
Abstract
In recent years, the growing attention to sustainable food production and animal welfare has positioned cultured meat technologies. A key technical requirement for implementing this approach is the development of culture media independent of fetal bovine serum (FBS). 3D organoids are composed of diverse stem and differentiated cells that replicate the structure and function of living tissues. Therefore, we hypothesized that the growth factors secreted from 3D organoids could replace the FBS. In this study, we evaluated the effects of conditioned medium prepared from organoid supernatants derived from various mouse organs, including the lung, gallbladder, spleen, kidney, and bladder, on myoblast proliferation. Among these, mouse bladder organoid-derived supernatant (MBOS) showed the most pronounced proliferative effect on C2C12 cells, a myoblast cell line. RNA sequencing and flow cytometry analyses revealed that MBOS-treated C2C12 cells upregulated cell cycle-related genes (CCNB1 and CDK1) and increased the proportion of cells in the G2/M phase. Although MBOS contained a higher concentration of insulin-like growth factor (IGF)-1 than the control medium, neutralization with IGF-1 antibody did not reduce the proliferative effect. MBOS significantly promoted the proliferation of primary bovine myoblasts, suggesting its cross-species applicability. These findings suggest that MBOS is a promising candidate for FBS-free culture media with the potential to produce cultured meat.
Similar content being viewed by others

Introduction
In recent years, cultured meat has gained global attention as a sustainable food production strategy to address critical issues such as climate change, protein security, and animal welfare1,2,3,4,5. Compared to conventional livestock farming, cultured meat offers multifaceted advantages, including reduced greenhouse gas emissions, conservation of land and water resources, and decreased reliance on antibiotics3,5. Consequently, interdisciplinary research spanning food science, cellular agriculture, ethics, and consumer acceptance has rapidly advanced1,2,6,7. Recently, perspectives involving life cycle assessment (LCA), as well as regulatory and cultural acceptability, have further enriched the discourse6,7.
A major technical challenge in the development of cultured meat is establishing a growth factor delivery system that supports cell proliferation and differentiation. Fetal bovine serum (FBS) is commonly used in cell culture systems. However, it is unsuitable for food applications owing to batch-to-batch compositional variability, risk of pathogen contamination, ethical concerns, and high environmental impact, as shown by LCA8,9,10,11,12. Considering these issues, the development of sustainable serum-free culture systems has become an urgent priority. Various alternatives to FBS, including human platelet lysate11,13,14,15,16, algal extracts17,18,19,20, insect-derived components21,22,23, and chemically defined media24,25, have been explored and compared for their effectiveness and limitations. These studies have elucidated the effectiveness and remaining challenges of chemically defined media, laying an important foundation for serum-free culture research in the field of cultured meat 26.
In recent years, increasing attention has been directed toward the use of conditioned media derived from three-dimensional (3D) organoid cultures as an alternative approach. Conditioned media are the culture supernatants collected after incubating living cells, which contain a complex mixture of secreted bioactive molecules, including growth factors, cytokines, and extracellular matrix components, that can influence the behavior of other cells. Unlike standard basal media, which provide only basic nutrients and lack these signaling factors, conditioned media are enriched with physiologically active molecules, such as cytokines and growth factors, which are naturally secreted by cells and may offer a culture environment that minimizes the need for externally added supplements27,28,29,30. Furthermore, studies on gastric and intestinal organoids have demonstrated that these systems can sustain themselves through autocrine signaling31,32, highlighting the potential of conditioned medium as a viable substitute for FBS.
In this study, we evaluated the proliferative effects of organoid-derived conditioned media on C2C12 mouse myoblasts under serum-free conditions as a potential alternative to FBS. To explore the broader applicability of this approach, we assessed its effects on primary bovine muscle-derived cells. Our results contribute to the advancement of serum-free culture strategies and highlight the potential of organoid-derived factors in cultured meat production.
Materials and methods
Chemicals and reagents
To generate organoids, primary cells isolated from the lungs, gallbladder, spleen, kidney, and bladder of 5-week-old male C57BL/6J mice (Sankyo Labo Service Corporation, Tokyo, Japan) were cultured in a stem cell-supporting medium, as previously described33,34,35. The components of the medium were as follows: Advanced Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 50% Wnt, Noggin, and R-Spondin conditioned medium; GlutaMax; B-27 supplement; 100 μg/mL Primocin (Thermo Fisher Scientific, Waltham, MA, USA); 1 mM N-Acetyl-L-cysteine; 10 mM nicotinamide (Sigma-Aldrich, St. Louis, MO, USA); 50 ng/mL mouse EGF (PeproTech, Rocky Hill, NJ, USA); 500 nM A83-01 (Adooq Bioscience, Irvine, CA, USA); 3 μM SB202190; and 10 μM Y-27632 (Cayman, Ann Arbor, MI, USA). The primary antibodies used were as follows: cytokeratin cocktail (AE1/AE3, Novus Biologicals, Centennial, CO, USA); vimentin (R&D Systems, Minneapolis, MN, USA); thyroid transcription factor-1 (TTF-1, Novus Biologicals, Centennial, CO, USA); cytokeratin 19 (CK19, Novus Biologicals); mucin 3 (MUC3, Bioss Inc., Woburn, MA, USA); cluster of differentiation 31 (CD31, Thermo Fisher Scientific); vascular endothelial growth factor receptor 2 (VEGFR-2, Santa Cruz Biotechnology, Dallas, TX, USA); paired box gene 8 (PAX8, GeneTex, Inc., Irvine, CA, USA); cytokeratin 5 (CK5; GeneTex, Inc); uroplakin IIIa (UPK3A, Novus Biologicals), myogenic factor 5 (Myf5, GeneTex, Inc), myoblast determination protein 1 (MyoD, GeneTex, Inc). The secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Cayman) and Dako Envision+ Dual Link System-HRP (Agilent Technologies Inc., Santa Clara, CA, USA). The fluorescent secondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 48 goat anti-mouse IgG (Thermo Fisher Scientific).
C2C12 culture and passage
C2C12 mouse myoblasts were obtained from the RIKEN BioResource Center (Tsukuba, Japan). Cells were cultured in low-glucose DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin–streptomycin (100 U/mL penicillin and 100 μg/mL streptomycin) at 37 °C in a humidified atmosphere of 5% CO₂. Cells were passaged every 3 days using TrypLE Express Enzyme solution (Thermo Fisher Scientific) at a split ratio of 1:2 to 1:4, as commonly practiced in routine cell cultures36.
