
Abstract
We developed a computational pathology pipeline to extract and analyze collagen disorder architecture (CoDA) features from whole slide images (WSIs) of 2,212 colon cancer (CC) patients across multiple institutions. CoDA features—capturing collagen fragmentation, bundling, anisotropy, density, and rigidity, were evaluated for associations with clinical variables (overall stage, T/N/M stage), molecular classifications (Consensus Molecular Subtypes [CMS1–4]), and genetic mutations (KRAS, BRAF, NRAS) using the Mann-Whitney U test with Bonferroni correction. These analyses revealed significant differences in CoDA feature distributions across multiple subgroups, suggesting that collagen architecture varies meaningfully with tumor stage, molecular subtype, and mutation status.To assess how well CoDA features could distinguish between these subgroups, we implemented a Random Forest classification framework. High mean AUC values (≥0.7) across several variables indicated strong discriminatory performance of CoDA features in separating clinically and biologically distinct groups.
For survival analysis, LASSO-Cox models were trained on the PLCO dataset to generate CoDA-based risk scores for overall survival (OS) and disease-free survival (DFS), which were used to stratify patients into high- and low-risk groups in a combined validation dataset (TCGA, UH, and Emory). Kaplan-Meier curves demonstrated significant survival differences across clinical stages, CMS subtypes, and KRAS mutation status. Multivariable Cox proportional hazards models further confirmed the independent prognostic value of CoDA features after adjusting for clinical, molecular, and genetic covariates. These findings highlight that CoDA features are significantly associated with key clinical and molecular characteristics, can distinguish relevant patient subgroups, and offer independent prognostic information, underscoring their potential utility in characterizing the tumor microenvironment and informing risk stratification in CC.
Similar content being viewed by others


Introduction
Colon cancer (CC) is the third most diagnosed cancer and the second leading cause of cancer-related deaths in the United States1. The American Joint Committee on Cancer (AJCC) TNM classification (Tumor, Node, Metastasis) is the primary prognostic tool for guiding treatment decisions2. The five-year survival rate for stages I–III CC can reach up to 80%, while it drops to just 13% for stage IV, which accounts for approximately 20% of all CC cases3. Among 37,769 AJCC stage I–III CC patients, Black patients had significantly lower survival than White patients (Hazard Ratio (HR) = 1.24; 95% Confidence Interval (CI): 1.14–1.35)4. Furthermore, a study of 52,882 metastatic CC patients found that younger women (18–44 years old) had better Overall Survival (OS) than their male counterparts, whereas older women (55 years and older) had significantly worse OS compared to older men5.
In metastatic CC, where the five-year OS is only 14%, Rat sarcoma virus (RAS) oncogene mutations, including alterations in Kirsten rat sarcoma virus (KRAS), neuroblastoma RAS viral oncogene homolog (NRAS), and B-Raf proto-oncogene (BRAF), are identified in about half of the patients and contribute to poorer disease prognosis6. A study on CC patients indicated resistance to therapies targeting the RAS/Rapidly Accelerated Fibrosarcoma (RAF)/ Mitogen-activated protein kinase kinase (MEK)/Extracellular Signal-Regulated Kinase (ERK) pathway, especially in cancer stem cells driven by SRC activation7. Analysis of 2250 CC tumors revealed a strong correlation between resistance to MEK inhibitors and SRC activation, which is linked to poor prognosis, epithelial-mesenchymal transition (EMT), and metastasis8. Moreover, CC has been classified into four Consensus Molecular Subtypes (CMS1–4), with CMS4, characterized by mesenchymal features such as high collagen content, stromal invasion, and angiogenesis, associated with worse outcomes8. These findings suggest that combining MEK and SRC inhibitors may be an effective strategy for targeting RAS-mutant CCs, especially in specific CMS subtypes.
Increasing attention is being paid to the tumor microenvironment (TME) in CC treatment, as interactions between cellular and structural components of the TME play a crucial role in cancer progression and metastasis9,10. The extracellular matrix (ECM) within the TME serves as a structural scaffold that coordinates signaling pathways essential for cancer cell behavior and survival11,12. The composition and remodeling of ECM components, particularly collagen, significantly influence the dynamics of the TME and impact cancer survival outcomes13. Collagen deposition, or desmoplasia, in CC is a recognized but difficult-to-quantify prognostic feature14. A TME collagen signature (CSTME) has been shown to be an independent prognostic risk factor in stage II and III CC patients15. Because TME quantification is not routinely performed by pathologists, a computational pathology framework could enhance risk stratification, prognosis prediction, and treatment planning16.
Collagen fiber (CF) architecture within the TME is emerging as a promising feature for correlating with survival outcomes in CC patients17. A study identified 294 genes upregulated in KRAS-mutated CC samples, with COL1A1 (Collagen type I alpha 1) being a key gene18. Inhibition of COL1A1 significantly reduced cell proliferation and invasion in KRAS-mutant cell lines, and higher COL1A1 expression was linked to serosal invasion and metastasis18. Collagen also plays a critical role in tumor progression, malignancy, and drug resistance19. A deeper exploration of CF features in the TME could improve predictions of drug response and identify patients likely to benefit from combined MEK and SRC inhibitor therapies8.
For CC patients, particularly those with metastasis, genomic profiling has proven valuable in identifying RAS mutations that are prognostic of survival and treatment response20. Genomic testing is commonly used to identify RAS mutations and inform treatment decisions, such as the use of anti–epidermal growth factor receptor monoclonal antibodies like cetuximab and panitumumab21. However, genomic testing can be time-consuming and costly, particularly in lower- and middle-income countries22. Computational tools, on the other hand, offer the advantage of detecting RAS mutations directly from histopathology slides, offering a more accessible and cost-effective approach23. Biomarkers, including tumor-infiltrating lymphocytes and components of the TME, are visually apparent in whole slide images (WSIs), and the use of computational pathology has become increasingly important for prognostic applications in CC24. Deep learning is a subfield of Artificial Intelligence (AI) that has been employed to not only diagnose CC directly from WSIs but also has been shown to predict the presence of KRAS, tumor protein p53 (TP53), and BRAF mutations on routine hematoxylin and eosin (H&E) images with an area under the receiver operating curve (AUC) ranging from 0.73 to 0.8625. However, a challenge with deep learning approaches is the lack of transparency regarding the representations derived from the networks, making it difficult to explain the predictions. While the lack of explainability might not be an issue for diagnostic decision making, these black box models might not be as appealing for prognostic or treatment response prediction applications26.
