
Abstract
To meet the requirements of the biopharmaceutical industry, improving the yield of recombination therapeutic protein (RTP) from Chinese hamster ovary (CHO) cells is necessary. The human cytomegalovirus (CMV) promoter is widely used for RTP expression in CHO cells. To further improve RTP production, we truncated the human CMV intron and further evaluated the effect of four synthetic introns, including ctEF-1α first, EF-1α first, chimeric, and β-globin introns combined with the CMV promoter on recombinant expression levels in transient and stably recombinant CHO cells. The results showed that the truncated, EF-1α first, chimeric, and β-globin introns can significantly improve stable transgene expression in CHO cells. The qPCR results indicated that the mRNA level of transgene increased through optimizing intron sequences combined with the CMV promoter. Transcriptomics analysis was performed and found that differential expression of genes involved in mRNA processing, RNA export from nucleus, cytoplasmic translation, transcriptional activation and cell cycle regulation. In conclusion, optimization of the intron sequences combined with the CMV promoter can achieve a higher yield of recombinant proteins in CHO cells. This will be valuable for generating CHO cell lines with high productivity for industrial applications.
Similar content being viewed by others


Since the launch of recombinant therapeutic protein (RTP) in 1982, biopharmaceuticals have played an important role in the global pharmaceutical industry1. More than 20 new products are approved by the FDA each year, most of which are RTP, including antibodies2. Mammalian cells are the main expression systems for producing RTP because the efficacy and immunogenicity of RTP are directly related to post-translational modifications (PTMs)3. At present, Chinese hamster ovary (CHO) cells are the main expression system for RTP manufacturing because of their capacity to perform proper protein folding, assembly, human-like PTMs, stable integration of exogenous genes, ability to scale up in suspension culture4,5,6,7. Currently, nearly 89% of approved recombinant monoclonal antibodies (mAbs) are produced in CHO cells8.
Over the past few decades, the yield of RTP, especially recombinant mAbs, has greatly increased through various efforts such as the advent of bioprocess engineering and serum-free suspension cultures, the design of sophisticated expression vectors, media development and metabolic engineering strategies9,10,11,12. Among these measures, the design of a high-yielding expression vector is one of the simplest and most effective measures13. The expression level of proteins of interest (POI) can be increased by optimizing the regulatory components of vectors including promoters, enhancers, and poly A14,15,16. Epigenetic elements can mitigate the silencing effect and enhance gene of interest (GOI) expression17,18,19.
The promoter can mediate the transcriptional initiation of genes, and cis-acting elements in the promoter can bind to transcription initiation factors to regulate gene expression20,21,22. Most existing commercial vectors are mediated by the human cytomegalovirus (hCMV) major immediate early promoter and its derivatives23. The DNA regulatory elements of hCMV gene contains an upstream regulatory sequence, nuclear factor-1 binding sites, a strong complex enhancer, and an RNA polymerase II promoter, and the first exon and intron A24. CpG island-rich promoter regions are easily affected by DNA methylation and histone modifications, which may inhibit gene transcription10. Expression vectors often combine the human CMV promoter with the first exon and intron A from CMV. Few studies have investigated the impact of intronic sequences on expression from the CMV promoter20,25,26.
Introns are involved in transcriptional regulation and some can act as enhancers in eukaryotes27. Introns can enhance the processing ability of RNA polymerase II, facilitate the interplay between splicing proteins and transcription proteins, and connect and facilitate multiple types of RNA processing mechanisms28,29. A promoter sequence with an intron can more effectively improve GOI expression than one without an intron30,31. Indeed, the activity of the CMV promoter is enhanced by introns31. In addition, intron A regulates the enhancer region of the CMV major immediate-early (IE) gene32. Here, we sought to improve RTP production by modifying the intron region combined with the CMV promoter. To that aim, we selected five synthetic introns, including the truncated CMV intron, C-terminal EF-1α (ctEF-1α) first, EF-1α first, chimeric, and β-globin introns according to literature reports31,33,34,35,36, and evaluated their capacity to enhance POI expression levels in transient and stably transfected CHO cells.
