
Abstract
RNA 5-methylcytosine (m5C) modification is a crucial epigenetic regulation, and aberrant m5C methylation is associated with the pathogenesis of certain cancers. However, the role and regulatory mechanisms of RNA m5C modification in multiple myeloma (MM) remain unclear. This study aimed to investigate the function and regulatory mechanisms of the primary m5C methyltransferase, NOP2/Sun RNA methyltransferase family member 2 (NSUN2), in MM. The results demonstrated NSUN2 overexpression in patients with MM, and higher NSUN2 levels were associated with poorer outcomes. In addition, elevated global RNA m5C levels were identified in specimens from MM patients, and NSUN2 knockdown decreased RNA m5C levels. Furthermore, NSUN2 knockdown suppressed cell proliferation, promoted apoptosis in vitro, and restrained the progression of xenograft tumors in vivo. Mechanistically, m5C methylated RNA immunoprecipitation (meRIP)-sequencing and RIP-quantitative polymerase chain reaction (RIP-qPCR) assays were applied to screen the candidate targets of NSUN2-mediated m5C modification and huntingtin interacting protein 1 (HIP1) was identified as the target. NSUN2-mediated m5C methylation upregulated HIP1 by enhancing HIP1 mRNA stability. Moreover, HIP1 overexpression counterbalanced the inhibitory effect of NSUN2 knockdown. In conclusion, we propose a novel mechanistic insight into the NSUN2/m5C-HIP1 signaling axis that contributes to the pathogenesis of MM. Thus, NSUN2 can be a novel prognostic biomarker in patients with MM and targeting NSUN2 may be a promising therapeutic strategy.
Similar content being viewed by others
Introduction
Multiple myeloma (MM) is the second most common malignant hematologic tumor, which is mainly caused by malignant proliferation of plasma cells and abnormal globulin secretion1. Although several effective treatment strategies, including immunomodulatory drugs, proteasome inhibitors, and new potent monoclonal antibodies, have improved clinical outcomes, MM still remains incurable and fatal2,3. Several studies have identified contributing factors in MM progression, such as genetic aberrations, epigenetic mechanisms4 and alterations in the bone marrow microenvironment5,6. However, the pathogenesis of MM remains unclear. Therefore, continuing the investigation of the mechanisms leading to MM onset and advancement and identifying potential targets for effective treatments is crucial.
RNA modifications such as N6–methyladenosine (m6A), m5C, and pseudouridine are critical in the posttranscriptional modulation of gene expression7,8. They are associated with the pathogenesis and progression of various diseases9,10. Several studies have revealed that MM progression is regulated by RNA m6A modifications11,12,13. However, the regulatory roles of m5C have not been fully clarified. Recently, m5C modification has been extensively studied in various RNA types, including mRNAs, transfer RNAs, ribosomal RNAs, and other non-coding RNAs14,15. Similar to m6A, m5C modification is a dynamic and reversible RNA process, primarily carried out by NOP2/Sun RNA methyltransferase family member 2 (NSUN2) and DNA methyltransferase 216. In contrast, ten-eleven translocation 2 (TET2) and TET3, which act as m5C demethylases, eliminate the methyl group from m5C-methylated bases17. Moreover, m5C can be identified by Aly/REF export factor (ALYREF) and Y-box binding protein 1 (YBX1), which have been verified as m5C-binding proteins18. The RNA m5C modification influences various biological processes, including early embryogenesis, tissue homeostasis, myelopoiesis, and tumorigenesis19. However, the functional importance of RNA m5C dysregulation in MM and its underlying regulatory mechanisms remain unclear.
NSUN2, a key m5C methyltransferase, plays critical roles in various biological processes, including increasing protein production20,21 promoting cellular proliferation22 and regulating cellular senescence23. Abnormal NSUN2 expression plays a crucial role in developing and progressing multiple types of cancers, such as lung, bladder, liver, kidney, breast, prostate, thyroid, and stomach cancers24. NSUN2-mediated m5C modifications primarily regulate cell proliferation25 influence cellular differentiation26 and facilitate tumor invasion27. Therefore, this study sought to investigate the molecular mechanisms underlying aberrant NSUN2-mediated mRNA m5C modifications that promote MM progression.
Results
NSUN2 upregulation enhances m5C hypermethylation in MM
We analyzed data from the GEO database (GSE80608) to assess NSUN2 expression in MM. Our findings revealed a significant overexpression of NSUN2 in MM patients compared with normal controls (Fig. 1A). The International Staging System (ISS) is a widely utilized prognostic evaluation frameworks in MM patients28. Notably, patients exhibiting elevated levels of NSUN2 expression are inclined to present with advanced ISS stages (GSE136400) (Fig. 1B). Additionally, the analysis of patient survival showed a correlation between higher NSUN2 expression and worse overall survival outcomes (Fig. 1C). Because NSUN2 predominantly catalyzes m5C modification of mRNA, we assessed m5C levels using dot blot assays. The results indicated that MM patients had higher levels of RNA m5C modification than healthy controls (Fig. 1D), suggesting that increased RNA m5C modification occurs in MM development. Immunohistochemistry (IHC) of bone marrow smears showed higher NSUN2 expression in MM patients compared with normal controls (Fig. 1E). Next, we applied RT-qPCR to quantify the expression levels of NSUN2 in MM patients, and the results indicated that NSUN2 expressions were remarkably upregulated in ten MM patients compared with those in four normal controls (Fig. 1F). The baseline characteristics of MM patients are summarized in Table 1. Collectively, NSUN2 upregulation may play a crucial role in promoting m5C RNA methylation and contributing to the pathogenesis of MM.

NSUN2 upregulation and RNA m5C hypermethylation in MM. (A) Expression of NSUN2 in patients with MM and healthy controls from GSE80608. (B) The relationship between the NSUN2 levels and ISS stages in GSE136400. (C) Relationship between NSUN2 expression and patient survival rate analyzed using Kaplan–Meier curves with dichotomized NSUN2 expression at the median. (D) Dot blot analysis displaying the RNA m5C hypermethylation pattern in three patients with MM and three healthy controls. (E) Expression of NSUN2 in bone marrow smears of normal control and MM patients examined by IHC. (F) NSUN2 mRNA expression in patients with MM and healthy controls detected by RT-qPCR. ***p < 0.001. MM, multiple myeloma; NSUN2, NOP2/Sun RNA methyltransferase family member 2; ISS, International Staging System; IHC, Immunohistochemistry; RT-qPCR, quantitative RT-PCR.
