
Abstract
Background
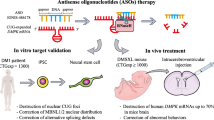
We developed the FORCETM platform to overcome limitations of oligonucleotide delivery to muscle and enable their applicability to neuromuscular disorders. The platform consists of an antigen-binding fragment, highly specific for the human transferrin receptor 1 (TfR1), conjugated to an oligonucleotide via a cleavable valine-citrulline linker. Myotonic dystrophy type 1 (DM1) is a neuromuscular disorder caused by expanded CUG triplets in the DMPK RNA, which sequester splicing proteins in the nucleus, lead to spliceopathy, and drive disease progression.
Methods
Multiple surrogate conjugates were generated to characterize the FORCE platform. DYNE-101 is the conjugate designed to target DMPK and correct spliceopathy for the treatment of DM1. HSALR and TfR1hu/mu;DMSXLTg/Tg mice were used as models of myotonic dystrophy, the latter expresses human TfR1 and a human DMPK RNA with >1,000 CUG repeats. Cynomolgus monkeys were used to determine translatability of DYNE-101 pharmacology to higher species.
Results
In HSALR mice, a surrogate FORCE conjugate achieves durable correction of spliceopathy and improves myotonia to a greater extent than unconjugated ASO. In patient-derived myoblasts, DYNE-101 reduces DMPK RNA and nuclear foci, consequently improving spliceopathy. In TfR1hu/mu;DMSXLTg/Tg mice, DYNE-101 reduces mutant DMPK RNA in muscle, thereby correcting splicing. Reduction of DMPK foci in cardiomyocyte nuclei accompanies these effects. Low monthly dosing of DYNE-101 in TfR1hu/mu;DMSXLWT/Tg mice or cynomolgus monkeys leads to a profound reduction of DMPK expression in muscle.
Conclusions
These data validate FORCE as a drug delivery platform and support the notion that DM1 may be treatable with low and infrequent dosing of DYNE-101.
Plain language summary
Oligonucleotides are small pieces of DNA or RNA that can be used to modify expression of genes. Myotonic dystrophy type 1 (DM1) is a severe disorder caused by an abnormal gene that affects multiple organs, including muscle. We developed the FORCE platform to deliver oligonucleotides to muscle. Here we evaluate the impact of this platform on muscle cells from people living with DM1, myotonic dystrophy mouse models, and healthy non-human primates. Our results show that FORCE can deliver oligonucleotides to muscle and provide beneficial effects in animal models of DM1. In the future, FORCE could potentially be used to treat people living with DM1.
Similar content being viewed by others

Introduction
Oligonucleotides are a powerful approach to treat genetic disorders; an example is the success achieved by intrathecal oligonucleotide infusion for spinal muscular atrophy1. Unfortunately, treatment of genetic disorders that require systemic oligonucleotide administration, including neuromuscular diseases, has been hampered by modest efficacy in target organs2. This limitation was overcome for select liver disorders by lipid nanoparticle formulations or N-acetyl galactosamine (GalNac) conjugation3. The latter enables asialoglycoprotein receptor-mediated uptake, indicating that receptor-mediated delivery may enhance uptake of oligonucleotides in tissues difficult to reach upon systemic administration. Antibody–drug conjugates (ADCs) that bind receptors to enhance delivery of therapeutics have been successful in oncology, and targeting the transferrin receptor (TfR)1 may enable receptor-mediated delivery to muscle4. This is because TfR1 is critical for muscle metabolism and internalization of receptor/transferrin complex is followed by rapid TfR1 recycling to the surface5,6.
Myotonic dystrophy type 1 (DM1) is a severe neuromuscular disease often lethal due to cardiorespiratory complications7. DM1 is caused by the expansion of CTG repeats in DMPK, which are transcribed as CUG triplets in the cognate RNA. The expanded CUG repeats sequester MBNL splicing factors into nuclear foci, thereby causing a spliceopathy, i.e., mis-splicing of multiple transcripts, believed to drive DM1 progression8. Although DMPK RNA downregulation with antisense oligonucleotide (ASO)s to induce release MBNL from nuclear foci and correct spliceopathy is a potential therapeutic approach for DM1, insufficient muscle efficacy has limited clinical translatability9,10,11.
We report the development of the FORCE™ platform and its applicability to DM1. The platform consists of a fragment antigen binding (Fab)–drug conjugate (FDC) with an anti-TfR1 Fab, conjugated to a payload via a cleavable valine–citrulline (Val-Cit) linker. ASOs with gapmer chemistry were chosen as payloads for DM1 because of their effectiveness against nuclear RNAs12. FDCs enhance ASO muscle efficacy in vivo and resolve myotonia in the HSALR model of myotonic dystrophy13. DYNE-101, an FDC under development for the treatment of DM1, corrects molecular defects in multiple DM1 models and achieves remarkable pharmacological effects after infrequent dosing in mice and cynomolgus monkeys.
Methods
Generation of mouse hybridoma cell lines
Novel monoclonal antibodies, cross-reactive against both the human and cynomolgus monkey TfR1, were developed using a traditional mouse hybridoma approach14. BALB/c, NZB/w, CD1, and B6;SJL mice (all from The Jackson Laboratory, Bar Harbor, ME) were immunized with the extracellular domain of human TfR1 and boosted with the extracellular domain of cynomolgus monkey TfR1. B cells from lymph nodes of the immunized animals were fused with myeloma cells to create stable hybridomas. Work was conducted in compliance with Curia Biologics (Albany, NY) Animal Care and Use Committee (IACUC) guidelines and local regulations under IACUC protocol LP-AD.
Hybridoma cloning and selection
Stable hybridomas were seeded at a one-cell per well density in 384-well plates, confirmed by microscopy, and expanded. Subsequently, supernatants were screened by ELISA against human TfR1. Recombinant human TfR1 extracellular domain (R&D Systems, Minneapolis, MN) at a concentration of 2 µg/ml in PBS was seeded on tissue culture plates overnight. Plates were blocked for 1 h with 1% BSA in PBS. Supernatants were added for 1 h, washed with tris-buffered saline, 0.1% Tween 20 (TBST), and monoclonal antibodies (mAbs) were detected using anti-mouse IgG conjugated to horseradish peroxidase. Positive clones were expanded for antibody sequencing and supernatants purified to determine TfR1 selectivity, species cross-reactivity, binding kinetics, baculovirus particle binding, and isotype.
Hybridoma supernatant purification
Supernatants were harvested and purified by affinity chromatography; mAbs with mIgG1 or mIgG2/3 isotypes were purified using protein G or protein A resin, respectively (ThermoFisher Scientific, Waltham, MA). Antibodies were eluted with 50 mM sodium acetate (pH 4.0), neutralized with 1 M Tris-HCl (pH 9.0), and buffer-exchanged into 20 mM sodium citrate, 150 mM sodium chloride (pH 6.0).
Kinetic surface plasmon resonance (SPR)
Experiments to establish the selectivity of antibodies to TfR1 over TfR2 and binding kinetics were performed on Carterra LSA (Carterra, Salt Lake City, UT) at 25 °C. An anti-mouse IgG antibody “lawn” was prepared on a HC30M chip by amine coupling and hybridoma antibodies for screening were captured. Dilution series of hTfR1, cynomolgus monkey TfR1, and hTfR2 were injected to the chip for binding (starting from 1000 nM, 1:3 dilution, 8 concentrations). Binding data were referenced by subtracting the responses from a buffer analyte injection and globally fitting to a 1:1 Langmuir binding model for an estimate of ka (association rate constant), kd (dissociation rate constant), and KD (affinity) using the CarterraTM Kinetics software. Five to six concentrations were used for curve fitting.
ASO sequences
The ASO1 and ASO2 gapmers are modifications of oligonucleotides 486,178 and 445,236 generated against the DMPK and ACTA1 RNAs, respectively9,10. The ASO3 gapmer is fully complementary to both human and cynomolgus monkey DMPK transcripts and identified following in vitro and in vivo potency assessments in rhabdomyosarcoma (RD) cells and TfR1hu/mu; DMSXLWT/Tg mice, respectively. Criteria set out by Gagnon and Corey15 and the GenScript Sequence Manipulation Suite16 were used to generate scrambled ASO control sequences. The ASO3 nucleotide sequence was scrambled to create gapmer ASO4 that lacks homology to human DMPK mRNA, as confirmed by BLAST analysis against the human reference RNA sequence database (refseq_rna; taxid 9606) (Supplementary Data 1).
FAB04 generation
Anti-picric acid monoclonal antibody, IgG2a (BioXCell #BE0089) was diluted v/v with 0.2 M sodium citrate to pH 3.5 to generate digestion buffer, which was incubated for 15 min before cleavage of the fragment crystallizable (Fc) domain. Pepsin-agarose (ThermoFisher Scientific) was washed with 2 × 10 resin bed volumes with digestion buffer and added to the antibody, followed by continuous agitation for 4 h at 37 °C. Solid-supported agarose was filtered and supernatant neutralized with 1 M sodium hydroxide. Digested Fc was removed using a 30-kDa spin filter (Sigma-Aldrich, St. Louis, MO) enabling buffer exchange of the resulting FAB04. FAB04 was incubated with cysteamine-hydrochloric acid 2.5 mM at 37 °C for 90 min, and buffer-exchanged into PBS (pH 7.2) 10 mM ethylenediaminetetraacetic acid by gel filtration.
Generation of antibody–drug conjugate (ADC)1
MAB01 mAb (Supplementary Data 1) was pH-adjusted to 7.5 with sodium hydroxide then mixed with a 2-molar excess of linker-payload for 18 h at room temperature (RT) prior to conjugation with the linker-payload. Reaction completion was confirmed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analytical size-exclusion chromatography. Unreacted mAb was removed using hydrophilic interaction chromatography (Butyl HP, Cytiva Marlborough, MA) with an ammonium sulfate gradient. Pooled fractions were buffer-exchanged into Dulbecco’s PBS (DPBS) and concentrated by amicon centrifugal filter tubes (Sigma-Aldrich). Removal of unconjugated linker-payload was achieved using solute exchange chromatography on AKTA system, HiLoad 16/600 Superdex 200 column, at 1 ml/min DPBS (pH 7.4). The ADC1 fractions were pooled and concentrated using 10 kDa tangential flow filtration (Vivaflow 200; Sartorius, Göttingen, Germany), followed by 0.22 µm sterile filtration (ThermoFisher Scientific). ADC1 (Table 1) concentration was measured by bicinchoninic acid assay (BCA) using the Rapid Gold BCA kit in accordance with the manufacturer’s recommendations (ThermoFisher Scientific). Endotoxin levels were measured using the Endosafe limulus amebocyte lysate kinetic chromogenic assay as recommended by the manufacturer (Charles River Laboratories, Wilmington, MA).
Lead clone humanization
Clone 4 humanization was achieved by matching the murine V sequence of the individual heavy and light chains against the closest human germline V sequence (IGHV4-59/IGKV1-39). Sequence segments across these regions were analyzed to predict liabilities such as post-translational modification sites and promiscuous CD4 + T-cell epitopes17. Site-directed mutagenesis of heavy and light chains was performed to remove predicted liabilities and increase sequence humanness. To select for optimal binding kinetics and maintain human TfR1 affinity, chain variants were selectively combined and transiently expressed to screen by SPR.