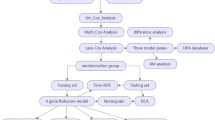
Recent studies across various cancer types have demonstrated that CF organization and orientation differ between benign and malignant tissues and are linked to disease stage, prognosis, and treatment response16,27. However, most prior studies have focused on collagen deposition or fibrosis, without quantitatively assessing CF architectural features within the TME17. Given the pivotal role of collagen in modulating tumor stiffness, cell motility, and drug resistance, a more detailed characterization of CF organization could offer valuable insights28,29,30. In this study, we introduce a computational pathology-based framework to extract novel collagen disorder architectural (CoDA) features from H&E-stained WSIs in CC patients. Unlike conventional methods based on molecular profiling or bulk collagen quantification, our approach analyzes CF fragmentation, bundling, anisotropy, density, and rigidity as potential biomarkers of cancer progression and treatment response. In this study, we investigated CoDA features in a multi-institutional cohort of 2212 CC patients, focusing on their associations with key clinical variables (overall stage, T/N/M stage classifications), molecular subtypes (Consensus Molecular Subtypes CMS1–4), and genetic mutations (KRAS, BRAF, NRAS). In addition to assessing statistical associations, we evaluated how well CoDA features could distinguish between these clinically and biologically relevant subgroups. Furthermore, we explored the prognostic relevance of CoDA features by analyzing their ability to stratify patients into high- and low-risk groups for overall survival (OS) and disease-free survival (DFS). Together, these analyses aim to improve our understanding of tumor–stroma interactions and demonstrate the potential of interpretable, image-derived biomarkers for enhancing risk stratification and guiding treatment decisions in CC. The workflow of this study, including CoDA feature extraction and statistical analysis, is illustrated in Fig. 1.

(1) CoDA FEATURE CALCULATION METHDOLOGY: (A) Whole Slide Image (WSI) from colon cancer patient (B) Tumor mask+HistoQC mask overlaid on WSI (C) Tiling of the WSI from the subtracted masked region (D) Example tiles (E) Collagen fibers within the stroma region of exmaple tile detected by a derivative-of-Gaussian (DtG) based model (F) Collagen fiber fragmentation (G) Collagen fiber bundling (H) Collagen fiber rigidity (I) Collagen fiber anisotropy. As the anisotropy values increase, the colormap transitions to green color at the highest end of the scale (corresponding to well aligned or anisotropic orientation). J Collagen fiber density. Hotter colors such as red and yellow represent higher density values while cooler colors such as blue and green represent lower density values in the density scale. K Cropped portion of Collagen fiber fragmentation (L) Cropped portion of Collagen fiber bundling (M) Cropped portion of Collagen fiber rigidity (N) Representation of tumor region tiles with example CoDA feature: Density, restitched back into their corresponding positions within the WSI. (2) STATISTICAL ANALYSIS: (A) Violin plots showing distribution across KRAS,BRAF and NRAS mutational variables for the individual CoDA features i.e., CF Fragmentation, CF Bundling, CF Anisotropy, CF Density and CF Rigidity. Significant differences between the groups (example: KRAS wild type vs KRAS mutated) were calculated by Mann–Whitney U test with Bonferroni corrected p values (*p) reported. Similar tests were done for clinical (Overall Stage, T Stage, N Stage and M Stage) and molecular variables (CMS1-4). Example Kaplan Meier plots showing risk stratified high and low risk groups, as derived using CoDA features using LASSO Cox proportional Hazards Model, for KRAS wild type for (B) Overall Survival (OS) and (C) Disease Free Survival (DFS). Risk stratification was done for other mutational variables (KRAS mutated, BRAF wild type and mutated and NRAS wild type and mutated) alongwith clinical and molecular variables. D To show CoDA as independent prognostic features, Multivariable Cox Proportional Hazards Analysis was done for OS and DFS for clinical, molecular and genetic variables alongwith variables like age, gender and race. Hazard ratios (HR) with corresponding 95% confidence intervals (CI) are presented.
Results
Quantative evaluation of CF and coda features
The accuracy of CF detection was visually assessed independently by an attending pathologist and resident pathologist on 200 random tiles from randomly selected WSIs. The pathologists assigned the tiles to one of four categories (excellent, good, fair, or poor) to describe the accuracy of the captured CF orientations. Inter-rater agreement was quantified using the Intraclass Correlation Coefficient (ICC), which measured the consistency between the pathologists’ ratings and yielded an ICC of 0.64, indicating moderate agreement and supporting the reproducibility of CF detection accuracy31,32. Examples of the four categories of CF detection accuracy by the two independent pathologists in example tiles are provided in Supplementary Fig. 1. A visual representation of how CoDA features differ across the KRAS mutational variable (i.e., KRAS wild type vs. KRAS mutant) is shown in the example tiles of Supplementary Fig. 2. Increased fragmentation (Supplementary Fig. 2C, H) and stiffness (Supplementary Fig. 2E, J) of the CF is seen in KRAS mutant as compared to KRAS wild type. In addition, thicker bundles (Supplementary Fig. 2D, I) along with denser aggregation (Supplementary Fig. 2G) and straight alignment (Supplementary Fig. 2F) of the CF were seen for the KRAS mutant than for the KRAS wild type.
Associations between Coda features and clinical, molecular, and genetic subgroups
The distribution of CoDA features was visualized using violin plots across groups defined by clinical variables, including detailed subgroup comparisons such as Stage I–II vs. Stage III–IV, T1 vs. T2, N0 vs. N + , and M0 vs. M + , molecular subtypes through all pairwise comparisons among CMS1, CMS2, CMS3, and CMS4 (i.e., CMS1 vs. CMS2, CMS3, CMS4; CMS2 vs. CMS1, CMS3, CMS4; CMS3 vs. CMS1, CMS2, CMS4; and CMS4 vs. CMS1, CMS2, CMS3), and genetic mutations based on the following comparisons-(KRAS-positive vs. KRAS-negative, BRAF-positive vs. BRAF-negative, and NRAS-positive vs. NRAS-negative). Statistical differences between groups were assessed using the Mann–Whitney U test33,34. The resulting p-values, along with Bonferroni-corrected p-values (*p)35,36,37, are presented in Table 1, demonstrating associations between CoDA features and each variable.
Figures 2–4 display these associations across the different categories: clinical variables in Fig. 2, molecular subtypes in Fig. 3, and genetic mutations in Fig. 4. Notable differences were observed, with all five CoDA features (CF fragmentation, CF bundling, CF anisotropy, CF density, and CF rigidity) showing significant variation across several clinical subgroup comparisons. Specific molecular subtypes were significantly associated with CF bundling and CF anisotropy, while CF rigidity showed notable differences related to genetic mutation status.

