
Abstract
Angelman syndrome (AS) is a debilitating neurodevelopmental disorder caused by loss of maternally-inherited UBE3A. In neurons, paternally-inherited UBE3A is silenced in cis by a long non-coding RNA called Ube3a-ATS. Here, we found that Neisseria meningitidis Cas9 with two mutations (D15A and H587A) in the nuclease domains (dNmCas9) can unsilence the dormant paternal Ube3a allele in mouse and human neurons when targeted to Snord115 snoRNA genes located in introns of Ube3a-ATS. Importantly, dNmCas9 disrupted Ube3a-ATS with a non-template bias and in the absence of a chromatin modifying domain, supporting a transcriptional interference mechanism. When packaged into an adeno-associated virus (AAV) vector, dNmCas9 exhibited dose-dependent Ube3a-ATS knock-down and paternal Ube3a unsilencing in vitro and in vivo. This vector also partially rescued the hind limb clasp phenotype when delivered to neonatal AS model mice. Collectively, our study underscores the potential of dCas9-based therapeutics without chromatin repression domains to mediate transcriptional downregulation.
Similar content being viewed by others

Introduction
Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by mutation or deletion of the maternally-inherited UBE3A allele (matUBE3A)1,2. In most cells, UBE3A is expressed from the maternal and paternal alleles; however, in neurons, the paternally-inherited UBE3A (patUBE3A) allele is silenced by a long-noncoding RNA commonly referred to as Ube3a-ATS3,4. Studies from our group and others indicate that interfering with U-ATS transcription is sufficient to unsilence patUBE3A including by small molecules5,6, antisense oligonucleotides (ASOs)7,8,9, or CRISPR/Cas910,11,12.
We previously found that an adeno-associated virus (AAV) vector delivering a Cas9 nuclease and guide RNA (gRNA) targeted to the Snord115 small nucleolar RNA (snoRNA) cluster within Ube3a-ATS could efficiently upregulate patUbe3a10. In this study, we packaged Cas9 from Staphylococcus aureus (SaCas9) alongside a guide RNA (gRNA) complimentary to approximately 75 Snord115 snoRNAs (Sajw33) into AAV9 particles. Injection of AAV9-SaCas9/Sajw33 into the lateral ventricles of AS model mice (Ube3am-/p+) at embryonic and early postnatal ages resulted in long-lasting patUbe3a unsilencing throughout the central nervous system (CNS) and improved behavioral phenotypes of the AS mouse model. Mechanistically, we found that the AAV genome integrated into double-strand breaks (DSBs) created by SaCas9, with vector-derived elements mediating premature transcriptional termination of Ube3a-ATS10. Subsequently, we found that AAV vector integration was the most frequent editing event at Cas9 induced DSBs in Ube3a-ATS and that vector-derived elements, such as the polyadenylation signal, can terminate Ube3a-ATS and unsilence patUbe3a11.
There are safety concerns associated with AAV vector genome integration at both Cas9 on- and off-target sites. Several groups demonstrated that genomic integration of vector-derived elements, such as promoters and enhancers, and viral genomes, including the AAV genome, increases risks of cancer and organ toxicity13,14,15,16,17,18. Furthermore, double-strand break activity in neurons is associated with neurodegeneration and neuroinflammation19,20.
CRISPR interference (CRISPRi) combines the DNA-targeting capabilities of dCas9 with the activity of chromatin repression domains like the Krüppel associated box (KRAB) to drive reductions in gene expression without permanent modifications of the genome21,22,23. Multiple groups demonstrated that targeted methylation by genome editors at the putative transcriptional start site of the Ube3a-ATS can increase patUbe3a levels as a potential therapeutic strategy for AS24,25,26. While these strategies unsilenced patUbe3a and improved behavioral phenotypes in a mouse model of Angelman syndrome, they simultaneously reduced the levels of Snord116 snoRNAs, which are located in the Prader-Willi syndrome critical region27,28,29.
Here, we sought to evaluate the extent to which dCas9 could disrupt Ube3a-ATS as an alternative therapeutic strategy with lower risks for genotoxicity and Snord116 dysregulation. Moreover, we sought to determine if dCas9 could disrupt Ube3a-ATS transcription in the absence of a chromatin modifying domain, as this would enable us to package both the editor and gRNA in a single AAV vector to improve efficiency of manufacturing and in vivo efficacy.
Results
dCas9 can disrupt Ube3a-ATS transcription and unsilence patUbe3a in the absence of a chromatin modifying domain
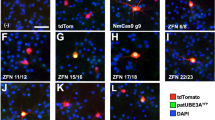
The Cas9 variant from Streptococcus pyogenes with two mutations in the RuvC and HNH nuclease domains (D10A, H840A; dSpCas9) is commonly used as a CRISPRi component because of its DNA-targeting capability21,30,31. Previous studies found that dCas9 can interfere with transcription21,30,32,33,34,35. Therefore, we hypothesized that targeting CRISPRi to Snord115 sequences could disrupt Ube3a-ATS and unsilence patUbe3a. To test this, we cloned the Spjw33 Snord115-targeting gRNA from our previous study, which directs dSpCas9 to the template strand of ~75 Snord115 sequences, into a plasmid containing dSpCas9 fused to the KRAB chromatin repression domain. The KRAB domain recruits repressive epigenetic machinery to shut down transcription at targeted loci22,36. We co-transfected the Spjw33 gRNA and a tdTomato transfection marker with either active SpCas9, dSpCas9-KRAB, or dSpCas9 lacking the KRAB domain into DIV3 primary mouse cortical neuron cultures prepared from Ube3am+/pYFP mice and measured the percentage of UBE3A-YFP positive transfected neurons (Fig. 1a–c)6,36. This mouse line enables quantification of UBE3A produced specifically from the paternal allele via measurement of UBE3A-YFP at both the protein and transcript level37. Reproducing our previous study, we found that SpCas9/Spjw33 significantly increased the percentage of UBE3A-YFP-positive neurons (Fig. 1c)10. We also found that dSpCas9-KRAB and dSpCas9 without the KRAB domain unsilenced patUbe3a compared to non-targeting gRNA negative controls (Fig. 1a–c).

a, b Representative images of UBE3A-YFP immunofluorescence in tdTomato positive, transfected Ube3am+/pYFP neurons from dSpCas9 negative control and Spjw33 conditions. Scale bar, 50 µm. c The percentage of transfected Ube3am+/pYFP primary mouse cortical neurons positive for UBE3A–YFP following transfection with each gRNA and SpCas9 variant indicated. n = 4 wells per condition. Error bars indicate standard error of the mean (SEM). Unpaired, two-tailed Student’s t-test comparing Spjw33 condition to non-target gRNA condition, *p < 0.05. d Expression of the indicated genes in Ube3am+/pYFP neurons 7 days after transduction with lentivirus delivering dSpCas9 and gRNA Spjw33 relative to dSpCas9 non-target gRNA control (dashed line). Expression normalized to Eif4a2. n = 2 wells per condition. e Schematic of non-template and template strand orientation relative to Ube3a-ATS transcription. Example palindromic gRNA target protospacer adjacent motifs are underlined. f, g Correlation of the percentage of transfected UBE3A-YFP positive neurons transfected with dSpCas9 and gRNAs targeting the (d) template or (e) non-template strand. The number of Snord115 target sites for each gRNA is shown. n = 8–12 wells per gRNA condition. h, i Correlation of UBE3A-YFP unsilencing when neurons were transfected with (f) dSpCas9 or (g) SpCas9 and sets of palindromic gRNAs that target the non-template versus template strand. n = 8–12. Hypergeometric means test, *p < 0.05.
