
Abstract
Transformation of cardiac fibroblasts into matrix-producing myofibroblasts plays an important role in tissue repair and fibrosis after myocardial injury. Tcf21 is a basic helix-loop-helix transcription factor that is essential for the development of cardiac fibroblasts and the epicardium. Myofibroblasts are derived from Tcf21 (+) residual fibroblasts; however, whether Tcf21 itself promotes fibrosis is still unknown. Since Tcf21 deficiency leads to perinatal lethality, cardiac fibroblasts were isolated from Tcf21-knockout mouse embryos, and mice that lack Tcf21 after birth were generated (inducible Tcf21 KO, iTcf21). In vitro analysis revealed that Tcf21 promoted cell proliferation and upregulated the expression of extracellular matrix genes. Notably, the Pdgfrb-Erk pathway was severely suppressed in Tcf21-knockout fibroblasts. Chromatin immunoprecipitation-sequencing assay demonstrated the direct binding of Tcf21 to COL1A1, COL3A1, IL6, and PDGFRB loci. Integrated analysis identified pathways involved in fibroblast activation, extracellular matrix production and cell proliferation. Moreover, iTcf21 mice were resistant to cardiac fibrosis induced by isoproterenol injection or metabolic overload with streptozotocin-induced diabetes and a high-fat diet. Our results demonstrate a critical role for Tcf21 in the transformation of cardiac fibroblasts into activated myofibroblasts. It directly binds to gene loci and works in a pro-fibrotic manner via Pdgfrb signaling.
Similar content being viewed by others


Introduction
Cardiac fibrosis is one of the main components of heart failure. It commonly occurs as a consequence of various cardiac injuries, such as pressure overload1, ischemia, and metabolic injuries including diabetic cardiomyopathy2,3. These injuries lead to the activation of cardiac fibroblasts, which proliferate and migrate to damaged sites and secrete extracellular matrix and inflammatory cytokines4. Various growth factors and cytokines, such as transforming growth factor beta (TGF-β) and platelet-derived growth factors (PDGF)5,6, play a role. However, the exact mechanisms and transcriptional regulation of fibroblast activation remain unclear.
Tcf21 is a cell type-specific basic helix-loop-helix (bHLH) transcription factor, which is a key regulator of organ development. It was first discovered as a mesoderm-specific protein expressed in mesenchymal cells and podocytes in the kidney7. Tcf21 is highly expressed in respiratory, urogenital, digestive, and cardiovascular cells and is essential for the development of the kidney, heart, and lung8,9,10,11. Additionally, cancer studies have revealed that Tcf21 functions as a tumor suppressor, and methylation of the Tcf21 promoter is associated with tumor invasion and metastasis12. In other studies, Tcf21 has been shown to play an important role in coronary artery disease13,14, diabetic kidney disease15, and adipose tissue inflammation16. Moreover, the Tcf21 allele has been identified as a risk locus for coronary artery disease in genome-wide association studies17.
In the heart, Tcf21 is expressed in the epicardium and cardiac fibroblasts. Tcf21-null mice show cardiac malformations similar to the Tetralogy of Fallot18 along with perinatal lethality. A recent analysis showed that Tcf21 is expressed in residual cardiac fibroblasts, and lineage tracing analysis using Cre recombinase at the Tcf21 locus showed that activated myofibroblasts are mainly derived from Tcf21(+) fibroblasts19. However, the precise role of Tcf21 in cardiac fibrosis remains to be elucidated.
In this study, we demonstrated that Tcf21 promotes cardiac fibrosis by modifying fibroblast proliferation and extracellular matrix expression. Based on RNA-sequencing (RNA-seq) and chromatin immunoprecipitation sequencing (ChIP-seq) analysis, we determined that Tcf21 binds to specific genomic regions related to PDGF signaling and collagen formation in cardiac fibroblasts. Overall, the results of this study show that Tcf21 is a novel regulator of fibroblast activation and that Tcf21 and its downstream targets may potentially be therapeutic targets for the modulation of cardiac fibrosis.
Methods
Mouse lines
The Tcf21-LacZ mouse is a conventional knockout (KO) mouse for Tcf21, which has been described earlier10. Tcf21-floxed mice were created previously15. Tcf21-LacZ and Tcf21-floxed mice were crossed with ROSA26-rtTA/tetO-Cre double transgenic mice to generate inducible whole-body KO of Tcf21 (ROSA26-rtTA/tetO-Cre/Tcf21lox/LacZ) by administration of doxycycline in drinking water as described previously. Briefly, KO mice were induced immediately after birth. For 3 weeks, mice were supplied with water containing doxycycline hydrochloride (5% sucrose, 2 mg Dox/mL), which was replaced three times a week. Littermates lacking tetO-Cre and LacZ were used as controls. We randomly chose to use mice, and there are no exclusion criteria for using mice. The primers used for genotyping are listed in the Supplemental Table 1.
Cardiac injury models
Isoproterenol (ISO; 200 mg/kg) dissolved in phosphate buffered saline (PBS) was intraperitoneally injected once at 8 weeks of age to generate the ISO-induced cardiac injury model. ISO dose was determined as the dose at which sufficient cardiac fibrosis was observed in the preceding dosing experiment. To generate the streptozotocin (STZ)- and high-fat diet (HFD)-induced cardiac injury model, mice were fed a diet containing 60% fat (Research Diet, New Brunswick, USA) for 6 weeks. STZ (50 mg/kg; Sigma-Aldrich, St. Louis, USA) dissolved in citrate buffer was intraperitoneally injected every day for 5 days starting at 8 weeks, as described by the Diabetic Complications Consortium sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases, USA. Blood glucose levels were checked after 2 weeks of STZ injection to confirm that it was > 250 mg/dL. Mice were sacrificed by cervical dislocation and subjected to the experiments according to the animal use protocol approved by Chiba University Ethics Committee (Chiba, Japan). All procedures were performed in the animal house. There are no excluded animals for data analysis. Mice were treated and measured in random order. Other confounders, such as cage location, were not controlled.
Immunostaining
For immunohistochemical staining, hearts were dissected and fixed in 10% formalin or 4% paraformaldehyde. Formalin-fixed tissues were embedded in paraffin and sliced into 5 µm thick sections. Before immunostaining, the sections were deparaffinized and boiled in citrate buffer for antigen retrieval. Paraformaldehyde-fixed tissues were fixed and cryoprotected in 30% sucrose overnight and embedded in Tissue-Tek O.C.T 4583 compound (Sakura Finetek Japan Co., Tokyo, Japan) at − 80 °C and sliced into 10 µm thick sections.
