
Abstract
Autoimmune diseases (ADs) affect almost 10% of the world population. They are also the leading cause of mortality among young and middle-aged women. Despite the available of therapeutic agents there is no definitive cure. The voltage-gated potassium channel (Kv1.3) plays a key role regulating various physiological processes, particularly immune responses, making it a promising target for immunomodulators. Moreover, the ability of Kv1.3 blockers to selectively modulate the activation of effector memory T-cells, without affecting other lymphoid subsets, make it a promising target to avoid fatal opportunistic infections encountered with broad spectrum immunosuppressants. Herein, a β-carboline-based lead (KMA) was developed through ligand-directed scaffold hopping based on the known Kv1.3 inhibitors, pyranoquinolinone and CP-339,818. Iterative structural derivatization and scaffold hopping produced piperidine and the hybrid analogs based on scaffolds of the potent Kv1.3 small inhibitors, UK-78,282 and PAP-1. Our biological assays produced two promising compounds; β-carboline-based (5c) and hybrid (13a). Notably, the hybrid compound 13a showed promising activity in relation to the Centruroides margaritatus scorpion short-chain toxin, margatoxin, which was used as positive control standard. Only compound 13a managed to decrease the Kv1.3 peak current amplitude by more than 80%, suggesting its viability as a lead compound for further investigations targeting ADs.
Similar content being viewed by others

Introduction
Autoimmune diseases (ADs) belong to a family of more than 100 clinically distinct chronic and usually disabling medical conditions1. Notably, the true cause of these diseases remains unknown even though both the genetic factors and environmental conditions play the major role within the individual’s susceptibility2,3,4. Common ADs comprise; type-1 diabetes, rheumatoid arthritis, psoriasis, lupus, multiple sclerosis, inflammatory bowel disease, and others1. Although each of the reported ADs is considered relatively rare, the global prevalence of collective ADs has significantly increased over the last 30 years affecting an approximate of 7.6–9.4% of the world population5. ADs are the leading cause of death among young and middle-aged women6. High incidences for several ADs have been estimated for Asia, Middle East and North Africa (MENA) regions collectively7,8,9,10,11,12.
Due to the chronic nature of ADs and the absence of a definitive cure for any of them, patients face a lifetime of illness and medical care where they often endure debilitating symptoms and loss of organ function. Therefore, ADs impose a heavy burden on health care system as well as on patients’ families and society in terms of medical expenses13. The direct annual health care costs for ADs exceeds 100 billion USD in the USA14. On the other hand, indirect burden includes both the loss of work productivity and diminished contributions to the community at large which impose more economic losses including disability, insurance, mortgages, and loans. As most of the ADs disproportionately afflict women, especially at childbearing age, these diseases can limit their ability to have children and to care for their families, both physically and financially14.
Current treatments available for ADs are typically based on modulating the complex and multi-orchestral pathophysiological pathways of such diseases. Treatment protocols generally vary according to the specific medical condition, nevertheless, most of such policies depend on the administration of immunosuppressants such as methotrexate, leflunomide, salazines or cyclosporin together with non-steroidal anti-inflammatory agents or corticosteroids. Such combination therapy limits the side effects of individual medication while reversing the pathological course of the ADs over long-term treatment15. Novel management of ADs uses intravenous synthetic anti-TNF biologics, which serve as a third line of defense16. Despite the diversity of the available therapeutic agents and established protocols, until now, there is no definitive cure for any of the ADs. Also, the prolonged use of the aforementioned therapeutic agents can impose serious complications and devastating long-term adverse effects17. For example, the intravenous monoclonal antibody, natalizumab (Tysabri®), has led to fatal opportunistic viral infections and progressive multifocal leukoencephalopathy within some patients18. Similarly, long-term usage of corticosteroids impose risk for infection and immunosuppression as well as gastrointestinal bleeding and bone damage16. Moreover, the prolonged use of NSAIDs cause significant gastrointestinal ulceration, renal toxicity, and cardiovascular risk, particularly with selective COX-2 inhibitors19,20. Concerning financial impact, the mean annual cost per patient is around 18,000 USD21. All such findings highlight the need for a novel safer as well as more effective, and economic therapeutic agents for ADs.
Over the past few decades, potassium voltage-gated (Kv) channels have been among the targets in the quest of new immunomodulating therapies22. The Kv1.3, as a delayed rectifier channel, possesses crucial biofunction in several organs, such as lung, brain, kidney, and olfactory bulbs. These channels are present at the brain’s cortex, cerebellum, and hippocampus sites contributing to the modulation of neuronal plasticity, firing frequency, excitability23. Additionally, central Kv1.3 channels play a significant role in neuroinflammatory responses linked to microglia cell activation24. Thus, altered Kv1.3 activity has been linked to seizures and neurodegenerative conditions including Alzheimer’s disease25,26. Kv1.3 location at olfactory sensory neurons and mitral cells being beneficial for regulating olfactory learning and odor discriminations27. Experimental animals with Kv1.3 knockout exhibited enhanced olfactory sensitivity and altered olfactory-directed behaviors28. Evidence further highlights the role of Kv1.3 in a few chronic respiratory inflammatory conditions being upregulated in asthma, bronchitis, and chronic obstructive pulmonary disease. Blocking Kv1.3 has been linked with reduced infiltration of inflammatory-immune cells and airway hyperresponsiveness29. In the kidney, Kv1.3 possess a significant role in renal disease pathogenesis modulating the immune cell infiltration into the kidney30. Evidence of Kv1.3 blockage has been related to the nephroprotective effects within pre-clinical animal models31. Though Kv1.3 possesses potential therapeutic usage over an array of medical conditions, the plethora of scientific literature has investigated the role of Kv1.3 in modulating immunological responses particularly for autoimmune diseases32,33,34,35,36,37,38,39. The Kv1.3 channels belong to the mammalian Shaker-related family of Kv transporters40. The distinct architecture of Kv1.3 tetramer, disclosed in 2021, showed the helices (S1–S6) of the transmembrane domain enclosing the potassium pore site with the conserved potassium-selectivity filter motif (TVGYG) (Fig. 1)41. Through electrophysiology studies, it became widely accepted that Kv1.3 regulates the chronically activated effector memory T (TEM) cell immune responses contributing to the pathogenesis of ADs32,42. This raised interest in developing efficient inhibitors of Kv1.3 channel as being beneficial in managing ADs, where chronic inflammatory pathogenesis is related to TEM cells. Interestingly, the central memory and Naïve T cells, protecting against infections and cancers, could circumvent the Kv1.3 blockage due to the up-regulation of the calcium-activated potassium channels, IKCa142. In these regards, developing specific Kv1.3 blockers could be a promising untapped approach for ameliorating ADs while avoiding adverse effects on normal immune response encountered with broad immunosuppressants.

Architecture of the Kv1.3 channel (PDB: 7EJ1) ; a) Stereo image of the human Kv1.3 channel tetramer viewed from the side, each subunit is displayed with different color (PDB: 7ej1; 3.20 Å resolution); b) A single Kv1.3 subunit viewed from the same view-point; helices are labelled numerically as S1–S6 and with P for the pore-helix; N- and C-mark the termini; the conserved potassium-selectivity filter motif (TVGYG) is indicated in red; c) A schematic diagram of the Kv1.3 subunit topology is shown with a red selectivity filter and a red arrow indicating the ion pathway (Images were prepared by PyMol v.2.0.6; Schrödinger, LLC).
Currently, both academia and the pharmaceutical industry are highly driven towards developing new peptide Kv1.3 blockers. The refined ShK-analog (Dalazatide) entered phase 1b-2a human trials showcasing durable pharmacological responses and more than 100-fold selectivity towards Kv1.3 over the neurotoxic hazardous Kv1.6 and Kv1.143. However, the development of small non-peptide Kv1.3 ligands remains uncharted territory. Small drug-like ligands are the cornerstone of drug discovery and development programs. Such ligands harbor the ease of structural alterations while maintaining optimized kinetic profiles, which allows their survival through the rigors of lead optimization and clinical development stages. Additionally, exhibiting drug-like properties favors the administration of small non-peptide ligands orally, the most convenient route44. Conversely, the molecular mass of peptide Kv1.3 toxins, combined with their characteristically high-pitched isoelectric points, causes their oral delivery to be unlikely45.
Several non-peptide small Kv1.3 blockers (MW 200-to-500 Da) have been reported (Fig. 2). The first of which is Correolide, the novel nor-triterpene extracted from the Costa-Rican trees, Spachea correa. Computational analysis predicts Correolide relevant binding into the lipophilic surface of the S6 α-helix, utilizing its hydrophobic moiety, while as chelating a permeating K+-cation via its hydrophilic acetyl moieties46. Correolide demonstrates selectivity (4-to-14 fold) for Kv1.3 over Kv1.x, without a significant inhibition of the neuronal sodium, or L-type calcium channels47. Nevertheless, an inhibitory activity of the cardiac Kv1.5, along with, the molecular complexity of Correolide have stalled medicinal chemistry attempts to recognize a drug-like analog for clinical development48. Several other small molecules have been introduced to circumvent the latter problem including Clofazimine, Progesterone, as well as derivatives of the dihydroquinoline, benzylpiperazine, benzamide, psoralen and khellinone-scaffolds. The khellinones represent the first class of small molecules being selective over the cardiac Kv1.5 channels49,50. Another interesting group having close similarity to the khellinone nucleus, is the psoralen category. The most recognized psoralen-based compound is PAP-1 which was reported to exhibit a preferential binding to the channel’s slow C-type state of inactivation. Under such a sustained depolarization state, a change in the outer vestibule and/or a localized constriction/collapse near the selectivity filter are expected. Notably, PAP-1 possesses up to 125-fold selectivity over Kv1.x and up to 7,500-fold selectivity over HERG, calcium, and sodium channels51. This was followed by the stereoselective handy chemical class, the benzamides, where their trans-isomers display some degrees of specificity for hKv1.3 over other Kv1.x channels52. Lastly, Clofazimine, the anti-lepromatous agent demonstrated a dual-mode blockage of Kv1.3, through a use-dependent and state-dependent manner53.

Diverse structures of literature reported small molecule Kv1.3 blockers.
This study aimed at developing small drug-like ligands that can effectively modulate Kv1.3 channel activity through a highly rationalized approach. Scaffold hopping was applied to design and optimize small drug-like ligands based on various reported Kv1.3 blockers. Chemical synthesis of three diverse chemical series (I, II, and III) was pursued and then the activity was tested using the patch-clamp functional assay in comparison to the highly potent scorpion short peptide toxin, Margatoxin. This work provided the foundations for developing small drug-like molecules as Kv1.3 inhibitors for management of autoimmune inflammatory diseases as potential alternatives to several currently used drug combinations, imposing a great socio-economic impact.