Stable cell line generation for recombinant Fab expression
Recombinant Fabs (Supplementary Data 1) were stably expressed in CHO-K1 cells (Genscript, Piscataway, NJ; Abzena, San Diego, CA). Cells were tested for mycoplasma with the MycoAlert kit (Lonza, Walkersville, MD) in accordance with the manufacturer’s recommendations, and validated for recombinant protein expression. Supernatants were harvested and purified by affinity chromatography (Protein G or CaptureSelect CH1-XL; ThermoFisher Scientific, Waltham, MA), eluted with 50 mM sodium acetate (pH 4.0), neutralized with 1 M Tris-HCl (pH 9.0), and buffer-exchanged into 20 mM sodium citrate, 150 mM sodium chloride (pH 6.0).
Accelerated stability assessments
Aggregation and binding studies were carried out to assess changes in FAB02 (Supplementary Data 1) properties following thermal and chemical stress. Thermal stress was evaluated by freeze/thaw cycles and high-temperature incubation. Five cycles of freezing at −80 °C and thawing at RT were performed after completion of cycles 1, 3, and 5. High-temperature stress was performed by incubating FAB02 at 50 °C for 1 week; samples were taken on days 1, 2, 4, and 7. Chemical stress using either 0.5% hydrogen peroxide or 1% ammonium bicarbonate (pH 8.0) was performed at RT, and samples were analyzed after a 24 h incubation.
Size-exclusion chromatography (SEC)
SEC analysis was carried out to determine aggregation of FAB02 under stress conditions. A 1260 Infinity II high-performance liquid chromatography (HPLC) system (Agilent Technologies, Inc., Santa Clara, CA) was used with an ACQUITY UPLC Protein BEH200 SEC column, 1.7 μm, 4.6 × 150 mm (Waters, Milford, MA). Ten-µL samples subjected to the heat and chemical stress conditions were injected onto the column. A 10 min isocratic elution at 30 °C with a 0.35 mL/min flow rate and mobile phase consisting of 0.2 M potassium phosphate, 0.2 M potassium chloride, and 5% isopropanol (pH 6.8) was used to separate any aggregate, product, and degraded fragments. Aggregate and impurity profiles were measured using 260 nm and 280 nm wavelengths to calculate the area under the curve and % peak area.
Generation of linker-payload intermediate
To pre-form the linker-payload intermediate, ASOs bearing a 5’-hexylamine (BioSpring GmbH, Frankfurt, Germany; Supplementary Data 1) were dissolved at 200 mg/mL in nuclease-free water and diluted 1:20 with dimethylformamide and 25 molar excess of tert-butylamine to adjust pH. The bifunctional linker was dissolved in N,N-dimethylacetamide at 30 mg/mL and mixed at a 4-molar excess to the ASO for 120 min at RT. Reaction completion of the linker-payload intermediate was monitored by HPLC. Excess linker reagents were removed by precipitating linker-payload in 15% v/v 3 M sodium chloride and 85% v/v isopropanol and washing the precipitate with dry acetonitrile. Linker-payload concentration was measured by spectrophotometry; purity was evaluated by reversed-phase HPLC.
Conjugation of Fab to linker-payload
Fabs were pH-adjusted to 7.5 with sodium hydroxide then mixed with a 2-molar excess of linker-payload for 18 h at RT prior to conjugation with the linker-payload. Reaction completion was confirmed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analytical size-exclusion chromatography. Unreacted Fab was removed using hydrophobic interaction chromatography (Butyl HP, Cytiva) with an ammonium sulfate gradient. Pooled fractions were diluted with nuclease-free water to reduce conductivity to <8 mS and purified by hydroxyapatite chromatography (Bio-Rad Laboratories, Hercules, CA) to remove unconjugated linker-payload. All FDCs (Table 1) were eluted directly into formulation buffer (100 mM disodium phosphate, 100 mM sodium chloride, pH 7.2) and concentrated using 10 kDa tangential flow filtration (Vivaflow 200; Sartorius, Göttingen, Germany), followed by 0.22-µm sterile filtration (ThermoFisher Scientific). FDC concentration was measured using the Rapid Gold BCA kit. Endotoxin levels were measured using the Endosafe limulus amebocyte lysate kinetic chromogenic assay as recommended by the manufacturer (Charles River Laboratories, Wilmington, MA).
TfR1 binding kinetics of FAB02 and DYNE-101
TfR1 binding kinetics were measured for FAB02 and DYNE-101, which is comprised of FAB02 and ASO3 (Tables 1 and 2), at 25 °C using a disposable microwell plate-powered SPR system (Alto SPR, Nicoya, Kitchener, ON, Canada). Alto sensors were normalized with Alto normalization solutions and cleaned with 10 mM hydrochloride acid according to the manufacturer's instructions. The carboxyl surface was activated by injection of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide for 300 s. One hundred µg/mL of FAB02 and DYNE-101 were prepared in 10 mM sodium acetate (pH 6.0) and oscillated on the active channels for 600 s to obtain mean immobilization levels of 1390 and 379 RU, respectively. One hundred µg/mL streptavidin control prepared in 10 mM sodium acetate (pH 5.0) was oscillated on the reference channels to provide similar mass between the active and reference channels. Sensor surface was blocked with 1 M ethanolamine for 300 s on all channels. Multiple kinetics single-sequence cycles were obtained using either recombinant hTfR1 (R&D Systems, Inc.), cynomolgus monkey TfR1, or hTfR2 (LakePharma, Burlington, MA) as the analyte. Analytes were prepared in PBS with 0.1% Tween (PBST) and loaded into the cartridge. Automated 5-points threefold serial dilutions were performed on-cartridge in PBST with 0.5% BSA from input concentrations (300 nM hTfR1 and cynomolgus monkey TfR1, 10 µM hTfR2). The analytes were passed over the ligands with an association time of 180 s, without dissociation or regeneration between concentrations. The last analyte concentration was followed by a 1000 s dissociation and 60 s regeneration with 10 mM glycine-hydrochloride (pH 1.5). Signals from the reference channels were used for background subtraction. The Nicoya Analyzer was used for 1:1 Langmuir kinetic evaluation.
FDC serum stability
Linker stability was analyzed by incubating FDC1 at 100 nM in PBS or fresh rat, mouse, cynomolgus monkey, or human serum at 37 °C. At selected intervals, serum was subjected to ELISA for FDC1 stability. Briefly, recombinant hTfR1 was seeded overnight at 2 µg/mL. Plates were then blocked for 1 h with 1% BSA in PBS. Serum was added directly to the plate for 1 h. After washing 3x with TBST, Quant-It RiboGreen (ThermoFisher Scientific) was added at 1:2000 dilution to detect intact payload. Relative fluorescence was measured using a Synergy plate reader (BioTek) at 495-nm excitation and 520-nm emission.
In vitro assessments
Cell culture conditions
HeLa cells homozygous null for TfR1 (TfR1−/−; Creative Biogene, Shirley, NY) and wild-type (WT) HeLa cells (ATCC, Manassas, VA) used for internalization assays were grown to 90% confluence in 24 or 96-well plates. RD cells (ATCC) to be used for internalization assays were grown to 90% confluence in 24-well plates. For ASO transfection, RD cells were grown in DMEM with 10% fetal calf serum and 1% penicillin/streptomycin (ThermoFisher Scientific) to 90% confluency in 96-wells. Unconjugated ASO3 or ASO4 were transfected into RD cells using Lipofectamine3000 at a concentration of 1 µM according to the manufacturer’s instructions (ThermoFisher Scientific). Cells exposed to transfection mix without ASOs served as controls.
Primary myoblasts from WT cynomolgus monkeys (Worldwide Primates, Miami, FL), or from a patient with DM1 harboring 380 CTG repeats (32 F; Cook MyoSite, Pittsburgh, PA), as well as immortalized myoblasts isolated from a patient with DM1 carrying 2600 CTG repeats (CL5; L’Institut national de la santé et de la recherche médicale (INSERM) Centre de Recherche en Myologie (CNRS), Institut de Myologie, Paris, France)18, or cardiomyocytes (PromoCell, Heidelberg, Germany) were used. Cells used for analysis of gene expression, or nuclear DMPK foci area measurements were seeded on tissue culture-treated plastic (ThermoFisher Scientific), or black-walled optically clear bottom 96-well plates (Perkin Elmer, Hopkinton, MA), respectively, at a density of 156,000 cells/cm2. Cells were grown in skeletal growth media with a supplementary mix (PromoCell) of 1% penicillin/streptomycin (ThermoFisher Scientific). After 24 h, fusion into myotubes was induced by replacing the culture medium with serum-free Dulbecco’s modified Eagle medium (ThermoFisher Scientific) supplemented with 0.1% insulin (Sigma-Aldrich). Primary cynomolgus monkey myoblasts, 32 F, and CL5 cells were exposed to conjugates immediately after transition to myotube differentiating conditions and harvested 10 days after treatment with no further culture medium replacements. Parallel cultures exposed to PBS served as negative controls. All cultures were conducted in tissue culture incubators (Fisher Scientific, Hampton, NH) at 37 °C in a humidified atmosphere with 5% CO2.
RD and HeLa cells were tested for mycoplasma with the MycoAlert kit (Lonza) in accordance with the manufacturer’s recommendations, whereas myoblasts and cardiomyocytes were not tested. RD, HeLa cells, and cardiomyocytes were not authenticated, whereas myoblasts were validated for their ability to form myotubes and expression of myogenic markers.
FAB02 and DYNE-101 labeling
For imaging experiments, FAB02 was labeled with AlexaFluor 647 (ThermoFisher Scientific). FAB02 was diluted in PBS to 5–10 mg/mL and the pH adjusted to 7.5–8.5 with 0.1 M sodium bicarbonate. An 8-molar excess of AlexaFluor 647 NHS ester was added and incubated at 300 rpm for 2 h at RT in the dark. After incubation, free dye was removed with 7 KDa Zeba desalting column (ThermoFisher Scientific) equilibrated with PBS. Concentrations were determined by measuring at 280 and 495 nm and using the equation: Abs (protein) = Abs (280 nm) – [Abs (494 nm) × (CF)]; CF A647 = 0.03 Abs (protein) × 0.56 = Protein (mg/mL); extinction coefficient of Fab = 0.56 (mg/mL)−1 cm−1. Degree of labeling for FAB02 was 1.59 (dye/Ab).
For flow cytometry experiments, FAB02 and DYNE-101 (Table 1) were labeled with Cypher5e mono NHS ester (Cytiva), a pH-sensitive dye that fluoresces at acidic pH. Briefly, FAB02 and DYNE-101 were diluted to 1 mg/mL in PBS and 0.5 M sodium carbonate (pH 8.3). A 10-molar excess of Cypher5e was added and the mixture incubated in the dark at RT for 1 h. After incubation, free dye was removed with 7 KDa Zeba desalting column (ThermoFisher Scientific) equilibrated with PBS. Concentrations were determined by measuring at 280 and 500 nm and using the equation: Abs (protein)=Abs (280 nm) – [Abs (500 nm) × (CF)]; CF Cypher5e = 0.16 Abs (protein) × 0.56 = Protein (mg/mL); extinction coefficient of Fab = 0.56 (mg/mL)−1 cm−1. The degree of labeling for DYNE-101 was 2.24 (dye/Ab).
In vitro TfR1-mediated internalization assessment by imaging
To confirm TfR1-selective internalization, 24 h after seeding, WT and TfR1−/− HeLa cells were incubated with 500 nM of FAB02-AlexaFlour 647 at 37 °C, 5% CO2 for 4 h. After a PBS wash, cells were fixed with 4% paraformaldehyde (PFA) for 15 min at RT. After three washes in PBS before the addition of 1 mg/ml of wheat germ agglutinin labeled with AlexaFluor (AF)-488 (ThermoFisher Scientific) and incubated at RT for 10 min.