Before immunostaining with the antibody, the internal peroxidase was blocked with 3% H2O2 at room temperature for 30 min. Sections were blocked with buffer (3% BSA and 0.1% Triton X-100 in PBS) for 1 h and then incubated with primary antibodies at 4 °C overnight. After washing several times with PBS, the sections were incubated with secondary antibodies for 1 h at room temperature. For immunofluorescence, Hoechst 33342 (Dojindo, Osaka, Japan) was used to stain the nuclei, and Acti-stain 488 fluorescent phalloidin (Cytoskeleton Inc., Denver, USA) was used to stain F-actin. For DAB staining, the VECTASTAIN Elite ABC Kit (Vector Laboratories, Inc., Burlingame, USA) and ImmPACT DAB Brown (Vector Laboratories, Inc., Burlingame, USA) were used according to the manufacturer’s instructions.
β-Galactosidase staining
β-Galactosidase staining of paraformaldehyde-fixed sections was performed using the X-Gal Staining Kit (Genlantis, San Diego, USA). The samples were fixed in 4% paraformaldehyde for 15–20 min. After cryoprotection, samples were embedded and stored in − 80°. Frozen sections were incubated with fixing buffer for 3 min at room temperature. After washing twice with PBS, the sections were incubated with X-Gal staining solution for 8 h at 37 °C. Immunostaining was performed using LacZ-stained sections, if necessary.
In situ hybridization
For in situ hybridization, we used RNAscope 2.5 HD Reagent Kit- BROWN (Advanced Cell Diagnostics, California, USA). Formalin-fixed paraffin-embedded sections were deparaffinized and stained according to the provided manual.
Histological analysis
Each slide was sectioned at the position of maximum ventricular diameter. Stained slides were imaged using a BZX-710 microscope (Keyence, Osaka, Japan). The images were analyzed using a BZ-X Analyzer (Keyence, Osaka, Japan) to estimate the stained areas. Fibrosis areas are calculated by detecting blue area of Masson trichrome staining.
Cell culture
Mouse cardiac fibroblasts were isolated using collagenase, as described previously20, with minor modifications. Briefly, embryonic hearts from embryonic day 18.5 mice were separated and minced into 1 mm3 pieces in ice-cold Hanks’ Balanced Salt Solution (HBSS). The minced tissues were incubated in HBSS with 0.2% collagenase type 2 (Worthington Biochemical, Lakewood, USA) for 10 min at 37 °C with agitation. The digestion buffer was collected and the procedure was repeated three times. The dispersed cells were incubated in 10 cm culture hard plastic dishes without coating for 80 min in DMEM supplemented with 10% FBS and 1% antibiotic–antimycotic (Gibco). Thereafter, the medium was changed to remove the non-fibroblast cells. Fibroblasts were cultured and passaged at a dilution of 1:3. All experiments were performed within five passages. Tcf21 KO fibroblasts were obtained from Tcf21LacZ/LacZ mice heart at embryonic day 18.5.
Human cardiac fibroblasts were obtained from PromoCell (C-12375) and cultured in fibroblast growth medium 3 (PromoCell, Heidelberg, Germany) with 1% antibiotic–antimycotic (Gibco). The cells were passaged at a dilution of 1:3.
Stimulation with cytokines was performed by the following procedure: fibroblasts were seeded in 6-well culture plates. Twenty-four hours before cytokine stimulation, the culture medium was replaced with 0.1% BSA-containing medium without FBS. Ten picograms per milliliter recombinant human TGF-β1 (R&D Systems, Minneapolis, USA; Catalog# 240-B) and 20 pg/mL recombinant human PDGF-BB (Oriental yeast, Tokyo, Japan; Catalog# 47083000) were used as stimulators. Fibroblasts were incubated with the stimulating medium for 15 min and then used for western blotting.
BrdU uptake assay
Fibroblasts were cultured in Lab-Tek II Chamber slides (154526; Nunc, NY, USA). Prior to the analysis, the cells were labeled with 10 µM 5-bromo-2-deoxyuridine (BrdU)-containing medium for 24 h. Thereafter, immunostaining was performed using an anti-BrdU antibody (Abcam, Cambridge, UK; ab6326). The average number of BrdU-positive cells was calculated from five fields per sample acquired from each animal.
RNA-sequencing analysis
Total RNA was extracted using the Pure Link RNA Mini Kit (Invitrogen, Carlsbad, CA, USA), and cDNA libraries were generated using an NEBNext Ultra RNA Library Prep Kit (New England BioLabs, Beverly, USA). Sequencing was performed using a HiSeq 1500 (Illumina) with a single-read sequencing length of 60 bp. The sequence data were aligned to the reference genome (GRCm38), and the expression values were calculated using RSEM (version 1.3.1; with default parameters) with Bowtie2 (version 2.3.4.2) as an aligner. The results were compared with those of DESeq2 to identify differentially expressed genes. Gene set enrichment analysis was performed using the DAVID and Metascape software.
ChIP-sequencing analysis
Approximately two million human cardiac fibroblasts overexpressing Flag-tagged TCF21 were crosslinked for 5 min in 1% formaldehyde. The cells were lysed for 10 min on ice in a buffer containing 50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100. The nuclei were then lysed on ice in a buffer containing 50 mM Tris–HCl (pH 8.0), 1% SDS, and 10 mM EDTA (pH 8.0). The cross-linked chromatin was sheared for 50 min using a Bioruptor (30 s on, 30 s off; Diagenode, Denville, USA). ChIP was performed for 6 h in ChIP dilution buffer (16.7 mM Tris–HCl (pH 8.0), 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA (pH 8.0), 167 mM NaCl) using 10 µg anti-Flag antibody (Sigma-Aldrich, St. Louis, USA; F-1804) and Protein G Magnetic Sepharose beads (GE Healthcare Bio-science AB, Uppsala, Sweden; 28944008). The beads were washed five times with a low-salt wash buffer (20 mM Tris–HCl (pH 8.0), 0.1% SDS, 1% Triton X-100, 2 mM EDTA (pH 8.0), 150 mM NaCl) and twice with a high-salt buffer (20 mM Tris–HCl (pH 8.0), 0.1% SDS, 1% Triton X-100, 2 mM EDTA (pH 8.0), 500 mM NaCl). The bound DNA was eluted from the beads with elution buffer (10 mM Tris–HCl (pH 8.0), 1% SDS, 5 mM EDTA (pH 8.0), 300 mM NaCl). RNase A and proteinase K reactions were performed after reversing the crosslinks. DNA was cleaned using the phenol–chloroform extraction method. cDNA libraries were generated using the NEBNext Ultra II DNA Library Prep with Sample Purification Beads (New England BioLabs, Beverly, USA). Sequencing was performed using a NextSeq 500 (Illumina) with a single-read sequencing length of 75 bp.