Results and discussion
Design rationale of our β-carboline lead compound
Based on our ligand design process, we focused on modifying the pharmacophoric features of reported Kv1.3 inhibitors. The aforementioned pyranoquinolinone-based compound has shown Kv1.3 inhibition at the low two-digit micromolar concentration (IC50 = 10 µM) and a 2-fold selectivity over Kv currents at neuroblastoma cell line (N1E-115)54. This compound possesses a significant scaffold rigidity owing to the tricyclic fused core skeleton. Higher conformational stability was depicted to a quinoline-based close analog, namely CP-339,818. The small molecules exhibited Kv1.3 inhibition profile (IC50 = 0.23 µM), yet with toxicity concerns owing to the blockage of neuronal sodium channels55. CP-339,818 has a N-benzyl substitution, which introduces relevant conformational maneuvers for the compound around the sp3 carbon of the benzylic scaffold. These compounds have comparable size and electrostatic properties where both are considered tricyclic with moderate hydrophobicity of XLogP 3.12 to 3.78, reflecting acceptable oral bioavailability and membrane permeation56. Topological polar surface areas (TPSA) of both compounds are quite comparable having values 40.46 Å2 and 17.29 Å2 for pyranoquinolinone and CP-339,818, respectively. Typically, the TPSA values above highlight balanced hydrophobic/hydrophilic properties for the compounds that are crucial for maintaining adequate membrane permeation and polar solvent dissolution/distribution, and in turn appropriate pharmacological profiles57,58.
To develop a relevant lead compound based on the reported Kv1.3 inhibitors, we initially used the scaffold hopping approach on the quinoline-based core of the above-mentioned compounds, while maintaining a tricyclic scaffold (Fig. 3). For synthetic feasibility, we selected the β-carboline ring system. This scaffold also exists in many compounds with diverse biological activities and acceptable pharmacokinetics59,60,61. Further, the β-carboline core represents a feasible bioisostere for the furanocoumarin scaffold (Bergaptol moiety) of the reported potent Kv1.3 inhibitor, PAP-151. This is a phenoxyalkoxypsoralen-based ligand exhibiting high efficacy for blocking Kv1.3 current at 2 nM, besides its recognized selectivity patterns for Kv1.3 over Kv1.5 as well as Kv2, Kv3, Kv4 and HERG channel members51. The adopted β-carboline synthon was suggested to allow the introduction of alkyl or aromatic substitutions at position-1 for conducting the SAR investigation corresponding to the N-substitutions of the reported compound’s quinoline core ring. Following this, we conducted similar optimizations on the other side of the quinoline ring, which corresponds to the tail part of PAP-1, while fixing the β-carboline core. We also replaced the PAP-1’s distal phenoxy group with different heterocyclic fused systems serving as ring isosteres. Ultimately, this led us to select KMA as a lead compound besides several β-carboline members as structurally diverse analogs at two different diversity sites, for subsequent synthesis and biotesting.

Structural design of our proposed β-carboline lead compound. The reported Kv1.3 small molecules, with areas marked for structural modifications and key pharmacophoric features: tricyclic core aromatic ring (red), aromatic substitution (green), and hydrophobic tail group (blue). The designed β-carboline lead compound, KMA, showing the three key structural features being introduced based on a scaffold hopping approach.
Feasible chemical synthesis of KMA and its analogs
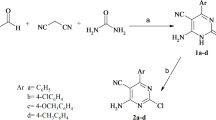
Preparation of KMA (5a) and several analogs bearing the β-carboline core scaffold is illustrated in Fig. 4, feasibly achieved through a parallel synthetic pathway. Pictet–Spengler condensation of the commercially available L-tryptophan (1) with different aldehydes62 followed by reaction with potassium dichromate in acidic medium furnished the 1-substituted β-carboline intermediates (2a-d). The structure of 2 was confirmed through 1H-NMR spectrum analysis highlighting the extra aromatic protons for the phenyl substitution as well as those of C3 and C4 of the β-carboline ring (δH ≈ 8.00 and 8.50 ppm). Parallel synthesis of the tail synthons (4a-d) was achieved through Mitsunobu cross-coupling of 4-bromobutanol with the appropriate phenols (3a-d)63. Finally, SN2 reaction between the furnished tail synthons and the free amino group of the substituted β-carboline intermediates (2a-d) provided the target compounds (5a-g)64. The presence of accurate number of aliphatic and aromatic protons within the 1H-NMR spectrum confirms the successful synthesis. Charts of compounds’ spectral data are presented in the Supplementary data.

Reagents and conditions: (a) (i) AcOH, aldehydes, reflux, 3 h; (ii) K2Cr2O7, AcOH, 100 °C, 20 min; (b) 4-bromobutanol, Ph3P, DIAD, THF, 0 °C-r.t, 16 h; (c) NaH, dry DMF, 60–80 °C, 48 h. Numbers in brackets denote; (1) L-tryptophan, (2a-d) β-carboline intermediates, (3a-d) corresponding phenols, (4a-d) tail synthons, (5a-g) target β-carboline analogs.
KMA exhibits promising activity at Kv1.3 patch clamp functional assay
With the synthesis and characterization of KMA (5a) molecule and analogs being completed, efforts were turned towards the biological evaluation of such compounds. A patch-clamp functional assay from stably transfected Chinese Hamster Ovary (CHO) K1 cells was adopted allowing specific channel activity to be studied at the single-cell level65. Kv1.3 currents were recorded from stably transfected cells, and the effect of a single concentration (10 µM) of the target compound were investigated by automated patch-clamping (Fig. 5). Notably, 5a showed highly promising results with the highest reduction of the Kv1.3 peak current amplitude (PCA) among all β-carboline analogs with a reduction value almost at 20% relative to the negative control sample (only vehicle; 0.1% DMSO). Regarding changes within channel’s inactivation kinetics, 5a illustrated moderate activity profile reducing the corresponding area under curve (AUC) parameter by more than 40% in relation to vehicle. Finally, the Kv1.3 inactivation time constant (ITC) was reduced by 5a by more than 30% as compared to vehicle control. Notably, the positive reference Kv1.3 inhibitor control, Margatoxin (3 nM)66, exhibited significant reduction of target peak current amplitude, area under the curve, and inactivation time constant at values of 4.44 ± 3.61, 5.16 ± 1.49, and 33.35 ± 11.98, respectively.
The synthesized β-carboline analogs depicted differential reduced activity of the Kv1.3 channel (Table 1). Fixing the p-methoxyphenyl synthon at the terminal aliphatic tail of the compounds while exploring different substitutions at β-carboline position 1, provided us with 5b and 5c. Notably, 5c with methyl at β-carboline position 1 showed marked reductions (above 80%) for both Kv1.3 AUC and RTC, whereas the channel peak amplitude current was of minimal alteration. Keeping the small methyl group at position 1, while exploring other terminal aromatic scaffolds like naphthalene and p-chlorophenyl moieties, was greatly tolerated by Kv1.3 with limited reductions of the Kv1.3 three investigated parameters. Further, values showed very limited change when altering the linker size (n = 4 to 2) as seen with the activity profiles of 5e and 5f mostly at the AUC values.
In addition, the latter observations highlight the importance of introducing hydrogen bond-capable functional group (methoxy as hydrogen acceptor) at the terminus of the hydrophobic tail to target the channel polar lining residues. In these regards, the best combination of substitutions would be a small-size methyl group at β-carboline position-1 and a terminal aromatic ring decorated with polar substitutions (hydrogen bond acceptors). This was further confirmed with the low activity patterns for compound 5 g harboring a terminal naphthyl tail group and a relatively large p-fluorophenyl ring at the β-carboline position 1 showing no significant differences from the vehicle readings over the three parameters.
Elaborating on the Kv1.3 current findings with compound 5c, its ability to slightly decrease peak current while profoundly decreasing the AUC implies that the main change imposed by this compound was shortening of the current duration. The latter suggests that compound 5c may accelerate inactivation or deactivation states. That is, the compound did not simply block the Kv1.3 pore instantaneously but also modulated the channel gating kinetics through either speeding the channel deactivation or stabilizing the inactivated channel state following the peak opening. A proposed mechanism of action could be related to the ability of 5c to impose enhanced inactivation mimicking the effects observed in the C-type inactivation of Kv1.3. In support of this explanation, a study by Bowlby et al. showed that exposure of Kv1.3-expressed cells to EGF highly accelerated the Kv1.3’s inactivation rates, without changing the kinetics of deactivation or activation67. Moreover, this suggests that the channel kinetics, as reflected in the AUC, can be independently changed without significant modulation of the peak current. Another study by Selvakumar et al. revealed through structural studies how a nanobody stabilized the inactivated conformation, leading to diminished sustained currents, even though the initial peak conductance is relatively preserved68. In terms of the immune context and physiological implication, the ability of compound 5c to stabilize the Kv1.3 inactivation state could (i) effectively suppress prolonged activation signals for mature T-cell proliferations, (ii) leave short bursts of activity intact for beneficial T-cell responses, and (iii) reduce the over-activation of T-cells that contributes to autoimmune diseases69,70.

Findings of patch clamp assay for inhibitory activity of KMA (5a) against Kv1.3 voltage conduction. (A) Comparative Kv1.3 current parameters (peak current amplitude, area under the peak, and inactivation time constant) for the developed β-carboline lead compound (KMA) in relation to vehicle and Margatoxin as control groups. All data were done in triplicates (n = 3) and presented as Mean ± SD. The mean of the vehicle condition is normalized to 100%. One-way analysis of variance (ANOVA) plus Dunnett’s post hoc test was used for statistical comparisons. Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001 for tested molecules against the vehicle samples; (B) The recorded Kv1.3 current amplitude in nano Amper (nA) across time in seconds (s) for KMA, vehicle, and Margatoxin. Experimental holding potential at -80 mV, Pulse / Duration at + 40 mV for 1000 ms, and Frequency at 0.2 Hz.
Iterative development of versatile molecules based on scaffold hopping and molecular hybridization
To explore other chemical synthons for developing Kv1.3 modulators with a more flexible aliphatic core, we shifted our sight towards the development of piperidine-based small molecules through a ligand-based approach. Members of this series were developed based on rational structural modifications of the well-known Kv1.3 blocker reported by Pfizer Inc. (UK-78282), which is also a good suppressor of the human T-lymphocyte activation71. This piperidine-based small molecule showed a sub-micromolar potency (IC50 ∼200 nM) and 1:1 stoichiometric blocking activity of the Kv1.3 currents through preferential binding to the channel’s C-type inactivated state. UK-78,282 depicted significant Kv1.3 selectivity over a band of closely related Kv channel members. Our strategy was to explore a linear aromatic head instead of the reported branched benzhydrol synthon of UK-78,282. The aim was to reduce the designed molecules’ bulkiness, the thing that was advantageous when adopting the position-1 methylation at the β-carboline series. Moreover, the tail part of UK-78,282 was replaced with different heterocyclic fused rings, guided by the results of the previous series, and serving as congruent isosteres of the methoxyphenyl group. The latter tail parts are linked to the central piperidine template through an amide linker to introduce some conformational restriction. The latter was suggested beneficial for minimizing the entropic expenditures associated with the compound adopting the target binding-preferred conformation72. Overall, this series possesses a higher fraction of sp3 carbon atoms (Fsp3), and hence drug-likeness, compared to the previous β-carboline series.
Synthesis of the piperidine-based series started with the commercially available alcohol; piperidin-4-ylmethanol (6), which represented a versatile chemical synthon for the introduction of both the head and tail parts subsequently through amide linkage formation and Mitsunobu coupling, respectively (Fig. 6). Thus, alcohol (6) was coupled with various heterocyclic carboxylic acids with the aid of EDCI to furnish the congruent amides (7a-d) carrying a free alcoholic group. Separately, Suzuki–Miyaura cross-coupling of 3-bromophenol (8) with the appropriate arylboronic acids provided the phenols (9a-b)73. Finally, Mitsunobu cross-coupling of such versatile intermediates, amides and phenols, furnished the target derivatives (10a-e) providing easiness and time/cost-effectiveness.

