
Abstract
Colorectal cancer (CRC) is an important global health challenge, with nearly 2 million diagnosed cases and over 900,000 deaths annually despite therapeutic advancements. The high morbidity and mortality rates underscore the need for more efficient therapies. Three-dimensional (3D) cell culture models have emerged as more physiologically relevant alternatives to traditional two-dimensional (2D) models for drug screening and mechanistic studies. However, generating consistent spheroids across different CRC cell lines presents technical challenges, and protocols remain inconsistent. This study evaluated different 3D culture methodologies, i.e. overlay on agarose, hanging drop, and U-bottom plates without matrix or with methylcellulose, Matrigel or collagen type I hydrogels, across eight CRC cell lines. Multicellular tumour spheroids (MCTS) morphology and cell viability were analysed. Co-cultures with immortalised colonic fibroblasts were explored to improve the physiological relevance of the tumour models. The study provided insights into the morphological and viability characteristics of 3D cultures across multiple CRC cell lines. A novel compact spheroid model using the SW48 cell line was successfully developed. Co-culture experiments with fibroblasts offered additional insights into tumour-stroma interactions in a 3D setting. This study contributes to the advancement of more physiologically relevant in vitro CRC models, potentially enhancing the accuracy of preclinical studies and drug screening processes. The successful 3D model of SW48 expands the repertoire of CRC cell lines available for 3D culture studies. The treatment of regular multi-well plates with anti-adherence solution allows to generate CRC spheroids at significantly lower cost than using cell-repellent multi-well plates. These findings may lead to improved preclinical models for CRC research and drug development.
Similar content being viewed by others


Background
Cancer is one of the leading causes of death globally, with colorectal cancer (CRC) ranking as the third most commonly diagnosed type and the second leading cause of cancer-related mortality when considered both sexes together1. Despite significant advancements in understanding the molecular basis of CRC initiation, progression, and metastasis processes, significant clinical challenges persist particularly for metastatic disease where the 5-years survival rate remains below 20%2,3,4,5. To unravel the molecular mechanisms underlying therapy resistance, it is crucial to advance the development of preclinical cancer models that facilitate deeper insights of tumour biology that ultimately lead to more effective therapies.
Two-dimensional (2D) cell cultures, in which cells grow as a flat monolayer on a dish, have been extensively employed in the initial phases of preclinical research. While 2D cultures have been instrumental to the successful development of numerous drugs currently employed in the treatment of CRC patients, they lack the intricate interactions found in native tumours, such as the tissue-specific architecture, cell-extracellular matrix interactions, spatial organisation, and diversity of cell types that are essential for tissue functionality6. In contrast, three-dimensional (3D) models provide a more comprehensive model of the natural tumour heterogeneity. They feature variations in cellular morphology and exposure to gradients of oxygen, nutrients, and environmental stresses, resulting in inner layers of non-proliferating and necrotic cells that partially recapitulate the cellular and histological differentiation of solid tumours. Unlike 2D cultures, 3D models better preserve tissue-specific architecture, support critical cell-matrix interactions, and maintain appropriate expression levels of essential proteins. These attributes significantly enhance their applicability in studying human tissue physiology and elucidating cancer pathophysiology7,8,9. Although more sophisticated in vivo models, i.e. animal models, are still essential in the latest stages of preclinical research, they present important challenges: low success rates, ethical concerns, much longer experimental times, and higher costs. In this context, preclinical 3D models have been profusely explored in the last years. In vitro 3D models are crucial for understanding tumor biology, advancing drug discovery, and promoting personalized therapeutic strategies. Budhwani et al. (2022) provided a comprehensive review of current cancer modeling platforms, including in vitro, in vivo, ex vivo, and in silico models, tracing their evolution and highlighting the growing need for integrated ‘cancer supermodels’, combining their strengths while mitigating their individual weaknesses10.
Historically, in vitro 3D experimental models encompassed a wide range of spherical cancer cell formations in culture. These models can be generated using diverse techniques, and incorporating different cell types, and consequently they display disparate characteristics. The nomenclature has also been very heterogeneous, complicating comparisons among studies. To better classify these models, a four-category system was proposed: multicellular tumour spheroids (MCTSs), tumorspheres, tissue-derived tumour spheres, and organotypic multicellular spheroids11. MCTSs are generated by aggregation and compaction of multiple cancer cells. Tumorspheres, in contrast, derive from clonal expansion of a single cancer cell. These two models can be generated from immortalised cancer cell lines and are easily expanded, greatly facilitating their use in high throughput analyses. On the other hand, tissue derived tumour spheres are derived from live tissues that, after partial cellular dissociation, are cultured in vitro facilitating the aggregation of the different cells but without preserving their original in vivo organisation. Finally, organotypic multicellular spheroids are formed by cutting small portions of tumour tissue that are cultured in non-adherent conditions, where they form rounded structures while more accurately preserving the original in vivo cellular organisation. Despite the latter two models more reliably mimic the in vivo physiology of tumours, they present important challenges: freshly resected tissues are required; the cancer cells are not easily manipulated with genetic tools; and they can be used for a limited time in culture because the non-tumoral cells hardly proliferate under in vitro conditions.
Among these models, MCTSs have become an essential in vitro model, easily generated and expanded, and allowing straightforward genetic manipulation, thus offering enhanced biological relevance for tumour biology studies and drug screening research7. MCTSs exhibit similarities to in vivo solid tumours in growth kinetics, metabolic rates, proliferation, invasion, and resistance to chemotherapy and radiotherapy12. MCTSs exhibit high cell density, facilitating strong intercellular and cell-extracellular matrix (ECM) communication13. The inhibition of cell attachment to the surface enhances cell-to-cell adhesion, facilitating the spontaneous formation of spheroids11,14. Several methods for producing MCTSs have been developed, including hanging drop (self-aggregation at the bottom of a droplet), liquid overlay (cell suspension on non-adherent surface), agitation-based methods (spinner flasks, rotating systems), scaffold-based methods, microfluidics, and external force methods (centrifugal, magnetic, electric, acoustic forces)15. The development of scaffold-based models has been a significant breakthrough in the field of 3D in vitro culture. Scaffold matrices for 3D culture are made from synthetic or naturally derived polymers that support cell growth and mimic extracellular matrix conditions12. Natural polymers, such as Matrigel, collagen, gelatin, and alginate, are preferred for their biocompatibility and formability16,17. Synthetic polymers, such as methylcellulose18, poly(lactic-co-glycolic) acid (PLGA), and polyethylene glycol (PEG), are also used for 3D scaffold fabrication due to their abundant availability, being metabolically neutral, uniform production, and ability to tailor for specific applications19,20. MCTSs can exhibit different morphologies, including single spheroids, multiple small spheroids, compact and loose aggregates, and single-cell suspensions21. While compact spheroids are often considered to be more representative of in vivo tumor structure and thus more biologically significant, their in vitro morphology heavily relies on the specific cell line and culture conditions employed. Single spheroids, typically formed in 96-well round-bottom plates, are generally more homogeneous in size and shape, making them ideal for high throughput drug screening. Multiple spheroids, usually formed in liquid overlay or hanging drop techniques, vary in size and may merge over time, leading to larger spheroids in advanced culture stages22. The main advantage of this approach is that a very large number of spheroids can be cultured in a single dish. However, single spheroids are more consistent and better suited for standardised experimental approaches23.