To study TfR1-mediated uptake and trafficking, 24 h after seeding, human cardiomyocytes were incubated with 100 nM of FAB02-AlexaFlour 647 in full media at 37 °C, 5% CO2 for 30 min. Cells were washed with PBS, fixed with 4% PFA for 15 min at RT, washed again with PBS, blocked with 5% BSA and incubated overnight at 4 °C with mouse anti-RAB5A (early endosome marker, Cell Signaling Technology, Danvers, MA). Cells were washed with PBS and incubated with AF488-goat anti-mouse secondary antibody (ThermoFisher Scientific).
After staining, following 3× washes in PBS, Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Newark, CA) was added, and images acquired using the Phenix High Content Imaging System with a 40x water-immersion lens (Perkin Elmer).
In vitro TfR1-mediated internalization of FAB02 and DYNE-101 by flow cytometry
WT and TfR1−/− HeLa cells were washed once with PBS and then treated with 100 nM of Cypher5e-labeled FAB02 or DYNE-101 in complete media for 24 h at 37 °C. Cypher5e is a pH-sensitive dye that only fluorescent at acidic pH, representing localization in endocytic compartments of the cell. Cells were washed once with PBS and harvested in StemPro Accutase (ThermoFisher Scientific). Collected cells were centrifuged at 500× g for 3 min in a V-bottom 96-well plate, washed twice in PBS, and resuspended in 100 µL of PBS for flow cytometry. Flow cytometry was performed using the Attune NxT flow cytometer (ThermoFisher Scientific).
Debris were excluded from gating and cells were gated using forward/side scatter plots and histograms. Single cells (singlets) were then selected on an FSC-A versus FSC-H plot, and fluorescent intensity at 645 nm excitation and 663 nm emission was analyzed (RL-1 channel, Fig. S1). Recorded cell events were ≥25,000 from each of the six replicates. Reported values are median fluorescent intensity normalized to the degree of labeling.
Genetically modified mice
HSALR mice
The HSALR mouse was created by insertion of an ACTA1 transgene harboring 220 CTG repeats within the sequence coding for the 3’-UTR of the ACTA1 mRNA. The repeats were inserted in the last exon, in a position that mirrors the location of the CTG repeats in the DMPK gene13. HSALR mice were generated at the University of Rochester (Rochester, NY) and maintained under IACUC protocol UCAR-2008-011E and Animal Welfare Assurance number D16-00188.
Human TfR1 knock-in mouse generation
To enable muscle uptake of a conjugate designed with a human TfR1-targeting Fab that does not cross-react with mouse TfR1, such as FAB02, a mouse expressing the human TfR1 from the murine Tfrc locus was generated by homologous recombination (Fig. S2). To this end, a gene-targeting vector was designed to insert the human TFRC cDNA in the region of the murine Tfrc locus coding for the start codon of the Tfrc transcript in exon 2, with the remainder of exon 2 plus a 436-bp fragment of the 5’-end of intron 2 deleted. The vector was constructed by cloning the human TFRC cDNA (NM_001128148) and SV40 polyadenylation (pA) sequences into a plasmid containing a neomycin selection cassette (PGK promoter-Neomycin-BGH pA) flanked by flippase-recognition target sites (pL453). The plasmid also contained a diphtheria toxin A negative selection cassette in the vector backbone. Homology arms of 3800 bp (5’) and 4574 bp (3’), generated by PCR using genomic DNA from C57BL/6N embryonic stem (ES) cells (Primogenix, St. Louis, MO), were cloned upstream of the TFRC cDNA and downstream of the neomycin selection cassette, respectively. The 5’- and 3’-homology arms correspond to genomic coordinates chr.16:32,428,209–32,432,008 and chr.16:32,432,481–32,437,054, respectively, according to mouse genome assembly GRCm39/mm39. The sequence of the final vector was confirmed by DNA sequencing. The vector was linearized with SalI (New England Biolabs, Ipswich, MA) and electroporated into C57BL/6N ES cells using the 4D-Nucleofection System with the X-unit platform (Lonza). ES cells were tested for mycoplasma with the IMPACT1 kit (IDEXX BioAnalytics, Westbrook, ME) in accordance with the manufacturer’s instructions. Colonies resistant to G418 were isolated and screened using long-range PCR assays across both the 5’ and 3’ homology arms. PCR-positive clones were validated by Southern blotting using internal and external probes to verify correct targeting. Correctly targeted ES cell clones were injected into blastocysts from B6(Cg)-Tyr c-2J/J mice (Charles River Laboratory, Wilmington, MA) for chimera production. Further authentication of ES cells was not deemed necessary given the ability of their ability to form chimeras following injection into blastocysts. Germline transmission of the targeted allele (TfR1hu) and removal of the neomycin selection cassette was achieved by mating male chimeras to female Rosa26-FlpO mice on a C57BL/6N genetic background (TransViragen, Research Triangle Park, NC). Generation of TfR1hu/mu mice was conducted in compliance with TransViragen IACUC and local regulations under IACUC protocol 15–278.0 and Animal Welfare Assurance number D16-00256. TfR1hu/mu mice were transferred and maintained at Biomere Biomedical Research Models (Biomere, Worcester, MA) in compliance with Biomere IACUC and local regulations under IACUC protocol 20-18 and Animal Welfare Assurance number D16-00632.
hTfR1;DMSXL mice
Mice expressing human TfR1 from one allele of the murine Tfrc locus (TfR1hu/mu) were crossed with DMSXL mice harboring a human DMPK locus containing a 1000 CTG expansion obtained from INSERM, CNRS, Institut de Myologie19. Although DM1 is an autosomal dominant disease in humans, DMSXL hemizygous mice (DMSXLWT/Tg) do not present with a DM1 phenotype despite expression of the mutant human DMPK. Conversely, homozygous female, but not male, mice (DMSXLTg/Tg) exhibit nuclear mutant human DMPK foci and splicing defects that partially overlap with those in DM1 patients20. Therefore, use of TfR1hu/mu;DMSXLWT/Tg mice of both sexes was deemed appropriate to investigate the ability of DYNE-101 to downregulate mutant human DMPK, whereas use of TfR1hu/mu;DMSXLTg/Tg female mice only was necessary to test whether DYNE-101 suppresses mutant human DMPK RNA expression, reduces DMPK foci area, and corrects splicing defects. TfR1hu/mu;DMSXLWT/Tg and TfR1hu/mu;DMSXLTg/Tg mice were maintained at Biomere (Worcester, MA) in compliance with Biomere IACUC and local regulations, under IACUC protocol 20-18 and Animal Welfare Assurance number D16-00632.
FDC2 and unconjugated ASO2 administration to HSALR mice
Male and female homozygous HSALR mice between ~6 and 10 weeks of age were randomized prior to tail intravenous (IV) injection with vehicle, 10 or 20 mg/kg ASO2, or 10 or 20 mg/kg ASO-equivalent doses of FDC2. Mice were euthanized 28 days after injection, and muscles were transferred into RNAlater (ThermoFisher Scientific) immediately after dissection and maintained at 4 °C for 48 h. Tissues were frozen in dry ice and stored at −80 °C. This study was conducted in compliance with the University of Rochester (Rochester, NY) IACUC and local guidelines under IACUC protocol UCAR-2008-011E and Animal Welfare Assurance number D16-00188.
In vivo studies in WT, hTfR1 hu/mu, TfR1 hu/mu;DMSXL WT/Tg, and TfR1 hu/mu;DMSXL Tg/Tg mice
To establish the antibody format of FORCE, WT male mice at ~5 weeks of age were administered by tail IV injection with 10 mg/kg ASO-equivalents of FDC4 or ADC1. The latter consists of MAB01 a commercially available anti-TfR1 antibody (BE0175, BioXCell) conjugated to ASO1. To determine whether FDCs are superior to unconjugated ASO and act via a TfR1-mediated uptake mechanism in vivo, WT male mice at ~5 weeks of age were administered by tail IV injection with 10 mg/kg ASO-equivalents of ASO1 or FDC5. Mice administered with vehicle served as controls for baseline Dmpk mRNA expression. All mice were sacrificed 14 days after injection to collect the gastrocnemius and tibialis anterior. To confirm that FORCE enhances efficacy of unconjugated ASOs, male and female hTfR1hu/mu mice between ~10 and 15 weeks of age were administered by tail IV injection on days 0 and 7 with 10 mg/kg of unconjugated ASO1 or with an ASO-equivalent dose of FDC1. Mice administered with formulation buffer served as controls for Dmpk expression. Mice were sacrificed 14 days after the first injection to collect heart, diaphragm, and tibialis anterior. This initial work aimed to establish whether TfR1 targeting is a viable approach for ASO delivery to muscle and impact on disease biology was not a consideration; therefore, use of male, but not female, mice was deemed appropriate to minimize the number of animals.
To determine whether DYNE-101 acts via a TfR1-mediated mechanism in vivo, male and female TfR1hu/mu;DMSXLWT/Tg mice between ~6 and 9 weeks of age were administered by tail IV injection on days 0 and 7 with 10 mg/kg of FDC6. Mice administered with formulation buffer served as controls for baseline DMPK mRNA expression. All mice were sacrificed 14 days after the first injection to collect the tibialis anterior and gastrocnemius muscles. To explore the ability of DYNE-101 to modify DM1 disease, TfR1hu/mu;DMSXLTg/Tg female mice between ~6 and 10 weeks of age were administered with vehicle or 10 mg/kg ASO-equivalent doses of DYNE-101 on days 0 and 7 by tail IV injection. Littermate-matched TfR1hu/mu female mice administered with vehicle were used to define baseline composite splicing index in muscle. Mice were euthanized one month after the first injection to collect selected muscles. To explore DYNE-101 dose regimens, TfR1hu/mu;DMSXLWT/Tg mice of both sexes between ~6 and 8 weeks of age received either a single 5 or 10 mg/kg ASO-equivalent dose of DYNE-101, or 4 monthly doses at 5 or 10 mg/kg ASO-equivalents. Sex-matched TfR1hu/mu;DMSXLWT/Tg littermates administered with vehicle served as controls. Mice were euthanized one month after the last injection to collect selected muscles. Dissected muscles were transferred into RNAlater immediately after dissection and maintained at 4 °C for 48 h. Tissues were frozen using dry ice and stored at −80 °C. These studies were conducted in compliance with Biomere IACUC and local regulations, under IACUC protocols 15-03, 13-08, or 20-18, and Animal Welfare Assurance number D16-00632.
In vivo studies in cynomolgus monkeys
To compare the efficacy of FDCs to unconjugated ASO delivery in higher species, unvaccinated male cynomolgus monkey (M. fascicularis) of Chinese origin between ~2 and 4 years of age were used. Cynomolgus monkeys do not manifest clear sexual dimorphism at 4 years of age or younger; therefore, to minimize the number of non-human primates, only males were used21,22. Prior to dosing, monkeys were acclimated to restraining chairs for 3 days, and food was withheld overnight. For the duration of the study, food was provided daily in amounts appropriate for the size of the animal, and tap water provided ad libitum via an automatic watering device; each holding cage was cleaned once daily. Conscious monkeys were infused under restraint into the saphenous vein with either vehicle, unconjugated ASO1 10 mg/kg, or a 10 mg/kg ASO-equivalent dose of FDC3. Infusions were well-tolerated with no reactions or notable clinical observations. Monkeys were euthanized 2 weeks after infusion in accordance with local regulation and procedures. Euthanized monkeys were subjected to cardiac perfusion with heparinized saline, and fragments ~40 mg in weight collected from the gastrocnemius, soleus, deep flexor, biceps, ileum, heart, diaphragm, masseter, quadriceps, jejunum, kidney, spleen, and liver. Work was conducted in compliance with Biomere IACUC and local regulations, under IACUC protocol 15-03, and Animal Welfare Assurance number D16-00632.
