
Abstract
Membranous nephropathy (MN) is a primary glomerular disease commonly causing adult nephrotic syndrome. Characterized by thickened glomerular capillary walls due to immune complex deposition, MN is a complex autoimmune disorder. Its pathogenesis involves immune deposit formation, complement activation, and a heightened risk of renal failure. Central to MN is immune system dysfunction, particularly the dysregulation of B and T cell responses. B cells contribute to renal injury through the production of autoantibodies, particularly IgG targeting the phospholipase A2 receptor (PLA2R) on podocytes, while T cells modulate immune responses that influence disease progression. Metabolic reprogramming alters lymphocyte survival, differentiation, proliferation, and function, potentially triggering autoimmune processes. Although the link between immune cell metabolism and MN remains underexplored, this review highlights recent advances in understanding immune metabolism and its role in MN. These insights may provide novel biomarkers and therapeutic strategies for MN treatment.
Similar content being viewed by others


Introduction
Membranous nephropathy (MN) is a primary glomerular disease commonly causing adult nephrotic syndrome, about 80% of MN cases don’t have clear secondary cause, called idiopathic membranous nephropathy (IMN) or primary membranous nephropathy (PMN), and about 20% of MN cases are secondary to autoimmune diseases, infections, malignant tumors, medicine use, and heavy-metal toxicity, called secondary membranous nephropathy(SMN), It presents as diffuse subepithelial immune complex deposition on the glomerular basement membrane (GBM) which leads to GBM thickening, the alteration of podocyte structure, causing the appearance of substantial proteinuria1,2.
To date, nearly 10 autoantigens have been identified in human MN, significantly advancing our understanding of its pathogenesis and clinical management. The most well-characterized autoantigens include phospholipase A2 receptor 1(PLA2R) and thrombospondin type 1 domain–containing 7A(THSD7A)3, which are implicated in the majority of IMN cases. Additionally, other autoantigens such as exostosin 1(EXT1), exostosin 2(EXT2), neural EGF-like-1 protein(NELL1), semaphorin 3B(Sema3B), and protocadherin 7(PCDH7) have been identified in specific subsets of patients, highlighting the heterogeneity of MN4. These discoveries have not only deepened our understanding of the disease mechanisms but also revolutionized clinical practice. For instance, several autoantibody assays targeting these antigens are now widely used in clinical settings, enabling more precise monitoring of disease progression and prediction of treatment response5.
While anti-PLA2R antibodies are the primary autoantigen involved in most cases of idiopathic MN, other autoantibodies have been identified in some patients, suggesting a more complex pathogenic landscape. This heterogeneity underscores the need for personalized approaches to diagnosis and treatment6. Furthermore, immune responses in MN are closely linked to metabolic alterations, a field that has gained increasing attention in recent years. Emerging evidence highlights the role of immunometabolism—metabolic reprogramming of immune cells in response to microenvironmental signals. Immune cells adapt nutrient uptake and utilization to meet functional demands, impacting signaling, differentiation, and function7,8. Although research has primarily focused on immune cell imbalances, the link between immune metabolism and MN remains underexplored9,10, In autoimmune diseases, metabolic reprogramming involves shifts from oxidative phosphorylation(OXPHOS) to aerobic glycolysis, driven by intrinsic and extrinsic signals11. This review explores key metabolic pathways (e.g., glucose, lipid, and amino acid metabolism) and their role in MN pathogenesis. Insights into immunometabolism, including its impact on immune cell development and function, may pave the way for novel biomarkers and targeted therapies.
Pathogenesis of immune cells in MN
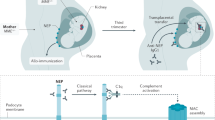
Anti-PLA2R antibodies produced by B cells, bind to PLA2R on the surface of podocytes, forming immune complexes and depositing them on the GBM. These immune complexes activate the complement system, leading to a localized inflammatory response that further damages the renal tubules and podocytes. Therapeutic strategies targeting B cells, such as rituximab, attenuates the formation of immune complexes by selectively removing B cells and reducing the production of anti-PLA2R antibodies12.
CD4+ T cells regulate the immune response by secreting cytokines that enhance antibody production by B cells and promote immune complex formation. In addition, T cells may also promote the exacerbation of immune injury through interaction with B cells. Targeting the T cell activation pathway or increasing the number of regulatory T (Treg) cells and suppressing overactive T helper (Th)17 cells can effectively reduce B cell activation and inhibit antibody production and inflammatory responses13.
Dendritic cells (DC) are key antigen-presenting cells that play a pivotal role in initiating and maintaining adaptive immune responses. In MN, DCs present podocyte antigens to naïve T cells, thereby activating T cell responses that promote autoantibody production by B cells. Dysregulated DC function can lead to the activation of pathogenic immune responses, exacerbating kidney injury14.
Macrophages are involved in the regulation of the immune response by recognizing deposited immune complexes through receptors on their surface, which in turn activate their effector function to produce inflammatory factors such as tumor necrosis factor α (TNF-α), interleukin (IL) -1β, IL-6, to promote inflammation15. By inhibiting the activation of M1-type macrophages or promoting the production of M2-type macrophages, immune injury in MN may be attenuated16.
Natural killer (NK) cells recognize and remove target cells through their surface receptors, and activate other immune cells at an early stage by releasing cytokines interferon γ (IFN-γ) to promote the immune response of B cells and T cells. It also exerts antibody-dependent cell-mediated cytotoxicity (ADCC) through the interaction of its Fc receptor with the immune complex bound by anti-PLA2R antibody17. Immunomodulation targeting NK cells may also become a new direction for therapy, for example, to improve the immune response in MN by enhancing the anti-tumor activity of NK cells or modulating their immune surveillance function18 (Fig. 1).

APCs can recognize PLA2R antigens and then present PLA2R fragments on MHC Class II molecules on their surface to Naïve T. Stimulated T cells differentiate into helper T (Th) cells and release cytokines, which are fed back to B cells, stimulating division and differentiation into plasma cells and antibody-producing and memory B cells. Autoantibodies begin to target PLA2R on the basal surface of podocytes and activate complement to form membrane attack complex (MAC). NK exerts antibody-dependent cell-mediated cytotoxicity (ADCC) by interacting with immune complexes via its Fc receptor. Macrophages cause kidney injury by producing the inflammatory factor TNF-α to promote inflammation. CR1 complement receptor 1, TCR T cell receptor, APCs antigen-presenting cells, NK Natural killer, DC Dendritic cells, GC-B germinal center B, IFN-γ cytokines interferon γ, TNF-α tumor necrosis factor This image was created by Figdraw.