Reagents and conditions: (a) Appropriate carboxylic acids, EDCI, HOBt, DCM, r.t, 16 h; (b) ArB(OH)2, (dppf)PdCl2, K2CO3, EtOH, reflux, 16 h; (c) Ph3P, DIAD, THF, 0 °C to r.t, 16 h. Numbers in brackets denote; (6) piperidin-4-yl methanol, (7a-d) corresponding amides, (8) 3-bromophenol, (9a-d) corresponding phenols, (10a-c) target piperidine analogs.
Biological testing of the synthesized piperidine-based analogs was also performed using the Patch-clamp functional assay. Interestingly, compounds with the polar-decorated aromatic scaffolds (10c-e) depicted the most significant reduction of Kv1.3 current activity, particularly in terms of AUC and RTC (Table 2). The most significant Kv1.3 blockage activity profile among the piperidine series was illustrated for compound 10c, carrying a terminal p-methoxyphenyl moiety, with almost 35% reductions of the Kv1.3 currents. It is worth noting that the same chemical fragment showed the best activity in the β-carboline series. Also, compounds with terminal benzodioxole moiety (10d and e) showed a significant reduction of the Kv1.3 activity only second to 10c. The reduction of the AUC and RTC parameters was around 25% and 30% for 10d and 10e, respectively. The rest of the prepared compounds with more hydrophobic tails (10a and b) depicted almost comparable activity profiles. Rigidification of the central spacer connecting the core piperidine and the terminal aromatic scaffold as in 10a resulted in an insignificant alteration of the Kv1.3 current in terms of PCA and RTC, yet almost 20% AUC reductions being statistically significant in relation to the vehicle.
In an attempt to combine the pharmacophores of each developed series, hybridization between the PAP-1 and UK-78,282 scaffolds was achieved to develop novel small molecules. Molecular hybridization has evolved as a highly promising strategy within the innovation pathway of developing new candidates with improved pharmacodynamic/kinetic profiles74. The benzhydrole head part of UK-78,282 was replaced with the furanocoumarin (Bergaptol) scaffold of PAP-1 molecule. Synthetic route of the designed hybrids started with the commercially available piperidine derivative (6). This compound was coupled with diverse commercially available propionic acid derivatives under the standard amide-coupling conditions (Fig. 7). The obtained amides (7c and e) underwent Mitsunobu reaction to be coupled with Bergaptol (12) to give the corresponding ethers (13a and b) as final products. Due to the unavailability of Bergaptol, we had to synthesize it starting with the commercially available 5-methoxypsoralen (Bergapten) (11). The latter was demethylated using boron tribromide (BBr3) in DCM at room temperature. Biological testing of the synthesized hybrids highlighted the superior activity profile of 13a as compared to its more hydrophobic congruent (13b) and even the other developed molecules of the β-carboline and the piperidine series. This top-active hybrid molecule showed more than 95% reduction of both the AUC and time constant for inactivation of the Kv1.3 currents (Table 3). Depicted 13a values were promising in relation to those of the positive control standard, Margatoxin, since the latter is considered as a highly potent selective Kv1.3 channel blocker. The peak current amplitude of Kv1.3 channel was also significantly reduced with 13a treatment showing almost 80% current loss. It is worth mentioning that this top-active hybrid molecule harbors the terminal aromatic scaffold of p-methoxyphenyl, which is present in the most active derivatives of the two previous series. Thus, the Kv1.3 blocking activity of our developed synthetic compounds could be correlated with the hydrogen-bond capability of the terminal aromatic scaffold across the three designed series. The representative current traces corresponding to the electrophysiological data presented in Tables 1, 2 and 3 are placed at the supplementary data (Figures S32-S47).
Safety profile of all synthesized compounds has been highlighted where chemical structures lack any of the potential PAINS structures (Pan-assay interference compounds) that could raise relevant issues regarding off-target/toxicity. The compounds do not incorporate any of the reactive electrophilic functionalities (e.g. Michael acceptor) which could potentially generate covalent adducts towards cellular proteins or DNA (nucleophiles) that in turn would promote high risk of mutagenesis, off-target toxicity, and/or even idiosyncratic toxicity due to heightened hypersensitivity immune reactions. Furthermore, the synthesized compounds were virtually screened against several biotargets and biological systems using the ProTox 3.0-Prediction Of Toxicity Of Chemicals webserver (https://tox.charite.de/protox3/#; Structural Bioinformatics Grp, Charite Medicine University, Berlin, Germany) for assessing their safety profiles75. This on-line server incorporates fragment similarity-based CLUSTERing cross-validation for predicting acute toxicities, toxicity on organs, molecular initiating events, toxicity endpoints, metabolic interaction, adverse events pathways, and toxicity on specific targets. Tested compounds predicted inactivity against several tested pathways as well as lacked binding towards various toxicity targets such as, AA2AR (Adenosine receptor A2a), ADRB2 (Beta-2 adrenergic receptors), ANDR (Androgen receptor), AOFA (Amine oxidase flavin A), CRFR-1 (Corticotropin-releasing factor receptor 1), DRD3 (D3-dopaminergic receptor), ESR1 & 2 (Estrogen receptor alpha and beta), GCR (Glucocorticoid receptor), HRH1 (Histamine-1 receptor), NR1I2 (Nuclear receptor subfamily-1 group-I), KOP (Kappa opioid receptor), MOP (Mu opioid receptor), PDE4D (Phosphodiesterase-4D), PGH1 (Prostaglandin G/H synthase-1), and PRGR (Progesterone receptor). ProTox 3.0 output charts are at the Supplementary data (Figures S48-61).

Reagents and conditions: (a) Appropriate acid, EDCI, HOBt, TEA, rt, 16 h; (b) BBr3, DCM, room temperature, 4 h; (c) Bergaptol, Ph3P, DIAD, THF, 0 °C to r.t, 16 h. Numbers in brackets denote; (6) piperidine derivative, (7c&e) corresponding amides, (11) 5-methoxypsoralen (Bergapten), (12) Bergaptol, (13a, b) target hybrid analogs.
Conclusion
In this study, we successfully developed novel β-carboline, piperidine, and hybrid-based analogs as non-peptide small-molecule blockers of the Kv1.3 ion channel. The main lead identification and development approach relied on a ligand-based strategy involving scaffold hopping and molecular hybridization. Following iterative SAR-guided optimization, we identified β-carboline-based compound 5c and hybrid analog 13a as potent Kv1.3 inhibitors in relation to the reference peptide toxin, Margatoxin. It is worth noting that small molecule blockers of the Kv1.3 are relatively few and have lower affinities compared to the peptidic toxin analogs. Thus, obtaining such promising activities for small molecules highlights their potential usefulness for therapeutic targeting of Kv1.3. Most interesting, is compound 5c’s impact in stabilizing the inactivation state of the Kv1.3 channel through slight decrease of peak current and profound AUC reduction. This potential of functional selectivity and reduced off-target excitability suppression, seemingly offers advantages over conventional immunosuppressants. As we move forward, the methodologies established in this study can serve as a framework for future research aimed at developing additional small-molecule modulators of Kv1.3. Specifically, we will employ long molecular dynamics simulations, pocket druggability protocols, and water mapping studies to fine-tune our compound designs. Iterative exploration of the channel kinetics should enable us to advance lead compound optimization. The insights gained from the present, and forthcoming work, not only contribute to the understanding of Kv1.3 modulation but also highlight the potential for innovative therapeutic strategies in treating conditions associated with dysregulated immune responses. Further optimization, channel kinetic modulation analysis, and in vivo evaluation of these scaffolds are planned to advance their therapeutic applicability.
Experimental section
Chemical synthesis
Reaction progress was monitored by thin layer chromatography (TLC) analysis on Merck silica gel 60 F254 plates. Visualization was with UV light (254 nm) or iodine. Yields are for purified compounds and were not optimized. Reagents and solvents were obtained from commercial sources and used without any further purification. For air and moisture sensitive reactions, solvents were dried using Fisher molecular sieves type 4 A grade 514 (8–12 mesh; beads effective pore size 4 Å) and solutions were transferred via syringe and introduced into reaction vessels through rubber septa (Aldrich Sure/Seal). Flash column chromatography was performed on Loba Chemie silica gel (200–400 mesh) as a stationary phase. Melting points (mp) were determined in open capillaries on an Electrothermal IA9100 melting point apparatus and were uncorrected. Low-resolution mass spectra (MS) were recorded with a Shimadzu QP2010-Plus gas chromatograph/mass spectrometer (GC/MS), electron impact (EI+) 70 eV maintained at 250 °C, equipped with Shimadzu workstation or with electrospray ionization (ESI+) in positive ion mode with a Waters Multi-mode ESCi mass detector. Infrared (IR) spectra were recorded on a Bruker Tensor 27 FT-IR spectrophotometer, driven by OPUSTM version 1.1 managing software (Vmax in cm-1, using KBr pellets) The proton and carbon nuclear magnetic resonance (1H and 13C-NMR) spectra were recorded on a Varian-MERCURY spectrometer or a Bruker Ascend Aeon spectrometer at room temperature. Chemical shifts (δ; ppm) are recorded using tetramethylsilane (TMS) as the internal standard and were referenced to the respective residual solvent or deuterated solvent peaks (δH = 7.24 and δC = 76.8 for CHLOROFORM, δH = 3.30 and δC = 49.0 for DMSO-d6). HRMS was done using LC-MS-MS-QTOF which run at an Agilent 1260 Infinity LC-6230TOF LC/MS spectrometer (LCMS) and software acquisition 6200 series TOF/6500 series Q-TOF B.09.00 (B9044.0) at the Natural product research lab, Faculty of Pharmacy Fayoum University.
General procedure A: synthesis of 1-(phenyl)-9 H-pyrido[3,4-b]indole (2a)
A mixture of L-tryptophan 1 (1 g, 4.90 mmol) and benzaldehyde (0.5 mL, 4.90 mmol) in 10 mL of glacial acetic acid was refluxed for 3 h under inert atmosphere. Afterward, the reaction mixture was allowed to cool to room temperature, and the pH was adjusted to approximately 6.0 using concentrated NH4OH. The resulting suspension was refrigerated for 2 h, and the precipitate formed, was collected by filtration. The solid was dried in an oven at 45 °C overnight, yielding a creamy white solid (1.2 g, 85%) and used in the next step without further purification. A mixture of 1-phenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid (1 g, 3.42 mmol) in 30 mL of distilled water was brought to a boil, followed by the slow addition of potassium dichromate (6 g) and acetic acid (7 mL). The brown suspension was heated for 20 min and then cooled under running tap water. The cold solution was treated with sodium sulfite, after which the mixture was turned into alkaline with sodium hydroxide. The solution was extracted with EtOAc (3 × 150 mL), and the extracts were dried over anhydrous sodium sulfate. After removing the solvent, the residue was crystallized from cold anhydrous EtOH, yielding the desired compounds as a brownish yellow solid (0.470 g, 57%). m.p. 245–250 °C. ESI-MS m/z: 244.10 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 7.27 (t, J = 7.61 Hz, 1 H), 7.50–7.58 (m, 2 H), 7.59–7.68 (m, 3 H), 8.01–8.06 (m, 2 H), 8.12 (d, J = 4.81 Hz, 1 H), 8.27 (d, J = 8.01 Hz, 1 H), 8.46 (d, J = 4.81 Hz, 1 H), 11.54 (s, 1 H).
