
Abstract
Subthreshold depression (StD) confers a high risk for major depression and is characterized by substantial individual clinical heterogeneity. However, the neurobiological substrates underlying this heterogeneity remain largely unknown. Using a large multisite resting-state functional MRI dataset including 1203 healthy participants and 197 individuals with StD, we constructed connectome-based normative models to identify individual brain deviations and biotypes in StD. We highlighted remarkable individual variability in the connectome deviations in StD, leading to the identification of two distinct biotypes. Subtype 1 exhibits severe positive deviations primarily in the default mode regions and negative deviations in the sensorimotor and ventral attention areas, while subtype 2 shows a moderate but opposite deviation pattern. The two subtypes differ significantly in depressive symptoms, gene expression profiles, and treatment responses to bright light therapy. These findings highlight the neurobiological underpinnings of the clinical diversity in StD, emphasizing the necessity for developing personalized interventions for this condition.
Similar content being viewed by others

Introduction
Subthreshold depression (StD), also known as subclinical or subsyndromal depression, is considered the prodromal stage of major depressive disorder (MDD) [1, 2]. Individuals with StD are at greater risk of functional impairment, suicide, and developing MDD than those without StD [1, 3]. The most widely used definition of StD is clinically relevant depressive symptoms or episodes that do not fulfill the diagnostic criteria for MDD according to the DSM-5 or ICD-10 [4]. StD can be characterized dimensionally on the basis of a defined cutoff value on a validated self-rating depression instrument or categorically on the basis of the presence of fewer than five symptoms of DSM-MDD [4]. The diversity of characterizations based on severity or distinct combinations of symptoms results in substantial clinical heterogeneity in individuals with StD. However, the neural mechanisms responsible for the clinical heterogeneity of StD remain unknown, which hampers early detection and effective intervention for this condition.
Resting-state functional magnetic resonance imaging (rs-fMRI) is an important technique for mapping spontaneous brain activity in healthy individuals and those with disorders [5, 6]. Group-level alterations in multiple local activity and functional connectivity were reported in individuals with StD; however, the findings are largely inconsistent across studies. For example, different studies have reported both higher or lower functional connectivity in the default mode network (DMN) [7,8,9] and limbic system (LIM) [10,11,12] in individuals with StD. These inconsistencies are partly due to sample heterogeneity and small effect sizes [13]. Furthermore, most rs-fMRI studies on StD used a case-control design, focusing on group averages and ignoring individual variability. As noted above, StD individuals are defined primarily by self-reported symptoms and thus have heterogeneous symptom profiles and different etiological pathways [4]. This heterogeneity raises the possibility that StD has distinct biological subtypes with different functional brain signatures. While MDD biotypes based on functional connectivity patterns have been reported [14], the neurobiological subtypes of StD have not been investigated. Therefore, quantitative assessment of the individual functional connectome features of StD and the identification of neurobiologically homogeneous StD subtypes holds promise for elucidating the neural mechanisms of clinical heterogeneity and guiding treatment strategies.
Neuroimaging-based normative modeling enables the quantification of individual-level deviations in brain phenotypes relative to a normative reference population [15]. In contrast to conventional case‒control analyses, normative modeling quantifies individual differences at the subject level by examining an individual’s deviation from typical trajectories [16]. Recently, neuroimaging studies have involved the construction of normative models of brain phenotypes with the aim of identifying individual deviations in gray matter morphology and structural and functional connectomes in psychiatric disorders [17], including schizophrenia [16, 17], bipolar disorder [16], and depression [14]. However, no studies have employed normative modeling to quantify individual deviations in the brain phenotypes of StD and to further identify StD biotypes.
If StD can be categorized into different biotypes, two key questions need to be addressed: Do these biotypes have distinct biological mechanisms, and do their therapeutic responses differ? First, the utility of transcriptome‒connectome association analyses bridges the link between the microscale transcriptome profile and the macroscale brain network. Therefore, it may be promising to ascertain whether different StD subtypes identified on the basis of brain connectome deviations are associated with specific gene expression profiles. Second, light therapy is a cost-effective, safe, and widely accepted nonpharmacological intervention for both seasonal and nonseasonal depression [18], comparable to traditional antidepressant therapy for StD [19]. Our previous study confirmed the efficacy of bright light therapy (BLT) in alleviating depression symptoms in StD [20]. Variations in the response of specific brain regions to light therapy, influenced by StD biotypes, may contribute to predicting treatment efficacy and serve as important indicators for exploring the value of the treatment response across different neurobiological subtypes.
To address these knowledge gaps, we employed connectome-based normative modeling to quantify the heterogeneity of individual brain phenotypes in StD, identify StD biotypes, and investigate the associations of these biotypes with gene expression signatures and treatment outcomes. Specifically, we first established normative models of the functional connectome using large-sample, multisite rs-fMRI data from healthy individuals (n = 1203). We subsequently reported individual variations in the functional connectome among participants with StD (n = 197) and identified neurobiological subtypes along with their associations with clinical features and cognitive function. Using data from the Allen Human Brain Atlas (AHBA), we examined whether connectome deviations in specific StD subtypes are associated with distinct gene expression profiles. Finally, we investigated whether StD biotypes differ in treatment response and the ability to predict responses to light therapy.
Methods
Imaging dataset and preprocessing
In this study, we used two imaging datasets with strict quality control. Dataset 1 included 1079 healthy participants (ages 13–81, Table 1, Supplementary Fig. 1) recruited from 9 research centers affiliated with DIDA-MDD [14]. Dataset 2 included 197 participants with StD and 124 healthy controls (ages 18–35, Table 2, Supplementary Fig. 1) from Jinan University. Within the StD cohort, 47 individuals completed an 8-week BLT regimen (Table 2) and underwent mood scale assessments and MRI scans at baseline and post-treatment. All rs-fMRI data were collected using 3.0 T MRI scanners (Supplementary Table 1) and preprocessed through a standardized pipeline [14, 21]. A detailed description of the datasets and preprocessing procedure is provided in the Supplementary Methods.