Results
Reporter gene transient expression
We first selected enhanced green fluorescent protein (EGFP) and secreted alkaline phosphatase (SEAP) as reporter genes and assessed their transient expression levels. At 48 h post-transfection, cells were digested and cultured in suspension with serum-free medium (SFM). Then cell pellet and supernatant were collected for EGFP and SEAP analysis, respectively. The results showed that compared with the control vector, the mean fluorescence intensity (MFI) of EGFP was significantly reduced by the pEGFP-I2 vector (ctEF-1α first intron) (P < 0.05); while other introns showed no significant difference (Fig. 1A,B). Similar, the SEAP activity was also decreased by pSEAP-I2 (P < 0.05), but pSEAP-I3 (EF-1α first intron) enhanced 1.49-fold (P < 0.05), while the pSEAP-I1, pSEAP-I4 and pSEAP-I5 vectors showed no difference versus the control intron (P>0.05, Fig. 1C).

Effect of different intron-containing vectors on the transient expression of reporter genes. (A) Vectors containing different introns were transfected into CHO cells and fluorescence intensity was imaged at 48 h post-transfection. (B) The cells were collected after 7 d suspension culture, and the mean fluorescence intensity (MFI) was measured. (C) The level of SEAP activity from transient expression (after 7 d suspension culture). Error bars indicate the standard deviation of triplicate data (*P < 0.05).
Reporter gene stable expression
Stable cell pools were established, and EGFP and SEAP expression was evaluated using SFM and feeds lasted for 12 days. The results showed that except for pEGFP-I2, the fluorescence intensity of EGFP greatly increased than that of the control intron (Fig. 2A). Flow cytometry showed that MFI of EGFP in the pEGFP-I1, pEGFP-I3, pEGFP-I4, and pEGFP-I5 vectors significantly increased by 2.02-, 3.33-, 2.36-, and 1.74-fold, respectively (P < 0.05, Fig. 2B). Similarly, the SEAP expression levels of the pSEAP-I1, pSEAP-I3, pSEAP-I4, and pSEAP-I5 vectors were 2.79-, 3.96-, 2.01-, and 2.29-fold higher (P < 0.05) versus the control, respectively. In contrast, the SEAP expression levels of the pSEAP-I2 vector were significantly reduced (P < 0.05, Fig. 2C).

Effect of different intron-containing vectors on the stable expression of reporter genes. The vectors were transfected into CHO cells and selected with BSD to establish stable cell pools. (A) Fluorescence images intensity of EGFP. (B) The mean fluorescence intensity of the EGFP cell pool. (C) The level of SEAP stable expression at day 12 suspension culture. Error bars indicate the standard deviation of triplicate data (*P < 0.05, **P < 0.01).
Transient expression of Human serum albumin and adalimumab
To explore the commercial production applications, the reporter gene was replaced with human serum albumin (HSA) and adalimumab (Ada) gene. At 48 h post-transfection, cells were digested and cultured in suspension with SFM and lasted for 7 days, and then supernatant were collected for further analysis. The results showed that there were no statistical difference in HSA and Ada expression among CMV promoters with different introns (P > 0.05, Fig. 3).

Analysis of HSA and Ada transient expression with different introns. At 48 h post-transfection, cells were digested and cultured in suspension with serum-free medium for about 7 days. Then supernatant were collected for further analysis. The expression was examined by Western blotting and the expression levels are expressed as relative changes by setting the values of the control vectors as 1. (A) Western blot analysis of HSA protein levels. (B) The HSA relative expression was analyzed by Image J. (C) Western blot results of Ada expression. (D) Ada expression normalized to that of the control. Error bars indicate the standard deviation of triplicate data.
Stable expression of HSA and Ada
Transient expression cannot fully elucidate the functions of CMV promoters with different synthetic introns. Therefore, stable cell pools expressing HSA and Ada were selected for further analysis. Different stable cell pools were inoculated with a density of 5 × 105 cells/mL in 125mL Erlenmeyer shake flasks for fed-batch suspension culture, and supernatant were collected on day 12 post suspension inoculation. Western blotting revealed that I1, I3, I4, and I5 enhanced the stable expression of HSA and Ada, which reached 1.39-, 1.45-, 1.29-, 1.24-fold, and 1.31-, 1.37-, 1.35-, 1.21-fold, respectively (P < 0.05,Fig. 4A–D).

