
Abstract
Hepatocellular carcinoma (HCC) is known for its high invasiveness, high fatality rate. Both hypoxia and senescence play crucial roles in the initiation and progression of cancer, yet their prognostic implications in HCC are yet to be fully understood. The hypoxia-senescence co-related genes (HSCRGs) were screened from public databases. Transcriptome data and clinical information were obtained from patients with HCC using the Cancer Genome Atlas, GSE76427, and International Cancer Genome Consortium (ICGC). The random forest tree algorithm was used to identify the characteristic genes of the disease, and the genes were verified by related experiments. SVM algorithm was used to classify HCC patients based on HSCRGs. The prediction model based on HSCRGs was established by LASSO, univariate and multivariate COX regression analysis. We used the ICGC for outside validation. The risk score model was analyzed from subgroup analysis, immune infiltration, and functional strength. The expression patterns of key prognostic genes in tumor microenvironment were decoded by single cell analysis. A total of 184 HSCRGs were identified. The expression pattern and functional characteristics of MLH1 gene in HCC were verified. Two HCC subtypes were identified based on HSCRGs. Then, a prediction model based on HSCRGs was established, and risk score was identified as an independent prognostic indicator of HCC. A new nomogram is constructed and shows good prediction ability. We further determined that the level of infiltration of immune cells and the expression of immune checkpoints are significantly affected by the risk score. The immune microenvironment was different between the two risk groups. The high-risk group was dominated by immunosuppressed cells, and the prognosis was poor. Single-cell analysis revealed the expression of seven key prognostic genes in the tumor microenvironment. Finally, qPCR results further verified the expression levels of seven prognostic genes. HSCRGs are of great significance in the prognosis prediction, risk stratification and targeted therapy of patients with HCC.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) accounts for nearly 80% of primary liver cancers and is the third most common cause of cancer-related deaths globally1,2. Owing to the early symptoms are not obvious, most patients with HCC are already in the advanced stage when diagnosed3. This makes the chance of surgical eradication small4. In recent years, with the development of immunotherapy, it plays a very important role in the treatment of tumors5, especially in the treatment of advanced HCC6, there is a consensus of the latest guidelines that atezolizumab plus bevacizumab has become the first-line preferred regimen for advanced HCC7. Despite advancements in therapeutic approaches for HCC, the overall 5-year survival rate for HCC patients still remains below 20%8. Therefore, the identification of molecular biomarkers is critically significant for the early diagnosis, survival prognosis, and recurrence monitoring of HCC9.
Hypoxia represents an inherent characteristic of solid tumors, arising from the imbalance between the proliferation rate of tumor cells and the vascular nutrient supply10. Existing research has recognized the crucial role of hypoxia in tumor angiogenesis, cell proliferation, cellular differentiation, and apoptosis11,12. However, the underlying regulatory mechanisms still remain unclear. Hypoxia-inducible factor (HIF), a key transcription factor expressed across nuclear cells, becomes upregulated under hypoxic conditions and serves a pivotal role in regulating cellular senescence13. Study also suggests that intermittent hypoxia is linked to oxidative stress and chronic inflammation, thus accelerating the cellular senescence process14.
Cellular senescence is defined as the gradual decline in the normal physiological functions and proliferative capacity of cells over time or in response to external stress, resulting in an irreversible or persistent cell cycle arrest process15,16. Notably, Carlos et al.17 identified 12 features of senescence, such as genomic instability and mitochondrial dysfunction, that are significantly associated with hypoxia, serving as either causes or consequences. The hypoxic microenvironment may protect tumors from natural antitumor immune responses by promoting immune evasion, thus modulating cellular senescence16. Furthermore, the incidence of cancer increases in the elderly population18. This indicates a potential connection between hypoxia, senescence, and cancer, where hypoxia contributes to both senescence and cancer. However, the prognostic role of hypoxia-induced senescence in HCC remains to be elucidated.
MutL homolog 1(MLH1), the protein encoded by this gene can heterodimerize with mismatch repair endonuclease PMS2 to form MutL alpha, part of the DNA mismatch repair(MMR) system19. The encoded protein is also involved in DNA damage signaling and can heterodimerize with DNA mismatch repair protein MLH3 to form MutL gamma, which is involved in meiosis20. This gene was identified as a locus frequently mutated in hereditary nonpolyposis colon cancer (HNPCC)19. Studies have shown that defects in the MMR genes, especially MLH1, can lead to genome-wide mutations and the development of cancer21,22,23. Most of the studies on MMR and MLH1 gene focus on intestinal tumors, but their studies in HCC are still very limited and need to be further elucidated.
The aim of this study was to explore the molecular subtypes and prognostic characteristics of hypoxia- senescence co-related genes (HSCRGs) in HCC through the development of molecular subtyping and prognostic risk models. The findings could potentially contribute to clinical risk stratification and personalized treatment for HCC patients, thereby improving the prognosis of HCC patients.
Materials and methods
Data collection and identification of HSCRGs
In this study, a total of 478 HCC samples from two high-throughput platforms were included: 363 samples from TCGA (https://tcga-data.nci.nih.gov/tcga/) and 115 samples from GEO (GSE76427) (http://www.ncbi.nlm.nih.gov/geo/). The mRNA expression data and clinical information from the Japan-HCC cohort were downloaded at International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/projects/LIRI-JP). Additional information, including somatic mutation data, copy number variations (CNV), clinical details, and survival data, was collected. All patients were required to have complete follow-up information and RNA-seq data. Patients with incomplete survival information were excluded from subsequent analyses. Subsequently, 308 senescence -related genes (SRGs) were obtained from the CellAge database (http://genomics.senescence.info/cells/, accessed on February 2, 2024). Additionally, a search with the keyword “hypoxia” in the GeneCards database (http://www.genecards.org/, accessed on February 2, 2024) under the filter condition of Relevance Scores > 1.5 yielded a total of 1603 hypoxia-related genes (HRGs). Intersection of these sets using Venn diagram resulted in 184 HSCRGs (Table S1). The single-cell RNA seq (scRNA-seq) data of 4 HCC patients were obtained from GSE146115 in the GEO database, with a total of 3200 cells obtained. Table S2 presents baseline characteristics of these cohorts.
Unsupervised clustering based on HSCRGs
The R package “ConsensusClusterPlus”24 was utilized for hierarchical agglomerative clustering of samples. Unsupervised analysis incorporated stability evidence to ascertain cluster count and membership. This iterative process was repeated 1,000 times to ensure clustering stability.
Gene enrichment analysis
The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) gene enrichment analysis was used to explore the relevant cytological functions and pathways of DEGs25,26,27. q-value < 0.05 was the cutoff criterion for determining whether a gene is significantly enriched.
Immune cell infiltration and immune function analysis
We employed the “GSVA” R package to perform single-sample gene set enrichment analysis (ssGSEA) for the quantitative assessment of immune cell infiltration and immune functionality in each sample. Additionally, 48 immune checkpoints from prior studies were incorporated28. The stromal/immune/estimate scores were computed using the “estimate” R package. Further analysis of the correlation between the expression of HLA-DPA1/CD4 in different immune cell types and marker genes was conducted using the Tumor Immune Estimation Resource (TIMER) database (timer.comp-genomics.org), accessed on February 22, 2024. The TIMER offers 6 main analysis modules that allow users to interactively explore the association between immune infiltration and a variety of factors, including gene expression, clinical outcomes, somatic mutations, and somatic copy number alterations29. Stemness Score (RNAss) was downloaded from the Pan-Cancer Atlas Center at https://pancanatlas.xenahubs.net.
Identification of differentially expressed genes between hypoxia and senescence co-related Subtypes
The “Limma” R package30was employed to detect differentially expressed genes (DEGs) among distinct HSCRGs subtypes, considering an adjusted p-value threshold of < 0.01(adjusted for multiple testing using the Benjamini–Hochberg correction method). The overlapping set of DEGs between the two groups was then identified, and their expressions across all samples were extracted for further analysis.
