
Abstract
N-acetylaspartate (NAA), the brain’s second most abundant metabolite, provides essential substrates for myelination through its hydrolysis1. However, the physiological roles of NAA in other tissues remain unknown. Here, we show that aspartoacylase (ASPA) expression in white adipose tissue (WAT) governs blood NAA levels for postprandial body temperature regulation. Genetic ablation of Aspa in mice resulted in systemically elevated NAA levels, and the ensuing accumulation in WAT stimulated pyrimidine production. Stable isotope tracing confirmed higher incorporation of glucose-derived carbon into pyrimidine metabolites in Aspa knockout cells. Additionally, serum NAA levels positively correlated with the abundance of the pyrimidine intermediate orotidine 5′-monophosphate, and this relationship predicted lower body mass index in humans. Using whole-body and tissue-specific knockout mouse models, we observed that fat cells provided plasma NAA and suppressed postprandial body temperature elevation. Moreover, unopposed NAA from adipocytes greatly enhanced whole-body glucose disposal exclusively in WAT. Exogenous NAA also increased plasma pyrimidines and lowered body temperature. These data place WAT-derived NAA as an endocrine regulator of postprandial body temperature and define broader roles for metabolic homeostasis.
Similar content being viewed by others


Main
Aspartoacylase (ASPA) hydrolyses N-acetylaspartate (NAA) to supply the acetate required for lipid synthesis in oligodendrocytes, the myelin-forming cells of the central nervous system. Consistent with this activity, ASPA loss-of-function mutations cause Canavan disease, a lethal condition associated with NAA accumulation and demyelination1. However, other functional relationships between NAA abundance and physiological outcomes remain unknown. We found that Aspa mRNA levels were fivefold greater in mouse adipose tissue depots relative to the whole brain (Extended Data Fig. 1a), confirming the expression patterns in the GTEx (Genotype–Tissue Expression) Portal and the Human Protein Atlas. We also measured Aspa gene expression during adipocyte differentiation in three in vitro human and mouse models. Similar to mature adipocyte marker genes (Adipoq, Fabp4, Pparg2), Aspa mRNA increased in mouse and human cells after differentiation (Extended Data Fig. 1b–d). Liquid chromatography (LC)–mass spectrometry (MS) analysis of steady-state metabolites revealed that NAA levels increased during 3T3-L1 adipocyte differentiation, along with most tricarboxylic acid (TCA) cycle intermediates (Extended Data Fig. 1e). NAA accrual was associated with higher levels of ASPA protein (Extended Data Fig. 1f), which was diffusely localized throughout the cytoplasm of adipocytes (Extended Data Fig. 1g). ASPA was firmly coexpressed with other adipocyte-specific genes in human white adipose tissue (WAT)2, including ADIPOQ before and after weight loss (Extended Data Fig. 1h).
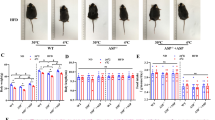
To explore how ASPA contributes to energy balance, we performed phenotyping of whole-body Aspa knockout mice (AspaKO)3. Aspa heterozygous breeders produced viable AspaKO mice born at a normal Mendelian ratio, but AspaKO mice failed to grow (Fig. 1a) and to achieve the body weights of wild-type (WT) littermates (AspaWT) (Fig. 1b). After weaning, AspaKO mice gained weight considerably faster (Extended Data Fig. 2a) than AspaWT mice yet remained smaller in size. Body composition studies revealed similar accumulation of fat and lean mass between genotypes when normalized to body mass (Fig. 1c). In addition to expected central nervous system phenotypes, the International Mouse Phenotyping Consortium4 identified fasting hyperglycaemia in AspaKO mice. Similarly, glucose tolerance tests (GTTs) and corresponding area-under-the-curve calculations showed that AspaKO mice cleared glucose more slowly compared with AspaWT mice (Extended Data Fig. 2b). Despite glucose intolerance, other gross insulin-resistant phenotypes were absent (Extended Data Fig. 2c–f).

AspaWT and AspaKO mice were fed a normal chow diet for 12 weeks and then subjected to metabolic phenotyping. a, Visual representation of AspaWT and AspaKO mice at 8 weeks of age. b, Body weight measurements of AspaWT and AspaKO mice over 12 weeks starting at 6 weeks of age (n = 6 WT, 10 KO mice per group). Data represent mean ± s.e.m. **P < 0.01 by an ordinary two-way ANOVA. c, Body composition by Echo magnetic resonance imaging, shown as a percentage of body weight (n = 11 WT, 9 KO mice per group). Data represent mean ± s.e.m., and statistical analysis was performed using an unpaired two-tailed Student’s t-test. d,e, Mice were individually housed and monitored in CLAMS cages during a 96-h period with measurements of food intake (d) and RER (e). Data represent mean ± s.e.m. Statistical analysis of d and e was performed within CalR and using ANCOVA with lean body mass as a covariate (n = 7 WT, 6 KO mice per group). f, ΔRER, defined as the difference between the average of the lowest 10% (light phase) and the highest 10% (dark phase) of RER values (n = 7 WT, 6 KO mice per group). *P < 0.05 by an unpaired two-tailed Student’s t-test. g, Tissue weights from WAT (scWAT and vWAT) depots, shown as a percentage of body weight (% BW) (n = 11 WT, 9 KO mice per group). h, Plasma leptin levels from ad libitum-fed mice (n = 8 WT, 9 KO mice per group). i,j, Representative histological analysis by H&E staining and mean adipocyte size (μm2) of scWAT (i) and vWAT (j) across three to five fields of view (n = 4 mice per group); scale bars, 100 μm. k–m, Relative abundance of NAA, glycolytic metabolites and pyrimidine metabolites in the scWAT (k), vWAT (l) and plasma (m) of AspaWT and AspaKO mice, measured by IC–MS targeted analysis. G6P, glucose-6-phosphate; F6P, fructose-6-phosphate. In c, d and f–j, data are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; centre line, median; whiskers, minimum and maximum values. In k–m, data are shown as the z score of log2-transformed values and relative changes denoted in the colour bar (n = 4 mice per group). In f–m, *P < 0.05, ****P < 0.0001 by an unpaired two-tailed Student’s t-test.
Despite the lower body weight and smaller size of AspaKO mice, food intake did not vary between genotypes (Fig. 1d). While AspaKO mice trended towards lower energy expenditure compared with AspaWT mice (Extended Data Fig. 2g), analysis of covariance (ANCOVA) performed in CalR5 suggested that any difference between the groups occurred not due to genotype but primarily due to the smaller body size and lower lean mass of AspaKO mice. Analysis of variance (ANOVA) identified that AspaKO mice exhibited a higher respiratory exchange ratio (RER) and a preference for carbohydrate oxidation during dark phases (Fig. 1e). The difference between the light (lower RER) and dark (higher RER) phases reflects metabolic adaptations to fuel preference and availability6. Based on RER changes, AspaKO mice deftly switch fuel sources during light-to-dark transitions (Fig. 1f).
Based on the observation that ASPA increased during adipocyte differentiation and knowing the proadipogenic effects of other proximal pathway lipogenic enzymes, such as acyl-CoA synthetase short-chain family member 2 (ACSS2) and ATP citrate lyase (ACLY)7,8,9, we hypothesized that Aspa depletion would cause fat cell loss. To test this hypothesis, we collected subcutaneous WAT (scWAT) and visceral WAT (vWAT) depots from AspaKO mice and littermate controls for immunohistochemistry and molecular analyses. While scWAT size trended lower, we recovered significantly less vWAT between genotypes (Fig. 1g). In agreement with reduced total WAT mass, serum leptin levels after ad libitum feeding were lower in AspaKO mice (Fig. 1h). Given that bigger fat cells, especially within scWAT depots, exhibit diminished competence for glucose disposal and lipogenesis10, we also speculated that AspaKO adipocytes would be larger in diameter than adipocytes from WT littermates, but this was not what we observed. Adipocytes from AspaKO mice were considerably smaller in scWAT (Fig. 1i) and vWAT (Fig. 1j). This phenotype appeared restricted to WAT because no differences were found in brown adipose tissue morphology (Extended Data Fig. 2h) or size (Extended Data Fig. 2i).
To characterize cell signalling events underpinning the smaller adipocyte size and energy balance in AspaKO mice, we applied the WAT samples to a reverse-phase protein array with broad pathway coverage (Extended Data Fig. 3). AspaKO tissues collected ad libitum showed altered activity of proteins involved in various pathways, including cell proliferation (MYC), insulin signalling (AKT) and mTORC1 (p70S6 kinase (p70S6K) T389). In parallel, we leveraged ion chromatography (IC)–MS to screen for metabolite concentrations in adipose tissue depots and plasma of AspaWT and AspaKO mice. As expected, NAA was higher in the scWAT and vWAT of AspaKO mice (Fig. 1k,l). We also found a higher abundance of glucose, glycolysis intermediates (glucose-6-phosphate, fructose-6-phosphate), and the pyrimidine metabolites carbamoyl aspartate (CarbAsp), dihydroorotate (DHOA), orotate and uridine in the WAT of AspaKO mice compared with controls. The build-up of nucleotides in AspaKO mice was also reiterated by the presence of more purine metabolites in WAT (Extended Data Fig. 3c,d). Constant regeneration of oxidized nicotinamide adenine dinucleotide (NAD+) is necessary for highly anabolic cells, such as adipocytes, to break down sugar and lipids while supporting nucleotide biosynthesis11. We observed that AspaKO scWAT favoured a more oxidized NAD status (higher NAD/NADH ratio) (Extended Data Fig. 3e), consistent with the production of more glycolysis intermediates (glucose-6-phosphate, fructose-6-phosphate, pyruvate) and pyrimidines. The plasma showed a similar accumulation of metabolites altered in WAT from AspaKO mice, including NAA and pyrimidine intermediates (CarbAsp, DHOA and orotate) (Fig. 1m). Taken together, discovery approaches performed on WAT formed the concept that NAA elevation from Aspa knockout promoted pyrimidine synthesis in coordination with anabolic and nutrient-sensing pathways.