Analysis of HSA and Ada stable expression levels. After establishing stable cell pools, cells were grown in fed-batch cultured with serum-free medium for about 12 days, and supernatant were collected for further analysis. (A) Western blot analysis of HSA expression levels. (B) Western bolt results of Adaexpression. (C) The analysisof HSA relative expression with Image J. (D) Ada expression normalized to that of the control. (E) ELISA analysis of HSA expression levels. (F) ELISA analysis of Ada expression levels. Error bars indicate the standard deviation of triplicate measurements (*P < 0.05).
Similar to Western blotting results, ELISA showed that I1, I3, I4, and I5 can increase HSA and Ada expression by 1.27, 1.37, 1.26, 1.23-fold and 1.36, 1.42, 1.37, 1.30-fold, respectively (P < 0.05, Fig. 4E,F). Moreover, there were no significant effects on viable cell density (VCD) and viability.
Intron impact promoter activity
Gene expression is closely related to the copy number and transcriptional activity of promoters. To further investigate whether intron modification can influence recombinant gene expression through enhancing the copy number and the mRNA level of the GOI, qPCR was further performed. The results showed that HSA mRNA in the vector containing modified intron increased (P < 0.05, Fig. 5A). and the mRNA levels of HC and LC in pAda-I1,3,4,5 also increased (P < 0.05, Fig. 5B), while the gene copy number did not increase (P > 0.05, Fig. 5C,D).

Relative copy number and mRNA expression levels of gene of interest. Relative copy number and mRNA expression levels were calculated by the 2−ΔΔCt method and normalized to the control vector, whose value was set to 1. (A) Relative mRNA expression levels of HSA. (B) Relative mRNA expression of HC/LC in stable pools. (C) Relative copy number of HSA. (D) Relative copy number of HC/LC. Error bars indicate the standard deviation of triplicate measurements (*P < 0.05).
Transcriptomics analysis
Based on the results of reporter gene and the POI stable expression, transcriptome analysis was performed on pHSA-I3 stable cell pools and control, a total of 48 different expressed genes were found (p-value < 0.05). Among them, 46 genes were upregulated and 2 genes were downregulated (Fig. 6; Table 1), including genes related to mRNA processing, RNA export from nucleus, cytoplasmic translation, transcriptional activation or inhibition processes, and cell cycle regulation. The differentially regulated genes included DEAH box polypeptide 9 (Dhx9) participating in RNA polymerase binding, regulation of mRNA processing, positive regulation of cytoplasmic translation and positive regulation of RNA export from nucleus; minichromosome maintenance complex component 5(Mcm5), proline rich 11 (Prr11) and chromatin licensing and DNA replication factor 1 (Cdt1) participating in regulation of cell cycle; myeloblastosis oncogene-like 2 (Mybl2) and early growth response 3 (Egr3) related to positive regulation of transcription by RNA polymerase II; thrombospondin 1 (Thbs1) participating in negative regulation of apoptotic process, and PHD finger protein 19 (Phf19) related to positive regulation of histone H3-K27.

Analysis of gene expression differences between high expression and control cell pools. (A) Heatmap of differentially expressed genes; (B) Expression correlation scatter plot, Group 2 and Group 1 represents highly expressed strain and control cell pools, respectively. The heatmap was created using the ‘pheatmap’ package in R, which can be found at https://mirrors.sjtug.sjtu.edu.cn/cran/web/packages/available_packages_by_name.html#available-packages-P. The scatter plot was generated using “ggplot2’, also available at the same link.
Discussion
The expression levels of GOI is largely determined by expression vectors37, and expression vectors gene construct components such as the promoter are key in regulating transcription38. The human CMV promoter is a strong constitutive promoter that involved in transgene expression at the initial stage of viral infection, is less dependent on tissue specificity, and is widely used in commercial applications for RTP production31,39. In this study, we modified the intron region combined with the CMV promoter to improve RTP production. We found that the truncated CMV, EF-1α first, chimeric, and β-globin introns can effectively increase stable GOI expression levels in recombinant CHO cells.