ChIP sequence data were aligned to the reference genome (GRCh37) using Bowtie2 (version 2.3.4.2) and visualized with the Integrative Genome Viewer (Broad Institute). MACS2 (version 2.1.1) was used to identify the peaks against an input DNA without ChIP, with the default setting for the q-value (< 1e−10). Two biological repeats, cross-linked and immunoprecipitated independently, were merged with HOMER2 (version 4.4). We utilized the Genomic Regions Enrichment of Annotations Tool (GREAT 3.0) to analyze the detected peaks of TCF21, with the parameter “Association rule settings” set to two nearest genes (within 20 kb). Motif analysis was performed using the HOMER2 findMotifGenome.pl script with default settings.
Luciferase assay
We made the luciferase construct using promoter and enhancer sequences of each gene. For Col1a1, −8432 to −7721 bp from TSS and −1962 to −1396 bp from TSS were used. For Col3a1, −5271 to −4634 bp from TSS and −344 to −1 bp from TSS were used. For Il6, −768 to + 434 bp from TSS were used. For Pdgfrb, −12,986 to −12,053 bp from TSS and −172 to + 419 bp from TSS were used. These loci were identified from the TCF21 binding sites by ChIP-seq and prediction of H3K4me3 and H3K27ac peaks from CHIP-Atlas.
We transferred the Tcf21 Mouse ORF Clone (OriGene, MR201596) to the pTRE3G vector and electroporated it into pEFa-Tet3G-stable expressing 3T3-L1 cells (Tet-On 3G Inducible Expression System, Clontech). We selected hygromycin-resistant cells and cloned them by the limiting dilution method. These cells overexpress Tcf21 with the addition of Doxycycline. These cells were plated in 24‐well dishes at 50% confluency and transfected with 3.25 μg of the luciferase construct plasmid per well using Lipofectamine LTX (Invitrogen). At 24 h after transfection, the medium was changed to DMEM supplemented with 10% FBS and with or without 1 ng/mL doxycycline, and collected after 24 h incubation. Luciferase activity in the cell lysate was measured using a 1420 ARVO SX multilabel counter (Wallac, Inc., Gaithersburg, MD) using the Dual-Glo Luciferase Assay System (Promega).
Echocardiography
Animals were placed in the supine position without anesthesia. Two-dimensional guided M-mode echocardiography was performed using an echocardiograph (Vevo 770 Imaging System; probe: RMV 707 B, Visualsonics, Toronto, Canada). Left ventricular diastolic posterior wall thickness (PWV), left ventricular diastolic dimension (LVDd), and left ventricular end-systolic dimension (LVDs) were measured. Left ventricular fractional shortening (% FS) was calculated as (LVDd–LVDs)/LVDd × 100. The operator performing echocardiography was not informed of the allocation of the mice.
Lentivirus infection
For FLAG-tagged Tcf21 overexpression, the pLVSIN-EF1α Pur vector was obtained from Takara (Tokyo, Japan). The 3xFLAG-TCF21-IRES-GFP genes were cloned into the multiple cloning site and then packaged into 293 T cells. For Tcf21 knockdown, the lentiCRISPR v2 vector (Addgene plasmid #52961) was obtained as a gift from Feng Zhang at the Broad Institute of MIT21. The designed oligonucleotides (Supplementary Table 1) were inserted and packaged into 293 T cells. Human cardiac fibroblasts were transfected with the particles. Three days after transfection, puromycin (1–2 µg/mL, Sigma-Aldrich, St. Louis, USA) was added to the culture medium for 7 days to obtain stably transfected cells.
RNA isolation and quantitative RT-PCR
Whole hearts were dissected and preserved at −80 °C after freezing in liquid nitrogen. They were homogenized using a micro-homogenizer (Microtec, Chiba, Japan) and a QIA shredder column (Qiagen, Hilden, Germany; 79656). Total RNA was extracted using the PureLink RNA Mini Kit (Invitrogen, Carlsbad, USA). Reverse transcription was performed using SuperScript III Reverse transcription (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Quantitative reverse-transcription PCR was performed with CFX Connect (Bio-Rad, Hercules, CA, USA) using FAST SYBR Green Master Mix (Applied Biosystems). Data were normalized using Hprt or B2m as internal controls. The primers used are listed in Supplementary Table 2.
Western blot analysis
Cardiac fibroblasts were lysed in SDS sample buffer (100 mM Tris–HCl (pH 6.8), 4% SDS, 20% glycerol, 0.02% bromophenol blue, 3% 2-mercaptoethanol) by heating at 95 °C for 10 min. The proteins were fractionated on 12.5% polyacrylamide gels (e-PAGEL; ATTO Corporation, Japan) and transferred onto polyvinylidene fluoride membranes (Immobilon-P Transfer Membrane; Millipore, Tullagreen, Ireland). The membranes were blocked with PBST containing 5% PhosphoBlocker blocking reagent (Cell Biolabs, San Diego, USA) for 1 h at room temperature to block nonspecific binding of the antibody and were then incubated with primary antibodies at 4 °C overnight. The primary antibodies used are listed in Supplementary Table 3. The blots were then washed and incubated with secondary antibodies, anti-rabbit IgG, HRP-linked whole antibody donkey (1:2500, GE Healthcare: NA934) or goat anti-mouse IgG-HRP (1:2500, Santa Cruz: sc-2055) for 1 h at room temperature. After washing several times, the bands were visualized using an ECL western blotting detection system (GE Healthcare: RPN2106). Densitometric analysis was performed using a ChemiDoc MP ImageLab PC system (Bio-Rad, Hercules, CA, USA).
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7.0. (GraphPad Software Inc., San Diego, USA). Comparisons between two groups were performed by Student’s unpaired two-tailed t-test when the Shapiro–Wilk normality test was passed; otherwise, the Mann–Whitney test was performed. For more than two groups, a one-way ANOVA and two-stage linear step-up procedure of benjamini krieger and yekutieli were used. Statistical significance was set at p < 0.05 and q < 0.05. The results are presented as means, and the error bars in all the figures represent SEM. Each point in the bar graph represents one sample.
Ethics statement/study approval
All experiments were performed in accordance with the institutional guidelines for animal studies. All animal experiments were approved by Chiba University Ethics Committee (Chiba, Japan). This study is conducted under ARRIVE guidelines.
Results
Tcf21 promotes proliferation of cardiac fibroblasts via Erk signaling and is required for the expression of extracellular matrix
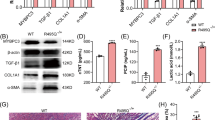
To clarify the role of Tcf21 in cardiac fibroblasts, we isolated fibroblasts from the hearts of WT and Tcf21 KO mice at embryonic day 18.5. These cells were spindle-shaped and the expression levels of fibroblast markers such as Col1a1, Pdgfra, Thy1, Ddr2, Fsp1 were equivalent to that of mouse embryonic fibroblasts (MEFs) and supported the validity of the fibroblasts. (Supplementary Fig. 1a,b). Upon culturing these cells, the proliferation rates seemed to be decreased in Tcf21 KO fibroblasts. To elucidate the difference of proliferation capacity, we performed BrdU-uptake assay using these cells. BrdU-uptake ratio decreased in Tcf 21 KO fibroblasts, which showed lack of Tcf21 impaired fibroblasts proliferation (Fig. 1a, b).