NAA accumulates in the rare inherited disorder Canavan disease due to loss-of-function ASPA mutations, but relationships among more common conditions remain unknown. The Atherosclerosis Risk in Communities (ARIC) study is a longitudinal study that measures associations of heart disease risk factors with atherosclerosis and the occurrence of cardiovascular disease12. The ARIC data13 allowed the discovery of factors correlated with NAA abundance and ASPA protein measurements in a large human cohort (Fig. 2a). Linear regression analyses revealed that NAA was negatively associated with body mass index (BMI) (β = −0.08, Plinear model = 1.94 × 10−6). Moreover, NAA levels were the highest in normal-weight groups relative to individuals with overweight and obesity, and these trends remained when the data were separated by sex (Fig. 2b). Although there were no changes in ASPA protein abundance among BMI groups in ARIC, we found that WAT14 from lean individuals had higher expression of genes encoding NAA synthesis (NAT8L) and hydrolysis (ASPA) compared with WAT from persons with metabolically unhealthy obesity (Fig. 2c).

a, Schematic for the analysis of the ARIC study dataset using an age-, sex-, race- and BMI-adjusted linear regression model. BMI (kg m−2) groups: normal, 18.5 < BMI ≤ 25; overweight, 25 < BMI ≤ 30; obesity, BMI > 30. b, Relative (Rel) NAA abundance among BMI groups and male and female participants (male + female: n = 1,308 normal, n = 2,088 overweight (Over), n = 1,790 obesity; male: n = 500 normal, n = 1,012 overweight, n = 740 obesity; female: n = 808 normal, n = 1,076 overweight, n = 1,050 obesity). Shown are mean ± 95% confidence interval. P < 0.05 (anormal versus obesity, bnormal versus overweight) by a two-way ANOVA with a Sidak multiple comparisons test. c, NAT8L and ASPA expression in the scWAT of persons with a metabolically normal lean (MNL) status and those with metabolically unhealthy obesity (MUO) (n = 5 per group). Data are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; centre line, median; whiskers, minimum and maximum values. *P < 0.05, **P < 0.01 by an unpaired two-tailed Student’s t-test. d, Estimated effect of OMP on NAA for each BMI category. The β coefficient is shown with the respective 95% confidence intervals and statistical significance from a linear regression model (male + female: n = 1,308 normal, n = 2,088 overweight, n = 1,790 obesity; male: n = 500 normal, n = 1,012 overweight, n = 740 obesity; female: n = 808 normal, n = 1,076 overweight, n = 1,050 obesity). e, Estimated effect of OMP on NAA for each BMI category and sex. The β coefficients for each BMI category are shown. Error bars in e represent s.d. The figure shows statistical assessments of the regression analysis, and P values (circles) summarize the strengths of the associations within each group. f,g, Relative abundance of NAA and pyrimidine metabolites in the medium of scWAT explants incubated for 24 h (f) and in scWAT explants (g) (n = 5 replicates per group). Relative abundance was normalized to an internal standard, followed by log10 transformation. Data are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; centre line, median; whiskers, minimum and maximum values. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by an unpaired two-tailed Student’s t-test.
To explore additional potential interactions of NAA abundance and ASPA with other proteins and metabolites, we performed a linear correlation analysis using MS data from ARIC, adjusted for sex, race, age and BMI. Among ARIC participants, we observed a significant association between circulating NAA and the pyrimidine intermediate orotidine 5′-monophosphate (OMP) across BMI categories (β = 0.722, Pmain = 3.866 × 10−26), and these correlations decreased with obesity (z = −4.3, Pinteraction = 1.74 × 10−5). Likewise, a stratified analysis identified significant stepwise declines in NAA–OMP correlations across BMI classes (Fig. 2d), which remained present in men and women (Fig. 2e). We did not find metabolites or proteins that correlated with circulating ASPA.
The greater abundance of pyrimidine metabolites in the plasma and WAT of AspaKO mice aligned with the ARIC analysis results, showing that NAA positively and uniquely correlated with OMP. Based on these findings, we predicted that ASPA governs NAA secretion from WAT for systemic effects. To explore how ASPA contributes to NAA release from WAT, we incubated AspaWT and AspaKO scWAT explants in DMEM; then, we performed MS analysis of the tissue and medium to assess differences in metabolite abundance. NAA was highly abundant in the medium of explants from AspaKO mice (Fig. 2f), detected at far higher levels than observed in explants from AspaWT mice, indicating that NAA from WAT is secreted into the surrounding microenvironment (Fig. 2g). We also detected higher levels of OMP and uridine in the medium and explants from AspaKO mice, strengthening the notion that elevated NAA directs pyrimidine synthesis in WAT.
Glucose supplies most of the carbon required for biosynthetic reactions in adipocytes15. Our data imply that NAA accrual in WAT directs the prioritized flux of carbon into pyrimidine synthesis. To assess pyrimidine synthesis directly, we performed stable isotope tracer analysis using uniformly labelled [13C6]glucose in adipocytes differentiated from stromal vascular fraction (SVF) cells, followed by targeted profiling of metabolites by IC–MS (Extended Data Fig. 4a). As expected, NAA abundance was eightfold higher in AspaKO cells than in controls (Extended Data Fig. 4b). To explore 13C assimilation into pyrimidine precursors, we focused on m + 2 and m + 3 isotopologues derived from the transamination of oxaloacetate to aspartate from either the TCA cycle or pyruvate carboxylase (Extended Data Fig. 4c). The incorporation of the label into m + 2 aspartate and pyrimidine precursors (DHOA and orotate) was greater in AspaKO cells. Similarly, Aspa knockout raised the delivery of m + 5 from ribose-5-phosphate into the pyrimidine intermediates OMP, uridine 5′-monophosphate (UMP), cytidine 5′-monophosphate (CMP) and the nucleotide uridine (Extended Data Fig. 4d). Altogether, these isotopomer enrichment patterns suggest that unopposed NAA forced glucose flux towards pyrimidine synthesis.
We also examined protein expression in SVF cells from AspaWT and AspaKO mice before and after adipocyte differentiation. As expected, ASPA was not present in preadipocytes, highly expressed in differentiated AspaWT cells and absent in AspaKO cells (Extended Data Fig. 4e). Mechanistically, mTORC1 activation of p70S6K allows the phosphorylation of CAD (carbamoyl phosphate synthetase II (CPSII), aspartate transcarbamylase (ATCase) and dihydroorotase) at S1859 (ref. 16) to catalyse de novo pyrimidine synthesis. In line with this idea, Aspa knockout resulted in sustained p70S6K T389 (1.7-fold) and 40% higher CAD S1859 phosphorylation in adipocytes. While AspaKO cells selectively engaged in pyrimidine synthesis, adipocyte differentiation was only nominally higher as measured by adiponectin (ADIPOQ; Extended Data Fig. 4e) and Oil Red O staining of neutral lipids (Extended Data Fig. 4f).
Pyrimidines interface with mitochondrial metabolism by producing DHOA for electron transfer in the respiratory complex and the TCA cycle17. Despite changes in pyrimidine synthesis, AspaKO and AspaWT adipocytes and their precursors showed indistinguishable levels of mitochondrial proteins (Extended Data Fig. 5a). To identify further the cellular changes associated with AspaKO adipocytes, we used high-throughput spinning disk confocal microscopy and CellProfiler18 to extract quantitative features informing cytoplasmic mitochondrial levels and lipid accumulation (Extended Data Fig. 5b). While Aspa knockout did not affect lipid features associated with the number of droplets or intensity, mitochondrial area and density were slightly diminished (Extended Data Fig. 5c), contributing to lower oxygen consumption rates than in controls (Extended Data Fig. 5d). Together, our data support a critical and selective role of ASPA in regulating the pyrimidine pool in adipocytes.
Pyrimidines secreted by adipocytes regulate postprandial body temperature19. Our results demonstrated that NAA elevation in adipocytes increased de novo pyrimidine availability, which in turn led us to predict that NAA has a role in postprandial body temperature regulation. To explore this prediction, we measured the rectal temperature of AspaWT and AspaKO mice after 16 h of fasting and in the 6-h refed state (Extended Data Fig. 6a). In both the fasted and refed states, AspaKO mice weighed 7–10 g less than AspaWT mice (Extended Data Fig. 6b), and we observed varied effects on vWAT and scWAT mass relative to body weight (Extended Data Fig. 6c,d). In line with the idea that NAA promotes a functional pool of pyrimidines, we found that the body temperature of AspaKO mice was almost 1 °C lower than that of AspaWT mice in the refed state (Extended Data Fig. 6e). Feeding also causes a drop in plasma pyrimidines to form an endocrine loop with leptin that affects energy balance19. Leptin levels were equivalent in the fasted state, but refeeding did not recover serum leptin abundance in AspaKO mice relative to controls (Extended Data Fig. 6f). Finally, we explored protein expression changes relative to the loading control (HSP90) in WAT from AspaWT and AspaKO mice during fasting and refeeding (Extended Data Fig. 6g). We saw greater phospho-CAD signals and phospho-CAD/total CAD ratios in AspaKO mice than in AspaWT mice in the refed state, suggesting that CAD and de novo pyrimidine synthesis remain unopposed when NAA levels are high. These data suggest that NAA levels regulate CAD, and Aspa deletion does not permit lower postprandial pyrimidine synthesis for body temperature homeostasis.