The tumour microenvironment is a complex and dynamic mixture of cancer cells, endothelial cells, immune cells, mesenchymal stromal cells, ECM, fibroblasts, and secreted substances, all playing significant roles in tumour development and response to chemo- and immunotherapy24. Incorporating tumour microenvironment components and interacting cell types can improve the relevance of MCTS models for drug screening25,26. Normal fibroblasts in the colorectal microenvironment can be activated by inflammatory and microbial cues into cancer-associated fibroblasts (CAFs), which influence tumor progression through paracrine signaling, direct cell–cell contact, ECM remodeling, immune modulation, and the promotion of therapy resistance27,28,29,30. Co-cultures of CRC organoids and immortalised CAFs significantly change the transcriptional profile of the cancer cells, recapitulating the histological and immunosuppressive characteristics of the very aggressive mesenchymal-like colorectal tumours31. In other cancer types, co-culture models incorporating fibroblasts have demonstrated that targeting the tumour microenvironment can enhance therapeutic efficacy32. All these data underscore the importance of developing more sophisticated 3D in vitro models to study the intricate interactions between cancer, stromal cells, and tumour microenvironment, that influence tumour progression and drug response.
A recent worldwide survey found that, despite over 80% of the researchers recognizing the importance of 3D models, the majority of them do not regularly implement them in their research mainly due to lack of experience and costs33. Establishing standardised and reproducible protocols for generating in vitro tumour models with consistent size, structure, and shape is essential to promoting their widespread use, increasing their clinical relevance in the future and reducing the use of animals for cancer research, thus aligning with the ethics of animal welfare and the 3Rs (replacement, reduction, and refinement of the inclusion of animals in research)33. Three dimensional models are still facing technical challenges, and not all methods reported in the literature are equally effective in producing spheroids12. Some cell types form loose cellular aggregates under conventional 3D culture conditions, and to generate compact spheroids they require additional additives or more sophisticated approaches34. As an example, SW48 CRC cells do not generate compact tumour spheroids and form just irregularly shaped cell aggregates in all tested culture conditions35.
In this study, we characterise the influence of the culture technique on the morphology of CRC tumour spheroids derived from 8 commonly used CRC cell lines, i.e. DLD1, HCT8, HCT116, LoVo, LS174T, SW48, SW480, and SW620. We also present novel straightforward and cost-effective experimental conditions to generate, for the first time, compact spheroids from CRC SW48 cells, and applicable to the other seven CRC cell lines.
Materials and methods
Cell culture
Human colorectal cancer cell lines DLD1 (CCL-221), HCT8 (CCL-244), HCT116 (CCL-247), LoVo (CCL-229), LS174T (CCL-188), SW48 (CCL-231), SW480 (CCL-228) and SW620 (CCL-227), along with the fibroblast cell line CCD-18Co (CRL-1459) were obtained from the American Type Culture Collection (ATCC). STR analysis was employed to authenticate the identity of all cell lines using the AmpFLSTR Identifiler Plus PCR Amplification Kit (Cat.#4486467, Thermo Fisher Scientific, Waltham, MA, USA) following the provided protocol. All cell lines were confirmed as mycoplasma-free using a PCR-based test before initiating experiments and throughout the experimental duration. A more detailed mutational profile of these cell lines, obtained from36, is shown in Supplementary Table 1.
All cell lines were cultured in DMEM/F12 medium (Gibco Cat. #11320033) supplemented with 10% fetal bovine serum (FBS, Gibco Cat # A525671), 2 mM L-glutamine (Gibco Cat. # 25030-024), 1 mM sodium pyruvate (NaPyr, Thermo FIsher Scientific Cat. # 11360070) and Antibiotic-Antimycotic 1x (Gibco Cat. # 15240-062). Cells were cultured in ø100-mm culture plates in a 37 °C incubator with a humidified atmosphere containing 5% CO2. Cell growth was monitored under a microscope, and passages were performed when cells reached 80–90% confluence.
CRC spheroid generation methods
Liquid overlay on agarose
The liquid overlay technique involved culturing cells over a thin layer of an inert, non-adherent substrate. Agarose was chosen for its superior non-adherent properties compared to agar14. A 1% (w/v) agarose solution was prepared in sterilised water, and 5 mL of this solution was poured into ø100-mm culture plates. After 20 min, to allow the agarose to solidify, cell suspensions with a concentration of 106 cells in 10 mL (100 cells/µL) in supplemented-DMEM/F12 medium were added to the plates. Culture plates were placed in a 37 °C incubator with a humidified atmosphere containing 5% CO2.
Hanging drop
To generate 3D cultures of the CRC cell lines using the hanging drop technique, cell suspensions were prepared at 100 cells/µL concentration in supplemented-DMEM/F12 medium. Subsequently, 30 µL drops from a 100 cells/µL suspension (3,000 cells), were dispensed onto the lid of a culture plate. The lid was carefully inverted and placed on top of the dish. 5 mL of PBS 1x was added to the dish to maintain humidity. Culture plates were placed in a 37 °C incubator with a humidified atmosphere containing 5% CO2. This setup allowed the cells to aggregate at the bottom of the drops, facilitating spheroid formation at the liquid-air interface.
Culture in U-bottom cell repellent plates
Greiner CELLSTAR U-bottom 96-well cell repellent plates (CR-plates, Greiner Bio One Cat.#650970) were used to generate MCTSs of homogeneous size per well. Cellular suspensions were prepared at 3 × 104 cells/mL in supplemented-DMEM/F12 medium and seeded 100 µL from a 30 cells/µL suspension (3,000 cells) per well. The initial amount of cells was identical in all experiments generating individual spheroids in multi-well plates and hanging drop methodologies. Culture plates were placed in a 37 °C incubator with a humidified atmosphere containing 5% CO2. In long-term experiments, the medium was changed by aspirating 50 µL of medium from each well and slowly adding 50 µL of fresh medium with hydrogel culture matrices every 3 days.
Hydrogel matrices
To promote the development of more compact 3D tumour cell structures, hydrogel culture matrices were added to the culture media. Methylcellulose (Sigma-Aldrich, Cat.#M7027) cellular matrix was prepared by filtering the stock solution through a 0.22 μm filter and mixed with supplemented-DMEM/F12 to a final 2.5 mg/mL concentration. Other concentrations of methylcellulose were tested on different cell lines (Supplementary Figs. 1 and 2). Matrigel (Corning, Cat.#356234) was thawed at 4 °C for four hours before use and then mixed with supplemented-DMEM/F12 to a final concentration of 2.5% (v/v) as previously reported6,13. Additional Matrigel percentages were tested on different cell lines (Supplementary Fig. 3). Collagen type I from rat tail (Corning, Cat.#354236) working solution (2 mg/mL) was prepared by mixing 670 µL of collagen type I stock, 100 µL PBS 10x, 17 µL NaOH 1 M and 213 µL H2O. After verifying that the pH was around 6.5–7.5, it was mixed with medium supplemented DMEM/F12 to a final concentration of 25 µg/mL as previously reported37. All hydrogel matrix mixtures were kept at 4 °C to prevent gelification. Hydrogel solutions were used to prepare cellular suspensions at 3 × 10⁴ cells/mL, from which 100 µL (3,000 cells) were seeded per well into U-bottom 96-well cell-repellent plates. Prior to pipetting, the cell suspension was thoroughly mixed to ensure homogeneity across cultures. The suspension was carefully dispensed against the well wall using a tilted pipette tip. Gentle handling of plates was crucial after seeding to maintain the structural integrity of the hydrogel-based cultures. Plates were placed in a 37 °C incubator that allowed hydrogel solidification with a humidified atmosphere containing 5% CO2.
SW48 compact spheroid model
Compact tumour spheroid models of the SW48 cell line were developed using SARSTEDT U-bottom 96-well microtest plates (Cat.#82.1582001) (MT-plate). To prevent cell adhesion, the plate wells were rinsed with 200 µL of STEMCELL Technologies Anti-Adherence Rinsing Solution (Cat.#07010) under sterile conditions at room temperature. The solution was incubated in the wells for 5 min, after which it was carefully and thoroughly aspirated, and the wells were washed with 200 µL of serum-free media to remove any residuals that could interfere with subsequent cell culture. Cell suspensions were prepared at 30 cells/µL in supplemented-DMEM/F12 containing 2.5 mg/mL methylcellulose, 2.5% Matrigel, or 25 µg/mL collagen type I. Then, 100 µL of cell suspension (3,000 cells) were dispensed in every well. Culture plates were maintained in a 37 °C incubator with a humidified atmosphere containing 5% CO2.
