
Abstract
Optineurin (OPTN) is an adaptor protein that plays a crucial role in many cellular pathways, including NF-κB signaling, programmed cell death, and vesicular trafficking. OPTN dysfunction has been implicated in the pathogenesis of several diseases, such as primary open angle glaucoma (POAG), amyotrophic lateral sclerosis (ALS). While mutations of OPTN seem to be predominantly loss-of-function in ALS, only gain-of-function mechanisms have been reported in POAG. Here, we demonstrate that OPTN knockout in the retina contributes to short-term astrogliosis, retinal ganglion cell (RGC) loss and long-term microglial activation. Moreover, OPTN loss of function does not exacerbate RGC death induced by ocular hypertension. Integrated bioinformatics and immunofluorescence analyses reveal that OPTN dysfunction leads to neuropeptide Y (NPY) downregulation and CHOP upregulation. Overexpression of wild-type OPTN in a hypertension glaucoma model prevents the RGC loss and attenuates microglial activation. Together, our findings highlight a neuroprotective role for OPTN as a key neuroimmune modulator.
Similar content being viewed by others

Introduction
Optineurin (OPTN) is an important adaptor protein involved in several critical cellular processes, including neuroimmune homeostasis1, protein trafficking, and organelle maintenance2. Mutations in OPTN have been implicated in the pathogenesis of several diseases, such as primary open-angle glaucoma (POAG), amyotrophic lateral sclerosis (ALS), frontotemporal lobar dementia, and Paget’s disease of bone3. Retinal ganglion cells (RGCs) are retinal neurons that transmit visual input from the eye to the brain. Degeneration of RGCs and their axons in the optic nerve are cardinal features of glaucoma and other optic neuropathy conditions, eventually leading to irreversible vision loss4,5,6,7. Importantly, among the genetic mutations associated with glaucoma, mutations in OPTN were found in 16.7% of families with hereditary POAG, including individuals with normal intraocular pressure (IOP) or normal tension glaucoma (NTG)8. Wild-type OPTN plays a crucial role in multiple cellular processes, including autophagy, endocytic trafficking, NF-κB regulation, neuroinflammation, and transcriptional activation9,10,11,12,13. In contrast to the disease-related loss-of-function OPTN mutations in ALS14, studies on glaucoma-associated OPTN mutations suggest a gain-of-function mechanism in optic neuropathies15. Intriguingly, in vitro studies have shown that suppression of OPTN expression in cultured primary RGCs and cell lines suppresses cell growth, increases apoptosis, and reduces neurotrophic support, suggesting a potential neuroprotective role of wild-type OPTN12,16. However, direct evidence related to the role of wild-type OPTN in RGC homeostasis and degeneration in vivo remains unclear. In addition, despite evidence supporting a potential neuroprotective function during RGC development17, it remains unclear whether OPTN also contribute to POAG via a loss-of-function mechanism. Therefore, a comprehensive understanding of the neurodegenerative signaling pathways associated with OPTN dysfunction would greatly facilitate the dissection of the shared and distinct mechanisms among multiple neurodegenerative conditions, particularly NTG, high-tension glaucoma (HTG), and ALS.
While the patterns of optic nerve damage and visual field impairment differ between NTG and HTG18,19, they share substantial pathological similarities, including activation of NF-κB signaling, elevated oxidative endoplasmic reticulum (ER) stress, RGC apoptosis, and gliosis20,21,22,23,24,25,26, all of which are closely associated with the physiological function of wild-type OPTN9,10,11,12,13,27. Meanwhile, it has been suggested that, in NTG, changes in RGC susceptibility may lead to their death within the normal range of IOP28,29. Consistently, lowering IOP is also beneficial for NTG to some extent30. On the other hand, even with significant IOP reduction, approximately 50% of POAG patients continue to experience progressive visual field loss31, highlighting the need to develop IOP-independent therapeutic strategies. As one of the most prevalent disease-associated genes in NTG, how OPTN loss of function contributes to RGC susceptibility and the pathogenesis of NTG and HTG is a critical but unanswered question.
In addition to the intrinsic susceptibility of RGCs to glaucomatous pathology, the responses of glial cells play a significant role in determining neuronal outcomes. Both astrocytes and microglia respond robustly to neurodegenerative stress in optic neuropathies32. Chronic neuroinflammation, resulting from the activation of these glial cells, is increasingly recognized as a key factor in optic neuropathies and various neurodegenerative conditions33,34,35. In a microbead occlusion model of glaucoma, astrocytes have been demonstrated to transform into a neurotoxic reactive state36,37. Microglia, the primary innate immune cells in the central nervous system (CNS), play a pivotal role in neuroimmune homeostasis38. Upon retinal or optic nerve damage, they may transition to an activated state and migrate to sites of injury, potentially contributing to RGC death by releasing neurotoxic factors39,40. Microglia activation is hypothesized as a significant contributing factor to retinal neurodegeneration, which might explain why IOP reduction cannot fully prevent progressive RGC degeneration in glaucoma41. Restoring microglial homeostasis through IGFBPL1 administration can alleviate neuroinflammation and prevent neurodegeneration and visual loss in glaucoma35. In addition, key transcription factors (TFs) involved in RGC degeneration, such as CHOP and ATF342,43,44, promote neurodegeneration through the activation of pro-inflammatory signaling pathways42,45,46,47,48. Therefore, targeting neuroinflammation and preserving neuroimmune homeostasis represent promising neuroprotective strategies for treating glaucoma and potentially other neurodegenerative conditions33. However, despite considerable attention given to the role of inflammation and the innate immune response49,50, it remains unclear whether OPTN plays a role in neuroimmune homeostasis.
Here, we asked whether wild-type OPTN is essential for regulating RGC homeostasis and vulnerability in neurodegeneration. Our findings demonstrate that wild-type OPTN plays a pivotal role in maintaining RGC survival and regulating neuroimmune homeostasis in the retina. This IOP-independent pathway mediated by OPTN may be shared between NTG and HTG. These results underscore the significance of restoring OPTN-mediated neuroimmune homeostasis to impede retinal neurodegeneration.
Results
OPTN C-terminal truncation leads to chronic RGC loss without IOP elevation
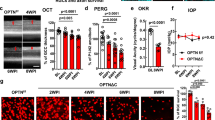
To examine the effects of OPTN deficiency in the retina, we performed the intravitreal injection of AAV2-Cre and AAV2-PLAP (control group) to 1-month-old OPTN floxed mice (Optntm1.1Jda/J; Jackson Labs; Stock# 029708), resulting in a truncated 470 amino acid protein that lacks the C-terminal 114 amino acids (Fig. 1a)51,52. Using immunofluorescent (IF) staining on retinal sections with the RGC marker RBPMS and OPTN C-terminus truncation protein, we detected that the percentage of OPTN expressing RGCs was significantly decreased at 1 month post intravitreal injection of AAV2-Cre, but not in AAV2-PLAP injected retinas (Supplementary Fig. 1a-b). IOP measurements were performed weekly after 4 weeks post injection. Retinas and optic nerves were collected at 1-, 3-, and 6-month post injection for IF analysis. To measure the number of RGCs with OPTN knockout, we quantified the number of RBPMS-positive RGCs in AAV2-Cre and AAV2-PLAP groups compared with those in the intact retina through sectioned and whole mount retinas (Fig. 1b, Supplementary Fig. 1c). RGC number progressively decreased at 1-, 3-, and 6-month post AAV2-Cre injection, but not in the AAV2-PLAP injected controls (Fig. 1c, Supplementary Fig. 2a, b). These results demonstrate that the wild-type OPTN plays a role in maintaining RGC survival. Moreover, knockout of OPTN in the retina did not cause any changes in IOP, consistent with its role in NTG (Fig. 1d).