Normative modeling of the functional connectivity strength
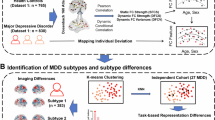
First, we calculated the whole-brain functional connectivity strength (FCS) map for each participant (Fig. 1a), based on a predefined functional parcellation [22], comprising 220 cerebral regions with qualified fMRI signals across all participants (Supplementary Methods). This metric has been demonstrated to exhibit high test-retest reliability and to have a strong association with regional cerebral blood flow and CMRGlucose [23]. Then, for each brain region, we established an age- and sex-based normative model of FCS using Gaussian process regression (GPR) [24] in 1203 healthy participants from both datasets (Fig. 1b, Supplementary Fig. 2). The inclusion of healthy participants from Jinan University enabled us to estimate and adjust for site effects in subsequent StD analyses. GPR, a Bayesian nonparametric interpolation method, not only provides point estimates, but also provides coherent measures of predictive confidence [25]. In addition to modeling potentially nonlinear predictions of brain features, it allows for regional estimates of the expected variation in the relationship between age and brain features (normative variance), along with uncertainty in this variance. To evaluate model generalizability, we initially estimated normative models in the healthy group using 10-fold cross-validation. Specifically, we randomly divided the data into 10 subsets, training the model on nine folds and testing its performance on the remaining fold. This procedure was repeated 10 times, ensuring each fold was used once as the test set. We subsequently applied the final normative models to the entire healthy dataset for subsequent StD deviation analyses.

a Calculation of the functional networks and FCS values for 1203 healthy participants and 197 participants with subthreshold depression. b Estimation of sex-specific normative models of FCS for each brain region via Gaussian process regression trained on the healthy group (females, red dots; males, blue dots). The solid line represents the predicted FCS values from the normative model. 10-fold cross-validation was performed to assess the generalizability of the models. c Characterization of FCS deviation for each brain region in participants with subthreshold depression (females, red triangles; males, blue triangles) on the basis of the normative models. d Identification of StD subtypes on the basis of individual FCS deviation patterns and characterization of their differences. FCS functional connectivity strength, F female, M male, StD subthreshold depression.
Estimating individual FCS deviations in normative models for StD
For each participant with StD, we mapped the FCS values of the brain regions onto the normative percentile chart derived from healthy participants to estimate individual deviations (Fig. 1c). By comparing observed FCS values against predicted FCS values from the normative model, we calculated a Z score to quantify deviation from the normative model for each brain region [24]. For a given StD participant (i), the deviation value (Z) of a brain region (j) was calculated as follows:
where \({y}_{{ij}}\) is the observed FCS value,\(\,{\hat{y}}_{{ij}}\) is the predicted FCS value, \({\sigma }_{{ij}}\) is the predictive uncertainty, and \({\sigma }_{{nj}}\) is the variance learned from the normative distribution (n). To detect extreme individual-level deviations in FCS, we applied a threshold to deviation maps using Z = ± 2.6 (corresponding to p < 0.005), as employed in prior studies [14, 16]. To quantify the overall extent of individual deviations, we calculated the number of brain regions exhibiting extreme deviations for each participant. To evaluate intersubject heterogeneity in deviations, we generated a spatial overlap map by determining the percentage of participants with extreme deviation (Z > 2.6 or Z < −2.6) in each brain region.
Identifying StD subtypes based on individual FCS deviations
We explored StD subtypes with distinct deviation patterns using a data-driven approach based on the k-means clustering algorithm (Fig. 1d). In this analysis, the deviation map of each participant with StD served as the clustering feature, where the distance between any two participants was defined as the Euclidean distance between their respective deviation maps. To minimize the influence of initial conditions, we executed the clustering algorithm 10 times with different random initial cluster centroids for each clustering number. The number of clusters varied from 2 to 10, and the optimal number was determined by a winner-take-all approach across 21 effective indices using the NbClust package [26].
Subtype differences in FCS deviations and clinical manifestations
To investigate the deviation pattern differences among StD subtypes, we generated mean deviation maps for each subtype and conducted network-level comparisons. Initially, we utilized a template consisting eight networks: visual, sensorimotor, dorsal attention, ventral attention, limbic, frontoparietal, and default mode networks, as defined by Yeo et al., [27] along with the subcortical system based on the Automated Anatomical Labeling (AAL) atlas [28]. The 220 brain regions were assigned to the network exhibiting the highest voxel attributions within each region. Next, we computed the mean deviation value for each network in participant with StD and compared these values between subtypes using two-sample t-tests. To assess subtype differences in the overall individual deviations, we calculated the number of extremely deviated regions, the sum of positive extreme deviations, and the sum of negative extreme deviations for each participant. These indices were then compared between subtypes via two-sample t-tests. Significance thresholds were adjusted for multiple comparisons using FDR correction with a q < 0.05. Furthermore, we conducted statistical comparisons on demographic and clinical disparities among subtypes, via two-sample t-tests or chi-square tests.