Establishment of mouse tissue-derived organoids
The mouse experiments conducted in this study were carried out according to the Institutional Animal Care and Use Committee of Tokyo University of Agriculture and Technology approval (Approval number: R06-131). All procedures were done in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. To generate organoids from different mouse tissues, the lungs, gallbladder, spleen, kidney, and bladder were disected from 5-week-old male C57BL/6J mice (20-25 g) after euthanasia under isoflurane anesthesia. Tissues were chopped, minced in Petri dishes using sterile scissors and scalps, and transferred into sterile Falcon tubes containing 5 mL of Advanced DMEM supplemented with Liberase TM (0.125 mg/mL; Roche Diagnostics, Basel, Switzerland)37. Enzymatic digestion was conducted in a thermostatic shaking water bath at 37 °C for 30 min. Subsequently, tubes were spun down, the digestion medium was aspirated, and the tissue fragment pellets were further treated with prewarmed (37 °C ) TrypLE™ Express solution and incubated in a water bath at 37 °C for 5 min. The resulting cell suspensions were passed through a 100 µm nylon mesh cell strainer (Falcon), centrifuged at 600 ×g for 5 min at 4 °C, and washed three times with phosphate-buffered saline (PBS). The obtained cell pellets were resuspended gently in Matrigel on ice and cultured in 24-well plates (40 µL per well). Plates were then kept in a CO₂ incubator at 37 °C for 20 min to polymerize the gel droplets, and a 500 µL/well of stem cell-nourishing culture medium was added and replaced three times weekly. After the organoids reached 80% confluence, they were passaged into new wells at a ratio of 1:3 to 1:4 using TrypLE™ Express solution38,39. All five types of organoids were fixed in buffered formalin and routinely processed to prepare paraffin blocks. Sections of 4 µm thickness were used for hematoxylin and eosin (H&E) staining and immunohistochemical analyses.
H&E staining
Paraffin-embedded sections from five types of organoids were prepared and stained with H&E as described before34,40. Images were captured using a camera attached to a microscope (BX-52; Olympus, Tokyo, Japan).
Immunohistochemical (IHC) staining
IHC staining of organoids was performed as described before37. Antigen retrieval was performed using 10 mM citrate buffer heated at 121 °C for 5 min. After cooling, the slides were treated with 1% hydrogen peroxide for 30 min at room temperature (RT) to stop endogenous peroxidase activity. After washing the slides with PBS, they were blocked with 10% normal goat serum (NGS) for 30 min at RT. After washing the slides with PBS, they were treated with primary antibodies (AE1/AE3 1:100, Vimentin 1:100, TTF-1 1:100, CK19 1:100, MUC3 1:200, CD31 1:100, VEGFR-2 1:100, PAX8 1:300, CK5 1:500, and UPK3A 1:500) and incubated at 4 °C overnight. After washing with PBS, the slides were incubated with a secondary antibody and visualized using DAB solution (Nacalai Tesque, Kyoto, Japan). After color change, the slides were counterstained with hematoxylin for one min. Images were captured using a camera attached to a microscope (BX-52; Olympus).
Preparation of conditioned medium from mouse tissue-derived organoids
Organoids derived from five mouse tissues (lung, gallbladder, spleen, kidney, and bladder) were established and maintained according to protocols previously reported by our laboratory [33,34,37,41]. Before collecting conditioned media, the organoids were immunostained with tissue-specific markers (e.g., CK5, UPK3A) to confirm their tissue identity, ensuring that the collected supernatants originated from organoids retaining their respective characteristics. To prepare the conditioned medium, culture supernatants were collected from five types of organoids (lung, gallbladder, spleen, kidney, and bladder) after two passages and upon reaching approximately 80% confluency. The stem cell-supporting medium was then removed, and each well, including the organoid surface, was gently rinsed with pre-warmed PBS (37 °C) to eliminate residual medium and unattached cells. Subsequently, 500 µL of serum-free low-glucose DMEM was added to each well and incubated for 48 h. The supernatants were collected and mixed at a 1:1 ratio with serum-free, low-glucose DMEM. The resulting mixtures were defined as mouse lung organoid supernatant (MLOS), mouse gallbladder organoid supernatant (MGOS), mouse spleen organoid supernatant (MSOS), mouse kidney organoid supernatant (MKOS), and mouse bladder organoid supernatant (MBOS). Conditioned media were used in subsequent experiments.
Cell proliferation assay
C2C12 cells were seeded at a density of 500 cells per well in 96-well plates. Cells were cultured under serum-free conditions in organoid-conditioned medium (MLOS, MGOS, MSOS, MKOS, and MBOS), serum-free low-glucose DMEM (negative control), or standard growth medium containing 10% FBS (positive control). After 72 h of incubation at 37 °C in a humidified atmosphere containing 5% CO₂, cell viability was examined using a PrestoBlue kit (Thermo Fisher Scientific) and a microplate reader (TECAN, Seestrasse, Switzerland). Data from the microplate reader were analyzed and plotted using SigmaPlot software (Systat Software, Inc., San Jose, CA., USA). To complement the metabolic PrestoBlue assay, a direct cell-counting assay using Trypan Blue exclusion was also performed. C2C12 cells were cultured under the same conditions described above, and viable cells were quantified after 72 h using a hemocytometer.
Comparative RNA sequencing analysis
Total RNA was extracted from C2C12 myoblasts using the NucleoSpin RNA Kit (TaKaRa Bio Inc.), and samples with RIN ≥ 8 were selected. Poly(A)+ RNA was enriched using the NEBNext Poly(A) Magnetic Isolation Module (NEB), and strand-specific libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit, which incorporates dUTP during second-strand synthesis. Libraries were barcoded, amplified (13 cycles), quantified using Qubit, and analyzed using TapeStation to determine fragment size. Sequencing was performed on a NovaSeq 6000 platform (Illumina) in 150 bp paired-end mode, generating ~20 million read pairs per sample. Adapter trimming and quality control were performed using Cutadapt v2.9. The reads were aligned to the Mus musculus reference genome (GRCm38, Ensembl release 101) using HISAT2 v2.2.0. Gene expression was quantified using RSEM v1.3.3 and normalized using the TMM method. Differential expressions were defined as FDR < 0.05 and >2-fold change. Volcano plots, heat maps, and Gene Set Enrichment Analysis (GSEA) (Broad GSEA v4.1.0 with MSigDB v7.4) were used to visualize transcriptomic changes and pathway enrichment analysis.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from C2C12 cells cultured under control (serum-free medium) and MBOS conditions using the NucleoSpin RNA kit (Takara Bio Inc., Japan), following the manufacturer’s instructions. First-strand cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Quantitative real-time PCR was performed using a QuantiTect SYBR I Kit (Qiagen) and StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The 2-ΔΔCq method and cycle threshold (Ct) values obtained during quantification were used to calculate the fold changes in mRNA abundance. Specific primer designs are presented in Table 1.
