
Abstract
Cardiovascular diseases (CVDs) and cerebrovascular diseases (CeVDs) are closely related vascular diseases, sharing common cardiometabolic risk factors (RFs). Although pleiotropic genetic variants of these two diseases have been reported, their underlying pathological mechanisms are still unclear. Leveraging GWAS summary data and using genetic correlation, pleiotropic variants identification, and colocalization analyses, we identified 11 colocalized loci for CVDs-CeVDs-BP (blood pressure), CVDs-CeVDs-LIP (lipid traits), and CVDs-CeVDs-cIMT (carotid intima-media thickness) triplets. No shared causal loci were found for CVDs-CeVDs-T2D (type 2 diabetes) or CVDs-CeVDs-BMI (body mass index) triplets. The 11 loci were mapped to 12 genes, namely CASZ1, CDKN1A, TWIST1, CDKN2B, ABO, SWAP70, SH2B3, LRCH1, FES, GOSR2, RPRML, and LDLR, where both GOSR2 and RPRML were mapped to one locus. They were enriched in pathways related to cellular response to external stimulus and regulation of the phosphate metabolic process and were highly expressed in endothelial cells, epithelial cells, and smooth muscle cells. Multi-omics analysis revealed methylation of two genes (CASZ1 and LRCH1) may play a causal role in the genetic pleiotropy. Notably, these pleiotropic loci are highly enriched in the targets of antihypertensive drugs, which further emphasizes the role of the blood pressure regulation pathway in the shared etiology of CVDs and CeVDs.

Similar content being viewed by others

Introduction
Vascular diseases encompass a broad range of disorders of the heart and circulation in other organs. Among them, cardiovascular diseases (CVDs) and cerebrovascular diseases (CeVDs) are the two main types that occur in the heart and brain, respectively, and account for more than 80% of total deaths1. CVDs mainly include coronary artery disease (CAD), myocardial infarction (MI), heart failure (HF), and atrial fibrillation (AF), while CeVDs mainly indicate stroke2. CVDs and CeVDs are closely related, caused by many shared cardiometabolic risk factors (RFs) and frequent coexistence3,4. Inherited DNA sequence variants play a role in conferring risk for CVDs and CeVDs5,6.
Previous studies, including family history studies, genome-wide association studies (GWAS), and cross-trait analyses, reveal that CVDs and CeVDs may share genetic architectures. For example, coronary heart disease (CHD) and ischemic stroke exhibit shared familial susceptibility, with a linkage between the family history of stroke to increased risks of CHD7,8,9. GWAS studies have shown three of the 42 genome-wide significant loci for CAD were related to ischemic stroke10. Chromosome 9p21, the most robust genetic marker of CHD, was also associated with stroke11. Other loci associated with both diseases include SH2B3, ABO, DDAH1, etc.10,12. In recent decades, the emergence of GWAS data with large sample sizes, along with advanced analysis technologies, has positioned research on genetic sharing across traits as a key area of focus. Consequently, numerous studies have examined the genetic links among cardiovascular diseases. For instance, several studies have uncovered shared genes between CVDs and CeVDs through methods such as meta-analysis13, network module analysis14, and across-trait association analysis10. Additionally, other research has quantified the degree of genetic correlation using approaches like Mendelian randomization (MR)15, linkage disequilibrium score regression (LDSC)16, and imaging connections17, with genetic correlations ranging around 0.4–0.5. Additional research has examined the genetic links between CVDs or CeVDs and RFs through MR18,19,20, genetic risk score-based methods21,22,23, across-trait association analyses24,25, and pleiotropy-informed methods26,27. However, previous studies have typically concentrated on two-trait pairs: CVDs-CeVDs, CVDs-RFs, and CeVDs-RFs, with few studies integrating all three aspects. As mentioned, CVDs and CeVDs not only exhibit comorbidity but also share common cardiometabolic RFs. Therefore, the full extent of shared genetics between CVDs and CeVDs remains to be fully elucidated and deserves further investigation through the CVDs-CeVDs-RFs triplet, which could help increasing analysis efficiency and facilitating mechanism interpretation. More importantly, the shared genetic bases and the relevant biological pathways identified from the triplet approach might affiliate the development of novel therapeutic and preventive strategies.
Recent methodologies have been developed to provide novel findings of genetic overlap beyond genetic correlation. The conditional/conjunctional false discovery rate (cond/conjFDR) method increased the yield of existing GWAS and aided the discovery of overlapping genomic loci between polygenic phenotypes28. The colocalization analysis allowed us to further ask whether two potentially related phenotypes share common genetic causal variant(s) in the given region29,30 and was extended to apply to multiple phenotypes31. Multi-omics association method enabled to prioritize molecular phenotypes that mediate the genetic effects for complex traits and further provide mechanistic interpretation of the pleiotropic signals32. Pleiotropy-driven approach for therapeutic target prioritization and evaluation was developed to leverage shared genetic and pathway architectures of related diseases to advance therapeutic discovery33. In our analysis, we queried, downloaded and harmonized the GWAS summary data of the European ancestry from four CVDs (CAD5, MI34, HF35, and AF36), two CeVDs (all stroke (AS)37 and all ischemic stroke (AIS)38), and seven RFs (systolic blood pressure (SBP)39, type 2 diabetes (T2D)40, body mass index (BMI)41, total triglyceride (TG)42, low-density lipoprotein cholesterol (LDL-C)43, high-density lipoprotein cholesterol (HDL-C)44, and carotid intima-media thickness (cIMT)45). Most of them resulted from meta-analyses comprising tens of thousands to millions of samples. Leveraging the GWAS summary data, in the first step, we quantified the genetic overlaps between CVDs and CeVDs using MiXeR44. Then, in the second step, the shared genetic components were further examined through their common RFs using pleiotropic association test28 and colocalization test29,30,31. Specifically, we identified pleiotropic loci for CVDs-RFs and CeVDs-RFs, respectively. In the third step, for the overlapped pleiotropic loci identified in step two, we identified the colocalized loci for the triplet combinations of CVDs-CeVDs-RFs, which approved the existence of the same causal variant driving CVDs-CeVDs-RFs triplet in the given pleiotropic region. Focusing on the colocalized loci for CVDs-CeVDs-RFs triplet, we also conducted multi-omics association analysis32 to explore the mediating role of other omics molecules in the causal chain from genes to disease phenotypes and conducted functional enrichment analysis to prioritize the most likely shared genes, biological pathways, tissue/cell types, and drug targets, which allowed us to gain a deeper understanding of the shared genetic etiology underlying both CVDs and CeVDs (Fig. 1).

A The overview of the study design. B The workflow of the study analyses. CVDs cardiovascular diseases, CeVDs cerebrovascular diseases, RFs risk factors, CAD coronary artery disease, MI myocardial infarction, HF heart failure, AF atrial fibrillation, AIS all ischemic stroke, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, T2D type 2 diabetes, BMI body mass index, cIMT carotid intima-media thickness.
Results
Polygenic overlaps between CVDs and CeVDs
The shared polygenic components estimated by MiXeR across CVDs and CeVDs were shown in the Venn diagrams (Fig. 2A, Supplementary Data 1). There were 1.5K and 1.0K variants that causally influenced CAD and AS, respectively. Among these variants, 0.9K is shared between the two disorders, presenting a high genetic correlation between them (rg = 0.59, P = 1.4E−226). Similarly, a large proportion of genetic causal variants overlapped between all other CVDs and CeVDs analyzed in this study, with genetic correlations ranging from 0.38 to 0.60 (P < 1.0E−50).