Screening of disease characteristic genes using the random forest algorithm
In this study, we employed the random forest model for gene selection31. To ensure the stability and convergence of the model, the number of trees (ntree) was set to 500. Additionally, other hyperparameters were configured as follows: the maximum number of features (max_features) was set to “sqrt”, meaning that the number of features considered at each split is the square root of the total number of features; the minimum number of samples required to split an internal node (min_samples_split) was set to 2; and the minimum number of samples required in a leaf node (min_samples_leaf) was set to 1. The importance score, measured by Mean Decrease Gini, was utilized to identify genes critical for disease classification. The parameter settings were determined based on default configurations and preliminary experimental results, ensuring optimal model performance while mitigating the risk of overfitting.
Construction of the HSCRGs-based prognostic risk model
A training dataset (n = 239) and a test dataset (n = 239) were randomly selected from a pool of 478 patients in a 1:1 ratio, so as to prevent overfitting, select the optimal model and parameters, and provide a reliable assessment of the generalization ability of the model. In the training cohort, we utilized the "survival," "glmnet," "survminer," and “timeROC” R packages to perform univariate and multivariate Cox regression analyses, and employed LASSO regression to construct the risk score. In the LASSO regression model, we utilized the R package “glmnet” for feature selection and model construction32. Specifically, we determined the optimal lambda value via tenfold cross-validation to ensure that the model avoided overfitting while retaining the most predictive genes. During the cross-validation process, the lambda value corresponding to the minimum cross-validation error was selected. Additionally, the alpha parameter was set to 1 to enforce the L1 regularization property of LASSO regression, enabling sparse feature selection. Initially, univariate Cox regression analysis was conducted to identify genes significantly associated with prognosis (P < 0.05, adjusted for multiple testing using the Benjamini–Hochberg correction method). Subsequently, these genes were subjected to LASSO regression analysis, with the optimal model parameters selected through tenfold cross-validation. Following this, multivariate Cox regression analysis was performed to establish the risk score, which was determined based on the minimum Akaike Information Criterion (AIC) value. A lower AIC value indicates a more accurate model33.The risk score was formulated as a predictive factor, represented by the sum of coefficients and associated genes: Risk score20,23,24 = ∑ni=1Coefi × Xi, Where Coefi is the correlation coefficient of the prognostic key genes, and Xi signifies the expression of the prognostic key genes. Lastly, multivariate Cox proportional hazards regression analysis was executed to identify crucial clinical phenotypes.
Validation of the HSCRGs-based risk score
To evaluate the generalizability of our model, we employed both a test set and an external validation dataset. Specifically, we validated the model’s generalizability by applying the risk score to the LIRI-JP dataset from the ICGC database, which comprises survival status and survival time data from 231 HCC patients. We reviewed prognostic signatures developed by various researchers in recent years, including anoikis-related gene signatures34, methylation-related gene signatures35, immune-related gene signatures36, and ferroptosis-related gene signatures37. Additionally, we conducted a C-index comparison, where the C-index ranges from 0.5 to 1, with values closer to 1 indicating higher predictive accuracy.
Quality control and single-cell RNA-seq analysis
To investigate tumor cell heterogeneity in HCC cells, we incorporated single-cell RNA sequencing data from GSE146115. Quality control (QC) procedures were conducted using the R package “Seurat”. Single cells were deemed low-quality and subsequently removed if they exhibited fewer than 50 unique molecular identifiers (UMIs) or if mitochondrial-derived UMI counts exceeded 5%. Patient batch effects were normalized using the integration function within “Seurat”. The top 1500 variable genes were selected for further analysis. Subsequently, principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) were employed to process the data, with major cell clusters visualized in two-dimensional t-SNE plots38. We employed the FindAllMarkers function to identify the markers of each cell cluster. The R package “SingleR” is used for the annotation of different cell types39. Finally, the R packages “CellChat”40 and “patchwork” were used for intercellular communication analysis and network visualization.
Human protein atlas (HPA)
Cancer tissue microarray sections were obtained from HPA (https://www.proteinatlas.org/) for immunohistochemical staining, and the corresponding slides were digitally scanned to generate images. All relevant immunohistochemistry results in this study are from the HPA database.
Cells culture and establishment of stably transfected cells
One strain of normal human liver cells (LO2) and three strains of human liver cancer cell lines (Hep 3B, HepG2, and Huh-7) were provided by the State Key Laboratory of Cellular Stress Biology at the School of Life Sciences, Xiamen University. Each cell line was cultured in its specific culture medium. MLH1 was knocked down with pPLK-CMV-MLH1-sh1-GFP-Puro plasmid (MLH1). pPLK-CMV-GFP-Puro plasmid was used as control (NC). The plasmids were purchased from GenePharma (Shanghai, China), and the whole constructs were verified by sequencing. Briefly, Hep 3B cells were placed in 6-well plates overnight and then transfected with pcDNA3.1/MLH1-sh1 and pcDNA3.1/MLH1-sh2 plasmids and pcDNA3.1/NC plasmid using LipoMax reagent (SUDGEN, China) according to the supplier’s instructions. Twenty-four hours after transfection, cells were selected with 1 μg/ml puromycin (Invitrogen, USA) for 14 days. Stable colonies were screened and stored in 1 μg/ml puromycin.
Quantitative real-time PCR
Total RNA extracts were prepared from cells and tissues using TRIzol reagent, followed by reverse transcription into cDNA for qRT-PCR. After obtaining the CT values for the target samples, the relative expression levels of the target genes were calculated using 2^(− ΔΔCt), with normal liver cells and adjacent tissues serving as controls. Human ACTB or GAPDH was utilized as the reference gene. The primer sequences were synthesized by QIAGEN Biotech Co., Ltd. (Xiamen Synthesis Department), as detailed in Table S3.
Western blot
The protein was transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA) following 10% SDS-PAGE electrophoresis. After blocking with 5% skim milk for 1 h, the membrane was incubated overnight at 4 °C with MLH1 antibody (diluted to 1:1000; #ab131924, Abcam, USA) and β-actin antibody (diluted to 1:1000; #ab115777, Abcam, USA). Subsequently, the membrane was incubated at room temperature for 1 h with secondary antibodies conjugated to enzymes. The blot signal was detected using an enhanced chemiluminescence reagent (Advansta, USA).
Cell counting Kit-8 assay
The growth of Hep 3B cells was assessed using the CCK-8 assay kit (APExBIO, USA). A total of 2000 cells were seeded per well in a 96-well plate with 100 μL of culture medium and incubated for a duration of 4 days.
Colony formation assay
Transfected Hep 3B cells are grown in 6-well plates (500 cells per well) and cultured for approximately 14 days. The medium was changed every 3 days. The cells were then fixed with 4% PFA and stained with 0.1% CVSS for 20 min. After rinsing with PBS, colony formation was photographed and counted.
Flow cytometry analysis of the cell apoptosis
Following a 48-h period of Hep 3B cells transfection and incubation, the cells were carefully washed and subsequently collected in flow cytometry tubes. Staining was performed in strict accordance with the instructions provided by the reagent manufacturer (YEASEN, #40305ES20, China), followed by detection using an advanced analytical flow cytometer (BD LSRFortessa™ X-20, USA).
Wound-healing assay
Hep 3B cells were seeded in a six-well plate and cultured overnight until confluence was achieved. Subsequently, the cells were gently scratched using a 200 μl pipette tip to create a wound, and the process of wound healing was monitored at both 0 and 48 h.