Tcf21 knockout (KO) fibroblasts show attenuated proliferation capacity and reduced Pdgfrb signaling. (a,b) 5-bromo-2′-deoxyuridine (BrdU) uptake decreased in Tcf21 KO fibroblasts (biologically n = 4). Scale bar, 200 µm. (c) Western blots of Smad2 and phosphorylated Smad2 (p-Smad2), with TGF-β (10 ng/mL) stimulation for 15 min (biologically n = 3). (d) Quantitative analysis of p-Smad2/Smad2. (e) Western blots of Erk and phosphorylated Erk (p-Erk), with PDGF-BB (20 ng/mL) stimulation for 15 min (biologically n = 3). (f) Quantitative analysis of p-Erk/Erk. (g) qRT-PCR, (h,i) Western blot analysis of Pdgfrb (biologically n = 3). (j) qRT-PCR analysis using human cardiac fibroblasts with TCF21 knockdown by CRISPR-CAS9 showing decreased expression of TCF21, POSTN, ACTA2, COL1A1, COL3A1, and PDGFRB (biologically n = 3).
Various signaling pathways have been implicated in the activation of cardiac fibroblasts, including those of TGF-β, PDGF, angiotensin II, CTGF, and other cytokines22. Upon TGF-β stimulation, levels of phosphorylated Smad2, the main signaling molecule downstream of the TGF-β receptor, did not differ between WT and Tcf21 KO fibroblasts (Fig. 1c,d). However, Erk1/2 phosphorylation was attenuated in Tcf21 KO fibroblasts after stimulation with PDGF-BB (Fig. 1e,f). Erk signaling stimulated by PDGF is known to enhance the proliferation of cardiac fibroblasts and inflammatory and fibrogenic responses23. Stat3 and Akt signaling were also reduced by the deletion of Tcf21 (Supplementary Fig. 2). In addition, expression of Pdgfrb was significantly attenuated in Tcf21 KO fibroblasts (Fig. 1g–i). This indicated that the loss of Tcf21 reduced Pdgfrb/Erk signaling and decreased the proliferation capacity of fibroblasts.
To confirm the changes in fibroblasts of Tcf21 KO mice and to exclude the possibility that the reduced expression of Pdgfrb is derived from the developmental defect of cardiac fibroblasts, we induced TCF21 gene deletion in a primary culture of human cardiac fibroblasts using the CRISPR/Cas9 system. The expression of TCF21 decreased to half that of the control fibroblasts (Fig. 1j). POSTN, ACTA2, and COL1A1 were significantly decreased by TCF21 knockdown, and COL3A1 and PDGFRB were strongly suppressed.
These results indicate that Tcf21 promotes proliferation and extracellular matrix production in cardiac fibroblasts by maintaining Pdgfrb expression and its downstream signaling.
Tcf21 modulates the expression of extracellular matrix genes
To gain further insights into the molecular basis of Tcf21 regulation in cardiac fibroblasts, RNA-sequencing (RNA-seq) was performed using isolated mouse cardiac fibroblasts. Gene ontology analysis of the data showed gene expression signatures related to fibroblast activation and cardiac fibrosis, such as extracellular matrix and proliferation (Fig. 2a,b). For instance, the levels of Col1a1, Col1a2, Col3a1, and Col6a1, which are extracellular matrix transcripts, were reduced in the absence of Tcf21 (Fig. 2c). The expression of activation markers of fibroblasts, Postin and Acta2, were also downregulated in Tcf21-deleted fibroblasts. In contrast, the expression of common fibroblast markers, Pdgfra and Vim, did not change in the presence or absence of Tcf21. RT-PCR also showed downregulation of Acta2, Postin, and Col1a1 expression in Tcf21 KO mice (Fig. 2d), suggesting the role of Tcf21 as a facilitator of cardiac fibrosis.

Tcf21 regulates extracellular matrix, proliferation, and inflammation-associated genes. (a,b) Results of gene expression cluster analysis obtained from DAVID 6.8 and Metascape showing altered expression of extracellular matrix and proliferation signaling genes in fibroblasts obtained from the heart of Tcf21 KO mice compared to those obtained from control mice (biologically n = 3). (c) Volcano plot shows that the expression of 440 genes was significantly altered by Tcf21. Upregulated genes in Tcf21 WT fibroblasts contain many extracellular matrix genes. Red dots represent significant genes upregulated in Tcf21 WT (P < 0.05). Blue dots represent significant genes downregulated in Tcf21 WT (P < 0.05). (d) Results of quantitative RT-PCR showing decreased expression of extracellular matrix and fibroblast activation genes in Tcf21 KO fibroblasts (biologically Control n = 15, iKO n = 24).
Tcf21 regulates extracellular matrix, Pdgfrb, and various other gene expression via direct binding to their DNA loci
For ChIP-seq, human cardiac fibroblasts were infected with lentivirus that overexpressed FLAG-tagged TCF21, and anti-FLAG antibody was used to detect TCF21 binding regions. The precipitated DNA accumulated around enhancers and transcription start sites (TSS), indicating that the DNA bound to the transcription factor was condensed appropriately (Fig. 3a). The total number of peaks was 52,562. Some TCF21 binding peaks were observed around the TSS of COL1A1, COL3A1, IL6, PDGFRB, and PDGFRA (Fig. 3b, Supplementary Fig. 3a). These regions have been reported to be enhancer sites.

Tcf21 bound to specific chromosomal regions related to extracellular matrix and PDGFRB. (a) DNA pulled down by chromatin immunoprecipitation (ChIP) was distributed around the transcription start site (TSS) and enhancer region. (b) Strong Tcf21 binding peaks were observed around the TSS of COL1A1, COL3A1, and IL6. (c) Result of motif analysis obtained using HOMER2 with a size parameter 200. Tcf21 majorly bound to the E-box domain (CAGCTG), and other DNA sequences, such as AP-1, TEAD4, and TWIST1 domains, were also detected as the Tcf21 binding motifs.
The bHLH transcription factors, including TCF21, form homo- or heterodimers to bind DNA sequences. ChIP-seq data revealed consensus sequences for TCF21 binding. The major identified sequence was the CAGCTG E-box motif, which was found in 70.7% of the peaks (P = 1e−14056) (Fig. 3c). This sequence was the same as that described in a previous report on coronary smooth muscle cells14. These data implicate another motif sequence, TGANTCA, which corresponds to the AP-1 motif. This motif was found in 21.8% of the peaks (P = 1e−2661). AP-1 has been shown to activate the collagen promoter and migration. Therefore, our results suggested that TCF21 interacts with AP-1 and maintains the production of extracellular matrix and migration of fibroblasts. TWIST and TEAD, transcription factors that regulate inflammation and cell proliferation, also scored highly. We performed another motif analysis with a small size (75 bp) from the peak center (Supplementary Fig. 3b). CAGCTG was the top hit motif, but other motifs were decreased in this analysis. The difference between these two motif analyses suggested that AP-1, TWIST, and TEAD bound around the E-box motif and interacted with TCF21.