a Schematic illustration of the OPTN ablation in retina. Created with BioRender.com and used with permission. b Representative IF images showing OPTN knockout induced RGC loss and astrogliosis 1 month post intravitreal injection. Scale bar, 50 μm. c Quantification of RGC survival with OPTN knockout 1, 3, and 6 months post intravitreal injection. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. Statistical analysis was performed using two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. d IOP of mice post intravitreal injection of AAV2-Cre or PLAP virus shows no significant difference between OPTN knockout and control mice. Data are shown as mean ± s.e.m. with n = 10 biologically independent samples. e Quantification of astrogliosis with OPTN knockout 1, 3, and 6 months post intravitreal injection. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. Statistical analysis was performed using two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons.
OPTN dysfunction results in short-term reactive astrogliosis in the retina
Astrocytes are reactivated in optic neuropathies, such as glaucoma36. In response to glaucoma, astrocytes upregulate the glial fibrillary acidic protein (GFAP), enlarge their cytoskeleton, and elongate their cellular processes, a process referred to as reactive astrogliosis32. Following OPTN deletion, we observed significant reactive astrogliosis, as quantified by astrocytic fiber count53 using GFAP immunolabeling, while astrogliosis was barely detectable in the control groups (Fig. 1b). This reactive astrogliosis following AAV2-Cre injection was reversible, with a peak at 1-month post injection that returned to levels comparable to the control group by 6-month post injection (Fig. 1b, e, Supplementary Fig. 2a, 2b). This suggests that OPTN dysfunction contributed to a short-term astrocytic response that resolved over time.
OPTN dysfunction leads to long-term microglia activation in the retina
Microglial activation is an early pathological hallmark of glaucoma-associated neurodegeneration and causes the release of pro-inflammatory cytokines, leading to damage in RGCs across various animal models of glaucoma38,41,54. To further identify the role of OPTN in retinal homeostasis, we examined the microglia activation (Fig. 2a, Supplementary Fig. 3a, 3b) and morphology (Fig. 2b) following OPTN knockout. CD68 was used to identify activated microglia in a phagocytic state55,56. We observed a basal level of microglia activation in the AAV2-PLAP injected retina, consistent with findings from a previous study57. However, following OPTN knockout, the proportion of activated microglia was significantly elevated compared to the control mice. This elevation persisted up to 6-month post injection (Fig. 2c). This suggests that OPTN knockout may induce persistent activation of microglia in the retina. The number of microglia (Fig. 2d), their cell body diameter (Fig. 2e) and primary process lengths (Fig. 2f) were systematically examined to assess the impact of OPTN dysfunction on their morphology and active state. The results of microglia count analysis indicate that OPTN loss of function did not significantly alter microglia cell numbers (Fig. 2d). Microglia had enlarged cell bodies, with shortened processes, suggesting that the microglia exhibited a transformation into activated, ameboid morphology following OPTN knockout. Together, these results demonstrate that OPTN deficiency leads to a persistent neuroinflammatory response.

a Representative IF images showing OPTN knockout induced microglia activation 6 months post intravitreal injection. Scale bar, 50 μm. b A lateral view of 3D-reconstituted section retina with OPTN knockout 3 months post intravitreal injection. Scale bar, 5 µm. c Quantification of percentage of Iba1-positive microglia that are also CD68-positive with OPTN knockout 1, 3, and 6 months post intravitreal injection. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. ***p < 0.001, ns, not significant, calculated by unpaired t-test. d–f Quantification of the number of Iba1-positive microglia (d), Iba1-positive microglia cell body diameter (e), and the average primary process length of Iba1-positive microglia (f) at 1, 3, and 6 months post intravitreal injection. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. Statistical analysis was performed using two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons.
OPTN dysfunction leads to microglia activation without astrogliosis in the optic nerve
To investigate whether glaucoma-related pathology in the retina with OPTN loss of function extends to the optic nerve, we conducted IF staining on the optic nerve section to detect neuroinflammation at 3-month post OPTN knockout. We found that OPTN knockout leads to activation of microglia (Fig. 3a, b) but no astrogliosis (Fig. 3c, d) in the optic nerve. Together, these findings imply that retinal OPTN may be involved in modulating the signaling pathways associated with microglia activation and that the neuroinflammatory response induced by OPTN knockout may be spread to the optic nerve.

a, c Representative IF images showing OPTN knockout induced microglia activation (a), and astrogliosis (c) in optic nerve. Scale bar, 50 μm (left) and 10 μm (right). b, d Quantification of microglia activation (b), and astrogliosis (d) in optic nerve 3 months post intravitreal injection. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. ***p < 0.001, ns not significant, calculated by unpaired t-test.
OPTN dysfunction does not further exacerbate RGC death in the ocular hypertension mouse model
The retinal OPTN knockout exhibited characteristic pathological features observed in NTG. We then introduced an HTG modeling method to analyze the role of the OPTN pathway in HTG pathology. Our recently developed viscobead injection model of ocular hypertension provides a more stable blockage of aqueous humor outflow with biodegradable viscoelastic beads, leading to sustained ocular hypertension58. We performed the viscobead injection 1 month after OPTN knockout, and retinas were collected 3 months post-OPTN knockout (Fig. 4a) for IF staining (Fig. 4b). Concentrated viscobeads were injected into the mouse anterior chamber to block aqueous drainage (Fig. 4c). During the 2 months following the viscobead injection, IOP remained significantly elevated, compared to the control group (Fig. 4d). IF results showed that RGC survival rates following viscobead injection were similar between the OPTN intact group and the OPTN knockout group, with no significant difference (Fig. 4b, e, Supplementary Fig 4a). The control group (PBS injected) showed no significant difference in RGC survival compared to intact, demonstrating that the injection surgery will not result in the glaucomatous phenotypes. OPTN loss of function did not exacerbate RGC death under induced ocular hypertension, which implies that elevated IOP and OPTN loss of function may affect an overlapping group of vulnerable RGCs. This observation aligns with clinical observations on the overlapping degeneration patterns in HTG and NTG59. It is also possible that there may be overlapping gene regulatory pathways regulating the optic neuropathies resulting from both ocular hypertension and OPTN loss of function.

a Schematic illustration of the viscobead injection model. Created with BioRender.com and used with permission. b Representative IF images showing viscobead (VB) injection induced RGCs loss in both OPTN knockout and control retina. Scale bar, 50 μm. c Representative photographs of viscobeads accumulated at mouse iridocorneal angle 5 min after injection. Arrows show that the white viscobeads were restricted at the iridocorneal angle. Scale bar, 1 mm. d IOP in the mice after the injection of viscobeads (n = 10 biologically independent samples) or PBS (n = 7 biologically independent samples) in OPTN knockout and control group. Data are shown as mean ± s.e.m. e Quantification of RGC survival in different groups of the viscobead injection model. Data are shown as mean ± s.e.m. with n = 6 biologically independent samples. *p < 0.05, ns, not significant. Statistical significance among multiple groups was determined using one-way ANOVA followed by Tukey’s multiple comparisons test.