A The Venn diagrams showed the shared (gray) and unique polygenic overlap between one specific CVD (red) and one specific CeVD (blue). The numbers in the Venn diagram represent the estimated quantity and standard error (in parenthesis) of shared and unique variants (in thousands). The size of the circles represents the degree of polygenicity. The bar under the Venn diagram represents the estimated genetic correlation (rg) (scaling from −1 to +1) between two traits. The orange bar on the right means the positive genetic correlation, otherwise, it means the negative correlation. All the results in the figure are based on MiXeR analysis. B The genetic correlations between CVDs or CeVDs (y-axis) and RFs (x-axis). Red indicates a positive genetic correlation, while blue represents a negative genetic correlation. The top left section of each square corresponds to the genetic correlation estimated using LDSC, while the bottom right section corresponds to the genetic correlation estimated using HDL. Genetic correlations that remain significant after Bonferroni correction (P < 0.05/21, where 21 = 6 × 7/2) are denoted by an asterisk (*). “−” indicates that no estimation was available from either LDSC or HDL. CVDs cardiovascular diseases, CeVDs cerebrovascular diseases, RFs risk factors, CAD coronary artery disease, MI myocardial infarction, HF heart failure, AF Atrial fibrillation, AIS all ischemic stroke, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, T2D type 2 diabetes, BMI body mass index, cIMT carotid intima-media thickness, rg genetic correlation, LDSC method linkage disequilibrium score regression, HDL method high-definition likelihood.
Shared genetic variants between CVDs (CAD, MI, HF, and AF) and CeVDs (AS and AIS) were also identified by the conjFDR method and represented by the look-up tables and the Manhattan plots. As shown in the look-up tables (Supplementary Fig. 1), considerable overlapped genetic variants existed in all CVDs-CeVDs pairs, especially, CAD/MI exhibited a higher number of shared genetic variant associations with CeVDs compared to HF/AF. Based on the Manhattan plot (Supplementary Fig. 2), aggregating significant shared genetic variants on chromosomes 1, 4, 7, 9, 10, 12, 13, 16, 17, and 19 were identified for various combinations of CVDs and CeVDs.
Pleiotropic variants and loci between CVDs/CeVDs and RFs
Genetic correlations between CVDs/CeVDs and RFs
Genetic correlations between CVDs/CeVDs and RFs were estimated using LDSC and HDL methods, respectively. Nearly all CVDs and CeVDs exhibited significant genetic correlations with all RFs, ranging from 0.11 to 0.61 (P < 2.4e−3 = 0.05/21). The strongest genetic correlations (above 0.4) existed in the pairs of CeVDs (AS, AIS) and SBP, CVDs (MI, HF) and T2D, and HF and BMI. All RFs, except HDL-C, displayed positive genetic correlations with the diseases, while HDL-C showed a negative genetic correlation (Fig. 2B, Supplementary Data 2).
Pleiotropic variants between CVDs/CeVDs and RFs
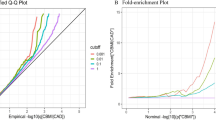
Using condFDR method, a large number of pleiotropic genetic variants were identified for all pairs of CVDs/CeVDs and RFs (Fig. 3A, Supplementary Data 3–8, Supplementary Figs. 3–9). The pleiotropy patterns between CVDs-RFs and CeVDs-RFs were different. First, the number and the position of pleiotropic variants and loci (aggregating from variants) between CVDs-RFs and CeVDs-RFs pairs were different. A large number of pleiotropic variants were detected for CVDs-RFs pairs (mainly for CAD/MI-RFs pairs) across the entire genome while only limited signals for CeVDs and RFs primarily on chromosomes 1, 4, 6, 7, 9, 10, 12, and 13 (Supplementary Data 9). Furthermore, the numbers of pleiotropic loci shared between CVDs/CeVDs with various RFs are different: CAD/MI had more pleiotropic variants with SBP and lipid traits than with T2D, BMI, and cIMT; CeVDs had more (nearly two times) pleiotropic variants with SBP than other RFs. Second, CVDs and CeVDs exhibited varying degrees of polygenic enrichment conditioned on different RFs (Fig. 3B). For SNPs with P values below 0.001, the fold enrichment for CAD/MI varied between 10 and 30 across most RFs, particularly, CAD/MI showed the highest enrichment for cIMT (~50 folds), followed by SBP (~30 folds). HF had the highest enrichment in T2D (~80 folds), and AF had low enrichment in all RFs. The primary enrichment for CeVDs was observed in SBP, reaching a fold enrichment of 50. For the other RFs, the enrichment for CeVDs ranged from 15 to 20 folds. Third, the pleiotropic variants with different RFs also accounted for various amounts of heritability for each vascular disorder (Fig. 3C). Heritability explained by pleiotropic variants was higher for CVDs (CAD and MI, 0.27–0.61, P < 2.4E−3 = 0.05/21) than CeVDs (AS and AIS, 0.02–0.11, P < 2.4E−3 = 0.05/21). Much higher heritability is explained by pleiotropic variants of SBP (CAD, prop.h2 (proportion of heritability) = 0.61, P = 1.3E−30; AS, prop.h2 = 0.11, P = 6.7E−6) than other RFs, regardless of CVDs or CeVDs (Supplementary Data 10).

A Number of pleiotropic loci conditioning on different RFs for CVDs and CeVDs. B The fold-enrichment plot illustrates the enrichment of CVDs (CAD, MI, HF, and AF) and CeVDs (AS and AIS) based on their association with cardiometabolic RFs (SBP, TG, LDL-C, HDL-C, T2D, BMI, cIMT). The fold enrichment is represented as the ratio of the −log10(P) cumulative distribution of SNPs conditional on a specific P threshold of 0.001 in one RF to the cumulative distribution for all SNPs of that RF. C Partitioned heritability for CVDs and CeVDs from pleiotropic variants with RFs. The number in each square represents the partitioned heritability as calculated by the number of pleiotropic SNPs of one vascular disease and one RF combination divided by all SNPs of that vascular disease. Different colors represent the heritability after standardizing the heritability of different RFs in each vascular disease. The redder the color, the stronger the association between the risk factor and the disease. CVDs cardiovascular diseases, CeVDs cerebrovascular diseases, RFs risk factors, CAD coronary artery disease, MI myocardial infarction, HF heart failure, AF atrial fibrillation, AIS all ischemic stroke, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, T2D type 2 diabetes, BMI body mass index, cIMT carotid intima-media thickness.
Colocalized loci between CVDs/CeVDs and RFs
Based on the pleiotropic variants identified from CVDs-RFs and CeVDs-RFs, colocalization analysis was further applied to prioritize the possible causal loci (posterior probability > 80%, Supplementary Data 11). For different pairs of vascular diseases and RFs, a certain amount of colocalized loci, as well as 95% credible variants and genes, were revealed (Table 1, Supplementary Figs. 10–16). Notably, there were significant disparities in the colocalized loci between CVDs-RFs and CeVDs-RFs in numbers and positions. A significantly larger number of loci were identified for CAD/MI-RFs than AS/AIS-RFs. For a given RF, the majority of colocalized loci identified with CVDs (denoted as CAD and MI) and CeVDs were found in distinct regions across the genome. More colocalized loci were identified for both CVDs and CeVDs with SBP and lipid levels than with T2D, BMI, and cIMT.
Shared genetic basis for CVDs and CeVDs with pleiotropy on their common RFs
By examining the pleiotropic variants for CVDs or CeVDs conditioned on RFs, we identified shared loci for CVD-CeVD-RF triplets (Table 2, Supplementary Figs. 17–23). The number of overlapping loci associated with both CVDs and CeVDs conditioning on SBP was much larger than conditioning on other RFs. Of these overlapping loci, 11 loci have been prioritized as the most possible causal genetic loci shared between CVDs and CeVDs conditioning on RFs by multiple-trait colocalization (MOLOC) analyses (posterior probability > 80%, Tables 2 and 3). The highest number of pleiotropic loci for the CVDs-CeVDs-RFs triplets was observed when conditioning on SBP, with a total of 7 loci located on chromosomes 1, 7, 9, 11, 12, 13, and 15. This was followed by conditioning on LDL-C, TG, HDL-C, and cIMT, where the numbers of pleiotropic loci for the CVDs-CeVDs-RFs triplets were 6, 4, 3, and 2, respectively. The shared loci conditioning on these blood lipid traits was found on chromosomes 1, 6, 9, 12, 13, 15, and 19, and the shared loci conditioning on cIMT was located on chromosome 19. No shared causal locus was found when conditioning on T2D or BMI. In addition, 5 loci (1p36.22, 9q34.2, 12q24.12, 13q14.13, and 15q26.1) showed pleiotropic effects on SBP and lipid traits simultaneously, and one locus (19p13.2) showed pleiotropic effects on cIMT and LDL-C simultaneously (Fig. 4, Table 3).