Metabolic regulation of B cells
The development and differentiation of B cells involve dynamic shifts between quiescent stages and those of rapid proliferation or increased protein secretion, necessitating rapid adjustments in nutrient uptake and metabolic pathway activity19. Naïve B cells, in a resting state, have minimal metabolic demands and remain non-proliferative20. Upon antigen stimulation, igniting the immune response, metabolic requirements elevate swiftly, facilitating metabolic reprogramming—from OXPHOS to aerobic glycolysis and tricarboxylic acid (TCA) cycling through the PI3K/AKT pathway8, cell proliferation and differentiation are driven primarily by antigens and are co-stimulated by secondary signaling from toll-like receptor (TLR) ligands. Upon antigen encounter, mature follicular B cells initiate immune responses within the germinal centers (GCs), these GC B cells exhibit an enhanced glucose uptake and increased mitochondrial content21. The rapid proliferation within the GC incurs significant metabolic demands. Notably, glycolytic activity within the GC is minimal, with GC B cells predominantly relying on the oxidation of long-chain fatty acids (LCFAs), both externally and internally sourced to generate substantial amounts of acetyl-CoA. This process is crucial for meeting their bioenergetic requirements, such as ATP production22,23, thus facilitating affinity maturation, clonal amplification, and positive selection24 (Fig. 2).

The stimulation of the B cell receptor (BCR) causes an upregulation of glycolysis, initiating germinal center (GC) responses. GC B cells exclusively rely on fatty acid oxidation (FAO) to fuel oxidative phosphorylation (OXPHOS) for ATP production. OXPHOS and glutamine are important for plasma cell (PC) differentiation. In contrast, the short-chain fatty acid (SCFA) butyrate and glycolysis contribute to the differentiation of regulatory B cells (Bregs). TCA tricarboxylic acid, ETC electron transport chain, AhR aryl hydrocarbon receptor, LLPC long-lived plasma cell, LDHA lactate dehydrogenase A. This image was created by Figdraw.
B cells leave the GC and eventually differentiate into plasma cells(PC) or memory B cells25, PC are categorized into two primary subsets, short-lived plasma cells (SLPCs) and long-lived plasma cells (LLPCs)26. In the case of LLPCs, the fundamental OXPHOS process is predominantly sustained by LCFAs27. In contrast to SLPCs, LLPCs exhibit a heightened reliance on amino acids to facilitate antibody production28. Glutamine was used for both mitochondrial respiration and anaplerotic reactions, yielding glutamate and aspartate for antibody synthesis. Key amino acids, such as leucine and arginine, could activate mammalian target of rapamycin complex (mTORC1) in PC to enhance antibody production. PC exhibit increased uptake of amino acids and glucose to meet the demands of high protein synthesis rates29, yet they primarily use glucose for antibody glycosylation rather than for energy30. Interestingly, LLPC have increased glucose uptake and utilize pyruvate catabolism in the mitochondria more effectively, demonstrating higher rates of oxygen consumption, provided that physiological levels of amino acids are present26. In contrast, memory B cells exhibit metabolic dormancy, often relying on OXPHOS, with their metabolic activity possibly governed by distinct signaling pathways.
A recent study of CD5 expressing B1 cells showed increased glycolysis and OXPHOS activity compared to B2 cells, which was supported by an upregulation of glycolytic and OXPHOS enzymes. These B1 cells display a metabolic adaptation to lipid-rich environments, such as the pleural and peritoneal cavities by enhancing lipid uptake, fat droplet synthesis for storage, and the use of autophagy to facilitate the beta-oxidation of lipids31.
Regulatory B cells (Breg) are recognized for their immunosuppressive role in autoimmune diseases, predominantly by secreting IL-1032. The aryl hydrocarbon receptor (AhR) transcription factor is pivotal in controlling the differentiation and functionality of Breg cells. Research has shown that the supplementation of SCFAs, particularly butyrate, stimulates the production of AhR ligands linked to tryptophan metabolism. This process leads to an increase in the transcription of the anti-inflammatory cytokine IL-10 in B cells33. Furthermore, Treg functions are linked to complex III, while OXPHOS and complexes I, IV, and V are essential for the proliferation and antibody production of B lymphocytes34.
Aberrant B cell metabolism in MN
The B cell tolerance checkpoint serves to limit the potential harm caused by an auto-reactive B cell repertoire and is interconnected with key metabolic pathways. Dysfunction of these checkpoints is a fundamental aspect of the pathogenesis of MN. The regulation and reprogramming of metabolic pathways are crucial for providing the necessary metabolic support for B cell function and immune responses. However, imbalances in immunometabolism can enable autoreactive B cells to bypass self-tolerance checkpoints, potentially initiating autoimmune diseases35. Several of these checkpoints, including CD40, are recognized for their role in modulating B cell metabolism during the activation process36.
Elevated B cell activating factor (BAFF) levels in MN could potentially cause abnormal B-lymphocyte activation and autoimmune dysregulation37, BAFF treatment leads to an increase in protein levels and the expression of genes that promote glycolytic metabolism. Compared to normal B cells, BAFF transgenic mouse-derived B cells have higher levels of glycolysis after lipopolysaccharide (LPS) stimulation38,39. BAFF is of great significance in autoimmune diseases, particularly in disrupting B cell glucose homeostasis. Prolonged exposure to BAFF activates the Erk1/2 pathway, which boosts glycolysis, increases cell proliferation, and rescues autoreactive B cells. The attachment of BAFF to the receptor activates the PI3K/AKT/mTOR pathway. An increase in BAFF plays a dual role in B cell metabolism. It not only stimulates an upregulation of glycolysis, a key process in cellular energy production, but also facilitates the entry of pyruvate into the mitochondria. This enhancement of pyruvate influx subsequently boosts oxidative metabolism, which is another critical pathway for energy generation within the cell38, Studies confirm that the above pathways exhibit higher activity in B cells from MN patients (Fig. 3).

Rapamycin complex (mTORC)-mediated glycolytic targets are enhanced by activation in MN B cells, leading to primitive B cell expansion. Glutaminolysis has also been found to contribute to primitive B differentiation in MN. Fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) in MN B cells, leading to expansion of plasma cells (PCs). BCR B-cell receptor. This image was created by Figdraw.
SCFAs serve as energy substrates and are converted to acetyl-CoA, which increases OXPHOS and fatty acid synthesis (FAS), and also boosts mitochondrial energy production and glycolysis40. B cells in autoimmunity exhibit different lipid metabolism patterns, which have been extensively studied in systemic lupus erythematosus (SLE)40,41,42. Metabolic reprogramming of lipid metabolism in SLE provides guidance for MN research. Studies have demonstrated that fatty acid metabolism is disrupted in MN, but the underlying mechanisms have not been further explored43.
Severe metabolic perturbations occur during the progression of MN. l-tryptophan, l-kynurenine, gamma-aminobutyric acid (GABA), indoleacetaldehyde, 5-hydroxyindoleacetylglycine, and N-alpha-acetyllysine, affecting amino acid metabolic pathways, the citric acid cycle, pantothenate and CoA biosynthesis, and hormone signaling pathways44. Tryptophan ameliorates MN by inhibiting the AhR pathway through the generation of indole metabolites45. Amino acid metabolic abnormalities may result from increased proteolytic metabolism or alterations in energy requirements during inflammatory states46.
Aberrant levels of citric, fumaric, and succinic acids are indicative of mitochondrial dysfunction, indirectly mirroring the oxidative stress observed in patients with MN, and serum citric acid levels are decreased in patients with MN who have more severely impaired renal filtration function47. Whether the metabolic reprogramming of amino acids in MN affects pathogenesis through B cells will be the focus of our studies. In conclusion, metabolic reprogramming drives autoreactive B cells in the MN.