Studies to determine the effects of DYNE-101 in higher species were conducted in cynomolgus monkeys of Chinese origin between ~1 and 1.5 years of age were conducted at Primate Products, LLC (Immokalee, FL). Prior to dosing, monkeys were acclimated to restraining chairs for three days, and food withheld overnight. Food was provided daily in amounts appropriate for the size of the animal and tap water provided ad libitum via an automatic watering device; each holding cage was cleaned once daily. Conscious monkeys were infused under restraint into the saphenous vein with either a single 5 or 10 mg/kg ASO-equivalent dose of DYNE-101, or 2 monthly doses at 5 or 10 mg/kg ASO-equivalents. Monkeys administered with vehicle served as controls. DYNE-101 was well-tolerated with no infusion reactions or notable clinical observations. Monkeys were euthanized one month after the last infusion. Euthanized monkeys were subjected to cardiac perfusion with heparinized saline, and ~40 mg tissue fragments collected from muscles of interest. Dissected muscle was transferred into RNAlater immediately after collection and maintained at 4 °C for 48 h. Tissues were then frozen using dry ice and stored at −80 °C. Studies were conducted in compliance with Primate Products, LLC IACUC and local regulations, under protocols DYT-2011 7116 or DYT2107 7172-21, and Animal Welfare Assurance number D16-00590.
Analysis of gene expression
RNA extraction and reverse transcription
Total RNA was extracted from cell monolayers with the PureLink Pro 96 Total RNA Purification kit in accordance with the manufacturer’s instructions (ThermoFisher Scientific). Dissected muscles from HSALR, TfR1hu/mu;DMSXLWT/Tg, and TfR1hu/mu;DMSXLTg/Tg mice, as well as cynomolgus monkeys administered with DYNE-101, and respective controls were homogenized in Qiazol on a refrigerated Precellys bead mill homogenizer (Bertin Technologies, Rockville, MD). Total RNA was subsequently extracted from Qiazol homogenates with the RNeasy 96 kit, in accordance with the manufacturer’s instructions (Qiagen). Dissected muscles from WT, TfR1hu/mu and cynomolgus monkeys administered with FDC3, ASO1, and respective controls were subjected to total RNA extraction with the Maxwell RSC simplyRNA Tissue Kit using the Maxwell RSC 48 instrument (Promega) in accordance with the manufacturer’s protocol. Purified total RNA was reverse-transcribed with Qscript complementary DNA SuperMix (Quantagene, Santa Monica, CA).
qPCR
Gene expression was analyzed by qPCR in cDNA generated from total RNA extracts with specific TaqMan assays (ThermoFisher Scientific; Table S1). Log-fold changes in RNA expression in human cells, TfR1hu/mu TfR1hu/mu;DMSXLTg/Tg and TfR1hu/mu;DMSXLWT/Tg mice, and cynomolgus monkeys were calculated with the 2-ΔΔCT method23 using PPIB as the reference gene. Primers and probe sets targeting ACTA1 were designed using the ThermoFisher Scientific custom TaqMan assay design tool (Table S2)24,25,26. Specificity was tested in skeletal muscle from WT and HSALR mice. Log-fold changes in RNA expression using Gtf2b as a reference were calculated to determine ACTA1 expression in HSALR mice.
A panel of qPCR assays (ThermoFisher Scientific) was used to assess the effect of DYNE-101 on splicing markers relevant to DM1 pathology in humans in patient-derived cells or TfR1hu/mu;DMSXLTg/Tg mice (Tables S3 and S4)19,20. We elected to use qPCR to assess changes in splicing due to the sensitivity of the technique, as previously reported27,28. Exon inclusion for splicing markers was expressed as log-fold changes in levels of mRNA isoform containing the mis-spliced exon, calculated according to the 2-ΔΔCT method using total transcript expression from the same gene as reference. Cells exposed to PBS and TfR1hu/mu mice treated with vehicle served as the controls for data analysis.
Composite splicing index calculations
Quantification of alternative splicing in the HSALR mice was performed as described29. Reads were aligned to reference sequences of inclusion and exclusion splice isoforms. Isoform-specific reads with unambiguous alignments were counted. Exon percent spliced-in was calculated for each event as the fraction of inclusion-isoform reads relative to all inclusion and exclusion-isoform reads. Overall splicing derangement was calculated using the mouse DM1 splicing index as previously described29.
Electromyography myotonia assessment
Myotonia was assessed in the skeletal muscle of HSALR mice by EMG and graded by a blinded examiner on a 4-point scale: 0, no myotonia; 1, myotonic discharge in <50% of needle insertions; 2, myotonic discharge in >50% of needle insertions 3, myotonic discharge with nearly every needle insertion30.
Taqman assays used for splicing analysis in TfR1 hu/mu;DMSXL Tg/Tg female mice
Inventoried TaqMan assays were purchased from ThermoFisher Scientific. Custom TaqMan assays for Ldb3 and Ttn exon inclusion were designed using the ThermoFisher Scientific custom TaqMan® assay design tool. The primer sets were designed to produce a product only if the target exon is included in the transcript. The primer and probe sets for Ldb3 and Ttn exon inclusion were tested in skeletal muscle from WT and TfR1hu/mu;DMSXLWT/Tg mice.
Only splicing events displaying variance between TfR1hu/mu and TfR1hu/mu;DMSXLTg/Tg mice were included in composite splicing index calculations (Table S5). To determine the composite splicing index, we calculated magnitude of relative changes in exon inclusion between TfR1hu/mu and TfR1hu/mu;DMSXLTg/Tg female mice treated with vehicle. To do so, the reciprocal of relative changes in splicing markers with reduced exon inclusion in the TfR1hu/mu;DMSXLTg/Tg female mice was calculated. Relative changes (x) were normalized according to this formula:
(x–[median expression in TfR1hu/mu mice])/([95th percentile in TfR1hu/mu;DMSXLTg/Tg female mice treated with vehicle]–[median expression in TfR1hu/mu mice])29. Composite splicing index was calculated as average of normalized relative splicing changes per tissue per mouse. When splicing defects are analyzed according to this technique, composite splicing indices close to 0 or 1 indicate the absence or presence of DM1 splicing defects, respectively.
Subcellular fractionation of muscle
Nuclear and cytoplasmic fractions were obtained from skeletal muscle of TfR1hu/mu;DMSXLWT/Tg mice one month after a single dose of DYNE-101 delivering 10 mg/kg of ASO by following a previously published protocol, with modifications31. Flash-frozen gastrocnemius was dissected and homogenized with a Cryolys Evolution homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) in sucrose Tris magnesium (STM) buffer consisting of 250 mM sucrose, 50 mM Tris-HCl (pH 7.4), 5 mM magnesium chloride, and Halt™ Protease inhibitor cocktail (ThermoFisher Scientific). Homogenates were incubated on ice for 30 min and then mixed for 15 s. Homogenates were centrifuged at 4 °C for 15 min at 1000× g to separate the nuclei from the cytoplasm. The nuclear precipitate was resuspended in cold STM buffer and mixed by agitation for 15 s. Nuclei were centrifuged at 500× g for 15 min at 4 °C to remove cell debris. The nuclear pellet was resuspended in cold STM buffer with protease inhibitors and nuclear purity assessed using trypan blue and a hemacytometer. Purified nuclei were centrifuged at 4 °C at 1000× g for 15 min and resuspended in cold STM buffer. Following centrifugation, the cytoplasmic supernatant was recovered into a separate 1.5 mL tube on ice. The cytoplasmic supernatant obtained after sedimentation of the nuclei from the crude homogenate was centrifuged at 800×g at 4 °C for 10 min. The supernatant was recovered and centrifuged at 4 °C at 11,000× g for 10 min then subjected to centrifugation at 4 °C at 12,000× g for 5 min to recover the cytoplasmic fraction. Purified fractions were stored at −80 °C prior to analysis.
Western blot analysis from subcellular fractions
The total protein content of each fraction was quantified using a Pierce™ rapid gold BCA kit (ThermoFisher Scientific) according to the manufacturer’s instructions. Six micrograms of total protein were heat denatured at 95 °C for 10 min in the presence of NuPAGE™ sample reducing agent (ThermoFisher Scientific) and 1× NuPAGE™ LDS sample buffer (ThermoFisher Scientific), followed by rapid cooling on ice for 2 min. Samples were loaded onto a NuPAGE™ 4–12%, Bis-Tris gel and separated by electrophoresis at 150 V for 45 min at RT. Proteins were transferred to PVDF membranes using an iBlot 2 gel transfer device (ThermoFisher Scientific) according to the manufacturer’s instructions. The membrane was blocked with Intercept® blocking buffer (TBS) (Li-Cor, Lincoln, NE) for 1 h at RT with continuous agitation. The blots were incubated in primary antibody (rabbit anti-histone H3; 1:1000 or rabbit anti-GAPDH; 1:1000) (Cell Signaling Technology) in blocking buffer at 4 °C with rocking overnight. The following day, the blots were washed at RT followed by a 1 h incubation in secondary antibody (anti-rabbit IgG, HRP-linked; 1:10,000) (Cell Signaling Technology) in blocking buffer at RT with continuous agitation. Blots were washed to remove excess secondary antibodies were developed using SuperSignal™ west femto maximum sensitivity substrate (ThermoFisher Scientific) and were imaged using the iBright imaging system (ThermoFisher Scientific).
DMPK foci detection and confocal microscopy
In vivo detection via in situ hybridization chain reaction (HCR)
In situ HCR was conducted with a probe set containing 30 complementary sequences for the human DMPK transcript (Molecular Instruments, Los Angeles, CA)32. Formalin-fixed, paraffin-embedded heart sections from TfR1hu/mu;DMSXLWT/Tg female mice were sectioned at a 7 µm thickness and mounted onto glass slides (HistoWiz, Brooklyn, NY). HCR was performed per the manufacturer’s protocol (Molecular Instruments). Stacks of confocal images collected as an average of 2 frames, scan zoom 0.5×, 16-bit, 2048 × 2048 pixels, were acquired on the Z-plane using the LSM800 confocal microscope with a plan-apochromat 20×/0.8 M27 objective (Zeiss, White Plains, NY) at a step size of 1 µm. Laser wavelengths of 405 nm and 640 nm and bandpass filters of 400–580 nm and 645–700 nm were used to acquire signals of DAPI and AlexaFluo-647, respectively. Laser power, detector gain, detector offset, and detector digital gain were kept consistent when imaging slides from the same staining session. Two random fields of view without empty space were acquired from each biological sample and both images used for foci analyses.
ImageJ 1.53c (Java 1.8.0_172 64 bit) was used for image processing and analysis. Maximum intensity z-projection images were split into single-channel images, DAPI for nuclei and AlexaFluo-647 for DMPK and binary images were then created using Otsu thresholding method. The threshold of DMPK images was adjusted to the criterion that 95% of foci particles are captured in the thresholded image. The “Analyze particle” function of ImageJ was used to measure the total area of foci, as well as the total area of nuclei. The “Image calculator” function was used to measure the relative foci area within the nuclear area.