Gene expression profiles associated with FCS deviations of each subtype
To identify genes associated with functional deviations of each subtype, we analyzed microarray expression data from six postmortem human brains provided by the AHBA [29]. For this analysis, gene expression data from the left hemisphere were utilized due to limited availability of right hemisphere data from only two donors. To generate transcriptional profiles for each brain region, regional microarray expression data were preprocessed using the recommended pipeline (Supplementary Methods) via the abagen toolbox [30]. This preprocessing yielded a regional gene expression matrix (105 brain regions × 12,639 gene expression levels). Subsequently, partial least squares (PLS) regression analysis was employed to explore the associations between transcriptional profiles and FCS deviations of each subtype. Here, the gene expression data of brain nodes (105 brain regions × 12,639 genes) served as predictor variables, while the mean FCS deviation map of each subtype (105 brain regions × 1 deviation value) acted as response variables. Several components were defined, each representing a linear combination of predictor variables that maximally explained variance in the response variables. The statistical significance of the variance explained by the PLS components was tested through a permutation test (10,000 times) incorporating spatial-autocorrelation correction (Supplementary Methods) [31]. For each PLS component map, spatial similarity between weighted gene expression and the mean FCS deviation map was quantified using Pearson correlation. The significance of the correlation was evaluated via permutation analysis (10,000 times), correcting for spatial-autocorrelation. For each significant component, a bootstrapping method (10,000 times) was then applied to estimate the error of each gene’s weight, normalizing the weights by dividing them by their estimated error [32]. Using univariate one-sample Z tests, normalized weights were ranked, and genes significantly contributing to positive (PLS + ) or negative (PLS-) weighting were identified (FDR-corrected q < 0.05). The significant genes thus identified were defined as a gene set and further analyzed to test gene specificity with the Gene Annotation using Macroscale Brain-imaging Association (GAMBA) Toolbox [33]. Specifically, this gene set was tested against two types of null models. The null-coexpressed-gene model includes genes with a similar co-expression level to the gene set of interest, providing a null distribution that accounts for co-expression structure. The null-brain-gene model generates null distributions exclusively from genes overexpressed in brain tissue and is thus more conservative than classical random-gene models. If the gene set remained significantly associated with the mean deviation map after both spatial and gene-specificity tests, it was subjected to Gene Ontology enrichment analysis using Metascape [34] to elucidate the functional significance of the associated genes. Three ontology categories-biological process, molecular function, and cellular component-were considered, with enrichment pathways threshold for significance at 0.05 after FDR correction.
Subtype differences in treatment response to BLT
Among individuals with available follow-up BLT outcomes, we assessed the treatment response associated with each subtype. First, we examined subtype differences in changes in clinical symptom severity using a two-way repeated-measures ANOVA on HDRS-24 total scores. The dependent variable was the HDRS-24 total score, with factors being treatment (pre-treatment vs. post-treatment) and subtype (subtype 1 vs. subtype 2). To further examine whether specific symptom items differed between subtypes, we conducted additional two-way repeated-measures ANOVAs on HDRS-24 items that showed significant baseline differences, using treatment and subtype as factors. For symptoms showing significant subtype-by-treatment interaction effect, post hoc paired-samples t-tests were performed to investigate treatment-induced changes within each subtype. To assess subtype differences in changes in brain functional deviations, we then conducted a two-way repeated-measures ANOVA for each brain region, with the dependent variable being the brain deviation value and the factors being treatment and subtype. Multiple comparisons across all brain regions were corrected using an FDR-corrected threshold of q < 0.05. Regions showing significant subtype-by-treatment interactions were grouped into sequentially ordered edge-connected clusters. Post hoc analyses of these clusters, performed via paired samples t-tests, further explored treatment-induced changes in brain deviations within each subtype. Finally, support vector regression (SVR) with a linear kernel was applied to examine whether baseline individual deviations could predict symptom improvements after BLT antidepressant treatment in each subtype. A 5-fold cross-validation strategy was adopted to estimate prediction accuracy (Supplementary Methods). The statistical significance of the prediction accuracy was assessed using a nonparametric permutation test (1000 times).
Validation analysis
To evaluate the reliability of our findings, we implemented a series of validation steps (Supplementary Methods). First, we recalculated whole-brain FCS values using alternative correlation thresholds (r = 0.15 and 0.25) and re-evaluated our results. Second, as no gold standard exists for threshold selection in extreme deviations, we tested the robustness of our results via an alternative threshold of Z = ± 2. Third, to assess the influence of head motion, we regressed mean framewise displacement (FD) from the FCS values and reassessed the findings. Moreover, we applied stricter quality control by excluding 148 participants with a mean FD exceeding 0.2 mm, and replicated all analyses. Fourth, to account for potential site effects, we employed hierarchical Bayesian regression [17] to estimate a normative model that accommodates both signal and noise variance in multisite data by estimating connected but distinct mean and variance components through shared prior distributions across sites. Fifth, to ensure the stability of our findings, we split the StD cohort into two subsets: one without longitudinal BLT treatment data, which was used to identify subtypes and analyze subtype differences in brain deviation, clinical characteristics, and gene expression; and another with longitudinal treatment data, serving as an independent group to validate the subtype classifications. Finally, to minimize potential biases arising from uneven sample distribution in the normative model estimation, we employed a balanced sampling strategy to ensure equitable participant distribution across age groups.
Results
Highly heterogeneous individual deviations of functional connectome in StD
The normative models revealed regionally heterogeneous growth trajectories of FCS for both sexes (Supplementary Fig. 3), consistent with our previous study [14]. Based on these models, a total of 69.04% (n = 136) of individuals with StD exhibited extreme FCS deviations in at least one brain region (Fig. 2a). Of these, 28.93% (n = 57) exhibited extreme positive deviations, and 58.38% (n = 115) exhibited extreme negative deviations. Extreme positive deviations in individuals with StD were mostly located in the medial prefrontal cortex, posterior cingulate gyrus, precuneus, angular gyrus, and insula, whereas extreme negative deviations were mainly concentrated in the lateral temporal cortex, medial occipital cortex, and medial sensorimotor cortex (Fig. 2b). However, for any single brain region, the percentage of individuals with extreme deviations from the normal range was very low for both positive (<3.05%) and negative (<3.55%) deviations (Fig. 2b). These findings indicate substantial heterogeneity in functional connectome alterations among individuals with StD.