Metabolic regulation of T cells
In the resting state, the metabolic demand of CD4+ and CD8+ naïve T cells is low, so they utilize a small amount of glucose for the TCA cycle and OXPHOS48. Upon activation, the increased energy demand triggers a metabolic switch that enables the rapid production of ATP from other metabolic pathways (including essential substances such as glucose and glutamine) to support rapid cell proliferation and increased differentiation rates. Additionally, the flux through glycolysis, the pentose phosphate pathway (PPP), and the hexosamine pathway also increases49. Activated effector T cells become metabolically and functionally active with increased rates of glycolysis and OXPHOS. Th1 and Th2 cells, in particular, rely on glycolysis mediated by mTORC1 and mTORC2 respectively50. Pro-inflammatory CD4+ T cells, including Th1 and Th2 cells, utilize glycolysis and glutaminolysis as their primary sources of energy51,52. The glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) can interfere with IFN-γ translation in Th1 cells when substrate levels are low, highlighting the importance of glycolysis in T cell function53. Compared to Th1 cells, T follicular helper (Tfh) cells exhibit lower rates of glycolysis and OXPHOS54, the costimulatory molecule ICOS activated mTORC1 and mTORC2 to drive glycolysis and lipogenesis, while inhibition of Myc or increased Glut1 metabolism promoted Tfh cell responses, Tfh cells might utilize this mixed metabolism to support both proliferation and cellular longevity while not depleting nutrients in GCs55. Memory T cells primarily depend on fatty acid oxidation (FAO) for their energy but also utilize glucose for OXPHOS56. Reduced glycolysis, AKT or mTOR activation, and lower mitochondrial membrane potential or cell size are characteristics that promote memory T cell formation57 (Fig. 4).

Naïve T cells are sustained in a dormant state by low levels of OXPHOS. Upon encountering an antigen, T cells experience significant metabolic reprogramming, altering their reliance on glycolysis to facilitate their activation and differentiation into effector T-cell. Conversely, memory T cells and regulatory T cells (Treg) rely on OXPHOS and FAO to support their survival and differentiation. TCR T cell receptor, ETC electron transport chain, IDO indoleamine 2,3-dioxygenase, mTOR mammalian target of rapamycin, FAS fatty acid synthesis, ACC1 acetyl-CoA carboxylase 1, TCA tricarboxylic acid cycle. This image was created by Figdraw.
T cell lipid metabolism is inactive. memory T cells primarily rely on FAO for their energy need. Th17 cell development depends on acetyl-CoA. The rate-limiting enzyme for FAS, acetyl-CoA carboxylase 1 (ACC1), undergoes AMPK-activated phosphorylation, which in turn regulates the differentiation of Th17 cells by modulating the binding of the transcription factor RORγt to its target genes. Inhibition of ACC1 impairs Th1 and Th2 development, indicating a common requirement for FAS in CD4+ effector T cells58. Tfh series cells exhibit a unique lipid metabolic programme, with the cytidine diphosphate (CDP)-ethanolamine pathway orchestrating CXCR5 expression and localization with Tfh cell responses and humoral immunity. The CDP-ethanolamine pathway is identified as a selective metabolic and post-transcriptional checkpoint to programme Tfh cell differentiation and to establish signalling functions from the synthesis of head phosphatidylethanolamine (PE). Treg relies on OXPHOS and FAO to produce ATP59. Upon activation, Treg cells upregulate the transcription factor myocyte enhancer factor 2 (Mef2), which induces genes necessary for OXPHOS60. The non-oxidative PPP regulates Treg function to prevent autoimmunity by integrating metabolism and epigenetics61. Glycolysis is essential for Treg function, and Foxp3 enhances fatty acid oxidative phosphorylation by suppressing Myc signaling and glycolysis and increasing the expression of electron transport chain (ETC) complexes62. peroxisome proliferator-activated receptor γ (PPARγ) has the potential to promote Treg cells by increasing FAO63.
Glutamine plays a crucial role in T cell differentiation and function, especially in Th1 and Th2 cells. Upon activation, effector T cells significantly increase their utilization of glutamine, which provides energy through its catabolism51. Glutaminase deficiency suppresses Th17 development while promoting Th1 cell differentiation and function through chromatin accessibility modulation64. Th1 and Th2 cells rely on glutaminolysis as a metabolic source of energy, and their differentiation is regulated by key enzymes in the glutamine synthesis pathway52. Degradation of tryptophan produces metabolic intermediates such as kynurenine, which binds to the AhR and induces Treg cell differentiation65. The binding of CTLA4 to CD80 and CD86, co-stimulatory molecules on antigen-presenting cells (APCs), induces the expression of the amino acid catabolic enzymes indoleamine 2,3-dioxygenase (IDO) and arginase 1 in Treg cells. The activation of these enzymes further promotes the expression of amino acid catabolic enzymes in Treg cells. Activation of these enzymes reduces the supply of amino acids such as tryptophan, arginine, histidine, and threonine to the surrounding T cells. This reduction inhibits PTEN-mediated mTOR signaling, suppresses effector T cell proliferation, and promotes Treg cell differentiation66.
In summary, the metabolic reprogramming of T cells influences their activation and proliferation and determines their lineage differentiation, involving various pathways and regulatory mechanisms in carbohydrate, lipid, and amino acid metabolism.
Aberrant T cell metabolism in MN
Emerging research indicates that abnormal T cell responses are instrumental in the pathogenesis of autoimmune diseases such as MN16,67. Metabolic pathways play a vital role in the function of T cells, The microenvironment of inflammatory diseases experiences shifts, including the availability of energy sources. These changes dynamically affect T cell metabolism, with energy supply molding their metabolic profiles68.
Lysine acetyltransferase 6 A (KAT6A) orchestrates the abundance of histone acetylation in chromatin where several glycolytic genes are located, thereby affecting glucose metabolic reprogramming and subsequent CD4+ T cell responses69. Inhibition of the histone H3K27 demethylase KDM6A/B leads to a global increase in inhibitory H3K27me3 histone labelling, resulting in the down-regulation of key transcription factors (myc) and subsequent metabolic reprogramming of Th17 cells, which leads to a reduction in mitochondrial biogenesis and concomitant anti-inflammatory effects70. These results suggest that the use of small molecule epigenetic and metabolic inhibitors provides a basis for understanding aspects of autoimmune disease, particularly MN, and offers a therapeutic approach71 (Fig. 5).

MN T cells exhibit enhanced glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) as well as the pentose phosphate pathway (PPP). ITA inhibits glycolysis and OXPHOS in Th17 and Treg polarised T-cells. MMF reduces the glycolytic activity and glutaminolytic fluxes of human CD4 + T cells activated in vitro. The loss of Pik3ip1 expression results in reduced OXPHOS and enhanced glycolysis in T cells. Epigenetic modifications resulting in metabolic reprogramming of Th17 cells, which leads to a reduction in mitochondrial biogenesis and concomitant anti-inflammatory effects. ITA Itaconic acid, MMF Mycophenolate mofetil, Pik3ip1 Phosphatidylinositide 3-kinase interacting protein 1. This image was created by Figdraw.