1-(thiophen-3-yl)-9H-pyrido[3,4-b]indole (2b)
This compound was synthesized according to the general procedure A from 1 and thiophene-3-carbaldehyde to afford a greenish yellow solid (0.515 g, 61%). m.p. 247–255 °C. ESI-MS m/z: 250.32 [M + H]+. 1H-NMR (500 MHz, DMSO-d6) δH ppm 7.27 (d, J = 7.61 Hz, 1 H), 7.57 (d, J = 7.61 Hz, 1 H), 7.69 (d, J = 8.01 Hz, 1 H), 7.78 (s, 1 H), 7.90 (d, J = 4.81 Hz, 1 H), 8.09 (d, J = 5.61 Hz, 1 H), 8.26 (d, J = 8.01 Hz, 1 H), 8.31 (d, J = 1.60 Hz, 1 H), 8.41 (d, J = 4.81 Hz, 1 H), 11.45 (s, 1 H).
1-methyl-9 H-pyrido[3,4-b]indole (2c)
This compound was synthesized according to the general procedure A from 1 and acetaldehyde to afford a golden yellow solid (0.390 g, 49%). m.p. 235–240 °C. ESI-MS m/z: 182.23 [M + H]+. 1H-NMR (500 MHz, DMSO-d6) δH ppm 2.76 (s, 3 H), 7.22 (s, 1 H), 7.53 (s, 1 H), 7.57–7.61 (m, 1 H), 7.92 (d, J = 5.61 Hz, 1 H), 8.14–8.26 (m, 2 H), 11.57 (s, 1 H).
1-(4-fluorophenyl)-9H-pyrido[3,4-b]indole (2d)
This compound was synthesized according to the general procedure A from 1 and 4-fluorobenzaldehye to afford a golden yellow solid (0.357, 42%). m.p. 235–240 °C. ESI-MS m/z: 262.29 [M + H]+. 1H-NMR (500 MHz, DMSO-d6) δH ppm 7.27 (s, 1 H), 7.44 (t, J = 8.82 Hz, 2 H), 7.56 (s, 1 H), 7.65 (d, J = 8.01 Hz, 1 H), 8.07 (dd, J = 8.41, 5.21 Hz, 2 H), 8.13 (t, J = 5.21 Hz, 1 H), 8.27 (d, J = 7.21 Hz, 1 H), 8.45 (d, J = 4.81 Hz, 1 H), 11.56 (s, 1 H).
General procedure B: synthesis of 1-(4-bromobutyloxy)-4-methoxybenzene (4a)
To a stirred and ice-bath cooled mixture of 4-methoxyphenol 3 (0.250 g, 2 mmol) in dried THF (10 mL) was added 4-bromobutanol (0.306 g, 2 mmol). The mixture was placed under nitrogen by evacuation and filling with nitrogen three times. Triphenylphosphine (0.577 g, 2.2 mmol) was added portion wise to this mixture, followed by diisopropyl azodicarboxylate (0.445 g, 2.2 mmol) in a strictly dropwise fashion. The reaction mixture was first stirred for 1 h at 0 °C and then overnight at room temperature. Then, the reaction mixture was concentrated under reduced pressure and the residue was partitioned between saturated aqueous NaHCO3 and EtOAc. The organic layer was washed with sodium chloride solution, dried over anhydrous MgSO4 and concentrated in vacuo affording a light-yellow oil. The crude oil was chromatographed on flash column with a mixture of Hexane/EtOAc (80:20) to yield 60% of the target ether 4a as a yellow oil. +ESI-MS m/z: 259.14 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.94 (m, 2 H), 2.03–2.14 (m, 2 H), 3.51 (t, J = 6.60 Hz, 2 H), 3.79 (s, 3 H), 3.97 (t, J = 6.60 Hz, 2 H), 6.85 (s, 4 H).
1-(4-bromobutoxy)-4-chlorobenzene (4b)
This compound was synthesized according to the general procedure B from 4-chlorophenol and 4-bromobutanol to afford yellow oil (0.323 g, 63%).+ESI-MS m/z: 263.56 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.97 (d, J = 8.56 Hz, 2 H), 2.03–2.14 (m, 2 H), 3.51 (t, J = 5.99 Hz, 2 H), 3.98 (t, J = 5.99 Hz, 2 H), 6.76–6.94 (m, 2 H), 7.17–7.36 (m, 2 H).
2-(4-bromobutoxy)naphthalene (4c)
This compound was synthesized according to the general procedure B from 2-naphthol and 4-bromobutanol to afford colorless oil (0.281 g, 0.58%).+ESI-MS m/z: 279.18 [M + H]+. 1H-NMR (500 MHz, DMSO-d6) δH ppm 1.86–1.95 (m, 2 H), 1.97–2.08 (m, 2 H), 3.64 (t, J = 6.81 Hz, 2 H), 4.12 (t, J = 6.81 Hz, 2 H), 7.16 (dd, J = 8.82, 2.40 Hz, 1 H), 7.29–7.36 (m, 2 H), 7.42–7.48 (m, 1 H), 7.77–7.84 (m, 3 H).
2-(2-bromoethoxy)naphthalene (4d)
This compound was synthesized according to the general procedure B from 2-naphthol and 4-chloroethanol to afford colorless oil (0.292 g, 0.67%).+ESI-MS m/z: 251.12 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 3.90 (t, J = 6.01 Hz, 2 H), 4.28–4.40 (m, 2 H), 7.11–7.23 (m, 2 H), 7.37 (s, 1 H), 7.47 (s, 1 H), 7.67–7.85 (m, 3 H).
General procedure C: synthesis of 9-(4-(4-methoxyphenoxy)butyl)-1-phenyl-9 H-pyrido[3,4-b]indole (5a) (KMA)
In a two-neck round-bottom flask under an inert argon atmosphere, 1-phenyl-9 H-pyrido[3,4-b]indole 2 (0.2 g, 0.82 mmol) was dissolved in 5 mL of anhydrous DMF, and the solution was cooled in an ice bath before the slow portion wise addition of sodium hydride (0.040 g, 1.6 mmol). The resulting mixture was stirred for one hour, followed by the addition of 1-(4-bromobutoxy)-4-methoxybenzene 4 (0.254 g, 0.98 mmol), which had been dissolved under an inert atmosphere in a separate flask containing 3 mL of DMF. The mixture was stirred under an inert atmosphere at 80 °C for 48 h. The reaction mixture was then extracted with water and EtOAc (3 × 50 mL), followed by a brine wash. The combined organic layers were dried over anhydrous sodium sulfate, and the solvent was evaporated under vacuum. The resulting residue was purified by flash chromatography on silica gel using a dichloromethane: methanol (98:2) eluent to yield (0.15 g; 73%) of the desired product KMA as a yellow solid. m.p. 250–255 °C. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.24–1.37 (m, 2 H), 1.51–1.68 (m, 2 H), 3.64 (t, J = 6.24 Hz, 2 H), 3.79 (s, 3 H), 4.00-4.14 (m, 2 H), 6.70–6.77 (m, 2 H), 6.81–6.87 (m, 2 H), 7.32–7.39 (m, 1 H), 7.47–7.57 (m, 4 H), 7.60–7.70 (m, 3 H), 8.06 (d, J = 5.14 Hz, 1 H), 8.23 (d, J = 7.83 Hz, 1 H), 8.57 (d, J = 5.14 Hz, 1 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 25.88, 25.97, 26.34, 44.39, 55.76, 67.55, 110.36, 114.07, 114.62, 114.66, 115.31, 115.47, 120.43, 121.14, 121.98, 128.48, 129.24, 129.46, 133.81, 152.81, 153.83. HRMS-ESI (m/z) [M + H]+ Calcd. for C28H26N2O2: 423.2064, found: 423.2084.
9-(4-(4-methoxyphenoxy)butyl)-1-(thiophen-3-yl)-9H-pyrido[3,4-b]indole (5b)
This compound was synthesized according to the general procedure C from 2b and 4a to afford a greenish yellow solid (0.277, 81%). m.p. 195–205 °C. ESI-MS m/z: 428.25 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.38–1.47 (m, 2 H), 1.59–1.69 (m, 2 H), 3.72 (t, J = 6.11 Hz, 2 H), 3.78 (s, 3 H), 4.12–4.18 (m, 2 H), 6.74–6.78 (m, 2 H), 6.81–6.83 (m, 1 H), 6.84–6.86 (m, 1 H), 7.36 (d, J = 7.46 Hz, 1 H), 7.42 (dd, J = 4.89, 1.22 Hz, 1 H), 7.46–7.53 (m, 2 H), 7.62–7.68 (m, 2 H), 8.06 (d, J = 5.38 Hz, 1 H), 8.22 (d, J = 7.46 Hz, 1 H), 8.55 (d, J = 5.38 Hz, 1 H). 13C-NMR (101 MHz, CHLOROFORM-d) δ ppm 25.97, 26.05, 26.50, 29.59, 44.35, 55.76, 62.55 (s, 1 C) 67.56, 68.47, 110.43, 114.30, 114.66, 115.33, 115.41, 115.47, 120.61, 121.01, 122.06, 126.29, 128.96, 129.50, 134.25, 142.76, 152.78, 153.82, 153.84. Anal. Calcd. for C26H24N2O2S: C, 72.87; H, 5.65; N, 6.54; Found: C, 72.97; H, 5.58; N, 6.53.
9-(4-(4-methoxyphenoxy)butyl)-1-methyl-9H-pyrido[3,4-b]indole (5c)
This compound was synthesized according to the general procedure C from 2c to 4a to afford a yellow solid (0.309, 78%). m.p. 193–203 °C. ESI-MS m/z: 360.46 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.80–1.95 (m, 2 H), 2.06 (d, J = 7.34 Hz, 2 H), 3.13 (s, 3 H), 3.78 (s, 3 H), 3.97 (t, J = 5.99 Hz, 2 H), 4.59–4.75 (m, 2 H), 6.77–6.96 (m, 4 H), 7.31 (d, J = 16.87 Hz, 1 H), 7.52 (d, J = 8.56 Hz, 1 H), 7.58–7.68 (m, 1 H), 7.92 (d, J = 5.38 Hz, 1 H), 8.16 (d, J = 7.82 Hz, 1 H), 8.35 (d, J = 5.38 Hz, 1 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 22.18, 26.64, 27.84, 44.76, 55.75, 67.80, 109.97 (s, 1 C) 113.45, 114.71, 115.36, 115.46, 120.29, 120.98, 121.88, 128.60, 129.09, 132.16, 134.78, 135.86, 140.33, 142.11, 152.78, 153.94. Anal. Calcd. for C23H24N2O2: C, 76.64; H, 6.71; N, 7.77; Found: C, 76.66; H, 7.16; N, 7.87.