a Distribution of the number of brain regions per StD participant showing extreme positive (red) and negative (blue) deviations. b Spatial overlap maps showing the percentage of StD participants with extreme deviations from the normative range in each brain region (left, extreme positive deviations; right, extreme negative deviations). c Determination of the optimal number of StD subtypes via the NbClust package and the intersubject similarity in FCS deviation patterns among all participants with StD. d Mean deviation maps for each subtype and their system-level differences. e Subtype differences in overall deviation indices. f Subtype differences in clinical manifestations. StD subthreshold depression, VIS visual network, SMN sensorimotor network, DAN dorsal attention network, VAN ventral attention network, LIM limbic network, FPN frontoparietal network, DMN default mode network, SUB subcortical regions, HDRS Hamilton Depression Rating Scale. *p < 0.05; **FDR-corrected q < 0.05.
Two StD subtypes characterized by distinct FCS deviations and clinical manifestations
We identified two neurophysiological subtypes based on individual deviation patterns. The optimal number of clusters was determined by 11 of 21 effective quality indices (Fig. 2c). Subtype 1 (35%, n = 68) exhibited severe deviations, with positive deviations observed in the DMN, LIM, and subcortical (SUB) regions and negative deviations evident in the sensorimotor (SMN), dorsal attention (DAN), and ventral attention (VAN) regions (absolute t > 2.17, FDR-corrected q < 0.05) (Fig. 2d, Supplementary Table 2). In contrast, subtype 2 (65%, n = 129) showed moderate deviations with a reversed pattern (Fig. 2d, Supplementary Table 2). All indices reflecting the overall extent of deviation, including the number of regions with extreme deviations and the sum of positive and negative extreme deviations, were higher in subtype 1 compared to subtype 2 (absolute t > 2.73, FDR-corrected q < 0.05) (Fig. 2e). In terms of demographic variables, no significant differences were observed in age (t = −0.49, p = 0.628) or sex (χ2 = 1.70, p = 0.193) distributions (Supplementary Table 3). With respect to clinical variables, subtype 1 exhibited more severe symptoms in the suicide item (t = 2.58, p = 0.012) of the HDRS and poorer performance in the speed of processing item of the MCCB (t = −2.14, p = 0.034) (Fig. 2f, Supplementary Table 3–5).
Subtype differences in gene expression profiles of FCS deviations
The gene associations with FCS deviations were found only in subtype 1 (Supplementary Table 6). The weighted gene expression pattern of the first PLS component (PLS1) accounted for the greatest spatial variance (25%) in FCS deviations (p = 0.003, spatial-autocorrelation corrected) (Fig. 3a, Supplementary Table 6). The PLS1 score map was spatially correlated with the FCS deviation pattern of subtype 1 (r = 0.50, p < 0.0001, spatial-autocorrelation corrected) (Fig. 3b). We ranked the normalized weights of PLS1 and identified 1390 PLS+ (Z > 2.48) and 1958 PLS- (Z < −2.48) significant genes (FDR-corrected q < 0.05) (Supplementary Data 1). Importantly, this combined gene set remained significant under both null models: p < 0.001 for the null-coexpressed-gene model and p = 0.003 for the null-brain-gene model. Gene Ontology enrichment analysis revealed that PLS+ genes were enriched in biological processes related to brain development, regulation of trans-synaptic signaling, and synapse assembly; cellular components related to dendrite and Golgi membrane; and molecular functions related to kinase binding, phosphoric ester hydrolase activity, and DNA-binding transcription activator activity, RNA polymerase II-specific (FDR-corrected q < 0.05) (Fig. 3c, Supplementary Table 7); PLS- genes were enriched in biological processes related to neuron projection development, vesicle-mediated transport in synapse, and cell junction organization; cellular components related to dendrite, axon, and presynapse; and molecular functions related to calcium ion binding and cell adhesion molecule binding (FDR-corrected q < 0.05) (Fig. 3d, Supplementary Table 7).

a Explained ratios for the first 10 components obtained from the partial least squares (PLS) regression analysis, with the significant PLS component marked by an asterisk. b Spatial patterns illustrating the mean deviation map of subtype 1 and the first PLS component (PLS1) scores in the left hemisphere. The deviation values are positively correlated with the PLS scores (10,000 permutation tests with spatial autocorrelation correction). The shaded area represents the 95% confidence interval. Each dot corresponds to a region. (c,d) Functional enrichment of the top-ranked PLS+ (c) and PLS- genes (d). Left: top ten ontology terms for PLS+ and PLS- genes, the size of the circles represents the number of genes involved in a given term. Right: Metascape enrichment network visualization depicting intra-cluster and inter-cluster similarities among enriched terms. Each term is represented by a circular node, with size proportional to the number of input genes included, and nodes of the same color belong to the same cluster. BP, biological processes; CC, cellular components; MF, molecular functions.
Subtype differences in treatment response to BLT
Among the 47 StD individuals with available follow-up BLT treatment outcomes, 15 belonged to subtype 1, and 32 belonged to subtype 2. Regarding subtype differences in total clinical symptom severity changes, a significant main effect of treatment was observed (F = 134.42, p < 0.001), while neither the main effect of subtype (F = 0.09, p = 0.762) nor the subtype-by-treatment interaction effect (F = 0.14, p = 0.709) was significant. These results revealed that the overall clinical symptoms of both subtypes were alleviated after BLT treatment (Fig. 4a). For the HDRS-24 item that differed at baseline, i.e., suicide item, we observed a significant subtype-by-treatment interaction (F = 5.52, p = 0.023, Fig. 4a). Post hoc analysis revealed a significant reduction in suicide symptoms in subtype 1 (t = 2.30, p = 0.029), but not in subtype 2 (t = 0.85, p = 0.399). These results suggest that while both subtypes improved globally, the symptom domains driving improvement differed.