Cell migration and invasion assays
The stably transfected cells (Hep 3B) were seeded in transwell chambers (8 mm pore size, Corning, USA) containing 100 μl of serum-free DMEM medium. The lower chamber was filled with 600 μl of medium supplemented with 20% FBS. To evaluate cellular invasion capability, the transwell chambers were pre-coated with matrigel and after a further incubation period of 48 h, the invaded cells that traversed the membrane were stained using a solution of 0.1% crystal violet and subsequently quantified.
Statistical analysis
Biological information analyses were conducted using R version 4.3.2. Data are expressed as mean ± SD of three independent tests. Differences between experimental and control groups were compared by Student’s t-test and one-way or two-way ANOVA. Survival rates were calculated by the K-M method. Results were analysed using GraphPad Prism 8.0. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
Results
Study workflow
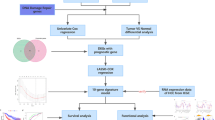
In this study, we integrated bioinformatics, machine learning, and experimental validation methodologies, as outlined in the accompanying flowchart (Fig. 1).

The flowchart of the entire study.
Genetic variation of HSCRGs in HCC
Using the “Venn” R package, 1603 hypoxia-related genes and 307 senescence -related genes were analyzed to identify 184 HSCRGs (Fig. 2a). Initially, widespread copy number variations (CNV) were observed on the chromosomes of HSCRGs (Fig. 2b). The analysis indicated that CNV is prevalent in HSCRGs associated with HCC. Over half of the HSCRGs exhibited copy number gains (Fig. S1), with SHC1 and TP73 being the most common genes associated with copy number gains and losses, respectively. Subsequently, we investigated the differential expression of these genes and identified 20 genes significantly upregulated in cancer tissues (Fig. 2c). Univariate COX and correlation analysis were employed to study the interaction, association, and impact of these DEGs on disease prognosis. 16 genes were identified as risk factors (HR > 1), 3 genes as a protective factor (HR < 1), and they exhibited a positive correlation, significantly influencing HCC prognosis (all P < 0.05) (Fig. 2d), suggesting that they may contribute to the occurrence and development of HCC.

(a) A total of 184 HSCRGs. (b) The location of CNV alterations of HSCRGs on different chromosomes. (c) The expression of different HSCRGs between normal and tumor tissues. (d) Prognostic network diagram of DEGs in HCC. The circle size represents the range of significance values of each DEG on the prognosis. The p-values were calculated by log-rank test. Green dots represent favorable factors for prognosis, and purple dots represent risk factors for prognosis. The lines linking DEGs represent their correlation. The thickness of the lines represents the strength of correlation between DEGs. Negative and positive correlations were marked with blue and red, respectively. HSCRGs, hypoxia-senescence co-related genes; CNV, copy number variation; HCC, hepatocellular carcinoma; DEGs, differentially expressed genes. *p < 0.05; **p < 0.01; ***p < 0.001.
MLH1 was identified as a disease characteristic gene
In order to further screen disease characteristic genes, we used the random forest algorithm to analyze the above 20 differential genes, and the results showed that MLH1 was the most important for disease (Fig. 3a). Survival analysis showed that HCC patients with high MLH1 expression had significantly lower survival rates (Fig. 3b). qPCR results further verified that MLH1 was highly expressed in liver cancer cell lines, while it was low expressed in normal liver cells (Fig. 3c). Western blot results showed that the expression of MLH1 in malignant tumor cells was higher than that in normal cells (Fig. 3d). It can be seen that MLH1 is highly expressed in Hep 3B cells, therefore, we will use Hep 3B cells for our subsequent experiments. In addition, we searched the immunohistochemical staining results of MLH1 from the Human Protein Atlas (HPA) database, and also found that MLH1 protein was highly expressed in tumor tissues compared to normal tissues (Fig. 3e,f). Similarly, single-cell sequencing also showed that MLH1 was highly expressed mainly in liver malignant cells (Fig. 3g). These results indicate that MLH1 gene, as a co-related gene of hypoxia and senescence, is highly expressed in HCC and also predicts poor survival prognosis.

(a) Random forests identifying key genes for disease. (b) Kaplan–Meier curves showed overall survival difference between high and low MLH1 expression groups in the TCGA-LIHC cohort (p = 0.037). (c, d) Real time-PCR and Western blot revealed that the expression of MLH1 was notably increased in HCC cell lines. (e, f) Immunohistochemical staining of MLH1 in tumor tissues and normal tissues of HCC patients in the HPA database (100 ×). (g) Expression pattern of MLH1 in the tumor microenvironment by single cell analysis. HCC, hepatocellular carcinoma; HPA, Human Protein Atlas.*p < 0.05, ** p < 0.01.
To elucidate the effect of MLH1 on HCC cells, we constructed two MLH1-targeting shRNA and transfected them into Hep 3B HCC cell lines. The efficiency of MLH1 knockdown was detected by qPCR and western blot (Fig. 4a,b). We found that MLH1 knockdown significantly inhibited the proliferation and clone formation capacity of Hep 3B cells in the CCK-8 and plate cloning experiments, respectively (Fig. 4c,d). The suppression of MLH1 expression, meanwhile, can effectively trigger apoptosis in Hep 3B cells (Fig. 4e). The results of wound healing assay showed that the migration ability of Hep 3B cells was significantly inhibited after knockdown of MLH1 (Fig. 4f). At the same time, the transwell assay also showed that MLH1 knockdown significantly inhibited the migration and invasion ability of Hep 3B cells (Fig. 4g). These results suggest that MLH1 may be an oncogene, as inhibiting the expression of MLH1 can suppress the proliferation, migration and invasion of HCC cells and induce apoptosis.

(a, b) The knockdown levels of MLH1 in Hep 3B cell line were detected by qPCR and western blot. β-actin was used as internal parameter. (c) Cell proliferation capacity was determined by CCK-8 analysis after knockdown of MLH1 in Hep 3B cells. (d) Effect of inhibition of MLH1 expression on colony formation of Hep 3B cells. (e) Effect of inhibition of MLH1 gene expression on apoptosis of Hep 3B cells (f) Wound healing assays revealed that knockdown MLH1 notably inhibited migration ability of Hep 3B cells (100 ×). (g) Transwell assays revealed that knockdown MLH1 notably inhibited migration and invasion of Hep 3B cells (100 ×). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The initial clustering: unique immune infiltration characteristics and biological behaviors of hypoxia and senescence co-related subtypes
We utilized the R package “ConsensusClusterPlus” to conduct a cluster analysis of the patients, based on the varied expression patterns of HSCRGs. The analysis delineated two hypoxia and senescence correlated phenotypes, designated as HSCRGs-clusters A and B (Fig. 5a). Furthermore, the principal component analysis (PCA) plot successfully differentiated the distribution patterns of HCC samples within clusters A and B of HSCRGs, demonstrating the efficacy of this clustering pattern (Fig. 5b). Among the two clusters, patients in HSCRGs-cluster A had a better survival prognosis, while patients in HSCRGs-cluster B had a relatively poor survival prognosis (P < 0.001) (Fig. 5c). A heatmap revealed elevated expression levels of HSCRGs in HSCRGs-cluster B, corresponding with advanced clinical stages (Fig. 5d). Additionally, we utilized the single-sample gene set enrichment analysis (ssGSEA) algorithm to assess the degree of immune infiltration between the two subgroups (Fig. 5e). The results revealed that HSCRGs-cluster B exhibited a markedly higher abundance of immune cells, including activated dendritic cells, MDSC and regulatory T cells. In contrast, HSCRGs-cluster A is enriched with only two types of cells, eosinophilia and neutrophils. To explore the biological distinctions between the two HSCRGs-clusters, we conducted GO and KEGG-related GSVAs (Fig. 5f,g). The results indicated that biological processes related to amino acid metabolism and signaling pathways associated with the metabolism of various bioactive substances were significantly enriched in cluster A. In contrast, the positive regulation of organelle tissue and the pathogenic Escherichia coli infection signaling pathway were significantly enriched in cluster B. These results showed that the two subgroups exhibited distinct characteristics in terms of survival prognosis, gene expression, and immune infiltration levels.