OPTN dysfunction and ocular hypertension induced similar patterns of astrogliosis and microglia activation in the retina
To determine the involvement of OPTN-mediated neuroinflammatory pathways in the pathogenesis of glaucoma with high IOP, we further analyzed the astrogliosis and microglia activation provoked by high IOP in both OPTN intact and OPTN knockout retinas. Quantification of astrogliosis and microglia activation was performed using the same methods described earlier to maintain consistency and scientific rigor. The PBS-injected control group exhibited comparable levels of astrogliosis (Fig. 5a, b) and microglia activation (Fig. 6a, b) to those observed in the OPTN intact control group (Figs.1d, 2b), indicating that neuroinflammatory responses triggered by PBS injection had resolved within 2 months. No significant differences in astrogliosis (Fig. 5a, b) and microglia activation (Fig. 6a, b) were observed between the OPTN knockout and intact groups following viscobead injection. These results are consistent with the observation that OPTN knockout does not further lead to RGC death in the ocular hypertension condition. Together, these findings imply that OPTN-mediated neuroinflammatory pathways might be shared between optic neuropathies in both normal tension and hypertension conditions.

a Representative IF images showing OPTN knockout induced astrogliosis. Scale bar, 50 μm. b Quantification of astrogliosis with OPTN knockout in retina. Data are shown as mean ± s.e.m. with n = 6 biologically independent samples. ***p < 0.001, ns, not significant. Statistical significance among multiple groups was determined using one-way ANOVA followed by Tukey’s multiple comparisons test.

a Representative IF images showing OPTN knockout and the high IOP model induced microglia activation. Scale bar, 50 μm. b Quantification of microglia activation with OPTN knockout and the high IOP model in retina. Data are shown as mean ± s.e.m. with n = 6 biologically independent samples. ***p < 0.001, ns, not significant. Statistical significance among multiple groups was determined using one-way ANOVA followed by Tukey’s multiple comparisons test.
OPTN promotes the expression of NPY to prevent RGC loss and maintain neuroimmune homeostasis in the retina
We have demonstrated that OPTN plays a role in neuroimmune modulation in RGC degeneration. It is still unclear what molecules in the OPTN mediated signaling pathways are directly involved in RGC vulnerability in optic neuropathy. Importantly, RGCs differ in their ability to survive from an injury related to optic neuropathies60,61. Using optic nerve crush (ONC), which is a well-established model of retinal neurodegeneration, previous single-cell transcriptomic analyses have offered an RGC subtype vulnerability roadmap following different time points after ONC injury60. By employing comprehensive bioinformatic analysis of the injury-associated RGC atlas, we found that OPTN is enriched in resilient RGCs in the ONC model and that its expression level significantly increases following optic nerve injury (Fig. 7a). This result aligns with the neuroprotective role of OPTN. To identify the most significantly associated genes by OPTN, we performed the differentially expressed gene (DEG) analysis between OPTN-positive and OPTN-negative RGCs (Fig. 7b, Supplementary Fig. 5a). Out of all DEGs, we found that chemokine FAM19A4 and neuropeptide Y (NPY), previously not recognized as downstream inflammatory modulators of OPTN, are significantly increased in the RGCs endogenously expressing OPTN (Fig. 7b). FAM19A4 encodes a chemokine that acts as a regulator of immune and nervous cells62, supporting the demonstrated role of OPTN in maintaining the neuroimmune homeostasis of the retina. NPY is a 36 amino acid peptide and is involved in many physiological processes, including stress response and cortical excitability63,64. It has been reported that activation of the NPY receptor could partially rescue the RGC death in glaucoma mouse model of ocular hypertension65, implicating that OPTN may activate NPY signaling to prevent RGC death from glaucomatous optic neuropathy. Our comprehensive bioinformatic analyses on open-sourced single-cell RNA sequencing (scRNA-seq) dataset of RGCs after ONC reveal that NPY is significantly enriched in OPTN-positive resilient RGCs, with increased expression after ONC injury in mice (Fig. 7c; Supplementary Fig. 5b-5e). In addition, both the proportion of cells expressing NPY and its expression level are markedly higher than those of FAM19A (Supplementary Fig. 5e). Of note, resilient and susceptible RGCs express different types of NPY receptors (Supplementary Fig. 5f).

a Single cell RNA-seq analysis of OPTN in intact and injured RGCs from ONC mouse model. Res, resilient RGCs; Sus, susceptible RGCs. Numbers on top of the violin plots show the percentage of RGCs expressing the analyzed genes. b Volcano plot showing differentially expressed genes between OPTN-positive and OPTN-negative intact RGCs from ONC mouse model. c Dot plot showing expression level and percentage expressed of NPY and FAM19A4 between Resilient and Susceptible RGCs from ONC mouse model. d Dot plot showing expression level and percentage expressed of OPTN, NPY, and FAM19A4 in control and NTG patients. ***p < 0.001, **p < 0.01, *p < 0.05, calculated by Wilcoxon rank-sum test. e Representative IF images of NPY fluorescent intensity after OPTN KO. Scale bar, 50 μm. f Quantification of NPY fluorescent intensity after OPTN KO. Data are shown as mean ± s.e.m. with n = 7 biologically independent samples. **p < 0.01, calculated by unpaired t-test.
To further examine whether OPTN expression level may be altered under glaucomatous conditions in human patients, we performed bioinformatic analysis on a recently published dataset integrating a cohort of 91 NTG patients and 92 healthy individuals66. Consistently, we found a statistically significant downregulation of OPTN, NPY, and FAM19A4 in RGCs from NTG patients (Fig. 7d, Supplementary Fig. 6a–d). In contrast, the average expression levels of OPTN in NTG patients exhibit a slight but insignificant reduction compared to those in control individuals (Supplementary Fig. 6e), implicating its cell-type-specific neuroprotective function in human NTG. Consistently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses between OPTN-positive and OPTN-negative RGCs indicate differential transcriptional and epigenetic programs associated with neurodegenerative disease-related pathways (Supplementary Fig. 7a–d).
To verify our hypothesis that the level of endogenous NPY may be affected by OPTN dysfunction, we further examined the expression level of NPY in ganglion cell layer (GCL) and inner plexiform layer (IPL) after OPTN knockout in the retina (Fig. 7e). The expression level of NPY is significantly decreased at 3-month post OPTN knockout (Fig. 7f). Although we cannot entirely exclude the possibility that NPY level in OPTN-deficient retina may be partially attributed to the reduced number of RGCs, this data implicates NPY as a potential downstream effector protein of OPTN-mediated neuroprotection.
