
Abstract
Atherosclerosis is the underlying cause of cardiovascular disease. Recent studies have shown that N6-methyladenosine (m6A) modification in macrophages is associated with atherosclerosis progression. However, there is a lack of systemic research on the role of m6A modification in macrophage differentiation and activation during atherosclerosis. Here we conducted multiomics analysis (MeRIP-seq and RNA-seq) of macrophages during their differentiation and activation to elucidate the regulatory network of the m6A spectrum at different stages. Western blot, quantitative PCR (qPCR), RNA-seq and RNA immunoprecipitation (RIP)–qPCR results demonstrated that m6A modification modulates KDM6B expression during macrophage activation. Through co-immunoprecipitation, RIP‒qPCR and genetic perturbation experiments, we revealed that Mettl3/Rbm15 regulates the stability of Kdm6b mRNA and that Kdm6b is required for interacting with and demethylating Jak1 to induce its phosphorylation-mediated macrophage activation. Next, through the analysis of single-cell RNA-seq data and coculture experiments, we revealed that Kdm6b-mediated macrophage activation promoted cytotoxic T lymphocyte cytotoxicity following atherosclerosis progression. Moreover, the systemic use of STM2457, a METTL3 inhibitor, revealed the importance of m6A modification in immune cell infiltration and plaque activation. Finally, we utilized macrophage-specific Kdm6b-knockout mice to determine whether Kdm6b facilitates macrophage and cytotoxic T lymphocyte activation and atherosclerosis. Our findings revealed that m6A modification plays a pivotal role in the upregulation of Kdm6b in response to IFN-γ stimulation, which is essential for the phosphorylation of Stat1-induced macrophage activation-mediated atherosclerosis development.
Similar content being viewed by others


Introduction
Atherosclerosis underlies cardiovascular diseases, including coronary heart disease and stroke, and their associated complications, making it the leading cause of death worldwide1. Recent studies have highlighted the critical role of immune-inflammatory responses, which are characterized by the infiltration of monocytes, macrophages and T cells, in accelerating atherosclerosis progression1. Upon engulfing oxidized lipoproteins via scavenger receptors, peripheral blood monocyte-derived macrophages become activated and secrete numerous proinflammatory factors, recruiting additional immune cells, including T cells, and thereby intensifying inflammation within atherosclerotic plaques2.
As a histone demethylase, KDM6B (also known as JMJD3) specifically targets the trimethylation of lysine 27 on histone H3 (H3K27me3), a repressive epigenetic marker associated with gene silencing3. By removing H3K27me3, KDM6B activates the transcription of genes involved in critical biological processes, including cell differentiation, immune responses and development4. Unlike its homolog UTX (KDM6A), KDM6B is activated by stimuli such as inflammation, infection or cellular stress, positioning it as a dynamic regulator of gene expression in response to environmental cues5,6. In macrophages, Kdm6b induces NF-κB-mediated inflammatory gene transcription, which is critical for amplifying local inflammation in abdominal aortic aneurysms7. Experimental knockout of Kdm6b in foam cells delays profibrotic gene transcription, suggesting that Kdm6b is closely connected with atherosclerosis progression8. Other studies also revealed that downregulation of Kdm6b shifts M1 polarization toward the M2 phenotype (characterized by CD163⁺ markers) of macrophages9. However, whether KDM6B is manipulated during macrophage activation and whether m6A modification regulates KDM6B expression in macrophages during atherosclerosis development are unknown.
N6-methyladenosine (m6A) represents the most prevalent epitranscriptional RNA modification in eukaryotic cells and affects various types of RNA, including mRNAs, tRNAs, noncoding RNAs and microRNAs10. m6A modifications on mRNAs are precisely regulated by a set of ‘writers’, such as METTL3, METTL14 and RBM15, and ‘erasers’, such as FTO and ALKBH511. Research has indicated that the methyltransferase Mettl14 enhances the NF-κB/IL-6 signaling pathway, driving an inflammatory response by modulating Myd88 mRNA stability in macrophages during atherosclerosis12. Furthermore, the RNA-binding protein Matr3 can regulate the depolymerization of the m6A modification complex, which includes Mettl3 and Mettl14, thereby mitigating the mitogen-activated protein kinase (MAPK)-mediated proinflammatory response in atherosclerotic macrophages13. Despite extensive research on the role of m6A modification and its associated factors in atherosclerotic macrophages14,15,16,17, a systematic investigation into m6A modification in monocyte-derived macrophages and its influence on their activation during atherosclerosis progression is still lacking. In this study, our team conducted methylated RNA immunoprecipitation (RIP) sequencing (meRIP-seq) on granulocyte–macrophage colony-stimulating factor (GM-CSF)-treated monocytes and IFN-γ-stimulated macrophages to provide a comprehensive understanding of the dynamic role of m6A modification in macrophage activation and its contribution to atherosclerosis.
Materials and methods
Cell culture
Primary bone marrow-derived monocytes (BMDMs) were isolated from wild-type 6- to 8-week-old C57BL/6 mice. Monocytes were cultured in complete medium supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin and 100 U/ml penicillin in 5% CO2 at 37 °C in a humidified incubator. For macrophage differentiation, GM-CSF (20 ng/ml, cat. no. 315-03-20UG, PeproTech) was added daily for a period of 5 days. For macrophage activation, interferon-gamma (IFN-γ) (20 μg/ml, cat. no. 315-05-100UG, PeproTech) was added for 12 h. Following these distinct stimuli, the cells were collected for further experiments.
Western blotting and co-IP
The cells were collected and lysed on ice. The protein concentration was determined using a BCA protein assay kit (cat. no. P0012, Beyotime), and 5 μg of protein was loaded onto 5–20% gradient sodium dodecyl sulfate–polyacrylamide gel. After transfer onto polyvinylidene fluoride membranes, the blots were incubated with primary antibodies at 4 °C overnight, followed by incubation with specific secondary antibodies (1:5000; goat anti-rabbit IgG H&L, cat. no. ab6721 and cat. no. ab205719, Abcam) for 1 h at room temperature. The immunoreactive signals were visualized using an enhanced chemiluminescence detection kit (cat. no. 32106, Pierce).