NSUN2 promotes tumorigenesis of MM cells in vitro and in vivo
We investigated the expression of NSUN2 in MM cell lines (RPMI 8226, MM1S, and U266) and found that NSUN2 mRNA expression was higher in RPMI 8226 and MM.1 S cells compared to U266 cells (Fig. 2A). To further explore the mechanism of NSUN2-mediated m5C methylation in MM development, NSUN2-knockdown RPMI 8226 cells and MM.1 S cells by lentiviral transfection were established. NSUN2 expression significantly decreased in RPMI 8226 cells and MM.1 S cells after transfection with short hairpin (sh) RNAs, as demonstrated by RT-qPCR and western blotting (Fig. 2B, C). Comparably, RNA m5C modification levels were significantly decreased following the knockdown of NSUN2 (Fig. 2D).

Knockdown of NSUN2 expression results in decreased proliferation and increased apoptosis. (A) NSUN2 expression level in MM cell lines detected using RT-qPCR and Western blot. (B) NSUN2 expression level in mock and NSUN2-knockdown RPMI 8226 cells assessed using RT-qPCR and Western blot. (C) NSUN2 expression level in mock and NSUN2-knockdown MM.1 S cells assessed using RT-qPCR and Western blot. (D) Dot blots show the m5C signal relative to the methylene blue signal in mRNA from RPMI 8226 cells and MM.1 S cells after NSUN2 knockdown. (E) Cell proliferation determined by CCK-8 assays. (F) Colony formation assays show proliferation of Mock-, shNUSN2-RPMI 8226 cells and shNUSN2-MM.1 S cells. (G) Downregulation of NSUN2 increases the apoptotic rate in RPMI 8226 cells and MM.1 S cells. The statistical analysis of apoptosis percentage in Mock and shNSUN2 RPMI 8226 cells and MM.1 S cells induced in the absence (0%) and presence (10%) of FBS detected using FACS analysis. Data are represented as the mean ± SEM (n = 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001. MM, multiple myeloma; NSUN2, NOP2/Sun RNA methyltransferase family member 2; RT-q-PCR, quantitative RT-PCR; CCK-8, Cell Counting Kit-8; SEM, standard error of the mean.
To further assess the impact of NSUN2 knockdown on MM cells, we measured the proliferation of RPMI 8226 cells and MM.1 S cells with Cell Counting Kit-8 (CCK-8) and colony formation analysis. Compared with the mock group, NSUN2-knockdown RPMI 8226 cells and MM.1 S cells showed significantly lower viability after 48 and 72 h (Fig. 2E) and fewer colonies (Fig. 2F). Furthermore, the percentage of apoptotic cells induced in both, with or without 10% fetal bovine serum (FBS), was increased after NSUN2 knockdown (Fig. 2G). Taken together, these results demonstrate that NSUN2-mediated RNA m5C hypermethylation may be an oncogenic regulator of MM progression and development.
NSUN2 regulates RNA m5C abundance and gene expression profiles in RPMI 8226 cells
Methylated RNA immunoprecipitation-sequencing (meRIP-seq) analysis was performed to map and contradistinguish m5C-methylated transcripts in NSUN2-knockdown RPMI 8226 cells and the mock group (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1244071). The results included both RNA-seq (transcriptome) and methylation data. In the RNA-seq data, a prominent decrease in NSUN2 expression was illustrated in the MM cells (Fig. 3A). 454 differentially expressed genes (DEGs) were gained, which included 223 overexpressed genes and 231 underexpressed genes (Fig. 3B-D). KEGG elucidated the affected various signaling pathways, including AMPK, PI3K-Akt, MAPK, cytokine-cytokine receptor interaction, Wnt, and NF-kappa B (Fig. 3E). Moreover, in the MeRIP-seq data, 6,661 m5C transcripts were differentially methylated, of which 3,591 were upregulated and 3,070 downregulated (Fig. 3F). Differentially methylated genes (DMGs) screened using m5C meRIP-seq were subjected to KEGG pathway enrichment analyses. The results showed the enrichment of these DMGs in 20 pathways, including the AMPK, apoptosis, Wnt, VEGF, and B cell receptor signaling pathways (Fig. 3G). Collectively, these results indicate that NSUN2-mediated m5C modification potentially modulates the expression of diverse RNA targets in the pathogenesis of MM.

m5C modification pattern and RNA expression in mock and NSUN2-knockdown RPMI 8226 cells. (A) Integrative Genomics Viewer (IGV) showing the expression of NSUN2 in RPMI 8226 cells following NSUN2 knockdown. (B) Histogram showing the number of overexpressed and underexpressed differentially expressed genes (DEGs). (C) Volcano plot after NSUN2 knockdown. (D) Heatmap expression profile of 454 significantly dysregulated RNAs between mock and shNSUN2-RPMI8226 cells. (E) Statistical analysis of KEGG pathway enrichment for the DEGs. (F) Histogram showing the number of upregulated and downregulated m5C-methylated transcripts in mock and shNSUN2-RPMI8226 cells. (G) Statistical analysis of KEGG pathway enrichment for the differentially methylated genes (DMGs). NSUN2, NOP2/Sun RNA methyltransferase family member 2; KEGG, Kyoto Encyclopedia of Genes and Genomes.
NSUN2 regulates the expression of HIP1
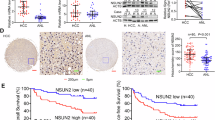
We analyzed the m5C meRIP-seq data to identify potential targets of NSUN2 involved in MM. We found 46 genes with significantly downregulated mRNAs and hypomethylated m5C levels. A significant decrease in HIP1 methylation level was observed when the NSUN2 gene was knocked down (Fig. 4A). Following this, we analyzed the expression levels of HIP1 in the GEO database (GSE80608), revealing that this gene exhibits markedly elevated expression in MM patients compared with normal controls (Fig. 4B). Further analysis indicated a positive correlation between the expression of NSUN2 and HIP1 (Fig. 4C). We then validated the elevated expression of HIP1 in MM patients compared to normal controls (Fig. 4D), and after knocking down NSUN2 in the MM cell line, the expression of HIP1 was also reduced (Fig. 4E). Next, we performed methylated RNA immunoprecipitation (MeRIP) followed by qRT-PCR to investigate how NSUN2 directly interacts with HIP1 mRNA. The results showed a substantial decrease in the m5C levels of HIP1 mRNA upon NSUN2 knockdown (Fig. 4F). Because HIP1 mRNA levels decreased following NSUN2 knockdown, we deduced that NSUN2 promotes HIP1 mRNA stability through m5C modification. Subsequently, when RPMI 8226 cells were treated with actinomycin D, the half-life of HIP1 mRNA was significantly reduced after NSUN2 knockdown (Fig. 4G). These data demonstrated that NSUN2-mediated RNA m5C methylation enhanced HIP1 mRNA stability and contributed to MM progression.