Fibroblasts co-cultures on CRC MCTS
Co-culture of CRC cell lines with CCD-18Co human fibroblasts were performed at 1:3, 1:1, and 3:1 ratios, in line with previous reports35,38. Cancer cell lines and fibroblasts were collected and counted to generate homogeneous cell suspensions in 2.5 mg/mL methylcellulose complete DMEM/F-12 medium. Cell numbers were adjusted to the specified ratios of cancer cells to fibroblasts in order to generate spheroids containing a total of 3,000 cells.
The 100 µL co-cultures cell suspensions were seeded in MT-plate and placed in a 37 °C incubator with a humidified atmosphere containing 5% CO2.
Cell labelling with vital fluorescent dyes
For co-culture and cell-death analysis, cells were labelled prior to spheroid generation with Cell Tracker Green CMFDA (Thermo Fisher Scientific, Cat.#C7025), BioTracker 555 Orange Cytoplasmatic Membrane Dye (Sigma-Aldrich, Cat. #SCT107), or BioTracker 400 Blue Cytoplasmatic Membrane Dye (Sigma-Aldrich, Cat. #SCT109) fluorescent dyes, following the protocol provided by the manufacturers. The dye stock was diluted 1:100 in serum-free DMEM/F12. Then, 106 cells were incubated in 500 µL of stain solution at 10 µM final concentration for 30 min at 37 °C, collected by centrifugation at 290 g for 5 min, and washed twice with 2 mL of fresh medium to remove the dye excess.
Microscopy analysis of 3D structures
3D culture morphology and cell death micrographs were obtained using a fluorescence microscope (LEICA DM1600B Microscope with Leica Application Suite X v.1.0.7 Software, https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/downloads), sing the 4x or 10x objectives. Brightfield images were used for the morphology analysis, while fluorescence images were used to study the cell death of the MCTS and the co-culture organisation. MCTS morphology was analysed using ImageJ 1.54j (https://imagej.net/)39 (Supplementary Fig. 4.1). Measures reported in this work were rescaled from pixels to µm based on the magnification used to take the pictures.
Morphological parameters were quantified using an automated ImageJ script to ensure objectivity and reproducibility of the analysis (Supplementary Fig. 4.2). This approach minimized evaluator-related subjectivity by eliminating manual measurement steps. Images included in the analysis were selected solely based on technical quality—such as appropriate focus and contrast—from two independent experiments. No selection criteria related to experimental outcomes were applied, in order to avoid selection bias and maintain the impartiality of the evaluation.
The following parameters were automatically analyzed by ImageJ: total area (in µm²), perimeter (in µm), the major and minor axes of a fitted ellipse (in µm), aspect ratio (the ratio of the major to minor axes), roundness (which indicates how closely the shape of the spheroid approaches a perfect circle), and circularity, which measures how close the relationship between a spheroid’s area and perimeter squared is to that of a perfect circle. Compared to roundness, circularity is more affected by an irregular spheroid surface because it directly incorporates the perimeter, which increases with the roughness and irregularities of the spheroid outline. In addition, we calculated the ellipticity-adjusted circularity, estimating a surface smoothness parameter valid for ellipses. Smoothness indicates how much the observed ratio of the area vs. perimeter squared of the spheroid deviates from that of its fitted ellipse, thus providing an estimation of the smoothness or roughness of the spheroid’s surface. Smoothness values near 1 indicate smooth-surfaced spheroids, with values decreasing as the spheroid’s surface becomes more irregular. Descriptions and formulas of the morphological parameters are detailed in Supplementary Table 2.
Also measured were the number of internal holes, their combined area (in µm²), and the percentage of cellular coverage (calculated as the difference between the total area and the area of the holes). The percentage of area covered by cells was considered a descriptor of the compactness of the spheroids. In loose-aggregate spheroids, some very refringent cells under phase contrast microscopy were difficult to differentiate from the background, which could lead to their misidentification as empty spaces and an underestimation of the area covered by cells. To mitigate this, holes below 200 µm2 were automatically filled and counted as regions covered by cells (Supplementary Fig. 4.3). Some compact spheroids - notably the elongated spheroids from DLD1 in Matrigel - exhibited very refringent regions that were not possible to differentiate from the background using color thresholding in ImageJ (Supplementary Fig. 4.4). In those cases, the area covered by cells was manually adjusted to 100% after the automatic ImageJ analysis. 3D micrograph reconstruction of DLD1 MCTSs (Supplementary Fig. 5) was performed using ReViSP40.
Confocal imaging
Confocal imaging was performed using an Abberior STEDYCON super-resolution module coupled to a fully motorised Olympus IX83 body equipped with QUAD-band epifluorescence illumination and 10x, 20x dry objectives plus 40x and 63x oil objectives. STEDYCON smart control software 9.0.815 (https://abberior.rocks/superresolution-confocal-systems/stedycon-smart-control/) was employed for obtaining and analysing the images, and SVI Huygens Pro 24.10 software (https://svi.nl/Huygens-Professional) was employed for image stitching. For spheroid image capturing, spheroids pre-labelled with fluorescent dyes were carefully transferred from the culture plates to bottom-glass µ-Slide 8 Well chambered slides (Ibidi Cat.No:80806), with the optimal refraction index for confocal imaging.
Evaluation of cell death by microscopy
Cell viability was assessed by DAPI staining in all cell lines under the different 3D-culture conditions. First, cells were stained with Cell Tracker Green CMFDA (Thermo Fisher Scientific, Cat.#C7025) before spheroid formation. After 72 h, spheroids were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, Cat.#D1306) at a final concentration of 1 µg/mL in PBS for 30 min in the dark at room temperature. Then, spatial distribution of the DAPI-stained cells was evaluated using a Leica DM6000B fluorescence microscope using a 4x or 10x objective. DAPI staining was chosen over the more common propidium iodide (PI) staining for cell death evaluation because its high tolerance to photobleaching enables a longer analysis time. We confirmed that DAPI and PI staining yielded concordant results (Supplementary Fig. 6).
Evaluation of cell death by flow cytometry
Cell death was quantified by staining with the Zombie Violet Fixable Viability Kit (BioLegend, Cat.#423113), following the manufacturer’s instructions. MCTS were collected with a pipette into separate 1.5 ml tubes at a rate of 16 wells per cell line per condition. The spheroids were decanted by centrifugation at 85 g and washed with PBS 1x to remove any remaining medium or matrix. Then, they were incubated with 1 mL 0.5% Trypsin-EDTA 1x (Gibco, Cat.#15400054) per tube for 15 min with gentle agitation to facilitate cell disaggregation. Cells were washed with PBS 1x and stained with 100 µL of Zombie Violet solution (1:500 dilution of the stock solution) at room temperature during 20 min. Stained cells were centrifuged at 290 g for 5 min, and the cell pellet was washed with 1 mL of PBS 1x. For cell fixation, 1 mL of 4% PFA for 20 min at room temperature was added to each tube. Fixed cells were centrifuged for 5 min at 290 g and washed with PBS 1x. Finally, the cell pellet was resuspended in 300 µL FAC Buffer (1% BSA in PBS 1x). Samples were analysed using a FACSCanto II Flow Cytometer (BD Biosciences, Franklin Lakes, New Jersey, USA), and the results were processed using FlowJo v10 software (https://www.flowjo.com/).