a Longitudinal changes in HDRS total and item scores between the two subtypes. b Subtype differences in changes of the brain functional deviations. Multiple comparisons were corrected across all brain regions using FDR-corrected q < 0.05. c Predictive ability of baseline individual deviations for treatment response in participants of subtype. The scatter plot presents the correlation between the observed HDRS score change rate after treatment and the predicted HDRS score change rate derived from the SVR. Each dot represents the data from one StD participant, and the dashes indicate the 95% prediction error bounds. The summed weights in 5-fold cross-validation were mapped onto the brain surface. The radar map represents the distribution of predictive power in different systems (red, positive; blue, negative). HDRS Hamilton Depression Rating Scale, pre pretreatment, post posttreatment, MPFC/DLPFC medial and dorsolateral prefrontal cortices, STG superior temporal gyrus, ROL Rolandic operculum, INS insula, SVR support vector regression, VIS visual network, SMN sensorimotor network, DAN dorsal attention network VAN ventral attention network, LIM limbic network, FPN frontoparietal network, DMN default mode network, SUB subcortical regions. *p < 0.05.
For the subtype differences in longitudinal changes in brain deviations, a significant subtype-by-treatment interaction effect (q < 0.05, FDR-corrected) was observed in the bilateral medial and dorsolateral prefrontal cortices (MPFC/DLPFC), the left superior temporal gyrus (STG)/Rolandic operculum (ROL)/insula (INS), and the right ROL/INS (Fig. 4b, Supplementary Table 8). Post hoc analyses showed that subtype 1 had reduced positive deviations in the MPFC/DLPFC and negative deviations in the left STG/ROL/INS and the right ROL/INS following treatment (|t | >3.35, p < 0.005) (Fig. 4b, Supplementary Table 9). Subtype 2 exhibited reduced negative deviations in the bilateral MPFC/DLPFC after treatment (t = 2.20, p = 0.036) (Fig. 4b, Supplementary Table 9). These results indicate that both StD subtypes exhibited normalization of brain deviations following BLT, regardless of positive or negative deviations.
SVR analysis revealed that the baseline deviation map significantly predicted the HDRS-24 change rate in subtype 1 (r = 0.41, p = 0.018; mean squared error = 0.06, p = 0.014; mean absolute error = 0.21, p = 0.007; R² = 0.17, p = 0.014; one-tailed permutation test) (Fig. 4c). The most positive contributions were from the visual network (VIS) (29.6%), DMN (21.0%), and LIM (20.1%), whereas the greatest negative contribution was from the VAN (18.6%) (Fig. 4c). In contrast, the baseline deviation map in subtype 2 was not predictive of HDRS-24 change rate (r = 0.23, p = 0.076; mean squared error = 0.08, p = 0.064; mean absolute error = 0.23, p = 0.068; R² = 0.05, p = 0.064; one-tailed permutation test).
Validation results
Overall, our main findings remained robust across various analytical choices (Supplementary Methods). Briefly, individual deviation patterns aligned with main results (r > 0.857), the two neurophysiological subtypes were consistently identified under different conditions with an overlap rate exceeding 96.45%, and the subtype differences in brain deviations, clinical manifestations, gene expression substrates, and treatment response to BLT largely persisted (Supplementary Fig. 4–24, Supplementary Table 10–24).
Discussion
In this study, we mapped the neurobiological heterogeneity of StD in reference to normative models of the functional connectome established from a large healthy cohort. We then identified two subtypes of StD that differ in functional connectome, clinical and cognitive characteristics, gene expression association, and treatment outcomes. Notably, the individuals with StD were treatment naïve, so treatment effects on FCS patterns and symptoms did not confound the data analysis. Our strategy offers an approach for identifying neurobiologically defined and data-driven subtypes of StD, providing valuable insights into the understanding of heterogeneous subclinical forms of depression and paving the way toward personalized diagnosis and treatment.
Using a normative model of the functional connectome from a large dataset of healthy participants, we found positive FCS deviations mainly in the DMN regions and negative FCS deviations in the sensorimotor, temporal and visual cortices in StD participants. The overlap rates among individuals with StD in these regions were minimal, suggesting significant inter-individual heterogeneity. These findings were comparable to our prior findings in MDD based on a large-sample dataset [14], but this work provides evidence for the prodromal stage of MDD. Several case-control studies in StD have documented increased functional connectivity within the DMN [7, 10], which is involved in social cognition, self-referential processes, and difficulties in disengaging from ruminative thought processes [35]. Meanwhile, dysfunctions in both the DMN and the sensorimotor network have also been observed in MDD [6, 36], implying that disrupted balance between advanced cognitive and primary sensorimotor functions may contribute to the typical symptoms of mood dysfunction and motor retardation in depression [37]. These findings suggest that a particular developmental pathophysiology involving regions of the DMN and primary sensorimotor networks may account for vulnerability to developing depression.