Based on the fact that dCas9 without the KRAB domain is smaller and hence would better fit into viral vectors with restrictive packaging limits, we next evaluated its efficacy at upregulating patUbe3a and effects on nearby genes when delivered virally37. To accomplish this, we transduced Ube3am+/pYFP primary mouse cortical neurons with lentivirus expressing dSpCas9 and gRNA Spjw33 and extracted RNA on DIV10 for RT-qPCR. We found that dSpCas9/Spjw33 upregulated patUbe3a and reduced the expression of Snord115 and the 3’ portion of Ube3a-ATS without disrupting the expression of Snord116 (Fig. 1d). Moreover, Snrpn and matUbe3a levels were unaffected (Fig. 1d). This contrasts with approaches that target the putative transcriptional start site of Ube3a-ATS, which downregulate Snrpn, Snord116 and Snord11524,25,27. These data indicate that dCas9 can selectively upregulate patUbe3a when targeted to Snord115 without dysregulating upstream Snord116 or Snrpn. Moreover, these data indicate that dCas9 can disrupt Ube3a-ATS transcription without a chromatin repression domain.
dCas9 transcriptional interference is sensitive to the number of Snord115 sequences targeted and exhibits strand-bias
We previously found that active Cas9 vectors which target multiple Snord115 sequences more effectively unsilence patUbe3a than vectors that target single unique regions11. We next sought to evaluate the extent to which dCas9-mediated transcriptional interference at the Ube3a-ATS locus was sensitive to the number of Snord115 sequences targeted. Additionally, others found that dCas9 blocks transcription with a non-template strand bias21,32,33,34,35,38, so we also evaluated strand bias at Ube3a-ATS. Taking advantage of the sequence-conserved nature of Snord115 repeats, we designed gRNAs that target a range of Snord115 repeats with specificity for either the template strand (TS) or the non-template strand (NTS) relative to Ube3-ATS transcription (Fig. 1e). Following co-transfection of these gRNAs into primary mouse cortical neurons along with dSpCas9, we found that the percentage of UBE3A-YFP positive neurons increased as the number of gRNA targets increased on both the TS and NTS (Fig. 1f, g). Moreover, NTS-targeted gRNAs generally outperformed TS-targeted gRNAs, as indicated by a greater proportion of gRNAs that unsilenced UBE3A-YFP in >50% of transfected neurons (62%, NTS; 21% TS). We also found that Staphylococcus aureus Cas9 with mutations in the RuvC (D10A) and HNH (N580A) nuclease domains (dSaCas9) could unsilence UBE3A-YFP when transfected into cortical neurons with gRNAs targeting different numbers of Snord115 repeats on the NTS (Fig. S1).
To rigorously evaluate dCas9 strand-bias, we designed and tested gRNA pairs with palindromic PAM sites in Snord115 such that dSpCas9 could be directed to identical positions on the TS and NTS (Fig. 1e). We quantified the percentage of UBE3A-YFP positive neurons following transfection with these gRNAs and plotted the performance of NTS-targeting gRNAs against the performance of the paired TS-targeting gRNAs. When co-transfected with dSpCas9, we found that most NTS-targeting gRNAs outperformed the corresponding TS-targeting gRNA (Fig. 1h). In contrast, active SpCas9 did not exhibit a strand bias with the same palindromic paired gRNAs (Fig. 1i). Collectively, these experiments support a NTS bias in dCas9-mediated transcriptional interference.
To further explore the mechanism by which dCas9 interferes with Ube3a-ATS transcription, we designed a Ube3a-ATS minigene reporter construct with a single Spjw33 gRNA target-site. This reporter contains a pair of highly spliced Ube3a-ATS exons surrounding this Spjw33 target site driven by the CMV promoter and positioned upstream of the red fluorescent protein dsRed (Fig. 2a, b). This vector also contains an independently transcribed GFP for normalization under the control of the Ubiquitin C promoter (Fig. 2b). When transfected into cells, pre-mRNA and mRNA levels can be quantified using RT-qPCR with the indicated amplicons (Fig. 2b). We previously developed and studied a similar Ube3a-ATS minigene, and found that RT-qPCR was more sensitive at detecting changes in expression relative to quantifying dsRed/GFP fluorescence11. We transfected the Spjw33 Ube3a-ATS minigene construct into HEK293T cells along with active SpCas9 and Spjw33 gRNA (positive control), dSpCas9 and Spjw33 (targets TS), or dSpCas9 and Spjw33nt (targets NTS). In all three conditions, we observed a reduction of the spliced exon, the intronic amplicon downstream of the target site, and downstream dsRed amplicon (Fig. 2c–e). NTS-targeted dSpCas9 more effectively reduced transcription. We also detected a decrease in intronic pre-mRNA upstream of the gRNA target site with both TS and NTS-targeted dSpCas9. In contrast, this 5’ intronic region was increased in cells transfected with catalytically-active SpCas9. These data collectively suggest that dSpCas9 more effectively terminates transcription from this minigene reporter when targeted to the NTS relative to the TS, and that nuclease activity and transcriptional interference have inherently different effects on mRNA processing.

a A 624 bp fragment containing a single Spjw33 target site and flanking exonic sequences (as indicated by read pileups) was cloned downstream of the CMV promoter and upstream of dsRed to create the (b) Ube3a-ATS minigene reporter plasmid. RT-qPCR amplicons indicated. c–e Quantification of reporter-derived transcripts by RT-qPCR following transfection of HEK293T cells with the Ube3a-ATS minigene reporter and (c) SpCas9/Spjw33, (d) template strand-targeted dSpCas9/Spjw33, or (e) non-template strand-targeted dSpCas9/Spjw33nt. Expression normalized to internal GFP control and negative control gRNA. n = 2 wells per condition.
We next reduced the amount of Cas9 plasmid transfected so that each condition had similar levels of dsRed knockdown (Fig. S2a). We found that NTS-targeted dSpCas9 reduced splicing efficiency compared to TS-targeted dSpCas9 at the Spjw33 target site as measured by the reporter exon/intron ratio (Fig. S2b). This was also true when targeting within the upstream exon (G1) and downstream within the intron (G2). These results suggest that NTS-targeted dCas9 also disrupts RNA processing and splicing.