Gene set enrichment analysis
Transcriptional data from seven CRC cell lines cultured in 2D and 3D conditions was obtained from GSE5744641. Data normalisation and differential gene expression analysis was performed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). Genes were ranked by the t-value of the comparison of their transcriptional level in 3D vs. 2D culture conditions. For genes interrogated by more than one probe, the probe with the larger absolute t-value was selected. Hallmark set enrichment analysis was performed using GSEA 4.4.0 (https://www.gsea-msigdb.org/)42 with the following parameters: weighted enrichment statistic, 1,000 permutations, and excluding gene sets with more than 500 or less than 15 genes. Network analysis of the up-regulated and down-regulated gene sets was performed using Cytoscape v3.10.3 (https://cytoscape.org/)43, including exclusively gene sets with FDR-adjusted p-value < 0.01 as nodes, and overlap coefficients larger than 0.5 as edges (connections) between nodes.
Statistical analyses
All experiments were replicated at least three times, with n ≥ 8 spheroids analysed in each independent experiment. Data are presented as the mean ± standard error of the mean. When analysing categorical variables, one-way or two-way analysis of variance (ANOVA) tests, followed by Tukey’s Honestly Significant Difference (HSD) test for pairwise comparisons of factors with more than 2 categories, were used to determine the statistical significance of morphological properties and cell death among different groups. In multivariate models combining continuous and categorical variables, multivariate linear model regressions were fitted. Then, estimated marginal means for each level of the categorical variables were computed, adjusting for the effect of the other variables in the model, using the R package emmeans (estimated marginal means)44. Pairwise comparisons between the levels of the categorical variables were assessed using the pairs() function from the emmeans package, which applies pairwise t-tests on the marginal means. By default, p-values were adjusted for multiple comparisons using Tukey’s method when appropriate. Deviation from normality was tested by Shapiro-Wilk’s test. Differences with p < 0.01 after multi-hypothesis correction were considered statistically significant. All statistical analyses were performed using R Environment for Statistical Computing 4.4.0 (https://www.r-project.org/)45.
Results
Morphological analysis of CRC MCTSs generated by different techniques
We investigated 3D growth characteristics of eight frequently used colorectal cancer cell lines, i.e. DLD1, HCT116, HCT8, LS174T, LoVo, SW480, SW48, and SW620, using three distinct methodologies to induce multicellular tumor spheroid (MCTS) formation via cell aggregation11: liquid overlay on agarose, hanging drop, and CELLSTAR U-bottom 96-well cell repellent plates (CR-plates), with or without the addition of different hydrogels (methylcellulose, Matrigel and collagen type I). Each method resulted in considerable differences in the morphology of the cell cultures (Fig. 1). This study included isogenic or related pairs of cell lines (DLD1⇔HCT8 and SW420⇔SW620) to explore possible differences in their 3D morphology despite their genotypic similarities.

Representative images of CRC cell lines spheroids generated using different techniques and extracellular matrices. The cell lines tested were DLD1, HCT8, HCT116, LoVo, LS174T, SW48, SW480, and SW620. Cells were seeded at 105 cells/mL for liquid overlay on 1% (w/v) agarose, 3,000 cells in 30 µL drops for hanging drop, and 3,000 cells/well in U-bottom 96-well cell repellent plates. The concentrations of the different extracellular matrices were 2.5 mg/mL methylcellulose, 2.5% Matrigel, and 25 µg/mL collagen type I. All pictures were taken 72 h after cell seeding using a Leica DM6000B microscope with 4X objective.
In liquid overlay on agarose, CRC cell lines exhibited diverse morphologies ranging from compact aggregates (DLD1, HCT8, HCT116, and LoVo) to loose aggregates (LS174T, SW48, and SW620) or single-cell suspensions (SW480). The hanging drop technique produced multiple compact aggregates in some cell lines (DLD1, HCT8, and HCT116) and loose aggregates in others (LoVo, LS174T, SW48, SW480, and SW620) (Fig. 1).
CR-plates without hydrogel additives enabled spheroid formation in DLD1, HCT8, HCT116, and LoVo, while LS174T, SW480, and SW620 formed loose aggregates, and SW48 grew as single-cell suspensions (Fig. 1). The addition of 2.5 mg/mL methylcellulose did not significantly alter the 3D morphology compared to hydrogel-free conditions in any of the cell lines (Fig. 1). The addition of 2.5% Matrigel or 25 µg/mL collagen type I, in contrast, resulted in the formation of single and compact spheroids in all cell lines except SW48, which continued to exhibit a single-cell suspension morphology, and failed to generate larger 3D structures by cell aggregation. Matrigel or collagen type I resulted in more compact morphologies for LS174T, SW480, and SW620 cells, which initially formed loose aggregates in CR-plates with ono matrix or methylcellulose (Fig. 1).
HCT8 cultures in CR-plates without a matrix or in the presence of methylcellulose exhibited a mixed morphology, with a central compact spheroid surrounded by cells that do not adhere to the main structure. This was not observed with Matrigel or collagen type I, where HCT8 cells formed single compact spheroids.
The quantitative morphological analysis revealed significant increase in compaction of LS174T, SW480 and SW620 cell lines when cultured in Matrigel or collagen type I cell matrices, reflected in a decrease in total area and an increase in cell-covered area of the MCTS (p < 1 × 10− 6, ANOVA followed by Tukey HSD test, Fig. 2). Similarly, HCT8 exhibited a more compact morphology in Matrigel and collagen type I, losing the mixed morphology observed without matrix or with methylcellulose. In general, methylcellulose did not significantly affect compaction compared to cultures without a matrix, showing similar cell-covered spheroid areas (p = 0.02) and total spheroid areas (p = 0.79). DLD1 and HCT116 consistently exhibited 100% cell-covered area and small total spheroid size when cultured in U-bottom cell-repellent plates, regardless of the presence or absence of matrices (Fig. 2). No major differences in other parameters such as circularity, smoothness, perimeter and roundness, were observed in any of the analysed cell lines when cultured in different matrices (Supplementary Fig. 7).

Compactness analysis of CRC MCTS cultured on different matrices. 3,000 cells/well were seeded in U-bottom cell-repellent plates for 72 h. The concentration of the extracellular matrices were: 2.5 mg/mL methylcellulose, 2.5% Matrigel, and 25 µg/mL collagen type I. Left) Compactness measurements of the micrographs were performed using ImageJ software. Right) The area covered by the cells was calculated by the difference between the total spheroid area and the sum of the area of the holes. Statistical analyses were performed by two-way ANOVA including cell line and matrix type as independent explanatory variables, followed by Tukey’s HSD tests for pairwise comparisons. Deviation from normality was tested using Shapiro-Wilk’s test. The comparisons of Matrigel vs. no-matrix and Collagen type I vs. no-matrix were statistically significant for the cell-covered area and total area (in all cases p < 10− 6). The comparison between methylcellulose vs. no matrix was borderline significant for cell-covered area (p = 0.02) and not significant for total spheroid area (p = 0.79).
Evaluation of CRC spheroids proliferation in long-term cultures
To assess the proliferative capacity of the MCTSs generated in our laboratory, the spheroids were cultured for one week in CR-plates with Matrigel 2.5%. Several morphology parameters of the spheroids (area, aspect ratio, roundness, circularity, %area covered by cells, and smoothness, see methods) were measured every 24 h. In all cell lines, the spheroids showed gradual and continuous growth without reaching plateau, indicating that the cells maintained their proliferative capacity for at least one week. In some cell lines we observed the presence of cells around the spheroid that had either not adhered to the spheroid (SW480) or formed satellite spheroids (DLD1 and HCT116) (Fig. 3, A). CRC spheroids from different cell lines showed similar growth rate, reaching 300–400% at seven days compared to day one. The exception was SW480, which exhibited an apparent delay in its growth curve. The most likely explanation is that, in contrast with the other cell lines, SW480 required two days to reach its maximum compaction, resulting in a smaller area at day 2 than at day 1 (Fig. 3, B).