This is the first study to identify StD biotypes using functional connectome data. Subtype 1 showed positive FCS deviations in the DMN, LIM, and SUB regions and negative deviations in the SMN and attention regions; subtype 2 displayed a moderate but reversed deviation pattern. These patterns are reminiscent of those observed in MDD, as described in our previous study [14], which may assist in identifying individuals with StD who are at risk of conversion to or experiencing more severe clinical presentations. Although methodological approaches vary, a growing consensus suggests that at least two to four distinct biological subtypes may exist in depression [38]. For instance, an rs-fMRI study identified two MDD subtypes with distinct connectivity patterns in the DMN using large multisite datasets [39]. Drysdale et al. (2017) defined four MDD subtypes by distinct patterns of dysfunctional connectivity in the limbic and frontostriatal networks [40]. Collectively, these findings indicate that the functional connectivity of the DMN and LIM areas, may serve as critical biomarkers for the neurophysiological subtyping of StD. Notably, subtype 1 had higher suicide, depersonalization and derealization item scores on the HDRS, as well as poorer processing speed compared to subtype 2. These clinical differences aligned with distinct patterns of brain abnormalities. For example, previous studies have associated increased functional connectivity and activity in the DMN, LIM, and SUB regions (including the orbitofrontal cortex, medial prefrontal cortex, cingulate cortex, and striatum) with suicidality [41,42,43,44]. Future studies should aim to subdivide individuals with StD into homogeneous groups to better understand how distinct brain functional patterns contribute to diverse clinical phenotypes and cognitive dysfunction.
To date, no genetic- or molecular-level studies have examined subtypes or biologically driven constructs of StD. Our results revealed associations between the spatial distribution of FCS deviations in subtype 1 and the expression of genes enriched in brain development, regulation of trans-synaptic signaling, neuron projection development, dendrites, calcium ion binding, and other related processes. However, no significant enrichment of genes was observed in subtype 2. These findings suggest a greater genetic vulnerability in subtype 1. A recent transdiagnostic study identified an enhanced expression of risk genes in the brain within the archetypal subtype, as opposed to the atypical subtype, based on the ALFF associated with major psychiatric disorders [6]. Our findings in subtype 1 are consistent with those of a previous study on MDD [21, 45]. Xia et al. (2022) reported that dysfunction in the connectome gradient of MDD patients was correlated with genes enriched in transsynaptic signaling and calcium ion binding [21]. Talishinsky et al. (2022) reported that alterations in functional connectivity within the DMN in MDD patients were correlated with regional gene sets which varied by sex and were enriched in depression, synapse function, immune signaling, and neurodevelopment [45]. Together, these findings suggest that subtype 1 might be a genetic and neurodevelopmental subtype of StD, indicating a high risk for transition to MDD.
BLT has been shown to alleviate depressive symptoms and prevent progression to MDD in StD adults [20]. Notably, our findings reveal that although both subtypes exhibited overall clinical and FCS improvement after 8 weeks of BLT, they differed in the specific symptom improvements, as well as in the brain regions involved and the directions of normalization. Furthermore, baseline FCS deviation patterns predicted treatment response only in subtype 1, with the most contributive features located in the VIS, DMN, and LIM regions. Previous animal studies have shown that visual circuits mediate the antidepressant effects of light exposure [46, 47], and a meta-analysis reported that treatment response in depression can be predicted by baseline DMN and LIM connectivity [48]. These networks may thus serve as potential biomarkers of BLT efficacy in subtype 1. In summary, while BLT is generally effective in alleviating depressive symptoms in StD, the underlying neurobiological mechanisms, symptom-specific improvements, and predictive biomarkers differ between subtypes, underscoring the importance of individualized, subtype-specific treatment strategies.
This study has several limitations. First, our study provides preliminary evidence of gene associations with FC subtypes. Future studies incorporating genetic data from individuals with StD are needed to more directly elucidate the factors underlying the different neurophysiological subtypes. Second, other modalities, including structural MRI, diffusion tensor imaging, and genetic and environmental data, need to be incorporated to parse the heterogeneity of StD. Third, we validated the main results by splitting the participants with StD into two subsets. To maximize generalizability and replicability and reduce bias in the model evaluation of our study, external validation in independent datasets from other populations and demographic groups is needed in future research.
Data availability
All data necessary to reproduce our findings are publicly available at https://github.com/Sun-Xiaoyi/subthreshold-depression-subtypes. The raw neuroimaging data from DIDA-MDD group and Jinan university are not available for public download, but access can be requested through the corresponding authors. The AHBA dataset is publicly available at https://human.brain-map.org.
Code availability
Software packages used in this manuscript include. SPM 12 (www.fil.ion.ucl.ac.uk/spm/), SeeCAT (www.nitrc.org/projects/seecat), PCNtoolkit 0.28 (https://github.com/amarquand/PCNtoolkit), NbClust 3.0 (https://www.rdocumentation.org/packages/NbClust), abagen 0.1.3 (https://github.com/rmarkello/abagen), BrainSMASH (https://brainsmash.readthedocs.io/en/latest/index.html), GAMBA (https://github.com/dutchconnectomelab/GAMBA-MATLAB), Metascape (https://metascape.org/gp/index.html#/main/step1), LIBSVM 3.3 (https://www.csie.ntu.edu.tw/~cjlin/libsvm/), Pattern_Regression (https://github.com/ZaixuCui/Pattern_Regression_Clean), BrainNet Viewer 1.62 (https://www.nitrc.org/projects/bnv), Matlab 2020a (https://www.mathworks.com/products/matlab.html), Python 3.8.18 (https://www.python.org), R 3.6.1 (https://www.r-project.org), and IBM SPSS Statistics 20 (https://www.ibm.com/spss). The codes used in this study are available at https://github.com/Sun-Xiaoyi/subthreshold-depression-subtypes.
References
Cuijpers P, Smit F. Subthreshold depression as a risk indicator for major depressive disorder: a systematic review of prospective studies. Acta Psychiatr. Scand. 2004;109:325–31.