AAV-dNmCas9 can dose-dependently disrupt Ube3a-ATS transcription and increase levels of paternal Ube3a in primary mouse cortical neurons
SpCas9 is too large to fit into a single AAV vector along with a gRNA and respective promoters. Smaller Cas9 variants have been identified, including those from Staphylococcus aureus (SaCas9) and Neisseria meningitidis (NmCas9), that can fit into a single AAV vector alongside a gRNA. We previously found that catalytically active SaCas9 can be delivered to neurons in vivo in a single AAV9 vector with Sajw33, a multitarget Snord115 gRNA10,11. While dSaCas9 can unsilence UBE3A-YFP when transfected into primary mouse cortical neurons with Sajw33, none of the SaCas9 gRNAs we tested performed better than dSpCas9/Sajw33 (Fig. S1). We thus next tested NmCas9, as it can also be packaged within a single AAV vector and be delivered to neurons in vivo11,39. NmCas9 also has a more restrictive PAM relative to SaCas9 which reduces risk of off-target edits40. Additionally, we found that SaCas9 with two nuclear localization signals (NLSs) is primarily cytoplasmic in neurons, while NmCas9 with four NLSs is nuclear. As we recently showed, the addition of multiple NLSs is important for Cas9 nuclear localization and increases targeting efficacy41. We confirmed that NmCas9 with D15A and H587A nuclease domain mutations (dNmCas9) exhibits primarily nuclear subcellular localization in neurons when packaged in and delivered by AAV2/1, a common capsid for in vitro neuronal transduction (Fig. 3a)42,43,44. We next designed and cloned NmCas9 gRNAs along the template or non-template strand with multiple targets within Snord115 and quantified the percentage of UBE3A-YFP positive neurons following transfection into Ube3am+/pYFP primary mouse cortical neurons. We identified several gRNAs that effectively unsilenced patUbe3a with both active NmCas9 and dNmCas9 (Fig. 3b). We also observed that the NTS-targeting gRNAs outperformed the TS-targeting gRNAs, suggesting that dNmCas9 also exhibits a non-template strand bias (Fig. 3b). In contrast, active NmCas9 appeared to exhibit no strand bias (Fig. 3b).

a Representative images of mouse primary mouse cortical neurons transduced by AAV2/1-dNmCas9 and immunostained for NmCas9 and NeuN. Scale bar, 10 µm. b The percentage of transfected Ube3am+/pYFP mouse neurons positive for UBE3A-YFP following transfection with the indicated Cas9 variant and gRNA. Number of Snord115 targets and strand (t template, n non-template) are indicated. n = 4 culture wells per condition. Error bars indicate SEM. c–f Relative expression of (c) patUbe3a, (d) Ube3a-ATS, (e) Snord115 snoRNAs, and (f) NmCas9 in neurons following dosing with AAV2/1-dNmCas9-Nmg15 or NmCas9-Nmg15 at the viral multiplicity of infection (MOI) indicated. Normalized to Eif4a2, n = 2–3 wells per condition per dose. Expression normalized to non-targeting gRNA virus (dashed line). Error bars indicate SEM. [inhibitor] or [agonist] vs normalized response non-linear regression analysis was used for curve fitting.
We then packaged dNmCas9 and NmCas9 into AAV2/1 vectors alongside one of the best-performing gRNAs identified in our screen, Nmg15. Notably, this gRNA has 112 putative targets within the Snord115 snoRNA cluster and has no predicted off-target sites with fewer than 4 mismatches outside of the Snord115 snoRNA locus (Supplementary Table 3). We transduced DIV3 cortical neuron cultures at several multiplicities of infection (MOI) and quantified patUbe3a, Ube3a-ATS, and Snord115 expression relative to non-targeting gRNA control on DIV10 by RT-qPCR. dNmCas9 and active NmCas9 achieved similar maximal effects of patUbe3a unsilencing and Ube3a-ATS knockdown at the two highest MOI tested; however, NmCas9 outperformed dNmCas9 at lower MOI (Fig. 3c, d). Additionally, NmCas9 disrupted Snord115 expression more than dNmCas9 at each MOI tested and dNmCas9 disrupted Snord115 expression only at the three highest MOI (Fig. 3e). We also confirmed that dNmCas9 and NmCas9 expression levels were not different (Fig. 3f). Together, these data suggest that higher levels of dNmCas9 than active NmCas9 are necessary per cell to achieve maximal patUbe3a unsilencing.
dNmCas9 can unsilence paternal UBE3A in human neurons
We next evaluated the extent to which dNmCas9 can unsilence patUBE3A in primary human neural progenitor-derived (phNPC) neurons when targeted to SNORD115 sequences. First, we designed a pair of overlapping dNmCas9 gRNAs that target multiple human SNORD115 sequences: NmhsaNT27 which has 27 targets on the non-template strand and NmhsaT28 which has 28 targets on the template strand (Fig. S3a). We subsequently transduced phNPC-derived neuron cultures with AAV2-hSYN1-eGFP and lentivirus delivering mCherry, dNmCas9, and either NmhsaNT27, NmhsaT28, or negative control gRNA. For these experiments, lentivirus was used to simultaneously delivery mCherry and dNmCas9 to enable collection of transduced neurons from differentiated cultures by fluorescence activated cell sorting (FACS). Following FACS, we isolated RNA and performed SNP-specific RT–qPCR utilizing a single nucleotide variant in UBE3A to quantify inferred maternal and paternal UBE3A alleles, as previously described10. We found that paternal UBE3A was significantly upregulated and expressed to near maternal UBE3A levels in neurons transduced with dNmCas9/NmhsaNT27 (Fig. S3b, c). In contrast, there was no upregulation of paternal UBE3A in neurons transduced with dNmCas9/NmhsaT28 or the negative control gRNA. This experiment demonstrates that dNmCas9 targeted to the non-template strand of SNORD115 can unsilence paternal UBE3A in human neurons.
dNmCas9 can disrupt Ube3a-ATS transcription in vivo
Reinstatement of Ube3a within the early postnatal period confers therapeutic benefit in mouse models of AS7,10,45,46,47. Therefore, we tested the extent to which our dNmCas9/Nmg15 vector could unsilence patUbe3a in neonatal AS model mice (Ube3am-/p+)48,49. We packaged dNmCas9/Nmg15 expressed from the neuron-specific hSyn1 promoter into AAV9-PHP.eB and performed bilateral intracerebroventricular (ICV) injections in postnatal day 1 (P1) AS model mice. We injected mice with 1.0 ×1010 vector genomes per ventricle and evaluated patUbe3a unsilencing by immunofluorescence staining for UBE3A at P30. We found that delivery of AAV-dNmCas9/Nmg15 unsilenced patUbe3a in neurons throughout the cerebral cortex, and sparsely in Purkinje neurons of the cerebellum, in accordance with previous studies using AAV9 and its derived capsids and the neuron-specific hSyn1 promoter (Fig. S4a, b, S5a, b, c)10,11,46,50,51.
We next evaluated the extent to which this dNmCas9/Nmg15 AAV vector could rescue behavioral phenotypes in the AS mouse model (Fig. 4a). We ICV injected AS and WT littermates at P1 with 1.0 × 1010 vector genomes per ventricle. Mice received either AAV-dNmCas9/Nmg15 or AAV-dNmCas9/NmhsaNT27 delivering the human gRNA (as a negative control). Male and female mice were included in behavioral studies which began at P30. We found that AS model mice injected with AAV-dNmCas9/Nmg15 exhibited significant recovery in the total time clasping in the hind limb clasp assay (Fig. 4b). We also found that AS mice injected with AAV-dNmCas9/Nmg15 demonstrated partial recovery in clasping severity score (Fig. 4c). AS model mice injected with AAV-dNmCas9/Nmg15 exhibited no improvements in rotarod coordination, open field hypoactivity, or marble burying deficits (Fig. 4d–f). Additionally, AS mice injected with AAV-dNmCas9/Nmg15 exhibited no improvements in microcephaly, as measured by brain weight, or body weight phenotypes (Fig. 4g–i).