We selected intracellular EGFP and secreted SEAP as reporter genes to detect the effects of recombinant protein production in CHO cells. In transient expression, the SEAP activity was increased by replacing full-length human CMV intron with a truncated intron, which was consistent with report of Quilici et al.31. In addition, the stable expression of EGFP and SEAP were significantly improved, which is consistent with the previous results33,40. However, the opposite trend was observed when the ctEF-1α first intron was used, even though it was truncated to the 367 bp C-terminal part and retained the major five Sp1 and a single Apl binding regions of the EF-lα gene33. The activity of EF-1α intron requires the co-operation of different promoter, and the only slightly increased in CHO cells when the EF-1α intron combined with hCMV promoter34, which was consistent with our study.
To provide industrially-relevant data, we chose HSA and Ada antibody as target genes. In transient expression, the Ada yield increased to 110% with the β-globin intron. Consistent with the results of the reporter gene, the truncated, EF-1α first, chimeric, and β-globin introns did not only improve the transient expression of HSA but also that of Ada. During transient expression system, the GOI is not been integrated into the host genome DNA, and its expression is highly affected by transfection efficiency. In a previous study, it has been found that introns can affect transfection efficiency, thereby affecting recombinant protein expression41. The expression of the GOI in stable systems is related to some factors, such as the promoter activity, gene silencing, translational regulation, and the activity of the promoter can be reflected in the stable expression system. In the present study, the increasing effect of the modified promoters was most significant. Overall, optimization of the intron of the CMV promoter construct was beneficial for enhancing GOI expression in stably recombinant CHO cells.
Promoters play important roles in the integration and processing of transcriptional signals21. The intron contribute to mRNA stability because splicing efficiency is inversely proportional to nonsense-mediated decay42. The mRNA expression of HSA increased with intron optimization, and the mRNA levels of LC increased by modification of the CMV promoter with the truncated and the chimeric introns. Additionally, there were slight improvements with the EF-1α first and β-globin introns. A previous study confirmed that more LC was beneficial for antibody assembly and yield43. Our findings also show no linear correlation between GOI expression and mRNA levels. This may be because efficient heterologous protein production in CHO cells is related to many factors such as transcription initiation, mRNA processing, ribosome recognition, and protein translation initiation44. In this study, we used cell pools rather than cell clones for transcriptomics analysis to avoid clone-specific differences between cell clones. Due to the different expression levels of recombinant proteins produced by promoters combining with different introns, there are differences in cellular stress and protein folding efficiency, which in turn cause changes in transcriptomic molecules profiles. Transcriptomics results showed that genes related to mRNA processing, RNA export from nucleus, cytoplasmic translation, transcriptional activation or inhibition processes, and cell cycle regulation were different, indicating that they may be related to the regulation of introns, and the underlying mechanisms need to be further investigated.
In summary, the yields of various GOI were increased in CHO cells by optimizing the intron element combined with the CMV promoter, which has the potential for industrial applications. However, only five introns have been studied; the scope of introns tested needs to be expanded in future studies. Moreover, the activities of expression vectors are not only related to individual element on the vector, but also to their synergistic effects. Further research should focus on optimizing the combination of different elements to achieve optimal functionality.
Materials and methods
Vector construction
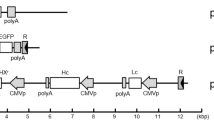
Five different intron regions were evaluated according to the previous studies31,36,38,43,44. These included the truncated intron that deleted 200 bp between the XcmI/BspEI sites in human CMV intron (I1), the ctEF-1α first (I2), EF-1α first (I3), chimeric (I4) and β-globin (I5) introns, all of which were artificially synthesized. The five synthetic introns replaced the natural intron region in the pCMV-EGFP and pCMV-SEAP plasmids (Fig. 7A,B). And the sequences of the hCMV original intron and the synthetic introns were provided in Fig. S1. The new vectors were named as pEGFP-I1, pEGFP-I2, pEGFP-I3, pEGFP-I4, and pEGFP-I5, and pSEAP-I1, pSEAP-I2, pSEAP-I3, pSEAP-I4, and pSEAP-I5, respectively. To further explore their effects on RTP expression, the introns that had a positive effect the reporter genes were used to replace the natural intron region in pCMV-HSA and pCMV-Ada vectors (Fig. 7C,D).