AspaKO mice are small in stature and develop macrocephaly3. Thus, whole-body knockout may influence metabolic phenotypes due to Aspa loss in other tissues, such as the brain. To determine the adipocyte-specific roles of NAA and ASPA in whole-body energy balance, we used CRISPR–Cas9 to generate conditional Aspa knockout alleles (Aspafl/fl; Extended Data Fig. 7a). The presence of loxP sites in the targeted regions was confirmed by Sanger sequencing and genotyping PCR of founder floxed alleles (Extended Data Fig. 7b). To examine the adipocyte-specific impact of ASPA on energy balance, we crossed Aspafl/fl mice with mice expressing Cre recombinase under the control of the ADIPOQ promoter (Adipoq-cre) to generate Aspa knockout in mature fat cells. Western blotting confirmed that ASPA was successfully depleted in the WAT of Aspafl/fl; Adipoq-cre (AspafKO) mice but not in Aspafl/fl littermates or in the liver of either genotype (Extended Data Fig. 7c).
After confirming adipocyte-specific Aspa knockout, we assessed energy balance parameters in ad libitum-fed AspafKO mice and littermate Aspafl/fl controls. Contrary to our expectations, weight gain (Extended Data Fig. 7d) and WAT mass (Extended Data Fig. 7e,f) were similar. AspafKO mice and littermate Aspafl/fl controls also displayed indistinguishable energy balance profiles (Extended Data Fig. 7g–i). Based on the AspaKO studies, we expected that AspafKO mice might harbour smaller fat cells in WAT. Indeed, adipocytes from AspafKO WAT were significantly smaller (Extended Data Fig. 7j), resembling AspaKO WAT adipocytes and favouring metabolic benefits associated with smaller fat cells within WAT depots10.
We then performed fasting–refeeding experiments to determine whether adipocyte-specific Aspa deletion could recapitulate the lower postprandial body temperature phenotypes observed in AspaKO mice. While the fasted body temperatures of AspafKO and Aspafl/fl mice were equivalent, AspafKO mice did not recover their postprandial body temperature to levels observed in littermate controls (Fig. 3a). Female AspafKO mice also showed lesser postprandial body temperature responses (Fig. 3b). These data confirm that NAA and ASPA in adipose tissue are important for body temperature regulation in male and female mice. We also measured leptin levels in male mice and observed decreased leptin in refed AspafKO mice (Fig. 3c). WAT depot mass and plasma insulin levels were not different (Extended Data Fig. 7k–m). As the mice were indistinguishable in body weight and size (Extended Data Fig. 7d), differences in leptin secretion from adipocytes are governed by NAA and ASPA, strengthening their distinct roles in the regulation of postprandial body temperature19,20.

a,b, Body temperature (temp) of male (a) and female (b) Aspafl/fl and AspafKO mice following fasting (n = 23 Aspafl/fl, 29 AspafKO males; n = 8 Aspafl/fl, 9 AspafKO females) and refeeding (n = 23 Aspafl/fl, 29 AspafKO males; n = 8 Aspafl/fl, 9 AspafKO females). For the change in body temperature (ΔTemp), in paired males (n = 23 Aspafl/fl, 29 AspafKO) and females (n = 8 Aspafl/fl, 9 AspafKO), data are mean ± s.e.m., and *P < 0.05 by an unpaired two-tailed Student’s t-test. c, Plasma leptin levels in male mice following fasting (n = 22 Aspafl/fl, 23 AspafKO) and refeeding (n = 18 Aspafl/fl, 22 AspafKO). d,e, Relative abundance of NAA and pyrimidine metabolites in the scWAT (d) and plasma (e) of Aspafl/fl and AspafKO mice in fasted and refed states measured by IC–MS analysis (n = 4 mice per group per condition) and shown as log10-transformed values normalized to Aspafl/fl fasted levels. f, Hyperinsulinaemic–euglycaemic clamp (n = 5 per group). Glucose infusion rate (GIR), glucose production rate (GPR) during basal (empty bars) and clamp (striped bars) conditions, glucose disposal rate (GDR), and glucose uptake in vWAT and gastrocnemius muscle. Data are mean ± s.e.m. In a–e, data are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; centre line, median; whiskers, minimum and maximum values. For a–e, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by an ordinary two-way ANOVA followed by Fisher least significant difference (LSD) tests. For f, *P < 0.05 by an unpaired Student’s t-test (glucose infusion rate, glucose disposal rate and glucose uptake). Glucose production was not different after an ordinary two-way ANOVA.
Mice with global Aspa loss produced systemic NAA and elevated plasma levels of pyrimidines and their intermediates. Given that fat cells secreted NAA, we hypothesized that adipocyte-specific Aspa knockout contributed to the accumulation of pyrimidines that lowered postprandial body temperature. To test this hypothesis, we prioritized MS analysis of scWAT and plasma from AspafKO and littermate controls. NAA levels were higher in the scWAT of AspafKO mice in the fasted and refed states (Fig. 3d). Similarly, CarbAsp and DHOA levels were also higher in AspafKO mice in both conditions. Other pyrimidines, such as uridine, trended higher in AspafKO mice only in the fasted state. Liver pyrimidine metabolites (Extended Data Fig. 8a) and triglycerides (Extended Data Fig. 8b,c) remained unchanged by Aspa deletion in adipocytes, except for small effect sizes on uridine disposal in the refed state. When we analysed plasma metabolite levels, we found that adipocyte-specific Aspa deletion was sufficient to raise NAA and CarbAsp levels systemically in AspafKO mice (Fig. 3e), predominantly in the refed state when ASPA expression is intact in Aspafl/fl mice but remains absent in AspafKO mice.
To determine how NAA from adipocytes contributes to whole-body insulin sensitivity and glucose fluxes, we performed hyperinsulinaemic–euglycaemic clamp procedures in unrestrained AspafKO and control mice. We observed increases in the glucose infusion rate required to maintain euglycaemia in AspafKO mice compared with littermate controls, indicative of better insulin sensitivity when NAA is raised in WAT in an unopposed manner (Fig. 3f). We did not detect any differences in insulin’s ability to suppress endogenous liver glucose production. The primary factor contributing to the increased glucose infusion rate in AspafKO mice was elevated whole-body glucose disposal, with pronounced effects observed in vWAT but not in skeletal muscle. These data establish that NAA abundance and endocrine actions in the circulation and outside the brain are entirely encoded by ASPA expression in adipocytes.
After discovering that adipocyte-specific Aspa deletion raises systemic NAA levels, lowers postprandial body temperature and increases the abundance of de novo pyrimidines, we asked whether acute NAA treatment in WT mice is sufficient to induce similar changes in body temperature (Fig. 4a). WT mice were fasted overnight for 16 h, followed by baseline rectal temperature measurements. We then randomized and gavaged mice with pH-balanced water or NAA and measured rectal temperature 15 min later. Finally, mice were refed for 1 h, followed by another round of body temperature measurements. We found that acute treatment with NAA was sufficient to induce a significant decrease in body temperature in WT mice 15 min following gavage, whereas there was no difference in body temperature in fasted and refed states (Fig. 4b). Using targeted IC–MS, we confirmed that gavage increased plasma NAA levels by nearly 100-fold (Fig. 4c), and the levels were considerably higher across the liver and WAT depots (Fig. 4d). Consequently, CarbAsp levels were also raised in the liver and scWAT (Fig. 4e). Moreover, acute NAA elevation was sufficient to raise the plasma levels of the pyrimidine metabolites CarbAsp, OMP and UMP in WT mice (Fig. 4f). These data strengthen the concept that higher NAA in the circulation affects systemic pyrimidine synthesis and contributes to body temperature regulation.

a, Schematic of the gavage experiment. Body temperature measurements and plasma collection in WT mice were performed following a 16-h fast, 15–30 min following gavage and after 1 h of refeeding. b, Body temperature measurements in fasted (n = 13 water, 13 NAA), post-gavage (n = 13 water, 13 NAA) and refed mice (n = 8 water, 8 NAA). *P < 0.05 by an ordinary two-way ANOVA followed by Fisher LSD tests. c, Plasma NAA levels of fasted (n = 4 water, 4 NAA), post-gavage (n = 9 water, 9 NAA) and refed mice (n = 4 water, 4 NAA). d, Tissue NAA levels in the liver, scWAT and vWAT after gavage (n = 4 mice per group per condition). e, Relative abundance of CarbAsp across the liver, scWAT and vWAT following gavage (n = 4 mice per group per condition). f, Relative abundance of plasma CarbAsp (n = 9 mice per group per condition), OMP (n = 9 mice per group per condition) and UMP (n = 4 mice per group per condition) following gavage. g, Immunoblot of scWAT from AspaWT mice following water or NAA gavage, probed for ASPA, phospho-CAD (pCAD) S1859, total CAD (tCAD), phospho-AKT S473, total AKT (tAKT), phospho-TSC2 T1462, total TSC2 (tTSC2), phospho-4EBP1 T37/46 and total 4EBP1 (t4EBP1). HSP90 served as the loading control (n = 3 mice per group per condition). h, Model for NAA’s mechanism of action in regulating de novo pyrimidine synthesis. NAA binds within the ATCase domain of CAD, promoting increased CAD activity. Carbamoyl-P, carbamoyl phosphate; N-carbamoyl-asp, N-carbamoyl aspartate; DHO, dihydroorotase; DHODH, DHOA dehydrogenase; PPi, pyrophosphate; PRPP, phosphoribosyl pyrophosphate; UMPs, UMP synthase; UDP, uridine 5′-diphosphate; UTP, uridine 5′-triphosphate; CTP, cytidine 5′-triphosphate; CDP, cytidine 5′-diphosphate; CMP, cytidine 5′-monophosphate; UCK, uridine cytidine kinase. In b–f, data are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; centre line, median; whiskers, minimum and maximum values. In c–f, data are shown as log10-transformed values. *P < 0.05, **P < 0.01, ****P < 0.0001 by an unpaired two-tailed Student’s t-test. NS, no significance. Panels a and h created with BioRender.com.