In vitro detection via in situ hybridization with peptide nucleic acid (PNA) probe
To measure the area of mutant DMPK nuclear foci in myotubes from DM1 patients, 32 F and CL5 cells seeded on optically clear plastic were fixed in 4% formalin, permeabilized with 0.1% Triton X 100 (Sigma-Aldrich) and hybridized at 70 °C with a cytidine-adenine-guanosine (CAG)5 PNA probe conjugated to a cyanine 5 (Cy5) fluorophore (PNA Bio, Thousand Oaks, CA) that hybridizes with the CUG repeats in the mutant DMPK RNA. Nuclei were counterstained with DAPI.
Fixed cells stained with the (CAG)5-Cy5 PNA probe and DAPI were imaged on an Opera Phenix spinning disc laser confocal imaging system. Nine fields per well were acquired at a magnification of ×400 with a water-immersion ×40 objective lens; a total of 6 optical slices at a thickness of 1 µm were collected for each field, so that final images captured an optical slice 6 µm thick. For each image, Cy5 and DAPI channels were acquired independently to avoid fluorophore cross-excitation. Excitation with a 405 nm 50 mW solid-state emission laser coupled with a 435–480 nm bandpass emission filter was used to image nuclei stained with DAPI, whereas excitation with a 640 nm 40 mW solid-state emission laser coupled with a 650–760 nm bandpass emission filter were used to image DMPK foci stained with the (CAG)5-Cy5 PNA probe.
Image analysis was conducted with Harmony software v 4.9 (Perkin Elmer) on a two-dimensional maximum projection of six individual images per field, nine fields were analyzed for each well. An analysis routine was developed to measure nuclear area in the DAPI channel and DMPK foci area in the Cy5 channel and calculate the ratio of foci area over nuclear area. Data are expressed as percent change compared to vehicle-treated cells.
ASO detection in muscles
Total ASO3 levels in muscle tissues were quantified by hELISA, as described33. The assay was performed by coating NeutrAvidin-coated plates with a capture probe specific to ASO3 (Integrated DNA Technologies, Coralville, IA). Collected tissues lysates were digested with proteinase K and subsequently incubated at 30 °C for 30 min with shaking to allow binding of total ASO3 to the capture probe. Plates were washed and the unbound capture probe was digested with micrococcal nuclease at 30 °C for 28 min with shaking. Plates were subsequently washed followed by the addition of a digoxigenin horseradish peroxidase (HRP)-conjugated antibody in Superblock buffer (ThermoFisher); plates were incubated at 30 °C for 30 min with shaking to bind to the intact capture probe. Plates were washed, then incubated with 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (R&D Systems, Inc., Minneapolis, MN) at 30 °C for 20 min with shaking in the dark. Stop solution was subsequently added to the plates and the absorbance at 450 nm by the 3,3’,5,5’-tetramethylbenzidine diamine generated by catalysis of TMB by HRP was quantified on a SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA). Quantification of total ASO3 concentrations was performed using a serial dilution of ASO3 at known concentrations from 0 to 580 pM, diluted in muscle tissue matrix (BioSpring Gmbh).
Statistics and reproducibility
The sample size for initial exploratory studies with FDC4 was established a priori based on available data on in vitro and in vivo gapmer ASO activity in cell lines and muscle tissue, respectively. Subsequent power analysis revealed that n = 3 was the minimum number of observations to detect a statistically significant (alpha = 0.05) with 95% power for a ~50% effect size. The statistical significance of differences in Cy5 intensity in RD cells treated with ASO1 or FDC1 was determined by unpaired t test. Statistical significance of differences in DMPK expression between vehicle-, FDC-, ADC-, or ASO-treated RD cells, and WT, TfR1hu/mu, as well as TfR1hu/mu;DMSXLWT/Tg mice, and cynomolgus monkeys was determined by one-way ANOVA followed by uncorrected Fisher’s LSD test. Statistical significance of differences in ACTA1 expression and overall splicing derangement between vehicle-, FDC-, ADC-, or ASO-treated HSALR mice was determined by one-way ANOVA followed by uncorrected Fisher’s LSD test. Statistical significance of differences in EMG myotonia between vehicle-, FDC-, ADC-, or ASO-treated HSALR mice was determined by Kruskal–Wallis test with Dunn’s multiple comparisons test. Statistical significance of differences in DMPK RNA expression and nuclear foci are between vehicle- and DYNE-101-treated groups in TfR1hu/mu;DMSXLTg/Tg female mice was determined by unpaired t test. Statistical significance of differences in splicing between vehicle- and DYNE-101-treated TfR1hu/mu and TfR1hu/mu;DMSXLTg/Tg female mice was determined by one-way ANOVA followed by uncorrected Fisher’s LSD test. Statistical significance of differences in DMPK expression and BIN1 exon 11 inclusion between vehicle- and DYNE-101-treated 32 F or CL5 cells was determined by one-way ANOVA followed by Dunnett’s post-hoc analysis. The statistical significance of differences in experimental groups as well as number and identity of observations is indicated in figure legends. Outlier analysis was conducted on ΔΔCt values of gene expression in HSALR mice as well as TfR1hu/mu and TfR1hu/mu;DMSXLTg/Tg female mice by robust regression and outlier removal (Q = 2%)34. All statistical analyses were conducted using GraphPad Prism v 9.2.0 (GraphPad Software, San Diego, CA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Murine anti-TfR1 mAb generation and FORCE delivery modality selection
Human and cynomolgus monkey TfR1 cross-reactive mAbs were generated using the mouse hybridoma technique. Antibody clones were screened by ELISA SPR for both TfR1 affinity and selectivity to exclude human TfR2 binding. Clones that did not compete with TfR1 ligands transferrin or HFE were prioritized based on human TfR1 binding affinity and cynomolgus monkey cross-reactivity. To determine the ability of prioritized antibody clones to deliver payloads to myoblasts and ensure that conjugation preserves payload activity, purified mAbs were conjugated to ASO1. This is a gapmer ASO that induces human, cynomolgus monkey, and murine DMPK RNA ribonuclease (RNase)H-mediated degradation10. Clone 4 was selected for humanization and immunogenic potential reduction based on the efficacy of the cognate FDC in DM1 patients or cynomolgus monkey myoblasts (Table S6).
To establish the format of FORCE, we compared FDC4 and ADC1 (Table 1) in WT mice. A single IV administration of ASO-equivalent doses of FDC4 or ADC1 led to a ~60–70% or ~25–45% reduction, respectively. Therefore, Fab was chosen as delivery modality. A single dose of FDC4 was then compared to ASO-equivalent doses of unconjugated ASO1 or non-TfR1-targeting FDC5 in mice. Only FDC4 reduced Dmpk RNA in skeletal muscle, demonstrating that TfR1-mediated delivery enhances ASO efficacy (Fig. 1).

WT male mice were administered by tail IV injection on day 0 with 10 mg/kg ASO-equivalents of a FDC4 (blue bars), ADC1 (green bars) or vehicle (gray bars), or b ASO1 (red bars), FDC5 (teal bars), and FDC4 (blue bars), and sacrificed on day 14 for analysis of gene expression. Total RNA extracted from the indicated muscles was reverse-transcribed and Dmpk RNA expression was measured by qPCR using Ppib as the reference gene. Expression in each muscle from mice injected with FDC4 or ADC1 was normalized to Dmpk expression in the corresponding muscle of mice administered with vehicle. Data are means + SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual mouse.
Humanization and developability assessments
To mitigate anti-drug antibody responses, Clone 4 was germline-matched to constant and framework regions of the human IGHV4-59/IGKV1-39 homolog, promiscuous major histocompatibility complex II binding was characterized in silico and both chains constant regions were mutated to mitigate putative CD4+ T-cell epitopes17. The resulting Fabs were prioritized to minimize predicted post-translational modifications and rescreened for TfR1 affinity and selectivity. FAB02 was selected (Table 2) and then subjected to chemical and thermal stresses (Table S7). FAB02 remained stable and only monomeric structures were detected by size-exclusion chromatography, demonstrating the absence of aggregation (Fig. 2). Consequently, FAB02 was chosen for FORCE.

Representative size exclusion-ultra performance liquid chromatography traces from 10 µL injections of 1.0 mg/ml FAB02 native or previously subjected to stressed conditions. Thermal stress was evaluated by freeze/thaw cycles (−80 °C to room temperature (RT)) (a) and to high temperature (50 °C) (b). Chemical stress consisted of forced oxidation with 0.5% hydrogen peroxide (c) and by exposure to 1% ammonium bicarbonate (d) at RT at the indicated time points.
Conjugation optimization
A multi-step process was developed to generate stable intermediates and allow flexibility in choice of payloads. A cathepsin-cleavable Val-Cit linker was chosen because of its serum stability and clinical use35,36. Titration of activated Val-Cit payload intermediate resulted in ~1 drug antibody ratio, with Fab conversion efficiencies >80% (Fig. S3). FDCs were purified by hydrophobic interaction chromatography followed by mixed-mode hydroxyapatite chromatography to remove unconjugated Fab and payload, respectively. Membrane-based filtration for formulation and concentration produced high yields and purity >95%.
Linker stability in vitro
Linker stability was evaluated in serum from mice, rats, cynomolgus monkeys, and humans, using FDC1 (Table 1). Intact and functional FDC1 was measured using a 2-site ELISA by controlling for TfR1 engagement and detection of payload as a surrogate measure of linker stability. The linker was ≥90% intact in serum across species over 72 h (Fig. S4). Since Fabs have a <2 h circulating half-life in humans, these data suggest that linker stability in circulation will not be a concern37.
FDCs enhance ASO activity in muscle via TfR1
Selectivity and high affinity for TfR1 are crucial to ensure muscle delivery while reducing targeting of TfR238. SPR revealed that FAB02 retained the nM affinity of the WT murine clone for cynomolgus monkey and hTfR1 (Table 2). TfR1 selectivity was maintained after humanization, with no appreciable hTfR2 binding (Table 2). Flow cytometry and confocal microscopy demonstrated that FAB02 is internalized in the endosomal pathway, as expected for TfR1-mediated uptake (Fig. 3)39.

Representative images of WT and TfR1−/− HeLa cells (a) treated for 4 h with FAB02 labeled with AlexaFluor 647 (pseudocolored to green). Cells were stained with wheat germ agglutinin labeled with AlexaFlour 488 (membrane, pseudocolored to red,) and DAPI (nuclei). White bars within images indicate size. b WT (blue bars) and TfR1−/− (black bars) HeLa cells were treated for 24 h with FAB02 labeled with a pH-sensitive dye (CypHer5E). Cells were harvested and analyzed by flow cytometry for median fluorescent intensity normalized to the degree of labeling (Dye/Ab). Data are medians + SD. Each circle represents a technical replicate from two independent experiments. c Representative images of human cardiomyocytes treated for 30 min with FAB02 labeled with AlexaFluor 647 (pseudocolored to green), fixed, stained with DAPI (nuclei) and RAB5 (30 min sample, red). The image with merged AlexaFluor 647, RAB5, DAPI signals is an enlargement of the white boxed area. White bars within images indicate size.
Preliminary in vivo studies with a FAB02-ASO1 conjugate (FDC1; Table 1) were conducted in mice heterozygous for the TfR1hu allele cloned into the Tfrc locus (TfR1hu/mu). TfR1hu/mu mice administered IV with FDC1 exhibited >70% reduction in Dmpk RNA in muscle compared to mice injected with vehicle. An ASO-equivalent dose of unconjugated ASO1 displayed minimal efficacy, demonstrating that FAB02 enhances oligonucleotide activity in muscle (Fig. 4).