Eaton WW, Badawi M, Melton B. Prodromes and precursors: epidemiologic data for primary prevention of disorders with slow onset. Am. J. Psychiatry. 1995;152:967–72.
Rodríguez MR, Nuevo R, Chatterji S, Ayuso-Mateos JL. Definitions and factors associated with subthreshold depressive conditions: a systematic review. BMC psychiatry. 2012;12:181.
Volz HP, Stirnweiß J, Kasper S, Möller HJ, Seifritz E. Subthreshold depression - concept, operationalisation and epidemiological data. a scoping review. Int. J. Psychiatry Clin. Pract. 2023;27:92–106.
Biswal B, Yetkin FZ, Haughton VM, Hyde JS. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med. 1995;34:537–41.
Chang M, Womer FY, Gong X, Chen X, Tang L, Feng R, et al. Identifying and validating subtypes within major psychiatric disorders based on frontal-posterior functional imbalance via deep learning. Mol. Psychiatry. 2021;26:2991–3002.
Hwang JW, Xin SC, Ou YM, Zhang WY, Liang YL, Chen J, et al. Enhanced default mode network connectivity with ventral striatum in subthreshold depression individuals. J. Psychiatr. Res. 2016;76:111–20.
Yin X, Yang J, Xiang Q, Peng L, Song J, Liang S, et al. Brain network hierarchy reorganization in subthreshold depression. Neuroimage Clin. 2024;42:103594.
Yokoyama S, Okamoto Y, Takagaki K, Okada G, Takamura M, Mori A, et al. Effects of behavioral activation on default mode network connectivity in subthreshold depression: a preliminary resting-state fMRI study. J. Affect. Disord. 2018;227:156–63.
Li R, Ma Z, Yu J, He Y, Li J. Altered local activity and functional connectivity of the anterior cingulate cortex in elderly individuals with subthreshold depression. Psychiatry Res. 2014;222:29–36.
Zhang B, Qi S, Liu S, Liu X, Wei X, Ming D. Altered spontaneous neural activity in the precuneus, middle and superior frontal gyri, and hippocampus in college students with subclinical depression. BMC Psychiatry. 2021;21:280.
Gao C, Wenhua L, Liu Y, Ruan X, Chen X, Liu L, et al. Decreased subcortical and increased cortical degree centrality in a nonclinical college student sample with subclinical depressive symptoms: a resting-state fMRI study. Front. Hum. Neurosci. 2016;10:617.
Button KS, Ioannidis JP, Mokrysz C, Nosek BA, Flint J, Robinson ES, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013;14:365–76.
Sun X, Sun J, Lu X, Dong Q, Zhang L, Wang W, et al. Mapping neurophysiological subtypes of major depressive disorder using normative models of the functional connectome. Biol. Psychiatry. 2023;94:936–47.
Rutherford S, Kia SM, Wolfers T, Fraza C, Zabihi M, Dinga R, et al. The normative modeling framework for computational psychiatry. Nat. Protoc. 2022;17:1711–34.
Wolfers T, Doan NT, Kaufmann T, Alnæs D, Moberget T, Agartz I, et al. Mapping the heterogeneous phenotype of schizophrenia and bipolar disorder using normative models. JAMA psychiatry. 2018;75:1146–55.
Segal A, Parkes L, Aquino K, Kia SM, Wolfers T, Franke B, et al. Regional, circuit and network heterogeneity of brain abnormalities in psychiatric disorders. Nat. Neurosci. 2023;26:1613–29.
Terman M, Terman JS. Light therapy for seasonal and nonseasonal depression: efficacy, protocol, safety, and side effects. CNS Spectr. 2005;10:647–63.
Geoffroy PA, Schroder CM, Reynaud E, Bourgin P. Efficacy of light therapy versus antidepressant drugs, and of the combination versus monotherapy, in major depressive episodes: a systematic review and meta-analysis. Sleep. Med. Rev. 2019;48:101213.
Jiang L, Zhang S, Wang Y, So KF, Ren C, Tao Q. Efficacy of light therapy for a college student sample with non-seasonal subthreshold depression: An RCT study. J. Affect. Disord. 2020;277:443–9.
Xia M, Liu J, Mechelli A, Sun X, Ma Q, Wang X, et al. Connectome gradient dysfunction in major depression and its association with gene expression profiles and treatment outcomes. Mol. Psychiatry. 2022;27:1384–93.
Shen X, Tokoglu F, Papademetris X, Constable RT. Groupwise whole-brain parcellation from resting-state fMRI data for network node identification. Neuroimage. 2013;82:403–15.
Liang X, Zou Q, He Y, Yang Y. Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proc. Natl Acad. Sci. USA. 2013;110:1929–34.
Marquand AF, Rezek I, Buitelaar J, Beckmann CF. Understanding Heterogeneity in Clinical Cohorts Using Normative Models: Beyond Case-Control Studies. Biol. Psychiatry. 2016;80:552–61.
Rasmussen CE, Williams CKI Gaussian Processes for Machine Learning. Gaussian Processes for Machine Learning2005.
Charrad M, Ghazzali N, Boiteau V, Niknafs A. NbClust: AnRPackage for determining the relevant number of clusters in a data set. J. Stat. Softw. 2014;61:1–36.
Yeo BT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 2011;106:1125–65.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–89.
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–9.
Markello RD, Arnatkeviciute A, Poline J-B, Fulcher BD, Fornito A, Misic B. Standardizing workflows in imaging transcriptomics with the abagen toolbox. elife. 2021;10:e72129.
Burt JB, Helmer M, Shinn M, Anticevic A, Murray JD. Generative modeling of brain maps with spatial autocorrelation. Neuroimage. 2020;220:117038.