(a) The consensus matrixes for all HCC samples displayed the clustering stability with 1000 iterations. All samples were clustered into an appropriate number of subtypes (k = 2). (b) Principal component analysis of the two subgroups. (c) Kaplan–Meier curves showed the overall survival difference between HSCRGscluster A and B (p < 0.001). (d) The heatmap demonstrates the expression of HSCRGs in different HSCRGsclusters. Heatmap colors indicate relative HSCRGs expression levels. (e) The abundance of each immune cell infiltration in HSCRGscluster A and B. (f, g) GO and KEGG related GSVA showing the activation status of biological behaviors in HSCRGscluster A and B.HCC, hepatocellular carcinoma; HSCRGs, hypoxia-senescence co-related genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes(www.kegg.jp/kegg/kegg1.html); GSVA, gene set variation analysis. *P < 0.05, ** P < 0.01, ***P < 0.001.
The secondary clustering: comprehensive analysis of differentially expressed genes based on hypoxia and senescence co-related subtypes in HCC
To further investigate the biological functions of HSCRGs subtypes in HCC, a differential analysis was conducted to identify the differentially expressed genes (DEGs) within HSCRGs clusters using the “limma” script. The consensus clustering analysis identified 831 DEGs, revealing three distinct gene cluster subtypes: 216 samples in gene cluster A, 186 samples in gene cluster B, and 76 samples in gene cluster C (Fig. 6a). Survival analysis showed that patients with gene cluster C had the worst prognosis, gene cluster A patients had the best prognosis, and patients with gene cluster B had a median prognosis (P < 0.001) (Fig. 6b). A heatmap illustrates that the majority of patients in gene clusters A and C are aligned with HSCRG clusters A and B, respectively (Fig. 6c). Expressions of all 184 HSCRGs varied significantly across the different gene clusters (Fig. 6d). Thus, the gene clusters corresponded well with HSCRGs-clusters. GO enrichment analysis revealed that the DEGs were involved in organic acid catabolic and carboxylic acid catabolic processes. Additionally, KEGG pathway enrichment analysis indicated that these DEGs were predominantly enriched in the cell cycle, ECM receptor interaction, and other signaling pathways (Fig. 6e,f).

(a) The consensus matrixes for TCGA-LIHC cohorts based on the DEGs among the 2 HSCRGs clusters. TCGA samples were clustered into an appropriate number of subtypes (k = 3). (b) Kaplan–Meier curves showed an overall survival difference between gene clusters (p < 0.001). (c)The heatmap shows the expression of the hypoxia and senescence co-related DEGs in different HSCRGs clusters and gene clusters. (d) The difference of HSCRGs expression in different gene clusters. (e, f) GO and KEGG enrichment analysis of hypoxia and senescence co-related DEGs. DEGs, differentially expressed genes; HSCRGs, hypoxia-senescence co-related genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes(www.kegg.jp/kegg/kegg1.html). *P < 0.05, **P < 0.01, ***P < 0.001.
Construction of a prognostic signature based on differentially expressed genes
An analysis was conducted to assess the prognostic value of differentially expressed genes (DEGs) based on HSCRGs in HCC patients. Specifically, univariate Cox analysis was executed, identifying 443 prognostic factors with p < 0.05, including 103 favorable factors and 340 risk factors. The figure displays the top 10 genes with the most significant prognostic impact (Fig. 7a). Using the “glmnet” package, LASSO analysis was performed, selecting 11 essential variables from the pool of 443 prognostic factors (Fig. 7b,c). Multivariate Cox analysis was employed to establish a prognostic risk model consisting of seven key prognostic genes and to calculate the risk score (RS) for HCC patients (Fig. 7d). The computation of the RS utilized the following formula: Risk score = (0.369) * KPNA2 + (0.271) * TRIB3 + (0.232) * ATP1B3 + (0.147) * EPO + (− 0.157) * CXCL9 + (− 0.139) * LINC01554 + (-0.132) * ANXA10. To validate the impact of these seven key prognostic genes on the survival and prognosis of HCC patients, the relationship between the expression levels of these genes and patients’ survival rates was analyzed. Findings revealed a significant association between the expression levels of these genes and patients’ survival prognosis. Specifically, patients exhibiting high expression of KPNA2, ATP1B3, TRIB3, and EPO demonstrated a significantly reduced survival rate, whereas those with elevated levels of CXCL9, LINC01554, and ANXA10 showed a significantly enhanced survival rate (Figs. 7e–k; S2a–g). These results emphasize the critical role of these seven genes.

(a)The univariate Cox analysis of hypoxia-senescence co-related DEGs. (b, c) Identification of feature prognostic variables via LASSO analysis. (d) The prognosis key genes coefficient via multivariate Cox regression analysis. (e–k) Survival curves of 7 key prognostic genes (p < 0.001 and 0.05). (l) The Sankey diagram shows the potential relationship of risk score and clinical survival outcome in HSCRGs-clusters and gene clusters. (m, n) Difference analysis of risk score in HSCRGs-clusters and gene clusters. DEGs, differentially expressed genes; HSCRGs, hypoxia-senescence co-related genes (p < 0.001).
The Sankey plot delineates the potential correlation between the risk score and clinical prognosis within HSCRGs clusters and gene clusters (Fig. 7l). HCC patients were initially classified into two HSCRGs clusters and then divided into three gene clusters. Contrary to gene cluster A, which is associated with more favorable prognoses, patients in gene cluster C, characterized by poorer prognoses, were grouped with the high-risk score group. Similarly, the majority of patients in gene cluster B, characterized by unfavorable prognoses, also were part of the high-risk score group. Further investigation into the relationship between the RS and the two clustering modes revealed significant differences in the RS across the diverse clustering modes. Notably, the median RS in HSCRGs-cluster B was significantly higher than in HSCRGs-cluster A (p < 0.001) (Fig. 7m). Moreover, the median RS of gene cluster C exceeded that of gene clusters A and B (p < 0.001) (Fig. 7n). Patients in HSCRGs cluster B and gene cluster C demonstrated a relatively worse prognosis, validating the consistency of the predictive efficacy. These results showed that the risk score functions as an independent indicator for prognostic prediction.
Development and validation of a risk model based on risk score
To explore the potential association between the risk score and clinical outcome prognosis for HCC samples, the samples were divided into a training cohort and a test cohort at a 1:1 ratio using the “caret” script. In addition, the ICGC-LIRI-JP cohort was utilized as our independent external validation cohort. Significant differences in survival rates between the low-risk and high-risk groups were observed across the entire cohort, training cohort, testing cohort, and ICGC cohort, with the poorest prognosis noted in the high-risk group across all four cohorts (p < 0.05) (Figs. 8a–c; S3a). Concurrently, Kaplan–Meier survival analysis indicated that the high-risk group experienced worse progression-free survival (PFS) compared to the low-risk group across all three cohorts (Fig. 8d–f). The area under the curve (AUC) from the ROC curve served to evaluate the model’s predictive efficacy. At one, three, and five years, the AUC of the ROC curve was 0.753, 0.728, and 0.706 for the entire cohort; 0.829, 0.833, and 0.786 for the training cohort; 0.690, 0.646, and 0.639 for the test cohort; 0.829, 0.760, and 0.503 for the ICGC cohort (Figs. 8g–i; S3b). This suggests that the risk score is more effectively utilized to predict patient prognosis. The nomogram illustrated that the risk score played a pivotal role in forecasting overall survival at 1, 3, and 5 years (Fig. 8j). Calibration curves indicated that the predictive lines closely approximated the ideal line at 1, 3, and 5 years (Fig. 8k). The nomogram also showed good predictive performance in the ICGC cohort (Fig. S3c,d). The areas under the ROC curves (AUCs) for the nomogram in the entire cohort, training cohort, test cohort, and ICGC cohort were 0.765, 0.780, 0.751, and 0.866, respectively. These AUC values were superior to other clinical features, thereby demonstrating the robust performance of this nomogram in predicting the survival of HCC patients (Figs. 8l; S3e–g). The expression patterns of the seven key prognostic genes across different risk groups, as well as the distribution of survival time in relation to the risk score within the training cohort, test cohort, and ICGC cohort were respectively displayed (Fig. S4a–c). These results showed that risk score has robust predictive effect.