Violin plots depict differences in collagen architectural features—CF Fragmentation, CF Bundling, CF Anisotropy, CF Density, and CF Rigidity—across clinical subgroups: (A) Overall Stage (I–II vs. III–IV), (B) T Stage (T1–2 vs. T3–4), (C) N Stage (N0 vs. N + ), and (D) M Stage (M0 vs. M1). Statistical significance was assessed with Bonferroni-adjusted p-values (*p). Significant differences were observed in CF Fragmentation, CF Anisotropy, and CF Rigidity for Overall Stage; CF Anisotropy, CF Density, and CF Rigidity for T Stage; all CoDA features for N Stage; and CF Fragmentation and CF Bundling for M Stage. These findings demonstrate a consistent association between collagen architectural features and clinical staging, highlighting the potential of CoDA features as non-invasive biomarkers of tumor progression and metastatic potential.

CoDA feature variations represented by violin plots are compared across molecular subgroups: (A) CMS1 vs. non-CMS1 (CMS2, CMS3, CMS4), (B) CMS2 vs. non-CMS2 (CMS1, CMS3, CMS4), (C) CMS3 vs. non-CMS3 (CMS1, CMS2, CMS4), and (D) CMS4 vs. non-CMS4 (CMS1, CMS2, CMS3). p-values were corrected for multiple testing using the Bonferroni method (*p). Significant differences were found in CF Fragmentation and CF Anisotropy for CMS1; CF Bundling for CMS2; CF Bundling, CF Density, and CF Rigidity for CMS3; and CF Anisotropy for CMS4. These results highlight distinct collagen architectural patterns associated with each CMS subtype, suggesting that CoDA features may reflect underlying biological differences between molecular subtypes of colon cancer.

Violin plots demonstrate differences in CoDA features among mutational subgroups defined by KRAS, BRAF, and NRAS mutation status: (A) KRAS wild type vs. mutated, (B) BRAF wild type vs. mutated, and (C) NRAS wild type vs. mutated. Multiple comparison corrections were applied via Bonferroni adjustment (*p). Significant differences were observed across all CoDA features between BRAF wild type and mutated groups. In KRAS, all features except CF Fragmentation differed significantly between wild type and mutant tumors. For NRAS, only CF Rigidity showed a significant difference. These findings suggest that specific collagen structural patterns captured by CoDA features are associated with mutation status, underscoring their potential relevance in characterizing tumor microenvironment differences linked to oncogenic mutations.
To further evaluate the ability of CoDA features to distinguish between these refined groups, we employed a Random Forest classification approach38,39 across 100 iterations and reported classification metrics including mean AUC values with standard deviations40, F1 scores41, and accuracy41. These results are summarized in Table 2, providing a comparative overview of how effectively CoDA feature patterns discriminate among patient subgroups. The observation of mean AUC values ≥ 0.7 across multiple variables indicates strong discriminative performance of CoDA features in differentiating clinical, molecular, and genetic subgroups.
These findings highlight the relevance of CoDA features in capturing distinct aspects of the tumor microenvironment that correspond to key clinical, molecular, and genetic characteristics.
Risk stratification using coda features across clinical, molecular and genetic subgroups
Risk stratification based on CoDA features was evaluated for both overall survival (OS) and disease-free survival (DFS) in the combined validation cohort (TCGA, UH, and Emory datasets). Risk scores were computed using LASSO-Cox proportional hazards models trained separately for OS and DFS in the PLCO dataset, with the median risk score used as the cutoff for defining high- and low-risk groups42,43.
Kaplan-Meier (KM) survival curves were generated to assess differences in OS and DFS between high- and low-risk groups across clinical variables (overall stage, T stage, N stage, M stage), molecular subtypes (CMS1–4), and genetic mutations (KRAS, BRAF, NRAS)44. The corresponding concordance indices (C-indices)45 were included in each KM plot to quantify the discriminative ability of the CoDA-derived risk scores.
Significant survival differences between risk groups were observed for clinical variables, particularly for overall stage and TNM staging, in both OS (Fig. 5) and DFS (Fig. 6). Notably, N+ stage showed no significant association with DFS. For molecular variables, CoDA-based risk stratification showed significant differences in OS across all CMS subtypes (Fig. 7); however, CMS3 was not significantly associated with DFS (Fig. 8). Among genetic alterations, KRAS mutations were significantly associated with both OS (Fig. 9) and DFS (Fig. 10), while BRAF mutations showed no significant association with either outcome. NRAS mutations also showed no significant association with DFS.

Kaplan–Meier (KM) curves showing overall survival (OS) stratified by CoDA-derived risk groups (high vs. low risk) within clinical subtypes: (A) Stage I–II, (B) Stage III–IV, (C) T1–T2, (D) T3–T4, (E) N0, (F) N + , (G) M0, and (H) M + . Significant survival differences were observed between risk groups across all clinical stages, underscoring the prognostic utility of CoDA features irrespective of tumor burden.

Kaplan–Meier (KM) plots presenting disease-free survival (DFS) stratified by CoDA-derived risk categories (high vs. low risk) across clinical subtypes: (A) Stage I–II, (B) Stage III–IV, (C) T1–T2, (D) T3–T4, (E) N0, (F) N + , (G) M0, and (H) M + . Notably, significant differences in DFS were detected in all groups except M+ (p = 0.13), highlighting the robust predictive capacity of CoDA features in most clinical settings.

Kaplan–Meier (KM) survival curves for overall survival (OS) stratified by CoDA-derived risk groups (high vs. low risk) within molecular subtypes: (A) CMS1, (B) CMS2, (C) CMS3, and (D) CMS4. Statistically significant OS differences between risk groups were identified across all molecular classifications, demonstrating the broad prognostic relevance of CoDA features.

Kaplan–Meier (KM) plots depicting disease-free survival (DFS) stratified by CoDA-derived risk groups (high vs. low risk) across molecular subtypes: (A) CMS1, (B) CMS2, (C) CMS3, and (D) CMS4. Significant DFS stratification was observed for all subtypes except CMS3 (p = 0.09), suggesting effective risk discrimination in most molecular contexts with some limitations for CMS3.

Kaplan–Meier (KM) curves illustrating overall survival (OS) stratified by CoDA-derived risk groups (high vs. low risk) within mutational subtypes: (A) KRAS wild type, (B) KRAS mutated, (C) BRAF wild type, and (D) BRAF mutated. Significant OS differences were observed in all mutational subgroups except BRAF-mutated cases (p = 0.60), indicating a potential reduced prognostic impact of CoDA features in this mutation subset.

Kaplan–Meier (KM) plots showing disease-free survival (DFS) stratified by CoDA-derived risk groups (high vs. low risk) across mutational subtypes: (A) KRAS wild type, (B) KRAS mutated, (C) BRAF wild type, (D) BRAF mutated, (E) NRAS wild type, and (F) NRAS mutated. Significant DFS differences between risk groups were identified in all mutational categories except BRAF-mutated (p = 0.10) and NRAS-mutated (p = 0.26) cases, indicating diminished predictive performance of CoDA features for these mutations.
These results highlight the prognostic relevance of CoDA features in stratifying patient risk across diverse clinical, molecular, and genetic contexts.