Phosphatidylinositide 3-kinase interacting protein 1 (Pik3ip1) is an upstream inhibitor of PI3K signaling72, the loss of Pik3ip1 expression results in reduced OXPHOS and enhanced glycolysis in T cells, which promotes inflammatory responses upon initial onset in a hypoxia-inducible factor 1α (HIF-1α)–dependent manner. Pik3ip1 as a key regulator of T cell metabolic homeostasis and highlights the importance of the Pik3ip1/Hif1α/glycolysis axis in autoimmune diseases73.
A key factor is the imbalance between Th17 and Treg cells. An increase in the number of Th17 cells and a decrease in Treg cells lead to an increase in the Th17/Treg, ratiometabolic reprogramming and external signals that modulate metabolic pathways can affect the Th17/Treg balance74. With the significant increase in the Th17/Treg ratio and the associated changes in cytokines and transcription factors, the reduction in Treg cells brings us closer to the early events in the pathogenesis of MN13.
Itaconic acid (ITA) is an endogenous metabolite derived from the mitochondrial TCA cycle. ITA inhibits glycolysis and OXPHOS in Th17 and Treg polarised T cells through interactions with key enzymes and metabolites, and may be a potential therapeutic agent for autoimmune diseases75.
Dehydroepiandrosterone sulfate (DHEAS) was identified as a significantly down-regulated differentially expressed metabolites (DEM) in the IMN group76, The disturbance of steroid hormones metabolism reduced the level of DHEA and DHEAS, thus inducing the Treg cells’ activity, further leading to Th1-type cytokines production and interfere with the synthesis of Th2-type cytokines77.
Current research indicates that metabolic reprogramming boosts the activation and differentiation of pro-inflammatory T cells in MN. Apart from ATP synthesis, metabolic reprogramming can exert multiple effects on T cells via intermediates. Studies on MN have also mainly focused on changes in the number of T cell subsets and pathways78, such as the effect of calmodulin phosphatase B1 subunit (CnB1) on calmodulin phosphatase-NFAT signaling-mediated T cell activation79. It’s still not been clearly whether the cellular metabolic imbalance is involved in the changes in T cell numbers, but the study of T cell metabolic imbalance in autoimmune diseases such as SLE and rheumatoid arthritis (RA) may provide ideas and base for us to study MN80,81.
Other immune cells metabolism in MN
DC, Macrophages, and NK cells also play an integral and important role in autoimmune diseases, and by modulating the metabolic pathways of these immune cells, MN could potentially be treated. For example, inhibition of the glycolytic pathway may attenuate pro-inflammatory cell function, whereas enhancement of FAO may help maintain anti-inflammatory cell activity.
Dendritic cells
DCs arise from precursor cells in the bone marrow can be further classified into conventional cDC1, cDC2, and plasmacytoid DCs (pDCs). The differentiation or survival of cDC1 is dependent on LKB1/AMPK/FAO, whereas cDC2 relies on reactive oxygen species (ROS) signaling82. Both cDC subsets undergo distinct metabolic changes and requirements during maturation and activation, including glycolysis, lipid metabolism, mitochondrial changes, and epigenetic modifications, which allow them to modulate the immune response83. DCs play a crucial role in driving autoimmunity through various mechanisms. DCs indirectly regulate autoantibody production through the crosstalk between Th cells and B cells84. Impaired DC uptake and clearance of apoptotic cells can release danger signals such as HMGB1 and expose autoantigens, inducing DC inflammatory responses and driving autoimmunity85. Stimulation of TLR activation causes DCs to switch their energy dependence from OXPHOS to glycolysis, allowing immediate access to the energy required for producing inflammatory mediators and antigen presentation86. For example, Activation of cDC in the RA synovium is accompanied by increased glycolysis and anabolism, while promoting catabolism can generate tolerogenic DCs in monocytes. The immunometabolism of DCs is a potential therapeutic target in RA86.
DCs in MN may also have impaired metabolism. TLR 9 activated pDCs produce IFN, which promotes FAO and OXPHOS, especially the oxidation of fatty acids needed for pDCs activation. IFN is crucial in autoimmune diseases87. However, the metabolic profile of these DCs with MN is far from clear, future research could be geared towards this research field.
Macrophages
Macrophages can be activated into M1 or M2 subtypes in response to different stimuli88. In response to pro-inflammatory signals, M1 macrophages undergo a phenotypic switch from OXPHOS to glycolysis, leading to renal injury in the early stages of kidney disease. In contrast, M2 macrophages maintain high OXPHOS and play a crucial role in the lupus nephritis and the chronic phase of kidney injury89,90,91. M1 macrophages, typically found in low-oxygen conditions, rely on glycolysis for energy production, a process driven by HIF1α. They produce ROS through the respiratory chain, utilizing NADPH from the pentose phosphate pathway to fuel NADPH oxidase. In contrast, M2 macrophages, which do not produce ROS, have a functional respiratory chain that supports FAO92,93. M2 macrophages primarily metabolize fat reserves and use their low ROS levels to protect tissues during repair. Changes in macrophage metabolic pathways in autoimmune disease-injured kidneys, also regulated by glutamine, lipids, and mitochondria94. Those factors influencing macrophage metabolism may disrupt M1/M2 homeostasis and exacerbate inflammation. Insulin-like growth factor 2 (IGF-2) effectively controls inflammation by preprogramming mature macrophages for OXPHOS Effects on experimental autoimmune encephalomyelitis (EAE)95. In addition, the immune complex (IC) can increase inflammatory reactions through the IRF5/NF-κB pathway. Furthermore, IRF5 plays a role in regulating proinflammatory cytokines for the development of Th1 and Th17 responses, which also stimulate immune responses. The mechanisms by which macrophage polarization is regulated in other autoimmune diseases have been studied96. However, whether changes in the metabolism of these cells play a role in the pathogenesis of MN is largely unknown.
Natural killer cells
NK cells primarily use glucose-fueled OXPHOS at rest and during activation, with glucose being the main fuel for receptor activation97. As NK cells mature and become active, metabolic changes take place. Immature NK cells boost the expression of glucose transporters and nutrient receptors. In the presence of IL-15 during in vitro experiments, these immature cells develop into mature NK cells. This maturation process is accompanied by an increase in FAO and OXPHOS97. The upregulated OXPHOS pathway is crucial for the production of IFN-γ in activation-induced NK cells. Additionally, the metabolic regulator mTOR is vital for IL-15 signaling during the development and activation of NK cells and the production of IFN-γ, with mTOR deficiency significantly impairing early-stage NK cell activation98. mTOR integrates cellular growth and metabolism by enhancing cell proliferation and glycolysis99. In autoimmune diseases, NK cells may enhance the immune response by producing cytokines that regulate immune function100, NK cells regulate autoimmune diseases by inhibiting autoreactive T cell proliferation and responses and by eliminating over-stimulated lymphocytes, macrophages, or immature DC101. Although the role of NK cellular metabolic changes in the pathogenesis of MN is largely unknown, NK cells may contribute to autoimmune disease pathogenesis by altering immune homeostasis through cytokine regulation or influencing interactions with other cells, which would be a potential therapeutic target for autoimmune diseases102. This provides a reference for understanding the molecular mechanisms of metabolism in MN.