The chromosome map illustrates the colocalized loci for CVDs-CeVDs-RFs triplets. The left part of each chromosome displays the loci name and the corresponding gene names, while the right part shows circles of various colors representing the different CVDs-CeVDs-RFs triplets when conditioned on a specific risk factor. CVDs and CeVDs combinations are linked using the symbol “&”. In cases where a locus is associated with CVDs and CeVDs conditioned on multiple RFs, multiple circles are observed in a single row. CVDs cardiovascular diseases, CeVDs cerebrovascular diseases, RFs risk factors, CAD coronary artery disease, MI myocardial infarction, HF heart failure, AF Atrial fibrillation, AIS all ischemic stroke, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, T2D type 2 diabetes, BMI body mass index, cIMT carotid intima-media thickness.
Gene prioritization and functional annotation
Functional annotation
For the 11 potential causal loci, we used Locus to Gene (L2G) and Variant to Gene (V2G) to prioritize the potential causal genes, namely CASZ1, CDKN1A, TWIST1, CDKN2B, ABO, SWAP70, SH2B3, LRCH1, FES, GOSR2, RPRML, and LDLR (Table 3). GOSR2 and RPRML are mapped into 17q21.31 by L2G and V2G, respectively. Open Targets Genetics (https://genetics-docs.opentargets.org/) database shows that all these genes have evidence of association with CVDs and CeVDs in previous association studies. Positional and functional annotations of the 11 causal loci are also shown in Table 3. Some of the loci showed potential biological functions. For example, rs2107595 (in the regulatory region of TWIST1 gene) has a Combined Annotation Dependent Depletion (CADD) score of 18.1 and is classified as 2b in RegulomeDB, providing strong evidence of functional effects such as DNase hypersensitivity, transcription factors binding sites and promoter sites. Rs1535791 (in the intron of the LRCH1 gene) also exhibits a high CADD score of 17.94. Rs35346340 (in the splice region of the FES gene), rs76774446 (in the intron region of the GOSR2/RPRML gene), and rs4135240 (in the intron region of CDKN1A gene) all have a RegulomeDB classification of 2b, indicating significant functional effects. All these genes are involved in the biological process of lipid metabolism (LDLR and ABO), regulation of immune cells (LRCH1 and FES), regulation of phosphatidylcholine catabolic process (LDLR and SWAP70), cell differentiation (CASZ1 for neuron cell, TWIST1 and CDKN1A for heart cell, CDKN2B for hematopoietic cell), etc., based on the Gene Ontology database (GO) (Supplementary Data 12). The expression of these genes in different tissues based on GTEx V8.0 was shown in Supplementary Fig. 24, particularly, LRCH1, RPRML/GOSR2, and CDKN1A genes were highly expressed in the brain. Annotation of pleiotropic variants and loci for CVDs-RFs and CeVDs-RFs pairs were shown in Supplementary Data 13. The majority of these variants have potential functional effects based on CADD score and RegulomeDB ranking. There are 787 and 37 genes annotated to the colocalized loci for pairs of CVDs-RFs and CeVDs-RFs separately (Supplementary Data 14).
Multi-omics pleiotropic associations for CVDs and CeVDs
Omics pleiotropic association (OPERA) analysis was used to uncover plausible molecular mechanisms for the 11 shared loci between CVDs and CeVDs with three types of omic data (GWAS, mQTL, and eQTL). In the pleiotropic loci located in the CASZ1 gene, there were 4 leading SNPs reported in original GWAS studies of vascular diseases and RFs (rs17035646 for AS and SBP, rs880135 for AF and TG, rs12046278 for LDL-C, rs4845953 for CAD), which exhibit strong linkage disequilibrium (LD) in one block. Two CpG sites specified in brain, blood, and artery tissues (namely cg15622917 and cg12760995) in this locus were colocalized with these leading SNPs in our analysis (PPA (posterior probability of associations) > 0.9 and P_HEIDI (P for heterogeneity in the dependent instrument test) > 0.01, Fig. 5, Supplementary Data 15). In the pleiotropic loci located in the LRCH1 gene, two CpG sites specified in brain tissue (cg11342437 and cg20294319, PPA > 0.9 and P_HEIDI > 0.01) were colocalized with the leading SNPs in original GWAS studies. Besides the above finding, we also identified multi-omics pleiotropic associations in CVDs or CeVDs alone, and most of them were two types of association signals (GWAS and mQTL, GWAS and eQTL), except that in AS, three types of molecular signals (GWAS, mQTL, and eQTL) were jointly associated with AS (Supplementary Data 15)46.

A Prioritizing DNA methylation sites at the CASZ1 locus for CAD, AF, and AS conditioned on SBP, TG and LDL-C. The top two tracks show the −log10(P) of the GWAS SNPs for vascular diseases (CAD (in orange), AF (in red) and AS (in green)) and related risk factors (SBP (in red), TG (in purple), LDL-C (in blue)), respectively. The next two tracks show −log10(P) of SNP associations for DNA methylation sites (cg12760995 and cg15622917) with OPERA marginal PPA > 0.9 and P-HEIDI > 0.01 (see the “Methods” section). The subsequent track shows the gene name and size of the flanking region within 500 kb of the 10.8 MB on chromosome 1. The track on the bottom shows 14 chromatin state annotations inferred from the 127 Roadmap Epigenomics Mapping Consortium samples at the 10.6 Mb to 11.0 Mb position of chromosome 1. B LD pattern of GWAS SNPs for CAD, AF, AS, SBP, TG, and LDL-C at the CASZ1 locus. The top six tracks show the −log10(P) of the GWAS SNPs for vascular diseases (CAD, AF, and AS) and related risk factors (SBP, TG, LDL-C) within 10 kb of the 10.8 MB on chromosome 1. In each single track, the red diamond with a black border represents the index SNP with the most strongly association in the region, and dots of different colors represent different LD scores with the index SNP. The track on the bottom shows LD heatmap, and the triangles with black borders represent the identified blocks defined by Gabriel et al.46. CAD coronary artery disease, AF Atrial fibrillation, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol.
Pathway enrichment and tissue specificity
The shared genes between CVDs and CeVDs were enriched in the GO terms related to cellular response to external stimulus, regulation of phosphate metabolic process, and heart development, and were enriched in the KEGG pathways of Cushing syndrome and cancer (Supplementary Fig. 25, Supplementary Data 16). Enrichment analyses for GO terms and KEGG pathways were conducted for colocalized loci of pairs of CVDs-RFs and CeVDs-RFs separately (Supplementary Data 17, 18). For the pairs of CVDs-RFs, enriched pathways also included cholesterol metabolism, pancreatic secretion, and AGE-RAGE signaling pathway in diabetic complications, etc.
The shared genes between CVDs and CeVDs are highly expressed in these organs (cardiovascular system and digestive system), tissues (esophagus, stomach, and artery), and cell types (endothelial cell, epithelial cell, smooth muscle cell) (Fig. 6A–C). The results also showed that colocalized genes for the CVDs-RFs pairs, particularly for SBP, were highly expressed in multiple systems, including the endocrine system (thyroid gland, mammary gland), sensory system (eye, retina), urinary system (bladder organ and kidney), respiratory system (lung and trachea) (Fig. 6D).