For co-immunoprecipitation (co-IP) assays, 1 × 107 macrophages were lysed with cell lysis buffer (cat. no. P0013C, Beyotime) on ice for 30 min. After preclearing by incubation with 5 μl of magnetic beads (cat. no. LSKMGAG02, MERCK) for 1 h, the cell lysates were incubated with anti-Jak1 antibody with rotation at 4 °C overnight. After incubation with 10 μl of magnetic beads for another 1 h at 4 °C with rotation, the magnetic beads were washed three times with cold cell lysis buffer, after which elution was performed using protein lysis buffer. Then, the magnetic beads were mixed with 10 μl of 5× loading buffer (cat. no. P0286, Beyotime) and boiled for 10 min for subsequent western blotting. The primary antibodies used were as follows: mouse anti-STAT1 (1:1000, cat. no. ab239360, Abcam), rabbit anti-phosphorylated STAT1 (p-STAT1, 1:1000, cat. no. 9167, CST), rabbit anti-KDM6B (1:1000, cat. no. 3457, CST), rabbit anti-Jak1 (1:1000, cat. no. 50996, CST), rabbit anti-p-Jak1 (1:1000, cat. no. 74129, CST), rabbit anti-Flag (1:1000, cat. no. 14793, CST), rabbit anti-Myc (1:1000, cat. no. 2276, CST), rabbit anti-panmethylation (1:1000, cat. no. 7315, Abcam), rabbit anti-NSD3 (1:1000, cat. no. 300489, Abcam), rabbit anti-DOT1L (1:1000, cat. no. ab239358, Abcam), rabbit anti-H3K79me3 (1:1000, cat. no. ab208189, Abcam), rabbit anti-H3K36me3 (1:1000, cat. no. 282596, Abcam), rabbit anti-H3K27me3 (1:1000, cat. no. 6002, Abcam), rabbit anti-H3 (1:1000, cat. no. ab1791, Abcam) and rabbit anti-GAPDH (1:1000, cat. no. ab8245, Abcam).
Reverse transcription quantitative polymerase chain reaction (RT–qPCR)
RT–qPCR was performed as briefly described: total RNA was extracted and reversed transcription was performed before qPCR was conducted using the SYBR Green mix kit (cat. no. B110031, Sangon Bio). Primers used were as follows: Kdm6b: F-5′ TGAAGAACGTCAAGTCCATTGTG-3′, R-5′ TCCCGCTGTACCTGACAGT-3′; GAPDH: F-5′ AGGTCGGTGTGAACGGATTTG-3′, R-5′ GGGGTCGTTGATGGCAACA-3′; Rbm15: F-5′ GCGAGTCCGCTGTGTGAAA-3′, R-5′ TCCCCACGAGAACTGGAGTC-3′.
Flow cytometry
Tissue-infiltrating lymphocytes were isolated from mice, and a single-cell suspension was prepared. First, the cells were incubated with an Fc receptor blocking solution (cat. no. 553141, BD) for 15 min at 4 °C to minimize nonspecific binding. Second, the cells were stained with the respective fluorochrome-conjugated antibodies against surface antigens for 30 min at 4 °C. After being washed with phosphate-buffered saline, the cells were incubated with permeabilization buffer (1×, BD) for 30 min and then stained with intracellular antigen-conjugated antibodies for 60 min at 4 °C. Finally, after being washed with permeabilization buffer, the cells were analyzed with a BD FACSCanto II. The data were analyzed via FlowJo X software (TreeStar). The antibodies used were as follows: FVS780 (cat. no. 565388, BD), V450 anti-CD45 (cat. no. 75-0451-U100, clone 30-F11, Tonbo), FITC anti-CD107a (cat. no. 121605, clone 1D4B, BioLegend), FITC anti-CD11b (cat. no. 101205, clone M1/70, BioLegend), PE anti-F4/80 (cat. no. 111603, clone W20065B, BioLegend), APC anti-CD20 (cat. no. 161403, clone QA18A73, BioLegend), BV510 anti-CD3e (cat. no. 100233, clone 17A2, BioLegend), Percp5.5 anti-CD8a (cat. no. 65-0081-U100, clone 53-6.7, Tonbo), PE-cy7 anti-PD1 (cat. no. 109110, clone RMP1-30, BioLegend), PE-cy7 anti-GZMB (cat. no. 25-8898-82, clone NGZB, Invitrogen), V450 anti-CD86 (cat. no. 560377, clone FUN-1, BD), V510 anti-CD80 (cat. no. 104741, clone 16-10A1, BioLegend), PE-cy7 anti-Ly6C (cat. no. 128017, clone HK1.4, BioLegend).
Gating strategies were as follows: (1) for macrophages, gating was based on FSC-A versus SSC-A, FSC-A versus FSC-H, FVS780 versus CD45 and F4/80 versus SSC-H; (2) for cytotoxic T lymphocytes (CTLs), gating was based on FSC-A versus SSC-A, FSC-A versus FSC-H, FVS780 versus CD45 and CD3e versus CD8a.
MeRIP-seq
RIP was conducted with a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (cat. no. 17-700, Millipore), as previously described18. The enriched RNA and input RNA (as controls) were purified and subjected to library preparation and high-throughput sequencing by Novo Co., Ltd. using the Illumina platform. Raw reads in FASTQ format were processed using fastp (v0.19.11) to remove adapter sequences, reads containing more than 10% unknown bases (poly-N) and low-quality reads (defined as those with >50% of bases having a Phred quality score ≤20). Quality metrics, including the Q20, Q30 and GC contents, were calculated. Clean reads were retained for downstream analyses. The mouse reference genome (GRCm38/mm10) and Gene Transfer Format (GTF) annotation files were downloaded from the Ensembl genome database. Reference index files were constructed using BWA (v0.7.12), and clean reads were aligned to the genome using the BWA-MEM algorithm with default parameters. Only uniquely mapped reads were retained for peak calling. m⁶A peaks were identified using the exomePeak R package (v2.16.0), with the matched input RNA serving as the control. Peaks with a q value <0.05 were considered significantly enriched. For motif analysis of m⁶A-enriched regions, HOMER (v4.9.1) was used to identify consensus m⁶A motifs. The identified peaks were annotated to genes using the ChIPseeker. Peaks located in exonic regions were assigned to their corresponding genes, which were considered m⁶A peak-associated genes for downstream functional enrichment analysis. Differential m⁶A peak analysis was performed using the exomePeak R package (v2.16.0). Differentially methylated peaks between experimental groups were identified with a P value <0.05 and a fold change >1 as significance thresholds. Peaks showing statistically significant changes in m⁶A enrichment were defined as differentially methylated regions.
RNA sequencing (RNA-seq)
The macrophages were pretreated with GSKJ1 (cat. no. HY-15648, MCE) before IFN-γ stimulation. Total RNA was extracted using TRIzol reagent (cat. no. 15596026CN, Invitrogen). The RNA samples were sent to Novo Co., Ltd. for sequencing using the Illumina Genome Analyser II platform (Illumina). Raw sequencing data in FASTQ format were first subjected to quality control. Adapter-containing reads, reads with more than 10% unknown nucleotides (N) and low-quality reads (defined as those in which more than 50% of the bases had Phred scores ≤20) were removed using Trimmomatic (v0.39). Quality metrics, including Q20, Q30 and GC content, were calculated, and the resulting clean reads were used for all downstream analyses. For alignment, clean reads were aligned to the mouse reference genome (GRCm38/mm10) using HISAT2 (v2.0.5). The aligned reads of each sample were assembled into transcripts using StringTie (v1.3.3b) in reference-guided mode. Gene expression quantification was performed using featureCounts (v1.5.0-p3) to calculate the number of reads mapped to each gene. Gene expression levels were normalized as fragments per kilobase of transcript per million mapped reads (FPKM) values. Differential expression analysis between two experimental conditions (with biological replicates) was performed via DESeq2 (v1.20.0). A negative binomial distribution model was applied to estimate dispersion and test for significance. The resulting P values were adjusted for multiple testing using the Benjamini‒Hochberg method. Genes with adjusted P values <0.05 were considered differentially expressed. Significant genes were defined on the basis of a corrected P value <0.05 and an absolute fold change ≥2.