a Timeline of behavioral assays performed following ICV injection of AAV-dNmCas9/Nmg15 or AAV-dNmCas9/NmhsaNT27 negative control. b–i Male and female mice are represented by blue and red points, respectively. All boxplot whiskers extend to the highest and lowest values and the middle line indicates the median. Unless otherwise indicated, n = 12–20 mice per group and assays were analyzed by One-way ANOVA with Tukey’s multiple comparisons tests. b, c Hind limb clasping assay (b) total time spent clasping and (c) clasping severity score. Clasp score was analyzed by Kruskal-Wallis test with Dunn’s multiple comparisons tests. d Latency to fall on the accelerating rotarod during trials 1–3 on day 1 and trials 4 and 5 run 48 h later. Error bars indicate SEM. Two-way ANOVA with Tukey’s multiple comparisons tests. e Total distance traveled in the open field during 30 min trial. f Number of marbles buried following 30 min marble burying trial. g Postmortem analysis of brain weight following dissection. h, i Body weights of treated (h) male and (i) female mice. n = 5–15 mice per group. Error bars indicate SEM. Two-way ANOVA with Tukey’s multiple comparisons tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We next quantified the degree of patUbe3a unsilencing and Ube3a-ATS knock-down in the brains of mice injected with AAV-dNmCas9/Nmg15 or NmhsaNT27. Following microdissection and RNA extraction from cerebral cortex, hippocampus, and cerebellum, we performed RT-qPCR to measure Ube3a, Ube3a-ATS, Snord115, and dNmCas9 expression. We detected significant Ube3a-ATS knock-down in the cortex and hippocampus from AS mice injected with AAV-dNmCas9/Nmg15 compared to AS mice injected with AAV-dNmCas9/hsaNT27, but no knockdown in the cerebellum (Fig. 5a–c). Additionally, we observed a significant increase in Ube3a expression in the cortex of AS mice injected with AAV-dNmCas9/Nmg15, but not in the hippocampus or cerebellum (Fig. 5d–f). Further, we observed significant reduction of Snord115 in the cortex from AS treated mice compared to AS control animals, but no change in the hippocampus or cerebellum of AS treated mice (Fig. 5g–i). This is consistent with the pattern of dNmCas9 mRNA expression among these tissues (Fig. S6a–c). Immunofluorescence staining for UBE3A in mice used in the behavioral study similarly showed modest patUbe3a unsilencing in the cortex and poor unsilencing in the hippocampus from AS mice injected with AAV-dNmCas9/Nmg15 compared to AS and WT mice injected with AAV-dNmCas9/NmhsaNT27 (Fig. 6a, b). These experiments confirm modest target engagement of our dNmCas9 vector in the cortex and hippocampus, brain regions that are well-transduced by AAV9-derived capsids following ICV administration10,46,51.

a–i RT-qPCR to quantify (a–c) Ube3a-ATS, (d–f) Ube3a, and (g–i) Snord115 expression levels in (a, d, g) cortical, (b, e, h) hippocampal, and (c, f, i) cerebellar samples from WT and AS mice injected with AAV-dNmCas9/Nmg15 or AAV-dNmCas9/NmhsaNT27. Normalized to Eif4a2 and WT control group. Male and female mice are represented by blue and red points, respectively. All boxplot whiskers extend to the highest and lowest values and the middle line indicates the median. n = 12–20 mice per group. One-way ANOVA with Tukey’s multiple comparisons tests, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.

a, b Representative images of UBE3A and NEUN immunofluorescence following the completion of behavioral testing in the (a) cortex and (b) dorsal hippocampus from WT control, AS control, and AS treated mice injected at postnatal day 1 with AAV-dNmCas9/NmhsaNT27 or Nmg15. Scale bar, 200 µm.
While mice were injected with similar total vector genomes and volumes, technical variability in injections can contribute to subtle differences in vector dose. This variability allowed us to assess correlations between expression of dNmCas9 and Ube3a-ATS, Ube3a, or Snord115 in brain samples from mice that were used in the behavioral study to test for dose-dependent effects of dNmCas9 expression. We found that dNmCas9 expression negatively correlated with Ube3a-ATS levels in the cortex and hippocampus of AS treated mice, but not the cerebellum (Fig. 7a–c). We also found that Ube3a expression positively correlated with dNmCas9 expression in the cortex and hippocampus of AS mice injected with AAV-dNmCas9/Nmg15, but not in the cerebellum or in any tissues collected from AS or WT mice injected with the negative control hsaNT27 vector (Fig. 7d–f). Snord115 expression negatively correlated with dNmCas9 expression in cortex of AS treated mice, but not in the hippocampus, cerebellum, or any tissues from AS and WT control mice (Fig. 7g–i). Lastly, we tested for a correlation between dNmCas9 expression and performance in the hind limb clasp assay. We did not observe a significant correlation between cortical, hippocampal, or cerebellar dNmCas9 expression with seconds clasped in the hind limb clasp assay (Fig. S7a–c). These experiments demonstrate the potential of AAV-dNmCas9/g15 to significantly disrupt Ube3a-ATS transcription and unsilence patUbe3a in the brains of AS model mice in a dNmCas9 dose-dependent manner.

a–i Correlation of (a–c) Ube3a-ATS, (d–f) Ube3a, and (g–i) Snord115 expression levels in (a, d, g) cortical, b, e, h hippocampal, and (c, f, i) cerebellar samples from WT and AS mice injected with AAV-dNmCas9/Nmg15 or AAV-dNmCas9/NmhsaNT27 relative to dNmCas9 expression. Relative dNmCas9 expression was normalized to Eif4a2. Ube3a-ATS, Ube3a, and Snord115 expression were normalized to Eif4a2 and WT control group. n = 12-20 mice per group. Lines of best fit (linear regression) are included for all groups, and goodness of fit (r2) and p are indicated for groups exhibiting a significant correlation. Pearson’s r correlation, two-tailed, p < 0.05.
Discussion
The unique imprinting of UBE3A in neurons enabled the development of several potentially disease-altering therapeutics for AS aimed at unsilencing paternal UBE3A by disrupting Ube3a-ATS. Previous studies implicate transcriptional collision of RNA polymerases as the mechanism by which Ube3a-ATS silences patUBE3A52. Here, we find that dCas9 targeted to Ube3a-ATS can unsilence patUBE3A by impeding Ube3a-ATS transcription. Importantly, we found that non-template strand bias, greater efficacy when using gRNAs to multiple regions within Ube3a-ATS, and dCas9 dose-dependency are key characteristics of dCas9-transcriptional interference of Ube3a-ATS transcription.
Compared to our previous study using active SaCas9/Sajw33, dNmCas9/Nmg15 was less efficacious at unsilencing patUbe3a in vivo10. Given this, it is not surprising that behavioral and anatomical recovery with dNmCas9/Nmg15 were minimal (Fig. 4). While differences in timing of patUbe3a reinstatement and total vector genomes delivered may contribute to these differences, our in vitro studies also demonstrate that active Cas9 outperforms dCas9 at identical viral doses and expression levels (Fig. 3). Previous studies similarly found that higher levels of dCas9 are necessary to potently disrupt transcription and linked saturation of dCas9 at its target site to its efficacy in disrupting transcription53. Indeed, we observed significant correlations between dNmCas9 expression and both Ube3a-ATS and Ube3a expression in the cortex and hippocampus, with Ube3a levels never approaching that of WT mice (Fig. 7). While use of dCas9 may offer safety advantages over active Cas9, an enzyme that generates DSBs, the limited efficacy at unsilencing patUbe3a and limited behavioral recovery suggest a need to explore other means of targeting Ube3a-ATS or increasing dCas9 expression for more complete disruption.