A–C Organ, tissue, and cell type enrichment for shared loci between CVDs and CeVDs based on all RFs, respectively. The y-axis in all three figures represents the -log (combined P value) which indicates the probability that the set of significantly expressed genes of a given size would have occurred by chance. The smaller the P value, the more likely that the pathway is significantly pathway identified. The x-axis represents different organs, tissues, and cell types in each figure, respectively. D Gene set size distribution and organ enrichment for pleiotropic loci shared by different CVDs or CeVDs and RFs combinations. The upper bar chart represents the number of gene sets shared by CVDs and RFs or CeVDs and RFs, in which different colors mean different CVDs or CeVDs and RFs combinations. In the lower chart, the left rectangles with different colors represent different organ systems, and each square with different colors from red to blue represents the −log (combined P value) of a certain CVDs/CeVDs-RFs combination in one specific organ system enrichment. Each column represents the enrichment of one specific CVDs-RFs or CeVDs-RFs combination, and each row represents the enrichment for different CVDs or CeVDs and RFs pairs in one specific organ system. CVDs cardiovascular diseases, CeVDs cerebrovascular diseases, RFs risk factors, CAD coronary artery disease, MI myocardial infarction, HF heart failure, AF atrial fibrillation, AIS all ischemic stroke, SBP systolic blood pressure, TG total triglyceride, LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, T2D type 2 diabetes, BMI body mass index, cIMT carotid intima-media thickness.
Druggability
We used the Genome for REPositioning drugs (GREP) database to quantify the enrichment of shared genes between CVDs and CeVDs in the target of clinical indication categories and found they were enriched in the targets of “antihypertensive drugs” (odds ratio = 26.7, Fisher’s exact P value = 0.05) and “antineoplastic agents” (odds ratio = 4.3, Fisher’s exact P value = 0.26). To support the robustness, we also conducted pleiotropy informing prioritization and evaluation (PIPE) analysis to advance therapeutic discovery. We used 21 shared causal variants for CVDs-CeVDs-RFs triplets in MOLOC results (Table 3) as pleiotropic association inputs and called six cardiometabolic disorders of interest, including cardiovascular disease (contains the CVDs and CeVDs defined in this study, hereafter as “CVDs & CeVDs”), hypertension, hyperlipidemia, obesity, diabetes mellitus, and internal carotid artery stenosis. Pleiotropy informing clinical therapeutics (PICT) was calculated to enable the identification of the specific disease in which pleiotropy can inform clinical therapeutics, and the highest score (PICT = 4.85, with a significant enrichment FDR = 1E−5) was observed for CVDs & CeVDs (Supplementary Data 19). Next, we elucidated inter-disease relationships inferred by the shared prioritization of therapeutic targets within six cardiometabolic disorders (Supplementary Fig. 26). As shown in Supplementary Fig. 27, based on the degree of shared prioritized target genes, the strongest relationship was observed between CVDs & CeVDs and hypertension. To visually represent the inferred relationships, we provided a side-by-side summary of clinical proof-of-concept targets for each disease of six cardiometabolic disorders (Supplementary Fig. 28).
Discussion
Relying on the largest European ancestry summary GWAS statistics to date, we investigate shared genetic components between CVDs, CeVDs, and their common cardiometabolic RFs. A total of 11 pleiotropic loci (12 genes: CASZ1, CDKN1A, TWIST1, CDKN2B, ABO, SWAP70, SH2B3, LRCH1, FES, GOSR2, RPRML, LDLR) were identified to be simultaneously associated with CVD, CeVD, and RFs. Among these loci, more than half of them were found to be related to SBP and blood lipids but none was related to CVDs-CeVDs-T2D and CVDs-CeVDs-BMI triplets. Among these 12 genes, two genes (SWAP70 and LRCH1) were newly discovered in recent large GWAS studies6,47. CASZ1 and LRCH1 have multi-omics evidence that differential DNA methylation sites were identified in these loci, and they showed brain, blood, and artery tissue specificity. All of these shared genes were found to be highly expressed in endothelial cells of the cardiovascular and digestive systems and were enriched in pathways related to response to external stimuli, regulation of metabolic processes, and cardiac development. These genes were enriched in genes targeted by antihypertensive drugs. Based on the shared prioritization of therapeutic targets, the strongest relationship was also observed between CVDs & CeVDs and hypertension. Our study suggested the use of enrichment of cardiometabolic RF-related genes could enhance the ability to identify sharing variants of diseases. Genetic causes discovered from pleiotropic variants associated with RFs also help to understand disease pathogenesis and propose population interventions.
We found significant positive genetic correlations between CVDs and CeVDs, and the genetic overlap accounted for the majority proportion of each disease, in line with previous findings5,16,37,48. Beyond genetic correlation, ConjFDR analyses also identified a large number of pleiotropic variants associated with both CVDs and CeVDs, most of which had been reported in previously two traits joint meta-analyses10. After introducing RFs into the analysis of genetic overlap between CVDs and CeVDs, we found both CVDs and CeVDs genetically correlated to all RFs, specifically, the pairs of HF-BMI, HF-T2D, AS-SBP, and AIS-SBP exhibit the highest genetic correlations (rg-HDL > 0.4), and a number of pleiotropic genetic variants were also identified for CVDs or CeVDs and RFs combinations, in line with the previous findings5,10,26,37,48,49,50,51,52. There were also some diversities, for example, CVDs seemed to have much more pleiotropic variants associated with RFs than CeVDs. Moreover, these pleiotropic variants contribute to a higher heritability for CVDs compared to CeVDs. Previous GWAS studies have also demonstrated a substantially larger number of genetic variants associated with CVDs in comparison to CeVDs5,6. There were several possible explanations for these findings. Firstly, the sample size of the GWAS studies utilized for identifying pleiotropic variants was considerably larger for CVDs compared to CeVDs. This discrepancy in sample sizes results in different statistical power for discovering significant pleiotropic variants associated with CVDs and CeVDs. Secondly, the total estimated heritability for CeVDs was lower than that of CVDs based on the same GWAS summary data when estimating from LDSC and HDL methods (Supplementary Data 20). This lower heritability may indicate a smaller number or weaker effect of genetic determinants for CeVDs, as a larger proportion of the variation is attributed to non-genetic factors. Thirdly, there was a considerably greater degree of phenotypic heterogeneity observed in CeVDs compared to any of CVDs. Taking AIS as an example, it is usually classified into different subtypes according to the well-known TOAST classification. This increased heterogeneity makes it more difficult to identify the genetic etiology of CeVDs. Fourthly, pleiotropic variants analyzed in our study primarily focused on SNPs. However, there may be many more additional types of genetic variants present for CeVDs, such as rare variants, copy number variants, and other structural variations53.