Multivariable cox proportional hazards analysis of coda features with clinical, molecular and genetic variables
To evaluate whether CoDA features provide prognostic information independent of established clinical, molecular, and genetic subgroups, we performed multivariable Cox proportional hazards regression analyses46,47 both OS and DFS. These models included the CoDA-derived risk score along with covariates such as age, gender, race, overall stage, TNM staging, consensus molecular subtypes (CMS1–4), and common genetic mutations (KRAS, BRAF, NRAS)44,45.
The results are presented in Table 3, which reports hazard ratios (HRs), 95% confidence intervals (CIs), and p-values for each variable. The CoDA risk score remained significantly associated with both OS and DFS after adjusting for all other covariates, indicating an independent association with survival outcomes.
Several clinical variables, including overall stage and N Stage, were also significantly associated with survival in the multivariable context. Among molecular subtypes, CMS4 was associated with better outcomes, while significant associations were observed for KRAS and BRAF mutation status. Race was significantly associated with both OS and DFS as well.
These findings suggest that CoDA features may capture aspects of the tumor microenvironment that are not fully explained by traditional clinical, molecular, or genetic variables.
Discussion
In addition to imaging collagen topology through traditional histological staining or advanced imaging methods, quantifying collagen fibers is essential for patient prognosis48. In a previous study, collagen bundles formed in the tumor-stroma region correlated with tumor progression and invasiveness49. Anisotropy of CF in Mueller matrix images has been previously associated with cervical pre-cancer detection50. A high density of collagen structure has been correlated with the metastatic behavior of breast cancer in microscopic fluorescent images of picrosirius red-stained sections51. Spatial collagen stiffening have been shown to be associated with collective breast cancer cell invasion52. Collagen density and stiffening were performed for the collective collagen structure and not for the individual fibers, as in the aforementioned studies. Our methodology examined and calculated novel collagen disorder architecture features in the TME of H&E-stained images using a computational pathology-based pipeline. We demonstrate that CoDA features can effectively distinguish between various clinical, molecular, and genetic mutational variables, all of which have implications for DFS and OS.
Although AI technologies have been useful in assessing TME components in previous studies, the deep learning models used in AI methodologies53 do not specifically consider domain-specific expert knowledge in their predictions. This limitation can sometimes lead to models learning incorrect features, such as unintended patterns, hallucinations, or visual artifacts present in the database, which can diminish the reliability of AI outcomes53. While a number of approaches have been proposed that directly use deep learning to predict outcomes, a lack of interpretability can engender distrust and skepticism among physicians53. Feature-based approaches can alleviate this anxiety by imbuing more interpretability into the prediction models, and several feature-driven approaches using biomarkers such as CD8 + T cells, tumoral spatial heterogeneity54, and tumor-infiltrating lymphocytes55 have been used for prognosis in CC. However, none of these has been associated with KRAS mutational status. Unlike other TME factors, CF content and architecture offer a clear and measurable parameter that can aid in risk stratification and treatment decision-making56. Research has shown that CF density, organization, and alignment can significantly affect tumor cell invasion, metastasis, and response to therapy57. Apart from colon cancer, collagen characteristics have been previously linked to prognosis in various cancer types, such as breast cancer and head and neck cancer16,58. Thus, the study of collagen architecture provides a promising avenue for improving prognostic models across different cancer types. TME assessments, in general, require high-resolution imaging and molecular profiling, which can be resource-intensive, both in terms of time and cost59. Considering the disadvantages of second-harmonic generation microscopy, such as high cost and difficult equipment assembly, the most common technique of CF imaging, liquid crystal-based polarization microscopy (LC-PolScope), has been used as an alternative60. However, a significant challenge in using LC-PolScope for collagen imaging is the proper interpretation of signals, as they may represent other biological structures60. These imaging technologies, although promising, are still largely in the research phase and therefore not scalable for clinical translation.
Our study differs from the previously mentioned studies in that it employed standard H&E images to identify and extract collagen-related features. Critically, our study’s findings directly address the clinical need for efficient risk-stratified therapies that assess TME characteristics in CC patients. Our analysis demonstrated that CoDA-based risk stratification is significantly associated with survival outcomes (OS and DFS) across multiple clinical, molecular, and genetic factors. Notably, significant differences in survival between high-risk and low-risk groups were observed for clinical variables, particularly overall stage, in both OS and DFS analyses (Figs. 5 and 6, respectively). Among molecular subtypes, significant associations were found with OS across all CMS groups (Fig. 7), except CMS3, which did not show a significant association with DFS (Fig. 8). For genetic mutations, KRAS was significantly associated with both OS (Fig. 9) and DFS (Fig. 10), whereas BRAF and NRAS mutations showed no significant association with DFS (Fig. 10).These results highlight the potential of CoDA-based risk stratification in identifying patient subgroups with differing survival outcomes. The ability of CoDA features to distinguish between clinical, molecular and genetic factors emphasizes the need for personalized treatment strategies that consider patient backgrounds and clinical presentations. Additionally, our findings demonstrate that the integration of clinical, molecular, and genetic variables significantly influences survival outcomes, reinforcing the necessity for comprehensive prognostic models that account for these diverse factors. The observed differences in survival outcomes among various CMS subtypes indicate that molecular profiling can be a crucial element in risk stratification for treatment planning, while the significant correlation between KRAS mutations and patient survival outcomes underscores the importance of incorporating genetic testing in treatment decision-making processes.
While CoDA feature associations with key histological characteristics i.e., tumor budding61 (quantitative count), mucinous component62 (present vs. absent), differentiation63 (moderately vs. poorly differentiated), and invasive front type64 (infiltrative vs. broad), were evaluated using a randomly selected subset of 100 cases from the UH dataset, the limited sample size restricts the statistical power and generalizability of these findings. Appropriate statistical methods were applied: Pearson’s correlation65 was used to assess associations between CoDA features and tumor budding (treated as a continuous variable), while Spearman’s rank correlation66 was used for the categorical or ordinal features, including mucinous component, differentiation, and invasive front type. The results of these analyses are included in Supplementary Section 3. Notably, such detailed histopathological annotations were either not available or could not be assessed in the PLCO, TCGA, and Emory datasets, limiting our ability to perform a comprehensive evaluation across all cohorts. Although tumor grade data67 were available in the PLCO and TCGA datasets, most colorectal cancer (CC) cases were classified as grade 3, further limiting statistical comparisons. Future studies involving larger, multi-institutional cohorts with comprehensive histopathological annotation will be essential to validate and expand upon these exploratory observations. Another notable limitation of our study is the heterogeneity in KRAS mutation status determination across datasets. TCGA and PLCO datasets incorporate sequencing data generated over many years using different platforms, including targeted gene panels and whole-genome sequencing, with varying coverage, quality control measures, and bioinformatics pipelines. This variability likely contributes to underestimation or misclassification of KRAS mutations, which could affect the observed associations. While TCGA provides access to raw sequencing data enabling harmonization, such data were not available or consistent across all datasets in this study. We therefore caution that these technical differences may influence the accuracy of mutation calls, and the generalizability of our findings related to KRAS mutation status. Future work incorporating standardized sequencing protocols or comprehensive raw data analysis will be needed to validate and refine these associations.