To investigate the obtained peaks, we assigned the peaks to genes using GREAT 3.0. A total of 31,167 peaks were used as inputs, and 8301 peaks were annotated to at least one gene. The obtained genes were summarized by gene ontology and pathway analyses (Table 1). These genes were significantly enriched in extracellular matrix production, migration, and angiogenesis in gene ontology analysis. Moreover, pathway analysis revealed that proliferation-associated pathways, such as PDGF and activation of the mitogen-activated protein kinase signaling pathway, were significantly enriched. IL6-mediated signaling events were also associated with TCF21 targets. In summary, our data indicated that Tcf21 regulates the proliferation and expression of extracellular matrix proteins in cardiac fibroblasts by directly binding to specific DNA loci.
In addition, we conducted luciferase reporter assays using the upstream regions of COL1A1, COL3A1, IL6, PDGFRB. Doxycycline-inducible overexpression of TCF21 led to a slight but statistically significant increase in luciferase activity for Col1a1 and Col3a1 (Supplementary Fig. 3c). In contrast, no significant changes in luciferase activity were observed for the upstream regions of Il6 and Pdgfrb. (Supplementary Fig. 3c). Thus, TCF21 has direct transactivation potential for Col1a1 and Col3a1, but the upregulation of Pdgfrb and Il6 may require additional cofactors or regulatory elements.
Deletion of Tcf21 after birth does not show an obvious phenotype
To dissect the role of TCF21 in cardiac fibrosis in vivo, we used a genetic approach. Since conventional Tcf21 KO mice die soon after birth, iTcf21 KO mice were generated using the Cre-loxP system. Doxycycline administration via drinking water for adult mice could attenuate Tcf21 expression, however, expression was not fully suppressed. Therefore, we used doxycycline from postnatal day 0 and Tcf21 gene was systemically deleted. iTcf21 KO mice grew as normally as WT mice, and there were no differences in body weight until 24 weeks of age (Supplementary Fig. 4a). At 8 weeks of age, no differences were observed in heart weight, cardiac histology, and blood pressure between controls and iTcf21 KO mice (Supplementary Fig. 4b–d). We confirmed that Tcf21 mRNA levels are almost undetectable in hearts of iTcf21 KO mice. (Supplementary Fig. 4e). On the other hand, the weight of the kidneys and spleen was reduced, and lung weight was increased, suggesting the role of Tcf21 in postnatal growth of these organs (Supplementary Fig. 4f). Tcf21 is important for ureteric bud branching and glomerular development, and deletion of Tcf21in embryonic stage leads to severe defects in branching morphogenesis and podocyte development11,15. The nephron induction begins at E10.5 and podocyte differentiation starts at around E14.5, therefore we successfully circumvented the critical stage that require Tcf21 so that kidney defect was minimized.
ISO-induced cardiac injury leads to increased Tcf21 expression
To analyze the role of Tcf21 in cardiac injury and subsequent fibrosis, we used high-dose ISO as the acute catecholamine-induced cardiac injury model (Fig. 4a). A single dose of 150 mg/kg ISO induced acute necrotic changes in cardiomyocytes within 24 h24. This change is caused by myocardial ischemia, accumulation of oxidative stress, and inflammatory reactions, and is subsequently accompanied by cardiac fibrosis. ISO injection at 8 weeks of age induced a significant increase in fibrosis area after 7 and 28 days (Fig. 4b). Tcf21 expression in the whole heart was increased in a time-dependent manner (Fig. 4c), and the Tcf21-expressing area positive for β-galactosidase staining in Tcf21LacZ/+ mice was also increased (Fig. 4d). In situ hybridization for Tcf21 following doxycycline induction, before and 7 days after ISO treatment also confirmed the increase of Tcf21 (Supplementary Fig. 5a). Tcf21-positive fibroblasts migrated into the Masson trichrome-positive area at the endocardium side of the left ventricular free wall (Fig. 4e,f). Immunostaining using Tcf21LacZ/+ hearts revealed that Postin, an activated myofibroblast marker, co-localized with Tcf21 in the fibrotic area (Fig. 4g), indicating that Tcf21-positive cardiac fibroblasts transformed into myofibroblasts. In addition, Ki67-positive cells were increased after 7 days of ISO injection (Fig. 4h,i, Supplementary Fig. 5b), and most of the Ki67-positive cells overlapped with Tcf21-expressing cells, as shown by β-galactosidase staining. Postn and Acta2, and two collagen genes Col1a1 and Col3a1, were markedly upregulated after ISO injection (Fig. 4j) as understood by qPCR. These results showed that ISO-induced injury increased the expression of Tcf21 and was associated with the upregulation of proliferation and transcription of extracellular matrix proteins.

Isoproterenol (ISO) injection leads to cardiac fibrosis and expansion of Tcf21-expressing cells. (a) Overview of the ISO injection schedule. (b) Cardiac fibrosis area of Masson trichrome-stained sections after ISO injection. Dunnett’s test was performed for post-hoc analysis (biologically n = 3). (c) Tcf21 expression before and after ISO injection assessed by qPCR. Dunnett’s test was performed for post-hoc analysis (biologically n = 3). (d) Tcf21-positive area calculated based on β-galactosidase staining (biologically n = 4). The detected area was normalized to the cardiac cross-sectional area. (e,f) Tcf21-positive cells accumulated in the fibrotic area after 3 or 7 days of ISO injection. Tcf21-positive cells were calculated in the Masson trichrome (MT) positive or negative area. Scale bar, 100 µm (biologically n = 3). (g) Immunostaining of a section of heart from Tcf21LacZ/+ transgenic mice. Tcf21 expression as assessed by β-galactosidase (green) and periostin (red) staining coexisted after 3 days of ISO injection. Scale bar, 100 µm. (h) Immunostaining of Ki67 (brown) and Tcf21 expression by β-galactosidase (blue). Ki67-positive cells accumulated in the Tcf21-positive area. Scale bar, 100 µm. (i) Proliferation capacity of Ki67-positive cells. Each sample was counted in a high-power microscopic field more than three times, and the average values were used for analysis. Unpaired two-tailed t-tests with Bonferroni correction were used for post-hoc analysis (biologically n = 4). (j) Fibroblast-associated gene expression increased after ISO injection. Dunnett’s test was performed for post-hoc analysis (biologically n = 3).