Due to its structural similarities with N-acetylglutamate, NAA can activate the urea cycle enzyme CPSI21. CPSII is one of the enzymatic domains of CAD and shares 70% sequence similarity with CPSI. Based on these structural properties, we hypothesized that NAA acts as an allosteric activator of CAD to promote the synthesis of pyrimidines. To test this hypothesis, we used AlphaFold22 to generate a three-dimensional structure and explore the interactions of NAA and CAD (Extended Data Fig. 9a). In the modelled CAD, we performed targeted docking and found that NAA bound CAD in the C-terminal region (amino acids 1918–2225) containing the ATCase domain (Extended Data Fig. 9b). In a similar fashion, aspartate occupies the ATCase domain and joins with carbamoyl phosphate to bring about CarbAsp. This led us to perform docking studies of aspartate using the predicted model as a control. As expected, aspartate bound to the ATCase domain, but with a binding energy estimate less than the predicted NAA affinity (Extended Data Fig. 9c). Gavage experiments corroborated the structural studies by demonstrating that NAA caused greater CAD activity as defined by phospho-CAD and CAD in scWAT (Fig. 4g) and pyrimidine accumulation in plasma. Phospho-4EBP1, as a surrogate of mTORC1 activity, remained unchanged by NAA gavage. We were also surprised to detect more phospho-AKT S473 (indicative of insulin sensitivity), extending the data that NAA caused more glucose disposal during hyperinsulinaemic–euglycaemic clamps. These data support a model describing how high NAA levels exert allosteric regulation of CAD and biases signalling towards pyrimidine synthesis for body temperature regulation (Fig. 4h).
The brain-enriched amino acid NAA carries acetate for the synthesis of acetyl-CoA and myelin lipids in oligodendrocytes. Thus, the presence of the NAA synthesis and hydrolysis machinery in fat cells predicts that ASPA provides lipogenic acetate for adipose tissue expansion. Instead, we present evidence demonstrating that NAA within WAT governs pyrimidine pools that contribute to body temperature regulation. There are some limitations to this study. In contrast to patients with Canavan disease, AspaKO mice do not exhibit a demonstrably shortened lifespan, but their smaller body size and macrocephaly may obscure effect sizes and metabolic responses unique to WAT. However, our studies performed in AspafKO mice clearly showed that unopposed NAA accumulation in WAT directs body-wide pyrimidine pools and postprandial body temperature. We acknowledge that future experiments that deplete NAA and, consequently, pyrimidines from WAT will be needed to identify precise signal integration sites in the central or peripheral nervous system for regulating body temperature homeostasis.
A major unanswered question is whether NAA is sensed by mTORC1 and CAD for stimulating pyrimidine synthesis. Anabolic signals activate the mTORC1-dependent phosphorylation of CAD by p70S6K to promote de novo pyrimidine synthesis16. Consistent with these studies, the higher p70S6K phosphorylation in AspaKO WAT helped us identify how NAA elevation tied into pyrimidine synthesis and the allosteric regulation of CAD. NAA also increased CAD levels, suggestive of additional post-translational regulation. Only p70S6K16 and MAPK23 perform the two known CAD post-translational modifications, and dedicated efforts will be necessary to identify other operant mechanisms regulating de novo pyrimidine synthesis when NAA levels are high in the cell. Alternatively, AlphaFold structures argue strongly that NAA docks to the ATCase domain of CAD, obviating direct communication with mTORC1. NAA gavage studies also showed biased signalling toward CAD and pyrimidine synthesis in the WAT. In these tissue contexts, mTORC1 signalling appeared nominally altered. Nevertheless, it remains possible that NAA trapping of aspartate and lesser interconversions to asparagine or glutamine alter amino acid sensing24 to mTORC1. It will now be important to determine how nutrient sensing in WAT governs the interactions of NAA with CAD for energy balance regulation. Lastly, the lower serum NAA levels in obesity and the lessened postprandial body temperature responses observed in this population25,26,27,28,29 suggest that this system is also operative in humans. Our findings justify future studies in humans to examine whether dietary interventions that raise NAA levels influence body weight.
Methods
Animal studies
Animal procedures were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine (BCM). Experimental animals were housed at 23 °C in a barrier-specific pathogen-free animal facility with a 12-h dark and 12-h light cycle and access to water and normal chow (Harlan Laboratories 2920X) unless otherwise specified. We used the C57BL/6N Aspatm1b(EUCOMM)Wtsi mouse strain with deletion of Aspa exon 2 provided by the European Mouse Mutagenesis Consortium (EUCOMM). Mice homozygous for the Aspa allele (AspaKO) have total gene knockout.
Aspafl/fl mice were generated by the Genetically Engineered Rodent Models Core at BCM using previously established methods. We used Cas9-initiated homology-directed repair using a pair of single guide RNAs (sgRNAs) coupled with a long single-stranded DNA (lssDNA) donor template harbouring short homology arms and loxP-flanked exons. The sgRNAs and the lssDNA donor were used to insert loxP sites 5′ and 3′ of exon 2 of the mouse Aspa gene. To minimize the probability of off-target events, only sgRNAs predicted to have off-target sites with three mismatches or more were used to target Cas9 endonuclease activity to intronic sequences flanking exon 2 (Supplementary Table 1). Two hundred C57BL/6J pronuclear-stage zygotes were co-injected with Cas9 mRNA, sgRNAs and lssDNA. Following microinjection, zygotes were transferred into pseudopregnant ICR recipient females at approximately 25–32 zygotes per recipient. Sanger sequencing of cloned loxP sites and founder line genotyping from mouse genomic DNA confirmed loxP insertions and sequence fidelity. Progeny generated from putative founders were sequence-confirmed for the fidelity of the loxP sequences and floxed exons. Aspafl/fl mice were crossed to Adipoq-cre mice (The Jackson Laboratory #028020) to generate adipocyte-specific Aspa knockout mice (AspafKO) and littermate controls (Aspafl/fl). Aspafl/fl mice are distributed on request.
Experiments were conducted using littermate-controlled mice maintained on a C57BL/6J background. At the end of each experiment, mice were killed by cervical dislocation under isoflurane anaesthesia. After killing, tissues were collected, fixed in 10% formalin or flash-frozen in liquid nitrogen, and stored at −80 °C until use. All experiments adhered to the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
In vitro studies
SVF cells were isolated from mouse scWAT. Fat depots were digested in PBS containing collagenase type I (100 U ml−1; Gibco #17100-017) and dispase II (2.4 U ml−1; Sigma #D4693) supplemented with 10 mM CaCl2 at 37 °C for 45–50 min. Primary cells were filtered twice through 70-µm strainers and centrifuged at 700g to collect the SVF. The SVF cell pellets were rinsed and plated. Adipocyte differentiation was induced by treating confluent cells in DMEM/F12 medium containing GlutaMAX (Thermo Fisher #10565-018), 10% dialysed FBS (Gibco #A3382001), 0.250 mM isobutylmethylxanthine (Sigma #13347), 1 μM rosiglitazone (Cayman Chemical #71740), 1 μM dexamethasone (Tocris Biosciences #1126), 850 nM insulin (Sigma I5500) and 1 nM T3 (Sigma #T-074). Three days after induction, cells were switched to the maintenance medium (same as above but without isobutylmethylxanthine). Experiments occurred 8–10 days after the induction of differentiation, and Oil Red O (Biovision K580) was used to assess lipid accumulation. Subcutaneous human preadipocytes (ZenBio #SP-F-SL) were differentiated as described above. For Seahorse studies, rates of oxygen consumption and extracellular acidification were measured in cultured adipocytes using an XF96e analyser (Agilent), and the cells were differentiated as stated above. For the assay, the medium was replaced with the following assay medium: 37 °C unbuffered DMEM containing 4.5 g l−1 glucose, sodium pyruvate (1 mM) and l-glutamine (2 mM). Basal respiration was defined before the sequential addition of oligomycin, FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone), rotenone and antimycin A.
Antibodies and western blotting
Tissue and whole-cell lysates were extracted using a protein extraction reagent (Thermo Fisher) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher). Immunoblotting was performed on 4–12% Bis–Tris NuPage gels (Life Technologies), and proteins were transferred onto Immobilon-P transfer membranes (Millipore), followed by antibody incubation. Immunoreactive bands were visualized by chemiluminescence using a ChemiDoc MP system (Bio-Rad) and Image Lab Touch Software v3.0.1 (Bio-Rad). The following antibodies were used: anti-ASPA (1:2,500, ab154503, Abcam), anti-ADIPOQ (1:500, GTX112777, GeneTex), anti-FABP4 (1:1,000, GTX116036, GeneTex), anti-PPARG (1:1,000, 2443, Cell Signaling), anti-phospho-CAD S1859 (1:500, 12662, Cell Signaling), anti-CAD (1:500, 11933, Cell Signaling), anti-phospho-p70S6K T389 (1:1,000, 9205, Cell Signaling), anti-p70S6K (1:1,000, 2708, Cell Signaling), anti-DHOA dehydrogenase (1:1,000, 14877-I-AP, Proteintech), anti-HSP90 (1:1,000, 4877, Cell Signaling), anti-phospho-TSC2 (1:1,000, 3617, Cell Signaling), anti-TSC2 (1:1,000, 4308, Cell Signaling), anti-phospho-4EBP1 T37/46 (1:1,000, 2855, Cell Signaling), anti-4EBP1 (1:1,000, 9644, Cell Signaling), anti-phospho-AKT S473 (1:1,000, 4060, Cell Signaling), anti-AKT (pan) (1:1,000, 4691, Cell Signaling) and Total OXPHOS Rodent WB Antibody Cocktail (1:1,000, ab110413, Abcam).