Previous genome-wide sequencing studies have demonstrated the activation of CHOP and ATF3 in retinal neurodegeneration, both of which target neuroinflammatory pathways42. Given that OPTN knockout induces neuroinflammation, we subsequently investigated whether it activates the CHOP and ATF3 expression (Supplementary Figs. 8a, 9a). Our results demonstrated that the CHOP signaling pathway is activated following OPTN loss-of-function but not ATF3 (Supplementary Figs. 8b, 9b). It implies that these injury-induced transcription factors may have different regulatory mechanisms in retinal neurodegeneration. These findings suggest that the OPTN-NPY signaling pathway may suppress the CHOP-mediated neuroinflammation to maintain neuroimmune homeostasis in the retina.
Overexpression of wild-type OPTN rescues the RGC loss and microglia activation in wild-type mice following ocular hypertension
To investigate the neuroprotective role of wild-type OPTN in the context of ocular hypertension-induced glaucoma, we overexpressed human wild-type OPTN in the retinas of 1-month-old C57BL/6 mice using AAV.CPP.16 (CPP16), a capsid-modified AAV vector previously validated for high transduction efficiency in the CNS of both mice and non-human primates67,68. The use of the CPP16 capsid enhances the translational potential of OPTN-based gene therapy from rodents to primates. We performed the viscobead injection 1 month after OPTN overexpression, and retinas were collected 3 months post-OPTN overexpression for IF analysis on RGC death, astrogliosis, and microglia activation. As a disease-relevant control, we also overexpressed the human E50K OPTN69, which is commonly associated with NTG, to evaluate whether this mutation exacerbates the pathophysiology of high IOP-induced glaucoma. Retinas transduced with PLAP served as controls for both normal and elevated IOP conditions following PBS or viscobead injection, respectively (Fig. 8a). As a result, our IF analysis revealed that wild-type OPTN overexpression significantly attenuated RGC loss (Fig. 8b, c) and microglia activation (Supplementary Fig. 10a, 10b) under ocular hypertension, although it did not reduce astrogliosis (Fig. 8b, d). These results suggest that wild-type OPTN may exert neuroprotective effects by suppressing microglia activation rather than modulating astrogliosis. Notably, overexpression of E50K OPTN did not exacerbate RGC loss, astrogliosis, or microglia activation, implying that E50K OPTN may share overlapping pathological mechanisms with high-IOP glaucoma. IOP measurements taken weekly after viscobead injection revealed no significant differences among viscobead-injected groups (Supplementary Fig. 10c), indicating that the protective effects of wild-type OPTN were not due to alterations in IOP. Collectively, these findings demonstrate that wild-type OPTN mitigates high IOP-induced RGC death via suppressing microglia activation, highlighting its therapeutic potential in glaucoma.

a Schematic illustration of the OPTN overexpression in the high IOP model. Created with BioRender.com and used with permission. b Representative IF images showing wild-type OPTN overexpression alleviates high IOP-induced RGC loss but not astrogliosis. Scale bar, 50 μm. c Quantification of RGC survival in high IOP model after OPTN overexpression. d Quantification of astrogliosis in high IOP model after OPTN overexpression. Data are shown as mean ± s.e.m. with n = 5 biologically independent samples. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant. Statistical significance among multiple groups was determined using one-way ANOVA followed by Tukey’s multiple comparisons test.
Discussion
One major finding of our study is the critical role of wild-type OPTN in RGC survival and retinal neuroimmune homeostasis. OPTN loss of function contributes to retinal gliosis, neuroinflammation, and RGC degeneration, which is complementary to the widely recognized gain-of-function mechanisms of OPTN. Gain-of-function OPTN mutations, such as E50K or M98K, have been demonstrated to induce RGC degeneration by enhancing the OPTN-TBK1 pathway in NTG70,71,72. Our results are consistent with the role of loss-of-function OPTN mutations in ALS73, implying that OPTN mutations may contribute to optic neuropathies through both loss-of-function and gain-of-function manner. Our GO enrichment analysis revealed that the involvement of OPTN-positive RGCs in distinct signaling pathways in intact and injured conditions (Supplementary Fig. 5c, e), suggesting that OPTN-related signaling may contribute to homeostatic regulation in response to injury. Consistent with this analysis, KEGG analysis indicated an enrichment of neurodegeneration-associated pathways in OPTN-positive RGCs (Supplementary Fig. 5d, f), implicating a potential role of OPTN in modulating neuronal survival and regeneration. Interestingly, recent studies investigating the role of endogenous OPTN dysfunction in neurodegeneration have yielded divergent findings. Consistent with our results, by employing the mouse γ-synuclein (mSncg) promoter to specific knockout OPTN in RGCs, one recent study has demonstrated that OPTN dysfunction results in IOP-independent RGC death and axon degeneration by decreasing axonal mitochondrial transport52. Given that AAV2 may also transduce amacrine cells74, we cannot entirely exclude the possibility of non-cell-autonomous RGC death mechanisms induced by OPTN loss-of-function. Intriguingly, another recent study has suggested a reduced total thickness of the ganglion cells and inner plexiform layers, while the RGC loss was not statistically significant in a mouse model with CMV-driven systemic knockout of OPTN from the first coding exon75. It should be noted that our study flanked exon 12 of OPTN with LoxP sites, resulting in a version of OPTN with C-terminus truncation, which has been found in both NTG8 and ALS76 patients. Intriguingly, the OPTN N-terminal coiled-coil region of OPTN, where gain-of-function mutations such as E50K and M98K have been identified, is responsible for the OPTN/TBK1 interaction71,77. Thus, an intriguing possibility is that mutations in different domains of OPTN may result in different pathological mechanisms. In addition, other binding partners of OPTN/TBK1 complex might also contribute to neuroprotection or neurodegeneration, which certainly merits future investigation.
Neuroinflammation is a critical hallmark of many neurodegenerative conditions, such as Alzheimer’s disease78, Parkinson’s disease79, Huntington’s disease80, and ALS81. In the case of optic neuropathies such as glaucoma, activation of microglia and astrocytes is recognized as a relatively early event of damage preceding RGC loss82,83,84. Our study suggests that OPTN loss-of-function may contribute to neuroinflammation that exacerbates RGC death in both HTG and NTG caused by OPTN mutations. This observation aligns with previous studies showing that inhibiting microglia activation can delay RGC death39,85. Using our optimized viscobead injection model, we found that OPTN loss of function does not further exacerbate RGC degeneration in ocular hypertension. This observation suggests that OPTN deficiency might affect an overlapping subset of vulnerable RGCs under ocular hypertension. Furthermore, by overexpressing human wild-type OPTN and the NTG-associated E50K mutant in the retina, we demonstrated that wild-type OPTN confers neuroprotection against high IOP-induced RGC degeneration, suppressing microglia activation but not astrogliosis. Notably, overexpression of E50K OPTN did not exacerbate RGC loss or neuroinflammatory responses under elevated IOP, implicating that a subset of vulnerable RGCs might be affected by certain overlapped pathways involved in both IOP-dependent and IOP-independent degeneration. This possibility is not fully explored in the current study and warrants further investigation. Our study has identified a shared OPTN-driven neuroprotective mechanism underlying retinal neurodegeneration in both NTG and HTG. Previous studies have identified CHOP and ATF3 as important injury-induced TFs that activate a set of neuroinflammation genes42,48,49,86. Our study has also revealed that OPTN loss of function leads to upregulation of CHOP, a major pro-apoptotic gene induced by ER stress46,48, which is consistent with in vitro evidence that OPTN regulates the ER stress-induced signaling pathways and cell death27. Intriguingly, ATF3, whose expression has been demonstrated to be drastically increased in acute neural injury42,86, was not activated in OPTN knockout condition (Supplementary Fig. 9). One possible explanation is that the gradual loss of RGCs in our normal tension optic neuropathy model did not reach the threshold for injury induced up-regulation of ATF3 that can be observed in ONC injury42,60,87.