MeRIP–qPCR
The meRIP assay was performed as described previously with slightly modifications19. In brief, 10–20 μg RNA was fragmented into 200–300-nt fragments via a 15-min incubation at 70 °C in fragmentation buffer (10 mM ZnCl2 and 10 mM Tris–HCl pH 7.0). For exogenous Kdm6b methylation analysis, RNA fragmentation was omitted to preserve full-length transcripts. Thereafter, the fragmented RNA was incubated with 2–3 μg m6A antibody (cat. no. 202003, Synaptic Systems) at 4 °C overnight, and then the complex was incubated with protein G beads (Invitrogen) overnight at 4 °C. After washing with meRIP lysis buffer (150 mM NaCl, 0.1% NP-40 and 10 mM Tris–HCl), the bound RNAs were recovered by proteinase K digestion, phenol–chloroform extraction and ethanol preparation. One-tenth of the fragmented RNA was used as an input control. The input or bound RNA was subjected to reverse transcription and subsequent qPCR. Relative enrichment was calculated as fold change relative to IgG controls after normalization to input RNA. Primers used were as follows: Kdm6b: F-5′ AATGGAAGAGCGGCGCGCTGCG-3′, R-5′ TCTGTACAGAACTGTAGCAGGACC-3′.
Immunofluorescence (IF) staining
IF staining was performed as previously described20. Primary antibodies, including rabbit anti-JAK1 (1:200, cat. no. ZRB1915, MERCK) and rabbit anti-KDM6B (1:200, cat. no. PA5-22974, Invitrogen), were used. The nuclei were stained with DAPI (cat. no. ab104139, Abcam) and examined with a fluorescence microscope (Olympus).
Oil Red O staining
The macrophages were subjected to pretreatment with GSKJ1 before oxidized low-density lipoprotein (oxLDL) (40 μg/ml, cat. no. YB-002, YiYuan) treatment for 24 h. After they were washed twice with phosphate-buffered saline, the macrophages were stained with Oil Red O working solution (cat. no. D027-1-2, JianCheng) for 10 min and compound staining solution for 10 min. All steps of the staining procedure were conducted at room temperature.
MiloR analysis
MiloR analysis, which is primarily used to identify phenotype-related perturbations at the single-cell level, was performed as previously described (https://github.com/MarioniLab/miloR). In brief, after the samples were refined to capture continuous trajectories, a count matrix from single-cell (sc)RNA-seq was used to perform differential abundance testing using the k-nearest neighbor graph method. The main parameters used are as follows: (1) k = 30 and d = 20 for the buildGraph function; (2) prop = 0.2, k = 30, d = 20 and refined = TRUE for the makeNhoods function; and (3) d = 20 for the calcNhoodDistance function.
Analysis of mRNA stability
Analysis of mRNA stability was performed as described previously21. To determine the half-life of endogenous Kdm6b mRNA, actinomycin D (2 μg/ml, cat. no. SBR00013, MERCK) was added to the cell culture medium after the cells had been transfected with Mettl3 or Rbm15 small interfering RNA for 48 h. Cells were collected at the indicated time points. The total RNA was extracted by TRIzol reagent and subjected to RT–qPCR analysis.
Animal experiments
Macrophage-specific Kdm6b-knockout (CKO) mice were generated via CRISPR–Cas9-based targeting and the cre-loxp (Csf1r-cre + Kdm6b-loxp) system on the Ldlr−/− background by Jicui Biosciences (China). Ldlr−/− mice and C57BL/6 mice were purchased from the Model Animal Research Center of Nanjing. All the mice were housed at the Ningbo University Laboratory Animal Center with controlled temperature and humidity and a 12:12 h dark‒light cycle and were provided water and mouse chow ad libitum.
Wild-type, Ldlr−/− and CKO mice were fed with a high-fat diet (Research diests, D12108c) for 16 weeks to induce atherosclerosis. STM2457 (cat. no. HY-134836, MCE) was administered by intraperitoneal injection at a daily dose of 10 mg/kg. The mice used in the in vitro experiments were euthanized humanely using CO2 gas. The aorta was meticulously dissected from the aortic arch to the iliac bifurcation, opened longitudinally, and stained with Oil Red O as described above. The hearts were then embedded in OCT compound (Tissue-Tek, Sakura), rapidly frozen, and from the aortic sinus, consecutive 5-μm-thick cryosections were meticulously cut. These sections underwent sequential staining with Oil Red O, Masson’s trichrome staining and hematoxylin–eosin staining as we described previously22. All acquired images were rigorously analyzed by a seasoned investigator who was blinded to the specific mouse genotypes; the representative micrographs were quantified for lesion area utilizing ImageJ software. The lesion area in the en face preparations was articulated as a percentage of the overall aortic surface area, consistent with prior methodology. Echocardiograms were obtained using a Vevo 3100 Ultrasound System as we described previously23. Mouse biochemical indicators in serum were analyzed using ADVIA 2120i system. All experimental procedures were approved by the Animal Care Ethics Committee of Ningbo University (application number 13720) and conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Statistical analysis
The data are presented as the means ± standard deviations. Statistical comparisons between two groups were performed using an unpaired two-tailed Student’s t-test, whereas comparisons among multiple groups were analyzed via one-way analysis of variance. A P value of <0.05 was considered statistically significant. All the experiments were repeated at least three times. Statistical analyses were performed with GraphPad Prism software.
Results
Dynamic changes in the m6A spectrum during macrophage differentiation
We initially cultured BMDMs with GM-CSF to induce their differentiation into macrophages (Supplementary Fig. 1a) and performed meRIP-seq at days (D) 0, 3 and 5. As expected, the m6A modification peaks were predominantly found surrounding the stop codon in the mRNA transcripts (Fig. 1a and Supplementary Fig. 1b). Interestingly, m6A modification-related enzymes were almost upregulated following GM-CSF stimulation (Supplementary Fig. 1c). Moreover, both the density of m6A modifications and the number of m6A-containing mRNAs increased following GM-CSF treatment (Fig. 1b–d). Inhibition of m6A modification suppressed macrophage differentiation (Supplementary Fig. 1d), which suggests that m6A modification is crucial for macrophage differentiation. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the differentially expressed genes (DEGs) between the 5D group and the 3D group or between the 3D group and the 0D group revealed that various pathways were regulated, including those related to immunity (antigen processing and presentation, FcγR-mediated phagocytosis and Toll-like receptor signaling), inflammation (JAK–STAT signaling and TNF signaling) and molecular processing (ubiquitin-mediated proteolysis, steroid biosynthesis, lysine degradation and N-glycan biosynthesis) (Fig. 1e). These findings indicate that m6A methylation may participate in the remodeling of immunological activities. A combined analysis of RNA-seq and meRIP-seq revealed a negative correlation between mRNA expression levels and m6A modification, which was particularly pronounced in the group cultured for 5 days (Fig. 1f–h). Notably, the mRNAs of Ccr1, Csf1, Hif1a and Tlr4 were significantly induced after 5 days of stimulation; however, the changes in m6A modifications within these mRNAs were markedly different (Fig. 1i), suggesting that different regulatory mechanisms are involved. Collectively, these results indicate that m6A modifications are involved in the process of macrophage differentiation.

