
Abstract
Quantifying the biological processes that drive cancer progression remains a key challenge in oncology. Although the hallmarks of cancer provide a foundational framework for understanding tumor behavior, existing diagnostic tools rarely measure these hallmarks directly. Here we present a neural multi-task learning-based framework that estimates hallmark activity using gene expression data from tumor biopsies. The model was trained on transcriptomic profiles from 941 tumors spanning 14 tissue types and tested on five independent datasets. It predicts the activity of ten cancer hallmarks simultaneously and with high accuracy. Additional validation on large-scale datasets including normal and cancer samples confirmed its sensitivity and specificity. Predicted hallmark activity was associated with clinical staging, suggesting biological relevance. A web-based tool was developed to facilitate integration into research and clinical workflows. This approach enables efficient analysis of transcriptomic data to inform understanding of tumor biology and support individualized treatment strategies.
Similar content being viewed by others


Introduction
Cancer is an inherently heterogeneous disease, yet it progresses through well-defined biological principles that govern its development and spread1,2. Hanahan and Weinberg introduced the concept of the “hallmarks of cancer,” a unifying framework that identifies the fundamental capabilities cancer cells acquire during tumorigenesis3. These core hallmarks include: (1) sustaining proliferative signaling to drive uncontrolled growth; (2) evading growth suppressors to bypass regulatory constraints; (3) resisting cell death to survive environmental and intracellular stress; (4) enabling replicative immortality to achieve limitless cell division; (5) inducing angiogenesis to ensure a continuous nutrient supply through neovascularization; and (6) activating invasion and metastasis to colonize distant tissues. This framework has been expanded to incorporate emerging hallmarks, such as (7) deregulating cellular energetics to sustain rapid proliferation, and (8) avoiding immune destruction by escaping immune surveillance. Enabling characteristics, including (9) genome instability and mutation, which accelerate tumor evolution, and (10) tumor-promoting inflammation, which supports a microenvironment conducive to malignancy, further illustrate the complexity of cancer biology4.
Despite the insights offered by these hallmark frameworks, current diagnostic approaches often fail to integrate these molecular underpinnings into routine clinical practice. Traditional methods, such as staging systems (e.g., AJCC and TNM) and grading scales (e.g., Gleason grading), primarily focus on macroscopic and microscopic tumor characteristics, overlooking the molecular heterogeneity that drives tumor behavior5,6. Consequently, patients with the same cancer type, stage, and grade may exhibit divergent outcomes, exposing the limitations of these approaches. Moreover, these approaches do not provide insights into the dynamic, micro-evolutionary molecular changes within tumors, limiting their capacity to guide personalized treatment strategies7. A hallmark-based diagnostic framework has the potential to address these limitations by integrating molecular data to illuminate the biological mechanisms underlying tumorigenesis—an essential step toward precision oncology, in which treatments are tailored to the individual tumor’s molecular profile8. Although multi-omics technologies, artificial intelligence, and real-time monitoring have advanced considerably, we still lack a single, unified method that can simultaneously annotate all hallmark activities in a tumor9,10.
To address this critical gap, we developed OncoMark, a high-throughput neural multi-task learning (N-MTL) framework designed to simultaneously quantify the activity of all cancer hallmarks using transcriptomic data from tumor biopsies. This is, to the best of our knowledge, the first computational tool specifically designed to predict all cancer hallmarks concurrently. Upon input, OncoMark calculates the probability of each hallmark’s activity, providing a detailed molecular profile of the tumor. The model underwent a rigorous validation process to ensure robustness and generalizability. Cross-validation demonstrated accuracy, precision, recall, and F1 scores exceeding 99%. Testing on five independent external datasets further confirmed its performance, consistently maintaining a minimum of 96.6% across these metrics. Additional validation on eight gold-standard datasets, comprising six cancer (TCGA, MET500, CCLE, TARGET, PCAWG, and POG570) and two normal (GTEx and ENCODE) datasets, verified the model’s high accuracy in distinguishing cancer and normal samples. Moreover, predicted hallmark activity demonstrated significant co-association with AJCC stages and TNM staging, with the strongest co-association observed at advanced stages of cancer progression. Building on its clinical utility, we have developed a user-friendly software platform, accessible at https://oncomark-ai.hf.space/, which seamlessly integrates hallmark activity profiling into research and clinical workflows.
Results
Overview of OncoMark
The hallmarks of cancer comprise a set of core biological capabilities acquired by tumor cells during malignant transformation and progression4. Despite their central role in oncogenesis, no experimental standard currently exists for annotating hallmark activity in clinical biopsy samples. While techniques such as immunohistochemistry (IHC) offer some capacity to infer hallmark states, their reliance on single biomarkers limits both specificity and sensitivity, as no single marker universally defines a given hallmark11. Consequently, hallmark-annotated biopsy datasets remain unavailable for model training.
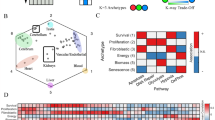
To address this gap, we developed a strategy to generate synthetic, hallmark-annotated bulk transcriptomic profiles by leveraging single-cell RNA sequencing (scRNA-seq) data12,13,14 (Fig.1 and Supplementary Fig. 1). We first curated gene sets corresponding to each hallmark of cancer and used these to compute digital hallmark activity scores for individual cells15. These scores were binarized to indicate the presence or absence of each hallmark, and cells with identical annotations were aggregated to create synthetic pseudo-bulk samples. Single-cell profiles were not used directly for model training, as their resolution captures only localized and often transient transcriptional activity, which does not reliably reflect the population-level hallmark patterns observed in clinical biopsies16. Our aggregation approach preserved biological realism while ensuring training labels were accurate and noise-free.