ER stress is closely linked to microglial activation88 and has been reported in both NTG and HTG models23,24. Our bioinformatic analyses and IF validation have revealed that NPY enriched in OPTN-expressing RGCs and OPTN knockout results in the downregulation of NPY, while NPY has been demonstrated to inhibit ER stress89,90. Furthermore, many other studies have demonstrated that NPY functions as an immunomodulator65,91,92. These findings suggest that OPTN dysfunction-induced downregulation of NPY may disinhibit ER stress and result in persistent microglia activation and progressive RGC death. Our study provides a possible OPTN-mediated mechanism by which NPY modulates CHOP-related ER stress, which may be crucial for maintaining neuroimmune homeostasis. This discovery highlights the OPTN-NPY pathway as a promising therapeutic target for NTG, HTG, and potentially other neurodegenerative conditions, especially for cases unresponsive to IOP-lowering interventions.
Notably, anti-inflammatory treatments for neurodegenerative conditions have yielded promising outcomes in pre-clinical and clinical studies, which at least effectively prevent CNS neuronal cell death in glaucoma93,94,95, Alzheimer’s Disease96, and ALS97. Nevertheless, different neuroimmune modulators targeting different components of the neuroinflammatory signaling networks resulted in inconsistent results, which potentially explains why some early trials of this drug category failed98,99,100,101. Further understanding of the intrinsic mechanisms underlying neuroimmune dysregulation in neurodegeneration is essential to optimize such therapeutic strategies. Here, we demonstrate an endogenous neuroprotective mechanism mediated by OPTN, showing that enhancement of the wild-type OPTN pathway can prevent RGC death in a hypertension glaucoma model by suppressing microglia activation. This targeted approach may help minimize off-target effects commonly associated with broad-spectrum neuroimmune modulation. Importantly, the neuroprotective effect by retinal overexpression of wild-type OPTN mediated by the CPP16 capsid AAV, which is highly efficient in transducing CNS cells in both mice and non-human primates67, further enhances the translational potential of this study. Furthermore, our study has suggested NPY, FAM19A, and CHOP as potential downstream components of the OPTN signaling pathway in neuroprotection. Importantly, a recent study has reported that NPY exerts significant neuroprotective effects in a glaucoma mouse model65. NPY has also been evaluated in clinical trials for various other neurodegenerative conditions. For example, a randomized controlled trial assessing intranasal NPY for major depressive disorder (MDD) reported a significant reduction in depression severity within 24 h post-treatment, suggesting rapid therapeutic effects102. This rapid therapeutic action is attributed to NPY’s ability to mitigate stress-induced damage to the CNS, which is consistent with the DEG and GO associated with OPTN. The OPTN-driven and IOP-independent neuroprotection and neuroimmune modulation mechanisms demonstrated by us offer a complementary strategy to existing glaucoma treatments. Endogenous neuropeptides such as NPY represent a promising target for neuroimmune modulation in neurodegenerative conditions.
Methods
Animals
All experimental procedures were performed in compliance with animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Beth Israel Deaconess Medical Center and Harvard Medical School in accordance with guidelines from the Office of Laboratory Animal Welfare (OLAW). The housing environment included controlled temperature (20–24 °C), humidity (40–60%), and a 12 h light/dark cycle. Animals had ad libitum access to food and water. Environmental enrichment, such as nesting materials and shelters, was provided to support animal welfare and natural behaviors. OPTN floxed mice or wild-type control mice aged 4 weeks were used for OPTN knockout through intravitreal AAV2 injection. Viscobead injection was conducted at the age of 8 weeks. Male and female mice were used in this study at ratios dependent on litter available and with equal distributions across experiments conducted extemporaneously. OPTN floxed mouse strain (Optntm1.1Jda/J) was obtained from Jackson Laboratory (Stock# 029708).
Intravitreal AAV injection
\For all surgical procedures, mice were anesthetized with ketamine and xylazine and received Buprenorphine as a postoperative analgesic. As previously described, intravitreal virus injection was performed at the age of 4 weeks. Briefly, a pulled-glass micropipette was inserted near the peripheral retina behind the ora serrata and deliberately angled to avoid damage to the lens. 2 μL of the AAV2/2-CAG-Cre virus was injected for OPTN fl/fl mice. For OPTN knockout injection, the titer of each AAV2/2-CAG-Cre was adjusted to 1 × 1012 genome copies/mL. For OPTN overexpression, we utilized the CPP16 capsid to deliver human wild-type OPTN and the E50K OPTN via intravitreal injection (2 μL per eye, viral titer ~3 × 1013 genome copies/mL). In all experiments, an AAV2 vector expressing placental alkaline phosphatase (AAV2-PLAP) was used as the control103,104,105.
Viscobead-induced experimental mouse glaucoma model
The elevation of IOP was induced by injection of viscobead to the anterior chamber of mouse eyes. The surgery procedures were modified by a well-established microbead occlusion model106. Briefly, by using a standard double emulsion method, poly-d,l-lactic-co-glycolic acid (PLGA) / polystyrene (PS) core-shell microparticles (viscobead) with 1–20 μm size distributed were first fabricated at a concentration of 30% (v/v) in saline. The corneas of anesthetized mice were gently punctured near the center using a 33 g needle (CAD4113, sigma). A bubble was injected through this incision site into the anterior chamber to prevent possible leakage. Then, 1 μL viscobead was injected into the anterior chamber. After 5 min when the viscobead accumulated at the iridocorneal angle, the mouse was applied antibiotic vetropolycin ointment (Dechra Veterinary Products, Overland Park, KS) and placed on a heating pad for recovery.
Intraocular pressure measurement
The IOP measurements were performed using a TonoLab tonometer (Colonial Medical Supply, Espoo, Finland) according to product instructions. Mice were first anesthetized with a sustained isoflurane (NDC 14043-704-05, Patterson Veterinary) flow (3% isoflurane in 100% oxygen). The administration of isoflurane was discontinued once the mice were anesthetized, and IOP measurements were conducted within a 3 min timeframe, in order to minimize the influence of isoflurane on IOP. Average IOP was generated automatically with five measurements after the elimination of the highest and lowest values.
Perfusions and tissue processing
For IF staining, animals were given an overdose of anesthesia and transcardiacally perfused with ice-cold PBS followed by 4% paraformaldehyde (PFA, sigma). After perfusion, retinas and optic nerves were dissected out and postfixed in 4% PFA overnight at 4 ⁰C. Tissues were cryoprotected by sinking in 30% sucrose in PBS for 48 h. Samples were frozen in Optimal Cutting Temperature compound (Tissue Tek) using dry ice and then sectioned at 14 μm for retinas and 12 μm for optic nerves.