Despite the aforementioned limitations, the methodology presented in this study could have a substantial impact in low- to middle-income countries (LMICs), where the majority of CC deaths occur68. Although population-based CC screening can be effective in LMICs, there are no published prospective studies testing screening methods, screening programs, or evidence-based screening guidelines using prospectively collected data69. Additionally, in Southeast Asia and Africa, cancer centers and departments are only available in 55% and 30% of the countries, respectively70. Given these challenges and the increased number of patients presenting with late-stage disease, there is an urgent need for effective interventions. Because of all these limitations and more patients presenting with late stage disease, efforts are being made to implement the recently published European Society for Medical Oncology consensus guidelines, of which molecular pathology and biomarkers comprise the first section71. One option is the use of broad-panel sequencing as an initial strategy for devising treatments; however, there are disadvantages of its availability and affordability in LMICs71. Next-generation sequencing is the current diagnostic gold standard for RAS mutational analysis; however, the standard procedures require extra cost, and the results can take up to 1–2 weeks on average72. Considering all these limitations, AI-based systems are increasingly being used in LMICs as a viable option, especially when there are limited resources73. The opaque nature of these models can raise concerns regarding their reliability for prognostic and treatment response predictions. In contrast, the digital pathology-based pipeline employed in our study offers an affordable and accessible assistive tool, specifically addressing the need for effective solutions in areas where genomic profiling facilities are lacking. By leveraging our computationally efficient, accurate, and inexpensive approach using novel CoDA features, we could significantly enhance rapid non-invasive risk stratification of high- and low-risk CC patients concerning clinical, molecular, and genetic variables.
Methods
Patient populations for the study
The study utilized multiple patient datasets to enhance the robustness and generalizability of findings related to CF characteristics in CC. H&E WSIs of patients with CC were collected from The Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO, n = 15488), The Cancer Genome Atlas (TCGA, n = 583), Emory University (EU, n = 759) and University Hospitals (UH, n = 619). The magnification of the WSIs were 20X and 40X.HistoQC, a quality control tool for digital pathology slides, was employed to identify and remove fat tissues and regions with artifacts (e.g., tissue folding, pen marking, and blurriness)74. Detailed information on the datasets, including inclusion and exclusion criteria, are presented in Fig. 11.

PLCO: (The Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial), TCGA: (The Cancer Genome Atlas), UH :(University Hospitals), EU (Emory University).
In this study, the dataset PLCO was used for model training while the remaining datasets (TCGA, Emory, and UH) were used for independent testing.
Automated detection of collagen fiber in tumor-stroma region on H&E slides
Tumor regions in 81 WSIs from the TCGA dataset and 50 WSIs from the UH dataset were manually annotated by a pathologist, and tumor masks were generated from the tumor-annotated regions using the digital pathology software QuPath75. Examples of TCGA WSIs and the corresponding tumor masks, generated using QuPath from the manually annotated tumor regions, are shown in Supplementary Fig. 1.Using these tumor masks as training set, Resnet18, a residual learning convolutional neural network, was used to generate the tumor masks for the rest of WSIs of TCGA and UH datasets and all the WSIs from EU and PLCO datasets76. The tumor masks and HistoQC masks were combined to form the final masks. For the CF detection process, image tiles (600×600 pixels) were first extracted from the tumor+HistoQC masked regions of individual WSIs. Morphological operations, such as image dilation, erosion, and hole-filling, have been used for epithelial-stroma segmentation16. Although CF is best highlighted by Masson’s trichome staining as opposed to H&E staining, they were not available for our study because these types of stained images are not standard77. CF in H&E slide images typically exhibit a linear phenotype due to the linear arrangement of type I collagen molecules16. A derivative-of-Gaussian (DtG) based model was applied to detect these linear structures in tumor-associated stroma tiles16. The DtG model classified each pixel into one of seven image structures: Flat, Slope, Blob (dark or light), Line (dark or light), and Saddle16. By isolating the pixels identified as the dark Line structure, the model successfully detected CF in the tiles. Following the calculation of CoDA features for each individual tumor-stroma tile, the average value of the features derived from all the tiles was calculated as the patient-level feature.
Collagen disorder architectural (Coda) features
The following CoDA features were calculated on the detected CFs: [1] Collagen fiber fragmentation measure, [2] collagen fiber bundling percentage, [3] collagen fiber rigidity measure, [4] collagen fiber anisotropy, and [5] collagen fiber density indices. The features are calculated as follows:
-
1.
Collagen fiber fragmentation measure: Collagen fiber fragmentation refers to the breakdown or degradation of CFs in the tumor-stroma region. The collagen fiber fragmentation for one tile was calculated using the following equation:
$${Collagen\; fiber\; fragmentation\; measure}=\frac{{{CF}}^{f}}{{{CF}}^{l}}$$(1)where CFf is the collagen fiber fragments and CFl is the collagen fiber length.
-
2.
Collagen fiber bundling percentage: Collagen fiber bundling refers to the process of CFs aligning and organizing into tight bundles within the tumor-stroma region. The percentage of collagen fiber bundling for one tile was calculated using the following equation:
$${Collagen\; fiber\; bundling\; percentage}=\frac{\,{{CF}}^{{ba}}\,}{{{CF}}^{b}}\times 100$$(2)where CFba is the collagen fiber bundling area, and CFb is the collagen fiber bundle area.
-
3.
Collagen fiber rigidity measure: Collagen fiber rigidity refers to the straightness, i.e., “non-curliness” of CFs within the tumor-stroma region. The collagen fiber rigidity for one tile was calculated using the following equation:
$${Collagen\; fiber\; rigidity\; measure}={{CF}}^{t}\times {{CF}}^{o}$$(3)where CFt is the thickness of the collagen fibers, and Cf° is the orientation of the collagen fibers.
-
4.
Collagen fiber anisotropy index: Collagen fiber anisotropy refers to the directional preference or arrangement of CFs along a certain direction within the tumor-stroma region. The collagen fiber anisotropy index for each tile was calculated using the following equation:
$${Collagen\; fiber\; anisotropy\; index}=\left(\,{{CF}}^{{dx}}\,{{CF}}^{{dy}}\right)$$(4)where CFdx and Cfdy are the collagen fiber changes in the x- and y-directions, respectively.
-
5.