Western blotting
Western blotting was performed as described before41,42. Cell lysis reagent containing 1% protease inhibitor cocktail (Sigma-Aldrich) was added to C2C12 cells cultured under serum-free control or MBOS conditions to prepare the protein lysates. After measuring the protein concentrations of these samples, equal amounts of protein (10 µg) were loaded onto a gel (SDS-PAGE 7.5%), separated via electrophoresis, and transferred to a nitrocellulose membrane (Wako). The membrane was blocked with a 0.5% skim milk solution at RT for one h and treated with primary antibodies (CDK1 1:100, CCNB1 1:100, and VCP 1:500) overnight at 4 °C. After washing the membranes with Tris-buffered saline containing Tween 20 (TBS-T), they were treated with secondary antibodies (HRP-bound anti-rabbit IgG 1:10,000, HRP-bound anti-mouse IgG 1:10,000) at RT for 1 h, and then washed again with TBS-T. Images were captured using an imaging device (LAS3000, Fujifilm) after treatment with the Immobilon Forte Western HRP substrate (Millipore). The expression levels of the captured images were quantified using the ImageJ densitometry analysis software.
Flow cytometric analysis of cell cycle
Fluorescence-activated cell sorting (FACS) analysis was performed to assess the cell cycle distribution of C2C12 cells cultured in serum-free control or MBO. Briefly, the cells were dissociated using TrypLE™ Express, fixed in cold 70% ethanol at 4 °C for at least 2 h, and stained with propidium iodide (PI; 50 µg/mL) in the presence of RNase A (100 µg/mL) in FACS buffer (0.1% BSA/3 mM EDTA in PBS). After incubation at RT in the dark for 30 min, the samples were filtered through a 144 µm nylon mesh and analyzed using a Guava easyCyte flow cytometer (Millipore). Cell populations in the G0/G1, S, and G2/M phases were quantified using integrated analysis software.
Enzyme-linked immunosorbent assays (ELISA) analysis of insulin-like growth factors (IGF)-1 and IGF-2 in MBOS
To quantify IGF-1 and IGF-2 levels in MBOS, ELISA was performed using the Mouse/Rat IGF1 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA, MG100) and Mouse IGF2 ELISA Kit (Abcam, Cambridge, UK, ab100696), respectively. MBOS samples were prepared according to the manufacturer’s protocol and added to 96-well plates. Absorbance was measured at 450 nm using a microplate reader, and the concentrations were calculated based on the standard curves provided in each kit. All measurements were performed in duplicate.
Culture of primary bovine muscle-derived cells
The bovine experiments were conducted according to the Institutional Animal Care and Use Committee of Tokyo University of Agriculture and Technology approval (Approval number: R07-36). Bovine samples were collected post-euthanasia. Euthanasia was performed by intramuscular administration of xylazine (Selactar® 2% injectable solution, 0.2 ml/kg) for sedation, followed by intravenous injection of an overdose of pentobarbital (60 mg/kg). All procedures were done in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Skeletal muscle tissue was collected from each cow at the end of its productive life. Cell pellets were prepared using a dissociation method similar to that used to process mouse organs. The cells were seeded in culture media, as described in Table 2. The following day, the culture supernatants were transferred to a gelatin-coated dish. Once the cells reached confluence, they were passaged using standard cell culture techniques and used in subsequent experiments.
Immunofluorescence staining
Immunofluorescence staining of the cells was performed as described before43. After dissociation of the cultured cells using TrypLE Express, the cells were fixed in 4% paraformaldehyde for 60 min at RT and directly seeded onto gelatin-coated glass slides. Following fixation, cells were blocked with 1.5% NGS in PBS at RT for 60 min and then incubated with primary antibodies against Myf5 and MyoD at 4 °C overnight. After washing, the cells were incubated with a secondary antibody for 60 min at RT. The protein expression levels of Myf5 and MyoD were visualized using a fluorescence microscope (BX-52; Olympus) and quantified using the ImageJ densitometry analysis software.
Statistical analysis
For all experiments except RNA sequencing, statistical comparisons were performed using an unpaired two-tailed Student’s t-test. Data are reported as mean ± standard deviation (SD), and P-values less than 0.05 were considered statistically significant.
Result
Generation of organoids derived from mouse lung, gallbladder, spleen, kidney, and bladder tissues
To generate organoids from mouse lung (MLO), gallbladder (MGO), spleen (MSO), kidney (MKO), and bladder (MBO) tissues, cells extracted from each tissue were seeded into 24-well plates containing Matrigel (Fig. 1A). All organoids exhibited three-dimensional structures within 1 week of seeding (Fig. 1B). H&E staining revealed solid-type organoid structures in MLO, MKO, and MBO and tubular-type organoid structures in MGO and MSO (Fig. 1B). IHC staining using epithelial (AE1/AE31, a keratin cocktail) and mesenchymal markers (vimentin) revealed that MLO, MGO, MKO, and MBO expressed epithelial markers, whereas none of the organoids expressed mesenchymal markers (Fig. 1B). MLO cells expressed the pulmonary alveolar epithelial marker TTF-1. MGO expressed the bile duct epithelial marker CK19 and the gallbladder mucosal epithelial marker MUC3. MSO cells expressed the vascular endothelial cell markers CD31 and VEGFR-2. MKO cells express the renal tubule cell marker PAX8. The MBO cells expressed the basal cell layer marker of the bladder epithelium CK5 and an umbrella cell marker UPK3A (Fig. 1B).

Schematic diagram of the production of mouse normal tissue-derived organoids and comparative analysis of the culture supernatants of tissue-derived organoids from each organ (A). At 5 weeks of age, male C57BL/6J mice were euthanized, and the lungs (MLO), gallbladder (MGO), spleen (MSO), kidneys (MKO), and bladder (MBO) were removed. Each tissue was digested, washed, mixed with Matrigel, and cultured as three-dimensional organoids. Each conditioned medium (CM) of the organoid was applied to C2C12 myoblasts to assess their proliferation activity in vitro. Brightfield microscopy (scale bar: 50 µm), hematoxylin and eosin (H&E) staining, and immunohistochemical staining (scale bars: 200 µm) of MLO, MGO, MSO, MKO, and MBO (B). Organoids were probed with the pan-epithelial marker AE1/AE3 and the mesenchymal marker vimentin to assess lineage composition. The following markers were confirmed as tissue-specific: thyroid transcription factor (TTF) for MLO; cytokeratin 19 (CK19) and mucin-3 (MUC3) for MGO; vascular endothelial growth factor receptor 2 (VEGFR-2) and CD31 for MSO; paired box protein 8 (PAX8) for MKO; and cytokeratin 5 (CK5) and uroplakin 3A (UPK3A) for MBO.