Therapeutics targeting immunometabolism in MN
Many drugs influence metabolism and the literature has provided comprehensive reviews on their role in other autoimmune diseases103,104. Utilizing metabolizing drugs for lymphocytes in MN holds promise as a relatively less toxic therapeutic option with a better understanding of the immunopathogenesis of MN.
Rituximab, a monoclonal antibody against the B cell surface antigen CD20, is used as a first-line therapeutic agent for MN, mediating mechanisms such as direct B cell clearance, ADCC and complement-dependent cytotoxicity (CDC) to reduce the number of B cells105. The energy supply of B cells was further inhibited by interfering with their glycolytic and fatty acid metabolic pathways during the antibody secretion and proliferation phases. Alterations in B cell metabolic pathways after removal of B cells may affect the persistence and intensity of the body’s immune response. Rituximab effectively depletes B cells in the peripheral blood, especially CD40+ memory B cells, switching memory B cells and PC, which has been suggested that part of the effect of rituximab may be mediated by an increase in the Treg fraction, while affects the mechanisms of peripheral B cell and T cell tolerance106,107. In diffuse large B cell lymphoma (DLBCL), pyruvate dehydrogenase kinase 4 (PDK4) mediates a metabolic shift from OXPHOS to glycolysis, resulting in rituximab resistance108.
Mycophenolate mofetil (MMF), a prodrug of mycophenolic acid, inhibits inosine monophosphate dehydrogenase type II, is a rate-limiting enzyme for guanosine synthesis and DNA synthesis109. Mycophenolic acid also reduces the glycolytic activity and glutaminolytic fluxes of human CD4+ T cells activated in vitro110. MMF monotherapy has almost no effect on the treatment of IMN111, but the effect of MMF plus corticosteroids is similar to that of cyclophosphamide (CTX) plus corticosteroids107. MMF can be considered as an alternative choice for patients with poor responses to conventional CTX and Calcium-NFAT inhibitors (CNIs) and patients who cannot tolerate rituximab.
The proteasome mediates the degradation of several proteins during cell growth and differentiation via the ubiquitin-proteasome pathway. Proteasome inhibitors ultimately lead to the accumulation of misfolded proteins that induce apoptosis112. In particular, it inhibits the degradation of key enzymes of glycolysis and affects the efficiency of glycolysis, as well as altering the OXPHOS process by affecting mitochondrial function113. For example, bortezomib, a selective and reversible inhibitor of the proteasome, induces apoptosis and PC depletion by targeting high turnover PC. In patients with refractory nephropathic MN, this results in the depletion of activated B cells and PC114,115.
Anti-CD38 drugs such as Daratumumab and Felzartamab (MOR202) are monoclonal antibodies that belong to the human IgG1κ class. They specifically target the CD38 protein, which is abundantly present on plasmacytoma cells as well as on both SLPC and LLPC Interferes with the NAD+ metabolic pathway by targeting and inhibiting CD38, affecting intracellular NAD+/NADH ratios and thus cellular energy metabolism and apoptosis116. The depletion of PC through these mechanisms is expected to be a promising direction for the treatment of MN117.
Leflunomide is a low-toxicity immunoregulatory drug that inhibits the T/B lymphocyte pyrimidine synthesis pathway by suppressing the activity of dihydroorotate dehydrogenase. This inhibition blocks the generation of immune products and complexes and their deposition in the glomeruli118. Glucocorticoids, like prednisone, could increase gluconeogenesis, leading to fat accumulation and having secondary metabolic effects on immune cells119. The combination of leflunomide and glucocorticoids is highly effective in PMN120.
CNIs, which include cyclosporin A (CsA) and Tacrolimus (FK506), affects the aerobic glycolysis and mitochondrial respiration metabolic activity of T cells by inhibiting NFAT-dependent gene expression through the blockade of the calmodulin-NFAT pathway121. CSA can restrain the production of IL-2, IL-3, and interferon-γ, as well as the translocation of nuclear factor-activated T cells, thereby inhibiting T-cell activity and reducing lymphocyte proliferation122. FK506 has a similar therapeutic mechanism to CsA, achieving immunosuppression by regulating inflammatory factor production and inhibiting T-cell activation and proliferation123. The KDIGO guidelines recommend treatment with CNIs for PMN patients who are intolerant to glucocorticoid-combined alkylator regimens or have contraindications to such treatment124. A study found that CNIs were more effective than cyclophosphamide in reducing the duration of nephropathy and the risk of nephrotic syndrome (NS)-related complications125. Therefore, CNIs serve as an alternative when cyclophosphamide is contraindicated. CSA has also been widely used in treating steroid-resistant PMN. For patients receiving CNI therapy, blood levels and renal function should be tested regularly during the drug administration period to ensure drug safety and long-term efficacy. CNIs are commonly used in autoimmune diseases and organ transplants to prevent rejection and are a cornerstone of MN therapy126.
Sodium-glucose cotransporter protein 2 (SGLT2) inhibitors as renin-angiotensin system (RAS) blockers play a major protective role against MN by inhibiting glucose and sodium reabsorption in the proximal tubules, improving glomerular hemodynamics, decreasing proteinuria, and delaying renal fibrosis in patients with chronic kidney disease127,128, Meanwhile, SGLT2 inhibitors have been reported in the literature to have immunomodulatory effects on renal diseases by reducing inflammation, enhancing autophagy, and reversing the imbalance of Th1/Th2 cells, which may play a secondary role in renal protection129,130. Canagliflozin affects the circulating metabolism of TCA through inhibition of mitochondrial glutamate dehydrogenase (GDH), affects ERK and mTORC1 activity and reduces c-Myc, resulting in metabolic abnormalities in T cells that impair their effector function in patients with SLE and RA. Empagliflozin appears to regulate CD4+ T cells through inhibiting the mTOR signal pathway, and T cell metabolism shifts from OXPHOS to glycolysis131,132.
With in depth research on the immunology of MN, the treatment options for MN will be more precise and diversified in the future. The development of novel immunosuppressants provide more possibilities for drug selection, aiming to reduce side effects and improve patients’ quality of life and long-term prognosis (Table 1).
Metabolomic studies in case of MN
In recent years, several metabolomics studies have focused on identifying markers in non-invasively collected samples, such as urine and serum. These studies have highlighted significant correlations between disease progression and processes including pyrimidine metabolism, nicotinamide-adenine dinucleotide (NAD) metabolism, fatty acid biosynthesis, dietary lipid digestion, and the transport of nucleosides and related molecules133. Patients with IMN exhibit disturbed metabolism of amino acids, nucleotides, and steroid hormones134. Serum and urine samples from these patients show significant increases in citric acid and four amino acids (L-asparagine, L-serine, L-threonine, and pyroglutamic acid). Additionally, elevated levels of dicarboxylic acids, phenolic acids, and cholesterol have been observed in the urine of patients with severe proteinuria135. Fatty acids, particularly palmitic and stearic acids, are upregulated in MN, with palmitic acid inducing endoplasmic reticulum (ER) stress in podocytes, leading to apoptosis136. Jo HA et al.137. High urinary fumarate levels have been shown to predict composite outcomes in PLA2R-associated MN. Fumarate hydratase, the enzyme responsible for hydrolyzing fumarate, may influence changes in podocyte phenotypic profiles following the development of PLA2R autoimmunity. Furthermore, several studies have reported decreased levels of gut microbiota-derived SCFAs in IMN. Fecal microbiota transplantation (FMT) has been used to treat patients with MN and chronic diarrhea, resulting in reduced symptoms, improved renal function, and lower levels of propionate and butyrate in feces compared to healthy controls137. Dysregulation of gut flora may lead to an imbalance between immune tolerance and immune response, promoting the production of autoantibodies and inflammatory factors, which contribute to kidney disease progression138.