Single-cell transcriptomic data from multiple cancer types undergo quality control to remove low-quality cells. Each cell is then scored for hallmark gene expression signatures, followed by binary annotation (Yes/No) indicating the presence or absence of each hallmark. These annotated single cells are aggregated to create synthetic pseudo-bulk datasets for each hallmark. A multi-task neural network (M-TNN) is trained on this synthetic data, learning a shared feature representation across all hallmarks, with hallmark-specific output layers enabling accurate prediction of hallmark presence.
However, real-world tumor samples frequently exhibit co-activation of multiple hallmarks, a complexity not present in our synthetic training data by design. If synthetic biopsies were constructed to include cells with diverse hallmark profiles, reliable binary annotation of hallmark presence or absence would become infeasible. Such ambiguity would increase label noise and hinder model performance, particularly in a supervised learning context where robust class separation is critical for effective training17. Moreover, the biological processes underlying different hallmarks are highly interdependent, with shared regulatory pathways and transcriptional programs18. To address both the lack of mixed hallmark states in the training data and the need to capture inter-hallmark dependencies, we adopted a multi-task learning (MTL) framework19.
Our MTL architecture is designed with shared backbone layers that learn generalized transcriptomic features across all hallmarks, paired with hallmark-specific output heads that refine these representations for individual predictions. This setup enables the model to leverage shared biological signals and learn hallmark coordination patterns implicitly—even though the training data contains only non-overlapping hallmark labels. (Supplementary Fig. 2) By doing so, the MTL framework mitigates the disconnect between our cleanly labeled synthetic data and the complex, interwoven hallmark states present in real tumors, allowing the model to generalize effectively to heterogeneous biopsy transcriptomes. The resulting predictions are biologically grounded, robust to hallmark co-activation, and aligned with the functional interplay of cancer hallmark processes. More details are provided in the Methods section.
Validation of hallmark annotations in synthetic training data
To assess the accuracy of hallmark annotations used for model training, we compared the gene expression profiles of hallmark-positive and hallmark-negative samples within the synthetic (pseudo-bulk) datasets. For each hallmark, differential expression analysis was performed20, and the results were visualized as heatmaps representing the log2 fold change and −log10 false discovery rate (FDR) for the associated gene sets (Fig. 2). We observed strong enrichment of hallmark genes in samples annotated as hallmark-positive, as indicated by deep blue signals in both the fold change and FDR panels. Notably, several gene sets displayed infinite FDR values (i.e., unadjusted p values of zero), represented as white or missing entries in the −log10 FDR heatmap, reflecting extremely significant associations. These findings confirm the reliability of our binary hallmark annotations and support the validity of the synthetic training data used for model development.

Heatmaps depict the log2fold change and −log10FDR values for hallmark gene sets, comparing hallmark-present versus hallmark-absent samples for each hallmark. Deep blue intensity in both the log2fold change and −log10FDR panels indicates strong and significant enrichment of the gene set in hallmark-positive samples, supporting accurate hallmark annotation. Blank areas in the −log10FDR heatmap indicate infinite FDR values (i.e., unadjusted p values of zero), corresponding to highly significant associations.
Robust performance across internal cross-validation
We evaluated our model performance in predicting cancer hallmarks using a five-fold cross-validation repeated twice. Our model performance in predicting AIM hallmark achieved an accuracy of 99.97% and an F1 score of 99.97%, with a balanced accuracy of 99.97%, tested on 729 patients (3364 positive and 4184 negative samples). Similarly, the DCE hallmark demonstrated an accuracy of 98.91% and an F1 score of 97.99%, achieving a balanced accuracy of 98.88% across 764 patients (3841 positive and 4249 negative samples). The EGS hallmark delivered exceptional performance with an accuracy of 99.95% and an F1 score of 99.92%, supported by a balanced accuracy of 99.96%, evaluated on 711 patients (3724 positive and 3168 negative samples). The GIM hallmark attained an accuracy of 98.73% and an F1 score of 98.68%, with a balanced accuracy of 98.74% across 395 patients (1282 positive and 846 negative samples). The RCD hallmark showed strong metrics, achieving an accuracy of 99.93%, an F1 score of 99.92%, and a balanced accuracy of 99.93%, tested on 652 patients (2590 positive and 2833 negative samples). The SPS hallmark exhibited perfect performance, achieving 100% accuracy, F1 score, and balanced accuracy, validated on 695 patients (3305 positive and 3796 negative samples). The AID hallmark achieved an accuracy of 99.71% and an F1 score of 99.96%, with a balanced accuracy of 99.74%, evaluated across 691 patients (2832 positive and 3671 negative samples). The IA hallmark recorded an accuracy of 99.92% and an F1 score of 99.87%, with a balanced accuracy of 99.91%, tested on 705 patients (2318 positive and 4334 negative samples). The ERI hallmark demonstrated an accuracy of 99.28% and an F1 score of 98.53%, alongside a balanced accuracy of 99.08%, across 816 patients (3205 positive and 7238 negative samples). Lastly, the TPI hallmark achieved an accuracy of 99.43% and an F1 score of 99.13%, with a balanced accuracy of 99.57%, tested on 730 patients (2326 positive and 4824 negative samples) (Fig. 3a and Table 1). The confusion matrix demonstrated strong classification performance, with clear separation between true positives and false predictions across hallmarks. (Supplementary Fig. 3) For the ROC curves, all cancer hallmark predictions achieved near-perfect true positive rates (TPR) across all thresholds, with AUROC values of 1.00 ± negligible standard deviations, indicating robust discrimination between positive and negative samples for each hallmark (Supplementary Fig. 4a). Similarly, the precision-recall curves confirm the models’ ability to maintain high precision at varying recall levels for each hallmark. The precision values remained consistently high, even as recall approached 1.0, highlighting the reliability of predictions in identifying positive cases without sacrificing precision (Supplementary Fig. 4b).