Effects of conditioned media from five mouse organoid types on C2C12 cell growth
C2C12 cells were cultured in conditioned media prepared by mixing organoid culture supernatants (MLO, MGO, MSO, MKO, or MBO) with low-glucose DMEM at a 1:1 ratio. Cell adhesion and proliferation were observed in both conditioned and growth media consisting of low-glucose DMEM supplemented with 10% FBS (Fig. 2A). Among the supernatants, MBOS significantly enhanced the proliferation of C2C12 cells and showed a growth-promoting effect comparable to that of FBS (Fig. 2A and B). Furthermore, a direct cell-counting assay using Trypan Blue exclusion also demonstrated a similar proliferative effect of MBOS, consistent with the results obtained from the PrestoBlue assay (Supplementary Fig. S1).

Effect of mouse organoid-derived CM on the proliferation of C2C12 cells Representative bright-field microscopic images of C2C12 myoblasts cultured in different media (A). Top row: Serum-free control medium (left) and growth medium containing 10% FBS (right). Bottom row: Representative images of cells cultured in CM prepared by mixing organoid culture supernatants at a 1:1 ratio with low-glucose DMEM. Scale bars: 100 µm. Quantitative analysis of C2C12 cell proliferation based on fluorescence intensity (B). Data are expressed as mean ± SEM. n = 6 for biological replicates. *P < 0.05 vs. Control.
To determine the optimal concentration of MBOS, we performed preliminary concentration–response experiments using MBOS-based media containing various proportions of mouse bladder organoid supernatant. MBOS maintained its proliferative activity even when diluted to 30% (Supplementary Fig. S2). To evaluate the reproducibility of MBOS preparation, independent batches were tested for their proliferative effects on C2C12 cells. Consistent activity was observed among batches, with a coefficient of variation (CV) of 9.7%, indicating high reproducibility and stability across preparations (Supplementary Fig. S3).
Mechanism by which MBOS promotes C2C12 cell growth
To investigate the mechanism by which MBOS promotes C2C12 cell proliferation, C2C12 myoblasts were cultured in a serum-free control medium, growth medium (with FBS), or MBOS-supplemented medium, and transcriptomic analysis was conducted. RNA sequencing data revealed substantial transcriptional alterations, with 3,748 genes significantly affected by FBS and 2,714 genes affected by MBOS. Of these, 2,317 genes were altered under both conditions (Fig. 3A). Hierarchical clustering of differentially expressed genes (DEGs) demonstrated condition-specific transcriptional landscapes (Fig. 3B). A volcano plot highlighted the significant DEGs between the control and MBOS-treated cells (Fig. 3C). The complete list of DEGs, including gene IDs, log₂ fold changes, and adjusted p-values, is provided in Supplementary Table S1. GSEA identified the enrichment of cell cycle-related pathways, including Hallmark E2F targets and G2/M checkpoint genes, in MBOS-treated cells compared to controls (Fig. 3D). GSEA revealed significant enrichment of the “HALLMARK_E2F_TARGETS” (NES = 3.65, FDR < 0.001) and HALLMARK_G2M_CHECKPOINT” (NES = 3.46, FDR < 0.001) pathways in MBOS-treated C2C12 cells, indicating strong activation of cell-cycle regulatory genes. The transcript levels of CCNB1 and CDK1 were markedly upregulated by MBOS treatment, as quantified by FPKM (Fig. 3E). These transcriptional changes were validated using qRT-PCR, which revealed elevated mRNA levels normalized to GAPDH (Fig. 3F). Western blotting confirmed the increased expression of CCNB1 and CDK1 proteins in MBOS-treated cells (Fig. 3G). These findings suggest that MBOS promotes cell cycle progression in C2C12 myoblasts, potentially enhancing their proliferative capacity by upregulating key regulators, such as CCNB1 and CDK1.

Mechanism of promotion of C2C12 cell proliferation by MBO supernatant (MBOS). RNA sequencing analysis of C2C12 myoblasts cultured in serum-free control medium, growth medium (FBS), and MBOS (A). Heatmap of differentially expressed genes (DEGs) in C2C12 cells cultured in control, FBS, and MBOS (B). Genes were selected based on an adjusted P-value < 0.05, and hierarchical clustering revealed distinct transcriptional profiles across conditions. Volcano plot illustrating the DEGs between C2C12 cells cultured in serum-free control medium and MBOS (C). Genes with adjusted P-value < 0.05 and absolute log₂ fold change > 1 were considered significant. Gene Set Enrichment Analysis (GSEA) comparing transcriptomic profiles under control and MBOS conditions (D). Transcript-level expression of CCNB1 and CDK1 in MBOS-treated cells, as measured by FPKM (E, n = 3) and quantified using PCR (F). The expression level of each gene was quantified based on the ratio of the expression level to GAPDH and is shown as a fold increase relative to the control. The results are expressed as mean ± SEM (n = 4). *P < 0.05 vs. Control. The expression of CCNB1 and CDK1 in C2C12 cells after MBOS treatment (24 h) was determined using western blot analysis (G). The expression levels were quantified and plotted based on the ratio of the expression level to that of the control (G, n = 5). The results are expressed as mean ± SEM. Equal protein loading was confirmed using the VCP antibody as a control. ∗ P < 0.05 vs. Control.
MBOS promotes cell cycle progression in C2C12 myoblasts
To investigate the effect of MBOS on cell cycle dynamics, flow cytometry was performed on C2C12 cells cultured under serum-free control and MBOS-supplemented conditions. Following culture, the cells were stained with PI to assess their DNA content and phase-specific distribution. Flow cytometry gates R2, R3, and R4 corresponded to the G1, S, and G2/M phases, respectively. The MBOS-treated cells exhibited a noticeable shift in the cell cycle profile, with a significant increase in the proportion of cells in the G2/M phase (R4), indicating enhanced cell cycle progression (Fig. 4A). Quantitative analysis confirmed a statistically significant increase in the G2/M cell population under MBOS conditions compared to the control, while the G1 and S phase distributions remained relatively unchanged (Fig. 4B). These findings suggest that MBOS facilitates C2C12 cell cycle progression, particularly by promoting the transition into and retention within the G2/M phase, which may contribute to the elevated proliferative activity observed after MBOS treatment.