The differential Cas9/dCas9 efficacy per vector genome we observed is consistent with the mechanisms by which active and dead Cas9 disrupt Ube3a-ATS transcription (Fig. 3). Active Cas9 disrupts transcription at Ube3a-ATS both by the creation of transient DSBs at the cut site and the integration of vector-derived elements10,11,52. In contrast, dCas9 disrupts transcription via sterically obstructing RNA polymerase II during transcriptional elongation and inducing R-loop formation which can further stall RNA polymerase II33,54,55. Importantly, dCas9 transcriptional roadblocks are transient, only locally disruptive, and occur without permanent modification to the genome56. The transience of these effects is highlighted by the greater reduction of Ube3a-ATS downstream of the dCas9 binding site in the Snord115 cluster (Fig. 1d, Fig. 3d, e, Fig. 5). We expect this effect will persist for only as long as dCas9 and the gRNA are expressed. This mechanism suggests the need for sustained dCas9 genome binding and high dCas9 expression to unsilence patUbe3a.
CRISPR/Cas9 has enabled the development of many potentially disease-altering therapeutics; however, its potential for CNS disorders has been severely hindered by its size and inefficient delivery to the brain. To overcome the large amount of genetic material that must be delivered, many proof-of-concept studies have relied on transgenic mice genetically expressing a gene editor, highlighting the potential of a therapeutic without any way to deliver it24,57. Additionally, delivery of Cas9-based machinery to neurons by lentivirus is useful for proof-of-concept studies, however its potential for insertional mutagenesis is a major safety concern58,59. Several groups used dual or split-AAV systems and performed local injections to specific brain regions60,61,62. While other delivery approaches such as non-viral or lipid nanoparticle based approaches are actively in development, adeno-associated viruses are currently the most reliable and effective tool for CNS gene therapy63,64.
ASOs for AS are currently in clinical trials, however, they must be regularly administered into the cerebrospinal fluid necessitating potentially life-long, invasive injections which themselves come with risks7,8,9,65. Therefore, a gene therapy for AS, whether it be gene replacement, genome editing, or epigenome-modifying, holds a major advantage over ASOs and small molecules in its potential for a one-time administration of a therapeutic. However, AAV-based gene therapies also carry risks of immunogenicity or toxicity especially following high vector dosages13,66. As there are several AAV-based gene therapy strategies in development, comparisons of efficacy per vector genome, such as our comparison of NmCas9 and dNmCas9, will be important to identify which vectors and payloads deliver maximal therapeutic effect at minimal vector doses. Compared to other gene editing or epigenetic-modifying therapeutic approaches for AS, our AAV-dNmCas9 approach reduces risks of off-target edits, AAV integration at on- and off-target DSBs, and dysregulation other genes such as Snord11624,25,27. Further, it is unknown whether Snord115 deletion carries deleterious effects in humans; however, we have not previously observed negative effects of Snord115 downregulation in mice10.
An important limitation of our dNmCas9 approach, and of all patUBE3A unsilencing approaches, is that many individuals with AS have deletions that remove multiple genes expressed in the brain in addition to UBE3A67. It is not yet known whether unsilencing patUBE3A in individuals with large deletions will be sufficient or have therapeutic value. While upregulation of other deleted genes may be important in these instances, an advantage of unsilencing-focused therapeutics, as opposed to gene replacement approaches, is that they mitigate risks of UBE3A overexpression which contributes to Dup15q syndrome46,68.
Collectively, these experiments highlight the potential of dCas9-based therapeutics without a chromatin repression domain to drive behaviorally-relevant reductions in gene expression in the brain. Our finding that dCas9 can significantly disrupt Ube3a-ATS transcription without a chromatin repression domain enabled the development and testing of a single-AAV based dNmCas9 gene therapy vector for the treatment of AS. To our knowledge, this is the first evidence that an untagged dCas9 can disrupt transcription in a behaviorally-impactful manner in vivo. Importantly, a dCas9-based strategy targeting Snord115 can unsilence patUbe3a without the formation of double-strand breaks and dysregulation of Snord116, key advantages over previous gene-editing approaches to treat AS.
Methods
Animal use
All animal procedures performed in this study were approved by the Institutional Animal Care and Use Committee and the University of North Carolina at Chapel Hill. Ube3am+/pYFP and Ube3am+/p− were maintained on the C57BL/6J background and genotyped using previously described genotyping procedures6.
Plasmids
pLenti-Camk2a:tdTomato vector was previously described69. Experiments using SpCas9 used plentiCRISPR v2 (Addgene plasmid #52961)70. Experiments using dSpCas9-KRAB used pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro (Addgene plasmid #71236)22. To generate untagged dSpCas9, we removed the KRAB domain from pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro using site-directed mutagenesis. Experiments using SaCas9 used pX601-AAV-hSyn::NLS-SaCas9-NLS 3xHA-bGHpA:U6::BsaI-sgRNA as previously described or with the D10A and N580A mutations introduced using site-directed mutagenesis10. Experiments using NmCas9 (Nme1Cas9) used pAAV-hSyn1-NmCas9-gRNA previously described or pAAV-dNmCas9-gRNA with the D15A and H587A mutations11, or lentiCRISPRv2 and lentiCRISPRv2-mCherry (Addgene plasmid #99154) with NmCas9 or dNmCas9 replacing SpCas9. gRNAs were cloned using either BsmBI (SpCas9), BsaI (for SaCas9), or SapI (for NmCas9). gRNA sequences are included in Supplementary Table 1. Negative control gRNAs for experiments in mouse and human cells do not target a known sequence in the respective mouse or human genome. For lentivirus production, we used psPax2 (Addgene plasmid #12260) and pMD2.G (Addgene plasmid #12259). For AAV production, we used pAAV2/1 (Addgene plasmid #112862) and pAdDeltaF6 (Addgene plasmid #112867).
Primary mouse cortical neuron cultures
Primary mouse cortical neuron cultures were prepared as previously described from E15.5 Ube3am+/pYFP embryos6,71. Neurons were maintained in neurobasal medium (Gibco, #12348017) supplemented with B-27 Plus (Gibco, #A3582801), GlutaMAX (Gibco, #35050061), and 5’-fluoro-2’-deoxyuridine (Sigma, #F0503) at DIV3.
For immunocytochemistry (ICC) transfection experiments, cortical neurons were plated on poly-D-lysine-coated 384-well plates at 3.5 × 104 cells per well. Neurons were transiently transfected in triplicate on day in vitro 3 (DIV3) with 50 ng of Cas9/gRNA plasmid and 50 ng of CamKII:tdTomato plasmid using Lipofectamine 2000 transfection reagent (11668027, Invitrogen). After 1 h, whole media was replaced with conditioned neurobasal medium. On DIV10, neurons were fixed with 4% phosphate-buffered paraformaldehyde (PFA) and stained with primary rabbit anti-GFP antibody (NB600-308, Novus), secondary donkey anti-rabbit IgG Alexa 647 (A31573, Thermo Fisher) and DAPI. Images were acquired using a GE IN CELL Analyzer 2200 high-content imager. Four fields of view per well were quantified and averaged for each replicate. CamKII:tdTomato was used to identify transfected neurons and quantification of YFP expression was performed in only tdTomato positive nuclei, marking transfected neurons, using a custom Cell Profiler pipeline. These data are included in the Supplementary Data file.