NSUN2 methylates HIP1 RNA and regulates its expression. (A) IGV tracks from m5C-meRIP-seq analysis showed m5C enrichment of HIP1. (B) Expression of HIP1 in patients with MM and healthy controls from GSE80608. (C) The correlation analysis of mRNA expression between NSUN2 and HIP1. (D) HIP1 mRNA expression in patients with MM and healthy controls detected by RT-qPCR. ***p < 0.001. (E) NSUN2 knockdown decreases the level of HIP1 mRNA in RPMI8226 cells. **p < 0.01. (F) Abundance of methylated HIP1 detected by RNA m5C RIP-qPCR from mock and shNSUN2 RPMI8226 cells. m5C RIP-qPCR with IgG antibody experiments are performed as negative controls. (G) Half-life of HIP1 mRNA measured after actinomycin D treatment. ***p < 0.001, **p < 0.01, *p < 0.5. NSUN2, NOP2/Sun RNA methyltransferase family member 2; IGV, Integrative Genomics Viewer; HIP1, huntingtin interacting protein 1; RT-PCR, reverse transcription PCR; m5C RIP-qPCR, m5C methylated RNA immunoprecipitation quantitative PCR.
HIP1 is essential for MM cell proliferation mediated by NSUN2
To confirm the synergetic carcinogenic effect of NSUN2 and HIP1, HIP1 was overexpressed in NSUN2-knockdown RPMI 8226 cells. The results showed that HIP1 overexpression reversed the decrease in HIP1 levels caused by NSUN2 knockdown (Fig. 5A, B). However, the expression of NSUN2 was not recovered (Fig. 5B, C). HIP1 overexpression largely neutralized the inhibition of MM cell proliferation resulting from NSUN2 knockdown (Fig. 5D). Furthermore, HIP1 partly rescued the increase in apoptosis induced by NSUN2 knockdown (Fig. 5E, F). A subcutaneous xenograft model was then created to assess the oncogenic impact of NSUN2 and HIP1 on MM advancement in vivo. Compared to the mock group, the volume and weight of xenograft tumors formed by NSUN2-knockdown RPMI 8226 cells were smaller, but the decreased subcutaneous xenograft tumor formation was rescued by HIP1 overexpression (Fig. 5G-I). These results were in line with the findings in vitro and further confirmed that NSUN2-mediated HIP1 methylation participates in MM cell proliferation and malignancy.

HIP1 is required for NSUN2-mediated MM cell growth. (A) mRNA expression of HIP1 in mock, shNSUN2, and shNSUN2 + oeHIP1 groups. (B) NSUN2 and HIP1 protein level in mock, shNSUN2, and shNSUN2 + oeHIP1 groups. (C) mRNA expression of NSUN2 in mock, shNSUN2, and shNSUN2 + oeHIP1 groups. (D) CCK8 assay is performed to assess the proliferation of NSUN2-knockdown RPMI8226 cells after HIP1 overexpression. (E) HIP1 rescues the increase of cell apoptosis induced by NSUN2 knockdown. Data are represented as the mean ± SEM (n = 3 independent experiments). (F) Apoptosis percentage of mock, shNSUN2, and shNSUN2 + oeHIP1 groups detected by FACS analysis. ***p < 0.001, ****p < 0.0001, ###p < 0.001, ####p < 0.0001 (* indicates mock vs. shNSUN2, # indicates shNSUN2 vs. shNSUN2 + oeHIP1). (G-I) The subcutaneous xenograft tumor formation in mice injected with Mock, shNSUN2 and shNSUN2 + oeHIP1 RPMI8226 cells. Macroscopic photographs (H). Statistical analysis of the tumor size (H) and weights (I). (J) p-AMPK and p-JNK protein level in Mock, shNSUN2, and shNSUN2 + oeHIP1 groups. NSUN2, NOP2/Sun RNA methyltransferase family member 2; HIP1, huntingtin interacting protein 1; AMPK, AMP-activated protein kinase; JNK, c-Jun Nterminal kinase; SEM, standard error of the mean.
Additionally, meRIP-seq in NSUN2-knockdown RPMI 8226 cells showed that m5C methylated genes are involved in multiple cancer-related signaling pathways, including AMPK signaling and MAPK signaling (Fig. 3E). Thus, we performed western blotting to test the expression level of AMPK and JNK (one of the MAPK family proteins). The results showed that the expression levels of p-AMPK and p-JNK were increased after NSUN2 knockdown. Furthermore, HIP1 overexpression could weaken shNSUN2-induced phosphorylation of AMPK and JNK (Fig. 5J). Taken together, NSUN2/m5C-HIP1 may promote tumor growth and inhibit apoptosis through AMPK signaling and MAPK signaling.
Discussion
Accumulating evidence indicates that posttranscriptional RNA modifications, such as m6A methylation, have been confirmed to participate in the development and advancement of MM11,12,13,29,30. In contrast, the functional significance of mRNA m5C modification, another critical epitranscriptomic marker, in MM tumorigenesis remains poorly characterized. Although Yu et al. recently reported that NSUN2-mediated m5C methylation promotes osteolytic bone destruction via exosome regulation in MM31. Our study establishes a novel oncogenic axis in MM progression: NSUN2-driven m5C modification of HIP1 mRNA. We demonstrate that NSUN2 is significantly overexpressed in MM and that its knockdown suppresses MM cell proliferation. Mechanistically, NSUN2 catalyzes m5C methylation of HIP1 mRNA, thereby accelerating MM development through RNA m5C-dependent oncogenic signaling. These findings position NSUN2-mediated m5C modification as a previously unrecognized driver of MM pathogenesis. While these insights were derived from bulk bone marrow (BM) analyses revealing robust NSUN2 and RNA m5C alterations in MM, this approach reflects a composite signal across heterogeneous marrow populations. The absence of CD138 + cell sorting precludes definitive attribution of these changes exclusively to malignant plasma cells or direct comparison with intratumoral normal counterparts. Future studies using fluorescence-activated cell sorting (FACS) or single-cell approaches will be essential to resolve cellular specificity within the BM microenvironment. Nevertheless, our functional validation in pure MM cell lines confirms the tumor cell-intrinsic role of NSUN2-m5C-HIP1 signaling, underscoring its biological relevance.