RNA isolation and qPCR
Total RNA was extracted using the RNeasy Mini Plus kit (in vitro cells) or RNeasy Lipid Tissue kit (tissues) (Qiagen). cDNA was synthesized from total RNA using qScript (QuantBio #95048-100). Relative mRNA expression was measured with SsoAdvanced Universal Probes Supermix reactions (Bio-Rad #175284) on the QuantStudio 3 real-time PCR system (Applied Biosystems). TATA box-binding protein (TBP) was the invariant control. Roche Universal Probe Gene Expression Assays (mouse) or individual TaqMan Gene Expression Assays (Thermo Fisher) were used (Supplementary Table 1).
Reverse-phase protein arrays
Protein lysates were prepared by the BCM Antibody-Based Proteomics Core for reverse-phase protein array assays. The Aushon 2470 Arrayer (Aushon BioSystems) with a 40-pin (185-µm) configuration was used to spot lysates onto nitrocellulose-coated slides (Grace Bio-Labs). Slides were probed with 244 antibodies to total and phosphorylated proteins using an automated slide stainer (Dako). Primary antibody binding was detected using a biotinylated secondary antibody, followed by the streptavidin-conjugated IRDye 680 fluorophore (LI-COR). Fluorescence-labelled slides were scanned using GenePix AL4200, and images were analysed using GenePix Pro 7.0 (Molecular Devices). The background-subtracted total fluorescence intensities of each spot were normalized for variation in total protein (SYPRO Ruby) and nonspecific labelling.
Genotyping
DNA was extracted from mouse ear clips and used in PCR reactions with primers designed to detect the WT (5′-TGTCCCTGATCCAGTAGTCAT, 3′-GAGTTAGTTCTAGGACAGCAGT, 426-bp band) and mutant (LAR3) (5′ primer was the same as for WT, 3′-CAACGGGTTCTTCTGTTAGTCC, 388-bp band) sequences in Aspa whole-body knockout mice. Primers were used to detect the 5′ loxP sequences (forward: CAGTGCACCATAACTACAAGCA, reverse: TCTGAAACCTCTGCAAATGACA) in the conditional mice. The PCR product was run on agarose gels. Whole-body knockout and Cre transgenic mice were genotyped according to protocols provided by EUCOMM and The Jackson Laboratory.
Indirect calorimetry and body temperature measurements
Mice were maintained on experimental diets and housed at room temperature in Comprehensive Laboratory Animal Monitoring Systems Home Cages (CLAMS-HC, Columbus Instruments). Oxygen consumption, CO2 emission, energy expenditure, food and water intake, and activity were measured for 6 days (BCM Mouse Metabolism and Phenotyping Core). Mouse body weight was measured, and body composition was examined by magnetic resonance imaging (Echo Medical Systems) before indirect calorimetry. Body temperature was measured using a rectal probe connected to a digital thermometer (BAT-12 Microprobe-Thermometer, Physitemp). All treatment and temperature measurements were performed at ambient room temperature (23–25 °C). Statistical analysis of energy expenditure was performed by ANCOVA with lean body mass as a covariate using the CalR web-based tool5.
Glucose and insulin tolerance tests
Mice were maintained on experimental diets for 10–18 weeks and then subjected to tolerance tests. For glucose tolerance assessment, mice were fasted for 16 h, and glucose was administered (1.5 g per kg body weight) by intraperitoneal injection. For insulin tolerance assessment, mice were fasted 4 h before intraperitoneal injection of insulin (1.5 U per kg body weight). Blood glucose levels were measured using a handheld glucometer.
Hyperinsulinaemic–euglycaemic clamp studies
Unrestrained mice were catheterized in the right jugular vein and allowed to recover for 4–7 days. After overnight fasting, mice received a primed dose of [3H]glucose (10 μCi), followed by constant-rate intravenous infusion (0.1 μCi min−1) of [3H]glucose using a syringe infusion pump for 90 min. Blood samples were collected to determine basal glucose production. After 90 min, mice were primed with a bolus injection of insulin, followed by 2 h of continuous insulin infusion. Simultaneously, 25% glucose was infused at an adjusted rate to maintain the blood glucose level at 100–140 mg dl−1. Blood glucose concentration was determined every 10 min using a glucometer. Blood was collected at 120 min to measure hepatic glucose production and peripheral glucose disposal rates. For tissue-specific uptake, we injected 2-deoxy-d-[1-14C]glucose (10 μCi) during the hyperinsulinaemic–euglycaemic clamp, 45 min before the end of the clamp procedure, and collected blood samples at 5, 10, 15, 25, 35 and 45 min. Following the clamp, mouse tissues were collected to evaluate glucose uptake. Glucose uptake was calculated from the plasma 2-[14C]deoxyglucose profile and tissue content of [14C]glucose-6-phosphate.
ELISAs and free fatty acid assays
Serum samples collected from fasted, refed and/or ad libitum-fed mice were used to measure insulin (Millipore #EZRMI-13K), leptin (Crystal Chem #90030), ADIPOQ (Thermo Fisher #KMP0041) and free fatty acids (ZenBio #sfa-1).
Hepatic triglycerides
Liver tissue samples were analysed for triglycerides (Triglyceride reagent TR22421; Thermo Scientific) using homogenates mixed with a 1:2 chloroform/methanol solution, followed by isolation of the lipid-rich chloroform layer (modified Folch method).
Histology
Formalin-fixed, paraffin-embedded adipose sections were stained with haematoxylin and eosin (H&E) by the BCM Human Tissue Acquisition and Pathology Core. Images were captured (20×) using a Nikon Ci-L brightfield microscope, and adipocyte morphometry was quantified using ImageJ.
Fluorescence microscopy
Human adipocytes were fixed in 4% paraformaldehyde. Glycine was used to quench autofluorescence derived from residual paraformaldehyde. Fixed cells were incubated with rabbit anti-ASPA (1:2,500, ab154503, Abcam) primary antibody at 4 °C overnight in 2% BSA/0.1% saponin/PBS, washed in 1× PBS three times (5 min per wash) and then incubated with Alexa Fluor 647-conjugated goat anti-rabbit IgG (1:1,000, A-21245, Life Technologies). DAPI (Sigma #D8417) and LipidTOX (Life Technologies #H34475) were used for nuclei and lipid droplet labelling, respectively. Imaging was performed with the Olympus IX83 epifluorescence deconvolution microscope (Olympus).
High-content analysis was performed using AspaKO and AspaWT SVF cells plated and differentiated in 96-well plates (PerkinElmer PhenoPlate). After differentiation, cells were stained live with MitoTracker Deep Red FM and BODIPY for 20 min at 37 °C. Stains were removed, and cells were fixed with 4% paraformaldehyde at room temperature for 20 min. Following fixation, cells were washed three times in PBS at room temperature for 3 min each. Finally, cells were stained with DAPI in PBS solution containing 1% BSA and 0.1% Triton X-100 for 20 min. Cells were then washed three times with PBS at room temperature for 3 min each. Cells were left in PBS to be imaged on a Yokogawa CV8000 spinning disk high-throughput confocal microscope, with maximum projection intensity images collected for each channel. A total of 15 fields were collected per well. Images were analysed using CellProfiler18, and intensity measurements were extracted to describe the mitochondrial and lipid intensity for each well. Measurements were calculated according to CellProfiler’s instructions (https://cellprofiler-manual.s3.amazonaws.com/CellProfiler-4.2.6/index.html).
Metabolomics
To determine the relative abundance of metabolites in tissues, we prepared extracts and analysed them by high-resolution MS. Briefly, individual tissue samples (~50 mg) were homogenized. Metabolites were extracted using ice-cold 0.1% trichloroacetic acid in 90:10 (v/v) acetonitrile/water. Then, extracts were centrifuged at 17,000g for 5 min at 4 °C, and supernatants were transferred to clean autosampler vials. In the end, a 10 μl volume of each sample was injected for analysis by LC–MS. The Thermo Vanquish LC system included a SeQuant ZIC-cHILIC column (3 μm particle size, 100 × 2.1 mm), with the column compartment kept at 30 °C. Mobile phase A (MPA; weak) was 95:5 (v/v) acetonitrile/ammonium acetate buffer (200 mM, pH 5.8), and MPB (strong) was 50:45:5 acetonitrile/water/ammonium acetate buffer (200 mM, pH 5.8). The autosampler tray was chilled to 4 °C. The mobile phase flow rate was 300 μl min−1, and the gradient elution programme was set as follows: 0–2 min, 20% MPB; 2–6 min, 20–80% MPB; 6–10 min, 80% MPB; 10–11 min, 80–20% MPB. The total run time was 15 min. Data were acquired using a Thermo Orbitrap Fusion Tribrid mass spectrometer under the electrospray ionization (ESI) positive ionization mode at a resolution of 240,000. Raw data files were imported to Thermo TraceFinder software for final analysis. The relative abundance of each metabolite was normalized by tissue weight.