These findings underscore the association between metabolite disorders and the pathogenesis of MN, suggesting that metabolites could serve as promising biomarkers for predicting MN. Nevertheless, the metabolomics of MN remains in its early stages and warrants further investigation.
Conclusions
While existing research on MN primarily examines the quantitative changes in T and B immune cells, it is important to consider the broader impact of metabolic imbalances on immune cells—imbalances that could potentially be rectified with targeted treatments. Current evidence points to cell-specific metabolic regulation, highlighting the prospect of leveraging these unique pathways to fine-tune immune responses and provide more precise, personalized treatment strategies. Cell energy metabolism uncovers a wealth of therapeutic targets, which are relatively uncharted, offer fewer side effects, and hold the promise of selectively neutralizing pathogenic immune cells. Nevertheless, environmental factors also play a role in cellular metabolism, presenting an additional layer of complexity to this budding treatment approach for MN. Autoimmune diseases are characterized by metabolic shifts that result in aberrant immune cell differentiation or hyperactivation. Each immune subset possesses a metabolic signature that supports its function, presenting an opportunity to direct disease course by manipulating key metabolic drivers. Despite this potential, appreciable gaps remain in our comprehension of lymphocyte metabolic irregularities in autoimmune conditions, warranting further investigation. Progress in decoding the multifaceted metabolic dynamics of immune cells will be instrumental in developing novel therapeutic interventions for metabolic pathways in immunity.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Couser, W. G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 12, 983–997 (2017).
Ronco, P. et al. Membranous nephropathy. Nat. Rev. Dis. Primers 7, 69 (2021).
van de Logt, A. E. et al. The anti-PLA2R antibody in membranous nephropathy: what we know and what remains a decade after its discovery. Kidney Int. 96, 1292–1302 (2019).
Sethi, S. New ‘Antigens’ in Membranous Nephropathy. J. Am. Soc. Nephrol. 32, 268–278 (2021).
Liu, W. et al. Course monitoring of membranous nephropathy: Both autoantibodies and podocytes require multidimensional attention. Autoimmun. Rev. 21, 102976 (2022).
Murtas, C. et al. Novel biomarkers and pathophysiology of membranous nephropathy: PLA2R and beyond. Clin. Kidney J. 17, sfad228 (2024).
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Pearce, E. L. et al. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).
Xu, L. et al. Follicular Helper T Cells and Follicular Regulatory T Cells Involved in Immune Disorders of Idiopathic Membranous Nephropathy. Indian J. Pediatr. 91, 702–708 (2024).
Deng, B. et al. Imbalance of T follicular helper cell subsets trigger the differentiation of pathogenic B cells in idiopathic membranous nephropathy. Inflamm. Res. 73, 485–498 (2024).
Wang, S., Yang, N. & Zhang, H. Metabolic dysregulation of lymphocytes in autoimmune diseases. Trends Endocrinol. Metab. 35, 624–637 (2024).
Kistler, A. D. & Salant, D. J. Complement activation and effector pathways in membranous nephropathy. Kidney Int. 105, 473–483 (2024).
Motavalli, R. et al. Altered Th17/Treg ratio as a possible mechanism in pathogenesis of idiopathic membranous nephropathy. Cytokine 141, 155452 (2021).
Coutant, F. & Miossec, P. Altered dendritic cell functions in autoimmune diseases: distinct and overlapping profiles. Nat. Rev. Rheumatol. 12, 703–715 (2016).
Funes, S. C. et al. Implications of macrophage polarization in autoimmunity. Immunology 154, 186–195 (2018).
Shi, M. et al. Single-cell RNA sequencing shows the immune cell landscape in the kidneys of patients with idiopathic membranous nephropathy. Front Immunol. 14, 1203062 (2023).
Vivier, E. et al. Functions of natural killer cells. Nat. Immunol. 9, 503–510 (2008).
Alexopoulos, E. et al. Immune mechanisms in idiopathic membranous nephropathy: the role of the interstitial infiltrates. Am. J. Kidney Dis. 13, 404–412 (1989).
Boothby, M. & Rickert, R. C. Metabolic Regulation of the Immune Humoral Response. Immunity 46, 743–755 (2017).
Weisel, F. J. et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 21, 331–342 (2020).
Jellusova, J. et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 18, 303–312 (2017).
Lunt, S. Y. & Vander Heiden, M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev. Cell Dev. Biol. 27, 441–464 (2011).
Ji, X. et al. Lipid metabolism in regulation of B cell development and autoimmunity. Cytokine Growth Factor Rev. 73, 40–51 (2023).
Chen, D. et al. Coupled analysis of transcriptome and BCR mutations reveals role of OXPHOS in affinity maturation. Nat. Immunol. 22, 904–913 (2021).
Mesin, L., Ersching, J. & Victora, G. D. Germinal Center B Cell Dynamics. Immunity 45, 471–482 (2016).
O’Connor, B. P., Cascalho, M. & Noelle, R. J. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 195, 737–745 (2002).
Kunisawa, J. Metabolic changes during B cell differentiation for the production of intestinal IgA antibody. Cell Mol. Life Sci. 74, 1503–1509 (2017).
Fu, Y. et al. Immunometabolism shapes B cell fate and functions. Immunology 166, 444–457 (2022).
Lam, W. Y. et al. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep. 24, 2479–2492.e6 (2018).
Lam, W. Y. et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity 45, 60–73 (2016).
Clarke, A. J. et al. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J. Exp. Med. 215, 399–413 (2018).
Rosser, E. C. & Mauri, C. Regulatory B cells: origin, phenotype, and function. Immunity 42, 607–612 (2015).
Rosser, E. C. et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 31, 837–851.e10 (2020).
Zotta, A., O’Neill, L. A. J. & Yin, M. Unlocking potential: the role of the electron transport chain in immunometabolism. Trends Immunol. 45, 259–273 (2024).
Barberis, M. & Rojas López, A. Metabolic imbalance driving immune cell phenotype switching in autoimmune disorders: Tipping the balance of T- and B-cell interactions. Clin. Transl. Med. 14, e1626 (2024).
Luo, W., Weisel, F. & Shlomchik, M. J. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 48, 313–326.e5 (2018).
Han, S. S. et al. BAFF and APRIL expression as an autoimmune signature of membranous nephropathy. Oncotarget 9, 3292–3302 (2018).
Caro-Maldonado, A. et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 192, 3626–3636 (2014).
Patke, A. et al. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J. Exp. Med. 203, 2551–2562 (2006).
Zhang, Y. et al. Inositol-Requiring Enzyme 1α-Mediated Synthesis of Monounsaturated Fatty Acids as a Driver of B Cell Differentiation and Lupus-like Autoimmune Disease. Arthritis Rheumatol. 73, 2314–2326 (2021).