a Bar plots summarize precision, recall, F1 score, accuracy, and balanced accuracy from two independent rounds of five-fold cross-validation. Individual dots represent performance on each fold, and error bars denote standard deviations. b OncoMark’s generalizability is assessed across synthetic data created from five independent external datasets, using the same evaluation metrics, highlighting robust and consistent predictive performance.
Generalizability across independent external cohorts
The model was validated on five external datasets, with performance metrics reported for OncoMark’s predictions. On the dataset from ref. 21, an accuracy of 97.26% and an F1 score of 96.19% were achieved across 13 patients (105 positive and 41 negative samples). Yost et al.22 achieved an accuracy of 99.64% and an F1 score of 99.42% on 23 patients (175 positive and 105 negative samples). The dataset from ref. 23 achieved an accuracy of 97.40% and an F1 score of 93.46%, validated on 45 patients (260 positive and 395 negative samples). Both Gao et al.24 and Nam et al.25 achieved perfect metrics with 100% accuracy and F1 scores on four patients (1 positive and 22 negative samples) and ten patients (65 positive and 50 negative samples), respectively (Fig. 3b and Table 1). The ROC curves show high AUC values for datasets from ref. 21 (0.98), ref. 22 (0.99), and ref. 23 (0.97), whereas datasets from Gao et al.24 and Nam et al.25 achieved perfect AUC scores of 1.0 (Supplementary Fig. 4c). Similarly, precision values remained close to 1.0 across recall levels for most datasets, with ref. 21 (0.99), ref. 22 (0.99), and ref. 23 (0.98) achieving excellent results, whereas Gao et al.24 and Nam et al.25 maintained perfect values (Supplementary Fig. 4d). Altogether, the external validation included 95 patients with 627 positive and 600 negative samples, highlighting the robust generalizability and performance of the models.
Superior discrimination of normal and malignant tissues
The hallmark-specific signature probabilities predicted by OncoMark across normal (GTEx and ENCODE) and cancer datasets (TCGA, CCLE, POG570, PCAWG, TARGET, and MET500) revealed a critical biological distinction underlying tissue homeostasis and malignancy (Fig. 4a and Supplementary Fig. 5). In normal datasets, hallmark activities exhibit tightly regulated density distributions, reflecting the stable, balanced biological processes essential for maintaining normal cellular function. In contrast, cancer datasets display a marked shift, with significantly elevated probabilities for hallmark-specific signatures, indicative of dysregulated pathways driving tumor progression.

a The left panel displays probability density distributions of hallmark predictions in normal tissue datasets (GTEx and ENCODE), revealing hallmark-specific variation in healthy tissues. The right panel contrasts this with hallmark activity in cancer datasets (TCGA, CCLE, POG570, PCAWG, TARGET, and MET500), showing pronounced shifts in predicted probabilities consistent with malignant transformation. b Density plot for the activating invasion and metastasis hallmark across primary tumor sites with known metastatic cases.
To contextualize OncoMark’s performance, we also compared it against several standard machine learning models, including logistic regression (LR), support vector classifier (SVC), decision tree (DT), random forest (RF), XGBoost, and multi-layer perceptron (MLP). (Supplementary Fig. 6a–f) Unlike OncoMark, these baseline models exhibited a strong bias toward assigning near-zero probabilities for hallmark signatures across all samples. This resulted in acceptable performance for normal tissues but consistently poor sensitivity for cancer samples, indicating that standard models failed to learn meaningful patterns associated with malignancy. Instead, they defaulted to predicting hallmark inactivity across all inputs, highlighting a lack of genuine discriminative learning.
The Kolmogorov–Smirnov (K–S) test further validated the hallmark distributional differences captured by OncoMark, with K–S statistic values exceeding 0.7 and p values effectively zero for all hallmarks (Table 2). These findings highlight both the biological significance of OncoMark’s hallmark activity profiles and its superior capacity to distinguish between healthy and malignant states—a performance not replicated by baseline approaches.
Detection of metastasis hallmark in primary tumors
To evaluate the ability of OncoMark to detect hallmark activity associated with metastatic progression, we focused on the Activating Invasion and Metastasis hallmark using datasets from Vareslija et al.26 and Cosgrove et al.27, comprising matched primary and metastatic tumor transcriptomes. Given that all patients had confirmed metastatic disease, we reasoned that the primary tumors should already exhibit transcriptional programs indicative of metastatic potential. We therefore applied OncoMark exclusively to primary tumor samples. The model predicted elevated activity for the Activating Invasion and Metastasis hallmark across these cases, suggesting that it captures biologically relevant signatures reflective of early metastatic competence (Fig. 4b).