Schematic representation of different types of GOI expression vectors. (A) pCMV-EGFP. (B) pCMV-SEAP. (C) pCMV-HSA. (D) pCMV-Ada. EGFP enhanced green fluorescent protein, SEAP secreted alkaline phosphatase, HSA human serum albumin, LC adalimumab light chain, HC adalimumab heavy chain, poly A SV40 polyadenylation signal, PGKp mouse phosphoglycerate kinase 1 promoter, BSD blasticidin, EF-1αp human elongation factor 1 alpha promoter, hCMVp human cytomegalovirus immediate early promoter.
Cell culture and transfection
CHO cells (#A11557-01; Carlsbad, CA, USA) were cultured in DMEM/F12 medium containing 10% fetal bovine serum (FBS) (ProteinEasy Biological Products Co., Ltd., Xinxiang, China), and cultivated in a 37 °C humidified incubator with 5% CO2. After passaging three times, the cells were inoculated into 12-well plates at 2.5 × 105 cells/well and cultured overnight. When the cells reached 80–90% confluence, 1.6 µg of the above-mentioned plasmids DNA was diluted with 4 µL Lipofectamine 2000 per well, and transfected into the cells (Gibco-BRL, Gaithersburg, MD). All experiments were performed in triplicates.
Transient expression analysis
At 48 h post-transfection, cells were digested and washed with PBS. And then, cells were inoculated into deep-well shaking 6-well plates at a density of 5 × 105 cells/well and cultured under suspension with SFM (ProteinEasy Biological Products Co., Ltd., Xinxiang, China) for batch culture. After 7 d of culture, the cell pellet and supernatant were collected for further analysis, respectively.
Stable expression analysis
The stably transfected cell pools were selected with 10 µg/mL blasticidin (BSD) (Sangon Biotech, SHH, China.) at 48 h after transfection. When the un-transfected cells were eradicated, the concentration of BSD was gradually decreased to 2.5 µg/mL for about 2 weeks with culture medium change every 2–3 d. Different cell pools were inoculated with a density of 5 × 105 cells/mL in 125mL Erlenmeyer shake flasks (Corning) at a working volume of 30 ml for fed-batch suspension culture. The cell pellets and supernatant were collected at day 12 post- inoculation for protein expression analysis.
Fluorescence microscopy and flow cytometry
At 48 h post-transfection, EGFP fluorescence intensity was inspected using an inverted fluorescence microscope. The cells were then digested with 0.25% trypsin and suspended at equal concentration. Seven days later, a total of 10,000 events per sample were acquired for EGFP cell signals and the analysis was maintained at an event rate of ≤ 600 per second using an emission filter of 530/30 nm and an excitation laser of 488 nm. The MFI was analyzed using flow cytometer (Invitrogen, Thermo Fisher Scientific, USA).
Analysis of SEAP expression
The enzymatic activity of SEAP was detected using an alkaline phosphatase kit (Beyotime Biotech Co., Ltd., Jiangsu, China) in 96-well plates. 50 µL sample was added per well after dilution according to the manufacturer’s instructions. Then the sample and chromogenic substrate were mixed, incubated at 37℃ for 10 min, and 100 µL reaction stop solution was added. The absorbance at 405 nm was determined to generate a standard curve and analyze the data.
Western blotting
The supernatant was collected, mixed with 5× same volume SDS sample loading buffer, and boiled for 10 min. Then, the same aliquots of the HSA and Adalimumab (Ada) samples were loaded onto 10% and 8% SDS-PAGE gels, respectively. The proteins were transferred to a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA) by electroblotting. HSA membranes were incubated with 1:3,000 dilutions of anti-His-tag mouse antiserum (Huada Protein Research and Development Co., Ltd., China), followed by a 1:4000 dilution of a secondary goat anti-mouse antibody conjugated to alkaline phosphatase (ZSGB-Bio, China). A horseradish peroxidase (HRP)-linked goat anti-human secondary antibody (1:8000 dilution, EARTHOX, USA) was incubated with the Ada membrane. Proteins were detected and Image J v2.1.4.7 software was used for calculations.