(a–c) Survival differences between the low-risk group and the high-risk group in the entire cohort, training cohort, and test cohort (p < 0.001 and 0.05). (d–f) PFS between low-risk group and high-risk group in the entire, training and test cohorts (p < 0.001 and 0.05). (g–i) ROC curves at 1, 3, and 5 years in the entire cohort, training cohort, and test cohort. (j) Predictive nomogram based on the risk score and other clinical features. (k) Calibration curves at 1, 3, and 5 years. (l) ROC curves for clinical parameters, risk score and nomogram in the entire cohort. PFS, Progression-free survival; ROC, receiver operating characteristic curve.
Finally, we performed a comprehensive and systematic comparison of the prognostic signatures associated with HCC that have been developed in recent years. The results demonstrated that the C-index of our model was significantly higher than those of the other four models, thereby providing stronger evidence for the superior predictive accuracy and overall performance of our model (Fig. S5).
Analyzing tumor immune microenvironment, functional strength and mutation characteristics in the two risk groups
Results from CIBERSORT showed higher infiltration levels of resting CD4 + memory T cells and Macrophages M1 in the low-risk group, while activated CD4 + memory T cells, Monocytes, and Neutrophils demonstrated elevated levels in the high-risk group (Fig. 9a,b). Findings from ssGSEA revealed that several immune functions, including cytolytic activity and type I IFN response, were heightened in the low-risk group (Fig. 9c). Furthermore, it was observed that expression levels of immune checkpoints, including CD44, CTLA4, and PDCD1, were significantly higher in the low-risk group than in the high-risk group (Fig. 9d). The immune cell bubble plot, generated by seven different software tools, predicted the correlation between various immune cells and risk scores (Fig. 9e). Correlations between the seven prognostic key genes and 22 immune infiltrating cells were also unfolded (Fig. 9f). Diverse immune infiltrating cells were significantly correlated with the seven prognostic key genes, such as CD8 T cells, Macrophages M1, and activated CD4 memory T cells, etc., indicating potential functional associations. The ESTIMATE algorithm analysis revealed that the stromal score and composite score were significantly higher in the low-risk group, suggesting that the tumor microenvironment in the low-risk group possessed elevated levels of stromal and immune cells, leading to reduced tumor cell infiltration (Fig. 9g). Accumulating evidence indicates that the elevated expression of stemness-related biomarkers in tumor cells correlates strongly with drug resistance and cancer recurrence. Therefore, the correlation between RNA stemness score (RNAss) and risk score was assessed, revealing a significantly positive correlation between the risk score and RNAss (Fig. 9h). These findings demonstrated that the low-risk group exhibited an immune-active profile, while the high-risk group was characterized by a relatively immune-cold status. Patients within the low-risk group could potentially derive greater benefits from immunotherapy, given their active tumor microenvironment.

(a) Estimation of 22 immune infiltrating cells between the low-risk group and the high-risk group by CIBERSOR. (b) Proportion of 22 immune infiltrating cells for each HCC sample by CIBERSOR. (c) Estimation of immune activities between the low-risk group and the high-risk group by ssGSEA. (d) The expression pattern of immune checkpoints between the low-risk group and the high-risk group. (e) Different software analyzed the correlation of immune cells with risk scores. (f) Correlations between 7 key genes and 22 immune infiltrating cells. (g) Differences in stromal, immune, and ESTIMATE scores in the two different risk groups. (h) The relationship between risk score and RNAss. HCC, hepatocellular carcinoma; ssGSEA, Single sample gene set enrichment analysis; RNAss, RNA stemness score.
We presented the top ten significantly enriched functional annotations for the two risk groups using GSEA (Fig. S6a–d). The results indicate that in the high-risk group, there is a strengthening of functional annotations related to the chromosome centromeric region, condensed chromosome centromeric region, and cell cycle, etc. Conversely, in the low-risk group, functional annotations related to cellular amino acid catabolic process, fatty acid catabolic process, and fatty acid metabolism, etc. were strengthened. Next, we presented the top 20 most frequently altered genes in both the low-risk and high-risk groups. CTNNB1 (25%) and TP53 (39%) were identified as the most frequently altered genes in the low-risk and high-risk groups, respectively. The most common mutation type observed in both groups was missense mutation (Fig. S6e,f).
Single-cell analysis
Single-cell analysis was conducted based on GSE146115. In this study, a total of 3200 single cells from 4 HCC patients were included. The quality control criteria are presented in the Materials and Methods section. Among them, 3198 single cells met the quality control standards (Fig. S7a–c). The heatmap was used to display the most significantly expressed genes in 11 clusters (Fig. S7d). We used tSNE to perform dimensionality reduction and clustering on the preprocessed scRNA-seq data, and finally obtained 11 clusters (Fig. 10a). Cell types were automatically annotated by the SingleR package, and these 11 clusters were clustered into 4 cell types, including Tumor cells, T cells, Macrophage, and NK cells (Fig. 10b). Subsequently, we assessed the expression patterns of the seven key prognostic genes. KPNA2 was expressed in both tumor cells and T cells; ATP1B3 was found in T cells and macrophages; and TRIB3, CXCL9, LINC01554, and ANXA10 were predominantly expressed in tumor cells (Fig. 10c,d). Cellchat, a computational tool, is designed to identify communication patterns and predict functions within less explored pathways40. Our analysis revealed that tumor cells function as the primary signal transducers within the cellular communication network, evidenced by both the number and strength of interactions (Fig. 10e,f). We then explored whether the seven key prognostic genes played roles in cell signaling pathways. Our findings demonstrate the enrichment of key genes in the SPP1 and MK signaling pathways. Further examination of these pathways revealed that tumor cells predominantly function as senders in the SPP1 signaling pathway, with major recipient cell subsets being NK cells, macrophages, and T cells. Additionally, tumor cells serve as senders, receivers, and influencers in the MK signaling pathway, indicating the potential for feedback loops (Fig. 10g–j).

(a, b) Identification of cell subgroups in the tumor microenvironment of hepatocellular carcinoma. (c, d) Expression pattern of the seven key genes in the tumor microenvironment. (e, f) Diagrams displaying the interaction number and strength in cell clusters. (g,h) associated with malignant cell subpopulation and their cell communication networks. The thicker the line represented, the more the number of interactions, and the stronger the interaction strength between the two cell types. (i,j) Primary originators and influencers of key signaling pathways.
Validation of the expression levels of the seven key prognostic genes
In further validation of the expression of the seven key genes, the immunohistochemical staining results for five genes (KPNA2, ATP1B3, TRIB3, EPO, and ANXA10) were obtained from the HPA database, revealing significant differences in the expression of related proteins between cancerous and normal liver tissues (Fig. S8). Furthermore, the mRNA expression levels of these genes were evaluated in normal hepatocytes and various HCC cell lines. Results indicated significant upregulation of KPNA2, ATP1B3, TRIB3, and CXCL9 in HCC cell lines, whereas EPO, LINC01554, and ANXA10 were markedly upregulated in normal hepatocytes (P < 0.05) (Fig. 11a–g). These results showed that the seven key prognostic genes could serve as a crucial foundation for evaluating the prognosis of HCC patients.