Monocytes were stimulated with GM-CSF to induce macrophage differentiation. MeRIP-seq was performed using monocytes incubated with GM-CSF for 0 days, 3 days and 5 days (0D, 3D and 5D, respectively). a, The distribution of m6A peaks in gene body regions, including the 5′ UTR, CDS and 3′ UTR, among the three groups. b, The density of differential m6A peaks was compared between the indicated groups. c, Bar plot showing the number of mRNAs containing m6A sites in the three groups. d, Venn diagram showing the number of shared genes detected between the indicated groups. e, KEGG pathway enrichment analysis of DEGs was compared between the indicated groups. f,g, Correlation analysis of gene expression levels and changes in m6A modifications in the 3D_vs_0D group (f) or in the 5D_vs_3D group (g). The x axis denotes the change in mRNA (RNA-seq); the y axis denotes the change in m6A peaks (RIP-seq). h, Venn diagram showing the number of shared genes detected from RNA-seq and RIP-seq between the indicated groups. i, Heatmap showing the differences in expression and m6A modifications between the 3D and 0D groups or between the 5D and 3D groups.
Proinflammatory pathways and epigenetic enzymes are specifically manipulated by m6A modification during macrophage activation
To investigate the alterations in the m6A modification landscape during macrophage activation, we conducted meRIP-seq on macrophages after stimulation with IFN-γ. Our findings revealed that, in contrast to the differentiation phase, macrophage activation did not significantly alter the overall density of m6A modifications (Fig. 2a,b and Supplementary Fig. 1e,f). However, m6A modification is also necessary for IFN-γ-induced macrophage activation (Supplementary Fig. 1g,h). KEGG enrichment analysis of DEGs between the IFN-γ and control groups revealed that the PI3K–Akt signaling and Toll-like receptor signaling pathways were upregulated, whereas the PD-1/PD-L1 checkpoint pathway and FcγR-mediated phagocytosis were downregulated (Fig. 2c). A correlation analysis integrating RNA-seq and meRIP-seq data indicated that m6A modification is positively correlated with mRNA expression levels during macrophage activation (Fig. 2d). Notably, cytokine‒cytokine receptor interactions, chemokine signaling and MAPK signaling pathways were consistently regulated at both the mRNA and m6A modification levels (Fig. 2e,f). However, IFN-γ stimulation also induced a specific cluster of genes distinct from those expressed by GM-CSF (Fig. 2g). KEGG enrichment analysis of these genes that were regulated only in the IFN-γ group revealed that the expression of macrophage maturity-related genes, such as Stat1, Cd86, Cxcl10 and Cd274, was specifically modulated during the activation process (Fig. 2h). Intriguingly, a substantial group of epigenetic modification enzymes, including Dot1l, Kdm6B, Kdm5b and Kmt2c, also appeared to be regulated in only the IFN-γ group (Fig. 2i). Because epigenetic modification enzymes regulate gene transcription at the initial stage, we focused on this cluster of genes. To determine the basal expression levels and specificity of these enzymes in macrophages, their expression was also examined via scRNA-seq data from a previous study24. After rigorous integration and reduced dimension analysis, we obtained 16 clusters of immune cells, including macrophages and T cells (Supplementary Fig. 2a–c). As atherosclerosis progresses, the proportions of macrophages and T cells increase (Supplementary Fig. 2d). Unexpectedly, with the exception of Kdm6b and Nsd3, the majority of these enzymes were scarcely expressed in immune cells derived from atherosclerotic plaques, with Kdm6b expression observed predominantly in myeloid cells (Supplementary Fig. 2e). Indeed, IFN-γ stimulation markedly enhanced m6A modification of Kdm6b mRNA and resulted in the upregulation of Kdm6b (Supplementary Fig. 3a–d). Interestingly, the induction of H3K27me3, H3K36me3 and H3K79me3 modifications did not markedly change after IFN-γ stimulation (Supplementary Fig. 3e). Overall, both proinflammatory pathways and the epigenetic enzyme Kdm6b are specifically manipulated by m6A modifications during macrophage activation, highlighting the complex regulatory network that governs immune responses.

Macrophages were stimulated with IFN-γ to induce macrophage activation. MeRIP-seq was performed using macrophages incubated with or without IFN-γ (control and IFN-γ groups). a, The density of differential m6A peaks was compared between the indicated groups. b, Bar plot showing the number of mRNAs containing m6A sites in the two groups. c, KEGG pathway enrichment analysis of DEGs between the indicated groups. d, Correlation analysis of differential genes and m6A modification changes between the IFN-γ group and the control group. The x axis denotes the change of mRNA (RNA-seq); the y axis denotes the change of m6A peaks (RIP-seq). e, KEGG pathway enrichment analysis of genes in the first quadrant of d. f, KEGG pathway enrichment analysis of genes in the third quadrant of d. g, Venn diagram showing the number of overlapping genes among the indicated groups. h, KEGG pathway enrichment analysis of genes detected only in IFN-γ-activated macrophages and not in GM-CSF-treated macrophages. i, Scatter plot showing the fold changes in the mRNA and m6A modification levels of the indicated epigenetic enzyme mRNAs.
KDM6B promotes STAT1 phosphorylation in a demethylase activity-dependent manner
To elucidate the role of Kdm6b in macrophages, we incubated cells with GSKJ1, a specific inhibitor of Kdm6b. Our results revealed that GSKJ1 significantly reduced lipid uptake and reactive oxygen species (ROS) formation in macrophages stimulated with IFN-γ (Fig. 3a,b). We subsequently conducted RNA-seq to assess the impact of IFN-γ and GSKJ1 on gene expression during macrophage activation. As anticipated, IFN-γ stimulation enhanced pathways related to antigen processing and presentation, the cytosolic DNA-sensing pathway and PPAR signaling (Supplementary Fig. 4a–c). However, compared with IFN-γ-only treatment, GSKJ1 treatment attenuated the IFN-γ-induced upregulation of genes involved in FcγR-mediated phagocytosis, dilated cardiomyopathy and extracellular matrix receptor interaction (Fig. 3c). In addition, the expression of molecules associated with antigen presentation (H2-DMa, Cd74 and Ciita), chemokine factors (Ccr5, Ccr2, Cxcl12 and Dock2) and collagen deposition (Col1a1, Col1a2 and Col3a1) decreased with GSKJ1 treatment, indicating a dampening effect on IFN-γ-mediated macrophage activation (Fig. 3d). Furthermore, we applied METAFlux analysis25 to examine metabolic gene changes during GSKJ1 incubation. Figure 3e shows that the IFN-γ-induced upregulation of acyl-CoA hydrolysis and the metabolism of various small molecules, including galactose, folate, glutathione, valine, leucine, isoleucine, lysine, pyrimidine, linoleate, β-alanine and retinol, as well as fatty acid activation and biosynthesis, were counteracted by GSKJ1. These findings suggest that the suppressive effect of GSKJ1 on IFN-γ signaling is not limited to a specific set of target genes but rather has a broad inhibitory effect. Accordingly, we investigated key events in the IFN-γ signaling cascade and discovered that either GSKJ1 or Kdm6b knockdown inhibited the phosphorylation of Stat1 (Fig. 3f and Supplementary Fig. 4d,e).