For lentivirus experiments, neurons were plated in poly-D-lysine-coated 12-well plates at 1.0 × 106 neurons per well and transduced on DIV3. RNA was extracted on DIV10 using Trizol. For AAV experiments, neurons were plated in poly-D-lysine-coated 24-well plates at 4.0 × 105 cells per well and transduced on DIV3 with AAV2/1. RNA was extracted on DIV10.
For dNmCas9 localization experiment, neurons were plated on poly-D-lysine-coated glass coverslips (Corning, #354087) at 2.5 × 105 cells per well. Neurons were transduced on DIV3 and fixed on DIV10 with 4% PFA in PBS. Following washing in PBS, coverslips were blocked in 10% NDS in TBSTX (0.2% Triton X-100) for 1 h and incubated with primary guinea pig anti-NEUN (Sigma, #ABN90P) and rabbit anti-NmCas9 (abcam, #ab202638) in 10% NDS/TBSTX overnight at 4 °C with gentle rocking. Cells were then washed three times with PBS and incubated with secondary donkey anti-guinea pig IgG Alexa Fluor 647 (Jackson ImmunoResearch Labs, #706-605-148), donkey anti-rabbit IgG Alexa Fluor 568 (Invitrogen, #A10042), and DAPI (Invitrogen, #D1306). Coverslips were mounted on glass slides using FluoroGel mounting media (Electron Microscopy Sciences, #17985-10). Images were acquired using a Zeiss LSM 710 Confocal Microscope.
Virus production
For lentivirus preparation, HEK293T cells were seeded in 6 well plates on DIV0 at 1.0 × 106 cells per well in DMEM (Gibco, #11995065) supplemented with 10% fetal bovine serum (FBS). Cells were transfected on DIV1 with pMD2.G, psPAX2, and lentivirus plasmid of the vector to be produced using Fugene 6 (Promega, #E2691) in Opti-MEM I reduced serum medium (Gibco, #31985062). 48 h following transfection, supernatant was collected, centrifuged (5000 x g, 1 min) through a 0.45 μM filter (Costar, #8162), and stored at −80 °C.
For in vitro experiments, AAV was produced using previously described methods to prepare small-scale AAV preps43. HEK293T cells were plated in DMEM (Gibco, #11995065) with 10% FBS on a 12 well plate at 4.0 × 105 cells per well. 24 h later, cells were transfected with 0.4 µg of AAV plasmid, 0.8 µg of helper plasmid pAD-DeltaF6, and 0.4 µg of pAAV2/1 Rep/Cap plasmid using Fugene 6 (Promega, #E2691) in Opti-MEM I reduced serum medium (Gibco, #31985062). 8–16 h later, HEK293T cells underwent whole media change into glutamine-free DMEM (Gibco, #11960044) supplemented with 10% FBS. AAV-containing supernatant was collected 48 h later and centrifuged (5000 x g, 1 min) through a 0.45 μm filter (Costar, #8162). Virus was stored at 4 °C and used within 5 days. Viral titers (genome copies/ml) were determined by qPCR.
For in vivo experiments, AAV9-PHP.eB virus was prepared by the BRAIN Initiative Viral Vector Core at the UNC Neuroscience Center. Virus was purified through three rounds of CsCl density gradient centrifugation and exchanged into 1X PBS, 5% D-Sorbitol, and 350 mM NaCl. Viral titers (genome copies/ml) were determined by qPCR.
RNA extractions and RT-qPCR
All RNA extractions were performed using TRIzol (Invitrogen, #15596026). Primary mouse cortical neurons were washed with PBS and collected on DIV10. phNPC-derived neurons were collected on DIV56. For in vivo experiments, cortical, hippocampal, and cerebellar samples were microdissected and dounce homogenized in TRIzol. All RNA samples were reverse transcribed using SuperScript IV VILO (Invitrogen, #11766050). RT-qPCR experiments were performed using SsoAdvanced Universal SYBR Green Supermix (Bio Rad, #1725271) on a QuantStudio5 (Applied Biosystems). Primers used are listed in Supplementary Table 2. All data were analyzed using the 2-ddCT method normalized to Eif4a2.
For allele-specific qPCR in phNPC-derived neurons, RNA was treated with Dnase I (NEB, #M0303L) and cDNA was generated from 100 ng total RNA using SuperScript III Reverse Transcriptase (Thermo Fisher, #18080044) and 2 pM of UBE3A-specific primer (5′-TCCTTTAGATCATACATCATTGG). Allele-specific expression levels were determined using TaqMan Universal Master Mix II (Thermo Fisher, #4440040), and TaqMan genotyping probes for rsID:61734190 (Applied Biosystems). Quantification of allele-specific expression was calculated by assessing the distance between Ct values for each allele within each replicate at ∆Rn = 0.5.
Ube3a-ATS minigene reporter experiments
A Spjw33 target region was amplified from C57BL/6J mouse gDNA and cloned into pHAGE-CMV-dsRed-UBC-GFP-W plasmid (Addgene plasmid #24526)72. The resulting plasmid, referred to as Ube3a-ATS/Spjw33 minigene, contains a DNA fragment with a single gRNA Spjw33 target site and flanking exons cloned between the CMV promoter and dsRed gene. Additionally, this plasmid contains an independently-transcribed Ubc-GFP that can be used for transfection normalization. HEK293T cells were plated at a density of 1.25 × 105 cells per well in DMEM (Gibco, #11995065) supplemented with 10% FBS and transfected 24 h later with the Ube3a-ATS/Spjw33 mini gene reporter and Cas9/gRNA plasmids using Fugene 6 (Promega, #E2691) in Opti-MEM I reduced serum medium (Gibco, #31985062). Cells were washed 48 h later with PBS and collected in TRIzol for RNA. RT-qPCR was performed as described above. Primers are listed in Supplementary Table 2.
Prediction of Off-Target Sites
Cas-OFFinder was used to identify potential off-target sites with fewer than 4 mismatches for gRNA Nmg15 in the mouse genome (mm10) with Cas9 from Neisseria meningitidis73. Notably, all predicted off-target sites with fewer than 4 mismatches are within the Snord115 snoRNA cluster, the highly repetitive target of Nmg15. The results of these analyses are summarized in Supplementary Table 3.
Primary human NPC-derived neuron experiments
Human fetal brain tissues were obtained from the UCLA Gene and Cell Therapy Core and phNPCs were grown and differentiated as previously described in refs. 10,74. Briefly, cells were thawed and plated in 10 cm plates in proliferation media (Neurobasal A supplemented with primocin, BIT9500, glutamax, heparin, EGF, FGF, LIF and PDGF). Cells were transferred into six-well plates after 2 passages and plated at 4 × 105 cells per. Media was replaced 24 h later with differentiation media (Neurobasal A, primocin, B27+, glutamax, NT3 and BDNF) and a 50% media change was performed every 2–3 days. On DIV14, 1.6 × 1010 AAV2 particles carrying hSyn1:eGFP (pAAV-hSyn1-eGFP, UNC Vector Core) were added to each well. On DIV42, lentivirus particles carrying dNmCas9:mCherry and gRNAs were added to cultures. On DIV56, cells were lifted, six wells for each gRNA were pooled, stained with DAPI for live/dead discrimination, and sorted using a FACSAria II cell sorter. GFP positive neurons were sorted into dNmCas9/gRNA positive and negative populations by mCherry signal and collected for RNA extraction.