Long-term cultured of CRC spheroids. (A) Representative images of CRC spheroids cultured over a week. All cell lines were cultured in U-bottom 96-well cell repellent plates with 2.5% Matrigel. Cells were seeded at 3,000 cells/well. All pictures were taken using a LEICA DM6000B microscope with 4X objective. (B) CRC spheroid area measurements using Image J from micrographs taken over one week. Every time point represents data from 8 individual spheroids. The grey shaded areas indicate the 95% confidence intervals of the Locally Estimated Scatterplot Smoothing (loess).
Effect of culture matrix on MCTS cell viability
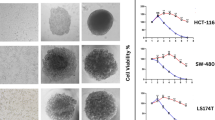
Cell viability in 3D cultures was assessed by microscopy and cytometry (see methods). The microscopy analysis revealed that dead cells were scattered throughout the spheroid structures, without concentrating in clear foci, in all cell lines with the exception of LS174T cells cultured with type I collagen, where cell mortality appeared to be localised in the central region of the spheroids (Fig. 4, A). The quantification by cytometry demonstrated a cell death rate of less than 15% in all cell lines and culture conditions. We found no statistically significant difference in cell viability in any of the tested matrices compared to no matrix (methylcellulose p = 0.92, Matrigel p = 0.11 and collagen type I p = 0.87, p-values calculated by ANOVA followed by Tukey’s HSD tests), with similar cell death values in 3D without matrix and 2D culture condition (p = 0.99). Slight differences in cell death were observed among cell lines, with SW620 having lower overall cell death, while HCT116 and LS174T exhibited higher proportions of dead cells (Fig. 4, B).

Effect of culture matrix on MCTS cell viability. (A) Representative cell viability images of the CRC MCTS. Cells were stained with Cell Tracker Green CMFDA (1 µM) and seeded at 3,000 cells/well in U-bottom 96-well cell repellent plates. The concentrations of the extracellular matrices were 2.5 mg/mL methylcellulose, 2.5% Matrigel, and 25 µg/mL collagen type I. After 72 h, the MCTS were stained with DAPI to label dead cells, and pictures were taken using a Leica DM6000B microscope with 4X or 10X. (B) Cell viability quantification of the MCTS cultured in different matrices. Cells were stained at 72 h with Zombie Violet (BioLegend), and cell death was analysed by flow cytometry. Statistical analyses were done independently for every cell line, using one-way ANOVA with culture condition as explanatory variable followed by Tukey’s HSD tests for pairwise comparisons (n = 3 independent replicates per group of 12 spheroids each). No statistical significant differences were found between cell death rate in absence of matrix vs. any of the tested matrices, or between culturing the cells in 2D vs. 3D.
Development of a novel compact SW48 spheroid model
The cell line SW48 did not form compact MCTSs under any tested conditions. Moreover, SW48 cells did not aggregate cultured in CR-plates with or without hydrogel matrices (Fig. 1), a limitation seemingly inherent to this cell line as no SW48 MCTS models were found in the literature. In our preliminary experiments, we found that methylcellulose enabled the formation of spheroids from certain CRC cell lines in SARSTEDT U-bottom 96-well microtest plates (MT-plates), which are not treated for cell culture and are substantially more economical than CR-plates. We evaluated whether supplementing MT-plates with methylcellulose, Matrigel, or collagen I would facilitate the generation of MCTSs in the eight cell lines. The results showed that, in the absence of hydrogels, as well as with Matrigel or collagen I, none of the cell lines formed MCTS in MT-plates. However, the addition of methylcellulose enabled MCTS aggregation in all cell lines (Supplementary Fig. 8), producing morphologies very similar to those observed in CR-plates but at a fraction of the cost. Notably, SW48 generated loose aggregates in MT-plates with methylcellulose.
Next, we evaluated the effect of treating the MT-plates with an anti-adherence rinsing solution from STEMCELL Technologies, where we cultured SW48 in absence or presence of methylcellulose. In parallel, we cultured two other cell lines, i.e. HCT116 and LS174T, as controls of compact and loose MCTSs, respectively (Supplementary Fig. 9). This experiment revealed that the treatment with the anti-adherence solution facilitated the formation of MCTSs in all three cell lines even in the absence of methylcellulose. We then investigated if the addition of Matrigel or collagen I to the rinsing solution treated MT-plates would lead to the formation of more compact SW48 MCTSs. These conditions allowed the generation of SW48 spheroids, exhibiting a more compact and spherical morphology in the presence of Matrigel and collagen type I compared to cultures in the absence of a matrix or cultured with methylcellulose. We monitored SW48 3D morphology in 7 days culture in anti-adherence-treated MT-plates revealed that SW48 cells formed smaller and more compact spheroids in Matrigel or collagen I, compared to cultures without hydrogels (Fig. 5). While similar in size (p = 0.99) and compactness (p = 0.36), Matrigel-cultured spheroids were notably less round (0.81 ± 0.09) and featured a smoother surface (0.35 ± 0.06) than those in collagen I (roundness: 0.86 ± 0.08, p = 0.0001; smoothness: 0.21 ± 0.07, p < 0.0001). All p-values were obtained from t-tests on marginal means, with adjustments for culture time and multiple comparisons (Tukey’s method) (Fig. 5). Cell viability measured at 72 h showed no statistical significant effect of using the anti-adherence solution treatment (Fig. 5).

SW48 compact spheroid models. (A) Representative images of MCTS generated in MT-plates treated with an anti-adherence rinsing solution. Cells were seeded at 3,000 cells per well in different conditions: no matrix, 25 µg/mL collagen type I, and 2.5% Matrigel. Pictures were taken using a LEICA DM6000B microscope with 4X objective, and analysed using an automated ImageJ script (Supplementary Fig. 4.2). The complete set of pictures are provided in the Supplementary File TimeSeries.zip. (B) Morphological analysis of SW48 spheroids cultured with no matrix, collagen I or Matrigel. The addition of collagen I or Matrigel generated smaller, rounder, smoother and more compact SW48 spheroids. P-values indicate the statistical significance of the effect of the matrix after correcting for time (multivariate linear regression, with time and matrix as explanatory variables). (C) Viability evaluation by DAPI staining of SW48 spheroids cultured for 72 h in anti-adherence treated MT-plates in absence of matrix, or with 2.5 mg/mL methylcellulose, 25 µg/mL collagen type I, or 2.5% Matrigel. MCTS were stained with DAPI (1 µg/mL) to label dead cells. Pictures were taken using a LEICA DM6000B microscope with 10X objective.
Applicability of Non cell-repellent plates to generate CRC spheroids
In view of the success in developing compact SW48 MCTS by treating MT-plates with anti-adherence rinsing solution, we explored the applicability of this method in other CRC cell lines. We performed a 7-day culture of the 8 cell lines, comparing their morphology in Matrigel using CR-plates vs. anti-adherence-treated MT-plates. Our results demonstrate that the morphology and growth dynamics of MCTSs cultured in anti-adherence treated MT-Plates with Matrigel was extremely similar to that in CR-Plates with Matrigel (Fig. 6), with the exception of SW48 that did not generate spheroids in CR-Plates.

Morphology of CRC MCTSs generated in CR-plates and anti-adherence treated MT-plates. (A) Representative images of MCTS from the 8 CRC cell lines in CR-Plates (left) and anti-adherence treated MT-Plates (right), in Matrigel. The complete set of pictures are provided in the Supplementary File TimeSeries.zip. (B) Morphological analysis of MCTSs shown in panel A. P-values indicate the statistical significance of the effect of the type of plate (CR-Plate vs. MT-Plate) after correcting for time (multivariate linear regression, with time and plate type as explanatory variables).