As an RNA m5C modification methyltransferase, NSUN2 and its mediated m5C modification are increasingly studied for contributing to the progression of multiple types of cancers. For instance, the upregulation of NSUN2 in gastric cancer (GC) could stimulate the proliferation and malignant metastasis of GC cells by directly interacting with a small ubiquitin-like modifier (SUMO)-2/332. In addition, NSUN2 stabilizes the mRNA of growth factor receptor-bound protein 2 (GRB2) through m5C modification, promoting the initiation and progression of esophageal squamous cell carcinoma (ESCC)33. Several recent investigations have revealed that NSUN2 and its mediated RNA m5C modification can drive tumor growth by increasing the stability of target gene mRNA26,34,35. In this study, we used m5C meRIP-seq to investigate the downstream mRNAs regulated by NSUN2 and identified HIP1 as a potential target. The findings from RT-qPCR, western blotting, and RNA stability analysis showed that NSUN2 stabilized HIP1 mRNA via m5C modification to promote the progression of MM. The following evidence supported this hypothesis: (1) NSUN2 knockdown reduced RNA m5C levels and, in turn, decreased HIP1 mRNA expression in MM cells. (2) m5C RIP-qPCR confirmed that knockdown of NSUN2 decreased the enrichment of HIP1 m5C modification in MM cells. (3) HIP1 overexpression in NSUN2-knockdown MM cells partially offset the increased cell apoptosis and inhibited cell proliferation. Therefore, our findings support the hypothesis that NSUN2 contributes to MM progression by stabilizing HIP1 mRNA. These results further indicate that NSUN2 has been identified as an oncogene that modulates the stabilization of target genes’ mRNA. Apart from NSUN2, we also detected 45 other demethylated genes with reduced expression levels. Some of these genes are closely related to the mechanisms of tumorigenesis. For instance, ITPKA is elevated in glioma and lung cancer and the heightened expression of ITPKA in tumor cells markedly enhances invasion and metastasis36,37. These genes’ relevance and potential implications in the context of NSUN2-mediated m5C modification need further investigation.
In addition, ALYREF and YBX1 are important m5C readers. They can increase the stability of various RNA targets38. For instance, ALYREF can interact with NSUN2, leading to the stabilization of phosphoribosyl formylglycinamidine synthase (PFAS) mRNA, which facilitates the progression of retinoblastoma39. Thus, further research is warranted to establish the complicated mechanism by which HIP1 mRNA translation is regulated by NSUN2 during MM development.
HIP1 is an endocytic protein overexpressed in various malignancies, including brain40, colon41, prostate41, breast42 and esophageal squamous cell carcinoma (ESCC)43. Overexpression of HIP1 transforms fibroblasts42 and promotes tumorigenesis in murine prostate cancer44. In addition, HIP1 overexpression is correlated with the malignant proliferation of ESCC, suggesting that it is a prognostic marker for ESCC patients43. Marghalani et al.45 showed that HIP1 serves as a marker in the pathological diagnosis of Merkel cell carcinoma. Ross et al.46 observed that overexpressed HIP1 in a brain tumor was associated with the expression of epidermal growth factor receptor(EGFR) and platelet-derived growth factor B receptor (PDGFβR), which are important in tumor cell invasion and proliferation through their respective signaling pathways40. In hematopoietic malignancies, HIP1 was first identified as an oncogenic HIP1/PDGFβ fusion protein resulting from a t(5;7) chromosomal translocation in a patient with chronic myelomonocytic leukemia. Additionally, HIP1 overexpression has been reported in lymphoid malignancies, and its upregulation was correlated with the progression of lymphoid neoplasms47. Thus, HIP1 might be a new oncogene in tumors. However, the mechanisms underlying epigenetic changes of HIP1 mRNA in the development of tumors have not been reported. In this study, we first demonstrated that HIP1 overexpression plays a crucial role in MM development and HIP1 mRNA is regulated by NSUN2 via m5C modification. This has opened up a new field of research on the role of epigenetic abnormalities of HIP1 in the pathogenesis of tumors.
AMP-activated protein kinase (AMPK) is an energy sensor that plays versatile roles in response to various stimuli, including glucose deprivation, caloric restriction, obesity, hypoxia, and ischemia48. Activated AMPK functions as an energy regulator, regulating numerous cellular processes including autophagy, proliferation, and apoptosis49. As a kinase with diverse functions, it plays a multifaceted role in tumor advancement, demonstrating both tumor-enhancing and tumor-inhibiting activities. C-Jun N-terminal kinase (JNK), a member of the MAPK family of proteins, plays a crucial role in apoptosis, metabolism, cytokine synthesis, and inflammation50. Our mechanistic studies revealed that NSUN2 knockdown elevates p-AMPK and p-JNK levels - phenomena linked to tumor suppression in hematologic contexts. Crucially, HIP1 overexpression reverses these phosphorylation events, indicating that the NSUN2/m⁵C-HIP1 axis promotes myeloma progression by restraining AMPK/JNK activation. Our conclusions are strongly supported by both published literature and mechanistic evidence: (1) Sustained AMPK signaling inhibits lipogenesis and protein translation, leading to irreversible nutrient depletion and apoptosis51. Compound C (AMPK inhibitor) rescue experiments confirm this causality in MM models52. (2) While transient AMPK activation induces protective autophagy, chronic AMPK hyperactivation exceeds homeostatic thresholds, redirecting cells toward apoptosis53. This aligns with observations in MM treated with AMPK-activating agents54. (3) JNK hyperactivation (via p-JNK elevation) directly triggers pro-apoptotic cascades through caspase-dependent pathways. This mechanism is reinforced by NSUN2-mediated stabilization of MALAT1, which promotes MAPKs/JNK activation to drive myeloma progression31. NSUN2 knockdown thus disrupts this pathological axis, liberating JNK-dependent tumor suppression. In a recent study, pseudolaric acid B (PAB) has been demonstrated to inhibit tumor growth and induce apoptosis in hepatocellular carcinoma by inducing the AMPK/JNK signaling pathway55. However, the exact NSUN2-AMPK/JNK regulatory mechanism warrants further study, particularly how m5C modifications tune HIP1-mediated kinase repression.
In conclusion, the NSUN2/m5C-HIP1 signaling axis represents a novel mechanism promoting MM pathogenesis. Our findings contribute to our comprehension of the development of MM and offer an opportunity to improve its therapeutic management by modulating NSUN2-mediated m5C modification.
Materials and methods
Bioinformatics analysis
Datasets GSE80608 and GSE136400 were downloaded from the GEO database (www.ncbi.nlm.nih.gov) and subsequently analyzed. In addition, transcriptome data from MMRF-COMMPAS and matching survival information from The Cancer Genome Atlas (TCGA) were obtained and analyzed. The survival package in R was used for survival analysis.