ICV Injections
Stock virus was diluted in sterile PBS with 1 mg/ml fast green (Sigma-Aldrich) immediately prior to use. Postnatal day 1 pups from Ube3am+/p− dams were placed on wet ice for 3 min to induce hypothermic anesthesia. Mice were bilaterally injected with virus following previously published protocols75. Briefly, 1 µl per ventricle was injected using a 10 μL syringe with a 32-gauge, 0.4-in-long sterile syringe needle (7803-04, Hamilton). Mice were injected with 2.0 × 1010 total vector genomes. Pups were placed on a heating pad for recovery and then returned to their dam.
Immunohistochemistry
Following extraction of whole brains from mice, brains were immersion fixed in phosphate-buffered 4% paraformaldehyde (PFA) for 72 h at 4 °C and stored in PBS. Brains were sectioned at a thickness of 100 µm on a vibratome (Thermo Fisher Scientific). Sections underwent antigen-retrieval for 30 min in Citrate buffer at 95 °C, several PBS washes, and were permeabilized with PBST (0.3% Triton X-100) and blocked with 10% normal donkey serum (NDS) in PBST for 1 h at room temperature. Sections were then immunostained with primary mouse IgG2A anti-UBE3A (Sigma, #SAB1404508, 1:600) and guinea pig anti-NEUN (Millipore, #ABN90P, 1:600) overnight at room temperature with gentle shaking. Following several washes in PBST, sections were blocked in 10% NDS in PBST for 30 min and incubated with goat anti-mouse IgG2A Alexa Fluor 488 (Invitrogen, #A211311, 1:250), donkey anti-guinea pig Alexa Fluor 647 (Jackson #706-605-148, 1:250), and DAPI (ThermoFisher Pierce #EN62248) for 6 h at room temperature. Following washes in PBST and PBS, sections were mounted, treated with TrueBlack Autofluorescence Quencher (Biotium #23007), 1:20 in 70% EtOH for 2 min, and thoroughly washed in PBS before coverslipping. Images were acquired using a Zeiss LSM 710 confocal microscope.
Behavioral Experiments
Paternal-null (Ube3am+/p−) female mice were bred with wild-type (Ube3am+/p+) males to obtain maternal-null (Ube3am−/p+) and wild-type littermates. Both male and female mice were included in behavioral experiments and sex was not considered as a biological variable, with the exception of body weight, based on previous studies demonstrating phenotypes are present in male and female Angelman syndrome model mice76. The treatment assignments were random, experimenter was blind to genotype and treatment, and sample sizes were based on power analyses performed by previous studies76. Only wild-type and AS mice injected with the AAV-dNmCas9/NmhsaNT27 negative control vector and AS mice injected with the AAV-dNmCas9/Nmg15 vector were included in behavioral phenotyping and individual animals were the experimental unit. Wild-type animals injected with AAV-dNmCas9/Nmg15 were identified upon genotyping at weaning and were not run or analyzed in the study. Mouse genotypes were confirmed following completion of the behavioral battery.
The hind limb clasp assay was run at P30. Briefly, mice were suspended approximately 10 inches (measured from tip of their tail) above a benchtop for 30 s held near the base of their tail. Each mouse received two trials with a 5-minute inter-trial interval. Clasping behavior was defined by curling of the hind limbs inwards towards the belly. Total time clasping is presented as the average of two trials. Hind limb clasp scores were scored from 0 to 3 by increasing severity: 0 = consistently splayed outwards, 1 = one hind limb pulled inward more than 50% towards the body, 2 = both hind limbs pulled inwards more than 50% towards the body, 3 = both hind limbs entirely retracted and touching the abdomen. Hind limb clasp score is presented as the average of 2 trials.
The rotarod assay was next performed with the rotating rod set to accelerate from 3 to 30 rpm over a maximum trial length of 5 min (Ugo Basile Biological Research Apparatus; #47600). Mice were given three trials on their first assessment day with a 10 min inter-trial period. Mice were re-tested 48 h later and were given 2 trials. Latency to fall or three consecutive passive rotations were recorded.
For the open field assay, mice were individually placed in a 45 × 45 cm open field arena and allowed to explore for 30 min. Total distance traveled was recorded and quantified using EthoVision software (Noldus Wageningen).
The marble burying assay was performed in open polycarbonate cages (50 × 26 × 18 cm) filled with 3 liters of bedding material (Lab Supply, #CC8-IRR, 1/8” Corn Cob Irradiated Bedding). On top of the bedding material, 20 black, glass marbles were arranged in a 5 × 4 grid. Mice were given access to the marbles for 30 min after which the number of marbles buried was determined. A marble was defined as buried if it was more than 50% covered by bedding.
Statistics and Reproducibility
Data are presented as mean ± SEM. For boxplots, whiskers extend to the highest and lowest values and the middle line indicates the median. Specific statistical tests used are included in figure legends. Generally, for experiments comparing two groups, a two-tailed unpaired Student’s t-test was used. For data comparing more than two groups with one varying factor, a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-hoc test was used. For data comparing more than two groups with two-varying factors, a two-way ANOVA with Tukey’s multiple comparisons post hoc test was used. Pearson correlation coefficients were calculated for correlations. Statistics were calculated in Graphpad Prism 10 (including assumptions of statistical tests), and were considered statistically significant if p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data are included in Supplementary Data source data file.
References
Dagli, A. I., Mueller, J. & Williams, C. A. Angelman Syndrome. in GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, Seattle, 1993) https://doi.org/10.1002/9781119432692.ch5.
Buiting, K., Williams, C. & Horsthemke, B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593 (2016).
Hsiao, J. S. et al. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl Acad. Sci. USA 116, 2181–2186 (2019).
Gonzalez Ramirez, C. et al. Regional and cellular organization of the autism-associated protein UBE3A/E6AP and its antisense transcript in the brain of the developing rhesus monkey. Front. Neuroanat. 18, 1410791 (2024).
Vihma, H. et al. Ube3a unsilencer for the potential treatment of Angelman syndrome. Nat. Commun. 15, 5558 (2024).
Huang, H.-S. et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189 (2011).
Meng, L. et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412 (2015).
Milazzo, C. et al. Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight 6, e145991 (2021).
Dindot, S. V. et al. An ASO therapy for Angelman syndrome that targets an evolutionarily conserved region at the start of the UBE3A-AS transcript. Sci. Transl. Med. 15, eabf4077 (2023).
Wolter, J. M. et al. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 587, 281–284 (2020).
Bazick, H. O., Mao, H., Niehaus, J. K., Wolter, J. M. & Zylka, M. J. AAV vector-derived elements integrate into Cas9-generated double-strand breaks and disrupt gene transcription. Mol. Ther. 32, 4122–4137 (2024).
Schmid, R. S. et al. CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J. Clin. Invest. 131, e142574 (2021).
Chandler, R. J., Sands, M. S. & Venditti, C. P. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 28, 314–322 (2017).
Chandler, R. J. et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 125, 870–880 (2015).
Rosas, L. E. et al. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. 20, 2098–2110 (2012).
Nault, J.-C. et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 47, 1187–1193 (2015).
Calabria, A. et al. Intrathymic AAV delivery results in therapeutic site-specific integration at TCR loci in mice. Blood 141, 2316–2329 (2023).
Dalwadi, D. A. et al. Liver Injury Increases the Incidence of HCC following AAV Gene Therapy in Mice. Mol. Ther. 29, 680–690 (2021).
Welch, G. M. et al. Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 8, eabo4662 (2022).
Dileep, V. et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell 186, 4404–4421.e20 (2023).