Collagen fiber density index: Collagen fiber density refers to the abundance of CFs within a specific volume or area of the tumor-stroma region. The collagen fiber density index for each tile was calculated using the following equation:
where Cfa is the area occupied by collagen fibers in the tumor stroma region and TSa is the total area of the tumor stroma region.
Statistical analysis
Differences in CoDA features were assessed across clinical, molecular, and genetic variables using all available datasets. The subgroup comparisons included: 1] Clinical variables: (A) Overall stage: Stages I–II vs. III–IV, (B) T stage: T1–2 vs. T3–4, (C) N stage: N0 vs. N + , (D) M stage: M0 vs. M + ;[2] Consensus Molecular Subtypes (CMS): Pairwise comparisons among CMS1, CMS2, CMS3, and CMS4; [3] Genetic mutations: Wild-type vs. mutant status for KRAS, BRAF, and NRAS.
The Mann–Whitney U test33,34 was applied to evaluate differences in CoDA features i.e., CF fragmentation, CF bundling, CF anisotropy, CF density, and CF rigidity, between each group. To ensure robustness, the analysis was repeated across 100 iterations, with 50 samples randomly selected from each group per iteration. Bonferroni correction35,36,37 was applied to the pooled p-values to account for multiple testing, using a significance threshold of 0.05 for both raw and corrected p-values(denoted by *p).
To further assess the association of CoDA features with these variables, a Random Forest classification framework38,39 with iterative random sampling (100 iterations) was implemented. In each iteration, class-balanced resampling was performed, followed by feature selection using permutation-based importance78,79. The classifier was trained using the selected features, and performance was evaluated via area under the ROC curve (AUC)40. Mean AUC and standard deviation across iterations were computed for each variable, and corresponding F1 scores41 and accuracy41 were also calculated to determine overall classification performance.
For survival analysis, separate LASSO-Cox models42,43 were trained on the PLCO dataset for OS and DFS endpoints using the CoDA features. Risk scores were generated for each patient, and the median risk score in the PLCO dataset was used as a cutoff to stratify patients into high-risk and low-risk groups. This cutoff was then applied to the external validation datasets (TCGA, UH, and Emory) to stratify patients in both OS and DFS cohorts. The concordance index (C-index)45 was calculated for each survival model to assess discriminative ability and is displayed alongside Kaplan-Meier (KM) curves44.
Risk stratification was further evaluated within subgroups defined by the same clinical, molecular, and genetic variables listed above. The resulting KM curves visualized survival differences between high- and low-risk groups across these variables.
Finally, multivariable Cox proportional hazards regression46,47 was employed to assess the independent prognostic significance of the CoDA as an independent prognostic while adjusting for covariates including clinical, molecular, and genetic factors. Hazard ratios, 95% confidence intervals, and p-values were reported. All tests were two-sided, and p ≤ 0.05 were considered statistically significant.
Ethical statement
This study was performed under the Emory University Institutional Review Board (IRB) protocol STUDY00005782, which was approved as a non-human study and all relevant ethical regulations were followed. De-identified human samples obtained from the other institution used in our study i.e., University Hospitals, were collected under the same IRB approved protocol STUDY00005782. All institutions collected specimens with participants informed consent. The other datasets used in our study i.e., Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO) and The Cancer Genome Atlas (TCGA) are publicly available datasets.
Data availability
The dataset and related clinical information for TCGA and PLCO datasets are available in https://portal.gdc.cancer.gov/projects/TCGA-COAD and https://cdas.cancer.gov/plco/ respectively. The CMS scores for TCGA dataset used in our analysis is available from https://bmccancer.biomedcentral.com/articles/10.1186/s12885-022-09344-3. Since the cases from the involved institutions are protected through institutional compliance, the clinical repository of cases can only be shared per specific institutional review board (IRB) requirements. This applies to the datasets from Emory University and University Hospitals. Upon reasonable request, a data sharing agreement can be initiated between the interested parties and the clinical institution following institution-specific guidelines. For inquiries or requests regarding data sharing, please contact corresponding author.
Code availability
HistoQC code is available from: https://github.com/choosehappy/HistoQC.Tumor segmentation code is available from: https://github.com/jnkather/MSIfromHE.The CoDA feature calculation code is uploaded in GitHub by author Reetoja Nag and is available in: https://github.com/nagreetoja/CoDA. CoDA feature calculation code was generated using MATLAB 2023b software.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Liu, Q., Luo, D., Cai, S., Li, Q. & Li, X. P-TNM staging system for colon cancer: combination of P-stage and AJCC TNM staging system for improving prognostic prediction and clinical management. Cancer Manag. Res. 10, 2303–2314 (2018).
Siegel, R. L. et al. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 70, 145–164 (2020).
White, A., Vernon, S. W., Franzini, L. & Du, X. L. Racial disparities in colorectal cancer survival: to what extent are racial disparities explained by differences in treatment, tumor characteristics, or hospital characteristics?. Cancer 116, 4622–4631 (2010).
Hendifar, A. et al. Gender disparities in metastatic colorectal cancer survival. Clin. Cancer Res. 15, 6391–6397 (2009).
Patelli, G. et al. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 6, 100156 (2021).
Yang, M. et al. An integrative gene expression signature analysis identifies CMS4 KRAS-mutated colorectal cancers sensitive to combined MEK and SRC targeted therapy. BMC Cancer 22, 256 (2022).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Lin, A., Zhang, J. & Luo, P. Crosstalk between the MSI status and tumor microenvironment in colorectal cancer. Front. Immunol. 11, 2039 (2020).
Whipple, C. A. Tumor talk: understanding the conversation between the tumor and its microenvironment. Cancer Cell Microenviron. 2, e773 (2015).
Neophytou, C. M., Panagi, M., Stylianopoulos, T. & Papageorgis, P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers (Basel) 13, 2053 (2021).
Werb, Z. & Lu, P. The role of stroma in tumor development. Cancer J. 21, 250–253 (2015).
Wang, Q. et al. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 12, 11149–11165 (2023).
Ueno, H. et al. Prognostic value of desmoplastic reaction characterisation in stage II colon cancer: prospective validation in a Phase 3 study (SACURA Trial). Br. J. Cancer 124, 1088–1097 (2021).
Dong, S. et al. Development and validation of a collagen signature to predict the prognosis of patients with stage II/III colorectal cancer. iScience 26, 106746 (2023).
Li, H. et al. Collagen fiber orientation disorder from H&E images is prognostic for early stage breast cancer: clinical trial validation. NPJ Breast Cancer 7, 104 (2021).
Zhang, Q., An, Z.-Y., Jiang, W., Jin, W.-L. & He, X.-Y. Collagen code in tumor microenvironment: Functions, molecular mechanisms, and therapeutic implications. Biomed. Pharmacother. 166, 115390 (2023).