ELISA
The HSA and Adalimumab production were measured by enzyme-linked immunosorbent assay (ELISA) (ELK Biotechnology, Wuhan, China) kit according to the manufacturer’s instructions.
Quantitative PCR
GOI relative copy numbers and mRNA expression levels were determined using stable transfected cells. Genomic DNA was isolated using a genomic DNA extraction kit (ProteinEasy Biological Products Co., Ltd.,Xinxiang, China).Total RNA was extracted using TRIzol (Beyotime Biotech Co., Ltd., Jiangsu, China). The RNA concentration was measured, and 1 µg total RNA was reverse transcribed into cDNA. Then, qPCR analysis was applied to elucidate changes in the DNA and mRNA level of HSA and Ada, and undertaken using NovoStart®SYBR qPCR SuperMix Plus (Novoprotein, China) on an ABI 7500 SYBR fluorescence quantitative PCR instrument (Applied Biosystems, Foster, CA, USA). The primers used for HSA, Ada heavy chain (HC), Ada light chain (LC), and β-actin are listed in Table 2. The relative copy number and mRNA expression level of the target gene was calculated using the 2−ΔΔCt method using β-actin for normalization. The quantitative data shown are presented as the means of triplicate experiments.
Transcriptome analysis
Three samples from pHSA-I3 high expression cell pools and the control cell pools were selected to transcriptome sequencing by Shanghai biotechnology corporation. RNA was extracted from three cell pools with MJzol Animal RNA Isolation Kit (Majorivd). Library generation with input of RNA was performed using the VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina® (Vazyme, China) according to the manufacturer’s recommendations. The libraries were pooled and sequenced on an Illumina NovaSeq6000 (Illumina, Inc., San Diego, US) in PE150 model. Gene expression levels were quantified based on algorithms of the Cufflinks Suite and standardized on FPKM values. Comparison of gene expression between two groups was considered as up- or downregulated if they result in a significant log2FC < -1 and > 1, respectively. Official gene symbols for identified proteins were entered into the STRING (https://string-db.org) and gene ontology (GO) databases DAVID (https://david.ncifcrf.gov).
Statistical analysis
All statistical analyses were carried using GraphPad Prism 8.0. The data were analyzed using independent sample t-tests and displayed as mean ± standard deviation. The statistical significance was set at P < 0.05.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Deshaies, R. J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 580, 329–338 (2020).
Grilo, A. L. & Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 37, 9–16 (2019).
Grindes, L. et al. Weak promoters to drive selection marker expression: improvement of cell line development process for therapeutic protein production in CHO-K1 cells. J. Biotechnol. 369, 43–54 (2023).
Stolfa, G. et al. CHO-omics review: the impact of current and emerging technologies on Chinese Hamster ovary based Bioproduction. Biotechnol. J. 13, e1700227 (2018).
Dumont, J., Euwart, D., Mei, B., Estes, S. & Kshirsagar, R. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit. Rev. Biotechnol. 36, 1110–1122 (2016).
Yi, D. D. et al. Construction of an expression vector mediated by the dual promoter for prokaryotic and mammalian cell expression system. Mol. Biol. Rep. 47, 5185–5190 (2020).
Wells, E. & Robinson, A. S. Cellular engineering for therapeutic protein production: product quality, host modification, and process improvement. Biotechnol. J. 12 (2017).
Walsh, G. & Walsh, E. Biopharmaceutical benchmarks 2022. Nat. Biotechnol. 40, 1722–1760 (2022).
Guo, X., Wang, C. & Wang, T. Y. Chromatin-modifying elements for recombinant protein production in mammalian cell systems. Crit. Rev. Biotechnol. 40, 1035–1043 (2020).
Katayama, S. & Andou, M. Editing of DNA methylation using CRISPR/Cas9 and a ssDNA template in human cells. Biochem. Biophys. Res. Commun. 581, 20–24 (2021).