a, Macrophages were pretreated with GSKJ1 for 30 min, stimulated with IFN-γ for 12 h and subsequently incubated with oxLDL for 24 h. Lipid uptake was evaluated via Oil Red O staining. Scale bars, 200 μm (top) and 100 μm (bottom). b, Macrophages were pretreated with GSKJ1 for 30 min and then stimulated with IFN-γ for 12 h. Mitochondrial ROS production was detected via a ROS assay kit. Green fluorescence indicates the intensity of the ROS. Scale bar, 100 μm. The macrophages were pretreated with GSKJ1 for 30 min and then stimulated with IFN-γ for 12 h, and total RNA was subsequently collected for RNA-seq. c, Cnet plot showing KEGG enrichment pathways identified from DEGs between the IFN-γ-treated group and the GSKJ1 + IFN-γ-treated group. d, Heatmap showing DEGs among the control, IFN-γ and GSKJ1 + IFN-γ groups. e, Heatmap showing the enriched metabolic pathways affected by GSKJ1 treatment in macrophages, with differentially regulated pathways marked in red. f, Left: western blot analysis of Stat1, p-Stat1 and Kdm6b in macrophages pretreated with GSKJ1 for 30 min before they were stimulated with IFN-γ at the indicated times, with Gapdh used as an endogenous control. Right: the quantified results. *P < 0.05.
KDM6B promotes JAK1 phosphorylation by demethylating JAK1
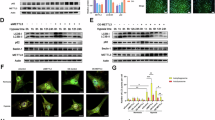
To confirm the role of Kdm6b in Stat1 phosphorylation, we assessed IFN-γ-induced Stat1 phosphorylation over a shorter time frame and obtained consistent results (Fig. 4a). However, because our data revealed no direct interaction between Kdm6b and Stat1 (Fig. 4b), we hypothesized that GSKJ1 might affect kinases upstream of the JAK–STAT signaling pathway. Our subsequent experiments confirmed that GSKJ1 indeed inhibits the phosphorylation of Jak1 induced by IFN-γ (Fig. 4c). Moreover, we found that IFN-γ significantly enhances the interaction between Kdm6b and Jak1 (Fig. 4d). Given that Kdm6b, an epigenetic enzyme, is primarily nuclear, its interaction with Jak1, which is predominantly cytoplasmic, raises questions about its cellular localization dynamics. To test this hypothesis, we performed colocalization staining and discovered that a subset of the Kdm6b protein indeed translocates to the cytoplasm and colocalizes with Jak1 upon IFN-γ stimulation (Fig. 4e). This observation led us to investigate whether Kdm6b demethylates Jak1 to promote its phosphorylation. To test this hypothesis, we conducted co-IP experiments in IFN-γ-stimulated macrophages. Our results revealed that the methylation of Jak1 is reduced and that the phosphorylation of Jak1 is induced after IFN-γ treatment (Fig. 4f), which suggests that this methylation is related to the phosphorylation of Jak1.

a, Macrophages were pretreated with GSKJ1 for 30 min and then stimulated with IFN-γ at the indicated times. Left: western blot analysis of Stat1, p-Stat1 and Kdm6b was performed, with Gapdh used as an endogenous control. Right: the quantified results. *P < 0.05. b, Macrophages were pretreated with GSKJ1 for 30 min and stimulated with IFN-γ at the indicated times. Co-IP assessment of the interaction between Stat1 and Kdm6b for the indicated times using an anti-Stat1 antibody. c, Macrophages were pretreated with GSKJ1 for 30 min and stimulated with IFN-γ at the indicated times. Left: western blot detection of Jak1, phosphorylated Jak1 (p-Jak1) and Kdm6b with Gapdh used as an endogenous control. Right: the quantified results. *P < 0.05. d, Macrophages were pretreated with GSKJ1 for 30 min and stimulated with IFN-γ at the indicated times. Left: co-IP analysis of the interaction between Jak1 and Kdm6b using an anti-Jak1 antibody. Right: the quantified results. *P < 0.05. e, Macrophages were stimulated with IFN-γ for 30 min. IF analysis of Jak1 and Kdm6b in macrophages was performed. Nuclei are visualized with DAPI. Scale bars, 50 μm (left) and 10 μm (right). f, Macrophages were stimulated with IFN-γ for 30 min with or without GSKJ1. Left: co-IP detection of the interaction between Jak1 and Kdm6b using an anti-Jak1 antibody. Right: the quantified results. *P < 0.05.
IFN-γ induces METTL3-dependent m6A modifications on KDM6B mRNA
To determine which m6A writers are involved in the modification of Kdm6b mRNA induced by IFN-γ, we conducted RIP-qPCR. As shown in Fig. 5a, both Mettl3 and Rbm15 interact with Kdm6b mRNA in macrophages under steady-state conditions. However, upon IFN-γ stimulation, there is an increase in the association of Mettl3 with Kdm6b mRNA and a corresponding decrease in the association of Rbm15. Inhibition of METTL3 but not knockdown of Rbm15 eliminated the IFN-γ stimulation-induced m6A modifications of Kdm6b mRNA (Fig. 5b,c and Supplementary Fig. 5a) and reduced its stability (Fig. 5d,e). Furthermore, to explore the downstream effects of m6A modification on Kdm6b mRNA, we performed RIP‒qPCR using antibodies against YTH domain-containing proteins (including Ythdf1/2/3 and Ythdc1/2) and Igf2bp1/2/3 proteins (Fig. 5f and Supplementary Fig. 5b). In conjunction with mRNA degradation assays, we discovered that Ythdc1/2 promotes the stability of Kdm6b mRNA in response to IFN-γ stimulation (Fig. 5g). Indeed, both the mRNA and protein levels of Kdm6b were reduced following m6A modification inhibition or Ythdc1 knockdown (Supplementary Fig. 5c,d). Given that these findings were based on in vitro cellular experiments, we questioned the relevance of m6A-mediated Kdm6b regulation in the context of atherosclerosis in vivo. We initially conducted miloR analysis26 across various time points, including the control, 4-week (w), 8w, 12w, 16w and 26w time points. We identified a significant phenotypic switch, primarily between the 16w/12w groups and the control/4w groups (Fig. 5h,i). We subsequently assessed the transcriptional changes in myeloid cells, which include monocytes and macrophages. As anticipated, we observed an increase in the expression of Mettl3 and Ythdc1/2, whereas the expression of Rbm15 decreased as pseudotime progressed (Fig. 5j).