After using stringent criteria to select pleiotropic loci through MOLOC analyses, a total of 11 possible causal loci were identified to be shared by CVDs and CeVDs based on RFs. The 11 loci were mapped to 12 genes, namely CASZ1, CDKN1A, TWIST1, CDKN2B, ABO, SWAP70, SH2B3, LRCH1, FES, GOSR2, RPRML, and LDLR. The pleiotropic associations between CVDs and CeVDs in these 11 loci have been reported in previous studies17,37. Particularly, the study by Bingxin Zhao et al. found genetic pleiotropy between multiple cardiovascular magnetic resonance imaging (CMR) traits and brain-related disorders (particularly AS) in 6p21.2 (corresponding to CDKN1A), 7p21.1 (corresponding to TWIST1), and 12q24.12 (corresponding to SH2B3) regions17. Rs2107595 in 7p21.1 was eQTLs of TWIST1 in human brain tissues54, suggesting that the heart-associated variants were known to affect gene expression in the human brain. LRCH1 and SWAP70 were recently identified novel genes associated with both CVDs and CeVDs6,47. Among these 11 loci, most loci were found in the analysis based on SBP and blood lipids, implying the shared genetic components of CVDs and CeVDs were most likely contributed by SBP and lipids traits. These results were also ascertained in the partitioned heritability analysis based on different RFs. Our study identified shared genetic loci for CVDs-CeVDs-BP/lipids but did not detect shared loci for CVDs-CeVDs-T2D/BMI, despite T2D/BMI being common shared environmental RFs for both CVDs and CeVDs. The GWAS sample sizes used to analyze shared genetic determinants were comparable between T2D/BMI and BP/lipid traits. Therefore, the lack of shared loci for CVDs-CeVDs-T2D/BMI is unlikely to be attributed to insufficient statistical power due to imbalanced sample sizes. Differences in effect sizes of loci across traits may contribute to this result. Moreover, these findings suggest that BP, lipids, T2D, and BMI may play distinct roles in the development of CVDs and CeVDs. Previous studies have demonstrated that hypertension preceding T2D is associated with a significantly higher risk of CVDs and CeVDs compared to T2D preceding hypertension55. The genetic predisposition to T2D/BMI may influence the risk of CVDs and CeVDs through other factors or pathological pathways, such as BP and lipid levels56,57. These results indicate that the relationship between BP/lipids and CVDs-CeVDs may be more direct and has a stronger effect compared to the relationship between T2D/BMI and CVDs-CeVDs. Furthermore, environmental factors may play a more significant role in the pathway linking T2D/BMI to CVDs and CeVDs. A recent cohort study has provided supporting evidence that obese adults determined by environment (obesity despite genetically predicted low BMI) have a higher cardiovascular risk compared to those who are obese due to genetic predisposition (genetically predicted high BMI)58. Although no shared genetic locus for CVDs-CeVDs-T2D/BMI was identified in this study, we found that CVDs and CeVDs separately shared some pleiotropic loci with T2D/BMI. These findings remain valuable for advancing our understanding of the pathogenic pathways underlying CVDs and CeVDs.
Extensive multi-omics analyses highlight two out of the 11 loci (1p36.22 and 13q14.13, corresponding to CASZ1 and LRCH1) for prioritization in epigenetic regulation. CASZ1 (Castor Zinc Finger 1) encodes a protein named zinc finger transcription factor involved in vascular assembly and morphogenesis through direct transcriptional regulation of epidermal growth factor-like domain 759. The methylation level of cg12760995 in CASZ1 was previously reported to be associated with SBP and AIS60. Polymorphism in LRCH1 has been reported to be associated with stroke37 and SBP61. The regulatory factor of LRCH1 was active in multiple arteries, tissues and blood, and gene expression of this gene was significantly associated with stroke47. In addition, cg11342437 in LRCH1 was found in our study to be causally linked to CVDs and CeVDs, which was consistent with the important molecular underpinnings of aging-related degenerative disorders with possible pathways in cellular-signaling events and transmission across chemical synapses62. Analyses on biological functions, cell type enrichments, and drug target enrichment for these loci in our study further supported the role of shared pathways related to blood pressure and lipid metabolism for CVDs and CeVDs.
In our analysis, the shared genes of CVDs and CeVDs showed significant enrichment in the target genes of antihypertensive drugs. Recently, several tools and methods have been developed using genetic variants that serve as proxies for drug effects to study corresponding drug side-effects and repurposing potential33,63,64,65, with implications for clinical practice of CVDs and CeVDs, subsequent trials as well as drug development of polypills6,66. Using a GWAS meta-analysis of 750,000 individuals, Dipender Gill et al. identified genetic variants that were significantly associated with SBP within the loci of the genes encoding the targets of β-blockers (BBs), calcium channel blockers (CCBs), and angiotensin-converting enzyme inhibitors67. As proxies for antihypertensive drug classes, these variants showed associations with the risk of CAD and stroke, which were comparable to those derived from clinical trials testing antihypertensive drugs against placebo67,68. Another study also showed that genetically proxied medications of antihypertensive drugs (mediated by targets of BBs or CCBs) had an effect on CAD and stroke66. These findings highlight antihypertensive drugs as a promising strategy for the prevention of both CVDs and CeVDs.
In this study, we also found some different genetic patterns for CVDs and CeVDs. For example, we found all RFs shared a similar number of pleiotropic loci with CVDs (CAD, MI, and AF), but SBP shared many more loci with CeVDs than other RFs. This finding was in line with the previous studies, that hypertension exhibited the strongest association with both family risk of stroke and MI, while T2D, dyslipidemia, and obesity displayed consistent and relatively high associations with MI (odds ratio range: 1.22–1.38) but much weaker risk of stroke (odds ratio range: 1.04–1.26)69. Another study also reported hypertension had clear genetic correlations with heart and brain health17. In addition, our results showed that pleiotropic loci for pairs of CVDs-RFs were widely distributed across the human autosomes, while pleiotropic loci for pairs of CeVDs-RFs were primarily concentrated on chromosomes 1, 4, 6, 7, 9, 10, 12, and 13. These findings were consistent with previous genetic studies on CVDs and CeVDs6,70, which further imply the specific pathologic pathways for CVDs or CeVDs.
Our study has several strengths. First, this study explored the overlapping genetic basis between vascular diseases in different organs through pleiotropy on common risk factors. It provides a novel strategy for understanding the shared genetic mechanisms existing in human complex diseases. Second, the study advances previous knowledge on the sharing genetic etiology for CVDs and CeVDs by providing insights into the potential biological mechanisms, which could facilitate the development of disease prevention and treatment strategies. However, our study also has some limitations. Given the consideration of sample size, only GWAS studies with European ancestry have been analyzed in this analysis. The power to detect causal signals will be improved if multi-ancestry research with large sample sizes is included. Furthermore, there are additional common RFs beyond the seven analyzed in this study. More genetic variants would be revealed incorporating additional RFs in further research. Nevertheless, our study presents a novel approach to understanding the shared genetic component between two types of diseases, which is applicable and can be utilized for other human complex traits.
Conclusion
In this study, we aimed to unravel the shared genetic etiology of two complex traits, CVDs and CeVDs, by disentangling the pleiotropic variants with their common RFs. We discovered that CVDs and CeVDs possess significant shared genetic components, primarily linked to blood pressure and lipid traits. Furthermore, these shared genetic components are likely to be regulated through epigenetic mechanisms and may potentially serve as targets for antihypertensive drugs. This research can provide valuable insights into the underlying mechanisms of these diseases and potentially contribute to the development of more targeted interventions and treatments.
Methods
GWAS datasets
Summary statistics for CVDs including CAD5, MI34, HF35, and AF36, CeVDs including AS37 and AIS37, and their RFs including SBP39, TG42, LDL-C42, HDL-C42, T2D40, BMI41, and cIMT45, were obtained from the largest published GWASs of European descent of that trait to date (March 2023). Sample size, ancestry, sources, and other information for the different GWAS were given in Supplementary Data 21, and the extensive quality control procedures were also described in detail in original publications. The location of SNPs was based on the Genome Reference Consortium Human Build 37 (GRCh37), and the MHC region was removed from the analysis mainly because of the complicated LD structure.
MiXeR analysis
We applied MiXeR44 to quantify the polygenic overlap between each CVD and each CeVD using GWAS summary statistics. A bivariate Gaussian mixture model was constructed to estimate the total number of shared and trait-specific causal variants, which jointly account for 90% of SNP heritability in each phenotype, represented as Venn diagrams of shared and unique polygenic components across traits. We used the 1000 Genomes Project phase 3 data71 as a genotype reference panel to calculate the LD score for the European populations.
Genetic correlation analysis
To estimate SNP-based genetic correlations (rg) between CVDs, CeVDs, and their RFs using GWAS summary statistics, we performed the LDSC (cross-trait linkage disequilibrium score) regression analysis72,73 and the HDL (high-definition likelihood) analysis74, we computed LD scores using the 1000 Genomes Phase 371 for LDSC and “1,029,876 QCed UK Biobank imputed HapMap3 SNPs” (https://github.com/zhenin/HDL/wiki/Reference) for HDL. In addition, we estimated the heritability of each phenotype, including CVDs, CeVDs, and their RFs, with the default parameter settings using both LDSC and HDL methods.