Shibafuji, Y., Nagao, N. & Yohda, M. Cystine and tyrosine feed reduces oxidative and ER stress in CHO cells. Biotechnol. J. 18, e2200638 (2023).
Schellenberg, J. et al. Stress-induced increase of monoclonal antibody production in CHO cells. Eng. Life Sci. 22, 427–436 (2022).
Matasci, M., Baldi, L., Hacker, D. L. & Wurm, F. M. The PiggyBac transposon enhances the frequency of CHO stable cell line generation and yields recombinant lines with superior productivity and stability. Biotechnol. Bioeng. 108, 2141–2150 (2011).
Li, Q. et al. Effects of different promoters and MAR combinations on transgene expression of recombinant CHO cells. Sichuan Da Xue Xue Bao Yi Xue Ban. 49, 18–23 (2018).
Marx, N. et al. Enhanced targeted DNA methylation of the CMV and endogenous promoters with dCas9-DNMT3A3L entails distinct subsequent histone modification changes in CHO cells. Metab. Eng. 66, 268–282 (2021).
Wang, X. Y. et al. Enhanced transgene expression by optimization of poly A in transfected CHO cells. Front. Bioeng. Biotechnol. 10, 722722 (2022).
Doan, C. C. et al. Enhancement of anti-TNFα monoclonal antibody production in CHO cells through the use of UCOE and DHFR elements in vector construction and the optimization of cell culture media. Prep. Biochem. Biotechnol. 52, 452–470 (2022).
Hoseinpoor, R., Kazemi, B., Rajabibazl, M. & Rahimpour, A. Improving the expression of anti-IL-2Rα monoclonal antibody in the CHO cells through optimization of the expression vector and translation efficiency. J. Biotechnol. 324, 112–120 (2020).
Sizer, R. E. & White, R. J. Use of ubiquitous chromatin opening elements (UCOE) as tools to maintain transgene expression in biotechnology. Comput. Struct. Biotechnol. J. 21, 275–283 (2023).
Brown, A. J., Sweeney, B., Mainwaring, D. O. & James, D. C. NF-κB, CRE and YY1 elements are key functional regulators of CMV promoter-driven transient gene expression in CHO cells. Biotechnol. J. 10, 1019–1028 (2015).
Román, R. et al. Enhancing heterologous protein expression and secretion in HEK293 cells by means of combination of CMV promoter and IFNα2 signal peptide. J. Biotechnol. 239, 57–60 (2016).
Dou, Y. et al. The CAG promoter maintains high-level transgene expression in HEK293 cells. FEBS Open. Bio. 11, 95–104 (2021).
Romanova, N. & Noll, T. Engineered and Natural promoters and chromatin-modifying elements for recombinant protein expression in CHO cells. Biotechnol. J. 13, e1700232 (2018).
Mariati, Ho, S. C., Yap, M. G. & Yang, Y. Evaluating post-transcriptional regulatory elements for enhancing transient gene expression levels in CHO K1 and HEK293 cells. Protein Expr. Purif. 69, 9–15 (2010).
Moritz, B., Woltering, L., Becker, P. B. & Göpfert, U. High levels of histone H3 acetylation at the CMV promoter are predictive of stable expression in Chinese hamster ovary cells. Biotechnol. Prog. 32, 776–786 (2016).
Soo, B. P. C. et al. Correlation between expression of recombinant proteins and abundance of H3K4Me3 on the enhancer of human cytomegalovirus major immediate-early promoter. Mol. Biotechnol. 59, 315–322 (2017).
Rojas-Sánchez, U., López-Calleja, A. C., Millán-Chiu, B. E., Fernández, F. & Loske, A. M., Gómez-Lim, M. A. enhancing the yield of human erythropoietin in Aspergillus Niger by introns and CRISPR-Cas9. Protein Expr. Purif. 168, 105570 (2020).
Shaul, O. How introns enhance gene expression. Int. J. Biochem. Cell. Biol. 91, 145–155 (2017).
Mishra, S. K. & Thakran, P. Intron specificity in pre-mRNA splicing. Curr. Genet. 64, 777–784 (2018).