Patients and clinical samples
The Ethics Committee of the Second Hospital of Shandong University approved this study (KYLL-2019(KJ)P-0125) in line with the Declaration of Helsinki. All patients agreed to participate in the study and had provided written informed consent. The healthy controls were selected based on the normal bone marrow aspiration results. Bone marrow specimens of newly diagnosed MM patients and healthy controls were sourced from the Department of Hematology at the Second Hospital of Shandong University. We collected bone marrow by puncture, then isolated total RNA and proteins for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blotting.
Cell culture and treatments
The MM cell lines, including MM.1R, MM.1 S, U266, and RPMI 8226, were acquired from the American Type Culture Collection based in Manassas, VA, USA (CRL-2975, CRL-2974, TIB-196, CCL-155, ATCC). They were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (11875093, Invitrogen, Carlsbad, USA) supplemented with 10% fetal bovine serum (FBS, 10100147, Gibco, USA) and 1% penicillin-streptomycin (P/S, 60162ES76, Yeasen, Shanghai, China). The cells were placed in a cell culture incubator and kept at a steady temperature of 37 °C with 5% CO2 during incubation.
Cell transfection
The lentiviral constructs were acquired from Shanghai GeneChem Co., Ltd., China. MM cells were seeded and treated with lentivirus to knock down NSUN2 (sh-NSUN2-1: GAGCGAUGCCUUAGGAUAUTT; sh-NSUN2-2: CCGAGAUCGU CAAGGAGAATT) or mock sequences (UUCUCCGAACGUGUCACGUTT) and HIP1 overexpression lentivirus (oeHIP1: NM_005338.7). Next, we used puromycin (2 µg/mL, 60210, Yeasen) or blasticidin (2 µg/mL, 60218, Yeasen) to select the stable transfected MM cells. Transfection efficiency was obtained using RT-qPCR and western blotting.
Apoptosis assay
MM cells were cultivated in a medium with either 10% FBS or no FBS (0% FBS) for 72 h and then subjected to the PE-Annexin V/7-AAD Apoptosis Detection Kit (AP104, MULTISCIENCES, Hangzhou, China) following the provided guidelines. Stained cells were subjected to flow cytometry (Beckman, Miami, FL, USA). The ratio of Annexin V + 7-AAD- and Annexin V + 7-AAD + cells was evaluated to calculate the apoptosis rate.
Cell viability assays
A CCK8 Cell Counting Kit (RM02823, ABclonal, Wuhan, China) was used to evaluate the cell viability. Ninety-six-well plates were filled with 1 × 104 cells, with each well containing 100 µL of complete medium. Each well received 10 µL of CCK8 solution at 0, 24, 48, and 72 h after seeding, followed by a 2-h incubation period. A microplate reader (BioTek, Vermont, USA) was applied to determine the absorbance at 450 nm.
Soft agar colony formation analysis
The sorted RPMI 8226 cells were seeded at a density of 1000 cells/well in 6-well plates with soft agar. The upper agar layer consisted of 0.6% agarose in RPMI Medium 1640 supplemented with 20% FBS, and the base layer was made up of 1.1% agarose. A total of 200 µl fresh medium was added to the cells twice a week. After 3 weeks of culture at 37 °C, colonies were examined using an inverted microscope and taken photos with a camera to record. The experiments were conducted in triplicate.
RNA extraction and Dot blot analysis
Total RNA from specimens was segregated using a SteadyPure Quick RNA Extraction Kit (AG21023; Accurate Biology, Hunan, China). The RNA was then denatured at 95℃ for 3 min and quickly cooled on ice. The same quantities of RNA were then applied onto nitrocellulose membranes (HATF00010, Merck, Billerica, MA, USA). After a short drying process, the membrane was crosslinked at 37℃ for 30 min and then washed to eliminate unbound RNA. The membrane was then soaked in 5% Skim Milk (232100, BD Difco, San Diego, USA) and subsequently exposed to mouse anti-human m5C monoclonal antibody (68301-1-Ig, Proteintech, Wuhan, China) at 4℃ for incubation. Next, the horseradish peroxidase (HRP)-conjugated with goat anti-mouse IgG (H + L) (AS003, Abclonal, Wuhan, China) was treated with the membrane for 1 h. Finally, the Azure Biosystems imaging system (Dublin, California, USA) was used for detection, and 0.2% Methylene Blue Solution (G1301, Solarbio, Beijing, China) served as a loading control.
Immunohistochemistry (IHC)
IHC analysis was conducted to ascertain the expression of NSUN2 utilizing the streptavidin-peroxidase (SP) technique. Initially, we extracted 0.1mL of bone marrow from two MM patients (tumor cell proportions of 84.5% and 85%) and two normal controls to create bone marrow smears. Subsequently, the bone marrow smears were treated with anti-NSUN2 (dilution 1:200; 20854-1-AP, Proteintech) antibody at 4 °C overnight. Following this, the bone marrow smears were incubated with the secondary antibody (Goat anti-Rabbit IgG [H + L] HRP; GB23303, Servicebio) for 50 min. Target signals were visualized using diaminobenzidine, and counterstaining was carried out with hematoxylin. The three most elevated immunoreactive areas were scrutinized at ×40 magnification by two independent assessors, unaware of the patient’s clinical information.
Quantitative RT-PCR (RT-qPCR)
Following the extraction of RNA, it was reversed transcribed into complementary DNA following the instruction of Evo M-MLV RT Kit (AG11707, Accurate Biology) for qPCR analysis. RT-qPCR analysis was performed by a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA), following the protocol of SYBR Green Pro Taq HS qPCR Kit (AG11701, Accurate Biology). The following primer sequences were utilized for RT-qPCR: NSUN2 (Forward 5′-GAACTTGCCTGGCACACAAAT-3′; Reverse 5′-TGC TAACAGCTTCTTGACGACTA-3′), HIP1 (Forward 5′-GCGGCTCATTCAGATCCC C-3′; Reverse 5′-GAGGTCATCCTTCTCTAGGACTG-3′), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Forward 5′-AACGGATTTGGTCGTATTGGG-3′, Reverse 5′-CCTGGAAGATGGTGATGGGAT-3′). The expression level of target genes was calculated by the 2−ΔΔCt method and standardized to the levels of the housekeeping gene GAPDH.