Retinal wholemount staining and quantification of RGC survival
Dissected retinas were rinsed in PBS and then blocked in PBS with 1% Triton X-100 and 5% horse serum (wholemount buffer) overnight at 4 °C. Retinas were then incubated with primary antibodies diluted in wholemount buffer for 2–4 days at 4 °C, followed by three rinses with PBS (10 min each time). Next, retinas were incubated with secondary antibodies (all with 1:500 dilution) diluted in PBS overnight at 4 °C. Finally, after five PBS washes (10 min each time), retinas were mounted with Fluoromount-G (Southern Biotech, Cat. No. 0100-01).
IF staining and imaging analysis
Cryosections were permeabilized and blocked in blocking buffer (0.5% Triton X-100 and 5% horse serum in PBS) for 1 hour at room temperature and overlaid with primary antibodies against OPTN, RBPMS, Iba1, CD68, and GFAP (OPTN, Cayman, No. 100,000, 1:100; RBPMS, Raygene, A008712, 1:500; Iba1, Novus, NB100-1028, 1:500; CD68, Biorad, MCA1957T, 1:500; GFAP, Dako, Z0334, 1:500) overnight at 4 °C. On the next day, the corresponding Alexa Fluor 488-, 555- or 647-conjugated secondary antibodies were applied (all secondary antibodies were purchased from Invitrogen). All stained sections were mounted with solutions with DAPI-containing mounting solution and sealed with glass coverslips. All immunofluorescence-labeled images were acquired using Zeiss 700 or Zeiss 710 confocal microscope. For each biological sample, 3–5 sections of each retina or optic nerve were imaged and were taken under 10x or 20x objectives for quantification. For optic nerve analysis, astrogliosis and microglial activation were quantified within the region located 1–2 mm distal to the optic nerve head. Images were processed using Fiji software (provided in the public domain, https://imagej.net/Fiji/). Briefly, GFAP quantification was achieved by counting GFAP-positive fibers within the inner nuclear layer (INL) layer and post the INL layer of the retina55. The number of CD68-positive microglia was counted using a multi-point tool in Fiji. The microglia number, diameter of Iba1-positive cell bodies and the process length were measured in Fiji. The expression level of NPY was evaluated by measuring the fluorescence intensity within the GCL and IPL of the retina.
scRNA-seq analysis—differential gene expression
The Homo sapiens eye dataset was obtained from Human Cell Atlas and only the normalized UMI matrix was used for further analysis66. The RGC dataset GSE137398 underwent a standard Seurat pipeline with the R package Seurat107. Differential expression analysis was conducted with the Bioconductor package DESeq2108. Statistical significance of differentially expressed genes (DEGs) was determined at padj < 0.05 and |Log2FC | > 1. Statistical significance between groups in scRNA-seq dataset was determined using the nonparametric alternative of the t-test, the Wilcoxon rank-sum test.
Statistics and reproducibility
The normality and variance similarity were measured by Microsoft Excel and R programming Language before we applied for any parametric tests. A two-tailed student’s t-test was used for the single comparison between the two groups. The rest of the data were analyzed using one-way or two-way ANOVA depending on the appropriate design. Post hoc comparisons were carried out only when the primary measure showed statistical significance. The P-value of multiple comparisons was adjusted by using Bonferroni’s correction. Error bars in all figures represent mean ± S.E.M. The mice with different litters, body weights and sexes were randomized and assigned to different treatment groups, and no other specific randomization was used for the animal studies. For all experiments, each group consists of a minimum of four biologically independent samples to ensure reproducibility.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files or are available upon reasonable requests to the authors.
References
Ames, J. et al. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat. Commun. 12, 5401 (2021).
Ryan, T. A. & Tumbarello, D. A. Optineurin: a coordinator of membrane-associated cargo trafficking and autophagy. Front. Immunol. 9, 1024 (2018).
Slowicka, K., Vereecke, L. & van Loo, G. Cellular functions of optineurin in health and disease. Trends Immunol. 37, 621–633 (2016).
Kwon, Y. H., Fingert, J. H., Kuehn, M. H. & Alward, W. L. Primary open-angle glaucoma. N. Engl. J. Med 360, 1113–1124 (2009).
Wiggs, J. L. & Pasquale, L. R. Genetics of glaucoma. Hum. Mol. Genet. 26, R21–R27 (2017).
Zhang, N., Wang, J., Li, Y. & Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: a meta-analysis and systematic review. Sci. Rep. 11, 13762 (2021).
Jonas, J. B. et al. Glaucoma. Lancet 390, 2183–2193 (2017).
Rezaie, T. et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295, 1077–1079 (2002).
Korac, J. et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 126, 580–592 (2013).
Sundaramoorthy, V. et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 3830–3846 (2015).
Sahlender, D. A. et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J. Cell Biol. 169, 285–295 (2005).
Sippl, C., Bosserhoff, A. K., Fischer, D. & Tamm, E. R. Depletion of optineurin in RGC-5 cells derived from retinal neurons causes apoptosis and reduces the secretion of neurotrophins. Exp. Eye Res. 93, 669–680 (2011).
Nagabhushana, A. et al. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol. 11, 4 (2010).
Ito, Y. et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016).
Morton, S., Hesson, L., Peggie, M. & Cohen, P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 582, 997–1002 (2008).
Li, H., Ao, X., Jia, J., Wang, Q. & Zhang, Z. Effects of optineurin siRNA on apoptotic genes and apoptosis in RGC-5 cells. Mol. Vis. 17, 3314–3325 (2011).
Wang, J. T. et al. Disease gene candidates revealed by expression profiling of retinal ganglion cell development. J. Neurosci. 27, 8593 (2007).
Kiriyama, N. et al. A comparison of optic disc topographic parameters in patients with primary open angle glaucoma, normal tension glaucoma, and ocular hypertension. Graefe’s. Arch. Clin. Exp. Ophthalmol. 241, 541–545 (2003).
Thonginnetra, O. et al. Normal versus high tension glaucoma: a comparison of functional and structural defects. J. Glaucoma 19, 151–157 (2010).
Xia, Q. & Zhang, D. Apoptosis in glaucoma: a new direction for the treatment of glaucoma (Review). Mol. Med Rep. 29, 82 (2024).
Trivli, A. et al. Normal‑tension glaucoma: pathogenesis and genetics (review). Exp. Ther. Med 17, 563–574 (2019).
Hernandez, H., Roberts, A. L. & McDowell, C. M. Nuclear factor-kappa beta signaling is required for transforming growth factor Beta-2 induced ocular hypertension. Exp. Eye Res. 191, 107920 (2020).
Zode, G. Increased endoplasmic reticulum stress in human glaucomatous trabecular meshwork cells and tissues. Investig. Ophthalmol. Vis. Sci. 56, 3260–3260 (2015).
Sayyad, Z. et al. A glaucoma-associated OPTN polymorphism, M98K sensitizes retinal cells to endoplasmic reticulum stress and tumour necrosis factor α. FEBS J. 290, 3110–3127 (2023).
Mi, X. S., Yuan, T. F. & So, K. F. The current research status of normal tension glaucoma. Clin. Inter. Aging 9, 1563–1571 (2014).
García-Bermúdez, M. Y. et al. Glial cells in glaucoma: friends, foes, and potential therapeutic targets. Front. Neurol. 12 (2021).