(a–g) The mRNA levels of the seven prognostic key genes in normal hepatocellular cell line and hepatocellular carcinoma cell lines (benign cell: LO2; malignant cells: Hep3B, HepG2, and Huh-7). *P < 0.05, **P < 0.01, ***P < 0.001, ns, non-significant.
Discussion
In this study, the identification of 184 HSCRGs was followed by the revelation of two distinct co-expression clusters within these genes. Significant differences in immune infiltration are exhibited by these two clusters. Analysis of immunoinfiltration reveals that cluster B of HSCRGs is characterized by active immune activity and significant immune cell infiltration within the tumor microenvironment. Within cluster B, MDSCs, activated dendritic cells, and Tregs are identified as playing crucial roles among the main immune cells. MDSCs exhibit strong immunosuppressive activity, inhibiting T cell and NK cell functions, thus facilitating immune escape41. It has been demonstrated that Tregs are recruited by tumor cells within the tumor microenvironment, undermining anti-tumor immunity42. Furthermore, systematic disruption of dendritic cell differentiation and development within the tumor microenvironment is regarded as a significant contributor to tumor immune escape43. These immunosuppressive characteristics might account for the adverse prognosis observed in cluster B of HSCRGs.
In recent years, significant progress has been made in molecular classification and precision oncology research for HCC. For example, studies based on genes related to anoikis, methylation, and immune subtypes have successfully divided HCC patients into different molecular subtypes and identified prognostic genes, providing valuable insights into the molecular mechanisms and treatment of HCC34,35,36. However, these studies primarily focused on individual factors such as anoikis, methylation, or immune subtypes, and did not comprehensively explore the interplay between hypoxia and senescence in HCC. Our study uniquely integrates HSCRGs to delineate molecular subtypes and construct a prognostic risk model. This approach not only captures the complex interactions between hypoxia and senescence but also provides a novel perspective on the tumor microenvironment and immune landscape in HCC44. By identifying two distinct HSCRGs clusters and developing a risk score model, we offer a more comprehensive understanding of HCC heterogeneity and potential therapeutic targets45. This distinguishes our work from previous studies and underscores the importance of considering multiple biological processes in HCC classification and prognosis prediction.
The external validation of our prognostic model using the ICGC cohort significantly strengthens the reliability and generalizability of our findings. The consistent performance of the nomogram across both our internal datasets and the independent ICGC cohort underscores the robustness of our risk stratification approach. This external validation is crucial as it demonstrates that our model can be effectively applied to diverse patient populations, enhancing its potential clinical utility46,47. The successful validation in the ICGC cohort48, which comprises patients from different geographical regions and treatment backgrounds49, further supports the notion that our HSCRGs signature captures fundamental biological processes in HCC progression that transcend specific patient cohorts.
MLH1 is a tumor suppressor gene located in the region of human chromosome 1p36.1, and the protein encoded by MLH1 gene is one of the key proteins involved in DNA mismatch repair pathway50. Studies have shown that the mutation of MLH1 gene is closely related to the incidence of various cancers, such as colorectal cancer, gastric cancer, and endometrial cancer51,52,53. Statistics show that MLH1 gene mutations occur in 8–21% of colorectal cancer patients54,55,56, 14.5% of gastric cancer patients, and 24–37% of endometrial cancers53,57,58. Loss, low expression of MLH1 gene, or mutations are important factors in the pathogenesis of these cancers59,60,61. However, the function of MLH1 in liver cancer, especially HCC, is unclear. In this study, we found that MLH1 is highly expressed in HCC, and the prognosis of HCC patients with high expression of MLH1 is significantly worse. In addition, we have verified the function of MLH1 at the cellular level for the first time, and knocking down MLH1 can significantly inhibit the proliferation, migration and invasion of cancer cells. This suggests that MLH1 may be an oncogene in HCC, and targeting MLH1 may be a new target for HCC treatment.
Subsequently, three clusters of DEGs related to hypoxia and senescence were identified, and these clusters exhibited significant associations with immune activity. We reiterated the significance of alterations in hypoxia and senescence in shaping distinct tumor immune microenvironment (TIME) landscapes. Additionally, we developed a scoring system, denoted as the RS (Risk Score), to quantify the hypoxia and senescence patterns in each HCC patient. This scoring system aims to facilitate individualized prognostic analysis and precise treatment planning for HCC. Previous studies have demonstrated that a prognostic nomogram based on RS can enhance the accuracy of survival rate estimation for patients with breast cancer62. Likewise, the risk score may be valuable in predicting prognosis and indicating immune infiltration in colon cancer63. In addition, the risk score can also be used to risk stratify patients with HCC and help individualize treatment for patients64. These analyses further suggest that the RS could serve as a potential and reliable prognostic marker in HCC.
Single-cell transcriptome data is sequenced from high-quality annotated cells, which makes it more accurate than ordinary bulk RNA sequencing data65. Therefore, it is widely used to dissect the tumor microenvironment to further understand tumor heterogeneity66. In this study, we dissected the expression patterns of seven key prognostic genes in the tumor microenvironment of HCC based on single-cell analysis. We found that tumor cells, T cells, and macrophages were the main cell subtypes, and TRIB3 was the most expressed in the tumor microenvironment among the 7 key genes, mainly expressed in tumor cells. TRIB3 has been found to reduce CD8( +) T cell infiltration and induce immune evasion by inhibiting the STAT1-CXCL10 axis in colorectal cancer67, which is consistent with our findings suggesting that TRIB3 may be a target for the treatment of cancer. Next, we identified that the ligand receptor CXCL9/SPP1 mediates cellular crosstalk between tumor cells and macrophages. It has been shown that changes in macrophage polarity, determined by the expression of two genes, CXCL9 and SPP1, are a simple but critical feature of the tumor microenvironment68. The CXCL9:SPP1 ratio can characterize the abundance of anti-tumor immune cells in tumors, the regulation of communication networks that indicate tumor control or progression, and the response to immunotherapy.
The present study had certain limitations. Primarily, in the experimental part, we did not combine clinical samples for further validation, and our findings have not been fully elucidated, and the biological function of the prognostic core gene has not been fully elucidated. Secondly, our findings suggest that these HSCRGs play different roles in HCC, such as IME landscape, drug sensitivity, and immunotherapy efficacy. However, the underlying molecular mechanisms have not been evaluated, and further studies are needed to elucidate the detailed mechanisms of HSCRGs in HCC. Thirdly, HSCRGs clustering and risk models can effectively distinguish the risk stratification, sensitivity of immunotherapy, and prognosis assessment of HCC patients. Therefore, further research is needed on whether these HSCRGs can be used as diagnostic markers or therapeutic targets for HCC to guide more effective immunotherapy strategies. Finally, in our study, we confirmed that MLH1 gene is a pro-cancer gene in HCC, but its potential molecular mechanism of action and whether it has clinical application value need to be further studied.
In addition, future research should explore the potential of drug repurposing and the use of various vitamins, such as vitamin D, as preventive measures. These interventions have been shown to have regulatory effects and could positively impact HCC outcomes. For instance, vitamin D has been reported to modulate immune responses and exert anti-tumor effects, which could be beneficial in the management of HCC69.
Conclusions
In summary, this study delineated subgroups correlated with hypoxia and senescence, constructing a prognostic risk model for HCC. The expression and specific function of MLH1 gene in HCC were identified. The results highlighted the identification of seven prognostic genes significantly associated with tumor occurrence and immune cell infiltration. The prognostic risk model exhibited robust and effective performance in predicting overall survival in HCC, as demonstrated by successful external validation in the ICGC cohort. This validation underscores the potential clinical utility of our model in improving risk stratification and guiding personalized treatment strategies for HCC patients. Overall, these findings have significant implications for exploring the molecular pathways and mechanisms in HCC and advancing treatments and prognostic approaches.