Western blot analysis
Protein from MM cells was extracted using a radioimmunoprecipitation assay buffer (RIPA, 20101ES60, Yeasen) and diverted onto polyvinylidene fluoride membranes (ISEQ00010, Millipore, Billerica, MA, USA). Subsequently, these membranes were exposed to rabbit anti-human antibodies against NSUN2 (20854-1-AP, Proteintech), HIP1 (90830, CST, Danvers, MA, USA), and GAPDH (5174, CST). Ultimately, the membranes were exposed to a goat anti-rabbit antibody (31460, Thermo, Waltham, MA, USA), followed by band detection utilizing Immobilon ECL Ultra Western HRP Substrate (WBULS0500, Millipore) and the AlphaView FluorChem Q system (ProteinSimple, San Francisco, CA, USA).
m5C RNA Immunoprecipitation (RIP)-qPCR
RNA was extracted and immunoprecipitated with magnetic beads precoated with 10 µg of anti-m5C antibody (ab214727, Abcam, Cambridge, MA, USA) or anti-IgG (ab172730, Abcam). The beads were then combined with Dynabeads Protein A beads and incubated at 4 °C for 3 h. The resuspended RNA was analyzed using RT-qPCR to gain the enrichment of m5C RNA-containing mRNA after washing. The following are the primer sequences for m5C RIP-qPCR: Forward 5′-CGGCTCAGCACCTTGGGCAGT-3′, Reverse 5′-CTCGGCTCC TCGCGGCGACAT-3′. IgG m5C RIP-qPCR was used as the negative control.
RNA stability assay
Actinomycin D (HY-17559, MCE, Monmouth Junction, NJ, USA) with a concentration of 5 µg/mL was utilized to treat MM cells for various time points of 0, 2, 4, 6, and 8 h. The cells were subsequently collected to extract total RNA. Following reverse transcription, mRNA levels were assessed through RT-qPCR analysis.
MeRIP-seq and data analysis
MeRIP-seq and data analysis were offered by Cloudseq Biotech Inc. (Shanghai, China). The NanoDrop ND-1000 (Thermo Fisher Scientific, Massachusetts, USA) was applied to assess the quality and quantity of total RNA extracted from all samples. Subsequently, RNA was fragmented randomly into fragments approximately 200 nt in length. The m5C antibody (NEB, Massachusetts, USA) was allowed to incubate with protein A/G magnetic beads at atmospheric temperature for 1 h. GenSeq® Low Input Whole RNA Library Prep Kit (GenSeq, Inc.) was then applied to analyze the immunoprecipitated RNA to generate RNA sequencing libraries. Additionally, Input libraries (a portion of the fragmented total RNA not subjected to immunoprecipitation) were also generated for each sample using the same kit. The quality of both IP and Input sequencing libraries was monitored using an Agilent 2100 Bioanalyzer.
High-throughput sequencing was performed on an Illumina NovaSeq 6000 sequencer to generate raw reads for both IP and Input libraries. Raw reads underwent quality control using Q30 metrics. Adapters and low-quality reads were removed using Cutadapt software (v1.9.3) to obtain high-quality clean reads for both IP and Input libraries. These clean reads were aligned to the UCSC hg38 reference genome using Hisat2 software (v2.0.4). Methylated regions (peaks) were identified from the immunoprecipitated (IP) data using MACS software (v2), utilizing the corresponding Input library as the control for normalization and background signal estimation. Differential methylated regions/sites (DMRs/DMSs) between NSUN2-knockdown and mock groups were pinpointed using diffReps software, again using Input libraries for comparison. Peaks (methylated sites) located on mRNA exons were identified and annotated. The genes harboring differentially methylated sites were subsequently analyzed for pathway enrichment using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database56,57,58. The biological processes of KEGG pathways enrichment analyses were carried out by cluster Profiler package of R. The figure was drawn by ggplot2. Statistical significance was defined as p < 0.05.
RNA-seq and data analysis
Transcriptome profiles were derived from the Input libraries generated during the MeRIP-seq procedure for both NSUN2-knockdown and mock-treated RPMI 8226 cells (refer to the MeRIP-seq methods section for detailed library preparation and sequencing protocols). HTSeq-count was used to count the number of reads mapped to each gene locus based on the UCSC hg38 reference genome annotation (GENCODE v38). DEGs between the NSUN2-knockdown group and the mock control group were identified using DESeq2 (with thresholds |log2 Fold Change| >0.58 and adjusted p-value < 0.05). KEGG pathway enrichment analyses were performed on the list of DEGs using enrich R package to identify biological pathways significantly impacted by NSUN2 knockdown. Statistical significance for enrichment was defined as p < 0.05.
Tumor xenograft model
Male NCD mice, aged 6–8 weeks (mice weight 18–20 g), were acquired from Gem Pharmatech Co., Ltd. (Nanjing, China). All mice were raised in specific-pathogen-free animal equipment with individual ventilation. The environment was kept at a constant temperature of 23℃ and humidity level, with a 12-h light-dark cycle. The mock or shNSUN2 MM cells (approximately 1 × 107) were collected and suspended in 100 µL phosphate-buffered saline. Each mouse (n = 5) received a subcutaneous injection of the cell suspension into their right foreleg. The gross volumes of tumors were assessed every 4 days with a caliper. The criterion used to calculate the tumor volume was V = LW2 × π/6 (L = length, W = width). Once the xenograft tumors reached a diameter greater than 15 mm (mice weight 20–23 g), we euthanized the mice using cervical dislocation and separated the xenograft tumors. The Ethics Committee of the Second Hospital of Shandong University approved the animal experiments (No. KYLL-2019(KJ)A-0181).
Statistical analysis
Statistical analyses were conducted using GraphPad Prism software 8.0.1 (GraphPad, La Jolla, CA, USA). Data were quantified and shown as mean ± standard error of the mean, and group variances were compared with an unpaired Student’s t-test, one-way analysis of variance (ANOVA), or two-way ANOVA. Differences were indicated at p < 0.05 (two-tailed).
Data availability
NSUN2 gene expression data in human MM bone marrow and the normal controls were derived from the GEO (GSE80608 and GSE136400) databases. The transcriptome data and the matching survival information of NSUN2 were derived from the TCGA database. The datasets utilized and/or analyzed in the present study can be obtained from the corresponding author on reasonable request.
References
Kumar, S. K. et al. Multiple myeloma. Nat. Reviews Disease Primers 3, 586. https://doi.org/10.1038/nrdp.2017.46 (2017).
Rajkumar, S. V. Multiple myeloma: every year a new standard? Hematol. Oncol. 37, 62–65. https://doi.org/10.1002/hon.2586 (2019).
Pinto, V. et al. Multiple myeloma: available therapies and causes of drug resistance. Cancers (Basel) 12, 526. https://doi.org/10.3390/cancers12020407 (2020).