Ramachandran, G., Moharir, S. C., Raghunand, T. R. & Swarup, G. Optineurin modulates ER stress-induced signaling pathways and cell death. Biochem. Biophys. Res. Commun. 534, 297–302 (2021).
Nemesure, B., Honkanen, R., Hennis, A., Wu, S. Y. & Leske, M. C. Incident open-angle glaucoma and intraocular pressure. Ophthalmology 114, 1810–1815 (2007).
Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Am. J. Ophthalmol. 126, 487–497 (1998).
Heijl, A. et al. Reduction of intraocular pressure and glaucoma progression: results from the early manifest glaucoma trial. Arch. Ophthalmol. 120, 1268–1279 (2002).
Shalaby, W. S., Ahmed, O. M., Waisbourd, M. & Katz, L. J. A review of potential novel glaucoma therapeutic options independent of intraocular pressure. Surv. Ophthalmol. 67, 1062–1080 (2022).
Tezel, G. Molecular regulation of neuroinflammation in glaucoma: current knowledge and the ongoing search for new treatment targets. Prog. Retinal Eye Res. 87, 100998 (2022).
Bariş, M. & Tezel, G. Immunomodulation as a neuroprotective strategy for glaucoma treatment. Curr. Ophthalmol. Rep. 7, 160–169 (2019).
Baudouin, C., Kolko, M., Melik-Parsadaniantz, S. & Messmer, E. M. Inflammation in Glaucoma: from the back to the front of the eye, and beyond. Prog. Retinal Eye Res. 83, 100916 (2021).
Pan, L. et al. IGFBPL1 is a master driver of microglia homeostasis and resolution of neuroinflammation in glaucoma and brain tauopathy. Cell Rep. 42, 112889 (2023).
Guttenplan, K. A. et al. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep. 31, 107776 (2020).
Yoo, H.-S., Shanmugalingam, U. & Smith, P. D. Harnessing astrocytes and müller glial cells in the retina for survival and regeneration of retinal ganglion cells. Cells 10, 1339 (2021).
Guo, L., Choi, S., Bikkannavar, P. & Cordeiro, M. F. Microglia: key players in retinal ageing and neurodegeneration. Front. Cell. Neurosci. 16, 804782 (2022).
Bosco, A. et al. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Models Mech. 8, 443–455 (2015).
Qu, J. & Jakobs, T. C. The time course of gene expression during reactive gliosis in the optic nerve. PLOS ONE 8, e67094 (2013).
Ishikawa, M. et al. Glaucoma and microglia-induced neuroinflammation. Front. Ophthalmol. 3, 1132011 (2023).
Tian, F. et al. Core transcription programs controlling injury-induced neurodegeneration of retinal ganglion cells. Neuron 110, 2607–2624.e2608 (2022).
Syc-Mazurek, S. B., Fernandes, K. A., Wilson, M. P., Shrager, P. & Libby, R. T. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol. Neurodegen. 12, 71 (2017).
Fang, F. et al. RGC-specific ATF4 and/or CHOP deletion rescues glaucomatous neurodegeneration and visual function. Mol. Ther. Nucleic Acids 33, 286–295 (2023).
Li, Y. et al. The multifaceted roles of activating transcription factor 3 (ATF3) in inflammatory responses—potential target to regulate neuroinflammation in acute brain injury. J. Cereb. Blood Flow. Metab. 43, 8–17 (2023).
Goodall, J. C. et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 107, 17698–17703 (2010).
Willy, J. A., Young, S. K., Stevens, J. L., Masuoka, H. C. & Wek, R. C. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol. Biol. Cell 26, 2190–2204 (2015).
Liu, X. et al. Microglia-derived IL-1β promoted neuronal apoptosis through ER stress-mediated signaling pathway PERK/eIF2α/ATF4/CHOP upon arsenic exposure. J. Hazard. Mater. 417, 125997 (2021).
Rolle, T., Ponzetto, A. & Malinverni, L. The role of neuroinflammation in glaucoma: an update on molecular mechanisms and new therapeutic options. Front. Neurol. 11, 612422 (2021).
Kaur, G. & Singh, N. K. The role of inflammation in retinal neurodegeneration and degenerative diseases. Int. J. Mol. Sci. 23, 386 (2022).
Munitic, I. et al. Optineurin insufficiency impairs IRF3 but Not NF-κB activation in immune cells. J. Immunol. 191, 6231–6240 (2013).
Liu, D. et al. Optineurin-facilitated axonal mitochondria delivery promotes neuroprotection and axon regeneration. Nat. Commun. 16, 1789 (2025).
Augustine, J. et al. IL-33 deficiency causes persistent inflammation and severe neurodegeneration in retinal detachment. J. Neuroinflamm. 16, 251 (2019).
Fan, W. et al. Retinal microglia: functions and diseases. Immunology 166, 268–286 (2022).
Fu, R., Shen, Q., Xu, P., Luo, J. J. & Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422–1434 (2014).
Kumari, A. et al. Single cell RNA sequencing confirms retinal microglia activation associated with early onset retinal degeneration. Sci. Rep. 12, 15273 (2022).
Bonilha, V. L. et al. Geographic atrophy: confocal scanning laser ophthalmoscopy, histology, and inflammation in the region of expanding lesions. Investig. Ophthalmol. Vis. Sci. 61, 15–15 (2020).
Hong, E. et al. Biologically driven in vivo occlusion design provides a reliable experimental glaucoma model. bioRxiv, 2024.2001.2018.576306 (2024).
Iester, M., De Feo, F. & Douglas, G. R. Visual field loss morphology in high- and normal-tension glaucoma. J. Ophthalmol. 2012, 327326 (2012).
Tran, N. M. et al. Single-cell profiles of retinal ganglion cells differing in resilience to injury reveal neuroprotective genes. Neuron 104, 1039–1055.e1012 (2019).
Duan, X. et al. Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron 85, 1244–1256 (2015).
Sarver, D. C., Lei, X. & Wong, G. W. FAM19A (TAFA): an emerging family of neurokines with diverse functions in the central and peripheral nervous system. ACS Chem. Neurosci. 12, 945–958 (2021).
Li, Q., Bartley, A. F. & Dobrunz, L. E. Endogenously released neuropeptide y suppresses hippocampal short-term facilitation and is impaired by stress-induced anxiety. J. Neurosci. 37, 23 (2017).
Bacci, A., Huguenard, J. R. & Prince, D. A. Differential modulation of synaptic transmission by neuropeptide Y in rat neocortical neurons. Proc. Natl. Acad. Sci. USA 99, 17125–17130 (2002).
Palanivel, V. et al. Neuropeptide Y receptor activation preserves inner retinal integrity through PI3K/Akt signaling in a glaucoma mouse model. PNAS Nexus 3, pgae299 (2024).
Daniszewski, M. et al. Retinal ganglion cell-specific genetic regulation in primary open-angle glaucoma. Cell Genomics 2, 100142 (2022).
Yao, Y. et al. Variants of the adeno-associated virus serotype 9 with enhanced penetration of the blood–brain barrier in rodents and primates. Nat. Biomed. Eng. 6, 1257–1271 (2022).