Data availability
All data generated or analyzed during this study are included in this published article and are available from the corresponding author on reasonable request.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- HSCRGs:
-
Hypoxia-senescence co-related genes
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- ICGC:
-
International Cancer Genome Consortium
- CNV:
-
Copy number variation
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ssGSEA:
-
Single-sample gene set enrichment analysis
- TIMER:
-
Tumor Immune Estimation Resource
- PCA:
-
Principal component analysis
- RS:
-
Risk score
- PFS:
-
Progression-free survival
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- HPA:
-
Human Protein Atlas
- qPCR:
-
Quantitative real-time PCR
- SDS-PAGE:
-
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- PVDF:
-
Polyvinylidene difluoride
- CCK-8:
-
Cell Counting Kit-8
- PFA:
-
Paraformaldehyde
- CVSS:
-
Crystal violet staining solution
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- FBS:
-
Fetal bovine serum
- ANOVA:
-
Analysis of variance
- K-M:
-
Kaplan–Meier
- MDSCs:
-
Myeloid-derived suppressor cells
- Tregs:
-
Regulatory T cells
- NK cells:
-
Natural killer cells
- RNAss:
-
RNA stemness score
- SPP1:
-
Secreted phosphoprotein 1
- MK:
-
Midkine
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Ali, N. A., Hamdy, N. M., Gibriel, A. A. & El Mesallamy, H. O. Investigation of the relationship between CTLA4 and the tumor suppressor RASSF1A and the possible mediating role of STAT4 in a cohort of Egyptian patients infected with hepatitis C virus with and without hepatocellular carcinoma. Arch. Virol. 166, 1643–1651. https://doi.org/10.1007/s00705-021-04981-8 (2021).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 7, 6. https://doi.org/10.1038/s41572-020-00240-3 (2021).
Youssef, S. S. & Hamdy, N. M. SOCS1 and pattern recognition receptors: TLR9 and RIG-I; novel haplotype associations in Egyptian fibrotic/cirrhotic patients with HCV genotype 4. Arch. Virol. 162, 3347–3354. https://doi.org/10.1007/s00705-017-3498-7 (2017).
Elanany, M. M., Mostafa, D. & Hamdy, N. M. Remodeled tumor immune microenvironment (TIME) parade via natural killer cells reprogramming in breast cancer. Life Sci. 330, 121997. https://doi.org/10.1016/j.lfs.2023.121997 (2023).
Eldosoky, M. A. et al. Diagnostic significance of hsa-miR-21-5p, hsa-miR-192-5p, hsa-miR-155-5p, hsa-miR-199a-5p panel and ratios in hepatocellular carcinoma on top of liver cirrhosis in HCV-infected patients. Int. J. Mol. Sci. 24, 3157. https://doi.org/10.3390/ijms24043157 (2023).
Cappuyns, S., Corbett, V., Yarchoan, M., Finn, R. S. & Llovet, J. M. Critical appraisal of guideline recommendations on systemic therapies for advanced hepatocellular carcinoma: A review. JAMA Oncol. 10, 395–404. https://doi.org/10.1001/jamaoncol.2023.2677 (2024).
Luo, P. et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 67, 662–675. https://doi.org/10.1002/hep.29561 (2018).
Abaza, T. et al. Emerging role of circular RNAs in hepatocellular carcinoma immunotherapy. Int. J. Mol. Sci. 24, 16484. https://doi.org/10.3390/ijms242216484 (2023).
Gray, L. H., Conger, A. D., Ebert, M., Hornsey, S. & Scott, O. C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 26, 638–648. https://doi.org/10.1259/0007-1285-26-312-638 (1953).
Li, Q. et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct. Target. Ther. 6, 76. https://doi.org/10.1038/s41392-020-00453-8 (2021).
Nishida, N. & Kudo, M. Oxidative stress and epigenetic instability in human hepatocarcinogenesis. Dig. Dis. 31, 447–453. https://doi.org/10.1159/000355243 (2013).
Alique, M. et al. Hypoxia-inducible factor-1α: The master regulator of endothelial cell senescence in vascular aging. Cells 9, 195. https://doi.org/10.3390/cells9010195 (2020).
Lv, R. et al. Pathophysiological mechanisms and therapeutic approaches in obstructive sleep apnea syndrome. Signal Transduct. Target Ther. 8, 218. https://doi.org/10.1038/s41392-023-01496-3 (2023).
Beauséjour, C. M. et al. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. Embo J. 22, 4212–4222. https://doi.org/10.1093/emboj/cdg417 (2003).
Prieto, L. I. & Baker, D. J. Cellular senescence and the immune system in cancer. Gerontology 65, 505–512. https://doi.org/10.1159/000500683 (2019).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: An expanding universe. Cell 186, 243–278. https://doi.org/10.1016/j.cell.2022.11.001 (2023).
Qi, J. et al. National and subnational trends in cancer burden in China, 2005–20: An analysis of national mortality surveillance data. Lancet Public Health 8, e943–e955. https://doi.org/10.1016/s2468-2667(23)00211-6 (2023).
De’ Angelis, G. L. et al. Microsatellite instability in colorectal cancer. Acta Biomed. 89, 97–101. https://doi.org/10.23750/abm.v89i9-S.7960 (2018).
Niv, Y. Microsatellite instability and MLH1 promoter hypermethylation in colorectal cancer. World J. Gastroenterol. 13, 1767–1769. https://doi.org/10.3748/wjg.v13.i12.1767 (2007).
Kolodner, R. D. & Marsischky, G. T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9, 89–96. https://doi.org/10.1016/s0959-437x(99)80013-6 (1999).
Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98. https://doi.org/10.1038/cr.2007.115 (2008).
Modrich, P. & Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133. https://doi.org/10.1146/annurev.bi.65.070196.000533 (1996).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672-d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Tang, Y. et al. Prognosis and dissection of immunosuppressive microenvironment in breast cancer based on fatty acid metabolism-related signature. Front. Immunol. 13, 843515. https://doi.org/10.3389/fimmu.2022.843515 (2022).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77, e108–e110. https://doi.org/10.1158/0008-5472.Can-17-0307 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Hu, J. & Szymczak, S. A review on longitudinal data analysis with random forest. Brief Bioinform. https://doi.org/10.1093/bib/bbad002 (2023).
Alhamzawi, R. & Ali, H. T. M. The Bayesian adaptive lasso regression. Math. Biosci. 303, 75–82. https://doi.org/10.1016/j.mbs.2018.06.004 (2018).
Bozdogan, H. Akaike’s information criterion and recent developments in information complexity. J. Math. Psychol. 44, 62–91. https://doi.org/10.1006/jmps.1999.1277 (2000).
Chen, Y., Huang, W., Ouyang, J., Wang, J. & Xie, Z. Identification of Anoikis-related subgroups and prognosis model in liver hepatocellular carcinoma. Int. J. Mol. Sci. 24, 2862. https://doi.org/10.3390/ijms24032862 (2023).
Li, D. et al. The m6A/m5C/m1A regulated gene signature predicts the prognosis and correlates with the immune status of hepatocellular carcinoma. Front. Immunol. 13, 918140. https://doi.org/10.3389/fimmu.2022.918140 (2022).
Chen, Z. D., Luo, J. Y., Ye, Y. P. & Dang, Y. W. Identification of immune-related genes and patient selection for hepatocellular carcinoma immunotherapy. Transl. Cancer Res. 12, 1210–1231. https://doi.org/10.21037/tcr-22-2304 (2023).
Wang, W. et al. Ferroptosis-related hub genes in hepatocellular carcinoma: Prognostic signature, immune-related, and drug resistance analysis. Front. Genet. 13, 907331. https://doi.org/10.3389/fgene.2022.907331 (2022).