Dimopoulos, K., Gimsing, P. & Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 4, e207. https://doi.org/10.1038/bcj.2014.29 (2014).
Corre, J., Munshi, N. & Avet-Loiseau, H. Genetics of multiple myeloma: another heterogeneity level? Blood 125, 1870–1876. https://doi.org/10.1182/blood-2014-10-567370 (2015).
Huang, X., Yang, Z., Li, Y. & Long, X. m6A methyltransferase METTL3 facilitates multiple myeloma cell growth through the m6A modification of BZW2. Ann. Hematol. 102, 1801–1810. https://doi.org/10.1007/s00277-023-05283-6 (2023).
Chen, X. et al. RNA methylation and diseases: experimental results, databases, web servers and computational models. Brief. Bioinform. 20, 896–917 (2017).
Pan, Y., Ma, P., Liu, Y., Li, W. & Shu, Y. Multiple functions of m(6)A RNA methylation in cancer. J. Hematol. Oncol. 11, 48. https://doi.org/10.1186/s13045-018-0590-8 (2018).
Nombela, P., Miguel-López, B. & Blanco, S. The role of m(6)A, m(5)C and Ψ RNA modifications in cancer: novel therapeutic opportunities. Mol. Cancer. 20, 18. https://doi.org/10.1186/s12943-020-01263-w (2021).
Xu, Y. et al. Role of main RNA methylation in hepatocellular carcinoma: N6-Methyladenosine, 5-Methylcytosine, and N1-Methyladenosine. Front. Cell. Dev. Biol. 9, 767668. https://doi.org/10.3389/fcell.2021.767668 (2021).
Jia, Y. et al. PRMT1 methylation of WTAP promotes multiple myeloma tumorigenesis by activating oxidative phosphorylation via m6A modification of NDUFS6. Cell. Death Dis. 14, 512. https://doi.org/10.1038/s41419-023-06036-z (2023).
Xu, A. et al. FTO promotes multiple myeloma progression by posttranscriptional activation of HSF1 in an m(6)A-YTHDF2-dependent manner. Mol. Ther. 30, 1104–1118. https://doi.org/10.1016/j.ymthe.2021.12.012 (2022).
Jia, C. et al. HNRNPA2B1-mediated m6A modification of TLR4 mRNA promotes progression of multiple myeloma. J. Transl Med. 20, 537. https://doi.org/10.1186/s12967-022-03750-8 (2022).
Trixl, L. & Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip Rev. RNA. 10, e1510. https://doi.org/10.1002/wrna.1510 (2019).
Heissenberger, C. et al. The ribosomal RNA m(5)C methyltransferase NSUN-1 modulates healthspan and oogenesis in caenorhabditis elegans. Elife 9, 856. https://doi.org/10.7554/eLife.56205 (2020).
Zhang, Q. et al. The role of RNA m(5)C modification in cancer metastasis. Int. J. Biol. Sci. 17, 3369–3380. https://doi.org/10.7150/ijbs.61439 (2021).
Shen, Q. et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554, 123–127. https://doi.org/10.1038/nature25434 (2018).
Yang, X. et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell. Res. 27, 606–625. https://doi.org/10.1038/cr.2017.55 (2017).
Blanco, S. & Frye, M. Role of RNA methyltransferases in tissue renewal and pathology. Curr. Opin. Cell. Biol. 31, 1–7. https://doi.org/10.1016/j.ceb.2014.06.006 (2014).
Zhang, X. et al. The tRNA methyltransferase NSun2 stabilizes p16INK⁴ mRNA by methylating the 3’-untranslated region of p16. Nat. Commun. 3, 712. https://doi.org/10.1038/ncomms1692 (2012).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990. https://doi.org/10.1038/s41556-019-0361-y (2019).
Wang, W. mRNA methylation by NSUN2 in cell proliferation. WIREs RNA. 7, 838–842. https://doi.org/10.1002/wrna.1380 (2016).
Li, Q. et al. NSUN2-Mediated m5C methylation and METTL3/METTL14-Mediated m6A methylation cooperatively enhance p21 translation. J. Cell. Biochem. 118, 2587–2598. https://doi.org/10.1002/jcb.25957 (2017).
Li, M. et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J. Transl Med. 20, 214. https://doi.org/10.1186/s12967-022-03427-2 (2022).
Sun, Z. et al. Aberrant NSUN2-mediated m(5)C modification of H19 LncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene 39, 6906–6919. https://doi.org/10.1038/s41388-020-01475-w (2020).
Yang, M. et al. NSUN2 promotes osteosarcoma progression by enhancing the stability of FABP5 mRNA via m(5)C methylation. Cell. Death Dis. 14, 125. https://doi.org/10.1038/s41419-023-05646-x (2023).
Chen, S. Y. et al. RNA bisulfite sequencing reveals NSUN2-mediated suppression of epithelial differentiation in pancreatic cancer. Oncogene 41, 3162–3176. https://doi.org/10.1038/s41388-022-02325-7 (2022).
Palumbo, A. et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J. Clin. Oncol. 33, 2863–2869. https://doi.org/10.1200/jco.2015.61.2267 (2015).
Alzrigat, M., Párraga, A. A. & Jernberg-Wiklund, H. Epigenetics in multiple myeloma: from mechanisms to therapy. Sem. Cancer Biol. 51, 101–115. https://doi.org/10.1016/j.semcancer.2017.09.007 (2018). https://doi:.
Szalat, R. & Munshi, N. C. Genomic heterogeneity in multiple myeloma. Curr. Opin. Genet. Dev. 30, 56–65. https://doi.org/10.1016/j.gde.2015.03.008 (2015).
Yu, M. et al. Aberrant NSUN2-mediated m5C modification of Exosomal LncRNA MALAT1 induced RANKL-mediated bone destruction in multiple myeloma. Commun. Biology. 7, 1249. https://doi.org/10.1038/s42003-024-06918-8 (2024).
Hu, Y. et al. NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation. Cell. Death Dis. 12, 842. https://doi.org/10.1038/s41419-021-04127-3 (2021).
Su, J. et al. NSUN2-mediated RNA 5-methylcytosine promotes esophageal squamous cell carcinoma progression via LIN28B-dependent GRB2 mRNA stabilization. Oncogene 40, 5814–5828. https://doi.org/10.1038/s41388-021-01978-0 (2021).
Chen, Y. et al. Upregulation of LRRC8A by m(5)C modification-mediated mRNA stability suppresses apoptosis and facilitates tumorigenesis in cervical cancer. Int. J. Biol. Sci. 19, 691–704. https://doi.org/10.7150/ijbs.79205 (2023).