9-(4-(4-chlorophenoxy)butyl)-1-methyl-9H-pyrido[3,4-b]indole (5d)
This compound was synthesized according to the general procedure C from 2c and 4b to afford a yellow solid (0.289, 73%). m.p. 195–205 °C. ESI-MS m/z: 364.87 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.92 (d, J = 8.31 Hz, 2 H), 2.07 (br. s., 2 H), 3.14 (s, 3 H), 3.97 (t, J = 5.99 Hz, 2 H), 4.55–4.77 (m, 2 H), 6.80 (q, J = 9.05 Hz, 2 H), 7.20–7.27 (m, 2 H), 7.34 (s, 1 H), 7.51 (t, J = 8.31 Hz, 1 H), 7.64 (s, 1 H), 7.93 (d, J = 5.38 Hz, 1 H), 8.16 (d, J = 7.82 Hz, 1 H), 8.35 (d, J = 5.38 Hz, 1 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 21.48, 26.46, 27.78, 44.77, 67.40, 110.01, 113.75, 115.63, 115.76, 120.66, 120.81, 122.11, 125.82, 129.30, 129.43, 129.63, 130.84, 134.62, 134.81, 139.76, 142.45, 157.19. Anal. Calcd. for C22H21ClN2O: C, 72.42; H, 5.80; N, 7.68; Found: C, 72.43; H, 5.82; N, 7.71.
1-methyl-9-(4-(naphthalen-2-yloxy)butyl)-9 H-pyrido[3,4-b]indole (5e)
This compound was synthesized according to the general procedure C from 2c to 4c to afford a brownish yellow solid (0.342, 82%). m.p. 200–210 °C. ESI-MS m/z: 380.49 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 1.98 (d, J = 8.01 Hz, 2 H), 2.11 (br. s., 2 H), 3.08 (s, 3 H), 4.12 (t, J = 6.01 Hz, 2 H), 4.59–4.75 (m, 2 H), 7.08–7.16 (m, 2 H), 7.29 (s, 1 H), 7.35 (s, 1 H), 7.45 (s, 1 H), 7.50 (d, J = 8.82 Hz, 1 H), 7.59 (s, 1 H), 7.69–7.79 (m, 3 H), 7.86 (d, J = 5.61 Hz, 1 H), 8.13 (d, J = 7.21 Hz, 1 H), 8.35 (d, J = 5.61 Hz, 1 H). 13C-NMR (126 MHz, CHLOROFORM-d) δC ppm 23.76, 26.77, 27.99, 44.80, 67.38, 106.71, 109.92, 113.22, 118.99, 119.87, 121.55, 121.76, 123.88, 126.62, 126.91, 127.87, 128.37, 129.18, 129.32, 129.70, 134.70, 135.29, 138.26, 141.44, 141.66, 156.85. Anal. Calcd. for C26H24N2O: C, 82.70; H, 6.36; N, 7.36; Found: C, 82.10; H, 6.19; N, 7.28.
1-methyl-9-(2-(naphthalen-2-yloxy)ethyl)-9H-pyrido[3,4-b]indole (5f)
This compound was synthesized according to the general procedure C from 2c and 4d to afford a brownish yellow solid (0.267, 69%). m.p. 188–208 °C. ESI-MS m/z: 352.44 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 3.18 (s, 3 H), 4.43–4.52 (m, 2 H), 5.09 (d, J = 5.61 Hz, 2 H), 6.99–7.03 (m, 2 H), 7.29–7.36 (m, 2 H), 7.39–7.43 (m, 1 H), 7.61–7.65 (m, 3 H), 7.68 (d, J = 9.62 Hz, 1 H), 7.73 (d, J = 8.01 Hz, 1 H), 7.87 (d, J = 5.61 Hz, 1 H), 8.14 (d, J = 8.01 Hz, 1 H), 8.37 (d, J = 5.61 Hz, 1 H). 13C-NMR (126 MHz, CHLOROFORM-d) δC ppm 23.63, 43.86, 66.30, 106.17, 109.66, 112.81, 118.23, 119.82, 121.32, 123.62, 126.22, 126.41, 127.35, 128.01, 128.82, 129.06, 129.28, 133.96, 137.99, 141.15, 141.33, 155.79. Anal. Calcd. for C24H20N2O: C, 81.79; H, 5.72; N, 7.95; Found: C, 81.87; H, 5.55; N, 8.17.
1-(4-fluorophenyl)-9-(4-(naphthalen-2-yloxy)butyl)-9H-pyrido[3,4-b]indole (5 g)
This compound was synthesized according to the general procedure C from 2d and 4c to afford a yellow solid (0.260, 74%). m.p. 205–215 °C. ESI-MS m/z: 460.55 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 1.36–1.50 (m, 2 H), 1.58–1.70 (m, 2 H), 3.81 (t, J = 6.41 Hz, 2 H), 4.04–4.17 (m, 2 H), 7.00 (d, J = 2.40 Hz, 1 H), 7.06 (dd, J = 8.82, 2.40 Hz, 1 H), 7.19 (t, J = 8.41 Hz, 2 H), 7.34 (td, J = 7.41, 4.41 Hz, 2 H), 7.40–7.52 (m, 2 H), 7.58–7.66 (m, 3 H), 7.69–7.82 (m, 3 H), 8.00 (d, J = 4.81 Hz, 1 H), 8.20 (d, J = 8.01 Hz, 1 H), 8.53 (d, J = 4.81 Hz, 1 H). 13C-NMR (126 MHz, CHLOROFORM-d) δC ppm 26.03, 26.45, 44.43, 67.10, 106.65, 110.40, 114.07, 115.42, 115.59, 118.92, 120.25, 121.91, 123.84, 126.61, 126.90, 127.84, 128.78, 129.12, 129.62, 130.86, 131.29, 131.36, 134.31, 134.70, 136.40, 138.67, 142.30, 143.22, 156.78, 162.06, 164.04. Anal. Calcd. for C31H25FN2O: C, 80.85; H, 5.47; N, 7.00; Found: C, 80.83; H, 5.49; N, 6.98.
General procedure D: synthesis of (E)-1-(4-(hydroxymethyl)piperidin-1-yl)-3-phenylprop-2-en-1-one (7a)
In a two-neck round-bottom flask under an argon atmosphere, a mixture of cinnamic acid (0.257 g, 1.73 mmol), EDCI (0.5 g, 2.6 mmol), and HOBt (0.352 g, 2.6 mmol) was dissolved in 6 mL of DMF and stirred for 10 min at room temperature. Subsequently, 4-piperidinemethanol (6) (0.2 g, 1.73 mmol) was added, followed by the addition of triethylamine (TEA). The reaction mixture was stirred at room temperature for 18 h. Upon completion, the reaction mixture was subjected to a standard work-up, involving partitioning between ethyl acetate and distilled water, followed by sequential washes with 2 M ammonium chloride solution and brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. Purification was performed using flash chromatography, eluting with 2% methanol in dichloromethane (DCM), yielding 0.383 g (90%) as colorless sticky oil. ESI-MS m/z: 245.32 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.06–1.25 (m, 3 H), 1.82 (m, 3 H), 2.62 (m, 1 H), 3.05 (m, 1 H), 3.45 (br. s., 2 H), 4.07 (d, J = 12.23 Hz, 1 H), 4.67 (d, J = 12.23 Hz, 1 H), 6.83 (d, J = 15.41 Hz, 1 H), 7.29 (d, J = 6.85 Hz, 3 H), 7.45 (d, J = 6.85 Hz, 2 H), 7.56 (d, J = 15.41 Hz, 1 H).
3-(4-fluorophenyl)-1-(4-(hydroxymethyl)piperidin-1-yl)propan-1-one (7b)
This compound was synthesized according to the general procedure D from 6 and 4-fluoropropioic acid to afford a colorless sticky oil (0.391, 85%). ESI-MS m/z: 265.33 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 0.97–1.21 (m, 2 H), 1.74–1.81 (m, 4 H), 2.50–2.65 (m, 3 H), 2.90–3.03 (m, 3 H), 3.50 (t, J = 6.48 Hz, 2 H), 3.83 (d, J = 13.20 Hz, 1 H), 4.67 (d, J = 12.96 Hz, 1 H), 6.98 (t, J = 8.56 Hz, 2 H), 7.13–7.23 (m, 2 H).
1-(4-(hydroxymethyl)piperidin-1-yl)-3-(4-methoxyphenyl)propan-1-one (7c)
This compound was synthesized according to the general procedure D from 6 and 4-methoxypropioic acid to afford a colorless sticky oil (0.423, 88%). ESI-MS m/z: 277.36 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.09 (br. s., 1 H), 1.65–1.81 (m, 3 H), 2.05 (s, 2 H), 2.56–2.73 (m, 3 H), 2.83–2.98 (m, 3 H), 3.49 (d, J = 5.62 Hz, 2 H), 3.80 (s, 3 H), 6.85 (t, J = 8.31 Hz, 2 H), 7.15 (t, J = 8.31 Hz, 2 H).
3-(benzo[d]1,3dioxol-5-yl)-1-(4-(hydroxymethyl)piperidin-1-yl)propan-1-one (7d)
This compound was synthesized according to the general procedure D from 6 and 3-(benzo[d]1,3dioxol-5-yl)propanoic acid to afford a colorless sticky oil (0.410, 81%). ESI-MS m/z: 291.35 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 0.99–1.20 (m, 2 H), 1.76 (br. s., 3 H), 2.57–2.62 (m, 2 H), 2.82 (s, 2 H), 2.86–2.95 (m, 2 H), 2.97 (s, 1 H), 3.50 (t, J = 6.72 Hz, 2 H), 3.84 (d, J = 13.20 Hz, 1 H), 4.68 (d, J = 13.20 Hz, 1 H), 5.93 (s, 2 H), 6.65–6.70 (m, 1 H), 6.71–6.79 (m, 2 H).
3-(4-chlorophenyl)-1-(4-(hydroxymethyl)piperidin-1-yl)propan-1-one (7e)
This compound was synthesized according to the general procedure D from 6 and 4-chloropropioic acid to afford a colorless sticky oil (0.490, 90%). ESI-MS m/z: 281.78 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.07 (br. s., 2 H), 1.27 (s., 1 H), 1.64–1.86 (m, 3 H), 2.11 (br. s., 2 H), 2.62 (t, J = 7.70 Hz, 2 H), 2.77–2.88 (m, 1 H), 2.95 (t, J = 7.70 Hz, 2 H), 3.49 (d, J = 5.62 Hz, 2 H), 4.63 (br. s., 1H), 7.16 (d, J = 8.31 Hz, 2 H), 7.26 (d, J = 8.31 Hz, 2 H).
General procedure E: synthesis of [1,1’-biphenyl]-3-ol (9a)
A solvent system consisting of toluene, methanol, and a 2 M aqueous solution of K₂CO₃ (20 mL: 10 mL: 2.5 mL, respectively) was prepared and purged with argon for 1 h. To this, 3-bromobiphenol (8) (0.2 g, 1.18 mmol), phenylboronic acid (0.180 g, 1.42 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.067 g, 0.058 mmol) were added. The reaction mixture was heated under reflux at 95–105 °C for 24 h. After completion, the reaction mixture was concentrated by heating without a condenser to reduce the solvent volume and ensure the oxidation of residual palladium. The pH was then adjusted to 4.0 using 1 N HCl, and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (3 × 50 mL), dried over anhydrous Na₂SO₄, and concentrated under reduced pressure. Purification was performed by column chromatography on silica gel, eluting with hexane: EtOAc (80:20) to give the desired product 9a as white crystalline solid (0.179 g, 90%). m.p. 145–150 °C. ESI-MS m/z: 170.21 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 6.86 (dd, J = 7.95, 2.32 Hz, 1 H), 7.10 (t, J = 1.96 Hz, 1 H), 7.21 (d, J = 7.82 Hz, 1 H), 7.31–7.42 (m, 2 H), 7.47 (t, J = 7.58 Hz, 2 H), 7.60 (d, J = 7.58 Hz, 2 H), 9.61 (s, 1H).