Hallmark activity increases with advancing clinical cancer stage
Given the critical role of clinical staging in hallmark progression, we extended the model’s application to the Cancer Genome Atlas (TCGA) data to further investigate hallmark co-occurrence patterns across various AJCC and TNM staging systems. We quantified co-occurrence using odds ratios (ORs) to assess the strength of associations between specific hallmarks and their corresponding clinical stages28. We found a dynamic progression of hallmark activity corresponding to different clinical cancer stages, offering valuable insights into the biological changes associated with tumor development and progression. In the AJCC stages, hallmark activities progressively increased from Stage I to Stage IV, with the most significant co-association observed at advanced stages, underscoring the critical role of hallmark pathways in promoting tumor aggressiveness (Fig. 5a). The metastasis stage (M0 to M1) revealed increased hallmark activity associated with metastatic potential, while the node stage (n0 to n2/3) demonstrated intensified activity with greater lymph node involvement, highlighting the role of hallmark pathways in tumor spread and metastasis (Fig. 5b, c). A similar trend is observed in tumor stages (t1 to t4), where hallmark activity is highest in t4, reflecting the elevated engagement of these pathways in advanced and aggressive tumors (Fig. 5d). Collectively, these findings highlight the dynamic regulation of hallmark pathways during cancer progression and their potential as biomarkers for disease staging and therapeutic intervention.

Heatmaps display associations between hallmark activity and key clinical staging metrics such as (a) AJCC stage, b metastasis stage, c lymph node involvement, and d tumor size. Color intensity reflects the strength of association, with darker hues indicating stronger relationships. Asterisks (*) denote statistically significant associations (p < 0.05).
Therapeutic associations of hallmark activity with patient survival outcomes
We examined the impact of cancer therapies on patient outcomes, specifically overall survival (OS), disease-free survival (DFS), and progression-free survival (PFS). Using TCGA data, we investigated how cancer drugs influence hallmark activity. Associations between hallmarks and drug treatments were quantified for all cancer patients and incorporated as features in a logistic regression model, with survival status as the dependent variable. The feature weights derived from the trained logistic regression model were interpreted as impact scores, offering a quantitative measure of each drug’s contribution to improving patient survival. These impact scores were used to rank the efficacy of different treatments in terms of their ability to alter hallmark pathways. For disease-free survival (DFS), hallmark ERI demonstrated strong associations with anastrozole, AIM with cyclophosphamide, TPI with radiation, and RCD with trastuzumab and vinorelbine, as indicated by higher impact scores. These findings suggest that these therapies effectively target and modify the underlying hallmarks that drive tumor recurrence (Fig. 6a). In progression-free survival (PFS), hallmark RCD was predominantly influenced by cyclophosphamide, trastuzumab, and vinorelbine, reflecting these drugs’ potential to suppress disease progression. The high impact scores of these treatments highlight their efficacy in disrupting hallmark-specific pathways crucial for tumor growth and metastasis (Fig. 6b). For overall survival (OS), hallmark ERI was significantly modulated by anastrozole and leuprolide, RCD by cyclophosphamide and trastuzumab, GIM and IA by irinotecan, and SPS by radiation, as reflected by high impact scores (Fig. 6c). These results underscore the capacity of these therapies to enhance patient survival by effectively targeting hallmark pathways central to tumor progression.

Heatmaps show the impact of various anticancer drugs on hallmark activity, alongside their associations with (a) disease-free survival, b progression-free survival, and c overall survival. Darker shades correspond to stronger drug–hallmark interactions, revealing key therapeutic contributions to improved patient outcomes.
Discussion
Tumor staging and grading are indispensable for cancer assessment, offering insights into tumor size, spread, and cellular differentiation5. However, these conventional metrics primarily reflect anatomical and morphological characteristics, often failing to capture the molecular mechanisms driving tumor behavior29. Cancer progression is governed not only by physical growth but also by hallmark biological processes4. Identifying and quantifying these hallmarks could provide a more nuanced understanding of tumor biology, uncovering therapeutic vulnerabilities and resistance mechanisms that traditional diagnostic methods overlook30,31. Routine cancer diagnostics, such as imaging and histopathology, are limited in their ability to assess molecular hallmarks due to their focus on observable features rather than the underlying gene expression profiles that drive hallmark activation. Advances in transcriptomics and machine learning offer the potential to bridge this gap32. By leveraging these tools, hallmark-specific molecular patterns can be identified and quantified, enabling more precise assessments of tumor biology. This approach may complement conventional methods, enhancing prognostic accuracy and supporting personalized therapeutic interventions.
OncoMark addresses these challenges by quantifying the activation states of cancer hallmarks using a multi-task neural network trained on synthetic biopsy transcriptomics data. By identifying hallmarks, OncoMark enables the stratification of tumors based on molecular profiles rather than solely anatomical characteristics. This molecular stratification may reveal biologically aggressive tumors that appear indolent through traditional grading and staging methods, improving risk assessment and early detection of relapse or resistance. Additionally, hallmark-driven profiling might uncover novel prognostic biomarkers, enhancing personalized care and disease outcome predictions11. Furthermore, OncoMark capacity to capture hallmark interdependencies offers a comprehensive view of tumor heterogeneity, which is critical for advancing precision oncology. Beyond prognosis, OncoMark offers significant potential in therapy design by identifying hallmark-specific vulnerabilities. For instance, tumors characterized by hallmark activation of angiogenesis may benefit from anti-angiogenic therapies, while those with genomic instability might respond to DNA damage repair inhibitors. OncoMark also ensures clinical relevance across diverse types of cancer, potentially supporting the development of tailored treatment strategies. Moreover, tracking hallmark states over time might help guide therapeutic adjustments and monitor treatment responses, which could contribute to improving patient outcomes31.