Conditional and conjunctional false discovery rate analysis
We used the conditional false discovery rate (condFDR) and conjunctional false discovery rate (conjFDR) method38 implemented in pleioFDR to identify pleiotropic variants, which allows us to re-adjust the GWAS statistics in a primary phenotype by leveraging pleiotropic enrichment with a GWAS in a secondary phenotype. For CVDs or CeVDs and their RFs, condFDR estimates were conducted to increase the discovery of disease-associated SNPs. For CVDs and CeVDs, conjFDR estimates were conducted to discover SNPs likely to be shared by two phenotypes simultaneously. Thresholds for significant condFDR and conjFDR associations were set at 0.05 and 0.01 as suggested. Furthermore, S-LDSC (stratified LD-score regression)75 was carried out to estimate the proportion of heritability explained by the disease-associated SNPs conditioning on a certain risk factor according to condFDR analysis.
Genomic loci definition
We defined independent genomic loci of condFDR and conjFDR results according to FUMA (functional mapping and gene annotation) protocol76. The independent significant SNPs were defined as SNPs with condFDR < 0.01 or conjFDR <0.05 and LD r2 < 0.6 with each other. Subsequently, a subset of lead SNP was obtained using the clumping threshold of LD r2 < 0.1. The border of the genetic loci was defined with candidate SNPs that have an LD r2 ≥ 0.6 with at least one independent significant SNP. Two loci were merged if they were closer than 250 kb. 1000 Genomes Project phase 371 was used as a reference panel to compute LD. Therefore, we defined (1) the pleiotropic loci for one CVD conditioning on an RF based on condFDR estimates, (2) the pleiotropic loci for one CeVD conditioning on an RF based on condFDR estimates, (3) If the pleiotropic loci for one CVD and one CeVD conditioning on the same RF overlap in genomic location, we combined them into one pleiotropic loci for CVD-CeVD-RF triplet. We considered loci not identified in the GWAS catalog (downloaded in January 2024) and in previous GWAS studies5,34,35,36,37,39,40,41,42,45 as novel risk loci. We inferred the effect directions of the pleiotropic loci by comparing the z-scores in the GWAS summary statistics corresponding to the phenotype.
Colocalization analysis
To estimate the probability that each pair of vascular disease and its RF shared the same causal variant(s) in a given pleiotropic locus, we conducted a Bayesian test for colocalization of two traits using SuSiE algorithm in the COLOC package29,30. COLOC-SuSiE requires a matrix of signed linkage disequilibrium (r) values, which we generated using LDlinkR77. This method also requires specifying prior probabilities for an SNP being associated with trait1 only (P1), trait2 only (P2), and with both traits (P12). We applied the default P values, with P1 and P2 set to 1 × 10−4, assuming that 1 in 10,000 SNPs are causal for either trait and P12 was set to 1 × 10−5. Evidence for colocalization was assessed using the posterior probability for hypothesis 4 (PPH4, indicating the two phenotypes share the same causal variant(s) at the locus). We used PPH4 > 80% as a threshold to suggest that associations were highly colocalized. To determine the 95% credible set of colocalized variants, we extracted the posterior probabilities of each SNP, conditioned on H4 being true.
Multiple-trait colocalization analysis
To estimate the probability of whether the same causal variant is driving CVDs-CeVDs-RFs triplet in a given pleiotropic region, we performed multiple-trait-colocalization analysis across three traits (CVDs, CeVDs, and RFs) using MOLOC31. The method requires specifying the prior probabilities for the association of a SNP with one (P1), two (P2), or three traits (P3), and the variance W of the normal prior. We set the prior probabilities to P1 = 1 × 10−4, P2 = 1 × 10−5, P3 = 1 × 10−6, as the posterior expectation of colocalization most closely resembled the prior expectation under this choice of priors in a sensitivity analysis using different priors. We set the W to average the Bayes Factors across three prior variances, 0.01, 0.1, and 0.5, as suggested78. MOLOC computed the evidence supporting the 15 possible configurations (H0…H14) of sharing of SNPs among three traits in the given genomic region. The posterior probability for hypothesis 14 (PPH14) represents the posterior probability that all three traits are associated and share the same causal variant, with 80% or higher being considered evidence of colocalization. The most likely causal SNP in this region with the highest posterior probability was also reported. A comprehensive overview of the colocalization analysis method is provided in Supplemental Method.
Gene prioritization
To assign conceivable causal genes for the 95% credible causal SNPs in COLOC analysis and the most likely causal SNPs in MOLOC analysis, we employed a V2G (Variant to Gene) analysis pipeline79, which provides a single aggregated score for each variant-gene prediction. This analysis combined four categories of predictive features: molecular phenotype quantitative trait loci experiments (e.g. eQTLs and pQTLs), Chromatin interaction experiments (e.g. Promoter Capture Hi-C), In silico functional predictions (e.g. Variant Effect Predictor from Ensembl80), and distance from the canonical transcript start site (TSS). To prioritize multiple genes at colocalized loci (PPH4 in COLOC analysis or PPH14 in MOLOC analysis > 0.8), we employed an L2G (Locus to Gene) analysis pipeline79, which provides a single aggregated score for each locus-gene prediction. The L2G pipeline combined data from three sources, including pathogenicity prediction, colocalization with molecular quantitative trait loci, and genomic distance and chromatin interaction data, to generate L2G predictive features. Genes with the highest V2G/L2G scores for each variant/locus, indicating the highest confidence variant/locus-gene assignment, were included in subsequent analyses.
Functional annotation
The 95% credible causal SNPs in COLOC analysis and the most likely causal SNPs in MOLOC analysis were then annotated for functional consequences on gene functions (based on Ensembl genes [build 85] using ANNOVAR81), deleteriousness score (combined annotation dependent depletion [CADD] score using VarNote82), and potential regulatory functions (RegulomeDB score using VarNote82).
The most likely causal genes assigned by V2G/L2G pipeline were annotated using Metascape43, encompassing 7 categories: gene descriptions (NCBI database), disease implications (DisGeNET database, GeDiPNet database), pathogenic loss-of-function (ClinVar database), GWAS (NHGRI-EBI database), drug target annotation (DrugBank database), protein function (Protein Atlas database), and pathway mapping (Gene Ontology [GO] database, KEGG database). Moreover, Normalized gene expressions (reads per kilobase per million, RPKM) of 54 tissue types of shared genes (those assigned in colocalized loci of CVD-CeVD-RF triplets or CVD-CeVD pairs) were obtained from Genotype-Tissue Expression Project version 8 (GTEx v8)83.
Pathway enrichment analysis
To identify risk pathways and gain mechanistic insights from genes of CVDs/CeVDs indicated by a specific RF, functional enrichment for GO terms84 and KEGG pathways85 was conducted using Metascape43. The integrated gene lists below served as inputs to ensure sufficient statistical power: (1) CVDs | RFs gene lists (e.g. CVDs | SBP gene list): consisting of the most promising genes at colocalized loci of all CVDs (CAD, MI, HF, and AF) and a certain risk factor. (2) CeVDs | RFs gene lists (e.g. CeVD | SBP gene list): consisting of the most promising genes at colocalized loci of all CeVDs (AS, AIS) and a certain RF. (3) CVDs and CeVDs gene list: consisting of the most credible genes at colocalized loci of all CVDs-CeVDs-RFs triplets. FDR < 0.05 was considered as significant. Besides, Metascape automatically clustered enriched terms into non-redundant groups and chose the most significant (lowest P value) term to represent the cluster.
Tissue/cell-type enrichment analysis
To understand the context-specific expression of genes of CVD/CeVD indicated by a specific RF in human tissue-cell types (TCs), enrichment analyses for TCs specificity were conducted using WebCSEA (Web-based Cell-type Specific Enrichment Analysis of Genes)86, with the same input lists as the pathway enrichment analysis. WebCSEA was conducted over 1355 TCs by leveraging single-cell RNA sequencing (scRNA-seq) data and generated combined P values to measure the TCs specificity without the bias from the number of signature genes between different TCs and input gene list size. We identified significant enrichment TCs for the gene sets at a significance level of combined P value < 0.01 (https://bioinfo.uth.edu/webcsea/tutorial.php).