TfR1hu/mu mice were injected via tail vein on days 0 and 7 with vehicle (gray bars), 10 mg/kg ASO1 (red bars), or FDC1 delivering 10 mg/kg ASO-equivalent of ASO1 (blue bars) and sacrificed 14 days later for analysis of gene expression. Total RNA extracted from the indicated muscles was reverse-transcribed and Dmpk RNA expression was measured by qPCR using Ppib as the reference gene. Expression in each muscle from mice treated with FDC1 or ASO1 was normalized to Dmpk expression in the corresponding muscle of mice treated with vehicle. Data are means + SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle or between ASO1 and FDC1 are indicated with exact P values. Each circle represents an individual mouse.
To explore the pharmacology of FDCs in higher species, cynomolgus monkeys were infused IV with vehicle or with ASO-equivalent doses of unconjugated ASO1 or FAB03-ASO1 conjugate (FDC3; Table 1). FAB03 has similar selectivity, affinity, and binding kinetics to TfR1 compared to FAB02 (Table S8). FDC3 led to ~35–70% DMPK suppression in muscle, whereas ASO1 displayed modest inconsistent effects. ASO1, but not FDC3, reduced DMPK expression in spleen and liver, suggesting that FORCE conjugation mitigates ASOs pharmacodynamics in off-target organs (Fig. S5).
Conjugation to TfR1-targeting Fab enhances the functional benefit of ASOs in HSALR mice
To determine whether conjugation to a TfR1-targeting Fab improves ASO efficacy in a model of myotonic dystrophy, HSALR mice with muscle-specific expression of a transgenic human ACTA1 RNA containing ~220 CUG repeats were used. HSALR mice are a well-established system to estimate benefit of DM1 therapeutic approaches13,29. Like in DM1, pathology in HSALR mice is driven by expanded CUG repeats that sequester MBNL1, leading to spliceopathy and myotonia in skeletal muscle13. Since FAB02 does not cross-react with murine TfR1 and the HSALR mouse phenotype is driven by ACTA1 RNA, we generated the surrogate FAB01-ASO2 conjugate (FDC2; Table 1 and Supplementary Data 1). FAB01 targets murine TfR1 with a similar affinity and kinetic profile to FAB02 and demonstrably enhances ASO efficacy in muscle40. ASO2 is a gapmer ASO that induces RNAseH-mediated degradation of ACTA1 and reverses the HSALR mouse phenotype upon frequent administration41.
HSALR mice were injected with single-ascending IV ASO-equivalent doses of FDC2 or unconjugated ASO2. FDC2 led to ~60% reduction of ACTA1 expression in muscle, whereas ASO2 only had modest effects. More robust ACTA1 downregulation by FDC2 translated into greater spliceopathy correction compared to unconjugated ASO2. Targeted RNAseq to measure 36 distinct splicing events simultaneously (Supplementary Data 2) revealed that FDC2 dose-dependently corrects spliceopathy in skeletal muscle. Accordingly, FDC2 achieved a dose-dependent correction of myotonia that was nearly complete at the highest dose, whereas unconjugated ASO2 provided minimal benefit (Fig. 5).

HSALR mice were injected IV with vehicle (black bars/circles), 10 or 20 mg/kg unconjugated ASO2 (red bars/circles), or 10 or 20 mg/kg ASO-equivalent doses of FDC2 (blue bars/circles) and sacrificed 28 days later. a ACTA1 RNA expression measured by RT-qPCR of total RNA from quadriceps (top) or gastrocnemius (bottom) using Gtf2b as the reference gene. Expression in each muscle was normalized to ACTA1 expression in the corresponding muscle of mice injected with vehicle. Data are means + SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual mouse. b Overall splicing derangement calculated using murine DM splicing index, horizontal lines are means ± SD, data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual mouse. WT splicing data (green line) previously published29. c Grading of EMG myotonic discharges. Data are means + SEM; data were analyzed by Kruskal–Wallis test with Dunn’s multiple comparisons test, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual mouse.
DYNE-101 is internalized via TfR1 and targets mutant human DMPK RNA
DYNE-101 consists of FAB02 and ASO3, a potent and specific gapmer ASO designed against human DMPK RNA (Table 1 and Fig. S6). SPR and flow cytometry confirmed TfR1-mediated binding of DYNE-101, respectively (Table 2 and Fig. S7). In complementary work, TfR1hu/mu;DMSXLWT/Tg mice that express human TfR1 and a mutant human DMPK RNA with expanded CUG repeats19 were administered IV with ASO-equivalent doses of DYNE-101 or control FAB05-ASO3 conjugate (FDC6; Table 1 and Supplementary Data 1). FAB05 is isotype-matched to FAB02 but does not interact with human TfR1 or known murine proteins42. DYNE-101 reduced mutant human DMPK expression by ~50% in skeletal muscle, whereas FDC6 had no effect, confirming that DYNE-101 acts via TfR1-mediated uptake (Fig. S8).
DYNE-101 improves molecular features of DM1
To determine whether DYNE-101 targets the mutant human DMPK RNA in skeletal muscle nuclei, TfR1hu/mu;DMSXLWT/Tg mice were administered IV once with vehicle or DYNE-101. After one month, the gastrocnemius was subjected to subcellular fractionation and fraction purity documented by the presence of histone H3 and GAPDH (Fig. S9). Nuclear and cytoplasmic fractions were enriched with the nuclear marker Malat1 or cytoplasmic markers Birc5 and Gapdh, respectively, confirming the nuclear localization of DMPK RNA43. DYNE-101 reduced DMPK levels by ~50% in nuclear fractions, demonstrating nuclear targeting of mutant DMPK (Fig. 6).

a TfR1;DMSXL mice enable assessment of DYNE-101 ability to target mutant human DMPK RNA via TfR1-mediated uptake. b DMPK, Malat1, Birc5, and Gapdh expression measured by RT-qPCR in total RNA from nuclear (N) and cytoplasmic (C) fractions from gastrocnemius of TfR1hu/mu;DMSXLWT/Tg mice. Data are means + SD. Each circle represents independent measurements except for analysis of Birc5 and GAPDH expression, where n = 1. c DMPK expression in fractions from gastrocnemius of TfR1hu/mu;DMSXLWT/Tg mice administered with 10 mg/kg ASO-equivalent of DYNE-101 (blue bars) relative to mice injected with vehicle (black bars). Data are means + SD. Each circle represents independent measurements. d DMPK expression measured by RT-qPCR in total RNA from muscles of TfR1hu/mu;DMSXLTg/Tg female mice administered weekly with 10 mg/kg ASO-equivalent of DYNE-101 (blue bars), 28 days after initial injection, relative to expression in corresponding muscles of mice injected with vehicle (black bars). Data are means + SD; data were analyzed by unpaired t test with Welch’s correction, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual mouse. e Representative images of nuclei (blue) and DMPK foci (red) in cardiomyocytes used to calculate foci area. Insets display myofibrils (gray) in greyscale. Total DMPK foci area in heart of TfR1hu/mu;DMSXLTg/Tg mice 28 days after initial injection. Data are means + SD, analyzed by unpaired t test: *P < 0.05. Each circle represents an individual mouse. f Composite splicing index in muscle of TfR1hu/mu mice injected with vehicle (gray circle) or TfR1hu/mu;DMSXLTg/Tg mice injected with vehicle (black circle) or DYNE-101 (blue circle), 28 days after initial injection. Horizontal lines are means ± SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from TfR1hu/mu mice injected vehicle or between TfR1hu/mu;DMSXLTg/Tg mice treated with vehicle and DYNE-101 are indicated with exact P values. Each circle represents an individual mouse.
To establish whether DYNE-101 corrects the DM1 phenotype in vivo, TfR1hu/mu female mice administered IV with vehicle were compared to sex-matched TfR1hu/mu;DMSXLTg/Tg littermates administered with vehicle or DYNE-101. Vehicle-treated mice served as controls to establish composite splicing index in the absence of mutant human DMPK. DYNE-101 suppressed DMPK expression in muscle, and analysis of heart sections stained by in situ HCR revealed a corresponding ~50% reduction in human DMPK foci area (Fig. 6). Evaluation of composite splicing index revealed that TfR1hu/mu;DMSXLTg/Tg female mice, like homozygous DMSXL mice, display a modest splicing phenotype19,20. DYNE-101 led to nearly complete splicing correction, so that except for the heart, no statistically significant differences were noted between TfR1hu/mu;DMSXLTg/Tg female mice treated with DYNE-101 and TfR1hu/mu controls (Fig. 6). Work in myotubes from DM1 patients with 380 or 2600 CTG repeats demonstrated that DYNE-101 reduces DMPK expression and nuclear foci area, thereby correcting BIN1 mis-splicing (Fig. S10). Collectively, these data demonstrate that DYNE-101 has the potential to address DM1 manifestations.
DYNE-101 monthly dosing in TfR1 hu/mu;DMSXL WT/Tg mice achieves durable mutant human DMPK suppression in muscle
To evaluate the dose-response relationship of DYNE-101 on mutant human DMPK expression, TfR1hu/mu;DMSXLWT/Tg mice were administered IV with escalating doses of DYNE-101, or vehicle. DYNE-101 led to a dose-dependent decrease in DMPK expression that achieved maximal levels of 46%, 42%, and 53% in the heart, gastrocnemius, and tibialis anterior, respectively (Fig. 7). Although not statistically significant, a maximal 51% DMPK downregulation was observed in the diaphragm.

Mutant human DMPK expression measured by RT-qPCR in muscle of TfR1hu/mu;DMSXLWT/Tg male and female mice a 1 month after a single IV injection with DYNE−101 (blue bars) or b 1 month following the last of 4 monthly doses at the indicated ASO-equivalent doses (blue bars), relative to expression in the same muscles of mice injected with vehicle (black bars). Data are means + SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle or between DYNE-101 dose levels are indicated with exact P values. Each circle represents an individual mouse.
The effects of prolonged monthly dosing with two dose levels of DYNE-101 were tested in TfR1hu/mu;DMSXLWT/Tg mice. DYNE-101 led to a statistically significant reduction in DMPK expression in all muscles analyzed. In heart and diaphragm, no dose-dependency was noted, whereas in the gastrocnemius and tibialis anterior, the highest dose regimen was most effective and attained a maximal ~60–65% DMPK suppression (Fig. 7). Hybridization ELISA (hELISA) revealed that DYNE-101 distribution in muscle was dose-dependent (Fig. S11).
DYNE-101 pharmacodynamics translate to higher species
To investigate the translatability of DYNE-101 pharmacology to higher species, cynomolgus monkeys were subjected to IV infusions with either vehicle or ascending DYNE-101 doses. DYNE-101 downregulated DMPK RNA to a similar extent in TfR1hu/mu;DMSXLWT/Tg mice of either sex, to reduce unnecessary use of non-human primates we elected to conduct these investigations only in male cynomolgus monkeys. DYNE-101 led to a statistically significant, dose-dependent reduction of DMPK RNA in the heart, and a trend toward reduced DMPK in the gastrocnemius and tibialis anterior. To explore the impact of repeat dosing, monkeys were infused with either vehicle or two ascending monthly DYNE-101 doses. DYNE-101 led to a maximal 32% reduction of DMPK RNA in the heart and ~60–70% suppression in skeletal muscles. A maximal ~50–70% DMPK suppression was measured in the esophagus and duodenum. The lowest dose of DYNE-101 reduced DMPK expression by ~20–55%, a magnitude consistent with the effect of a single higher dose (Fig. 8). DYNE-101 achieved dose-dependent ASO concentrations in muscle (Fig. S12).