Larson, M. H. et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 (2013).
Thakore, P. I. et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015).
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Liu, Y. et al. Epigenetic editing alleviates Angelman syndrome phenotype in mice by unsilencing paternal Ube3a. Cell Discov. 10, 97 (2024).
Bailus, B. J. et al. Protein delivery of an artificial transcription factor restores widespread ube3a expression in an angelman syndrome mouse brain. Mol. Ther. 24, 548–555 (2016).
Landers, M. et al. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res 32, 3480–3492 (2004).
O’Geen, H. et al. Transcriptional reprogramming restores UBE3A brain-wide and rescues behavioral phenotypes in an Angelman syndrome mouse model. Mol. Ther. 31, 1088–1105 (2023).
Helwak, A., Turowski, T., Spanos, C. & Tollervey, D. Roles of SNORD115 and SNORD116 ncRNA clusters during neuronal differentiation. Nat. Commun. 15, 10427 (2024).
Bieth, E. et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur. J. Hum. Genet. 23, 252–255 (2015).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Bikard, D. et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41, 7429–7437 (2013).
Zukher, I., Dujardin, G., Sousa-Luís, R. & Proudfoot, N. J. Elongation roadblocks mediated by dCas9 across human genes modulate transcription and nascent RNA processing. Nat. Struct. Mol. Biol. 30, 1536–1548 (2023).
Pinto, B. S. et al. Impeding transcription of expanded microsatellite repeats by deactivated cas9. Mol. Cell 68, 479–490.e5 (2017).
Hall, P. M. et al. Polarity of the CRISPR roadblock to transcription. Nat. Struct. Mol. Biol. 29, 1217–1227 (2022).
Mandegar, M. A. et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553 (2016).
Luther, D. C., Lee, Y. W., Nagaraj, H., Scaletti, F. & Rotello, V. M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges. Expert Opin. Drug Deliv. 15, 905–913 (2018).
Pacesa, M. et al. R-loop formation and conformational activation mechanisms of Cas9. Nature 609, 191–196 (2022).
Lee, C. M., Cradick, T. J. & Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. 24, 645–654 (2016).
Moreb, E. A. & Lynch, M. D. Genome dependent Cas9/gRNA search time underlies sequence dependent gRNA activity. Nat. Commun. 12, 5034 (2021).
Major, R. M. et al. Exploring the Cytoplasmic Retention of CRISPR-Cas9 in Eukaryotic Cells: The Role of Nuclear Localization Signals and Ribosomal Interactions. CRISPR J. 8, 120–136 (2025).
Hammond, S. L., Leek, A. N., Richman, E. H. & Tjalkens, R. B. Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS ONE 12, e0188830 (2017).
Gao, Y. et al. Plug-and-Play Protein Modification Using Homology-Independent Universal Genome Engineering. Neuron 103, 583–597.e8 (2019).
Burger, C. et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 10, 302–317 (2004).
Silva-Santos, S. et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Investig. 125, 2069–2076 (2015).
Judson, M. C. et al. Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight 6, e144712 (2021).
Rotaru, D. C., Mientjes, E. J. & Elgersma, Y. Angelman syndrome: from mouse models to therapy. Neuroscience 445, 172–189 (2020).
Jiang, Y. H. et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811 (1998).
Judson, M. C., Sosa-Pagan, J. O., Del Cid, W. A., Han, J. E. & Philpot, B. D. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 522, 1874–1896 (2014).
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179 (2017).
Kim, H. et al. Rescue of behavioral and electrophysiological phenotypes in a Pitt-Hopkins syndrome mouse model by genetic restoration of Tcf4 expression. eLife 11, e72290 (2022).
Meng, L. et al. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 9, e1004039 (2013).
Vigouroux, A., Oldewurtel, E., Cui, L., Bikard, D. & van Teeffelen, S. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 14, e7899 (2018).
Clarke, R. et al. Enhanced Bacterial Immunity and Mammalian Genome Editing via RNA-Polymerase-Mediated Dislodging of Cas9 from Double-Strand DNA Breaks. Mol. Cell 71, 42–55.e8 (2018).
Whinn, K. S. et al. Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci. Rep. 9, 13292 (2019).
Widom, J. R., Rai, V., Rohlman, C. E. & Walter, N. G. Versatile transcription control based on reversible dCas9 binding. RNA 25, 1457–1469 (2019).
Platt, R. J. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014).
Ranzani, M. et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat. Methods 10, 155–161 (2013).
Savell, K. E. et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro 6, ENEURO.0495-18.2019 (2019).
Truong, D.-J. J. et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 43, 6450–6458 (2015).
Zhi, S. et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 30, 283–294 (2022).
Matharu, N. et al. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 363, eaau0629 (2019).
Mendell, J. R. et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Lee, D. et al. Antisense oligonucleotide therapy rescues disturbed brain rhythms and sleep in juvenile and adult mouse models of Angelman syndrome. eLife 12, e81892 (2023).
Hinderer, C. et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 29, 285–298 (2018).
Tan, W.-H. et al. Angelman syndrome: Mutations influence features in early childhood. Am. J. Med. Genet. A 155A, 81–90 (2011).
Punt, A. M. et al. Molecular and behavioral consequences of Ube3a gene overdosage in mice. JCI Insight 7, e158953 (2022).
Mabb, A. M. et al. Topoisomerase 1 Regulates Gene Expression in Neurons through Cleavage Complex-Dependent and -Independent Mechanisms. PLoS ONE 11, e0156439 (2016).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
King, I. F. et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62 (2013).
Wilson, A. A. et al. Sustained expression of alpha1-antitrypsin after transplantation of manipulated hematopoietic stem cells. Am. J. Respir. Cell Mol. Biol. 39, 133–141 (2008).
Bae, S., Park, J. & Kim, J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Stein, J. L. et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86 (2014).
Kim, J.-Y., Grunke, S. D., Levites, Y., Golde, T. E. & Jankowsky, J. L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp. 51863 https://doi.org/10.3791/51863 (2014).
Sonzogni, M. et al. A behavioral test battery for mouse models of Angelman syndrome: a powerful tool for testing drugs and novel Ube3a mutants. Mol. Autism 9, 47 (2018).
Acknowledgements
This work was supported by grants to M.J.Z. from the Angelman Syndrome Foundation, the Simons Foundation (SFARI, award ID 631904), and the National Institute of Neurological Disorders and Stroke (NINDS; 1R01NS109304). J.M.W. and H.M. were supported by an NIH T32 postdoctoral training grant through the Carolina Institute for Developmental Disabilities (NIH T32HD040127). L.M.J. was supported by a fellowship from the UNC Royster Society of Fellows. Microscopy was performed at the UNC Neuroscience Microscopy Core, supported, in part, by funding from the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant P50 HD103573.
Author information
Authors and Affiliations
Contributions
J.M.W., L.M.J., S.L.B., H.M., E.S.M., D.F.R., G.F., and B.T.B. performed experiments. J.M.W. and L.M.J. analyzed the data. J.L.S. and M.J.Z. provided resources. M.J.Z. supervised experiments. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Mirko Luoni, Kevin Nash, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Yuhong Cao and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wolter, J.M., James, L.M., Boeshore, S.L. et al. AAV-dCas9 vector unsilences paternal Ube3a in neurons by impeding Ube3a-ATS transcription. Commun Biol 8, 1332 (2025). https://doi.org/10.1038/s42003-025-08794-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08794-2
