022).
Douangamath, A., Fearon, D., Gehrtz, P., Krojer, T. & Lukacik, P. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 11, 5047 (2020).
Abraham, M. J. et al. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX GROMACS, 1, 19–25 (2015).
Huang, J. & MacKerell, A. D. Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 (1983).
Darden, T., York, D. & Pedersen, L. Particle mesh ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Singh, K. & Briggs, J. M. Impact of lymphoma-linked Asn11Tyr point mutation on the interaction between Bcl-2 and a BH3 mimetic: insights from molecular dynamics simulation. Chem. Biol. Drug Des. 95, 435–450 (2020).
Berendsen, H., Postma, J., van Gunsteren, W., DiNola, A. & Haak, J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J Comp. Chem 18 (1997).
Humphrey, W., Dalke, A. & Schulten, K. VMD - Visual molecular dynamics. J. Mol. Graphics. 14, 33–38 (1996).
Pettersen, E. et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 (2004).
Schrödinger Release 2022-4. Protein Preparation Wizard; Epik, Schrödinger (LLC, 2022).
Banks, J. L. et al. Integrated modeling program applied chemical theory (IMPACT). J. Comp. Chem. 26, 1752–1780 (2005).
Schrödinger Release 2022-4. Glide (Schrödinger, LLC, 2022).
Schrödinger Release 2022-4: LigPrep. (Schrödinger, LLC, 2022).
Li, J. et al. The VSGB 2.0 model: a next-generation energy model for high-resolution protein structure modeling. Proteins 79, 2794–2812 (2011).
Cappelli, C. I., Manganelli, S., Lombardo, A., Gissi, A. & Benfenati, E. Validation of quantitative structure-activity relationship models to predict water-solubility of organic compounds. Sci. Total Environ. 463–464, 781–789 (2013).
Dearden, J. C. In Silico prediction of aqueous solubility. Expet Opin. Drug Discov. 1, 31–52 (2006).
Acknowledgements
We express our sincere gratitude to Charlie Burns and Samantha Roloff for their dedicated efforts in procuring the phytochemicals for testing. We also thank Reid Powell, Mary Sobieski, and Nghi Nguyen from Texas A&M for their assistance with the cytotoxicity analyses. Finally, we acknowledge Kong Vo for providing exceptional technical support throughout the duration of this project.
Author information
Authors and Affiliations
Contributions
K.S. carried out the in-silico portion of the research, including MD simulation, docking, MM-GBSA. and ADMET prediction. She analyzed the results and wrote the manuscript. J.P. conducted and analyzed the results of the viral replication assay. A.D. analyzed the cytotoxicity assay data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, K., Patten, J.J., Dimet-Wiley, A. et al. Unlocking the potential of phytochemicals in inhibiting SARS-CoV-2 MPro protein - an in silico and cell-based approach. Sci Rep 15, 22840 (2025). https://doi.org/10.1038/s41598-025-05907-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05907-z