Zhang, G. et al. NSUN2 stimulates tumor progression via enhancing TIAM2 mRNA stability in pancreatic cancer. Cell. Death Discov. 9, 219. https://doi.org/10.1038/s41420-023-01521-y (2023).
Küster, L. et al. The actin bundling activity of ITPKA mainly accounts for its migration-promoting effect in lung cancer cells. Biosci. Rep. 43, BSR20222150. https://doi.org/10.1042/BSR20222150 (2023).
Luo, X. et al. ITPKA phosphorylates PYCR1 and promotes the progression of glioma. Heliyon 10, 856. https://doi.org/10.1016/j.heliyon.2024.e35303 (2024).
Chen, Y. S., Yang, W. L., Zhao, Y. L. & Yang, Y. G. Dynamic transcriptomic m5C and its regulatory role in RNA processing. WIREs RNA. 12, 562. https://doi.org/10.1002/wrna.1639 (2021).
Zuo, S. et al. NSUN2-mediated m(5) C RNA methylation dictates retinoblastoma progression through promoting PFAS mRNA stability and expression. Clin. Transl Med. 13, e1273. https://doi.org/10.1002/ctm2.1273 (2023).
Bradley, S. V. et al. Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor. Cancer Res. 67, 3609–3615. https://doi.org/10.1158/0008-5472.CAN-06-4803 (2007).
Rao, D. S. et al. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J. Clin. Invest. 110, 351–360. https://doi.org/10.1172/jci15529 (2002).
Rao, D. S. et al. Altered receptor trafficking in Huntingtin interacting protein 1-transformed cells. Cancer Cell. 3, 471–482. https://doi.org/10.1016/S1535-6108(03)00107-7 (2003).
Sun, Y. et al. Abnormally high HIP1 expression is associated with metastatic behaviors and poor prognosis in ESCC. Oncol. Lett. 21, 79. https://doi.org/10.3892/ol.2020.12340 (2021).
Bradley, S. V. et al. Serum antibodies to Huntingtin interacting Protein-1: a new blood test for prostate cancer. Cancer Res. 65, 4126–4133. https://doi.org/10.1158/0008-5472.CAN-04-4658 (2005).
Marghalani, S., Feller, J. K., Mahalingam, M. & Mirzabeigi, M. Huntingtin interacting protein 1 as a histopathologic adjunct in the diagnosis of Merkel cell carcinoma. Int. J. Dermatol. 54, 640–647. https://doi.org/10.1111/ijd.12454 (2015).
Ross, T. S., Bernard, O. A., Berger, R. & Gilliland, D. G. Fusion of Huntingtin interacting protein 1 to Platelet-Derived growth factor β receptor (PDGFβR) in chronic myelomonocytic leukemia with t(5;7)(q33;q11.2). Blood 91, 4419–4426. https://doi.org/10.1182/blood.V91.12.4419 (1998).
Bradley, S. V. et al. Aberrant Huntingtin interacting protein 1 in lymphoid malignancies. Cancer Res. 67, 8923–8931. https://doi.org/10.1158/0008-5472.CAN-07-2153 (2007).
Penugurti, V., Manne, R. K., Bai, L., Kant, R. & Lin, H. K. AMPK: the energy sensor at the crossroads of aging and cancer. Sem. Cancer Biol. 106–107, 15–27. https://doi.org/10.1016/j.semcancer.2024.08.002 (2024).
Hsu, C. C., Peng, D., Cai, Z. & Lin, H. K. AMPK signaling and its targeting in cancer progression and treatment. Semin Cancer Biol. 85, 52–68. https://doi.org/10.1016/j.semcancer.2021.04.006 (2022).
Ouyang, H., Zhou, E. & Wang, H. Mst1-Hippo pathway triggers breast cancer apoptosis via inducing mitochondrial fragmentation in a manner dependent on JNK-Drp1 axis. Onco Targets Ther. 12, 1147–1159. https://doi.org/10.2147/ott.S193787 (2019).
Ruppin, H. [Motility inhibitors and secretion inhibitors]. Schweiz. Med. Wochenschr Suppl. 17, 23–28 (1984).
Mei, H. et al. Pterostilbene inhibits nutrient metabolism and induces apoptosis through AMPK activation in multiple myeloma cells. Int. J. Mol. Med. 42, 2676–2688. https://doi.org/10.3892/ijmm.2018.3857 (2018).
Banerjee, A., Koziol-White, C. & Panettieri, R. Jr. p38 MAPK inhibitors, IKK2 inhibitors, and TNFα inhibitors in COPD. Curr. Opin. Pharmacol. 12, 287–292. https://doi.org/10.1016/j.coph.2012.01.016 (2012).
Li, X. et al. Matrine induces autophagic cell death by triggering ros/ampk/mtor axis and apoptosis in multiple myeloma. Biomed. Pharmacother. 175, 116738. https://doi.org/10.1016/j.biopha.2024.116738 (2024).
Liu, Z., Wang, N., Meng, Z., Lu, S. & Peng, G. Pseudolaric acid B triggers cell apoptosis by activating AMPK/JNK/DRP1/mitochondrial fission pathway in hepatocellular carcinoma. Toxicology 493, 153556. https://doi.org/10.1016/j.tox.2023.153556 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Acknowledgements
We would like to give our sincere gratitude to the reviewers for their constructive comments.
Funding
This study was supported by the Shandong Provincial Natural Science Foundation (ZR2020QH092) and the Key Research and Development Program of Shandong Province (No. 2021CXGC011101) supported this work.
Author information
Authors and Affiliations
Contributions
C.S. designed and supervised the study and prepared the manuscript. Y.J. drafted the study, wrote the manuscript, and implemented most molecular mechanism assays and animal experiments. J.S. supervised the study and prepared the manuscript. Y.C. conducted most cellular function experiments, including cell proliferation and apoptosis experiments. L.C. performed western blotting, RT-qPCR, and dot blot analysis. S.F. and Y.W. finalized the figures and tables.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Ethics Committee of the Second Hospital of Shandong University approved this study (KYLL-2019(KJ)P-0125) in line with the Declaration of Helsinki. The Ethics Committee of the Second Hospital of Shandong University approved the animal experiments (No. KYLL-2019(KJ)A-0181).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, Y., Sun, J., Chen, Y. et al. NSUN2-mediated RNA m5C modification drives multiple myeloma progression by enhancing the stability of HIP1 mRNA. Sci Rep 15, 27888 (2025). https://doi.org/10.1038/s41598-025-13695-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13695-9