DLD1 exhibits elongated morphology when cultured in matrigel
DLD1 cells exhibited a very unique morphology when cultured in Matrigel (Fig. 6)l. After just 48 h of culture, DLD1 MCTSs developed protrusions that extended into highly elongated structures. We investigated this morphology over a 7-day period, monitoring the growth and morphological characteristics of DLD1 spheroids every 24 h in anti-adherence solution-treated MT-plates under three conditions: without matrix, with Matrigel, and with collagen I. The results revealed that all DLD1 MCTSs cultured in Matrigel formed extremely elongated structures, but not when cultured in collagen I or in absence of hydrogels (Supplementary Fig. 5 and Supplementary File TimeSeries.zip). This morphology was unique to DLD1, and not observed in MCTSs from the isogenic HCT8 cell line (Fig. 6).
Effect of co-culture with colonic fibroblasts on the morphological characteristics of CRC MCTS
In view of the morphological changes observed when the CRC cells were cultured in Matrigel or collagen I matrices (Fig. 1), we investigated if the co-culture with colonic fibroblasts, which are key components of the tumor microenvironment and extracellular matrix in vivo, would also affect the in vitro spheroid morphology. We selected CC18-Co, an immortalized cell line widely used as a colonic fibroblast model38, and four CRC cell lines: LS174T and SW480, which did not generate compact spheroids in methylcellulose, and HCT116 and DLD1, which did form compact spheroids. CRC cells and CC18-Co cells were mixed at a 1:1 ratio, and seeded together in MT-Plates with 2.5 mg/mL methylcellulose. Additional tumor cells : fibroblast ratios, i.e. 1:3, 1:1, and 3:1, selected based on prior literature35, were also tested and yielded similar results (Supplementary Fig. 10). To distinguish both cell types, fibroblasts and CRC cells were previously stained with different vital dyes (see methods). The dyes were swapped to study possible differences in cell distribution or morphology depending on the dye used. In HCT116 and DLD1, fibroblasts formed a single, dense, and spherical structure at the centre of the spheroids. In contrast, when co-cultured with LS174T or SW480, fibroblasts aggregated in the centre of the spheroids, and in several smaller foci scattered throughout the spheroids (Fig. 7). Compaction analysis after 48 h of co-culture revealed that DLD1 and HCT116 spheroids maintained their 100% compaction after adding fibroblasts. LS174T and SW480 cultures, which exhibited ~ 95% compaction in the absence of fibroblasts, became more compact when CCD-18Co cells were added. The increase in compaction was particularly evident in LS174T, where it increased from 94.8 ± 2.0% without fibroblasts to 99.8 ± 0.2% with fibroblasts (p < 10− 6, two-way ANOVA with dye and fibroblasts as factors). SW480 co-cultures with fibroblasts showed a smaller increase in compaction, from 93.8 ± 1.9% to 95.7 ± 2.1, although it was still statistically significant (p = 0.002, two-way ANOVA with dye and fibroblasts as factors) (Fig. 7).

Morphology, distribution and compactness of CRC spheroids cultured with colonic fibroblasts. (A) CRC cells stained with Biotracker 400 Blue and CCD-18Co cells stained with Biotracker 555 Orange (first column), or these cells with the dyes swapped (second column), were mixed at 1:1 proportions. Pictures were taken after 72 h of co-culture. The scale bar represents 75 μm. (B) The compaction of the cancer cells within the spheroids was measured after subtracting the area covered by the fibroblasts, since the fibroblasts formed dense foci that would increase the total spheroid compaction. (C) Percentage of the area covered by cancer cells in spheroids co-cultured with and without fibroblasts. Colours indicate the dye used to label the cancer cells (Biotracker 400 Blue or Biotracker 555 orange). Light blue or orange colours represent the CRC spheroids without CCD-18Co, and dark blue and orange indicate the co-cultures with fibroblasts. *** p-value < 0.01 (two-way ANOVA with dye and fibroblasts as explanatory variables, applied independently for each cell line).
Discussion
Cancer cell lines cultured in 2D have been fundamental pre-clinical models for the development of virtually all the drugs currently used in cancer treatments46. However, cells cultured in 2D monolayers are not representative of cells in the complex microenvironment of a tumour tissue. 3D tumour cell cultures provide a more comprehensive model of the natural tumour heterogeneity, better preserving tissue-specific architecture, support critical cell-matrix interactions, and maintain more physiologically relevant protein expression levels. However, developing 3D models is still not well standardised and presents certain limitations for widespread implementation12. One of the challenges faced by these models is the variation of the morphology of the cultures, depending on the employed methodology. Other researchers have studied the methodology impact in the morphology of breast cancer spheroids13, but to the best of our knowledge this has not been explored in depth in spheroids derived from CRC cell lines.
In this work, we have compared several of the most commonly used techniques to develop multicellular tumour spheroids from CRC cell lines: liquid overlay on agarose, hanging drop, and culture in U-bottom cultures plates, with or without the addition of hydrogel matrices. The liquid overlay on agarose technique allows anchorage-free culture on a large scale, which is particularly valuable for assays requiring substantial amounts of biological material. Hanging drop technique allows to generate hundreds of independent spheroids in a very short time and with minimal expense. However, the agarose liquid overlay and hanging drop techniques failed to generate single compact spheroids in most of the tested CRC cell lines. Ultra-low adherence U-bottom plates offer an advantage by facilitating cell proximity and adhesion, resulting in more compact structures. Additionally, these plates are well-suited for screening experiments, as each well can be used to test different conditions or drugs independently. Our results indicated that the addition of cell-matrix additives to the culture medium increases its consistency and promotes the compactness of the cell cultures6,13. Our results demonstrate that Matrigel and collagen type I are superior matrices to generate compact spheroids in LS174T, SW620 and SW480, and to a lesser extent in HCT8 and DLD1. In contrast, HCT116 and DLD1 do not require the addition of a hydrogel matrix to form compact spheroids. Although methylcellulose has been described in the literature as an additive that enhances 3D cultures in astrocyte spheroids47, in our hands it did not affect the degree of compaction of CRC cultures compared to cultures without an additional matrix. This is likely explained by the fact that it is biologically inert and may not have cell-cell adhesion enhancing effect beyond the increased density of the medium. Nevertheless, the addition of methylcellulose prevents cell adhesion to the culture plates, allowing to generate spheroids even in regular U-bottom plates, thus offering an economical alternative to low-binding plates which can result in a significant economic factor for high-throughput experiments. Of note, we found differences in morphology between DLD1 and HCT8, particularly when cultured without matrix or with methylcellulose (Fig. 1), despite the fact these two cell lines are isogenic and derived from the same CRC (Supplementary Fig. 1). Moreover, DLD1 but not HCT8 generated very irregular and elongated MCTSs when cultured in Matrigel (Fig. 6 and Supplementary Fig. 5). Similarly, SW480 and SW620, isolated from the same cancer patient, exhibited substantial morphological differences when cultured in Matrigel (Figs. 3 and 6). Thus, transcriptional differences not necessarily driven by specific gene mutations, might also influence the morphology of CRC cells cultured in 3D.
In contrast with 2D cultures, cells cultured in 3D are subjected to a gradient of oxygen and nutrients from the spheroid surface towards the innermost part, which could affect cellular growth rates and viability48,49. Other researchers have analysed transcriptional changes associated with 3D culture of CRC cell lines41. Although Luca et al. did not mention hypoxia in their paper, the GSEA reanalysis of their data confirmed hypoxia-response genes as the top upregulated gene set, and down-regulation of many gene sets related to DNA duplication and cell division (Supplementary Fig. 11).