Druggability analysis
To support the currently approved drugs and discover potentially repositionable drugs for other indications, functional enrichment for targets of drugs classified according to Anatomical Therapeutic Chemical Classification System (ATC) codes was conducted using GREP (genome for REPositioning drugs)65. GREP performed a series of Fisher’s exact tests to examine whether the gene set is enriched in genes targeted by drugs in a clinical indication category by ATC and further outputted the names of the drugs targeting the gene set. The drug-target database was collected from two major drug databases, Drug Bank87 and Therapeutic Target Database88, which made information on 22,300 drugs and 2029 genes of their target in total.
We also performed pleiotropy informing prioritization and evaluation (PIPE) analysis to leverage shared genetic and pathway architectures of CVDs-CeVDs-RFs triplets to advance therapeutic discovery33. The shared causal variants for CVDs-CeVDs-RFs triplets in MOLOC results with a uniformly assumed association p-value of 1e−8 were used as pleiotropic association inputs. PIPE features two components—prioritization and evaluation. Firstly, using default settings, we established the link from pleiotropic loci, including those located within the non-coding genome, to candidate target genes, which was made possible by harnessing the value of multimodal regulatory genomic datasets (genomic evidence) and leveraging knowledge of protein interactions (network evidence). Subsequently, we quantified the tendency of prioritized genes to be clinical proof-of-concept targets for several diseases of interest, including cardiovascular disease (contains the CVDs and CeVDs defined in this study), hypertension, hyperlipidemia, obesity, diabetes mellitus, and internal carotid artery stenosis in the ChEMBL database (https://www.ebi.ac.uk/chembl/) via leading prioritization analysis (LPA). Specially, LPA was performed to assess the extent to which clinical proof-of-concept targets are enriched at the “leading prioritization” of the entire prioritized gene list and reports three pieces of information on the enrichment results: normalized enrichment score, enrichment significance (false discovery rate), and the fraction of clinical proof-of-concept targets recovered at the leading prioritization (coverage). All three results were factored into the calculation of pleiotropy informing clinical therapeutics (PICT). For each disease, we defined its member genes as clinical proof-of-concept targets at the leading prioritization. The relationship between any two diseases was inferred by the extent of sharing their member genes.
Multi-omics association analysis
To uncover plausible molecular mechanisms underlying shared loci of CVDs and CeVDs, we jointly analyzed v8 release of the GTEx eQTL (gene expression quantitative trait loci) summary data83 and mQTL (DNA methylation quantitative trait loci) summary data in peripheral blood89,90 with GWAS summary data using OPERA (Omics PlEiotRopic Association)32. OPERA is a Bayesian extension of the summary-data-based Mendelian randomization (SMR) and HEIDI (heterogeneity in dependent instruments) approach91 to a multi-omics model, where the molecular phenotypes are considered as exposures, and the complex trait is considered as the outcome. This tool can, therefore, be used to prioritize molecular phenotypes that mediate the genetic effects for complex traits and further provide mechanistic interpretation of the GWAS signal. We implemented OPERA analysis in each colocalized loci of CVDs-CeVDs-RFs triplet with CVDs and CeVDs GWAS summary data separately, using default parameter settings (posterior probability of associations (PPA) > 0.9 and P_HEIDI (P for heterogeneity in the dependent instrument test) > 0.01).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets analyzed during the current study are freely available for download from the following URLs: GWAS summary data for CAD (https://www.ebi.ac.uk/gwas/studies/GCST90132314); GWAS summary data for MI (https://www.ebi.ac.uk/gwas/publications/33532862); GWAS summary data for HF (https://www.ebi.ac.uk/gwas/publications/31919418); GWAS summary data for AF (https://www.ebi.ac.uk/gwas/publications/29892015); GWAS summary data for AS and AIS (https://cd.hugeamp.org/dinspector.html?dataset=GWAS_MEGASTROKE_eu); GWAS summary data for SBP (https://www.ebi.ac.uk/gwas/publications/30224653); GWAS summary data for TG, LDL-C, HDL-C (https://csg.sph.umich.edu/willer/public/glgc-lipids2021/); GWAS summary data for T2D (https://diagram-consortium.org/); GWAS summary data for BMI (https://www.ebi.ac.uk/gwas/publications/30239722); GWAS summary data for c-IMT (https://www.ebi.ac.uk/gwas/publications/34852643). LD scores and reference panel derived from 1000 Genomes phase 3, https://data.broadinstitute.org/alkesgroup/LDSCORE/; LD scores and reference panel derived from 1,029,876 QCed UK Biobank imputed HapMap3 SNPs (https://github.com/zhenin/HDL/wiki/Reference).
Code availability
All code used in this project is available at https://doi.org/10.5281/zenodo.1427997292.
References
Organization, W. H. Cardiovascular Diseases (CVDs) Fact Sheet https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1.
Heart Research Institute, N. Cardiovascular Disease: Impacts and Risks https://www.hri.org.nz/health/learn/cardiovascular-disease/cardiovascular-disease-impacts-and-risks.
Gallacher, K. I., Jani, B. D., Hanlon, P., Nicholl, B. I. & Mair, F. S. Multimorbidity in stroke. Stroke 50, 1919–1926 (2019).
Buddeke, J. et al. Comorbidity in patients with cardiovascular disease in primary care: a cohort study with routine healthcare data. Br. J. Gen. Pract. 69, e398–e406 (2019).
Aragam, K. G. et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 54, 1803–1815 (2022).
Mishra, A. et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611, 115–123 (2022).
Calling, S., Ji, J., Sundquist, J., Sundquist, K. & Zöller, B. Shared and non-shared familial susceptibility of coronary heart disease, ischemic stroke, peripheral artery disease and aortic disease. Int. J. Cardiol. 168, 2844–2850 (2013).
KHAW, K.-T. & BARRETT-CONNOR, E. Family history of stroke as an independent predictor of ischiemtc heart disease in men and stroke in women. Am. J. Epidemiol. 123, 59–66 (1986).
Banerjee, A. et al. Familial history of stroke is associated with acute coronary syndromes in women. Circulation: Cardiovasc. Genet. 4, 9–15 (2011).
Dichgans, M. et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke 45, 24–36 (2014).
Holdt, L. M. & Teupser, D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler. Thromb. Vasc. Biol. 32, 196–206 (2012).
Ding, H. et al. A novel loss-of-function DDAH1 promoter polymorphism is associated with increased susceptibility to thrombosis stroke and coronary heart disease. Circ. Res. 106, 1145–1152 (2010).
Bentley, P., Peck, G., Smeeth, L., Whittaker, J. & Sharma, P. Causal relationship of susceptibility genes to ischemic stroke: comparison to ischemic heart disease and biochemical determinants. PLoS ONE 5, e9136 (2010).
Zhang, Y. et al. Significant overlapping modules and biological processes between stroke and coronary heart disease. CNS Neurol. Disord. Drug Targets 13, 652–660 (2014).
Frerich, S. et al. Cardiac risk factors for stroke: a comprehensive Mendelian randomization study. Stroke 53, e130–e135 (2022).
Cai, H. et al. Genetic correlations and causal inferences in ischemic stroke. J. Neurol. 267, 1980–1990 (2020).
Zhao, B. et al. Heart-brain connections: phenotypic and genetic insights from magnetic resonance images. Science 380, abn6598 (2023).
Georgakis, M. K. et al. Genetic architecture of stroke of undetermined source: overlap with known stroke etiologies and associations with modifiable risk factors. Ann. Neurol. 91, 640–651 (2022).
Wang, K. et al. Mendelian randomization analysis of 37 clinical factors and coronary artery disease in East Asian and European populations. Genome Med. 14, 63 (2022).
Hindy, G. et al. Role of blood lipids in the development of ischemic stroke and its subtypes: a Mendelian Randomization Study. Stroke 49, 820–827 (2018).