Flow cytometric analysis of cell cycle progression in C2C12 cells cultured with MBOS. C2C12 myoblasts cultured under serum-free control conditions and MBOS were stained with propidium iodide (PI) to evaluate DNA content and cell cycle phase distribution using flow cytometry (A). Gates R2, R3, and R4 correspond to the G1, S, and G2/M phases, respectively. Quantification of cell populations in the G1 (R2), S (R3), and G2/M (R4) phases under control and MBOS conditions (B). Data are reported as mean ± SEM,n = 6,∗P < 0.05 vs. Control.
Effect of growth factors in MBOS on C2C12 cell proliferation
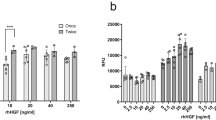
To elucidate the mechanisms underlying MBOS-induced cell proliferation, the concentrations of insulin-like growth factors IGF-1 and IGF-2 were measured in the serum-free control medium and MBOS-conditioned medium using ELISA. IGF-1 concentration was significantly higher in the MBOS medium than in the control, whereas IGF-2 concentration did not change (Fig. 5A). To assess whether IGF-1 production mediates MBOS-induced cell proliferation, anti-IGF-1 antibodies were added to the MBOS-conditioned medium, and C2C12 cell proliferation was subsequently evaluated. No significant reduction in cell proliferation was observed following antibody treatment, indicating that IGF-1 alone did not affect the proliferative effects of MBOS (Fig. 5B). These findings suggest that additional factors or signaling networks may be involved in mediating the full growth-promoting capacity of MBOS.

Effect of growth factors in MBOS on C2C12 proliferation. The concentrations of insulin-like growth factors (IGF)1 and IGF2 were measured in serum-free control and MBOS-conditioned media using ELISA (A, n = 5). Data are reported as mean ± SEM.∗P < 0.05 vs. Control. Effect of IGF1 inhibition on MBOS-induced C2C12 cell proliferation. Neutralizing anti-IGF1 antibody (0.01–1 μg/mL) was added to the MBOS medium for 72 h, and cell proliferation was evaluated (B, n = 6). Data are reported as mean ± SEM. ∗P < 0.05 vs. Control.
Effects of MBOS on proliferation of bovine muscle-derived primary myoblast cells
To determine whether MBOS exerts a similar proliferative effect on bovine muscle-derived primary myoblast cells, primary bovine muscle-derived cells were isolated from cows at the end of their productive life and cultured under MBOS-supplemented conditions (Fig. 6A). The isolated myoblasts showed increased cell density after seeding (Fig. 6B). We confirmed the expression of myogenic markers Myf5 and MyoD in the isolated myoblast cells (Fig. 6C). Cell count analysis demonstrated a significant increase in the total cell number in MBOS-treated samples compared to that in the serum-free controls (Fig. 6D). These findings show that MBOS promotes the proliferation of bovine primary muscle cells, indicating that its growth-promoting effects extend beyond established murine cell models.

Effect of MBOS on the proliferation of bovine muscle-derived primary myoblast cells. Schematic diagram illustrating the experimental design: primary cells were isolated from a cow and cultured to evaluate the proliferative effects of MBOS and the expression of myogenic markers (A). Bright-field images of bovine muscle-derived primary cells (B). Scale bar: 200 µm. Immunofluorescence staining of myogenic markers Myf5 and MyoD in bovine muscle-derived primary cells (C). Scale bar: 100 µm. Effect of MBOS on the proliferation of bovine muscle-derived primary myoblast cells (D, n=6). Cell proliferation was evaluated on day 3. Data are reported as mean ± SEM.∗P < 0.05 vs. Control.
Discussion
In this study, we aimed to contribute to the development of cultured meat by exploring a sustainable and effective alternative source of growth factors to replace FBS, which is one of the major challenges in the field. As a potential solution, we developed a conditioned medium derived from organoids of mouse bladder origin, referred to as MBOS. The main findings of this study are as follows: organoids were generated from five mouse organs: the lung, gallbladder, spleen, kidney, and bladder (Fig. 1). Conditioned medium was prepared by mixing the culture supernatants of these organoids with a basal medium devoid of serum components, and its effect on C2C12 myoblast proliferation was evaluated. All five types of conditioned media significantly promoted cell proliferation compared to the control, suggesting their potential as alternatives to FBS, with MBOS exhibiting the most pronounced effect (Fig. 2). Transcriptomic analysis of C2C12 cells cultured with MBOS revealed a significant upregulation of the cell cycle-related genes CCNB1 and CDK1. This increase was confirmed at the mRNA and protein levels using western blot analysis (Fig. 3). Furthermore, flow cytometric analysis showed a significantly higher proportion of cells in the G2/M phase in the MBOS-treated group than in the control group, suggesting that MBOS may activate cell cycle progression in C2C12 cells (Fig. 4). Although IGF-1 was present at significantly higher levels in MBOS cells, proliferation assays conducted with IGF-1-neutralizing antibodies did not show a significant suppression of cell growth (Fig. 5). MBOS treatment significantly promoted bovine myoblast proliferation (Fig. 6). These results demonstrate that MBOS can enhance the proliferation of both C2C12 cells and primary bovine myoblasts, suggesting its potential as a novel medium component to replace FBS.
In this proof-of-concept study, murine-derived organoids were employed because they provided a well-characterized and experimentally tractable model system, supported by extensive background information and available analytical tools. This approach enabled the establishment and optimization of the conditioning protocol with high reproducibility. In contrast, bovine tissues were not continuously available at the laboratory scale, making murine organoids the most practical model at the early stage of this research. Future studies will apply this approach to species-specific organoids derived from various animal sources to enhance physiological relevance and reduce cross-species variability.
Among the organoids tested, bladder organoids consistently showed the strongest and most stable proliferative activity with relatively low batch-to-batch variation (quantified as coefficient of variation; Supplementary Figure S3). This may be attributed to the unique characteristics of the bladder epithelium, which is constantly exposed to a chemically harsh urinary environment and has evolved to maintain barrier integrity under such stress conditions. Such intrinsic tolerance and stable secretory characteristics could contribute to the consistent biological activity observed in MBOS. In addition, bladder organoids are known to exhibit a stratified epithelial architecture containing CK5⁺ basal cells and UPK3A⁺ umbrella cells, both of which play important roles in epithelial regeneration and barrier maintenance. These cellular and functional features may partially underlie the stable proliferative effects of MBOS observed in this study.