4’-methoxy-[1,1’-biphenyl]-3-ol (9b)
This compound was synthesized according to the general procedure E from (8) and 4-methoxyphenylboronic acid to afford an off-white crystalline solid (0.199 g, 87%). m.p. 153–158 °C. ESI-MS m/z: 200.14 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 3.80 (s, 3 H), 6.66–6.78 (m, 1 H), 6.91–7.10 (m, 4 H), 7.23 (t, J = 7.95 Hz, 1 H), 7.48–7.62 (m, 2 H), 9.49 (s, 1H).
General procedure F: synthesis of (E)-1-(4-(((4’-methoxy-[1,1’-biphenyl]-3-yl)oxy)methyl)piperidin-1-yl)-3-phenylprop-2-en-1-one (10a)
In a one-neck round-bottom flask under an argon atmosphere, a mixture of 1-(4-(hydroxymethyl)piperidin-1-yl)-3-phenylprop-2-en-1-one 7a (0.100 g, 0.407 mmol), 4’-methoxy-[1,1’-biphenyl]-3-ol 9b (0.082 g, 0.407 mmol), and triphenylphosphine (0.160 g, 0.611 mmol) was dissolved in 5 mL of tetrahydrofuran (THF) and stirred for 10 min. The reaction mixture was then cooled in an ice bath, followed by the slow dropwise addition of diisopropyl azodicarboxylate (DIAD, 0.120 mL, 0.611 mmol). The reaction was allowed to reach room temperature and stirred for 24 h. Upon completion, THF was evaporated under reduced pressure, and the reaction mixture was subjected to a standard work-up by partitioning between ethyl acetate and distilled water (twice), followed by a single wash with brine. The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification was carried out using flash chromatography, eluting with 20% EtOAc in dichloromethane (DCM), affording the product as a sticky oil (0.113 g, 65% yield). ESI-MS m/z:427.54 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.22–1.35 (m, 3 H), 1.90-2.00 (m, 3 H), 2.17 (br. s., 2 H), 3.75–3.80 (m, 4 H), 4.73 (d, J = 11.62 Hz, 2 H), 6.93 (d, J = 8.44 Hz, 1 H), 6.83–6.93 (m, 4 H), 7.15 (d, J = 12.86 Hz, 1 H), 7.09 (s, 1 H), 7.23–7.34 (m, 3 H), 7.45 (d, J = 12.86 Hz, 1 H), 7.57 (t, J = 7.75 Hz, 2 H), 7.61 (d, J = 8.36 Hz, 2 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 25.10, 28.22, 31.43, 34.16, 41.42, 44.60, 54.45, 71.25, 112.33, 112.94, 114.32, 115.02, 116.43, 118.82, 120.54, 128.32, 129.65, 129.94, 130.19, 132.54, 136.24, 138.27, 142.72, 143.46, 159.11, 160.23, 162.45, 171.42. Anal. Calcd. for C28H29NO3: C, 78.66; H, 6.84; N, 3.28; Found: C, 78.74; H, 6.90; N, 3.34.
3-(4-fluorophenyl)-1-(4-(((4’-methoxy-[1,1’-biphenyl]-3-yl)oxy)methyl)piperidin-1-yl)propan-1-one (10b)
This compound was synthesized according to the general procedure F from 7b and 9b to afford colorless sticky oil (0.97 g, 53%). ESI-MS m/z: 447.55 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.20–1.32 (m, 3 H), 1.90 (t, J = 10.51 Hz, 2 H), 2.07 (br. s., 1 H), 2.55–2.70 (m, 3 H), 2.91–3.07 (m, 3 H), 3.84–3.90 (m, 5 H), 4.73 (d, J = 12.72 Hz, 1 H), 6.84 (d, J = 8.31 Hz, 1 H), 6.96–7.03 (m, 4 H), 7.08 (s, 1 H), 7.14–7.24 (m, 3 H), 7.34 (t, J = 7.95 Hz, 1 H), 7.54 (d, J = 8.56 Hz, 2 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 28.60, 29.62, 30.73, 35.15, 35.17, 36.35, 41.70, 45.50, 55.37, 72.05, 112.56, 113.10, 114.20, 115.12, 115.33, 119.43, 128.18, 129.75, 129.84, 129.92, 133.54, 137.04, 137.07, 142.46, 159.28, 159.32, 162.66, 170.32. Anal. Calcd. for C28H30FNO3: C, 75.14; H, 6.76; N, 3.13; Found: C, 75.17; H, 6.88; N, 3.16.
1-(4-(((4’-methoxy-[1,1’-biphenyl]-3-yl)oxy)methyl)piperidin-1-yl)-3-(4-methoxyphenyl) propan-1-one (10c)
This compound was synthesized according to the general procedure F from 7c and 9b to afford colorless sticky oil (0.116 g, 62%). ESI-MS m/z: 459.59 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.80 (d, J = 12.96 Hz, 2 H), 1.97 (br. s., 2 H), 2.14 (br. s., 2 H), 2.27 (br. s., 1 H), 2.57 (t, J = 7.83 Hz, 3 H), 2.73–2.98 (m, 3 H), 3.69 (s, 3 H), 3.73–3.81 (m, 5 H), 6.69–6.80 (m, 3 H), 6.90 (t, J = 8.80 Hz, 2 H), 6.98 (s, 1 H), 7.07 (d, J = 8.56 Hz, 3 H), 7.21–7.29 (m, 1 H), 7.44 (t, J = 8.80 Hz, 2 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 25.91, 26.45, 31.40, 33.44, 35.31, 42.34, 45.01, 55.83, 74.62, 110.32, 113.82, 114.21, 115.82, 119.5, 128.79, 129.80, 130.11, 133.62, 134.11, 141.43, 146.32, 157.82, 159.53, 175.31. Anal. Calcd. for C29H33NO4: C, 75.79; H, 7.24; N, 3.20; Found: C, 75.85; H, 7.26; N, 3.13.
3-(benzo[d]1,3dioxol-5-yl)-1-(4-(((4’-methoxy-[1,1’-biphenyl]-3-yl)oxy)methyl)piperidin-1-yl)propan-1-one (10d)
This compound was synthesized according to the general procedure F from 7d and 9b to afford colorless sticky oil (0.129 g, 67%). ESI-MS m/z: 473.57 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.16–1.33 (m, 3 H), 1.69 (br. s., 1 H), 1.83–1.95 (m, 2 H), 2.07 (br. s., 1 H), 2.62 (t, J = 7.83 Hz, 3 H), 2.82–2.96 (m, 2 H), 3.02 (br. s., 1 H), 3.82–3.90 (m, 4 H), 4.72 (br. s., 1 H), 5.92 (s, 2 H), 6.67–6.72 (m, 1 H), 6.73–6.78 (m, 2 H), 6.84 (d, J = 7.82 Hz, 1 H), 6.99 (t, J = 8.31 Hz, 2 H), 7.08 (br. s., 1 H), 7.16 (d, J = 7.58 Hz, 1 H), 7.31–7.39 (m, 1 H), 7.54 (t, J = 8.31 Hz, 2 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 28.61, 29.63, 31.35, 35.39, 36.34, 41.68, 45.53, 55.37, 72.11, 100.81, 108.29, 108.98, 112.56, 113.11, 114.20, 119.42, 121.22, 128.18, 129.74, 133.55, 135.24, 142.45, 147.66, 159.30, 159.32, 170.48. Anal. Calcd. for C29H31NO5: C, 73.55; H, 6.60; N, 2.96; Found: C, 73.68; H, 6.62; N, 3.00.
1-(4-(([1,1’-biphenyl]-3-yloxy)methyl)piperidin-1-yl)-3-(benzo[d]1,3dioxol-5-yl)propan-1-one (10e)
This compound was synthesized according to the general procedure F from 7d and 9a to afford colorless sticky oil (0.110 g, 61%). ESI-MS m/z: 443.34 [M + H]+. 1H-NMR (400 MHz, CHLOROFORM-d) δH ppm 1.26–1.34 (m, 2 H), 1.91 (t, J = 11.49 Hz, 2 H), 2.08 (br. s., 1 H), 2.56–2.70 (m, 3 H), 2.89–2.97 (m, 2 H), 2.98–3.09 (m, 1 H), 3.81–3.96 (m, 3 H), 4.74 (d, J = 12.96 Hz, 1 H), 5.93 (s, 2 H), 6.67–6.73 (m, 1 H), 6.73–6.79 (m, 2 H), 6.90 (d, J = 7.58 Hz, 1 H), 7.13 (br. s., 1 H), 7.21 (d, J = 7.58 Hz, 1 H), 7.34–7.41 (m, 2 H), 7.43–7.50 (m, 2 H), 7.58–7.64 (m, 2 H). 13C-NMR (101 MHz, CHLOROFORM-d) δC ppm 28.61, 29.62, 31.36, 35.41, 36.34, 41.68, 45.53, 72.13, 100.83, 108.30, 108.98, 113.20, 113.48, 119.82, 121.24, 127.20, 127.48, 128.76, 129.81, 135.23, 141.03, 142.85, 145.88, 147.66, 159.29, 170.50. Anal. Calcd. for C28H27NO4: C, 76.17; H, 6.16; N, 3.17; Found: C, 76.19; H, 6.18; N, 3.14.
4-hydroxy-7 H-furo[3,2-g]chromen-7-one (12)
To a solution of bergapten (11) (0.5 g) in dichloromethane (20 mL), boron tribromide (BBr₃, 20 mL, 1 M in DCM) was added dropwise under an argon atmosphere at room temperature. The reaction mixture was stirred for 4 h, after that, slowly poured into a saturated NaHCO3 solution (150 mL), leading to the precipitation of a yellowish solid. The mixture was stirred for an additional 30 min, and the resulting solid was collected by filtration, washed with cold water and diethyl ether, and dried under high vacuum. The product was obtained as an off-white solid (0.45 g, 96%). m.p. 275–280 °C. +ESI-MS m/z: 202.17 [M + H]+. IR (KBr): 3305, 3143, 1700, 1697, 1313 cm− 1. 1H-NMR (500 MHz, DMSO-d6) δH ppm 6.24 (d, J = 9.77 Hz, 1 H), 7.13 (s, 1 H), 7.20 (dd, J = 2.21, 0.95 Hz, 1 H), 7.89 (d, J = 2.21 Hz, 1 H), 8.26 (d, J = 9.77 Hz, 1 H), 11.49 (br., s., 1 H). 13C-NMR (101 MHz, DMSO-d6) δC ppm 91.29, 104.27, 105.24, 111.27, 113.04, 140.35, 145.35, 148.70, 153.20, 157.56, 160.93.