For ex vivo adipose tissue studies, fat pads were excised and cut into ~50 mg pieces in PBS on ice. At least two tissue pieces per mouse were allocated to each treatment group. Each tissue was placed in a well of 24-well plates containing 0.5 ml DMEM/F12. After overnight incubation at 37 °C and 5% CO2, tissues were snap-frozen in liquid nitrogen and the medium was snap-frozen on dry ice. Tissue samples were first pulverized; then, 10–30 mg tissue was weighed and used for extraction with 1 ml ice-cold 80% methanol/water. Samples were homogenized in TissueLyser II and incubated on ice for 10 min, followed by centrifugation at 20,000g at 4 °C for 10 min. The supernatant was collected, dried in a vacuum concentrator and stored at −80 °C until reconstitution for metabolomic analysis. For medium samples, 10 μl was collected for extraction with 500 μl ice-cold 80% methanol/water. Samples were vortexed and centrifuged at 20,000g for 10 min at 4 °C. The supernatant was collected, dried in a vacuum concentrator and stored at −80 °C. All dried samples were reconstituted into 30 µl of sample solvent (water/methanol/acetonitrile, 2:1:1, v/v/v) and centrifuged at 20,000g at 4 °C for 3 min. The supernatant was transferred to LC vials for analysis.
Chromatographic separations were carried out using a hydrophilic interaction LC system with an Xbridge amide column (100 × 2.1 mm internal diameter, 3.5 µm; Waters) on the Vanquish Horizon ultra-high-pressure LC system. The column temperature was maintained at 40 °C, the autosampler at 4 °C and the injection volume at 3 µl. The column was used with 5 mM ammonium acetate in water (pH 9.0 with ammonium hydroxide) as MPA and 100% acetonitrile as MPB. The linear gradient was set as follows: 0 min, 85% MPB; 1.5 min, 85% MPB; 5.5 min, 35% MPB; 10.5 min, 35% MPB; 10.6 min, 10% MPB; 14 min, 10% MPB; 14.5 min, 85% MPB and 24 min, 85% MPB. The flow rate was 0.3 ml min−1. MS analysis was performed on an Orbitrap Exploris 480 mass spectrometer equipped with a heated ESI probe. For polar metabolites, the relevant parameters were as follows: heater temperature, 120 °C; sheath gas, 30; auxiliary gas, 10; sweep gas, 3; spray voltage, 3.6 kV for positive mode and 2.5 kV for negative mode. The capillary temperature was set at 320 °C, and S-lens was 55. A full scan range at 60–900 mass-to-charge ratio (m/z) was set. The resolution was set at 60,000 (at 200 m/z). Customized mass calibration was performed before data acquisition using Xcalibur. Metabolite identification and peak integration were carried out using Skyline software (v23.1.0). The integrated peak intensity was used for further data analysis. Relative peak areas for MS data are provided in Supplementary Table 2.
Isotope tracing and IC–MS analysis
The SVF-derived fibroblasts were induced to mature into white adipocytes following the protocol described above. Experiments were performed in a medium containing 10% dialysed FBS and 100% glucose (13C6, Cambridge Isotope Laboratories) isotopomers. DMEM lacking glucose and pyruvate was supplemented with labelled glucose, and all reconstituted experimental media contained 11.1 mM glucose. After 24 h of incubation, the medium was aspirated, and cells were washed twice with ice-cold PBS followed by liquid nitrogen. To determine the incorporation of labelled carbon into intracellular intermediates, we prepared extracts and analysed them by high-resolution MS.
Metabolites from SVF-derived adipocytes (and 3T3-L1 cells) were extracted in 1 ml ice-cold 80:20 (v/v) methanol/water containing 0.1% ammonium hydroxide. Samples were then vortexed for 2 min and centrifuged at 17,000g for 5 min at 4 °C, and supernatants were transferred to clean tubes and evaporated to dryness under nitrogen. Samples were reconstituted in deionized water; then, 10 μl was injected into a Thermo Scientific Dionex ICS-5000+ capillary IC system containing a Thermo IonPac AS11 250 × 2 mm 4-μm column. The IC flow rate was 360 μl min−1 (at 30 °C), and the gradient conditions were as follows: initial 1 mM KOH, increased to 35 mM at 25 min, then to 99 mM at 39 min and maintained at 99 mM for 10 min. The total run time was 50 min. To assist desolvation and increase sensitivity, we delivered methanol through an external pump and combined it with the eluent in a low-dead-volume mixing tee.
Data were acquired using a Thermo Orbitrap Fusion Tribrid mass spectrometer under the negative ESI mode. The heated ESI source and global MS parameters for the acquisition were as follows: scan range = 80–800 m/z; multiplex ions = false; isolation mode = quadrupole; detector type = Orbitrap; Orbitrap resolution = 240,000 (at 200 m/z); radiofrequency lens = 50%; automatic gain control target = 200,000; injection ions for all available parallelizable time = true; maximum injection time = 100 ms; microscans = 1; data type = profile; polarity = negative; source fragmentation = disabled; use EASY-IC = true; include start and end times = false; scan cycle time = 3 s. Raw data files were imported to Thermo TraceFinder software for final analysis. The relative abundance of an isotopologue was calculated as the isotopologue area divided by (normalized by) the sum of all isotopologue peak areas. Relative peak areas for MS data are provided in Supplementary Table 2.
Human WAT samples
scWAT RNAs were obtained from individuals with obesity2 before and after 5% weight loss. Other WAT RNAs were collected as described previously14, and the following inclusion criteria were applied for each of the two groups: (1) persons with a BMI of 18.5–24.9 kg m−2, plasma triglyceride concentration of <150 mg dl−1, fasting plasma glucose concentration of <100 mg dl−1, 2-h oral GTT plasma glucose concentration of <140 mg dl−1, haemoglobin A1c value of ≤5.6% and intrahepatic triglyceride content of <4% were considered to have a metabolically healthy lean status; (2) persons with a BMI of 30–49.9 kg m−2, prediabetes (fasting plasma glucose concentration of ≥100 mg dl−1, 2-h oral GTT plasma glucose concentration of ≥140 mg dl−1 and/or haemoglobin A1c value of ≥5.7%) and intrahepatic triglyceride content of ≥5% were considered to have metabolically unhealthy obesity. Written informed consent was obtained from all individuals before their participation.
ARIC data
Data from ARIC study visit 5 were downloaded to explore interactions between BMI and metabolites in serum from 5,238 participants, after removing individuals with missing data. Untargeted metabolomic profiling was performed by Metabolon using gas chromatography–MS- and LC–MS-based protocols. More than 790 metabolites were analysed using a linear regression model to evaluate interactions of BMI and metabolites with sex (female, male), race (Black, white) and age (continuous) adjusted in the model. The significance threshold was calculated by dividing 0.05 by the number of tests to adjust for multiple comparisons. Interactions with P values less than the threshold were reported as significant. R program 4.3.1 was used for statistical analysis.
AlphaFold prediction modelling
AlphaFold prediction modelling was performed by Creative Proteomics. The CAD sequence, including the CPSII, dihydroorotase and ATCase domains, was downloaded from the UniProt database (PYR1_HUMAN), and NAA structures were downloaded from the Chemical Entities of Biological Interest database. For the analysis, unstructured and low-confidence loop regions were removed from the AlphaFold predicted unrelaxed model. The predicted model was simulated for 1 ns using the GROMACS simulation package. The simulated structure was used in docking studies with NAA. Blind or unbiased docking was performed, followed by targeted, high-stringency docking, using an algorithm set to predict at least nine docking poses. Unbiased docking with aspartate served as the positive control.
Statistical analysis and reproducibility
No statistical methods were used to predetermine sample sizes, and the sample size was determined based on the availability of animals generated from breeding strategies. Data distribution was assumed to be normal, but this was not formally tested. Metabolomic samples were randomized before analysis. WT mice were randomly assigned to water or NAA treatment. All measurements were taken from distinct biological samples. Most but not all investigators were blinded to conditions and underlying genotypes. Analyses of whole-body energy balance and carbohydrate disposal, metabolites and reverse-phase protein arrays were performed by experienced Core research staff who did not know the genotypes. All samples were treated equally, analysed by objective and quantitative methods (adipocyte size, ELISA, image analysis, MS), and compared with controls. Unless otherwise noted, all statistical analyses were performed using GraphPad Prism (version 9) and tests described in the figure legends. All data are presented as mean ± s.e.m. unless otherwise noted. Statistical significance is shown as #P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the findings of this study can be found within the article, extended data or supplementary tables. Source data are provided with this paper.
Code availability
This paper does not report original code.
References
Matalon, R. et al. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am. J. Med. Genet. 29, 463–471 (1988).
Magkos, F. et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 23, 591–601 (2016).
Mersmann, N. et al. Aspartoacylase-LacZ knockin mice: an engineered model of Canavan disease. PLoS ONE 6, e20336 (2011).
Groza, T. et al. The International Mouse Phenotyping Consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 51, D1038–D1045 (2023).
Mina, A. I. et al. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666 (2018).
Wernstedt Asterholm, I., Mundy, D. I., Weng, J., Anderson, R. G. W. & Scherer, P. E. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 15, 171–185 (2012).
Fernandez, S. et al. Adipocyte ACLY facilitates dietary carbohydrate handling to maintain metabolic homeostasis in females. Cell Rep. 27, 2772–2784 (2019).
Huang, Z. et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl Acad. Sci. USA 115, E9499–E9506 (2018).
Zhao, S. et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 17, 1037–1052 (2016).
Hammarstedt, A., Gogg, S., Hedjazifar, S., Nerstedt, A. & Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 98, 1911–1941 (2018).
Luengo, A. et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707 (2021).