Despite its promise, the widespread adoption of OncoMark faces several challenges. The high cost of transcriptomic profiling and the need for specialized infrastructure limit accessibility in many healthcare systems, particularly in resource-limited settings. Integration into clinical workflows will require significant adaptations, including updates to diagnostic guidelines and training for healthcare providers. Moreover, validation of the OncoMark model on real-world data was not possible due to the unavailability of ground truth hallmark data, and comprehensive benchmarking could not be conducted in the absence of tools capable of annotating these hallmarks. However, these challenges may diminish with technological advancements. The declining cost of sequencing, development of portable sequencing devices, and emergence of cloud-based analytical platforms could democratize access to transcriptomic analysis33. Additionally, the growing integration of electronic health records and data-sharing initiatives may facilitate the incorporation of hallmark-based diagnostics into clinical practice.
In conclusion, OncoMark is, to our knowledge, the first computational tool to predict all hallmarks of cancer simultaneously. By bridging the gap between molecular biology and clinical practice, OncoMark has the potential to enhance prognostication, improve therapeutic targeting, and support the transition toward precision oncology. With continued advances in technology and infrastructure, hallmark-based diagnostics may become a routine component of personalized cancer management, offering patients more accurate and effective care.
Methods
Data overview
We used a comprehensive dataset of 3.1 million single-cell transcriptomes from 14 tumor sites (Head and neck, lung, liver/biliary, kidney, prostate, sarcoma, brain, breast, pancreas, neuroendocrine, colorectal, ovarian, skin, and hematologic), collected from 941 patients across 56 studies worldwide as part of the Weizmann 3CA repository, to generate synthetic (pseudo-bulk) datasets. (Supplementary Table 1) For model training and cross-validation, we used 90% of the studies (encompassing 846 patients from 51 studies). For external validation, we used the remaining 10% of studies, which were excluded from the training set. These five excluded studies—refs. 2125—included a total of 95 patients. Additionally, we collected real-world bulk transcriptomic data from publicly available sources, including The Cancer Genome Atlas (TCGA, n = 6679), MET500 (n = 868)34, POG570 (n = 570)35, Cancer Cell Line Encyclopedia (CCLE, n = 1019)36, therapeutically applicable research to generate effective treatments (TARGET, n = 734), Pan-cancer analysis of whole genomes (PCAWG, n = 1210)37, and normal datasets from genotype-tissue expression (GTEx, n = 8228)38, and the encyclopedia of DNA elements (ENCODE, n = 329)39,40. These datasets were used to evaluate the sensitivity and specificity of OncoMark in predicting hallmark activity in both synthetic and real-world settings. The detailed methodology for processing and sampling real-world bulk transcriptomic data were outlined in the Supplementary Methods. All datasets were publicly available, de-identified, and obtained with appropriate consent from participants in their respective studies. Institutional permissions for data use were secured by respective studies, negating the need for further ethics approvals.
Synthetic data construction
Single-cell RNA-seq data processing and quality control
Prior to synthetic data generation, raw single-cell RNA sequencing (scRNA-seq) data underwent rigorous quality control (QC) to ensure reliability and biological relevance of downstream analyses41,42. The following QC criteria were applied:
• Mitochondrial content filtering: Cells with mitochondrial transcript content exceeding 15% were excluded to eliminate stressed or dying cells, which typically exhibit elevated mitochondrial RNA levels due to leakage.
• Transcript count thresholding: Cells expressing fewer than 200 or more than 6000 genes were discarded to remove low-quality cells and potential multiplets. These thresholds ensured the retention of biologically meaningful cells with appropriate RNA content.
Importantly, we did not filter cells based on cell type, nor did we restrict the analysis to malignant cells. This approach preserved the cellular heterogeneity characteristic of real tumor biopsies, including stromal and immune infiltration. Additionally, we intentionally refrained from applying batch correction to the source transcriptomes to retain technical variability introduced by differing sequencing platforms, library preparation protocols, and contributing centers. This design choice allowed the synthetic data to emulate the complexity of real-world transcriptomic profiles.
Gene set curation for cancer hallmarks
Gene sets associated with cancer hallmarks were curated from multiple databases, incorporating only genes identified in at least two independent sources43,44,45,46,47,48. To evaluate their prognostic relevance, we applied a Cox proportional hazards model using patient-level expression and survival data from The Cancer Genome Atlas (TCGA)49. Genes with hazard ratios (HR) <1.05 were excluded to enrich for features with a stronger association to adverse clinical outcomes and tumor progression. The resulting gene sets were further refined via manual literature curation to ensure inclusion of genes mechanistically implicated in hallmark-related biological processes. (Supplementary Data 1)
Computation of digital hallmark scores
Digital hallmark scores were computed across 3.1 million single cells using UCell, a robust method for quantifying gene signature enrichment in scRNA-seq data15. UCell calculates gene signature scores for single-cell RNA sequencing (scRNA-seq) data based on the Mann–Whitney U-statistic. Given a gene expression matrix \(X\) of size \({G\; \times\; C}\), where \(G\) is the number of genes and \(C\) is the number of cells, UCell first constructs a relative rank matrix \(R\) by ranking the expression values within each column (i.e., each cell). Due to the sparse nature of scRNA-seq data—characterized by many zero values—low-ranking genes tend to form a long, uninformative tail. To address this, all ranks greater than a predefined threshold rmax (default = 1500) are set to rmax + 1, thereby mitigating the influence of this tail. For each of the hallmark signatures consisting of n genes, the UCell score \({U}_{j}^{{\prime} }\) for each cell j in X is computed:
Where \({U}_{j}\) is the Mann–Whitney U statistics computed by:
Threshold determination and binary label assignment
To assign binary hallmark labels to single cells, we benchmarked three thresholding algorithms: Otsu50, Yen51, and Isodata. Given the tissue-specific expression patterns of hallmark-associated genes, we computed digital score thresholds independently for each tissue site and hallmark. Threshold performance was evaluated based on the separation of bimodal hallmark score distributions. Otsu’s method consistently outperformed others and was adopted for subsequent analyses (Supplementary Fig. 7).