4-((1-(3-(4-methoxyphenyl)propanoyl)piperidin-4-yl)methoxy)-7 H-furo[3,2-g]chromen-7-one (13a)
This compound was synthesized according to the general procedure F from 12 to 7c to afford colorless gummy solid (0.110 g, 48%). ESI-MS m/z: 461.51 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 1.56 (s, 2 H), 1.83 (d, J = 12.93 Hz, 2 H), 2.01–2.12 (m, 1 H), 2.56 (td, J = 7.80, 2.36 Hz, 2 H), 2.82–2.88 (m, 2 H), 2.91–2.99 (m, 1 H), 3.69 (s, 3 H), 3.79–3.87 (m, 1 H), 4.15 (dd, J = 8.83, 6.62 Hz, 1 H), 4.22 (dd, J = 8.83, 5.99 Hz, 1 H), 4.71 (d, J = 13.24 Hz, 1 H), 6.23 (d, J = 9.77 Hz, 1 H), 6.75–6.77 (m, 2 H), 6.85 (dd, J = 2.52, 0.95 Hz, 1 H), 7.06–7.07 (m, 1 H), 7.09 (s, 1 H), 7.37–7.42 (m, 1 H), 7.53 (d, J = 2.52 Hz, 1 H), 7.60 (ddd, J = 11.98, 8.20, 1.26 Hz, 1 H), 8.03 (dd, J = 9.77, 0.63 Hz, 1 H). 13C-NMR (126 MHz, CHLOROFORM-d) δC ppm 28.49, 29.47, 30.77, 35.39, 37.07, 41.54, 44.93, 55.30, 94.26, 104.95, 106.75, 112.92, 113.33, 113.96, 128.58, 129.46, 132.10, 133.36, 138.94, 145.01, 148.69, 158.10, 161.11, 170.74. Anal. Calcd. for C27H29NO5: C, 72.46; H, 6.53; N, 3.13; Found: C, 72.35; H, 6.51; N, 3.11.
4-((1-(3-(4-chlorophenyl)propanoyl)piperidin-4-yl)methoxy)-7 H-furo[3,2-g]chromen-7-one (13b)
This compound was synthesized according to the general procedure F from 12 and 7e to afford colorless gummy solid (0.117 g, 51%). ESI-MS m/z: 465.93 [M + H]+. 1H-NMR (500 MHz, CHLOROFORM-d) δH ppm 1.56 (br. s., 2 H), 1.85 (d, J = 12.61 Hz, 2 H), 2.02–2.11 (m, 1 H), 2.50–2.59 (m, 3 H), 2.87–2.92 (m, 2 H), 2.93-3.00 (m, 1 H), 3.82 (d, J = 13.87 Hz, 1 H), 4.14–4.18 (m, 1 H), 4.19–4.24 (m, 1 H), 4.70 (d, J = 13.56 Hz, 1 H), 6.22 (d, J = 9.77 Hz, 1 H), 6.81–6.87 (m, 1 H), 7.09 (d, J = 8.51 Hz, 3 H), 7.16–7.20 (m, 2 H), 7.53 (d, J = 2.52 Hz, 1 H), 8.03 (d, J = 9.77 Hz, 1 H). 13C-NMR (126 MHz, CHLOROFORM-d) δC ppm 28.50, 29.47, 30.88, 34.84, 37.06, 41.59, 45.38, 94.27, 104.95, 106.75, 112.91, 113.33, 128.62, 129.94, 131.97, 138.94, 139.81, 145.03, 148.65, 152.72, 158.28, 161.12, 170.24. Anal. Calcd. for C26H26ClNO4: C, 69.10; H, 5.80; N, 3.10; Found: C, 69.12; H, 6.87; N, 2.93.
Patch clamp assay
Testing highlights the usage of CHO K1 cells stably expressing the human isoform of KV1.3 (KCNA3) channel: Parental cell line: Chinese Hamster Ovary cells (ATCC, CCL-61) expressed gene: NCBI Reference Sequence: NM_002232. Patch-clamp experiments performed in whole-cell configuration using a QPatch 16X or QPatch HTX automated patch-clamp system and QPatch Assay Software version 5.6. In general, cells are cultured long term in flasks containing complete medium (culture medium plus selection antibiotics) and passaged at a confluence of about 50 to 80%. Cells harvested at a confluence of about 80–90% from sterile culture flasks containing culture complete medium. Cells are transferred as suspension in serum free medium to the QPatch 16X or QPatch HTX system to the centrifuge / washer directly. Standard Laboratory Conditions. Cells are incubated at 37 °C in a humidified atmosphere with 5% CO2 (rel. humidity about 95%). The cells are continuously maintained in and passaged in sterile culture flasks containing nutrient mixture HAM/F-12 (1x, liquid, with L-Glutamine) supplemented with 10% fetal bovine serum and 1.0% Penicillin/Streptomycin solution. The complete medium as indicated above is supplemented with 200 µg/ml Hygromycin B.
Cells transferred as suspension in serum-free medium to the QPatch HTX system and kept in the cell storage tank/stirrer during experiments. All solutions applied to cells including the bath (external) and intracellular (internal) solution are maintained at room temperature (19-to-30 °C). The composition of the external solution; 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1mM MgCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10 mM D-glucose, pH(NaOH) 7.40. On the other hand, the composition of the intracellular solution involved 125 mM KCl, 3.2 mM MgCl2, 5 mM K2-ATP, 10 mM KF, 5 mM HEPES, 5 mM Ethylene glycol-bis(β-aminoethyl ether)-N, N,N′,N′-tetraacetic acid (EGTA) at pH(KOH) 7.20 ± 0.05. After formation of a Gigaohm seal between the patch electrodes and individual cells (seal resistance range: > 1GΩ) the cell membrane is ruptured to assure electrical access to the cell interior (whole-cell patch-configuration). In case the quality of the seal is poor, the process of seal formation is repeated with a different cell. As soon as a stable seal could be established, currents were measured upon pulses described below. If current density is judged to be too low for measurement, another cell will be recorded. Once control recordings have been accomplished, cells will be continuously perfused with a bath solution containing the test items. During wash-in of the test item the voltage protocol indicated above will be run continuously again until the steady-state level of block will be reached (Table 4). Notably, the tested compounds were added to the external side of the cells.
The software files generated during recording are initially stored on the computer’s hard disk. Values (in A) of the peak amplitudes of currents and charge (area under curve = parameter to determine changes in inactivation kinetics) are generated for each voltage step. These data are printed for compilation and analysis. The recorded current amplitudes at the steady state level of current inhibition or stimulation are compared to those from control conditions measured in the pre-treatment phase of the same cell. The amount of current block or stimulation is calculated as a percentage of control. To determine whether the observed current inhibition/stimulation is due to a test item interaction with one of the channels or due to current rundown, these residual currents are compared to those measured in vehicle-treated cells. The standard monoexponential equation was adapted to fit the inactivation kinetics of ionic currents and determine the inactivation constants as following: It = I0⋅e−τ/t+I∞ ; where It is the current at time , I0 is the amplitude of the component that decays, τ is the time constant of decay describing how quickly the current inactivates, and I∞ is the steady-state or residual current (the plateau current that remains after inactivation). Data from at least 3 individual experiments are collected and the corresponding mean values and standard deviations are calculated.
Statistical analysis
All data were done in triplicates (n = 3) and presented as Mean ± SD. One-way analysis of variance (ANOVA) was adopted for statistical comparison among multiple treated groups. Dunnett’s post hoc test was performed to directly compare each treated group to the control one. Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001 for tested molecules against the vehicle samples. GraphPad Prism V.7.0 was used for carrying out the analysis.
Data availability
The authors declare that the data supporting the findings of this study are available within the manuscript and its Supplementary Information files. Should any data files be needed in another format they are available from the corresponding authors (K.M.D. and A.A.; email: [khaled_darwish@pharm.suez.edu.eg](mailto: khaled_darwish@pharm.suez.edu.eg); [khaled.darwish@gu.edu.eg](mailto: khaled.darwish@gu.edu.eg); [abdelwaly@temple.edu](mailto: abdelwaly@temple.edu)) upon reasonable request.
References
Lleo, A. et al. Definition of human autoimmunity–autoantibodies versus autoimmune disease. Autoimmun. Rev. 9 (5), A259–A266 (2010).
Ceccarelli, F., Agmon-Levin, N. & Perricone, C. Genetic Factors of Autoimmune Diseases 2017. J. Immunol. Res. 2017, 2789242. (2017).
Burek, C. L. & Talor, M. V. Environmental triggers of autoimmune thyroiditis. J. Autoimmun. 33 (3–4), 183–189 (2009).
Sawalha, A. H. Editorial: the innate and adaptive immune response are both involved in Drug-Induced autoimmunity. Arthritis Rheumatol. 70 (3), 330–333 (2018).
A, L. The world incidence and prevalence of autoimmune diseases is increasing. Int. J. Celiac Disease. 3 (4), 151–155 (2015).
Cooper, G. S. & Stroehla, B. C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2 (3), 119–125 (2003).
Sawcer, S., Franklin, R. J. & Ban, M. Multiple Scler. Genet. Lancet Neurol., 13(7): 700–709. (2014).
Group, D. P. Incidence and trends of childhood type 1 diabetes worldwide 1990–1999. Diabet. Med. 23 (8), 857–866 (2006).
Griffiths, L., Dyson, J. K. & Jones, D. E. The new epidemiology of primary biliary cirrhosis. Semin Liver Dis. 34 (3), 318–328 (2014).
Kang, J. Y. et al. Systematic review: worldwide variation in the frequency of coeliac disease and changes over time. Aliment. Pharmacol. Ther. 38 (3), 226–245 (2013).
Lisnevskaia, L., Murphy, G. & Isenberg, D. Systemic Lupus Erythematosus Lancet, 384(9957): 1878–1888. (2014).
McInnes, I. B. & Schett, G. The pathogenesis of rheumatoid arthritis. N Engl. J. Med. 365 (23), 2205–2219 (2011).
McInnes, I. B. & Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 389 (10086), 2328–2337 (2017).
Committee, T. A. D. C. Progress in Autoimmune Disease Research. Report To Congress (The Autoimmune Diseases Coordinating Committee, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 2005).
Hussain, Y. & Khan, H. Immunosuppressive Drugs, in Encyclopedia of Infection and Immunity. Copyright © 2022 Elsevier Inc. All rights reserved. pp. 726 – 40. (2022)
Li, P., Zheng, Y. & Chen, X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to Anti-TNF biologics. Front. Pharmacol. 8, 460 (2017).
Pazetti, R., Pêgo-Fernandes, P. M. & Jatene, F. B. Adverse effects of immunosuppressant drugs upon airway epithelial cell and mucociliary clearance: implications for lung transplant recipients. Drugs 73 (11), 1157–1169 (2013).
Hoepner, R. et al. Efficacy and side effects of natalizumab therapy in patients with multiple sclerosis. J. Cent. Nerv. Syst. Dis. 6, 41–49 (2014).
Marcum, Z. A. & Hanlon, J. T. Recognizing the risks of chronic nonsteroidal Anti-Inflammatory drug use in older adults. Ann. Longterm Care. 18 (9), 24–27 (2010).
DeBisschop, M. What are the risks of long-term NSAIDs and COX-2 inhibitors? J. Fam Pract. 52 (3), 199–200 (2003).
Bonafede, M. M. et al. Cost per treated patient for etanercept, adalimumab, and Infliximab across adult indications: a claims analysis. Adv. Ther. 29 (3), 234–248 (2012).