The ARIC investigators.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am. J. Epidemiol. 129, 687–702 (1989).
Wright, J. D. et al. The ARIC (Atherosclerosis Risk In Communities) Study: JACC Focus Seminar 3/8. J. Am. Coll. Cardiol. 77, 2939–2959 (2021).
Cifarelli, V. et al. Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J. Clin. Invest. 130, 6688–6699 (2020).
Green, C. R. et al. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 12, 15–21 (2016).
Ben-Sahra, I., Howell, J. J., Asara, J. M. & Manning, B. D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013).
Jones, M. E. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu. Rev. Biochem. 49, 253–279 (1980).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 433 (2021).
Deng, Y. et al. An adipo–biliary–uridine axis that regulates energy homeostasis. Science 355, eaaf5375 (2017).
Perry, R. J. et al. Leptin mediates postprandial increases in body temperature through hypothalamus–adrenal medulla–adipose tissue crosstalk. J. Clin. Invest. 130, 2001–2016 (2020).
Forman, H. J., Waddell, R., Hamilton, P. B. & Grisolia, S. Activation of carbamoyl phosphate synthase by N-acetyl-l-aspartate. Biochem. J. 143, 63–66 (1974).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Graves, L. M. et al. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328–332 (2000).
Goul, C., Peruzzo, R. & Zoncu, R. The molecular basis of nutrient sensing and signalling by mTORC1 in metabolism regulation and disease. Nat. Rev. Mol. Cell Biol. 24, 857–875 (2023).
Blaak, E. E. et al. Impaired fat-induced thermogenesis in obese subjects: the NUGENOB study. Obesity (Silver Spring) 15, 653–663 (2007).
Katzeff, H. L. et al. Metabolic studies in human obesity during overnutrition and undernutrition: thermogenic and hormonal responses to norepinephrine. Metabolism 35, 166–175 (1986).
Lanzola, E., Tagliabue, A., Bozzi, G. & Meroni, G. Obesity, diet and body temperature. Ann. Nutr. Metab. 35, 274–283 (1991).
Segal, K. R. et al. Independent effects of obesity and insulin resistance on postprandial thermogenesis in men. J. Clin. Invest. 89, 824–833 (1992).
Segal, K. R., Blando, L., Ginsberg-Fellner, F. & Edaño, A. Postprandial thermogenesis at rest and postexercise before and after physical training in lean, obese, and mildly diabetic men. Metabolism 41, 868–878 (1992).
Acknowledgements
This work was funded by National Institutes of Health (NIH) grants (F31DK130596 to J.B.F.; R01DK139397, R01DK138018, R01DK126042 and R01DK114356 to S.M.H.). The Reverse-Phase Protein Array Core is supported by the Cancer Prevention and Research Institute of Texas (CPRIT) (RP170005), National Cancer Institute (NCI) Cancer Center Support Grant P30CA125123 and intramural funds from the Dan L. Duncan Cancer Center. Other BCM Core services received support from NCI P30CA125123: Genetically Engineered Rodent Models, Human Tissue Acquisition and Pathology, Metabolomics, and Integrated Microscopy. Additional funding for the Integrated Microscopy Core at BCM was provided by the NIH (grants P30DK56338, P30ES030285, S10OD030414) and CPRIT (RP150578, RP170719). The Metabolomics Core Facility (P.L.L.) is supported by NCI Cancer Center Support Grant P30CA016672. We also acknowledge funding support from the NIH/NCI (R00 CA237618) and a CPRIT Scholar award (PR210056) to X.G. We also want to acknowledge expert input on the energy balance studies and their interpretation provided by A. Banks (Harvard) and R. Perry (Yale).
Author information
Authors and Affiliations
Contributions
J.B.F. and S.M.H. conceptualized the study and designed the research plan. Y.L. and N.S. performed statistical analyses to explore the interactions between BMI and metabolites using ARIC data. J.B.F., P.K.S., E.L.d.G., R.S., E.S.A. and S.M.H. conducted experiments. H.Q., L.T., P.L.L. and X.G. performed mass spectrometry studies. S.K. provided white adipose tissue samples for retrospective analysis and helped interpret metabolic phenotypes in humans and mice. L.A., C.M.B. and C.I.A. provided resources and critical discussion. J.B.F. and S.M.H. wrote the manuscript with editorial input from all authors. S.M.H. is the guarantor of this work and, as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Revati Dewal, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Aspa is highly expressed in differentiated adipocytes and across adipose tissue depots.
a, Aspa gene expression across mouse tissues (n = 3 mice/tissue). Data represent mean ± s.e.m. *P < 0.05, **P < 0.01, ****P < 0.0001 by ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test of brain vs different tissues. Expression of Aspa and adipogenic genes (Adipoq, Fabp4, and Pparg2) in pre-adipocytes (Pread) and mature adipocytes (Adipo) in mice: (b) SVF-derived adipocytes, (c) 3T3-L1 adipocytes (n = 3 biological replicates/group). Data represent mean ± s.e.m. d, Expression of ASPA and adipogenic genes (ADIPOQ, FABP4, and PPARG2) in human primary pre-adipocytes (Pread) and mature adipocytes (Adipo) (n = 3 biological replicates/group). Data represent mean ± s.e.m. e, Relative abundance of NAA and TCA metabolites in 3T3-L1 pre-adipocytes (Pread) and mature adipocytes (Adipo) (n = 5 biological replicates/group). Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values. f, Immunoblot of 3T3-L1 cells and human primary cells probed for ASPA, ADIPOQ, and PPARγ before (-d) and after adipocyte differentiation (+d). HSP90 served as the loading control. g, Immunofluorescent staining of ASPA (red) and co-labeling of lipid droplets (green, LipidTox), and nuclei (blue, DAPI) of primary human adipocytes before and after adipocyte differentiation. h, Correlation between expression of ADIPOQ and ASPA in WAT of people before and after weight loss (WL) (n = 15 samples/group). P-value for Pearson’s r (r) <0.001 by one-sided t-test performed on the linear regression. b-e, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by unpaired two-tailed Student’s t-test. f,g representative of 3 independent experiments.
Extended Data Fig. 2 AspaKO mice display decreased glucose tolerance and plasma leptin levels while maintaining insulin sensitivity.
AspaWT and AspaKO mice were fed NCD for 12 weeks. a, Body weight gain of AspaWT and AspaKO mice over 12 weeks starting at 6 weeks of age (n = 6 WT,10 KO mice/group), shown as a percentage of initial body weight. Data represent mean ± s.e.m. ***P < 0.001 by ordinary two-way ANOVA. b, Glucose tolerance test (GTT) (n = 11 WT,9 KO mice/group) and (c) insulin tolerance test (ITT) (n = 10 WT,9 KO mice/group) with corresponding area-under-curve (AUC) measurements. Data represent mean ± s.e.m. *P < 0.05, **P < 0.01 by mixed-effects model followed by Sidak’s multiple comparisons test. AUC: **P < 0.01 by unpaired two-tailed Student’s t-test. d, Plasma insulin levels following 4 h fast (n = 10 WT,9 KO mice/group). Statistical analysis by unpaired two-tailed Student’s t-test. e, Representative histological analysis by H&E of ad-libitum fed liver across 3-5 fields of view (n = 4 mice/group); scale bars, 100μm. f, Ad-libitum fed liver triglyceride levels (n = 5,4 mice/group). Statistical analysis by unpaired two-tailed Student’s t-test. g, Mice were individually housed and monitored in CLAMS cages during a 96 h period with measurements of energy expenditure (EE). Data represent mean ± s.e.m. Statistical analysis was performed within CalR by ANCOVA with lean body mass as a covariate (n = 7 WT,6 KO mice/group). h, Representative histological analysis by H&E of ad-libitum fed BAT across 3-5 fields of view (n = 4 mice/group); scale bars, 100μm. i, BAT depot tissue weight, shown as a percentage of body weight (n = 11 WT,9 KO mice/group). b-d,f,i Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values.
Extended Data Fig. 3 Broad metabolic pathway alterations occur in WAT from AspaKO mice.
AspaWT and AspaKO mice were fed NCD for 12 weeks and tissues were used for reverse-phase protein array (RPPA) analysis of (a) scWAT and (b) vWAT from AspaWT and AspaKO mice. Relative abundance of (c) AMP and (d) GMP in scWAT and vWAT of AspaWT and AspaKO mice, measured by IC/MS targeted analysis (n = 4 mice/group). Data are mean ± s.e.m. e, Ratio of NAD/NADH across scWAT (n = 8 WT,6 KO mice/group) and vWAT (n = 8 WT,9 KO mice/group). Data are mean ± s.e.m. a-b Data shown as z-score of log2 transformed values and relative changes denoted in the colour bar (n = 4 mice/group). a-e #P < 0.1, *P < 0.05 by unpaired two-tailed Student’s t-test.