Several serum substitutes, such as human platelet lysate, algae-derived extracts, and insect hydrolysates, have been developed and have greatly contributed to the advancement of serum-free and cultured-meat research. These alternatives effectively promote cell proliferation and play an important role in reducing FBS dependency. Although we did not perform direct experimental comparisons with these established materials, our goal was not to replace them but to complement existing strategies by introducing a new concept: the use of organoid-derived conditioned medium as a biologically active and sustainable supplement. We have positioned MBOS as a reproducible and physiologically relevant supplement that may broaden the options available for serum-free culture media design.
Combination experiments using conditioned media from multiple organoid types (Supplementary Fig. S4) showed slight additive effects on C2C12 proliferation, although no synergistic or optimal combination was identified.
Furthermore, combining multiple organoids can improve the physiological relevance of the secretory environment44, and research using composite organoid systems has advanced our understanding of how inter-organ interactions influence cellular functions45. Secreted factors derived from different organs may act cooperatively in cell cycle progression, metabolism, and differentiation, and the physiological significance of inter-organ crosstalk is suggested to play a crucial role in regulating cellular functions46. In the current study, we examined the proliferative effect of mixed conditioned media derived from five different organs on C2C12 cells; however, we could not identify the optimal combination. Nevertheless, even under conditions of stepwise dilution of MBOS, a certain level of proliferation was maintained, suggesting that optimizing the composition of conditioned medium may enable the design of cost-effective culture media. These findings suggest that optimizing the composition and mixing ratios of secreted factors may be an important subject for future investigations into the development of FBS-free culture media.
Several growth factors regulate both the proliferation and differentiation of skeletal muscles, including fibroblast growth factor-247,48,49,50,51, IGFs19,47,51,52, PDGF47,49,53, and LIF54. In the current study, IGF-1 was significantly enriched in the MBOS group compared to the control group; however, the proliferative effect observed could not be fully explained by IGF-1 alone. This suggests that multiple growth factors are required to cooperatively support skeletal muscle proliferation and regeneration. Further elucidation of the mechanisms underlying the proliferative effects of MBOS, including its interactions with the extracellular matrix and the coordinated action of multiple growth factors, may provide valuable insights into its potential applications. However, complex formulations such as MBOS likely contain unidentified bioactive components that could not be explained by IGF-1 alone. Therefore, future studies, including proteomic profiling of MBOS and investigation of IGF-binding proteins (IGFBPs), will be essential to identify the core active factors and establish a more complete mechanistic link between MBOS composition and myoblast cell-cycle regulation.
To ensure that the conditioned media used in this study were derived from authentic tissue-specific organoids, we confirmed organ identity in all five organoid types through immunostaining with organ-specific markers. Future studies that incorporate analyses of organoid maturation, such as ultrastructural and transcriptional profiling, may further clarify tissue-specific differences in secretome composition and deepen our understanding of organoid-derived factors. Independent MBOS batches were also tested for their proliferative effects on C2C12 cells to evaluate preparation reproducibility. The coefficient of variation (CV) among batches was 9.7%, indicating high preparation stability and consistency in biological activity (Supplementary Fig. 3).
While every effort was made to minimize potential carryover from the growth medium during the preparation of MBOS, complete elimination of animal-origin components remains technically challenging in conventional organoid culture systems. Trace contamination from basal media such as Advanced DMEM/F12 or protein adsorption on the organoid and matrix surfaces could not be entirely excluded. This limitation is important for interpreting the biological activity of MBOS as being truly organoid-derived rather than medium-derived. Future studies will include quantitative evaluation of residual albumin or other bovine proteins (e.g., by ELISA or proteomic analysis) and the development of improved culture systems using food-grade, animal-component-free media and matrices, which will further enhance the scientific rigor and biosafety transparency of MBOS-based applications.
Although the purity of primary bovine myoblasts was ensured through enzymatic dissociation, pre-plating, and collagen-coated culture, following previously validated protocols, quantitative flow cytometric verification (e.g., MyoD⁺/CD56⁺) was not performed in this study. Future studies incorporating such quantitative analyses will further strengthen the reproducibility and methodological rigor of primary cell experiments. Furthermore, while cell proliferation in this study was primarily evaluated using the metabolic PrestoBlue assay, the results were consistent with direct cell-counting data obtained in our preliminary comparison. Nonetheless, combining both metabolic and direct quantification approaches in future studies will further enhance the robustness of proliferation assessment and methodological reproducibility.
The present study mainly focused on the short-term proliferative activity of MBOS, since efficient myoblast proliferation is a key factor for the development of serum-free media applicable to cell-based meat production. Future research topics include whether proliferation-promoting effects and differentiation control remain achievable during long-term culture. Nevertheless, our findings in this study highlight that MBOS represents a complex yet reproducible bioactive system, whose multifactorial interactions offer valuable insights into the design of future serum-free culture media.
Data availability
The raw data from RNA sequencing are available in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) repository, (BioProject ID: PRJNA1310213, BioSample accessions: SAMN50770538, SAMN50770539, SAMN50770540, SAMN50770541, SAMN50770542, SAMN50770543, SAMN50770544, SAMN50770545, SAMN50770546).
References
Good Food Institute. 2023 State of the Industry Report-Cultivated meat and seafood, < https://gfi.org/wp-content/uploads/2024/04/2023-State-of-the-Industry-Report-Cultivated-meat-and-seafood.pdf> (2024).
Kumar, P. et al. In-vitro meat: a promising solution for sustainability of meat sector. J. Anim. Sci. Technol. 63, 693–724. https://doi.org/10.5187/jast.2021.e85 (2021).
Lynch, J. & Pierrehumbert, R. Climate impacts of cultured meat and beef cattle. Front. Sustain. Food Syst. https://doi.org/10.3389/fsufs.2019.00005 (2019).
Treich, N. Cultured meat: promises and challenges. Environ. Resour. Econ. (Dordr) 79, 33–61. https://doi.org/10.1007/s10640-021-00551-3 (2021).
Tuomisto, H. L. & de Mattos, M. J. Environmental impacts of cultured meat production. Environ. Sci. Technol. 45, 6117–6123. https://doi.org/10.1021/es200130u (2011).