Oliveira, I. S. et al. Scorpion toxins targeting Kv1.3 channels: insights into immunosuppression. J. Venom. Anim. Toxins Incl. Trop. Dis. 25, e148118 (2019).
Zhou, Y. et al. Effects of Kv1.3 knockout on pyramidal neuron excitability and synaptic plasticity in piriform cortex of mice. Acta Pharmacol. Sin. 45 (10), 2045–2060 (2024).
Di Lucente, J. et al. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 66 (9), 1881–1895 (2018).
Bhoi, R. et al. Role of ion channels in alzheimer’s disease pathophysiology. J. Membr. Biol. 258 (3), 187–212 (2025).
Zhang, X. et al. Blockade of Kv1.3 potassium channel inhibits Microglia-Mediated neuroinflammation in epilepsy. Int. J. Mol. Sci., 23 (23), 14693 (2022).
Tucker, K. et al. The olfactory bulb: A metabolic sensor of brain insulin and glucose concentrations via a Voltage-Gated potassium channel. Results Probl. Cell. Differ. 52, 147–157 (2010).
Fadool, D. A. et al. Kv1.3 channel gene-targeted deletion produces Super-Smeller mice with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron 41 (3), 389–404 (2004).
Kazama, I., T. Tamada, and M. Tachi, Usefulness of targeting lymphocyte Kv1.3-channels in the treatment of respiratory diseases. Inflamm. Res. 64, 753-65 (2015).
Kazama, I. Roles of lymphocyte Kv1.3-Channels in the pathogenesis of renal diseases and novel therapeutic implications of targeting the channels. Mediat. Inflamm. 2015 (1), 436572 (2015).
Hyodo, T. et al. Voltage-gated potassium channel Kv1.3 blocker as a potential treatment for rat anti-glomerular basement membrane glomerulonephritis. Am. J. Physiol. Ren. Physiol. 299 (6), F1258–F1269 (2010).
Wulff, H. et al. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J. Clin. Invest. 111 (11), 1703–1713 (2003).
Panyi, G. et al. Colocalization and nonrandom distribution of Kv1.3 potassium channels and CD3 molecules in the plasma membrane of human T lymphocytes. Proc. Natl. Acad. Sci. U S A. 100 (5), 2592–2597 (2003).
Panyi, G. et al. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc. Natl. Acad. Sci. U S A. 101 (5), 1285–1290 (2004).
Beeton, C. et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. U S A. 103 (46), 17414–17419 (2006).
Nicolaou, S. A. et al. The Ca(2+)-activated K(+) channel KCa3.1 compartmentalizes in the immunological synapse of human T lymphocytes. Am. J. Physiol. Cell. Physiol. 292 (4), C1431–C1439 (2007).
Nicolaou, S. A. et al. Localization of Kv1.3 channels in the immunological synapse modulates the calcium response to antigen stimulation in T lymphocytes. J. Immunol. 183 (10), 6296–6302 (2009).
Ghanshani, S. et al. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 275 (47), 37137–37149 (2000).
Beeton, C. et al. Selective Blockade of T lymphocyte K(+) channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc. Natl. Acad. Sci. U S A. 98 (24), 13942–13947 (2001).
Gutman, G. A. et al. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57 (4), 473–508 (2005).
Liu, S. et al. Structures of wild-type and H451N mutant human lymphocyte potassium channel K(V)1.3, Cell Discov. 7 (1), (2021).
Cahalan, M. D. & Chandy, K. G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 231 (1), 59–87 (2009).
Tarcha, E. J. et al. Durable Pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease. J. Pharmacol. Exp. Ther. 342 (3), 642–653 (2012).
Leeson, P. D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6 (11), 881–890 (2007).
Chandy, K. G. & Norton, R. S. Peptide blockers of K. Curr. Opin. Chem. Biol. 38, 97–107 (2017).
Bruhova, I. & Zhorov, B. S. Monte Carlo-energy minimization of correolide in the Kv1.3 channel: possible role of potassium ion in ligand-receptor interactions. BMC Struct. Biol. 7, 5 (2007).
Felix, J. P. et al. Identification and biochemical characterization of a novel nortriterpene inhibitor of the human lymphocyte voltage-gated potassium channel, Kv1.3. Biochemistry 38 (16), 4922–4930 (1999).
Remillard, C. V. et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell. Physiol. 292 (5), C1837–C1853 (2007).
Harvey, A. J. et al. A new class of blockers of the voltage-gated potassium channel Kv1.3 via modification of the 4- or 7-position of khellinone. J. Med. Chem. 49 (4), 1433–1441 (2006).
Baell, J. B. et al. Khellinone derivatives as blockers of the voltage-gated potassium channel Kv1.3: synthesis and immunosuppressive activity. J. Med. Chem. 47 (9), 2326–2336 (2004).
Schmitz, A. et al. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol. Pharmacol. 68 (5), 1254–1270 (2005).
Miao, S. et al. Benzamide derivatives as blockers of Kv1.3 ion channel. Bioorg. Med. Chem. Lett. 13 (6), 1161–1164 (2003).
Faouzi, M., Starkus, J. & Penner, R. State-dependent blocking mechanism of Kv 1.3 channels by the antimycobacterial drug Clofazimine. Br. J. Pharmacol. 172 (21), 5161–5173 (2015).
Butenschön, I., Möller, K. & Hänsel, W. Angular Methoxy-Substituted Furo- and pyranoquinolinones as blockers of the Voltage-Gated potassium channel Kv1.3. J. Med. Chem. 44 (8), 1249–1256 (2001).
Wanner, S. G. et al. WIN 17317-3, a new High-Affinity probe for Voltage-Gated sodium channels. Biochemistry 38 (34), 11137–11146 (1999).
Soliman, K. et al. Predicting the membrane permeability of organic fluorescent probes by the deep neural network based lipophilicity descriptor DeepFl-LogP. Sci. Rep. 11 (1), 6991 (2021).
Prasanna, S. & Doerksen, R. J. Topological Polar surface area: a useful descriptor in 2D-QSAR. Curr. Med. Chem. 16 (1), 21–41 (2009).
Fernandes, J. & Gattass, C. R. Topological Polar surface area defines substrate transport by multidrug resistance associated protein 1 (MRP1/ABCC1). J. Med. Chem. 52 (4), 1214–1218 (2009).
Szabó, T., Volk, B. & Milen, M. Recent advances in the synthesis of β-carboline alkaloids. Molecules 26 (3), 663 (2021).
Ueda, S. et al. Identification of biosynthetic genes for the β-carboline alkaloid Kitasetaline and production of the fluorinated derivatives by heterologous expression. J. Ind. Microbiol. Biotechnol. 46 (5), 739–750 (2019).
Bueno, M. L. P. et al. β-Carboline derivatives are potent against acute myeloid leukemia in vitro and in vivo. Pharmacol. Rep. 76 (4), 838–850 (2024).
Dalpozzo, R. The chiral pool in the Pictet-Spengler reaction for the synthesis of β-Carbolines. Molecules, 21 (6), 699 (2016).
Guo, H. M. et al. Synthesis of acyclic nucleosides with a chiral amino side chain by the Mitsunobu coupling reaction. J. Org. Chem. 75 (11), 3863–3866 (2010).
Rao, R. N., Maiti, B. & Chanda, K. Application of Pictet-Spengler reaction to Indole-Based alkaloids containing Tetrahydro-β-carboline scaffold in combinatorial chemistry. ACS Comb. Sci. 19 (4), 199–228 (2017).
Teisseyre, A., Środa-Pomianek, K. & Palko-Labuz, A. The Patch-Clamp Technique and its application in studies on Voltage-Gated potassium channels Kv1. 3 in normal and cancer cells. IntechOpen . On-line chapter at https://www.intechopen.com/chapters/1190637 (2024).
Bartok, A. et al. Margatoxin is a non-selective inhibitor of human Kv1. 3 K + channels. Toxicon 87, 6–16 (2014).
Bowlby, M. R. et al. Modulation of the Kv1.3 potassium channel by receptor tyrosine kinases. J. Gen. Physiol. 110 (5), 601–610 (1997).
Selvakumar, P. et al. Structures of the T cell potassium channel Kv1.3 with Immunoglobulin modulators. Nat. Commun. 13 (1), 3854 (2022).
Levite, M. et al. Extracellular K(+) and opening of voltage-gated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. J. Exp. Med. 191 (7), 1167–1176 (2000).
Zhu, J. et al. T-lymphocyte K(v)1.3 channel activation triggers the NLRP3 inflammasome signaling pathway in hypertensive patients. Exp. Ther. Med. 14 (1), 147–154 (2017).
Hanson, D. C. et al. UK-78,282, a novel piperidine compound that potently blocks the Kv1.3 voltage-gated potassium channel and inhibits human T cell activation. Br. J. Pharmacol. 126 (8), 1707–1716 (1999).
Fang, Z. et al. Conformational restriction: an effective tactic in ‘follow-on’-based drug discovery. Future Med. Chem. 6 (8), 885–901 (2014).
Düfert, M. A., Billingsley, K. L. & Buchwald, S. L. Suzuki-Miyaura cross-coupling of unprotected, nitrogen-rich heterocycles: substrate scope and mechanistic investigation. J. Am. Chem. Soc. 135 (34), 12877–12885 (2013).
Mohassab, A. M. et al. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAF(V600E) kinases. Bioorg. Chem. 106, 104510 (2021).
Banerjee, P. et al. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 52 (W1), W513–w520 (2024).
Acknowledgements
This paper is based upon work supported by the US-Egypt Collaborative Research Grant Cycle 20, National Academy of Science (NAS), US-Science, Technology, and Innovation Funding Authority (STDF), Egypt, under Project ID: 45896, PI: Dr. Khaled M. Darwish. Invaluable high performance computing resources were provided to M.L.K. and A.M.A. by the U.S. National Science Foundation via Major Research Infrastructure grant numbers: 1625061 and 2216289. All synthesized compounds are covered under the Egyptian Intellectual Property Authority (EGIPA) Patent Office, innovation/patent ID: EG/P/2024/1088 (date: 18 Aug, 2024).
Funding
This work has been funded by grant number 45896, US-Egypt Collaborative Research Grant Cycle 20, National Academy of Science (NAS), US-Science, Technology, and Innovation Funding Authority (STDF), Egypt; granted to principal investigator Dr. Khaled M. Darwish, October 26th, 2021.
Author information
Authors and Affiliations
Contributions
Conceptualization, K.M.D. and M.A.H.; Data curation, K.M.D., M.A.H., and A.A.; Formal analysis, all authors; Funding acquisition, K.M.D.; Investigation, A.A., M.F., and H.A.; Methodology, M.A.H., K.M.D., and A.A.; Project administration, M.A.H. and K.M.D.; Resources, K.M.D., M.A.H., and M.L.K.; Software, A.A. and M.L.K.; Visualization, K.M.D. and A.A.; Roles/Writing - original draft, all authors; and Writing - review & editing, all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abdelwaly, A., Helal, M.A., M. Fathy, M. et al. Design and synthesis of novel small molecules targeting the Kv1.3 voltage-gated potassium ion channel. Sci Rep 15, 32756 (2025). https://doi.org/10.1038/s41598-025-18086-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18086-8