Ibanez, L. et al. Overlap in the genetic architecture of stroke risk, early neurological changes, and cardiovascular risk factors. Stroke 50, 1339–1345 (2019).
Xu, K. et al. The combined effects of cardiovascular disease related SNPs on ischemic stroke. J. Neurol. Sci. 388, 141–145 (2018).
Malik, R. et al. Multilocus genetic risk score associates with ischemic stroke in case-control and prospective cohort studies. Stroke 45, 394–402 (2014).
Franceschini, N. et al. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat. Commun. 9, 5141 (2018).
Kochunov, P. et al. Whole brain and regional hyperintense white matter volume and blood pressure: overlap of genetic loci produced by bivariate, whole-genome linkage analyses. Stroke 41, 2137–2142 (2010).
LeBlanc, M. et al. Identifying novel gene variants in coronary artery disease and shared genes with several cardiovascular risk factors. Circ. Res. 118, 83–94 (2016).
Andreassen, O. A. et al. Identifying common genetic variants in blood pressure due to polygenic pleiotropy with associated phenotypes. Hypertension 63, 819–826 (2014).
Smeland, O. B. et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum. Genet. 139, 85–94 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Wallace, C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 16, e1008720 (2020).
Giambartolomei, C. et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34, 2538–2545 (2018).
Wu, Y. et al. Joint analysis of GWAS and multi-omics QTL summary statistics reveals a large fraction of GWAS signals shared with molecular phenotypes. Cell Genom. 3, 100344 (2023).
Bao, C. et al. A cross-disease, pleiotropy-driven approach for therapeutic target prioritization and evaluation. Cell Rep. Methods 4, 100757 (2024).
Hartiala, J. A. et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. hHeart J. 42, 919–933 (2021).
Shah, S. et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 11, 163 (2020).
Roselli, C. et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet 50, 1225–1233 (2018).
Malik, R. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet 50, 524–537 (2018).
Andreassen, O. A. et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am. J. Hum. Genet. 92, 197–209 (2013).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet 54, 560–572 (2022).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174 (2019).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Frei, O. et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun. 10, 2417 (2019).
Yeung, M. W. et al. Twenty-five novel loci for carotid intima-media thickness: a Genome-Wide Association Study in >45 000 individuals and meta-analysis of >100 000 individuals. Arterioscler. Thromb. Vasc. Biol. 42, 484–501 (2022).
Gabriel, S. B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Yang, J. et al. Integrative analysis of transcriptome-wide association study and gene expression profiling identifies candidate genes associated with stroke. PeerJ 7, e7435 (2019).
Torgersen, K. et al. Shared genetic architecture between neuroticism, coronary artery disease and cardiovascular risk factors. Transl. Psychiatry 11, 368 (2021).
Morris, A. P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990 (2012).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Speliotes, E. K. et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 42, 937–948 (2010).
Goodarzi, M. O. & Rotter, J. I. Genetics insights in the relationship between type 2 diabetes and coronary heart disease. Circ. Res. 126, 1526–1548 (2020).
Meschia, J. F. Effects of genetic variants on stroke risk. Stroke 51, 736–741 (2020).
de Klein, N. et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain-related diseases. Nat. Genet. 55, 377–388 (2023).
Yen, F. S., Wei, J. C., Chiu, L. T., Hsu, C. C. & Hwu, C. M. Diabetes, hypertension, and cardiovascular disease development. J. Transl. Med 20, 9 (2022).
Van Gaal, L. F. & Maggioni, A. P. Overweight, obesity, and outcomes: fat mass and beyond. Lancet (Lond., Engl.) 383, 935–936 (2014).
Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration (BMI Mediated Effects) et al. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. The Lancet 383, 970–983 (2014).
Ojalehto, E. et al. Genetically and environmentally predicted obesity in relation to cardiovascular disease: a nationwide cohort study. EClinicalMedicine 58, 101943 (2023).
Charpentier, M. S. et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev. Cell 25, 132–143 (2013).
Mo, X. B., Zhang, H., Wang, A. L., Xu, T. & Zhang, Y. H. Integrative analysis identifies the association between CASZ1 methylation and ischemic stroke. Neurol. Genet. 6, e509 (2020).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019).
Tan, Q. et al. Epigenetic drift in the aging genome: a ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 45, 1146–1158 (2016).
Walker, V. M., Davey Smith, G., Davies, N. M. & Martin, R. M. Mendelian randomization: a novel approach for the prediction of adverse drug events and drug repurposing opportunities. Int J. Epidemiol. 46, 2078–2089 (2017).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021).
Sakaue, S. & Okada, Y. GREP: genome for REPositioning drugs. Bioinformatics 35, 3821–3823 (2019).
Li, Y. et al. Association of genetic variants related to combined lipid-lowering and antihypertensive therapies with risk of cardiovascular disease: 2 × 2 factorial Mendelian randomization analyses. BMC Med. 22, 201 (2024).
Georgakis, M. K. & Gill, D. Mendelian randomization studies in stroke: exploration of risk factors and drug targets with human genetic data. Stroke 52, 2992–3003 (2021).
Gill, D., Walker, V. M., Martin, R. M., Davies, N. M. & Tzoulaki, I. Comparison with randomized controlled trials as a strategy for evaluating instruments in Mendelian randomization. Int. J. Epidemiol. 49, 1404–1406 (2020).
Kennedy, R. E. et al. Association between family risk of stroke and myocardial infarction with prevalent risk factors and coexisting diseases. Stroke 43, 974–979 (2012).
Tcheandjieu, C. et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 28, 1679–1692 (2022).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Ning, Z., Pawitan, Y. & Shen, X. High-definition likelihood inference of genetic correlations across human complex traits. Nat. Genet. 52, 859–864 (2020).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Myers, T. A., Chanock, S. J. & Machiela, M. J. LDlinkR: an R package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front. Genet. 11, 157 (2020).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Ghoussaini, M. et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic acids Res. 49, D1311–d1320 (2021).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids Res. 38, e164 (2010).
Huang, D. et al. Ultrafast and scalable variant annotation and prioritization with big functional genomics data. Genome Res. 30, 1789–1801 (2020).
The, G. C. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids Res. 28, 27–30 (2000).
Dai, Y. et al. WebCSEA: web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Res. 50, W782–W790 (2022).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–d1082 (2018).
Yildirim, M. A., Goh, K. I., Cusick, M. E., Barabási, A. L. & Vidal, M. Drug-target network. Nat. Biotechnol. 25, 1119–1126 (2007).
McRae, A. F. et al. Identification of 55,000 replicated DNA methylation QTL. Sci. Rep. 8, 17605 (2018).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Ding, K. Shared Genetic Etiology of CVDs and CeVDs through RFs https://doi.org/10.5281/zenodo.14279972 (2024).
Acknowledgements
This work was supported by the High-performance Computing Platform of Peking University. This research was funded by the National Natural Science Foundation of China (No. 82073642).
Author information
Authors and Affiliations
Contributions
Yiqun Wu and Xueying Qin designed the study. Kexin Ding undertook data processing and conducted data analysis. Yiqun Wu, Xueying Qin, and Kexin Ding drafted the manuscript. Tao Wang, Huairong Wang, and Kun Wang contributed to the manuscript writing. Xiaoying Kang and Tao Wang provided support for the data analysis. Yao Yu, Yang Liu, Haiying Gong, Tao Wang, Xiaoying Kang, Tao Wu, Dafang Chen, and Yonghua Hu revised the manuscript. Yiqun Wu, Xueying Qin, and Kexin Ding were responsible for interpreting the data that the manuscript is based on. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Kaoru Ito and Dario Ummarino.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, K., Qin, X., Wang, H. et al. Identification of shared genetic etiology of cardiovascular and cerebrovascular diseases through common cardiometabolic risk factors. Commun Biol 7, 1703 (2024). https://doi.org/10.1038/s42003-024-07417-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-07417-6