Zeng, Q. et al. Spleen fibroblastic reticular cell-derived acetylcholine promotes lipid metabolism to drive autoreactive B cell responses. Cell Metab. 35, 837–854.e8 (2023).
Li, J. et al. B cell metabolism in autoimmune diseases: signaling pathways and interventions. Front Immunol. 14, 1232820 (2023).
Fujita, T. et al. Comparison of lipid and fatty acid metabolism between minimal change nephrotic syndrome and membranous nephropathy. Vivo 20, 891–893 (2006).
Qu, Y. et al. Role of metabolomic profile as a potential marker to discriminate membranous nephropathy from IgA nephropathy. Int Urol. Nephrol. 56, 635–651 (2024).
Miao, H. et al. Lactobacillus species ameliorate membranous nephropathy through inhibiting the aryl hydrocarbon receptor pathway via tryptophan-produced indole metabolites. Br. J. Pharm. 181, 162–179 (2024).
Thurman, J. M. Triggers of inflammation after renal ischemia/reperfusion. Clin. Immunol. 123, 7–13 (2007).
Gao, X. et al. Systematic variations associated with renal disease uncovered by parallel metabolomics of urine and serum. BMC Syst. Biol. 6, S14 (2012).
van der Windt, G. J. & Pearce, E. L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 249, 27–42 (2012).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Procaccini, C. et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity 44, 406–421 (2016).
Gerriets, V. A. et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 125, 194–207 (2015).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Ray, J. P. et al. The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity 43, 690–702 (2015).
Zeng, H. et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 45, 540–554 (2016).
Ma, R. et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 20, 21–27 (2018).
Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 50, 37–50 (2019).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 20, 1327–1333 (2014).
Kempkes, R. W. M. et al. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front Immunol. 10, 2839 (2019).
Beier, U. H. et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. Faseb j. 29, 2315–2326 (2015).
Liu, Q. et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat. Metab. 4, 559–574 (2022).
Howie, D. et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2, e89160 (2017).
Angelin, A. et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 25, 1282–1293.e7 (2017).
Johnson, M. O. et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795.e19 (2018).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Cobbold, S. P. et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. USA 106, 12055–12060 (2009).
Hoxha, E. T-cell epitopes of PLA2R1 in membranous nephropathy: another step toward antigen-based immunotherapies. Kidney Int 103, 466–469 (2023).
Chang, C. H. & Pearce, E. L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 17, 364–368 (2016).
Fu, J. Y. et al. Lysine acetyltransferase 6A maintains CD4(+) T cell response via epigenetic reprogramming of glucose metabolism in autoimmunity. Cell Metab. 36, 557–574.e10 (2024).
Cribbs, A. P. et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc. Natl. Acad. Sci. USA 117, 6056–6066 (2020).
Chávez, M. D. & Tse, H. M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front Immunol. 12, 703972 (2021).
He, X. et al. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res 68, 5591–5598 (2008).
Xie, W. et al. Regulation of autoimmune disease progression by Pik3ip1 through metabolic reprogramming in T cells and therapeutic implications. Sci. Adv. 8, eabo4250 (2022).
Lee, G. R., The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci.19, 2018.
Aso, K. et al. Itaconate ameliorates autoimmunity by modulating T cell imbalance via metabolic and epigenetic reprogramming. Nat. Commun. 14, 984 (2023).
Ye, M. et al. Serum metabolomics analysis reveals metabolite profile and key biomarkers of idiopathic membranous nephropathy. PeerJ 11, e15167 (2023).
Liu, Q. et al. Novel Biomarkers in Membranous Nephropathy. Front Immunol. 13, 845767 (2022).
Shi, X. et al. The diversity of peripheral blood T cells and myeloid-derived suppressor cells in patients with idiopathic membranous nephropathy. Clin. Nephrol. 101, 287–297 (2024).
Koga, T. et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J. Clin. Invest 124, 2234–2245 (2014).
Mondal, S., Saha, S. & Sur, D. Immuno-metabolic reprogramming of T cell: a new frontier for pharmacotherapy of Rheumatoid arthritis. Immunopharmacol. Immunotoxicol. 46, 330–340 (2024).
Muñoz-Urbano, M., Quintero-González, D. C. & Vasquez, G. T cell metabolism and possible therapeutic targets in systemic lupus erythematosus: a narrative review. Immunopharmacol. Immunotoxicol. 44, 457–470 (2022).
Pelgrom, L. R. et al. LKB1 expressed in dendritic cells governs the development and expansion of thymus-derived regulatory T cells. Cell Res 29, 406–419 (2019).
Møller, S. H., Wang, L. & Ho, P. C. Metabolic programming in dendritic cells tailors immune responses and homeostasis. Cell Mol. Immunol. 19, 370–383 (2022).
Thomson, A. W. & Robbins, P. D. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann. Rheum. Dis. 67, iii90–iii96 (2008).
Lleo, A. et al. The consequences of apoptosis in autoimmunity. J. Autoimmun. 31, 257–262 (2008).
Krawczyk, C. M. et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010).
Crow, M. K. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 12, S5 (2010).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Zhao, L. et al. Regulation of macrophage polarization by targeted metabolic reprogramming for the treatment of lupus nephritis. Mol. Med 30, 96 (2024).
Anders, H. J. & Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int 80, 915–925 (2011).
Odegaard, J. I. & Chawla, A. Alternative macrophage activation and metabolism. Annu Rev. Pathol. 6, 275–297 (2011).
Galván-Peña, S. & O’Neill, L. A. Metabolic reprograming in macrophage polarization. Front Immunol. 5, 420 (2014).
Steinberg, G. R. & Schertzer, J. D. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol. Cell Biol. 92, 340–345 (2014).
Li, J. et al. Metabolic signatures of immune cells in chronic kidney disease. Expert Rev. Mol. Med 24, e40 (2022).
Du, L. et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-inflammatory Properties. Cell Metab. 29, 1363–1375.e8 (2019).
Peng, Y. et al. Regulatory Mechanism of M1/M2 Macrophage Polarization in the Development of Autoimmune Diseases. Mediators Inflamm. 2023, 8821610 (2023).
Keppel, M. P. et al. Activation-specific metabolic requirements for NK Cell IFN-γ production. J. Immunol. 194, 1954–1962 (2015).
Marçais, A. et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 15, 749–757 (2014).
Shaw, R. J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. (Oxf.) 196, 65–80 (2009).
Shahrabi, S. et al. Flip-flops of natural killer cells in autoimmune diseases versus cancers: Immunologic axis. J. Cell Physiol. 234, 16998–17010 (2019).
Deniz, G. et al. Regulatory NK cells suppress antigen-specific T cell responses. J. Immunol. 180, 850–857 (2008).
Gianchecchi, E., Delfino, D. V. & Fierabracci, A. Natural Killer Cells: Potential Biomarkers and Therapeutic Target in Autoimmune Diseases? Front Immunol. 12, 616853 (2021).
Rhoads, J. P., Major, A. S. & Rathmell, J. C. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 13, 313–320 (2017).
Iwata, S. & Tanaka, Y. Therapeutic perspectives on the metabolism of lymphocytes in patients with rheumatoid arthritis and systemic lupus erythematosus. Expert Rev. Clin. Immunol. 17, 1121–1130 (2021).