WT DMPK expression in muscle of male cynomolgus monkeys a 4 weeks after a single IV infusion with DYNE−101 (blue bars) or b or 4 weeks following the last of 2 monthly infusions (blue bars), relative to expression in the same muscle of monkeys infused with vehicle (gray bars), shown for each ASO-equivalent dose. Data are means + SD; data were analyzed by one-way ANOVA followed by uncorrected Fisher’s LSD test, statistically significant differences from vehicle are indicated with exact P values. Each circle represents an individual monkey.
Discussion
Oligonucleotides are a promising approach to treat genetic disorders; however, their development has been hampered by ineffective tissue delivery. Oligonucleotides are endocytosed in a non-specific manner when delivered in unconjugated form or via mechanisms that require membrane destabilization, leading to a narrow therapeutic index44,45. Antibody–oligonucleotide conjugates are being explored as alternatives to unconjugated strategies46,47. We selected the Fab format for the FORCE platform because of the superior efficacy of FDC compared to ADC in skeletal muscle. The absence of effects on TfR1 trafficking, stability under stressed conditions, blunted immunomodulation, and tissue penetration are further advantages of Fab over mAb5,48,49,50.
The choice of TfR1 to enable myocyte uptake is justified by its expression in muscle and internalization kinetics that allow for quick re-expression at the cell surface via endosomal recycling5,6. Initial work with FDCs in murine systems supports the feasibility of this approach40. FAB02 was selected as the lead Fab for FORCE based on high affinity, specificity, and ability to target human and cynomolgus monkey TfR1 without interfering with iron uptake. FAB02 binding kinetics to TfR1 were developed to optimize delivery to muscle, but it remains to be determined whether the kinetic profile of FAB02 also enables access to other organs51,52,53. FAB02 specificity, binding kinetics to TfR1, and internalization mechanisms were maintained in DYNE-101, demonstrating that the conjugation process preserves Fab function.
The Val-Cit linker was developed for Fab to ASO conjugation based on its favorable safety profile in humans, serum stability, and ability to release the therapeutic payload within the cell upon endosomal cleavage35,54,55. The Val-Cit linker is more stable than the hydrazone linker and cleavage begins in early endosomes prior to trafficking into the lysosome, mitigating the risk of depurination of the oligonucleotide payload in the acidic environment of the lysosome35,56,57. Importantly, the Val-Cit linker facilitates controlled conjugation of diverse payload chemistries, including neutral and charged steric blocking ASOs, and gapmers40. This flexibility enables rational selection of payloads with appropriate mechanisms of action to address the underlying cause of the disease of interest40,58. Other oligonucleotide conjugation strategies have been employed to enhance tissue uptake44. Conjugates using GalNac have been successfully developed for hepatic delivery; however, distribution to extra-hepatic tissues remains a challenge44,59,60,61,62,63,64.
Observations with multiple FDC surrogates and DYNE-101 demonstrated that the biochemical and biophysical profile of FDCs enables TfR1-mediated uptake of oligonucleotides in muscle. In vivo data demonstrated that FORCE leads to superior DMPK reduction in muscle compared to unconjugated ASOs or control FDCs that do not target TfR1. Moreover, Fab conjugation attenuated the pharmacology of ASO in clearance organs such as the liver and kidney, and in the spleen. These data are congruent with the known pharmacological profile of ASOs65, the reported expression profile of TfR1 in cynomolgus monkeys, and with the notion that TfR2, but not TfR1, regulates liver iron metabolism66. In HSALR mice a surrogate FDC led to a strong correction of myotonic dystrophy manifestations in skeletal muscle, whereas unconjugated ASO2 had no meaningful impact. The ability of DYNE-101 to target the mutant DMPK RNA in myonuclei was demonstrated by analysis of muscle nuclear and cytoplasmic fractions from TfR1hu/mu;DMSXLWT/Tg mice. Accordingly, TfR1hu/mu;DMSXLTg/Tg mice administered with DYNE-101 displayed a substantial reduction of mutant human DMPK expression that was associated with a commensurate decrease of DMPK nuclear foci area in the heart. A similar analysis was not conducted in skeletal muscle because the low number of foci detectable in a muscle cross-section of TfR1hu/mu;DMSXLTg/Tg mice did not provide a sufficient dynamic range to assess the effect of DYNE-101 on foci area. Regardless, DYNE-101 corrected the splicing phenotype in both cardiac and skeletal muscle, and this is indicative of restoration of MBNL1 function after degradation of the nuclear mutant DMPK RNA. Findings in mice are consistent with observations in DM1 patient-derived myotubes in vitro, confirming that DYNE-101 targets the mutant human DMPK RNA in the nucleus. Collectively, these data support the notion that DYNE-101 can improve the underlying spliceopathy in individuals with DM1 and provide functional benefit.
Homozygous DMSXL mice express low levels of mutant human DMPK and display a marginal splicing phenotype in muscle, minimal and sporadic myotonia, and require pharmacological challenge to induce modest heart and respiratory defects10,19,67,68. Consequently, it is difficult to directly correlate myotonia improvement to changes in DMPK RNA levels in this model. Therefore, use of the surrogate FDC in HSALR mice was necessary to establish the potential of TfR1-targeting FDCs to improve functional deficits. MBNL1 sequestration by expanded CUG repeats drives spliceopathy in HSALR mice and splicing defects in TfR1hu/mu;DMSXLTg/Tg mice. Therefore, association of spliceopathy with myotonia in HSALR mice and the relationship between splicing and mutant DMPK expression in TfR1hu/mu;DMSXLTg/Tg mice can be correlated to estimate the level of DMPK reduction that may lead to functional benefit. Based on these considerations and the reported data, one can speculate that a 30–50% DMPK downregulation may drive meaningful splicing correction and consequent functional benefit. Of interest, a similar estimate is obtained by analyzing the phenotype of Mbnl1 and Mbnl2 compound-null mice69.
In TfR1hu/mu;DMSXLWT/Tg mice, repeat monthly administration of DYNE-101 reduced mutant human DMPK expression in muscle to a magnitude associated with splicing correction in TfR1hu/mu;DMSXLTg/Tg mice. Repeat monthly dosing was more effective, at a lower dose level, compared to a single dose of DYNE-101, indicating that chronic dosing might be necessary to realize the full therapeutic potential of DYNE-101. Of note, dose-dependency was observed in most muscles except the heart and diaphragm, suggesting that DYNE-101 displays different uptake and clearance kinetics across muscle types. Variable DMPK RNA dynamics across muscles may also explain these observations, given that muscle ASO concentrations were dose-dependent. Although cynomolgus monkeys do not express a disease-causing DMPK RNA, the pharmacology of DYNE-101 appears to translate to higher species. In primates, DMPK downregulation in the heart was not as pronounced as in skeletal muscle. This is likely secondary to the reduced number of doses and shorter time on drug compared to the prolonged dosing regimen experienced by TfR1hu/mu;DMSXLWT/Tg mice. Besides, the magnitude of DMPK RNA suppression in cynomolgus monkey skeletal muscle was comparable to the level of DMPK reduction that corrected DM1 molecular features in TfR1hu/mu;DMSXLTg/Tg mice. Collectively, these observations indicate that DYNE-101 repeat monthly administration has the potential to lead to functional benefit in DM1.
This work has potential limitations. There is no single rodent model of myotonic dystrophy that allows studying the effects of full-length mutant human DMPK downregulation on cardiac or skeletal muscle manifestations of the disease. Although multiple genetically modified mice were developed to explore DM1 pathology in cardiac muscle, the truncated mutant DMPK RNA forms that drive disease in these models are not recognized by the ASO payload in DYNE-10170,71,72,73. The potential benefit of DYNE-101 on DM1 manifestations could not be assessed directly but was only estimated by extrapolating findings in HSALR mice administered with a surrogate FDC. An additional shortcoming of this work is the limited duration of studies in cynomolgus monkeys dosed with DYNE-101. This hindered our ability to establish with certainty whether the blunted DMPK downregulation observed in cardiac muscle was due to short time on drug, or secondary to pharmacodynamic differences between heart and skeletal muscle.
In conclusion, conjugation to a TfR1-targeting Fab enhances ASO efficacy in muscle, and DYNE-101 holds promise as a treatment for DM1.
Data availability
Data for Fig. 1 are available in Supplementary Data 3, data for Fig. 3b are available in Supplementary Data 4, data for Fig. 4 are available in Supplementary Data 5, data for Fig. 5 are available in Supplementary Data 6, data for Fig. 6 are available in Supplementary Data 7, data for Fig. 7 are available in Supplementary Data 8, data for Fig. 8 are available in Supplementary Data 9, data for Table 2 are available in Supplementary Data 10, data for Fig. S4 are available in Supplementary Data 11, data for Fig. S5 are available in Supplementary Data 12, data for Fig. S6 are available in Supplementary Data 13, data for Fig. S7 are available in Supplementary Data 14, data for Fig. S8 are available in Supplementary Data 15, data for Fig. S10 are available in Supplementary Data 16, data for Fig. S11 are available in Supplementary Data 17, data for Fig. S12 are available in Supplementary Data 18, data for Table S5 are available in Supplementary Data 19, data for Table S6 are available in Supplementary Data 20, data for Table S8 are available in Supplementary Data 21. Source data for additional figures and tables will be made upon reasonable request. Some authors are inventors on patents and/or patent applications owned by Dyne Therapeutics, Inc. Dyne Therapeutics, Inc. has material transfer agreements covering materials/methods included in the paper.
References
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. New Engl. J. Med. 377, 1723–1732 (2017).
Dzierlega, K. & Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 27, 407–416 (2020).
Wooddell, C. I. et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 21, 973–985 (2013).
Sheyi, R., de la Torre, B. G. & Albericio, F. Linkers: an assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 14, 396 (2022).
Barrientos, T. et al. Metabolic catastrophe in mice lacking transferrin receptor in muscle. EBioMedicine 2, 1705–1717 (2015).
Ciechanover, A., Schwartz, A. L., Dautry-Varsat, A. & Lodish, H. F. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J. Biol. Chem. 258, 9681–9689 (1983).
Bird, T. D. Myotonic Dystrophy Type 1. GeneReviews, (eds Adam, M. P. et al.) (University of Washington, Seattle, 1993 - 2025).
Thornton, C. A. Myotonic dystrophy. Neurol. Clin. 32, 705–719 (2014).
Pandey, S. K. et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharm. Exp. Ther. 355, 329–340 (2015).
Jauvin, D. et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids 7, 465–474 (2017).
Thornton, C. A. et al. Antisense oligonucleotide targeting DMPK in patients with myotonic dystrophy type 1: a multicentre, randomised, dose-escalation, placebo-controlled, phase 1/2a trial. Lancet Neurol. 22, 218–228 (2023).
Crooke, S. T., Wang, S., Vickers, T. A., Shen, W. & Liang, X. H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 35, 230–237 (2017).
Mankodi, A. et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773 (2000).
Kohler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
Gagnon, K. T. & Corey, D. R. Guidelines for experiments using antisense oligonucleotides and double-stranded RNAs. Nucleic Acid Ther. 29, 116–122 (2019).
Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 (2000).
Perry, L. C., Jones, T. D. & Baker, M. P. New approaches to prediction of immune responses to therapeutic proteins during preclinical development. Drugs R. D. 9, 385–396 (2008).
Arandel, L. et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model Mech. 10, 487–497 (2017).