a, Macrophages were stimulated with IFN-γ for 12 h. An RIP assay was performed to assess the interaction between Kdm6b mRNA and the indicated antibodies in macrophages stimulated with IFN-γ. *P < 0.05. b, Macrophages were pretreated with STM2457 and stimulated with IFN-γ for 12 h. MeRIP-qPCR was subsequently performed. KDM6B mRNA levels were quantified by real-time qPCR. *P < 0.05. c, Macrophages transfected with or without si-Rbm15 were stimulated with IFN-γ for 12 h. MeRIP-qPCR was subsequently performed. KDM6B mRNA levels were quantified by real-time qPCR. *P < 0.05. d, Macrophages were pretreated with STM2457 and stimulated with IFN-γ for 12 h. The RNA stability of Kdm6b mRNA was detected. *P < 0.05. e, Macrophages transfected with or without si-Rbm15 were stimulated with IFN-γ for 12 h. The RNA stability of Kdm6b mRNA was detected. *P < 0.05. f, Macrophages were stimulated with IFN-γ for 12 h. An RIP assay was performed to assess the association between Kdm6b mRNA and the indicated antibodies in IFN-γ-treated macrophages. *P < 0.05. g, Macrophages transfected with or without si-Ythdc1 were stimulated with IFN-γ for 12 h. The RNA stability of Kdm6b mRNA was detected. *P < 0.05. h, Neighborhood assignment map using Louvain clustering. The dashed line encloses the region of neighborhood groups that show distinct clustering patterns of plaque scRNA-seq. i, Beeswarm plot showing the distribution of log fold changes in neighborhoods containing cells from various cell subsets. Neighborhoods with differential abundance at a false discovery rate of 10% are highlighted in color. j, Heatmap representation of DEGs among differentially abundant neighborhoods in myeloid cell subsets isolated from atherosclerotic plaques in mice.
KDM6B-dependent macrophage activation is necessary for CTL cytotoxicity
While CTLs are known to promote atherosclerosis27, we investigated the transcriptional changes in CTLs associated with phenotypic transitions. As anticipated, we observed an upregulation of T cell receptor signaling molecules (Pdcd1, Il2ra, Itm2a, Lck, Cd27 and Cd8a), cytotoxicity-related molecules (Ifngr2, Gzmk, Klrd1, Prf1 and Ifng) and exhaustion markers (Cd160, Eomes, Lag3 and Cd244), which suggests continuous antigen stimulation of CTLs within the plaque during progression (Fig. 6a). Differential gene expression analysis revealed that the antigen-presenting machinery, including Ciita, H2-K1, B2m and H2-D1, was upregulated in a macrophage subset referred to as MC1 (Fig. 6b). Notably, the expression of Ifngr1, the ligand for IFN-γ secreted by CTLs, was also increased (Fig. 6b), which suggests a potential regulatory role of CTLs in this macrophage subset. Connectome analysis28 supported our hypothesis by revealing an increased association between MC1 macrophages and CTLs during the progression of atherosclerosis (Fig. 6c). To explore the role of Kdm6b–IFN-γ signaling in this interaction, we conducted in vitro coculture experiments. We found that IFN-γ-activated macrophages significantly enhanced the cytotoxicity of CTLs through direct coculture with CTLs; however, this effect was reversed by the inhibition of Kdm6b or the suppression of Stat1 phosphorylation (Fig. 6d,e and Supplementary Fig. 6a,b). Furthermore, the ability of Kdm6b to promote macrophage-induced CTL cytotoxicity was dependent on both Stat1 phosphorylation and CD80 signaling (Fig. 6f,g). These results suggest that the interaction between macrophages and CTLs is crucial for the development of atherosclerosis.

a, Heatmap showing the DEGs among differentially abundant neighborhoods in T subsets isolated from atherosclerotic plaques in mice. b, Heatmap showing the DEGs in immune cell subsets isolated from atherosclerotic plaques in mice. c, Chord plot depicting the interactions among immune cell subsets as analyzed by the ComPath method. d, Macrophages were pretreated with GSKJ1 for 30 min and stimulated with IFN-γ for 12 h before being cocultured with CTLs. Flow cytometry analysis of the activated CTL ratio (PD-1+GZMB+ subset). *P < 0.05. e, Macrophages were pretreated with Flu (a STAT1 phosphorylation inhibitor) for 30 min and stimulated with IFN-γ for 12 h before being cocultured with CTLs. Flow cytometry measurement of the activated CTL ratio. *P < 0.05. f, Macrophages were pretreated with both Flu and GSKJ1 for 30 min and stimulated with IFN-γ for 12 h before being cocultured with CTLs. Flow cytometry measurement of the activated CTL ratio. *P < 0.05. g, Macrophages were pretreated with IFN-γ for 6 h, followed by αCD80 and GSKJ1 treatment for 6 h before being cocultured with CTLs. Flow cytometry detection of the activated CTL ratio. *P < 0.05.
Macrophage-specific KDM6B deficiency delays atherosclerosis
To delineate the role of Kdm6b in macrophage-induced atherosclerosis, we generated macrophage-specific Kdm6b-knockout mice on an LDL receptor-deficient (Ldlr−/−) background and used Csf1r-cre to drive recombination in macrophages in Kdm6b-floxed mice. Heart function and major biochemical indicators, with the exception of triglycerides, were comparable among the four groups (Supplementary Fig. 7a,b). En face aorta staining and histological evaluation with hematoxylin and eosin revealed a reduction in the atherosclerotic lesion area in the CKO group compared with both the Ldlr−/− group and the flox control group (Fig. 7a,b). Furthermore, we observed a decrease in the accumulation of collagen fibers (Fig. 7c,d) and in macrophage infiltration (Fig. 7e) in the CKO group. To characterize the phenotype of CTLs within atherosclerotic plaques further, we isolated the plaques and conducted cytometric analysis. As depicted in Fig. 7f,g, both the total ratio and the activation status of CTLs decreased after the macrophage-specific knockout of Kdm6b. Interestingly, systemic use of m6A modification inhibitors also delayed plaque formation, collagen accumulation and macrophage and CTL infiltration (Supplementary Fig. 8a–h). These findings suggest that KDM6B in macrophages contributes to the progression of atherosclerosis partly by promoting the activation of CTLs and IFN-γ-dependent macrophage activation.