Lee, D. S. W., Rojas, O. L. & Gommerman, J. L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20, 179–199 (2021).
Sentís, A. et al. Kinetic analysis of changes in T- and B-lymphocytes after anti-CD20 treatment in renal pathology. Immunobiology 222, 620–630 (2017).
Rojas-Rivera, J. E., Carriazo, S. & Ortiz, A. Treatment of idiopathic membranous nephropathy in adults: KDIGO 2012, cyclophosphamide and cyclosporine A are out, rituximab is the new normal. Clin. Kidney J. 12, 629–638 (2019).
Jiang, D. et al. Pyruvate dehydrogenase kinase 4-mediated metabolic reprogramming is involved in rituximab resistance in diffuse large B-cell lymphoma by affecting the expression of MS4A1/CD20. Cancer Sci. 112, 3585–3597 (2021).
He, X. et al. Mycophenolic acid-mediated suppression of human CD4+ T cells: more than mere guanine nucleotide deprivation. Am. J. Transpl. 11, 439–449 (2011).
Fernández-Ramos, A. A. et al. A comprehensive characterization of the impact of mycophenolic acid on the metabolism of Jurkat T cells. Sci. Rep. 7, 10550 (2017).
Dussol, B. et al. Mycophenolate mofetil monotherapy in membranous nephropathy: a 1-year randomized controlled trial. Am. J. Kidney Dis. 52, 699–705 (2008).
Mateos, M. V. & San Miguel, J. F. Bortezomib in multiple myeloma. Best. Pr. Res Clin. Haematol. 20, 701–715 (2007).
Zaal, E. A. et al. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 5, 7 (2017).
Ochoa, T. A. et al. The Proteasome Inhibitor Bortezomib Induces p53-Dependent Apoptosis in Activated B Cells. J. Immunol. 212, 154–164 (2024).
Salhi, S. et al. Bortezomib plus dexamethasone for rituximab-resistant PLA2R(+) membranous nephropathy. Kidney Int. 100, 708–709 (2021).
Becherini, P. et al. CD38-Induced Metabolic Dysfunction Primes Multiple Myeloma Cells for NAD(+)-Lowering Agents. Antioxidants(Basel) 12, (2023).
Verhoeven, D., et al., B-cell targeting with anti-CD38 daratumumab: implications for differentiation and memory responses. Life Sci. Alliance 6. (2023).
Sanders, S. & Harisdangkul, V. Leflunomide for the treatment of rheumatoid arthritis and autoimmunity. Am. J. Med Sci. 323, 190–193 (2002).
Konishi, A. et al. Glucocorticoid imprints a low glucose metabolism onto CD8 T cells and induces the persistent suppression of the immune response. Biochem Biophys. Res Commun. 588, 34–40 (2022).
Guo, Y. et al. Efficacy of leflunomide combined with prednisone for the treatment of PLA2R-associated primary membranous nephropathy. Ren. Fail 42, 122–130 (2020).
Schlöndorff, J. et al. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol. 296, C558–C569 (2009).
Fervenza, F. C. et al. A Multicenter Randomized Controlled Trial of Rituximab versus Cyclosporine in the Treatment of Idiopathic Membranous Nephropathy (MENTOR). Nephron 130, 159–168 (2015).
Ramachandran, R. et al. Two-Year Follow-up Study of Membranous Nephropathy Treated With Tacrolimus and Corticosteroids Versus Cyclical Corticosteroids and Cyclophosphamide. Kidney Int. Rep. 2, 610–616 (2017).
Cybulsky, A. V. et al. Canadian Society of Nephrology Commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis: management of glomerulonephritis in adults. Am. J. Kidney Dis. 63, 363–377 (2014).
Waldman, M. & Austin, H. A. 3rd, Controversies in the treatment of idiopathic membranous nephropathy. Nat. Rev. Nephrol. 5, 469–479 (2009).
Lee, J. U., Kim, L. K. & Choi, J. M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front Immunol. 9, 2747 (2018).
Palmer, B. F. & Clegg, D. J. Kidney-Protective Effects of SGLT2 Inhibitors. Clin. J. Am. Soc. Nephrol. 18, 279–289 (2023).
Vallon, V. & Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu Rev. Physiol. 83, 503–528 (2021).
Zhao, X. Y. et al. SGLT2 inhibitors alleviated podocyte damage in lupus nephritis by decreasing inflammation and enhancing autophagy. Ann. Rheum. Dis. 82, 1328–1340 (2023).
Lv, X. et al. Canagliflozin reverses Th1/Th2 imbalance and promotes podocyte autophagy in rats with membranous nephropathy. Front Immunol. 13, 993869 (2022).
Jenkins, B. J. et al. Canagliflozin impairs T cell effector function via metabolic suppression in autoimmunity. Cell Metab. 35, 1132–1146.e9 (2023).
Qin, J. et al. Empagliflozin modulates CD4(+) T-cell differentiation via metabolic reprogramming in immune thrombocytopenia. Br. J. Haematol. 198, 765–775 (2022).
Taherkhani, A. et al. Metabolomic Analysis of Membranous Glomerulonephritis: Identification of a Diagnostic Panel and Pathogenic Pathways. Arch. Med. Res. 50, 159–169 (2019).
Miao, H. et al. Membranous nephropathy: Systems biology-based novel mechanism and traditional Chinese medicine therapy. Front Pharm. 13, 969930 (2022).
Jiang, S. et al. The primary glomerulonephritides: a systems biology approach. Nat. Rev. Nephrol. 9, 500–512 (2013).
Sieber, J. et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Ren. Physiol. 299, F821–F829 (2010).
Jo, H. A. et al. Fumarate modulates phospholipase A2 receptor autoimmunity-induced podocyte injury in membranous nephropathy. Kidney Int. 99, 443–455 (2021).
Chen, Y. Y. et al. Microbiome-metabolome reveals the contribution of gut-kidney axis on kidney disease. J. Transl. Med. 17, 5 (2019).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82001656 and 82271761), Fundamental Research Program of Shanxi Province (202303021224012), China Postdoctoral Science Foundation (2022M722008), Shanxi Province Clinical Research Center for Dermatologic and Immunologic Diseases (Rheumatic diseases, 2023G2RZ00), Research and Innovation Team Project for Scientific Breakthroughs at Shanxi Bethune Hospital (2024ZHANCHI09), and Shanxi Province Higher Education “Billion Project” Science and Technology Guidance Project (BYJL053).
Author information
Authors and Affiliations
Contributions
X.M.D., X.L.: Organize literature and original draft writing, search literature and writing editing; Y.N.Z., Y.K.J.: Conception and writing review; W.X.C., Y.F.Z., Y.H., H.N.L.: writing and revising of the manuscript. All authors contributed to the writing and revising of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Wenbin Liu and Corrado Murtas for their contribution to the peer review of this work. Primary Handling Editors: Joao Valente. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, X., Lv, X., Wang, X. et al. Impact of immune cell metabolism on membranous nephropathy and prospective therapy. Commun Biol 8, 405 (2025). https://doi.org/10.1038/s42003-025-07816-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07816-3