Zhang, Z. et al. COL1A1: A potential therapeutic target for colorectal cancer expressing wild-type or mutant KRAS. Int. J. Oncol. 53, 1869–1880 (2018).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Gutierrez, M. E. et al. Genomic profiling for KRAS, NRAS, BRAF, microsatellite instability, and mismatch repair deficiency among patients with metastatic colon cancer. JCO Precis. Oncol 3, 1–9 (2019).
Allegra, C. J. et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 27, 2091–2096 (2009).
Elsayed, I. et al. Evaluating utility and feasibility of mismatch repair testing of colorectal cancer patients in a low-middle-income country. Sci. Rep. 12, 10998 (2022).
Chen, M. et al. Classification and mutation prediction based on histopathology H&E images in liver cancer using deep learning. NPJ Precis. Oncol. 4, 14 (2020).
Viswanathan, V. S., Toro, P., Corredor, G., Mukhopadhyay, S. & Madabhushi, A. The state of the art for artificial intelligence in lung digital pathology. J. Pathol. 257, 413–429 (2022).
Bousis, D. et al. The role of deep learning in diagnosing colorectal cancer. Prz. Gastroenterol. 18, 266–273 (2023).
Bera, K., Schalper, K. A., Rimm, D. L., Velcheti, V. & Madabhushi, A. Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 16, 703–715 (2019).
Ouellette, J. N. et al. Navigating the collagen jungle: the biomedical potential of fiber organization in cancer. Bioengineering (Basel) 8, 17 (2021).
Popova, N. V. & Jücker, M. The functional role of extracellular matrix proteins in cancer. Cancers (Basel) 14, 238 (2022).
De Martino, D. & Bravo-Cordero, J. J. Collagens in cancer: structural regulators and guardians of cancer progression. Cancer Res. 83, 1386–1392 (2023).
Cui, G. et al. Overcoming the tumor collagen barriers: a multistage drug delivery strategy for DDR1-mediated resistant colorectal cancer therapy. Adv. Sci. (Weinh.) 11, e2402107 (2024).
Visser, M. et al. Inter-rater agreement in glioma segmentations on longitudinal MRI. Neuroimage Clin. 22, 101727 (2019).
Pinto, B. L., Stankovic, T., Frost, D. M. & Beach, T. A. C. Adjacent joint restriction differentially influences intra- and inter-rater reliability and agreement of goniometric measurements. Int. J. Sports Phys. Ther. 17, 276–285 (2022).
Rosner, B. & Grove, D. Use of the Mann–Whitney U-test for clustered data. Stat. Med. 18, 1387–1400 (1999).
Tai, K. Y., Dhaliwal, J. & Balasubramaniam, V. Leveraging Mann–Whitney U test on large-scale genetic variation data for analysing malaria genetic markers. Malar. J. 21, 79 (2022).
Sedgwick, P. Multiple hypothesis testing and Bonferroni’s correction. BMJ 349, g6284 (2014).
Armstrong, R. A. When to use the Bonferroni correction. Ophthalmic Physiol. Opt. 34, 502–508 (2014).
Kanning, J. P. et al. Prescribed drug use and aneurysmal subarachnoid hemorrhage incidence: a drug-wide association study. Neurology 102, e209479 (2024).
Kapsiani, S. & Howlin, B. J. Random forest classification for predicting lifespan-extending chemical compounds. Sci. Rep. 11, 13812 (2021).
Safia, N. S. Prediction of breast cancer through random forest. Curr. Med. Imaging 19, 1144–1155 (2023).
Nahm, F. S. Receiver operating characteristic curve: overview and practical use for clinicians. Korean J. Anesthesiol. 75, 25–36 (2022).
Attia, Z. I. et al. An artificial intelligence-enabled ECG algorithm for the identification of patients with atrial fibrillation during sinus rhythm: a retrospective analysis of outcome prediction. Lancet 394, 861–867 (2019).
Saha-Chaudhuri, P. & Juwara, L. Survival analysis under the Cox proportional hazards model with pooled covariates. Stat. Med. 40, 998–1020 (2021).
Chen, Y. et al. A novel immune-related gene signature to identify the tumor microenvironment and prognose disease among patients with oral squamous cell carcinoma patients using ssGSEA: a bioinformatics and biological validation study. Front. Immunol. 13, 922195 (2022).
Hess, A. S. & Hess, J. R. Kaplan-Meier survival curves. Transfusion 60, 670–672 (2020).
Pencina, M. J. & D’Agostino, R. B. Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat. Med. 23, 2109–2123 (2004).
Corredor, G. et al. Spatial architecture and arrangement of tumor-infiltrating lymphocytes for predicting likelihood of recurrence in early-stage non-small cell lung cancer. Clin. Cancer Res. 25, 1526–1534 (2019).
Corredor, G. et al. An imaging biomarker of tumor-infiltrating lymphocytes to risk-stratify patients with HPV-associated oropharyngeal cancer. J. Natl. Cancer Inst. 114, 609–617 (2022).
Fang, M., Yuan, J., Peng, C. & Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 35, 2871–2882 (2014).
Gong, X., Kulwatno, J. & Mills, K. L. Rapid fabrication of collagen bundles mimicking tumor-associated collagen architectures. Acta Biomater. 108, 128–141 (2020).
Zaffar, M. & Pradhan, A. Assessment of anisotropy of collagen structures through spatial frequencies of Mueller matrix images for cervical pre-cancer detection. Appl. Opt. 59, 1237–1248 (2020).
Shea, M. P., O’Leary, K. A., Wegner, K. A., Vezina, C. M. & Schuler, L. A. High collagen density augments mTOR-dependent cancer stem cells in ERα+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 433, 1–9 (2018).
Koorman, T. et al. Spatial collagen stiffening promotes collective breast cancer cell invasion by reinforcing extracellular matrix alignment. Oncogene 41, 2458–2469 (2022).
Viscaino, M., Torres Bustos, J., Muñoz, P., Auat Cheein, C. & Cheein, F. A. Artificial intelligence for the early detection of colorectal cancer: a comprehensive review of its advantages and misconceptions. World J. Gastroenterol. 27, 6399–6414 (2021).
Gong, C. et al. Quantitative characterization of CD8+ T cell clustering and spatial heterogeneity in solid tumors. Front. Oncol. 8, 649 (2018).
Jakab, A., Patai, ÁV. & Micsik, T. Digital image analysis provides robust tissue microenvironment-based prognosticators in patients with stage I-IV colorectal cancer. Hum. Pathol. 128, 141–151 (2022).
Jansen, K. A. et al. The role of network architecture in collagen mechanics. Biophys. J. 114, 2665–2678 (2018).
Baldari, S., Di Modugno, F., Nisticò, P. & Toietta, G. Strategies for efficient targeting of tumor collagen for cancer therapy. Cancers (Basel) 14, 4706 (2022).