Li, X., Zhong, F. & Qi, S. Modification of pGH cDNA using the first intron and adenovirus-mediated expression in CHO cells. Chin. Med. J. (Engl.). 116, 1267–1269 (2003).
Quilici, L. S. et al. A minimal cytomegalovirus intron a variant can improve transgene expression in different mammalian cell lines. Biotechnol. Lett. 35, 21–27 (2013).
Colston, J. M., Bolinger, B., Cottingham, M. G., Gilbert, S. & Klenerman, P. Modification of antigen impacts on memory quality after adenovirus vaccination. J. Immunol. 196, 3354–3363 (2016).
Lee, C. P. et al. Regulatory elements in vectors containing the ctEF-1α first intron and double enhancers for an efficient recombinant protein expression system. Sci. Rep. 8, 15396 (2018).
Kim, S. Y., Lee, J. H., Shin, H. S., Kang, H. J. & Kim, Y. S. The human elongation factor 1 alpha (EF-1 alpha) first intron highly enhances expression of foreign genes from the murine cytomegalovirus promoter. J. Biotechnol. 93, 183–187 (2002).
Hermening, S., Kügler, S., Bähr, M. & Isenmann, S. Increased protein expression from adenoviral shuttle plasmids and vectors by insertion of a small chimeric intron sequence. J. Virol. Methods. 122, 73–77 (2004).
Dong, W., Li, C., Yang, Y., Wang, T. & Wang, F. Increasing transgenic expression in recombinant Chinese hamster ovary cells using introns in different directions. Sheng Wu Gong. Cheng Xue Bao. 35, 1071–1078 (2019).
Li, Y. M. et al. Effects of different 2A peptides on transgene expression mediated by tricistronic vectors in transfected CHO cells. Mol. Biol. Rep. 47, 469–475 (2020).
Lam, C. K. C. & Truong, K. Engineering a synthesis-friendly constitutive promoter for mammalian cell expression. ACS Synth. Biol. 9, 2625–2631 (2020).
Campos-da-Paz, M. et al. Production of recombinant human factor VIII in different cell lines and the effect of human XBP1 co-expression. Mol. Biotechnol. 39, 155–158 (2008).
Palmiter, R. D., Sandgren, E. P., Avarbock, M. R., Allen, D. D. & Brinster, R. L. Heterologous introns can enhance expression of transgenes in mice. Proc. Natl. Acad. Sci. U S A. 88, 478–482 (1991).
Xu, D. H. et al. SV40 intron, a potent strong intron element that effectively increases transgene expression in transfected Chinese hamster ovary cells. J. Cell. Mol. Med. 22 (4), 2231–2239. https://doi.org/10.1111/jcmm.13504 (2018).
Roos, D. & de Boer, M. Mutations in cis that affect mRNA synthesis, processing and translation. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166166 (2021).
Schlatter, S. et al. On the optimal ratio of heavy to light chain genes for efficient recombinant antibody production by CHO cells. Biotechnol. Prog. 21, 122–133 (2005).
Sharker, S. M. & Rahman, A. A review on the current methods of Chinese Hamster ovary (CHO) cells cultivation for the production of therapeutic protein. Curr. Drug Discov. Technol. 18, 354–364 (2021).
Funding
This research was supported by the National Natural Science Foundation of China (No. U23A20270), the Project of Technology Innovation Leading Talent in Central Plain, China (234200510003), the Natural Science Foundation of Henan, China (232300421115).
Author information
Authors and Affiliations
Contributions
T.Y.W. designed the experiments and contributed reagents and materials. T.T.X., Y.F.W., X.Y.W. and C.L.M. performed the experiments. T.Y.W., T.T.X., X.Y.W. and S.L.G. drafted the manuscript, T.T.X ,J,J,Y and X.Y.W prepared Figs. 1, 2, 3, 4, 5, 6 and 7. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, TT., Wang, YF., Yuan, JJ. et al. Optimization of the intron sequences combined with the CMV promoter increases recombinant protein expression in CHO cells. Sci Rep 15, 3732 (2025). https://doi.org/10.1038/s41598-025-87941-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87941-5