Whitaker KJ, Vértes PE, Romero-Garcia R, Váša F, Moutoussis M, Prabhu G, et al. Adolescence is associated with genomically patterned consolidation of the hubs of the human brain connectome. Proc. Natl Acad. Sci. USA. 2016;113:9105–10.
Wei Y, de Lange SC, Pijnenburg R, Scholtens LH, Ardesch DJ, Watanabe K, et al. Statistical testing in transcriptomic‐neuroimaging studies: a how‐to and evaluation of methods assessing spatial and gene specificity. Hum. Brain Mapp. 2021;43:885–901.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523.
Wang X, Öngür D, Auerbach RP, Yao S. Cognitive vulnerability to major depression: view from the intrinsic network and cross-network interactions. Harv. Rev. Psychiatry. 2016;24:188–201.
Gong J, Wang J, Qiu S, Chen P, Luo Z, Wang J, et al. Common and distinct patterns of intrinsic brain activity alterations in major depression and bipolar disorder: voxel-based meta-analysis. Transl. Psychiatry. 2020;10:353.
Qiu S, Chen F, Chen G, Jia Y, Gong J, Luo X, et al. Abnormal resting-state regional homogeneity in unmedicated bipolar II disorder. J. Affect. Disord. 2019;256:604–10.
Beijers L, Wardenaar KJ, van Loo HM, Schoevers RA. Data-driven biological subtypes of depression: systematic review of biological approaches to depression subtyping. Mol. Psychiatry. 2019;24:888–900.
Liang S, Deng W, Li X, Greenshaw AJ, Wang Q, Li M, et al. Biotypes of major depressive disorder: Neuroimaging evidence from resting-state default mode network patterns. Neuroimage Clin. 2020;28:102514.
Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med. 2017;23:28–38.
Auerbach RP, Pagliaccio D, Allison GO, Alqueza KL, Alonso MF. Neural correlates associated with suicide and nonsuicidal self-injury in youth. Biol. Psychiatry. 2021;89:119–33.
Jollant F, Lawrence NL, Olie E, Guillaume S, Courtet P. The suicidal mind and brain: a review of neuropsychological and neuroimaging studies. World J. Biol. Psychiatry. 2011;12:319–39.
Zhang H, Chen Z, Jia Z, Gong Q. Dysfunction of neural circuitry in depressive patients with suicidal behaviors: a review of structural and functional neuroimaging studies. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;53:61–6.
Chen Z, Xia M, Zhao Y, Kuang W, Jia Z, Gong Q. Characteristics of intrinsic brain functional connectivity alterations in major depressive disorder patients with suicide behavior. J. Magn. Reson. Imaging. 2021;54:1867–75.
Talishinsky A, Downar J, Vertes PE, Seidlitz J, Dunlop K, Lynch CJ, et al. Regional gene expression signatures are associated with sex-specific functional connectivity changes in depression. Nat. Commun. 2022;13:5692.
Huang L, Xi Y, Peng Y, Yang Y, Huang X, Fu Y, et al. A visual circuit related to habenula underlies the antidepressive effects of light therapy. Neuron. 2019;102:128–142.e128.
Li X, Li X. The antidepressant effect of light therapy from retinal projections. Neurosci. Bull. 2018;34:359–68.
Long Z, Du L, Zhao J, Wu S, Zheng Q, Lei X. Prediction on treatment improvement in depression with resting state connectivity: a coordinate-based meta-analysis. J. Affect. Disord. 2020;276:62–8.
Acknowledgements
This work was supported by the STI 2030-Major Projects (2022ZD0211500), National Natural Science Foundation of China (82472057, 82172530, 82071998, 82021004, and 82402368), Science and Technology Projects in Guangzhou (2024B03J1299), and Beijing Natural Science Foundation (JQ23033).
Author information
Authors and Affiliations
Consortia
Contributions
Guanmao Chen, Xiaoyi Sun, Mingrui Xia, Yong He, Qian Tao, and Ying Wang designed research; Guanmao Chen, Pan Chen, Zhangzhang Qi, Shu Zhang, Chao Chen, Zixuan Guo, Xinyue Tang, Ruoyi Chen, Qian Tao, and Ying Wang collected the clinical scales and imaging dataset from Jinan university; Xiaoyi Sun, Xiaoqin Wang, Dongtao Wei, Yuan Chen, Bangshan Liu, Chu-Chung Huang, Yanting Zheng, Yankun Wu, Taolin Chen, Yuqi Cheng, Xiufeng Xu, Qiyong Gong, Tianmei Si, Shijun Qiu, Ching-Po Lin, Jingliang Cheng, Yanqing Tang, Fei Wang, Jiang Qiu, Peng Xie, Lingjiang Li, Yong He, and Mingrui Xia collected the imaging dataset from DIDA-MDD group; Xuan Bu and Mingrui Xia provided the methodological instruction; Xiaoyi Sun performed the data analysis; Guanmao Chen and Xiaoyi Sun wrote the paper; Guanmao Chen, Xiaoyi Sun, Mingrui Xia, Yong He, and Ying Wang revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods in this study were performed in accordance with the relevant guidelines and regulations, including the Declaration of Helsinki. This study was approved by the ethics committee of the First Affiliated Hospital of Jinan University (reference 2019/028). Informed consent was obtained from all the participants. For the publication of MR images, consent was expressly obtained from all participants, who also signed informed consent forms for the scan itself.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, G., Sun, X., Chen, P. et al. Connectome-based growth models reveal individual heterogeneity and neurophysiological subtypes of subthreshold depression. Mol Psychiatry (2026). https://doi.org/10.1038/s41380-026-03457-y
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41380-026-03457-y