Rather than applying a single cutoff, we defined upper and lower thresholds per hallmark and tissue type. Cells with scores above the upper threshold were labeled as hallmark-positive, while those below the lower threshold were labeled as hallmark-negative. Cells with intermediate scores were excluded to minimize label ambiguity.
Synthetic biopsy generation for model training and validation
Following label assignment, synthetic pseudo-bulk datasets were created by aggregating hallmark-positive or hallmark-negative single cells12,13,14,52. For each tumor site (n = 14), we selected 200 cells per sample (where available) from either class of each hallmark and summed their gene expression values to form a pseudo-bulk profile. This aggregation strategy enabled the modeling of hallmark-specific transcriptomic signals while retaining biological heterogeneity. The choice of 200 cells balanced representational depth with the need for adequate sample diversity for training.
For validation, we generated synthetic datasets from five independent scRNA-seq studies. To mitigate inductive bias, we reused the previously established hallmark thresholds but adopted a modified aggregation strategy. Specifically, we included all available hallmark-positive or negative cells per patient, applying only a minimum count threshold of 100 cells. This resulted in variable pseudo-bulk sizes, better reflecting the inherent heterogeneity and compositional imbalance of real tumor samples. These datasets were used exclusively for testing the generalizability and robustness of our model, across unseen conditions.
Feature selection and gene filtering
To identify the most informative genes while accounting for missing data, we implemented a variance-based feature selection approach53. Gene-wise variance was computed across all pseudo-bulk samples. Genes with undefined or zero variance were excluded. The top 10,000 genes with the highest valid variance were retained. To ensure consistency across hallmark-positive and -negative datasets, we intersected the top variable genes from both classes, yielding a final set of 9326 features for model training.
Data normalization and preprocessing
To ensure uniform feature representation, missing gene values were imputed as zeros. All datasets—including both synthetic and external validation sets—were transformed into rank space to minimize batch effects and negate the effects of diverse data normalization techniques across diverse datasets54. Expression values were subsequently log2-transformed and z-score standardized (mean = 0, standard deviation = 1) across samples. These normalization steps were done to improve model convergence.
Model architecture
The OncoMark framework leverages a deep learning approach to predict hallmark activity in biopsy samples, emphasizing both hallmark-specific precision and the biological interplay among hallmarks. The model architecture is structured as a multi-task neural network, consisting of a shared base layer and task-specific output layers19. The shared base layer processes the input features \(x\in {R}^{d}\) to extract pan-hallmark characteristics that are universal across all cancer hallmarks, resulting in a latent representation \(h={f}_{{shared}}\left(x\right)\). The task-specific output layers then refine these shared representations by focusing on hallmark-specific features, enabling the model to capture the nuanced interplay between hallmarks. Each output layer produces an independent probability \(\hat{{y}_{i}}={{\rm{\sigma }}}({f}_{{{\mbox{task}}}_{i}}(h)),{{\mbox{where}}} \,{{\mbox{i}}}\in \{1,\ldots ,10\}\) and \(\hat{y}=\left[\hat{{y}_{1}},\hat{{y}_{2}},\ldots ,\hat{{y}_{10}}\right]\) \(\in {\left[{\mathrm{0,1}}\right]}^{10}\) represents the model’s prediction of hallmark presence across all ten hallmarks. This architectural design reflects the interconnected nature of hallmark activities observed in tumor biology and ensures that predictions are biologically meaningful. By combining a shared representation with hallmark-specific refinement, the framework achieves accurate predictions that align with the cooperative and dynamic behavior of hallmarks in cancer progression. The detailed model’s architecture, and training methodology are elaborated in the supplementary methods (Supplementary Figs. 2, 8).
Model training and hyperparameter optimization
The model was trained on a balanced dataset comprising 67,930 samples with 9326 input features representing gene expression profiles. Of these, 57,735 samples (85%) were used for training, and 10,195 samples (15%) were used for validation, with data splitting performed separately for each hallmark using the train-test split method to ensure balanced representation across both sets. To prevent catastrophic forgetting (a phenomenon where a model forgets previously learned information when trained on new tasks), data from all hallmark tasks were merged and shuffled randomly during training, avoiding hallmark-specific batches and ensuring uniform exposure of the model to all hallmark datasets. The Adam optimizer, with a learning rate of 0∙0001, was employed to minimize the binary cross-entropy loss, which was calculated independently for each hallmark prediction task and combined into a weighted average using task-specific indicators to ensure balanced learning. Early stopping, with a patience of six epochs, was implemented to prevent overfitting by halting training when validation loss showed no improvement, while a learning rate scheduler further adjusted the learning rate by reducing it by 0∙5 after three consecutive stagnant epochs, with a minimum threshold set at 1e-6. Training was conducted for 50 epochs with a batch size of 256. Although validation losses consistently improved, gains became marginal in later epochs, leading to the decision to halt training after 50 epochs before full convergence (Supplementary Fig. 9 and Supplementary Table 2).