Sangster, M. L. et al. A blood-brain barrier-penetrant AAV gene therapy improves neurological function in symptomatic mucolipidosis IV mice. Mol. Ther. Methods Clin. Dev. 32, 101269 (2024).
Shim, M. S. et al. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci. Rep. 6, 33830 (2016).
Adi, V. et al. Longitudinal age effects of optineurin E50K mutation and deficiency on visual function. Investig. Ophthalmol. Vis. Sci. 62, 2385–2385 (2021).
Sirohi, K., Kumari, A., Radha, V. & Swarup, G. A glaucoma-associated variant of optineurin, M98K, activates Tbk1 to enhance autophagosome formation and retinal cell death dependent on Ser177 phosphorylation of optineurin. PLOS ONE 10, e0138289 (2015).
Minegishi, Y. et al. Enhanced optineurin E50K–TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet. 22, 3559–3567 (2013).
Toth, R. P. & Atkin, J. D. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front. Immunol. 9, 1017 (2018).
Zhang, Y. et al. Elevating growth factor responsiveness and axon regeneration by modulating presynaptic inputs. Neuron 103, 39–51.e35 (2019).
Su, C.-C., Liu, C., Adi, V., Chan, K. C. & Tseng, H. C. Age-related effects of optineurin deficiency in the mouse eye. Vis. Res. 224, 108463 (2024).
Goldstein, O. et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 86, 446–453 (2016).
Li, F. et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun. 7, 12708 (2016).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Pierce, S. & Coetzee, G. A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLOS ONE 12, e0175882 (2017).
Silvestroni, A., Faull, R. L. M., Strand, A. D. & Möller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. NeuroReport 20 (2009).
Jara, J. H. et al. Evidence for an early innate immune response in the motor cortex of ALS. J. Neuroinflammation 14, 129 (2017).
Vernazza, S., Tirendi, S., Bassi, A. M., Traverso, C. E. & Saccà, S. C. Neuroinflammation in primary open-angle glaucoma. J. Clin. Med. 9, 3172 (2020).
Neufeld, A. H. & Liu, B. Glaucomatous optic neuropathy: when glia misbehave. Neuroscientist 9, 485–495 (2003).
Soto, I. & Howell, G. R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med 4, a017269 (2014).
Bosco, A. et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 49, 1437–1446 (2008).
Renthal, W. et al. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 108, 128–144.e129 (2020).
Jacobi, A. et al. Overlapping transcriptional programs promote survival and axonal regeneration of injured retinal ganglion cells. Neuron 110, 2625–2645.e2627 (2022).
Jing, G., Wang, J. J. & Zhang, S. X. ER stress and apoptosis: a new mechanism for retinal cell death. J. Diab Res. 2012, 589589 (2012).
Palanivel, V. et al. Neuroprotective effects of neuropeptide Y on human neuroblastoma SH-SY5Y cells in glutamate excitotoxicity and ER stress conditions. Cells 11, 3665 (2022).
Lee, D. Y. et al. Neuropeptide Y mitigates ER stress–induced neuronal cell death by activating the PI3K–XBP1 pathway. Eur. J. Cell Biol. 97, 339–348 (2018).
Santos-Carvalho, A., Elvas, F., Alvaro, A. R., Ambrosio, A. F. & Cavadas, C. Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 4, e636 (2013).
Palanivel, V., Basavarajappa, D., Gupta, V. & Graham, S. Protective effects of neuropeptide Y treatment on retina in a mouse model of experimental glaucoma. Acta Ophthalmol. 102 (2024).
Sim, R. H., Sirasanagandla, S. R., Das, S. & Teoh, S. L. Treatment of glaucoma with natural products and their mechanism of action: an update. Nutrients 14, 534 (2022).
Nakazawa, T. et al. Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26, 12633 (2006).
Roh, M. et al. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF- α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLOS ONE 7, e40065 (2012).
Kitazawa, M. et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 187, 6539–6549 (2011).
Mizwicki, M. T. et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am. J. Neurodegener. Dis. 1, 305–315 (2012).
Group*, A. R. Cognitive Function Over Time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): Results of a Randomized, Controlled Trial of Naproxen and Celecoxib. Arch. Neurol. 65, 896–905 (2008).
Küchlin, S. et al. Treatment with erythropoietin for patients with optic neuritis. Neurol. Neuroimmunol. Neuroinflammation 10, e200067.
Rootman, D. B., Gill, H. S. & Margolin, E. A. Intravitreal bevacizumab for the treatment of nonarteritic anterior ischemic optic neuropathy: a prospective trial. Eye 27, 538–544 (2013).
Ahmadzadeh, A., Kessel, L., Schmidt, B. S., Kolko, M. & Bach-Holm, D. Steroids and/or non-steroidal anti-inflammatory drugs as postoperative treatment after trabeculectomy—12-month results of a randomized controlled trial. J. Clin. Med. 13, 887 (2024).
Mathé, A. A., Michaneck, M., Berg, E., Charney, D. S. & Murrough, J. W. A randomized controlled trial of intranasal neuropeptide Y in patients with major depressive disorder. Int. J. Neuropsychopharmacol. 23, 783–790 (2020).
Arcuri, J., Hegarty, S., He, Z. & Bhattacharya, S. K. Lipidomics dataset of PTEN deletion-induced optic nerve regeneration mouse model. Data Brief. 34, 106699 (2021).
Nawabi, H. et al. Doublecortin-like kinases promote neuronal survival and induce growth cone reformation via distinct mechanisms. Neuron 88, 704–719 (2015).
Norsworthy, M. W. et al. Sox11 expression promotes regeneration of some retinal ganglion cell types but kills others. Neuron 94, 1112–1120.e1114 (2017).
Yang, Q. et al. Microbead-induced ocular hypertensive mouse model for screening and testing of aqueous production suppressants for glaucoma. Investig. Ophthalmol. Vis. Sci. 53, 3733–3741 (2012).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We sincerely thank Dr. Zhigang He from Boston Children’s Hospital for constructive suggestions. This work was supported by grants from the National Institutes of Health (EY032181).
Author information
Authors and Affiliations
Contributions
Qinglong Wang, Y. W., R.D., and F.T. conceived, and Qinglong Wang, Y.W., Y.D.D.J., G.C. performed the experiments and analyzed the data. Qinglong Wang, Y.W., and G.C. performed sample collection. Y.D.D.J. and W.Y. performed bioinformatic analyses. Z.L. and J.L. constructed the plasmids for CPP16 virus package. Qinglong Wang, Y.W., R.D., Y.D.D.J., G.C., W.Y., H.G., J.H., Y.L., F.B., Qianbin Wang, and F.T. prepared the manuscript with the input from all authors.
Corresponding author
Ethics declarations
Competing interests
F.T. and D.J. are co-founders of Regenerative AI LLC. F.B. is a founder of and scientific advisor to Brave Bio Inc. F.B. is an inventor of AAV.CPP.16 used in this study. The other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ghanshyam Swarup, Stefania Vernazza, and the other anonymous reviewer for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Wang, Y., Jiang, Y.D.D. et al. OPTN protects retinal ganglion cells and ameliorates neuroinflammation in optic neuropathies. Commun Biol 8, 1475 (2025). https://doi.org/10.1038/s42003-025-08534-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08534-6