Sok, J. C. et al. Collagen type XI α1 facilitates head and neck squamous cell cancer growth and invasion. Br. J. Cancer 109, 3049–3056 (2013).
Head, T. & Cady, N. C. Monitoring and modulation of the tumor microenvironment for enhanced cancer modeling. Exp. Biol. Med (Maywood) 247, 598–613 (2022).
Keikhosravi, A. et al. Quantification of collagen organization in histopathology samples using liquid crystal based polarization microscopy. Biomed. Opt. Express 8, 4243–4256 (2017).
El Agy, F. et al. Prognostic impact of tumor budding on Moroccan colon cancer patients. Int. J. Surg. Oncol. 2022, 9334570 (2022).
Maeda, Y., Sadahiro, S., Suzuki, T., Haruki, Y. & Nakamura, N. Significance of the mucinous component in the histopathological classification of colon cancer. Surg. Today 46, 303–308 (2016).
Zweibaum, A. [Differentiation of human colon cancer cells: a new approach to colon cancer]. Bull. Acad. Natl. Med. 177, 63–71 (1993).
Harada-Kagitani, S. et al. Keratin 6A is expressed at the invasive front and enhances the progression of colorectal cancer. Lab. Invest. 104, 102075 (2024).
Iguacel, I. et al. Associations between dietary amino acid intakes and blood concentration levels. Clin. Nutr. 40, 3772–3779 (2021).
Ma, Y. et al. Aberrant single-subject morphological cerebellar connectome in chronic insomnia. Neuroimage Clin. 39, 103492 (2023).
Hestetun, K. E. et al. Mismatch repair phenotype determines the implications of tumor grade and CDX2 expression in stage II-III colon cancer. Mod. Pathol. 34, 161–170 (2021).
Arhin, N. et al. Age-standardised incidence rate and epidemiology of colorectal cancer in Africa: a systematic review and meta-analysis. BMJ Open 12, e052376 (2022).
Alatise, O. I. et al. A symptom-based model to predict colorectal cancer in low-resource countries: Results from a prospective study of patients at high risk for colorectal cancer. Cancer 124, 2766–2773 (2018).
Ralaidovy, A. H., Gopalappa, C., Ilbawi, A., Pretorius, C. & Lauer, J. A. Cost-effective interventions for breast cancer, cervical cancer, and colorectal cancer: new results from WHO-CHOICE. Cost. Eff. Resour. Alloc. 16, 38 (2018).
Maurel, J.-M. et al. Practical notes on the current ESMO consensus guidelines for the management of patients with metastatic colorectal cancer in resource-constrained environments of low- to middle-income countries. Oncology 101, 5–8 (2023).
Mahdi, Y. et al. Correlation between KRAS and NRAS mutational status and clinicopathological features in 414 cases of metastatic colorectal cancer in Morocco: the largest North African case series. BMC Gastroenterol. 23, 193 (2023).
Ciecierski-Holmes, T., Singh, R., Axt, M., Brenner, S. & Barteit, S. Artificial intelligence for strengthening healthcare systems in low- and middle-income countries: a systematic scoping review. npj Digit. Med. 5, 162 (2022).
Janowczyk, A., Zuo, R., Gilmore, H., Feldman, M. & Madabhushi, A. HistoQC: an open-source quality control tool for digital pathology slides. JCO Clin. Cancer Inform. 3, 1–7 (2019).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Kather, J. N. et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 25, 1054–1056 (2019).
Wei, B. et al. Human colorectal cancer progression correlates with LOX-induced ECM stiffening. Int. J. Biol. Sci. 13, 1450–1457 (2017).
Altmann, A., Toloşi, L., Sander, O. & Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics 26, 1340–1347 (2010).
Kaneko, H. Cross-validated permutation feature importance considering correlation between features. Anal. Sci. Adv. 3, 278–287 (2022).
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute under award numbers R01CA268287A1, U01CA269181, R01CA26820701A1, R01CA249992-01A1, R01CA202752-01A1, R01CA208236-01A1, R01CA216579-01A1, R01CA220581-01A1, R01CA257612-01A1, 1U01CA239055-01, 1U01CA248226-01, 1U54CA254566-01, National Heart, Lung and Blood Institute 1R01HL15127701A1, R01HL15807101A1, National Institute of Biomedical Imaging and Bioengineering 1R43EB028736-01, VA Merit Review Award IBX004121A from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program (W81XWH-19-1-0668), the Prostate Cancer Research Program (W81XWH-20-1-0851), the Lung Cancer Research Program (W81XWH-18-1-0440, W81XWH-20-1-0595), the Peer Reviewed Cancer Research Program (W81XWH-18-1-0404, W81XWH-21-1-0345, W81XWH-21-1-0160), the Kidney Precision Medicine Project (KPMP) Glue Grant and sponsored research agreements from Bristol Myers-Squibb, Boehringer-Ingelheim, Eli-Lilly and Astrazeneca. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. Department of Veterans Affairs, the Department of Defense, or the U.S. Government.
Author information
Authors and Affiliations
Contributions
Reetoja Nag, Joseph E. Willis and Anant Madabhushi conceived and designed the study. Reetoja Nag, Chuheng Chen and Haojia Li developed the methodology. Aya Aqeel, Chuheng Chen and Haider Mejbel helped in generation of intermediate data. Pingfu Fu supervised statistical analysis. Tilak Pathak, and Mojgan Mokhtari did quality check of the generated data. Reetoja Nag, German Corredor, Sirvan Khalighi and Anant Madabhushi analyzed and interpreted the data. Tilak Pathak, Krunal Pandav, Mojgan Mokhtari, Haider Mejbel, Michelle Dian Reid and Alyssa M. Krasinskas and Joseph E.Willis provided clinical insights into the project. Joseph E.Willis provided WSIs and clinical information the UH dataset. Haider Mejbel, Michelle Dian Reid and Alyssa M. Krasinskas provided WSIs and clinical information the EU dataset. Krunal Pandav curated all the datasets. The paper was written primarily by Reetoja Nag, Joseph E.Willis and Anant Madabhushi with support from all authors. The final version of the paper was reviewed, edited and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
Dr. Madabhushi is an equity holder at Picture Health, Elucid Bioimaging, and Inspirata Inc. Currently, he serves on the advisory board of Picture Health and SimBioSys. He currently consults Takeda Inc. He also sponsored research agreements with AstraZeneca and Bristol Myers Squibb. His technology has been licensed for Picture Health and Elucid Bioimaging. He is also involved in two different R01 grants from Inspirata, Inc. He also served as a member of the Frederick National Laboratory Advisory Committee. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nag, R., Chen, C., Mejbel, H. et al. Collagen disorder architecture features are associated with clinical, molecular, genetic factors and survival outcomes in colon cancer. npj Precis. Onc. 9, 304 (2025). https://doi.org/10.1038/s41698-025-01098-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-01098-y