We have found no substantial differences in cell viability in any of the tested matrices, nor an evident accumulation of dead cells in the nucleus of the spheroids in the 3D models tested. The cell death rates of each cell line were similar in 2D and 3D, and not influenced by the level of compaction of the spheroids. Moreover, all MCTSs maintained their proliferative potential for at least 7 days, with no evidence of growth limitation. Other authors have analysed the 3D growth dynamics and drug resistance of 26 cancer cell lines from the NCI60 panel, including seven CRC cell lines, in liquid-overlay culture for 7 days50. While their study shares important similarities with ours—such as the time-course morphological analysis of CRC cell line MCTSs—a direct comparison of morphology and growth dynamics is not possible. This is because while our MCTSs were generated from a fixed number of cells at time zero, they initiated their spheroids with varying cell numbers depending on the cell line, aiming to standardize spheroid size at 370–400 μm in diameter at 96 h. Additionally, they generated the spheroids in absence of hydrogel matrices, while according to our results the addition of hydrogels (particularly Matrigel or collagen I) has a pronounced effect on spheroid morphology, including size. This effect is especially notable in cell lines that do not form compact spheroids in the absence of Matrigel or collagen I, and is most evident in DLD1, which generates very elongated morphologies in Matrigel (Fig. 6 and Supplementary Fig. 5).
To the best of our knowledge, compact spherical structures of SW48 have not previously been achieved using conventional approaches35. Our experiments revealed that standard CR-plates for in vitro 3D model generation were unsuitable for SW48 spheroid formation (Fig. 1). However, by treating regular MT-plates with an anti-adherence solution, and adding hydrogels, we have been able to consistently generate SW48 spheroids, offering a novel 3D model of this cell line. Compared to other 3D culture models, this approach allowed the establishment of a new model for this cell line highlighting its versatility, ease of work and applicability. Furthermore, this technique can also be used to generate MCTSs from other tested cell lines, providing a significantly more cost-effective alternative to CR-plates, which could be advantageous for large-scale screening projects.
Our co-culture results between CRC cell lines and fibroblasts show a localized spatial distribution of cell types, with fibroblasts concentrated at the center of the spheroid (Figs. 7 and 8). This phenomenon has been previously described in similar 3D models. The central positioning of fibroblasts may be influenced by differential adhesion, migration, and proliferation rates between the two cell types. Cancer cells often exhibit higher motility and proliferative capacity, allowing them to migrate toward the periphery of the spheroid, while the less motile fibroblasts remain inward. This phenomenon is supported by time-lapse imaging and cell tracking studies, which show that either cancer cells migrate outward or fibroblasts actively migrate toward the center during spheroid formation51. Furthermore, the initial seeding ratio of cancer cells to fibroblasts is a critical determinant of spatial organization. When fibroblast numbers are low relative to cancer cells, central clustering of fibroblasts is more likely to occur. Increasing the proportion of fibroblasts leads to a more even distribution throughout the spheroid52. Cancer associated fibroblasts play an essential role in tumour progression53, modulating CRC cell proliferation, apoptosis, and drug responses38. Our results indicate a compaction promoting effect when CRC cells are co-cultured with colonic fibroblasts. A similar effect has also been described in pancreatic cancer cells54. This highlights the important role of fibroblasts in cancer and their implementation in vitro tumour cell cultures to achieve more biologically representative models.

Cellular organization of CRC spheroids cultured with colonic fibroblasts. (Top) MCTSs of DLD1 (left) or HCT116 (right) stained with Cell Tracker Green CMFDA (green) co-cultured with CCD-18Co fibroblasts stained with BioTracker 555 Orange Cytoplasmatic Membrane Dye (red). Images were taken after 48 h of culture in MT-Plates with 2.5 mg/mL methylcellulose, using Images were taken in a LLEICA DM1600B fluorescence microscope with 4X objective. (Middle) Stitched images of spheroids collected, decanted, and transferred to bottom-glass 8-well chambered slides (three spheroids per chamber), imaged using an Abberior STEDYCON super-resolution confocal microscope. Despite random orientation after transfer, CCD-18Co fibroblasts (red) consistently localize to the spheroid core. Yellow triangles indicate spheroids selected for Z-stack analysis. (Bottom) Composite Z-stack images of a DLD1/CCD-18Co MCTS (left) and a HCT116/CCD-18Co MCTS (right), with slices separated by 10 μm. CCD-18Co fibroblasts (red) occupy the central region, surrounded by cancer cells (green). Fluorescence signal intensity notably decreases beyond 50 μm depth.
Conclusions
This study presents a comprehensive analysis of different methods for establishing multicellular colorectal tumour spheroids of widely used colorectal cancer cell lines, identifying substantial morphological differences depending on the employed methodology. These differences emphasise the importance of standardised 3D in vitro culture systems in cancer research. Our results measure the effect of the 3D technique, presence of hydrogels, type of plate, culture additives, and co-cultures with colonic fibroblasts on the morphology and viability of in vitro 3D CRC models. We describe experimental conditions to generate compact SW48 spheroids, which had not been described before. We also identified a particular morphology of DLD1 cells cultured in Matrigel, where they generated very elongated 3D structures. Of note, those structures were not observed in the isogenic HCT8 cell line.
This work contributes to the development of reliable and reproducible 3D tumour models to enhance our understanding of CRC biology. Despite their limitations, cell-line-derived MCTSs offer an interesting alternative for fast, inexpensive, flexible, and scalable generation of in vitro models for screenings and functional studies. Future research should focus on refining these models to better simulate human tumour characteristics, ultimately contributing to more effective cancer therapies. As an improvement of the cell line-derived 3D MCTS presented in this work, expanding co-culture systems to include not only fibroblasts but also immune and endothelial cells would more accurately mimic the in vivo tumor microenvironment, and will enable more precise drug screening.
Data availability
Morphological and viability measurements raw data, as well as unprocessed individual pictures of all the spheroids analysed in this work are available upon request to Sergio Alonso (salonsou@igtp.cat).
Abbreviations
- CAF:
-
cancer-associated fibroblasts
- CR-plate:
-
CELLSTAR U-bottom 96-well cell repellent plates
- CRC:
-
colorectal cancer
- CSC:
-
cancer stem cells
- DAPI:
-
4′,6-diamidino-2-phenylindole
- MCTS:
-
multicellular tumour spheroid
- MT-plate:
-
SARSTEDT U-bottom 96-well microtest plate (not treated for cell culture)
- NSF:
-
non-spheroid forming
- PBS:
-
Phosphate-buffered saline
- PEG:
-
polyethylene glycol
- PFA:
-
paraformaldehyde
- PI:
-
propidium iodide
- PLGA:
-
poly(lactic-co-glycolic) acid
- TIC:
-
tumour-initiating cells
References
Morgan, E. et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut 72, 338–344 (2023).
Schmoll, H. J. et al. ESMO consensus guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making. Ann. Oncol. 23, 2479–2516 (2012).
Van Cutsem, E. et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 27, 1386–1422 (2016).
Araghi, M. et al. Colon and rectal cancer survival in seven high-income countries 2010–2014: variation by age and stage at diagnosis (the ICBP SURVMARK-2 project). Gut 70, 114–126 (2021).
Gmeiner, W. H. Recent advances in therapeutic strategies to improve colorectal cancer treatment. Cancers 16, 1029 (2024).
Badea, M. A. et al. Influence of matrigel on single- and multiple-spheroid cultures in breast cancer research. SLAS Discov. 24, 563–578 (2019).
Pampaloni, F., Reynaud, E. G. & Stelzer, E. H. K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell. Biol. 8, 839–845 (2007).
Langhans, S. A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 9, 6 (2018).
Manduca, N., Maccafeo, E., De Maria, R., Sistigu, A. & Musella, M. 3D cancer models: one step closer to human studies. Front. Immunol. 14, 1175503 (2023).
Budhwani, K. I., Patel, Z. H., Guenter, R. E. & Charania, A. A. A hitchhiker’s guide to cancer models. Trends Biotechnol. 40, 1361–1373 (2022).
Weiswald, L-B., Bellet, D. & Dangles-Marie, V. Spherical Cancer Models Tumor Biology Neoplasia ;17: 1–15. (2015).
Han, S. J., Kwon, S. & Kim, K. S. Challenges of applying multicellular tumor spheroids in preclinical phase. Cancer Cell. Int. 21, 1–19 (2021).