Lack of Tcf21 ameliorates cardiac fibrosis in two different disease models
Finally, to clarify whether Tcf21 promotes cardiac fibrosis in vivo, we injected ISO into iTcf21 KO mice (Fig. 5a). Compared to control mice, iTcf21 KO mice showed a smaller fibrosis area 7 and 28 days after ISO injection (Fig. 5b,c, Supplementary Fig. 5c). Periostin positive cells were also decreased in iTcf21 KO mice (Supplementary Fig. 6a,b). The number of Ki67-positive cells decreased in iTcf21 KO mice (Fig. 5d,e). These positive cells were found in periostin positive cells (Supplementary Fig. 6b). Additionally, to elucidate gene expression changes in tissue during cardiac injury, we performed in situ hybridization. In the control group, strong expression of Col1a1 and Col3a1 was observed at the site of cardiac injury, whereas their expression was markedly reduced in the iTcf21 KO group (Supplementary Fig. 7). Furthermore, the expression of Pdfgrb was decreased in both fibrotic and non-fibrotic areas in the iTcf21 KO group compared to controls. No significant changes were observed in Acta2 expression, while a decrease in Il6 expression was confirmed in the fibrotic regions (Supplementary Fig. 7).

Lack of Tcf21 suppresses cardiac fibrosis and maintains cardiac function. (a) Overview of isoproterenol (ISO) injection schedule. (b) Cardiac fibrosis observed at each time point. Masson trichrome staining was perfoemed. Before ISO injection and 3, 7, and 28 days after the injection. Scale bar, 1 mm. Yellow arrowheads indicated fibrotic area. (c) Fibrosis area from Masson trichrome staining normalized with cardiac cross-sectional area (biologically n = 3 ~ 4). (d) Immunostaining for Ki67. Scale bar, 50 µm. Arrows indicate Ki67-positive cells. (e) Change in the number of Ki67-positive cells, 7 days after ISO injection (biologically n = 5). (f) M-mode echocardiographic view, one week after ISO injection. (g) Changes in left ventricular fractional shortening (FS) after ISO injection. Dunnett’s multiple comparison test was performed to compare baseline data (biologically Control n = 14, iKO n = 7). (h) Protocol for hyperglycemic and hyperlipidemic injury. (i) Masson trichrome-stained heart Sections 16 weeks after diabetes induction with HFD. Arrows show fibrosis. Scale bar, 50 µm. (j) Fibrosis area calculated from Masson trichrome staining normalized by the cardiac cross-sectional area (biologically n = 4 and 3). Student’s t-test was performed.
Two-dimensional guided M-mode echocardiography was performed to calculate the fractional shortening (FS). Before ISO injection, there were no differences in FS between the control and iTcf21 KO mice. After 1 week of ISO injection, when cardiac fibrosis was observed, FS was decreased in the control mice compared to that in the mock-injected group. Unlike in control mice, cardiac dysfunction was not observed in iTcf21 KO mice after 1 week of ISO injection (Fig. 5f,g). In addition, the ejection fraction (EF) was decreased in the control but not in the iKO mice; however, PWV, LVDd, and LVDs were not significantly different (Supplementary Fig. 8a–d). In addition, as a model of cardiac fibrosis in diabetic heart disease, mice were fed with a HFD from 6 weeks of age, and diabetes was induced by streptozotocin at 8 weeks of age (Fig. 5h). Sixteen weeks after diabetes induction and HFD feeding, control mice showed expansion of the interstitial fibrosis area; however, this was not observed in iTcf21 KO mice (Fig. 5i,j). Using qPCR to compare gene expression, we observed an upregulation of fibrosis-related and inflammation related genes following HFD/STZ treatment. Notably, this upregulation tended to be suppressed in the iTcf21 KO group. (Supplementary Fig. 9). These results indicated that iTcf21 KO ameliorated fibroblast activation and cardiac fibrosis in an ISO-induced cardiac fibrosis model and a diabetes/HFD-induced cardiac fibrosis model.
Discussion
Despite various pathways and transcription factors, including TGFB/SMAD5, PDGFB/ERK, WNT/b-CATENIN, and YAP/TAZ, being reported to be involved in the activation of cardiac fibroblasts6,25,26,27,28, the details of these mechanisms remain unclear. Although activated cardiac fibroblasts are derived from Tcf21(+) resident fibroblasts19,29,30, the function of Tcf21 itself in myofibroblast transformation has not been elucidated. Our data, for the first time, introduced Tcf21 as a pro-fibrotic transcription factor in cardiac fibrosis. Tcf21 is essential for Pdgfrb expression; therefore, it may function as a major determinant of cardiac fibroblast cell number, since PDGF signaling is the strongest activator of fibroblast proliferation31.
TCF21 coordinates with the transcription factor JUN, a member of activation protein-1 (AP-1), and regulates the expression of SMAD3 and CDKN-2B AS132,33 by modulating H3K27Ac status. In Xenopus cardiac mesenchyme, Tcf21 binds to histone deacetylase 2 (HDAC2) and C-terminal binding protein 2 (CtBP2), forms a transcriptional repressor complex, and modulates histone acetylation34. Our ChIP-seq data also found TGAGTCAT, the target motif for FOS-JUN, to be a TCF21-enriched locus, suggesting an interaction between TCF21 and JUN in cardiac fibroblasts. These previous studies and our data strongly suggest that JUN-AP1 axis is regulated by TCF21 at the transcriptional and epigenetic level, and is a major downstream effector of TCF21.
Recent single cell analyses have revealed the heterogeneity of cardiac fibroblasts before and after injury, and four phases have been proposed: basal, expansion, activation, and resolution. Tcf21 is expressed in the basal and resolution phase, but not in the expansion and activation phases where periostin and type1 collagen (Col1) are expressed35. However, in the present study, fetal mouse and human cardiac fibroblasts that lack Tcf21 showed decreased expression of Col1a1, periostin, Col3a1 and Pdgfrb. These results appear to contradict previous reports. TCF21 binds to the TSS and enhancer regions of these genes. These results may suggest that TCF21 in the basal phase is required for subsequent expression of Col1 and periostin etc.in expansion/activation phase. Since Tcf21 plays a role in epigenetic regulation via HDACs, as mentioned above, it may open the chromatin status of Col1 and periostin within basal phase, it may define the transformation capacity of cardiac fibroblasts as a pioneer factor.
In addition, this hypothesis may also explain the limited or absent increase in luciferase activity upon Tcf21 induction. One possibility is that promoter–enhancer regulation is highly complex, and the sequences cloned into the reporter constructs may only partially recapitulate the native regulatory context. Another hypothesis is that Tcf21 functions as a pioneer transcription factor, modulating chromatin accessibility at the locus, and full transcriptional activation may require cooperative interactions with additional transcriptional regulators.