Extended Data Fig. 4 Aspa−/− adipocytes shunt glucose-derived carbon into pyrimidines.
a, Schematic of metabolite profiling of SVF-derived differentiated adipocytes generated from AspaWT and AspaKO mice. Glucose incorporation into metabolite pools was measured by incubating cells with U-13C glucose. Isotopolog distribution for indicated metabolites was measured after 24 h using IC-MS targeted profiling. b, Relative abundance of intracellular NAA (n = 3 biological replicates/group). Data represent mean ± s.e.m. ****P < 0.0001 by unpaired two-tailed Student’s t-test. c, Schematic showing contribution of 13C6-labeled glucose tracing into early pyrimidines via aspartate (m + 2 and m + 3 isotopologs), and R5P and downstream pyrimidines (m + 5 isotopologs). G6P, glucose-6-phosphate; 6-PG, 6-phosphogluconate; R5P, ribose-5-phosphate; F6P, fructose-6-phosphate; 3PG, 3-phosphoglycerate. d, Fractional labeling of aspartate, early and later pyrimidines, and the pentose phosphate pathway metabolite, R5P. DHOA, dihydroorotate; OMP, orotidine 5’-monophosphate; UMP, uridine monophosphate. Isotopolog data are corrected for 13C natural abundance (n = 3 replicates/group). Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired two-tailed Student’s t-test. e, Immunoblot of AspaWT and AspaKO primary SVF cells ± differentiation, probed for ASPA, phospho-CAD (S1859), total CAD, phospho-S6K (T389), total S6K, and ADIPOQ. HSP90 served as the loading control. Representative of 3 independent experiments. f, Representative Oil-Red O (ORO) staining of lipid accumulation in differentiated AspaWT and AspaKO SVF adipocytes. Scale bar is 0.783 cm. Panels a and c created with BioRender.com.
Extended Data Fig. 5 AspaKO adipocytes show altered mitochondrial metabolism profiles.
Profiling of mitochondria from differentiated AspaWT and AspaKO primary SVF cells. a, Immunoblot of AspaWT and AspaKO primary SVF cells ± differentiation, probed for markers of mitochondrial complexes I-V (CI-CV). HSP90 served as the loading control. Representative of 2 independent experiments. b, Representative images from high-throughput microscopy (HTM) following immunofluorescent labeling of lipid droplets (green, BODIPY), mitochondria (red, MitoTracker), and nuclei (blue, DAPI) of primary SVF-derived adipocytes (n = 8 replicates/group). Imaging was performed with a 60x objective (2K x 2K pixels2). c, High-content analysis of differentiated adipocytes from HTM: (top) nuclei count, mitochondrial area, mitochondrial fluorescence intensity, (bottom) number of big lipids (denoted as lipid count), big lipid area, and mean fluorescence intensity (n = 8 replicates/group). Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values. *P < 0.05, **P < 0.01, by unpaired two-tailed Student’s t-test. d, Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) with corresponding area-under-curve (AUC) measurements. (n = 12 replicates/group/experiment, 2 independent experiments). Data for kinetic assessments of OCR and ECAR are mean ± s.e.m. AUC measurements are represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median.
Extended Data Fig. 6 ASPA expression governs postprandial body temperature.
a, Schematic of fast/re-feeding experiment. Body temperature measurements and plasma of AspaWT and AspaKO male mice were taken following 16 h fast and after 6 h of refeeding. Necropsy data from fasted and refed mice: (b) Body weight, (c) vWAT and (d) scWAT depot weights, shown as % of body weight. Fasted (n = 5 WT,5 KO mice/group), refed (n = 6 WT,9 KO mice/group). e, Body temperature measured by rectal probe. Fasted (n = 10 WT,12 KO mice/group) and refed (n = 10 WT,12 KO mice/group). For ΔT (n = 10 WT,12 KO mice/group), data are mean ± s.e.m.,*P < 0.05 by unpaired two-tailed Student’s t-test. f, Plasma leptin levels. Fasted (n = 9 WT,7 KO mice/group), refed (n = 8 WT,6 KO mice/group). g, Immunoblot of scWAT from AspaWT and AspaKO mice following fasting and refeeding, probed for phospho-CAD (S1859), total CAD (tCAD), DHODH, phospho-S6K (T389), total S6K (tS6K), and ADIPOQ. HSP90 served as the loading control (n = 3 mice/group/condition). b-f Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by ordinary two-way ANOVA followed by Fisher LSD tests. Panel a created with BioRender.com.
Extended Data Fig. 7 Generation of the mouse model for conditional Aspa gene deletion.
a, Aspafl/fl mice were generated using CRISPR/Cas9 gene editing. Exon 2 of the Aspa gene was targeted by sgRNAs designed complementary to intronic sequences flanking the exon, followed by insertion of loxP sequences by DNA donor oligonucleotides. b, Validation of floxed allele by genotyping. PCR analyses of floxed alleles at the targeted loci in genomic DNA extracted from ear clips of wild-type (+/+), fl/+, and fl/fl mice. PCR products were run on agarose gels with expected band sizes: wild-type (+) 217 bp and loxP allele (fl) 251 bp. Representative of 5 independent experiments. c, Immunoblot validation of adipocyte-specific Aspa deletion in scWAT vs liver, probed for ASPA, PPARγ, and HSP90 (n = 3 mice/group). d, Body weight measurements of Aspafl/fl and AspafKO male mice fed NCD over 12-weeks starting at 6 weeks of age; shown as percent weight gain (n = 7 Aspafl/fl,10 AspafKO mice/group). Data represent mean ± s.e.m. No significant difference across genotypes, determined by mixed-effects model. e, vWAT and (f) scWAT depot tissue weights, shown as a percentage of body weight (n = 7 Aspafl/fl,6 AspafKO mice/group). Statistical analysis by unpaired two-tailed Student’s t-test. Mice were individually housed and monitored in CLAMS cages during a 96 h period with measurements of: (g) Energy expenditure (EE); (h) Respiratory exchange ratio (RER); (i) Food intake n = 5 Aspafl/fl,5 AspafKO mice/group. Statistical analyses of (g, h, i) were performed by ANCOVA with lean body mass as a covariate. j, Representative histological analysis by H&E and mean adipocyte size (μm2) of vWAT and scWAT across 3-5 fields of view (n = 4 mice/group); scale bars, 50 μm. ****P < 0.0001 by unpaired two-tailed Student’s t-test. Necropsy data from fasted and refed Aspafl/fl and AspafKO male mice: (k) vWAT and (l) scWAT, shown as % of body weight. Fasted (n = 13 Aspafl/fl,11 AspafKO mice/group and refed (n = 10 Aspafl/fl,10 AspafKO mice/group. m, Plasma insulin levels in fasted and refed Aspafl/fl and AspafKO male mice (n = 6 mice/group). k-m Statistical analyses by ordinary two-way ANOVA followed by Fisher LSD tests. No significant difference across genotypes or after fasting or re-feeding. e,f,i-m Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values.
Extended Data Fig. 8 Adipose tissue Aspa deletion does not affect liver pyrimidines.
a, Relative abundance of NAA and pyrimidine metabolites in the liver of Aspafl/fl and AspafKO mice in fasted and refed states. Metabolites were measured by IC-MS analysis (n = 4 mice/group/condition) and shown as log10 transformed values normalized to Aspafl/fl fasted levels. b, Representative histological analysis by H&E of livers of Aspafl/fl and AspafKO mice in fasted and refed states across 3-5 fields of view (n = 4 mice/group/conditon); scale bars, 100μm. c, Fasted and refed liver triglyceride levels (n = 4 mice/group/condition). Data represented as box-and-whisker plots using the Min-to-Max method in GraphPad Prism: box limits, 25th to 75th percentiles; center line, median; whiskers, minimum and maximum values. *P < 0.05, **P < 0.01, ****P < 0.0001 by ordinary two-way ANOVA followed by Fisher’s LSD tests.
Extended Data Fig. 9 Alphafold modeling docks NAA into the ATCase domain of CAD.
a, AlphaFold prediction of entire CAD protein structure based on theoretical modeling using information from UniProt ID PYR1_HUMAN. b, Aspartate transcarbamylase (ATCase) domain of CAD (green, 1918-2225 region) and NAA (deprotonated form) in predicted binding site. c, Binding energy of NAA and aspartate calculated using high-stringency docking studies.
Supplementary information
Supplementary Table 1
qPCR assays and strategy for inserting loxP at exon 2 of the mouse Aspa gene.
Supplementary Table 2
Peak areas for mass spectrometry data.
Source data
Source Data Fig. 1
Statistical source data for Fig. 1.
Source Data Fig. 2
Statistical source data for Fig. 2.
Source Data Fig. 3
Statistical source data for Fig. 3.
Source Data Fig. 4
Statistical source data for Fig. 4.
Source Data Fig. 4
Unprocessed western blot for Fig. 4g.
Source Data Extended Data Fig. 1
Statistical source data for Extended Data Fig. 1.
Source Data Extended Data Fig. 1
Unprocessed western blot for Extended Data Fig. 1e.
Source Data Extended Data Fig. 2
Statistical source data for Extended Data Fig. 2.
Source Data Extended Data Fig. 3
Statistical source data for Extended Data Fig. 3.
Source Data Extended Data Fig. 4
Statistical source data for Extended Data Fig. 4.
Source Data Extended Data Fig. 4
Unprocessed western blot for Extended Data Fig. 4e.
Source Data Extended Data Fig. 5
Statistical source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 5
Unprocessed western blot for Extended Data Fig. 5a.
Source Data Extended Data Fig. 6
Statistical source data for Extended Data Fig. 6.
Source Data Extended Data Fig. 6
Unprocessed western blot for Extended Data Fig. 6g.
Source Data Extended Data Fig. 7
Statistical source data for Extended Data Fig. 7.
Source Data Extended Data Fig. 7
Unprocessed western blot for Extended Data Fig. 7c.
Source Data Extended Data Fig. 8
Statistical source data for Extended Data Fig. 8.
Source Data Extended Data Fig. 9
Statistical source data for Extended Data Fig. 9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Felix, J.B., Saha, P.K., de Groot, E.L. et al. N-acetylaspartate from fat cells regulates postprandial body temperature. Nat Metab 7, 1524–1535 (2025). https://doi.org/10.1038/s42255-025-01334-6
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s42255-025-01334-6
This article is cited by
-
Much ado about NAA-thing
Nature Metabolism (2025)