Floor, J. & Manlosa-Kirk, A. Exploring the sustainability narratives of cultured meat. J. Clean. Prod. 412, 137302 (2025).
Tavan, M., Smith, N. W., McNabb, W. C. & Wood, P. Reassessing the sustainability promise of cultured meat: a critical review with new data perspectives. Crit. Rev. Food Sci. Nutr. https://doi.org/10.1080/10408398.2025.2461262 (2025).
Duarte Rojas, J. M., Restrepo Munera, L. M. & Estrada Mira, S. Comparison between platelet lysate platelet lysate serum, and fetal bovine serum as supplements for cell culture expansion, and cryopreservation. Biomedicines https://doi.org/10.3390/biomedicines12010140 (2024).
Lee, D. Y. et al. Review of the current research on fetal bovine serum and the development of cultured meat. Food Sci. Anim. Resour. 42, 775–799. https://doi.org/10.5851/kosfa.2022.e46 (2022).
Pilgrim, C. R. et al. A review of fetal bovine serum in the culture of mesenchymal stromal cells and potential alternatives for veterinary medicine. Front. Vet. Sci. 9, 859025. https://doi.org/10.3389/fvets.2022.859025 (2022).
Subbiahanadar Chelladurai, K. et al. Alternative to FBS in animal cell culture - An overview and future perspective. Heliyon 7, e07686. https://doi.org/10.1016/j.heliyon.2021.e07686 (2021).
Urzi, O., Olofsson Bagge, R. & Crescitelli, R. The dark side of foetal bovine serum in extracellular vesicle studies. J. Extracell. Vesicles 11, e12271. https://doi.org/10.1002/jev2.12271 (2022).
Barro, L. et al. Human platelet lysates for human cell propagation. Platelets 32, 152–162. https://doi.org/10.1080/09537104.2020.1849602 (2021).
Jafar, H. et al. hPL promotes osteogenic differentiation of stem cells in 3D scaffolds. PLoS One 14, e0215667. https://doi.org/10.1371/journal.pone.0215667 (2019).
Palombella, S. et al. Systematic review and meta-analysis on the use of human platelet lysate for mesenchymal stem cell cultures: comparison with fetal bovine serum and considerations on the production protocol. Stem Cell Res. Ther. 13, 142. https://doi.org/10.1186/s13287-022-02815-1 (2022).
Talpan, D. et al. Cytoprotective effects of human platelet lysate during the xeno-free culture of human donor corneas. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24032882 (2023).
Eisenberg, H., Hutker, S., Berger, F. & Lang, I. Native proteins from Galdieria sulphuraria to replace fetal bovine serum in mammalian cell culture. Appl. Microbiol. Biotechnol. 109, 119. https://doi.org/10.1007/s00253-025-13507-0 (2025).
Sibincic, N. et al. Screening algal and cyanobacterial extracts to identify potential substitutes for fetal bovine serum in cellular meat cultivation. Foods https://doi.org/10.3390/foods13233741 (2024).
Yamanaka, K., Haraguchi, Y., Takahashi, H., Kawashima, I. & Shimizu, T. Development of serum-free and grain-derived-nutrient-free medium using microalga-derived nutrients and mammalian cell-secreted growth factors for sustainable cultured meat production. Sci. Rep. 13, 498. https://doi.org/10.1038/s41598-023-27629-w (2023).
Mekhaimar, A. S., El Asely, A. M., Negm, S. & Shaheen, A. Evaluation of antibacterial properties of Laurencia obtusa and Cystoseira barbata against some bacterial fish pathogens. Benha Vet. Med. J. 46, 97–101. https://doi.org/10.21608/bvmj.2024.266909.1781 (2024).
Batish, I., Zarei, M., Nitin, N. & Ovissipour, R. Evaluating the potential of marine invertebrate and insect protein hydrolysates to reduce fetal bovine serum in cell culture media for cultivated fish production. Biomolecules https://doi.org/10.3390/biom12111697 (2022).
Liu, L. et al. Systematic evaluation of sericin protein as a substitute for fetal bovine serum in cell culture. Sci. Rep. 6, 31516. https://doi.org/10.1038/srep31516 (2016).
Park, S. H. et al. Effects of black soldier fly larvae hydrolysate on culture of primary myogenic and adipogenic cells isolated from broilers for cultured meat development. Foods https://doi.org/10.3390/foods14040678 (2025).
Marigliani, B., Balottin, L. B. L. & Augusto, E. F. P. Adaptation of mammalian cells to chemically defined media. Curr. Protoc. Toxicol. 82, e88. https://doi.org/10.1002/cptx.88 (2019).
van der Valk, J. et al. Optimization of chemically defined cell culture media–replacing fetal bovine serum in mammalian in vitro methods. Toxicol. In Vitro 24, 1053–1063. https://doi.org/10.1016/j.tiv.2010.03.016 (2010).
Rafnsdóttir, Ó. B. et al. A new animal product free defined medium for 2D and 3D culturing of normal and cancer cells to study cell proliferation and migration as well as dose response to chemical treatment. Toxicol. Rep. 10, 509–520. https://doi.org/10.1016/j.toxrep.2023.04.001 (2023).
Buss, L. F. et al. Conditioned media from human pulp stem cell cultures improve bone regeneration in rat calvarial critical-size defects. J. Funct. Biomater. https://doi.org/10.3390/jfb14080396 (2023).
Kim, M. H. et al. Galectin-1 from conditioned medium of three-dimensional culture of adipose-derived stem cells accelerates migration and proliferation of human keratinocytes and fibroblasts. Wound Repair Regen. 26(Suppl 1), S9–S18. https://doi.org/10.1111/wrr.12579 (2018).
Kim, M. H. et al. Conditioned medium from the three-dimensional culture of human umbilical cord perivascular cells accelerate the migration and proliferation of human keratinocyte and fibroblast. J. Biomater. Sci. Polym. Ed. 29, 1066–1080. https://doi.org/10.1080/09205063.2017.1340045 (2018).
Miceli, V. et al. Comparison of immunosuppressive and angiogenic properties of human amnion-derived mesenchymal stem cells between 2D and 3D culture systems. Stem Cells Int. 2019, 7486279. https://doi.org/10.1155/2019/7486279 (2019).
Gunther, C., Winner, B., Neurath, M. F. & Stappenbeck, T. S. Organoids in gastrointestinal diseases: from experimental models to clinical translation. Gut 71, 1892–1908. https://doi.org/10.1136/gutjnl-2021-326560 (2022).