a, Representative images and quantification of Oil Red O staining of aorta en face lesions isolated from the NC (Ldlr−/− mice fed with a chow diet), Ldlr−/−, flox and CKO groups (n = 6 per group). *P < 0.05. b, Hematoxylin and eosin staining images and quantification of the necrotic core area in aortic sections from the NC, Ldlr−/−, flox and CKO groups (n = 6 per group). *P < 0.05. c–e, Histological analysis with Masson’s trichrome staining (c), Sirius red staining (d) and MOMA2 (macrophage/monocyte monoclonal antibody) staining (antigen: mouse lymph node stroma) (e) of the aortic necrotic core area in the NC, Ldlr−/−, flox and CKO groups (n = 6 per group). *P < 0.05. f,g, Flow cytometry analysis of immune cells isolated from atherosclerotic plaques of the flox and CKO groups (n = 6 per group), showing the CTL ratio (f) and the ratio of activated CTLs (defined as PD1+CD107a+) (g). *P < 0.05.
Discussion
m6A is recognized as the most prevalent posttranscriptional modification of mRNAs and is known to regulate various aspects of macrophage function, including development, activation, inflammation12,13, polarization29, aging30, pyroptosis, lipid uptake31 and cholesterol efflux32. Atherosclerosis is a chronic inflammatory disease driven by lipid accumulation and immune dysregulation. METTL3, a m6A writer, is upregulated in M1 macrophages within atherosclerotic plaques. It stabilizes hepatoma-derived growth factor mRNA via m6A modification, reprogramming energy metabolism to increase glycolysis and suppress mitochondrial respiration, which promotes plaque progression29. However, the increase in Mettl3-mediated AKT1S1 reduces foam cell formation by activating macrophage autophagy33. These studies revealed the bidirectional regulatory role of METTL3 following atherosclerosis progression. Moreover, knockout of Mettl14, another m6A writer, significantly reduces the inflammatory response of macrophages by decreasing Myd88 mRNA stability and the development of atherosclerotic plaques34. Genome-wide analyses revealed m6A peaks enriched in coding sequences (CDSs) and 3′ untranslated regions (UTRs) of macrophage polarization-related genes (for example, STAT6 and PI3K/AKT pathway components), with consensus DRACH motifs35. The above studies indicate that m6A modifications in macrophages are strongly associated with atherosclerosis. Despite these insights, systematic studies tracking the dynamic changes in m6A modifications throughout macrophage differentiation and activation are lacking. To fill this gap, we conducted meRIP-seq on monocyte-derived and IFN-γ-stimulated macrophages in this study. Integrated analysis with RNA-seq revealed that, during macrophage differentiation, pathways involved in inflammation, innate immunity, small molecule metabolism and protein processing are extensively regulated by m6A modification. After differentiation, an increase in the number of mRNAs containing m6A modifications highlights its pivotal role. Furthermore, a cluster of genes, including myeloid cell markers and immunological factors, continued to be modulated by m6A. However, during macrophage activation, cytokine- and inflammation-related genes are positively regulated by m6A. These findings highlight the precise and stage-specific regulation of m6A modifications on mRNAs during macrophage differentiation and activation. The molecular machinery governing this regulation remains elusive and merits further investigation.
The JAK–STAT pathway is a critical signaling cascade that orchestrates macrophage polarization and inflammatory responses. Upon the binding of cytokines (for example, IFN-γ and IL-6) to their receptors, JAK kinases (JAK1, JAK2 and TYK2) are activated and phosphorylate STAT1. Phosphorylated STAT1 (p-STAT1) dimerizes, translocates to the nucleus and drives the transcription of proinflammatory genes, including IRF1, STAT1 itself, CXCL9/10 and NOS2 (inducible nitric oxide synthase, iNOS), thereby promoting M1-like macrophage polarization36. Persistent STAT1 activation in M1 macrophages drives plaque instability in atherosclerosis37. Meanwhile, activation of cytokine signaling (for example, via the IL-6–gp130 complex, CSF2 (GM-CSF) or oncostatin M receptor) had been reported to phosphorylate STAT3, driving macrophages toward an M2-polarized phenotype in multiple cancers38,39,40. Conversely, application of a STAT3 inhibitor has been shown to alleviate atherosclerosis by suppressing endothelial dysfunction, macrophage differentiation and CD4+ T effector activation41. In this study, our group demonstrated that the JAK–STAT1 pathway is regulated by the histone demethylase KDM6B and connects this epigenetic enzyme with JAK–STAT1 signaling in atherosclerotic macrophages. Moreover, the unspecific role of KDM6B during JAK–STAT1 activation was interpreted, which contributes to the understanding of the nonhistone demethylation activities of KDM6B. However, the overall process by which KDM6B demethylates JAK1 needs to be further explored.
Furthermore, during atherosclerosis progression, environmental factors such as oxidized lipids, inflammatory cytokines and cholesterol crystals also induce epigenetic heterogeneity in macrophages42. The histone demethylase KDM6B has been implicated in NF-κB-mediated inflammatory activation of macrophages in abdominal aortic aneurysms7. Interestingly, myeloid cell-specific deficiency of Kdm6b has been associated with the promotion of advanced atherosclerotic plaques43, a finding that contrasts with our results. In our opinion, three factors may contribute to these discrepancies: (1) Kdm6b deletion changes monocyte-derived macrophage differentiation and subsequent myeloid cell subset infiltration; (2) the interaction protein spectrum of Kdm6b in monocytes and macrophages differs, resulting in differential interaction targets during IFN-γ stimulation; and (3) cell type-specific Kdm6b deletion changes the interaction relationship between myeloid cells and other immune cells or stromal cells. More experiments should be carried out on myeloid cell subset-specific Kdm6b deletion mice to elucidate the reasons. In addition, KDM6B mRNA regulation by m6A modification was observed during macrophage activation but not during differentiation. However, KDM6B is crucial for the differentiation of monocytes into macrophages, which suggests that myeloid cell-specific KDM6B deficiency might impair this process.
Our research presents a comprehensive mapping of the m6A modification landscape throughout macrophage differentiation and activation for the first time. We revealed that m6A modification plays a pivotal role in the upregulation of KDM6B in response to IFN-γ stimulation, which is essential for the phosphorylation of STAT1.
References
Libby, P. The changing landscape of atherosclerosis. Nature 592, 524–533 (2021).
Ruiz-León, A. M., Lapuente, M., Estruch, R. & Casas, R. Clinical advances in immunonutrition and atherosclerosis: a review. Front. Immunol. 10, 837 (2019).
Ye, L. et al. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 11, 50–61 (2012).
Tran, N., Broun, A. & Ge, K. Lysine demethylase KDM6A in differentiation, development, and cancer. Mol. Cell Biol. https://doi.org/10.1128/MCB.00341-20 (2020).
Das, A. et al. Proteomic changes induced by histone demethylase JMJD3 in TNF alpha-treated human monocytic (THP-1) cells. Mol. Immunol. 56, 113–122 (2013).
Das, N. D. et al. Gene networking and inflammatory pathway analysis in a JMJD3 knockdown human monocytic cell line. Cell Biochem. Funct. 30, 224–232 (2012).
Davis, F. M. et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J. Exp. Med. https://doi.org/10.1084/jem.20201839 (2021).