The function of Tcf21 has been analyzed in development10,11,15, cancer36,37,38,39, atherosclerosis40, and organ fibrosis. During development, Tcf21 facilitates mesenchymal-to-epithelial transformation in the mesenchyme of the kidney, lung, spleen, and testis10,18,41,42. Recently, Tcf21 has been reported as a regulator of residual somatic mesenchymal stem cells in the testis43. In cancer, Tcf21 suppresses invasion, metastasis, and epithelial-to-mesenchymal transformation (EMT)38,44,45. Promoter methylation of Tcf21 and downregulation of Tcf21 expression have been reported in lung46,47, urogenital48,49, breast50, and adrenocortical cancers. In atherosclerotic plaques, smooth muscle cells undergo fibroblast-like phenotypic modulation. Tcf21 is essential for this transformation, which leads to plaque stabilization40,51. Accordingly, TCF21 commonly contributes to drive mesenchymal cells to terminally differentiate in development, malignant tumors, and atherosclerosis. Our present study also indicated that TCF21 modulates phenotypes of cardiac fibroblasts after injury.
In terms of organ fibrosis, liver cirrhosis in a mouse model was suppressed by Tcf2152, while Tcf21 promoted fibrosis in endometriosis53. In the present study, Tcf21 promoted the proliferation and extracellular matrix expression of cardiac fibroblasts. Recent study showed that LncRNA-Tcf21 antisense RNA inducing demethylation (TARID) suppressed mouse and porcine models of myocardial infarction through upregulation of Tcf2154. This is different from our results, and these differential effects of Tcf21 on cell proliferation and fibrosis could be due to the binding partners of Tcf21 or to epigenetic regulation of the genes downstream of Tcf21. Tcf21 may act as a growth suppressor in mesenchymal cells and fibroblasts in adults and as a growth promoter in fibroblasts in young and smooth muscle cells that are terminally differentiated from mesenchymal cells.
In addition, TCF21 is reported to be expressed in adult cardiac fibroblasts and to be downregulated in response to myocardial infarction55. However, in our experiments, Tcf21 expression increased in parallel with fibroblast expansion. The previous study utilized myocardial infarction (MI), which represents a model of severe ischemia, whereas our study employed isoproterenol (ISO), which induces cardiac injury through sympathetic stimulation and is associated with milder ischemia and inflammation. We hypothesize that Tcf21 expression is downregulated under conditions of severe ischemia, but upregulated in response to inflammation.
This study has two limitations. First, the identification of fibroblast subpopulations was not addressed. Previous studies have reported that Tcf21 knockout (Tcf21KO) mice lack fibroblasts of epicardial origin. In the present study, we observed no differences in heart weight or morphology following postnatal Tcf21 deletion. Furthermore, the distribution of Col1a1 in non-fibrotic regions was comparable, suggesting that significant changes in fibroblast numbers are unlikely. Nevertheless, we cannot exclude the possibility of a shift in fibroblast subpopulation. Identifying fibroblasts of endocardial and epicardial origin in in vivo sections remains technically challenging. Second, we were unable to confirm whether the cells analyzed were originally Tcf21-expressing fibroblasts or derived from other populations. To address this, a lineage tracing system using Tcf21-Cre is necessary for future studies. However, it should be noted that mouse cardiac fibroblasts with Tcf21 KO, human cardiac fibroblasts with Tcf21 KD, and cardiac tissue changes with iTcf21 KO all show results in a certain direction, suggesting reduced Tcf21 expression induces functional changes within the fibroblasts.
In summary, Tcf21 is a novel factor that regulates fibroblast activation following cardiac injury and plays an important role in accelerating cardiac fibrosis by promoting fibroblast proliferation and extracellular matrix expression. In addition, many genes related to fibroblast function are regulated by the direct binding of Tcf21 to their promoter and enhancer regions. Modulation of Tcf21 expression or its downstream targets could provide novel therapeutic options for fibrotic heart diseases.
Data availability
All sequencing data that support the findings of this study have been deposited in the DDBJ Sequenced Read Archive under the accession numbers DRA009326 and DRA009327. All other published sequencing data have been cited in the main text. All other relevant data are available from the authors upon request to Yusuke Baba.
Abbreviations
- bHLH:
-
Basic helix-loop-helix
- BrdU:
-
5-Bromo-2′-deoxyuridine
- ChIP:
-
Chromatin immunoprecipitation
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- FS:
-
Fractional shortening
- HF:
-
Heart failure
- HFD:
-
High-fat diet
- ISO:
-
Isoproterenol
- iTcf21 KO:
-
Inducible Tcf21 knockout
- KO:
-
Knockout
- LVDd:
-
Left ventricular diastolic dimensions
- LVDs:
-
Left ventricular end-systolic dimensions
- MAPK:
-
Mitogen-activated protein kinase
- NF-κB:
-
Nuclear factor-kappa B
- PDGF:
-
Platelet-derived growth factor
- PWV:
-
Left ventricular diastolic posterior wall thickness
- RNA-seq:
-
RNA-sequencing
- STZ:
-
Streptozotocin
- TSS:
-
Transcription start site
References
Grossman, W., Jones, D. & McLaurin, L. P. Wall stress and patterns of hypertrophy in the human left ventricle. J. Clin. Invest. 56(1), 56–64 (1975).
Depre, C. & Taegtmeyer, H. Metabolic aspects of programmed cell survival and cell death in the heart. Cardiovasc. Res. 45(3), 538–548 (2000).
Talman, V. & Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 365(3), 563–581 (2016).
Kawaguchi, M. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123(6), 594–604 (2011).
Khalil, H. et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 127(10), 3770–3783 (2017).
Frangogiannis, N. G. Cardiac fibrosis. Cardiovasc. Res. 117(6), 1450–1488 (2021).
Quaggin, S. E., Vanden Heuvel, G. B. & Igarashi, P. Pod-1, a mesoderm-specific basic-helix-loop-helix protein expressed in mesenchymal and glomerular epithelial cells in the developing kidney. Mech. Dev. 71(1–2), 37–48 (1998).
Hidai, H. et al. Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech. Dev. 73(1), 33–43 (1998).
Robb, L. et al. Epicardin: A novel basic helix-loop-helix transcription factor gene expressed in epicardium, branchial arch myoblasts, and mesenchyme of developing lung, gut, kidney, and gonads. Dev. Dyn. 213(1), 105–113 (1998).
Quaggin, S. E. et al. The basic-helix-loop-helix protein pod1 is critically important for kidney and lung organogenesis. Development 126(24), 5771–5783 (1999).
Ide, S. et al. Transcription factor 21 is required for branching morphogenesis and regulates the gdnf-axis in kidney development. J. Am. Soc. Nephrol. 29(12), 2795–2808 (2018).
Jiang, X. & Yang, Z. Multiple biological functions of transcription factor 21 in the development of various cancers. Onco. Targets Ther. 11, 3533–3539 (2018).