Froehlich, K. et al. Generation of multicellular breast cancer tumor spheroids: comparison of different protocols. J. Mammary Gland Biol. Neoplasia. 21, 89–98 (2016).
Ryu, N. E., Lee, S. H. & Park, H. Spheroid culture system methods and applications for mesenchymal stem cells. Cells 8 https://doi.org/10.3390/cells8121620 (2019).
Lee, S. W. et al. In vitro lung cancer multicellular tumor spheroid formation using a microfluidic device. Biotechnol. Bioeng. 116, 3041–3052 (2019).
Benien, P. & Swami, A. 3D tumor models: history, advances and future perspectives. Future Oncol. 10, 1311–1327 (2014).
Nunes, A. S., Barros, A. S., Costa, E. C., Moreira, A. F. & Correia, I. J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 116, 206–226 (2019).
Longati, P. et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 13, 95 (2013).
Naz, A., Cui, Y., Collins, C. J., Thompson, D. H. & Irudayaraj, J. PLGA-PEG nano-delivery system for epigenetic therapy. Biomed. Pharmacother. 90, 586–597 (2017).
Izquierdo, R. et al. Biodegradable PCL scaffolds with an interconnected spherical pore network for tissue engineering. J. Biomed. Mater. Res. A. 85A, 25–35 (2008).
Vinci, M. et al. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 10, 29 (2012).
Achilli, T-M., Meyer, J. & Morgan, J. R. Advances in the formation, use and Understanding of multi-cellular spheroids. Expert Opin. Biol. Ther. https://doi.org/10.1517/14712598.2012.707181 (2012). [cited 26 Jun 2024].
Mehta, G., Hsiao, A. Y., Ingram, M., Luker, G. D. & Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control Release. 164, 192–204 (2012).
Yau, J. N. N. & Adriani, G. Three-dimensional heterotypic colorectal cancer spheroid models for evaluation of drug response. Front. Oncol. 13, 1148930 (2023).
Kenny, H. A. et al. Erratum: Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat. Commun. 7, 10649 (2016).
Majety, M., Pradel, L. P., Gies, M. & Ries, C. H. Fibroblasts influence survival and therapeutic response in a 3D Co-Culture model. PLoS One. 10, e0127948 (2015).
Sahai, E. et al. A framework for advancing our Understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 20, 174–186 (2020).
Biffi, G. et al. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Calon, A. et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 22 https://doi.org/10.1016/j.ccr.2012.08.013 (2012).
Han, Y., Zhang, Y., Jia, T. & Sun, Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour Biol. 36, 1385–1394 (2015).
Strating, E. et al. Co-cultures of colon cancer cells and cancer-associated fibroblasts recapitulate the aggressive features of mesenchymal-like colon cancer. Front. Immunol. 14, 1053920 (2023).
Keller, F., Rudolf, R. & Hafner, M. Towards optimized breast cancer 3D spheroid mono- and co-culture models for Pharmacological research and screening. J. Cell. Biotechnol. 5, 89–101 (2019).
Martinez-Pacheco, S. & O’Driscoll, L. Pre-clinical in vitro models used in cancer research: results of a worldwide survey. Cancers 13, 6033 (2021).
Ivascu, A. & Kubbies, M. Diversity of cell-mediated adhesions in breast cancer spheroids. Int. J. Oncol. 31, 1403–1413 (2007).
Zoetemelk, M., Rausch, M., Colin, D. J., Dormond, O. & Nowak-Sliwinska, P. Short-term 3D culture systems of various complexity for treatment optimization of colorectal carcinoma. Sci. Rep. 9 https://doi.org/10.1038/s41598-019-42836-0 (2019).
Berg, K. C. G. et al. Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol. Cancer. 16, 116 (2017).
Calori, I. R., Alves, S. R., Bi, H. & Tedesco, A. C. Type-I collagen/collagenase modulates the 3D structure and behavior of glioblastoma spheroid models. ACS Appl. Bio Mater. 5, 723–733 (2022).
Koh, B., Jeon, H., Kim, D., Kang, D. & Kim, K. R. Effect of fibroblast co-culture on the proliferation, viability and drug response of colon cancer cells. Oncol. Lett. 17, 2409 (2019).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to imageJ: 25 years of image analysis. Nat. Methods. 9, 671–675 (2012).
Piccinini, F., Tesei, A., Arienti, C. & Bevilacqua, A. Cancer multicellular spheroids: volume assessment from a single 2D projection. Comput. Methods Programs Biomed. 118, 95–106 (2015).
Luca, A. C. et al. Impact of the 3D microenvironment on phenotype, gene expression, and EGFR Inhibition of colorectal cancer cell lines. PLoS One. 8, e59689 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550 (2005).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Lenth, R. V. Emmeans: estimated marginal means, aka Least-Squares means. CRAN: contributed packages. R Foundation. https://doi.org/10.32614/cran.package.emmeans (2017).
R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. (2019). Available: https://www.R-project.org/
Shoemaker, R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 6, 813–823 (2006).
Tao, F. et al. Rapid and stable formation method of human astrocyte spheroid in a high viscous Methylcellulose medium and its functional advantages. Bioengineering 10, 349 (2023).
Kamatar, A., Gunay, G. & Acar, H. Natural and synthetic biomaterials for engineering multicellular tumor spheroids. Polymers 12 https://doi.org/10.3390/polym12112506 (2020).
Jensen, C. & Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 7, 33 (2020).
Friedrich, J., Seidel, C., Ebner, R. & Kunz-Schughart, L. A. Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324 (2009).
Murphy, R. J., Gunasingh, G., Haass, N. K. & Simpson, M. J. Formation and growth of Co-Culture tumour spheroids: new Compartment-Based mathematical models and experiments. Bull. Math. Biol. 86, 1–34 (2023).
Yakavets, I. et al. Advanced co-culture 3D breast cancer model for investigation of fibrosis induced by external stimuli: optimization study. Sci. Rep. 10, 1–11 (2020).
Lotti, F. et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 210, 2851–2872 (2013).
Kuen, J., Darowski, D., Kluge, T. & Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One. 12, e0182039 (2017).
Acknowledgements
The authors sincerely thank Gerard Requena-Fernández from the IGTP Cytometry Unit for his expert guidance on cell viability experiments. They are also indebted to Jakub Chojnacki, head of the IGTP Microscopy Unit, for his assistance in acquiring and analyzing images using the Leica DM6000B microscope, the Abberior STEDYCON super-resolution module, and the Huygens image analysis software. The authors also express their gratitude to the entire Lab Managing team of the IGTP for their valuable assistance and experimental support provided at the institution.
Funding
This work has been supported by grants to SA from the Fundación Mutua Madrileña (AP174232020) and Instituto de Salud Carlos III Spanish Ministry of Science, Innovation and Universities (FIS PI21/01766). CMS contract was funded by Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR), with funds from the INVESTIGO programme (100008CT2), European Regional Development Funds (FEDER). AFC received a summer-internship fellowship from the Spanish Association Against Cancer (AECC).
Author information
Authors and Affiliations
Contributions
BG, CMS, GdMG and IMC set up the 3D in vitro models using different methodologies. CMS and BG performed proliferation, viability, matrix comparison and SW48 model development assays. GdMG and BG. carried out the co-culture assays with fibroblasts. SA, BG, and CMS performed the micrograph analysis, ImageJ scripting, and statistical tests. BG and GdMG generated the co-culture MCTSs, and SA and AFC took and analysed the confocal images. SA, BG and CMS designed the experimental strategy. SA and BG supervised the work. CMS, BG and SA interpreted the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Consent for publication
All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mateos-Sánchez, C., González, B., de Miguel-García, G. et al. Comparative analysis of 3D-culture techniques for multicellular colorectal tumour spheroids and development of a novel SW48 3D-model. Sci Rep 15, 27687 (2025). https://doi.org/10.1038/s41598-025-13588-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13588-x