Kuhn, M. R., Wolcott, E. A. & Langer, E. M. Developments in gastrointestinal organoid cultures to recapitulate tissue environments. Front. Bioeng. Biotechnol. 13, 1521044. https://doi.org/10.3389/fbioe.2025.1521044 (2025).
Elbadawy, M. et al. Establishment of an experimental model of normal dog bladder organoid using a three-dimensional culture method. Biomed. Pharmacother. 151, 113105. https://doi.org/10.1016/j.biopha.2022.113105 (2022).
Elbadawy, M. et al. Establishment of a novel experimental model for muscle-invasive bladder cancer using a dog bladder cancer organoid culture. Cancer Sci. 110, 2806–2821. https://doi.org/10.1111/cas.14118 (2019).
Usui, T. et al. Establishment of a dog primary prostate cancer organoid using the urine cancer stem cells. Cancer Sci. 108, 2383–2392. https://doi.org/10.1111/cas.13418 (2017).
Abugomaa, A. et al. Establishment of a direct 2.5D organoid culture model using companion animal cancer tissues. Biomed. Pharmacother. 154, 113597. https://doi.org/10.1016/j.biopha.2022.113597 (2022).
Elbadawy, M. et al. Efficacy of primary liver organoid culture from different stages of non-alcoholic steatohepatitis (NASH) mouse model. Biomaterials 237, 119823. https://doi.org/10.1016/j.biomaterials.2020.119823 (2020).
Nagashima, Y. et al. Establishment of an experimental model of canine apocrine gland anal sac adenocarcinoma organoid culture using a three-dimensional culture method. Sci. Rep. 15, 6108. https://doi.org/10.1038/s41598-025-90623-x (2025).
Kanaya, A. et al. Generation and characterization of feline colorectal adenocarcinoma organoids as a preclinical model. J. Vet. Med. Sci. 87, 752–762. https://doi.org/10.1292/jvms.24-0499 (2025).
Abugomaa, A. et al. Establishment of a direct 2.5D organoid culture model using companion animal cancer tissues. Biomed. Pharmacother. 154, 113597. https://doi.org/10.1016/j.biopha.2022.113597 (2022).
Shiota Sato, Y. et al. Derivation of a new model of lung adenocarcinoma using canine lung cancer organoids for translational research in pulmonary medicine. Biomed. Pharmacother. 165, 115079. https://doi.org/10.1016/j.biopha.2023.115079 (2023).
Elbadawy, M. et al. Evaluation of the efficacy of mitochondrial fission inhibitor (Mdivi-1) using non-alcoholic steatohepatitis (NASH) liver organoids. Front. Pharmacol. https://doi.org/10.3389/fphar.2023.1243258 (2023).
Usui, T. et al. Establishment of a novel three-dimensional primary culture model for hippocampal neurogenesis. Physiol. Rep. https://doi.org/10.14814/phy2.13318 (2017).
Chen, Z., Sugimura, R., Zhang, Y. S., Ruan, C. & Wen, C. Organoids in concert: engineering in vitro models toward enhanced fidelity. Aggregate 5, e478. https://doi.org/10.1002/agt2.478 (2024).
Ni, B., Ye, L., Zhang, Y., Hu, S. & Lei, W. Advances in humanoid organoid-based research on inter-organ communications during cardiac organogenesis and cardiovascular diseases. J. Transl. Med. 23, 380. https://doi.org/10.1186/s12967-025-06381-x (2025).
Denechaud, P. D. & Rabhi, N. Editorial: Intra and inter organ cross-talk and cellular communication. Front. Endocrinol. (Lausanne) 14, 1209436. https://doi.org/10.3389/fendo.2023.1209436 (2023).
Doumit, M. E., Cook, D. R. & Merkel, R. A. Fibroblast growth factor, epidermal growth factor, insulin-like growth factors, and platelet-derived growth factor-BB stimulate proliferation of clonally derived porcine myogenic satellite cells. J. Cell Physiol. 157, 326–332. https://doi.org/10.1002/jcp.1041570216 (1993).
Greene, E. A. & Allen, R. E. Growth factor regulation of bovine satellite cell growth in vitro. J. Anim. Sci. 69, 146–152. https://doi.org/10.2527/1991.691146x (1991).
McFarland, D. C., Pesall, J. E. & Gilkerson, K. K. The influence of growth factors on turkey embryonic myoblasts and satellite cells in vitro. Gen. Comp. Endocrinol. 89, 415–424. https://doi.org/10.1006/gcen.1993.1049 (1993).
Shima, A., Itou, A. & Takeuchi, S. Cell fibers promote proliferation of co-cultured cells on a dish. Sci. Rep. 10, 288. https://doi.org/10.1038/s41598-019-57213-0 (2020).
Wilkie, R. S., O’Neill, I. E., Butterwith, S. C., Duclos, M. J. & Goddard, C. Regulation of chick muscle satellite cells by fibroblast growth factors: interaction with insulin-like growth factor-I and heparin. Growth Regul. 5, 18–27 (1995).
Oksbjerg, N., Gondret, F. & Vestergaard, M. Basic principles of muscle development and growth in meat-producing mammals as affected by the insulin-like growth factor (IGF) system. Domest Anim. Endocrinol. 27, 219–240. https://doi.org/10.1016/j.domaniend.2004.06.007 (2004).
Yablonka-Reuveni, Z. & Seifert, R. A. Proliferation of chicken myoblasts is regulated by specific isoforms of platelet-derived growth factor: evidence for differences between myoblasts from mid and late stages of embryogenesis. Dev. Biol. 156, 307–318. https://doi.org/10.1006/dbio.1993.1079 (1993).
Spangenburg, E. E. & Booth, F. W. Multiple signaling pathways mediate LIF-induced skeletal muscle satellite cell proliferation. Am. J. Physiol. Cell Physiol. 283, C204-211. https://doi.org/10.1152/ajpcell.00574.2001 (2002).
Author information
Authors and Affiliations
Contributions
Yuko Nagashima, Haru Yamamoto, Mohamed Elbadawy, Yoshiko Naito, and Amira Abugomaa performed the experiments. Tatsuya Usui and Kazuaki Sasaki designed the study, analyzed, and interpreted the data. Masahiro Kaneda, Tatsuya Usui, Mohamed Elbadawy, and Kazuaki Sasaki wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nagashima, Y., Yamamoto, H., Elbadawy, M. et al. Bladder organoid conditioned media enhances myoblast proliferation under serum free conditions. Sci Rep 16, 7582 (2026). https://doi.org/10.1038/s41598-026-38603-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-38603-7