Lu, X. et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat. Genet. 44(8), 890–894 (2012).
Sazonova, O. et al. Characterization of TCF21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 11(5), e1005202 (2015).
Maezawa, Y. et al. Loss of the podocyte-expressed transcription factor Tcf21/Pod1 results in podocyte differentiation defects and FSGS. J. Am. Soc. Nephrol. 25(11), 2459–2470 (2014).
Akama, T. & Chun, T. H. Transcription factor 21 (TCF21) promotes proinflammatory interleukin 6 expression and extracellular matrix remodeling in visceral adipose stem cells. J. Biol. Chem. 293(17), 6603–6610 (2018).
Schunkert, H. et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43(4), 333–338 (2011).
Acharya, A. et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139(12), 2139–2149 (2012).
Kanisicak, O. et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260 (2016).
Takeda, N. et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Invest. 120(1), 254–265 (2010).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11(8), 783–784 (2014).
Travers, J. G. et al. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 118(6), 1021–1040 (2016).
Zhao, W. et al. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J. Mol. Cell. Cardiol. 51(5), 830–838 (2011).
Allawadhi, P. et al. Isoproterenol-induced cardiac ischemia and fibrosis: Plant-based approaches for intervention. Phytother. Res. 32(10), 1908–1932 (2018).
Frangogiannis, N. G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 65, 70–99 (2019).
Bomb, R. et al. Myofibroblast secretome and its auto-/paracrine signaling. Expert Rev. Cardiovasc. Ther. 14(5), 591–598 (2016).
Takeishi, Y. et al. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: Comparison with acute mechanical stretch. J. Mol. Cell Cardiol. 33(9), 1637–1648 (2001).
Esposito, G. et al. Induction of mitogen-activated protein kinases is proportional to the amount of pressure overload. Hypertension 55(1), 137–143 (2010).
Tallquist, M. D. & Molkentin, J. D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 14(8), 484–491 (2017).
Oka, T. et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 101(3), 313–321 (2007).
Zhao, T. et al. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 304(12), H1719–H1726 (2013).
Zhao, Q. et al. TCF21 and AP-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 11(1), 23 (2019).
Iyer, D. et al. Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 14(10), e1007681 (2018).
Tandon, P. et al. Tcf21 regulates the specification and maturation of proepicardial cells. Development 140(11), 2409–2421 (2013).
Tallquist, M. D. Cardiac fibroblast diversity. Annu. Rev. Physiol. 82, 63–78 (2020).
Wang, S. S. et al. Perivenous stellate cells are the main source of myofibroblasts and cancer-associated fibroblasts formed after chronic liver injuries. Hepatology 74(3), 1578–1594 (2021).
Hussain, A. et al. Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J. Exp. Med. 217(8), e20191094 (2020).
Varankar, S. S. et al. Functional balance between Tcf21-Slug defines cellular plasticity and migratory modalities in high grade serous ovarian cancer cell lines. Carcinogenesis 41(4), 515–526 (2020).
Duan, H. X. et al. TCF21 inhibits tumor-associated angiogenesis and suppresses the growth of cholangiocarcinoma by targeting PI3K/Akt and ERK signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 316(6), G763–G773 (2019).
Nurnberg, S. T. et al. Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS Genet. 11(5), e1005155 (2015).
Cui, S. et al. Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development 131(16), 4095–4105 (2004).
Cui, S., Schwartz, L. & Quaggin, S. E. Pod1 is required in stromal cells for glomerulogenesis. Dev. Dyn. 226(3), 512–522 (2003).
Shen, Y. C. et al. TCF21(+) mesenchymal cells contribute to testis somatic cell development, homeostasis, and regeneration in mice. Nat. Commun. 12(1), 3876 (2021).
Lotfi, C. F. P., Passaia, B. S. & Kremer, J. L. Role of the bHLH transcription factor TCF21 in development and tumorigenesis. Braz. J. Med. Biol. Res. 54(5), e10637 (2021).
Gardi, N. L. et al. Discrete molecular classes of ovarian cancer suggestive of unique mechanisms of transformation and metastases. Clin. Cancer Res. 20(1), 87–99 (2014).
Richards, K. L. et al. Methylation of the candidate biomarker TCF21 is very frequent across a spectrum of early-stage nonsmall cell lung cancers. Cancer 117(3), 606–617 (2011).
Smith, L. T. et al. Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc. Natl. Acad. Sci. USA 103(4), 982–987 (2006).
Mokkapati, S. et al. TCF21 promotes luminal-like differentiation and suppresses metastasis in bladder cancer. Mol. Cancer Res. 18(6), 811–821 (2020).
Gooskens, S. L. et al. TCF21 hypermethylation regulates renal tumor cell clonogenic proliferation and migration. Mol. Oncol. 12(2), 166–179 (2018).
Bakshi, D. et al. MassARRAY-based single nucleotide polymorphism analysis in breast cancer of north Indian population. BMC Cancer 20(1), 861 (2020).
Wirka, R. C. et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 25(8), 1280–1289 (2019).
Nakano, Y. et al. A deactivation factor of fibrogenic hepatic stellate cells induces regression of liver fibrosis in mice. Hepatology 71(4), 1437–1452 (2020).
Ganieva, U. et al. Involvement of transcription factor 21 in the pathogenesis of fibrosis in endometriosis. Am. J. Pathol. 190(1), 145–157 (2020).
Zhu, D. et al. Intrapericardial long non-coding RNA-Tcf21 antisense RNA inducing demethylation administration promotes cardiac repair. Eur. Heart J. 44(19), 1748–1760 (2023).
Fu, X. et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 128(5), 2127–2143 (2018).
Acknowledgements
We are grateful to A. Watanabe and N. Tomoda for technical support to H. Ginya and K. Tujimura for their experimental advice. We also thank Dr. Sue Quaggin and Dr. Tuncer Onay in Northwestern University for generating and providing the Tcf21 floxed mice.
Funding
This work was supported by JSPS KAKENHI grant 17H1558 to K. Y. and 17K09687, 20H03572 and 24K10549 to Y. M.
Author information
Authors and Affiliations
Contributions
Y.M., Y.K., and K.Y. designed the study; Y.B., N.K., T.M., S.I, K.I., Y.E., K.A,, M.M., T. S., Y. Y., T. K. carried out experiments; N.Teramoto., A.Y., H.Kaneko., S.F., H.Kato., M.S., R.I, M.Koshizaka., M.Kanda., analyzed the data; Y.B., M.T., Y.M. made the figures; Y.B., Y.M., N.Takayama., K.Y. drafted and revised the paper; all authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baba, Y., Maezawa, Y., Kondo, N. et al. Tcf21 modulates fibroblast activation and promotes cardiac fibrosis after injury via Pdgfrb signaling. Sci Rep 15, 28260 (2025). https://doi.org/10.1038/s41598-025-13102-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13102-3