Evaluation protocol and performance metrices
Our methodology incorporated a five-fold cross-validation repeated twice to ensure robust model assessment. In each iteration, four folds were used to train the model, while the fifth fold was divided into two equal parts: one part served as a validation set for model selection and performance monitoring, and the other part was reserved for independent performance evaluation. (Supplementary Table 3) Moreover, the model was also validated on five independent datasets. Model performance was assessed using the F1 score, accuracy score, precision score, recall score, balanced accuracy, confusion matrix, area under the precision-recall curve (AUC-PR) and the receiver operating characteristic curve (AUC-ROC). (Supplementary Data 2) We report the mean and standard deviation (SD) of these AUC values across all repetitions of cross-validation to capture variability and reliability. Additionally, the model was applied to two normal datasets, Genotype-Tissue Expression (GTEx) and The Encyclopedia of DNA Elements (ENCODE), as well as six cancer datasets from The Cancer Genome Atlas (TCGA), MET500, POG570, Cancer Cell Line Encyclopedia (CCLE), Therapeutically Applicable Research to Generate Effective Treatments (TARGET), and Pan-Cancer Analysis of Whole Genomes (PCAWG). Probability density distributions of hallmark predictions were plotted to assess the model’s ability to identify hallmark occurrences in cancer datasets, with no hallmark predictions made for normal samples. To determine whether the probability distributions between cancer and normal samples differ significantly, the Kolmogorov–Smirnov (K–S) test was conducted, providing statistical insights into these differences55,56.
Statistics and reproducibility
Python v3.11.0 with numpy v2.0.2 and pandas v2.2.3 was used for data analysis, and matplotlib v3.9.2 and seaborn v0.13.2 for visualization. Tensorflow v2.18.0 was used for training the model. Differential expression analysis was conducted using PyDESeq2 v0.5.1. Scipy v1.14.1 was used for conducting all the statistical tests. All p values resulting from the corresponding statistical tests were used without any adjustments or modifications, except for differential expression analysis, where p values were transformed using −log10.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All synthetic data generated in this study is publicly available at https://doi.org/10.5061/dryad.zw3r228jc57. The source data for all the figures in the manuscript is provided in the Supplementary Data 3. The single-cell data used in this study is publicly available at https://www.weizmann.ac.il/sites/3CA/. MET500 and TARGET data are publicly available at https://xenabrowser.net/datapages/. The TCGA data can be obtained from https://gdac.broadinstitute.org/. The PCAWG data were downloaded from https://www.cbioportal.org/. GTEx data were publicly available at https://www.gtexportal.org/home/. CCLE data were publicly available at https://sites.broadinstitute.org/ccle/datasets. ENCODE data were publicly available at https://www.encodeproject.org/. POG570 data were publicly available at bcgsc.ca/downloads/POG570/.
Code availability
All codes generated in this study are publicly available at https://github.com/SML-CompBio/OncoMark. The web server can be accessed here: https://oncomark-ai.hf.space/. The Python package is available here: https://pypi.org/project/OncoMark/. The comprehensive documentation for using OncoMark is available at https://oncomark.readthedocs.io/en/latest/.
References
Lenz, G. et al. The origins of phenotypic heterogeneity in cancer. Cancer Res. 82, 3–11 (2022).
Swanton, C. et al. Embracing cancer complexity: hallmarks of systemic disease. Cell 187, 1589–1616 (2024).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Brierley, J. et al. Global consultation on cancer staging: promoting consistent understanding and use. Nat. Rev. Clin. Oncol. 16, 763–771 (2019).
Greene, F. L. & Sobin, L. H. The staging of cancer: a retrospective and prospective appraisal. CA Cancer J. Clin. 58, 180–190 (2008).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680 (2020).
Su, X. et al. The promising role of nanopore sequencing in cancer diagnostics and treatment. Cell Insight 4, 100229 (2025).
Granja, J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465 (2019).
Gao, S. et al. Identification and construction of combinatory cancer hallmark–based gene signature sets to predict recurrence and chemotherapy benefit in stage II colorectal cancer. JAMA Oncol. 2, 37–45 (2016).
Zhou, Y. et al. Tumor biomarkers for diagnosis, prognosis and targeted therapy. Signal Transduct. Target. Ther. 9, 1–86 (2024).
Murphy, A. E. & Skene, N. G. A balanced measure shows superior performance of pseudobulk methods in single-cell RNA-sequencing analysis. Nat. Commun. 13, 7851 (2022).
Menden, K. et al. Deep learning–based cell composition analysis from tissue expression profiles. Sci. Adv. 6, eaba2619 (2020).
Chen, Y. et al. Deep autoencoder for interpretable tissue-adaptive deconvolution and cell-type-specific gene analysis. Nat. Commun. 13, 6735 (2022).
Andreatta, M. & Carmona, S. J. UCell: robust and scalable single-cell gene signature scoring. Comput. Struct. Biotechnol. J. 19, 3796–3798 (2021).
Kiselev, V. Y., Andrews, T. S. & Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 20, 273–282 (2019).
Frenay, B. & Verleysen, M. Classification in the presence of label noise: a survey. IEEE Trans. Neural Netw. Learn. Syst. 25, 845–869 (2014).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Caruana, R. Multitask learning. Mach. Learn. 28, 41–75 (1997).
Muzellec, B., Teleńczuk, M., Cabeli, V. & Andreux, M. PyDESeq2: a python package for bulk RNA-seq differential expression analysis. Bioinformatics 39, btad547 (2023).
Dong, R. et al. Single-cell characterization of malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell 38, 716–733.e6 (2020).