Neele, A. E. et al. Macrophage Kdm6b controls the pro-fibrotic transcriptome signature of foam cells. Epigenomics 9, 383–391 (2017).
Xun, J. et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B. Theranostics 11, 6847–6859 (2021).
Jiang, X. et al. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target Ther. 6, 74 (2021).
Meyer, K. D. & Jaffrey, S. R. Rethinking m6A readers, writers, and erasers. Annu Rev. Cell Dev. Biol. 33, 319–342 (2017).
Zheng, Y. et al. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell Mol. Life Sci. 79, 311 (2022).
Sun, Z. et al. Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin. Immunol. 245, 109176 (2022).
Fu, J. et al. The role of m6A ribonucleic acid modification in the occurrence of atherosclerosis. Front. Genet. 12, 733871 (2021).
Dong, G. et al. N6-methyladenosine methyltransferase METTL3 promotes angiogenesis and atherosclerosis by upregulating the JAK2/STAT3 pathway via m6A reader IGF2BP1. Front. Cell Dev. Biol. 9, 731810 (2021).
Xu, Z., Lv, B., Qin, Y. & Zhang, B. Emerging roles and mechanism of m6A methylation in cardiometabolic diseases. Cells https://doi.org/10.3390/cells11071101 (2022).
Yang, Q. et al. Exercise mitigates endothelial pyroptosis and atherosclerosis by downregulating NEAT1 through N6-methyladenosine modifications. Arterioscler. Thromb. Vasc. Biol. 43, 910–926 (2023).
Huangfu, N. et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int. Immunopharmacol. 83, 106432 (2020).
Mao, W. et al. TRIM21-mediated METTL3 degradation promotes PDAC ferroptosis and enhances the efficacy of anti-PD-1 immunotherapy. Cell Death Dis. 16, 240 (2025).
Huangfu, N. et al. DHX9 strengthens atherosclerosis progression by promoting inflammation in macrophages. Inflammation 46, 1725–1738 (2023).
Jian, D. et al. METTL3-mediated m6A modification of ISG15 mRNA regulates doxorubicin-induced endothelial cell apoptosis. J. Cell Mol. Med. 29, e70339 (2025).
Jian, D. et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 10, 8939–8956 (2020).
Jian, D. et al. Nsun2 controls cardiac homeostasis and hypertrophic response by regulating PRKACA expression. Theranostics 15, 2393–2412 (2025).
Pan, H. et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation 142, 2060–2075 (2020).
Huang, Y. et al. Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux. Nat. Commun. 14, 4883 (2023).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–25 (2022).
Tyrrell, D. J. et al. Clonally expanded memory CD8+ T cells accumulate in atherosclerotic plaques and are pro-atherogenic in aged mice. Nat. Aging 3, 1576–1590 (2023).
Raredon, M. S. B. et al. Computation and visualization of cell–cell signaling topologies in single-cell systems data using Connectome. Sci. Rep. 12, 4187 (2022).
Zheng, L. et al. RNA-m6A modification of HDGF mediated by Mettl3 aggravates the progression of atherosclerosis by regulating macrophages polarization via energy metabolism reprogramming. Biochem. Biophys. Res. Commun. 635, 120–127 (2022).
Zhao, W. et al. m6A plays a potential role in carotid atherosclerosis by modulating immune cell modification and regulating aging-related genes. Sci. Rep. 14, 60 (2024).
Li, Q. et al. METTL3 (methyltransferase like 3)-dependent N6-methyladenosine modification on Braf mRNA promotes macrophage inflammatory response and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 43, 755–773 (2023).
Zhu, X., Tang, H., Yang, M. & Yin, K. N6-methyladenosine in macrophage function: a novel target for metabolic diseases. Trends Endocrinol. Metab. 34, 66–84 (2023).
Yu, X. et al. Leonurine improves atherosclerosis by activating foam cell autophagy and metabolic remodeling via METTL3-mediated AKT1S1 mRNA stability modulation. Phytomedicine 134, 155939 (2024).
Zheng, Y. et al. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-kappaB/IL-6 signaling pathway. Cell Mol. Life Sci. 79, 311 (2022).
Pinello, N. et al. A multiomics dataset for the study of RNA modifications in human macrophage differentiation and polarisation. Sci. Data 11, 252 (2024).
Yang, X. et al. Cell volume regulation modulates macrophage-related inflammatory responses via JAK/STAT signaling pathways. Acta Biomater. 186, 286–299 (2024).
Ivashkiv, L. B. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Lee, Y. et al. Hyaluronic acid–bilirubin nanomedicine-based combination chemoimmunotherapy. Nat. Commun. 14, 4771 (2023).
Cai, H., Zhang, Y., Wang, J. & Gu, J. Defects in macrophage reprogramming in cancer therapy: the negative impact of PD-L1/PD-1. Front. Immunol. 12, 690869 (2021).
Xiao, C. et al. OSMR induces M2 polarization of glioblastoma associated macrophages through JAK/STAT3 signaling pathway. Front. Oncol. 15, 1538649 (2025).
Chen, Q. et al. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 9, 6424–6442 (2019).
Kuznetsova, T., Prange, K. H. M., Glass, C. K. & de Winther, M. P. J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17, 216–228 (2020).
Neele, A. E. et al. Myeloid Kdm6b deficiency results in advanced atherosclerosis. Atherosclerosis 275, 156–165 (2018).
Acknowledgements
We thank Z. Sun for kindly providing suggestions on the discussion of the JAK–STAT3 pathway in macrophages and atherosclerosis. This work was supported by Zhejiang Provincial Natural Science Foundation of China (MS25H020020), the National Natural Science Foundation of China (82200478, 82200489 and 82100437), Ningbo Young Scientific and Technological Innovation Leading Talent Program (2024QL027), Key Laboratory of Precision Medicine for Atherosclerotic Diseases of Zhejiang Province, China (2022E10026), Key Research and Development Project of Zhejiang Province, China (2021C03096), Key Laboratory of Precision Medicine for Atherosclerotic Diseases of Zhejiang Province, China (grant no. SZKF2023D003), Henan Province Medical Science and Technology Tackling Program Joint Co-Construction Project (project no. LHGJ20230154) and Natural Science Foundation of Henan Province (grant no. 24230421281).
Author information
Authors and Affiliations
Contributions
D.J., W.Z., D.Q. and L.J. conceptualized and supervised the study. N.H., F.L., C.W., S.J., X.Z. and Y.Y. performed the main experiments, acquired data and wrote the first draft of the manuscript. Y.W. and T.F. analyzed experimental data and bioinformatic data. J.S. conducted the animal experiments. All authors have read and approved the final version of this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experimental procedures were approved by the Animal Care Ethics Committee of Ningbo University (application number 13720) and conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huangfu, N., Li, F., Wang, C. et al. METTL3/RBM15 augments the stability of Kdm6b mRNA and promotes STAT1-mediated macrophage activation and atherosclerosis. Exp Mol Med 57, 2916–2929 (2025). https://doi.org/10.1038/s12276-025-01594-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01594-y