Kobak, D. & Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 10, 5416. https://doi.org/10.1038/s41467-019-13056-x (2019).
Wen, Z. & Ablimit, A. Comprehensive analysis of scRNA-Seq and bulk RNA-Seq reveals ubiquitin promotes pulmonary fibrosis in chronic pulmonary diseases. Sci. Rep. 14, 21195. https://doi.org/10.1038/s41598-024-70659-1 (2024).
Jin, S. et al. Inference and analysis of cell-cell communication using Cell Chat. Nat. Commun. 12, 1088. https://doi.org/10.1038/s41467-021-21246-9 (2021).
Ostrand-Rosenberg, S. & Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 182, 4499–4506. https://doi.org/10.4049/jimmunol.0802740 (2009).
Shang, B., Liu, Y., Jiang, S. J. & Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 5, 15179. https://doi.org/10.1038/srep15179 (2015).
Fu, C. & Jiang, A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 9, 3059. https://doi.org/10.3389/fimmu.2018.03059 (2018).
Gao, H. et al. Role of hypoxia in cellular senescence. Pharmacol. Res. 194, 106841. https://doi.org/10.1016/j.phrs.2023.106841 (2023).
Yeo, E. J. Hypoxia and aging. Exp. Mol. Med. 51, 1–15. https://doi.org/10.1038/s12276-019-0233-3 (2019).
Ma, D. et al. Development and validation of prognostic risk prediction models for hepatocellular carcinoma patients treated with immune checkpoint inhibitors based on a systematic review and meta-analysis of 47 cohorts. Front. Immunol. 14, 1215745. https://doi.org/10.3389/fimmu.2023.1215745 (2023).
Liu, Y. et al. Development and validation of a hypoxia-immune-based microenvironment gene signature for risk stratification in gastric cancer. J. Transl. Med. 18, 201. https://doi.org/10.1186/s12967-020-02366-0 (2020).
Lu, J. et al. A novel prognostic model based on single-cell RNA sequencing data for hepatocellular carcinoma. Cancer Cell Int. 22, 38. https://doi.org/10.1186/s12935-022-02469-2 (2022).
Chen, X. et al. Identification and in vitro and in vivo validation of the key role of GSDME in pyroptosis-related genes signature in hepatocellular carcinoma. BMC Cancer 23, 411. https://doi.org/10.1186/s12885-023-10850-1 (2023).
Boland, C. R. & Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073-2087.e2073. https://doi.org/10.1053/j.gastro.2009.12.064 (2010).
Gutierrez-Angulo, M., Ayala-Madrigal, M. L., Moreno-Ortiz, J. M., Peregrina-Sandoval, J. & Garcia-Ayala, F. D. Microbiota composition and its impact on DNA methylation in colorectal cancer. Front. Genet. 14, 1037406. https://doi.org/10.3389/fgene.2023.1037406 (2023).
Park, S. E. et al. Low PARP-1 expression level is an indicator of poor prognosis in patients with stage II and III gastric cancer. J. Cancer 13, 869–876. https://doi.org/10.7150/jca.65145 (2022).
Post, C. C. B. et al. Prevalence and prognosis of lynch syndrome and sporadic mismatch repair deficiency in endometrial cancer. J. Natl. Cancer Inst. 113, 1212–1220. https://doi.org/10.1093/jnci/djab029 (2021).
Cunningham, J. M. et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am. J. Hum. Genet. 69, 780–790. https://doi.org/10.1086/323658 (2001).
Kuismanen, S. A., Holmberg, M. T., Salovaara, R., de la Chapelle, A. & Peltomäki, P. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am. J. Pathol. 156, 1773–1779. https://doi.org/10.1016/s0002-9440(10)65048-1 (2000).
Thibodeau, S. N. et al. Microsatellite instability in colorectal cancer: Different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 58, 1713–1718 (1998).
Bischoff, J. et al. hMLH1 promoter hypermethylation and MSI status in human endometrial carcinomas with and without metastases. Clin. Exp. Metastasis 29, 889–900. https://doi.org/10.1007/s10585-012-9478-0 (2012).
Resnick, K. E. et al. Mismatch repair status and outcomes after adjuvant therapy in patients with surgically staged endometrial cancer. Gynecol. Oncol. 117, 234–238. https://doi.org/10.1016/j.ygyno.2009.12.028 (2010).
Lv, K. et al. Lymphocyte-activation gene 3 expression associates with poor prognosis and immunoevasive contexture in Epstein–Barr virus-positive and MLH1-defective gastric cancer patients. Int. J. Cancer 148, 759–768. https://doi.org/10.1002/ijc.33358 (2021).
Ran, X., Jing, H. & Li, Z. The clinical features and management of Lynch syndrome-associated ovarian cancer. J. Obstet. Gynaecol. Res. 48, 1538–1545. https://doi.org/10.1111/jog.15273 (2022).
Scarpa, M. et al. MLH1 deficiency down-regulates TLR4 expression in sporadic colorectal cancer. Front. Mol. Biosci. 8, 624873. https://doi.org/10.3389/fmolb.2021.624873 (2021).
Ye, Y. et al. Leveraging senescence-oxidative stress co-relation to predict prognosis and drug sensitivity in breast invasive carcinoma. Front. Endocrinol. 14, 1179050. https://doi.org/10.3389/fendo.2023.1179050 (2023).
Zhang, H. C. et al. Identification and validation in a novel quantification system of ferroptosis patterns for the prediction of prognosis and immunotherapy response in left- and right-sided colon cancer. Front. Immunol. 13, 855849. https://doi.org/10.3389/fimmu.2022.855849 (2022).
Yuan, W. et al. Cellular senescence-related genes: Predicting prognosis in hepatocellular carcinoma. BMC Cancer 23, 1001. https://doi.org/10.1186/s12885-023-11288-1 (2023).
Bartoschek, M. et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150. https://doi.org/10.1038/s41467-018-07582-3 (2018).
Ding, S., Chen, X. & Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. (Lond) 40, 329–344. https://doi.org/10.1002/cac2.12078 (2020).
Shang, S. et al. TRIB3 reduces CD8(+) T cell infiltration and induces immune evasion by repressing the STAT1-CXCL10 axis in colorectal cancer. Sci. Transl. Med. 14, eebf0992. https://doi.org/10.1126/scitranslmed.abf0992 (2022).
Bill, R. et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381, 515–524. https://doi.org/10.1126/science.ade2292 (2023).
Chen, Y. C. et al. Effect of vitamin D supplementation on primary dysmenorrhea: A systematic review and meta-analysis of randomized clinical trials. Nutrients 15, 2830. https://doi.org/10.3390/nu15132830 (2023).
Acknowledgements
We would like to express our sincere gratitude to Professor Zhou Jianyin for his invaluable guidance, assistance, and unwavering support throughout this research endeavor. Additionally, we extend our heartfelt appreciation to the team of Academician Lin Shengcai at Xiamen University’s School of Life Sciences for providing us with access to their exceptional experimental platform and various forms of assistance.
Funding
This study was supported by Fujian Provincial Natural Science Foundation (No.2024J011331) and Xiamen Natural Science Foundation(No.3502Z20227269).
Author information
Authors and Affiliations
Contributions
FC: Methodology, Formal Analysis, Software, Visualization, Validation, Writing–original draft, Writing–review and editing. YC, XC, and CC: Data curation, Formal Analysis, Visualization, Writing–original draft, Writing–review and editing. QF, JL, and YZ: Software, Validation , Writing–review and editing. JZ: Funding acquisition, Project administration, Writing–review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, F., Cai, Y., Chen, X. et al. The role of hypoxia-senescence co-related molecular subtypes and prognostic characteristics in hepatocellular carcinoma. Sci Rep 15, 12390 (2025). https://doi.org/10.1038/s41598-025-97604-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97604-0