Yost, K. E. et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019).
Pal, B. et al. A single‐cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 40, e107333 (2021).
Gao, R. et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 39, 599–608 (2021).
Nam, A. S. et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature 571, 355–360 (2019).
Varešlija, D. et al. Transcriptome characterization of matched primary breast and brain metastatic tumors to detect novel actionable targets. J. Natl Cancer Inst. 111, 388–398 (2019).
Cosgrove, N. et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat. Commun. 13, 514 (2022).
Cornfield, J. A method of estimating comparative rates from clinical data; applications to cancer of the lung, breast, and cervix. J. Natl Cancer Inst. 11, 1269–1275 (1951).
Bello, D. M., Russell, C., McCullough, D., Tierno, M. & Morrow, M. Lymph node status in breast cancer does not predict tumor biology. Ann. Surg. Oncol. 25, 2884–2889 (2018).
Hanahan, D. Hallmarks of cancer: a 2012 perspective. Ann. Oncol. 23, ix23 (2012).
Bailón-Moscoso, N., Romero-Benavides, J. C. & Ostrosky-Wegman, P. Development of anticancer drugs based on the hallmarks of tumor cells. Tumor Biol. 35, 3981–3995 (2014).
Wei, L. et al. Artificial intelligence (AI) and machine learning (ML) in precision oncology: a review on enhancing discoverability through multiomics integration. Br. J. Radiol. 96, 20230211 (2023).
Loose, M., Malla, S. & Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 13, 751–754 (2016).
Robinson, D. R. et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017).
Pleasance, E. et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat. Cancer 1, 452–468 (2020).
Ghandi, M. et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019).
Aaltonen, L. A. et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
THE GTEX CONSORTIUM. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Kazachenka, A. et al. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell 175, 1259–1271.e13 (2018).
Luecken, M. D. & Theis, F. J. Current best practices in single‐cell RNA‐seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Heumos, L. et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 24, 550–572 (2023).
Menyhárt, O. et al. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta 1866, 300–319 (2016).
Zhang, D. et al. CHG: a systematically integrated database of cancer hallmark genes. Front. Genet. 11, 29 (2020).
Iannuccelli, M. et al. CancerGeneNet: linking driver genes to cancer hallmarks. Nucleic Acids Res. 48, D416–D421 (2020).
Liang, P.-I. et al. Curation of cancer hallmark-based genes and pathways for in silico characterization of chemical carcinogenesis. Database 2020, baaa045 (2020).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Menyhart, O., Kothalawala, W. J. & Győrffy, B. A gene set enrichment analysis for the cancer hallmarks. J. Pharm. Anal. 15, 101065 (2024).
Cox, D. R. Regression models and life-tables. J. R. Stat. Soc. Ser. B 34, 187–202 (1972).
Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybernetics 9, 62–66 (1979).
Yen, J.-C., Chang, F.-J. & Chang, S. A new criterion for automatic multilevel thresholding. IEEE Trans. Image Process. 4, 370–378 (1995).
Crowell, H. L. et al. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 11, 6077 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Qiu, X., Wu, H. & Hu, R. The impact of quantile and rank normalization procedures on the testing power of gene differential expression analysis. BMC Bioinformatics 14, 124 (2013).
Smirnov, N. Table for estimating the goodness of fit of empirical distributions. Ann. Math. Statist. 19, 279–281 (1948).
Shiryayev, A. N. in Selected Works of A. N. Kolmogorov (ed. Shiryayev, A. N.) Ch. 15 (Springer, 1992).
Priyadarshi, S. Synthetic bulk RNA-Seq transcriptomic profiles representing 10 cancer hallmarks. Dryad https://doi.org/10.5061/dryad.zw3r228jc (2025).
Acknowledgements
We sincerely thank Dr. Amarnath Pal and Dr. Milan Patra for their valuable feedback on the initial manuscript draft, which helped us improve it. Our gratitude extends to the S.N. Bose National Centre for Basic Sciences and Ashoka University for their generous support and funding. S.P., B.N., and D.G. are grateful to the Mphasis F1 Foundation for their support. The funders were not involved in the study’s design, data collection and analysis, decision to publish, or manuscript preparation. The views expressed in this article are those of the authors and do not necessarily reflect the policies, decisions, or perspectives of their employers or associated institutions. The results shown here are in whole or part based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and Therapeutically Applicable Research to Generate Effective Treatments (https://www.cancer.gov/ccg/research/genome-sequencing/target) initiative, phs000218.
Author information
Authors and Affiliations
Contributions
S.P. conceived and designed the study, developed the whole framework, performed the analyses, and interpreted the results. S.H. and D.G. were responsible for overall project supervision. S.P., C.M., and S.B. contributed to data collection. S.P. and C.M. prepared the initial manuscript draft. S.P. and C.M. contributed to figure design. D.C. and B.N. provided invaluable insights throughout this study. B.N. and C.M. cross-checked the study for accuracy and consistency. All authors contributed to the review of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests related to the publication of this work. However, a patent application titled “CANCER HALLMARK ACTIVITY QUANTIFICATION SYSTEM” has been filed based on the methods and tools developed in this study.
Peer review
Peer review information
Communications Biology thanks Zixuan Zhao, Ragothaman Yennamalli and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Priyadarshi, S., Mazumder, C., Neekhra, B. et al. OncoMark: a high-throughput neural multi-task learning framework for comprehensive cancer hallmark quantification. Commun Biol 8, 1434 (2025). https://doi.org/10.1038/s42003-025-08727-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08727-z