Huguet, A. et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8, e1003043 (2012).
Nakamori, M. et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 74, 862–872 (2013).
Li, X. et al. Age- and sex-related changes in body weights and clinical pathology analytes in cynomolgus monkeys (Macaca fascicularis) of Mauritius origin. Vet. Clin. Pathol. 51, 356–375 (2022).
Drevon-Gaillot, E., Perron-Lepage, M. F., Clement, C. & Burnett, R. A review of background findings in cynomolgus monkeys (Macaca fascicularis) from three different geographical origins. Exp. Toxicol. Pathol. 58, 77–88 (2006).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Livak, K. J., Flood, S. J., Marmaro, J., Giusti, W. & Deetz, K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. Genome Res. 4, 357–362 (1995).
Vallone, P. M. & Butler, J. M. AutoDimer: a screening tool for primer-dimer and hairpin structures. Biotechniques 37, 226–231 (2004).
Lazaruk, K. et al. The design process of quantitative TaqMan gene expression analysis tools. Applied Biosystems White Paper (Applied Biosystems, 2014).
Freeman, W. M., Walker, S. J. & Vrana, K. E. Quantitative RT-PCR: pitfalls and potential. Biotechniques 26, 124–115 (1999).
Harvey, S. E. & Cheng, C. Methods for characterization of alternative RNA splicing. Methods Mol. Biol. 1402, 229–241 (2016).
Tanner, M. K., Tang, Z. & Thornton, C. A. Targeted splice sequencing reveals RNA toxicity and therapeutic response in myotonic dystrophy. Nucleic Acids Res. 49, 2240–2254 (2021).
Kanadia, R. N. et al. A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 (2003).
Dimauro, I., Pearson, T., Caporossi, D. & Jackson, M. J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes 5, 513 (2012).
Choi, H. M. et al. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 28, 1208–1212 (2010).
Efler, S. M., Zhang, L., Noll, B. O., Uhlmann, E. & Davis, H. L. Quantification of oligodeoxynucleotides in human plasma with a novel hybridization assay offers greatly enhanced sensitivity over capillary gel electrophoresis. Oligonucleotides 15, 119–131 (2005).
Motulsky, H. J. & Brown, R. E. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinforma. 7, 123 (2006).
Doronina, S. O. et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 21, 778–784 (2003).
Baah, S., Laws, M. & Rahman, K. M. Antibody-drug conjugates—a tutorial review. Molecules 26, 2943 (2021).
Bazin-Redureau, M. I., Renard, C. B. & Scherrmann, J. M. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J. Pharm. Pharm. 49, 277–281 (1997).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Trischler, M., Stoorvogel, W. & Ullrich, O. Biochemical analysis of distinct Rab5- and Rab11-positive endosomes along the transferrin pathway. J. Cell Sci. 112, 4773–4783 (1999).
Desjardins, C. A. et al. Enhanced exon skipping and prolonged dystrophin restoration achieved by TfR1-targeted delivery of antisense oligonucleotide using FORCE conjugation in mdx mice. Nucleic Acids Res. 50, 11401–11414 (2022).
Wheeler, T. M. et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115 (2012).
Ferrari, G. et al. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J. Virol. 85, 7029–7036 (2011).
Hutchinson, J. N. et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39 (2007).
Benizri, S. et al. Bioconjugated oligonucleotides: recent developments and therapeutic applications. Bioconjug Chem. 30, 366–383 (2019).
Agrawal, S., Temsamani, J., Galbraith, W. & Tang, J. Pharmacokinetics of antisense oligonucleotides. Clin. Pharmacokinet. 28, 7–16 (1995).
Dugal-Tessier, J., Thirumalairajan, S. & Jain, N. Antibody-oligonucleotide conjugates: a twist to antibody-drug conjugates. J. Clin. Med. 10, 838 (2021).
Malecova, B. et al. Targeted tissue delivery of RNA therapeutics using antibody-oligonucleotide conjugates (AOCs). Nucleic Acids Res. 51, 5901–5910 (2023).
Lesley, J., Schulte, R. & Woods, J. Modulation of transferrin receptor expression and function by anti-transferrin receptor antibodies and antibody fragments. Exp. Cell Res. 182, 215–233 (1989).
Vermeer, A. W. & Norde, W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 78, 394–404 (2000).
Li, Z. et al. Influence of molecular size on tissue distribution of antibody fragments. MAbs 8, 113–119 (2016).
Yu, Y. J. et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 6, 261ra154 (2014).
Kanodia, J. S. et al. Prospective design of anti-transferrin receptor bispecific antibodies for optimal delivery into the human brain. CPT Pharmacomet. Syst. Pharm. 5, 283–291 (2016).
Megan, A. et al. Presidential symposium and presentation of top abstracts. Mol. Ther. 31, 1–794 (2023).
Vaklavas, C. & Forero-Torres, A. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther. Adv. Hematol. 3, 209–225 (2012).
Francisco, J. A. et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 102, 1458–1465 (2003).
Tsui, C. K. et al. CRISPR-Cas9 screens identify regulators of antibody-drug conjugate toxicity. Nat. Chem. Biol. 15, 949–958 (2019).
An, R. et al. Non-enzymatic depurination of nucleic acids: factors and mechanisms. PLoS ONE 9, e115950 (2014).
Sugo, T. et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control Release 237, 1–13 (2016).
Debacker, A. J., Voutila, J., Catley, M., Blakey, D. & Habib, N. Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol. Ther. 28, 1759–1771 (2020).
Matsuda, S. et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 10, 1181–1187 (2015).
Rajeev, K. G. et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem 16, 903–908 (2015).
Cui, H., Zhu, X., Li, S., Wang, P. & Fang, J. Liver-targeted delivery of oligonucleotides with N-acetylgalactosamine conjugation. ACS Omega 6, 16259–16265 (2021).
Wang, Y., Yu, R. Z., Henry, S. & Geary, R. S. Pharmacokinetics and clinical pharmacology considerations of GalNAc3-conjugated antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 15, 475–485 (2019).
Yu, R. Z. et al. Disposition and pharmacokinetics of a GalNAc3-conjugated antisense oligonucleotide targeting human lipoprotein (a) in monkeys. Nucleic Acid Ther. 26, 372–380 (2016).
Geary, R. S., Norris, D., Yu, R. & Bennett, C. F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 87, 46–51 (2015).
Kawabata, H. et al. Regulation of expression of murine transferrin receptor 2. Blood 98, 1949–1954 (2001).
Panaite, P. A., Kuntzer, T., Gourdon, G., Lobrinus, J. A. & Barakat-Walter, I. Functional and histopathological identification of the respiratory failure in a DMSXL transgenic mouse model of myotonic dystrophy. Dis. Model Mech. 6, 622–631 (2013).
Algalarrondo, V. et al. Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul. Disord. 25, 308–320 (2015).
Lee, K. Y. et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 5, 1887–1900 (2013).
Mahadevan, M. S. et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 38, 1066–1070 (2006).
Wang, G. S., Kearney, D. L., De Biasi, M., Taffet, G. & Cooper, T. A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 117, 2802–2811 (2007).
Tylock, K. M., Auerbach, D. S., Tang, Z. Z., Thornton, C. A. & Dirksen, R. T. Biophysical mechanisms for QRS- and QTc-interval prolongation in mice with cardiac expression of expanded CUG-repeat RNA. J. Gen. Physiol. 152, e201912450 (2020).
Rao, A. N. et al. Reversible cardiac disease features in an inducible CUG repeat RNA-expressing mouse model of myotonic dystrophy. JCI Insight 6, e143465 (2021).
Acknowledgements
The authors acknowledge Iva Ivanovska Holder, Ph.D., Jennifer Johnson, and John W. Davis, Ph.D. of Dyne Therapeutics, Inc., for assistance in the preparation of this manuscript, histology work, HCR support, and helpful discussions, respectively; Alice Liang and Margaret Wong, Ph.D. of LakePharma for execution of the TfR1 hybridoma campaign and primary screens; Rafael Jiang of Genscript for recombinant expression and cell line development of selected clones; Duncan Brown, Ph.D. for support with design of ASO3; Geneviève Gourdon, Ph.D., Denis Furling, Ph.D., and Vincent Mouly, Ph.D. of INSERM, CNRS, Institut de Myologie for sharing the DMSXL mice and CL5 cells; Amanda Clark, Ph.D. and Kristin Sapp, M.S. of Biomere Biomedical Research Models for the execution of mouse studies and management of mouse breeding colonies, respectively; Alan W. LaRochelle, B.S. of Biomere Biomedical Research Models and Mario Arciniega of Primate Products LLC for the execution of cynomolgus monkey studies. The study was funded by Dyne Therapeutics, Inc.
Author information
Authors and Affiliations
Contributions
Experimental design: T.W., T.P., N.H., T.E., N.Y., C.D., M.Q., S.H., J.N., C.A.T., R.S., O.I.-B., and S.Z. Data acquisition: T.W., T.P., B.Q., S.S., P.-Y.S., Q.Q., B.V., L.S., T.E., P.T., K.O., B.L., R.R., C.D., R.V., Y.C., J.C., A.W., M.Y., N.H., Z.T., M.K.T., C.A.T., and S.Z. Data analysis: T.W., T.P., B.Q., S.S., P.-Y.S., Q.Q., B.V., L.S., R.R., Y.C., J.C., A.W., M.Y., N.H., T.E., P.T., K.O., B.L., J.H., C.D., M.Q., S.H., J.N., Z.T., M.K.T., C.A.T., R.S., O.I.-B., and S.Z. Data interpretation: T.W., T.P., N.H., T.E., K.O., B.L., C.D., M.Q., S.H., J.N., C.A.T., R.S., O.I.-B., and S.Z. Writing—original draft: T.W. and S.Z. Writing—review and editing: T.W., T.P., B.Q., S.S., P.-Y.S., Q.Q., B.V., L.S., R.R., Y.-A.C., J.C., A.W., M.Y., N.H., T.E., B.L., P.T., N.Y., K.O., B.L., J.H., C.D., M.Q., S.H., J.N., Z.T., M.K.T., C.A.T., R.S., O.I.-B., and S.Z.
Corresponding author
Ethics declarations
Competing interests
T.W., T.P., S.S., P.-Y.S., Q.Q., B.V., L.S., R.R., Y.C., J.C., M.Y., T.E., K.O., N.Y., J.H., C.D., S.H., J.N., O.I-B., and S.Z. are full-time employees of Dyne Therapeutics. B.Q., A.W., N.H., M.Q., P.T., B.L., and R.S. were employed by Dyne Therapeutics at the time the work for this study was conducted and may hold stock and/or stock options. C.A.T. is a member of the Scientific Advisory Board for Dyne Therapeutics, Inc. Z.T. and M.K.T. declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Mohamed Boutjdir and Rajesh Khanna for their contribution to the peer review of this work. Peer review reports are available.
Additional information
This manuscript is dedicated to the memory of Brendan Quinn, Ph.D.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Weeden, T., Picariello, T., Quinn, B. et al. FORCE platform overcomes barriers of oligonucleotide delivery to muscle and corrects myotonic dystrophy features in preclinical models. Commun Med 5, 22 (2025). https://doi.org/10.1038/s43856-025-00733-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-00733-w
This article is cited by
-
Model selection in preclinical nucleic acid therapeutics research
Communications Biology (2026)
-
The current and future landscape of RNA-based therapies and diagnostics
Communications Medicine (2025)
-
Molecular genetics of myotonic dystrophy and the evolution of therapeutic approaches
Journal of Human Genetics (2025)
